Embed Size (px)
Citation preview

Universidade Federal do Rio Grande do Norte
Departamento de Química
Programa de Pós-Graduação em Química
PREPARO E CARACTERIZAÇÃO DE MEMBRANAS DE
QUITOSANA MODIFICADAS COM POLI(ÁCIDO ACRÍLICO)
Maria do Socorro Pereira de Lima
Dissertação realizada sob a orientação da Profa.
Dra. Márcia Rodrigues Pereira, apresentada ao Pro-
grama de Pós-Graduação em Química do Depar-
tamento de Química da UFRN em preenchimento
parcial dos requisitos para a obtenção do grau de
Mestre em Química.
Natal, novembro de 2006.

Dissertação de mestrado
PREPARO E CARACTERIZAÇÃO DE MEMBRANAS DE
QUITOSANA MODIFICADAS COM POLI(ÁCIDO ACRÍLICO)
Maria do Socorro Pereira de Lima
Profa. Dra. Márcia Rodrigues Pereira

Divisão de Serviços Técnicos
Catalogação da Publicação na Fonte. UFRN / Biblioteca Central Zila Mamede
Lima, Maria do Socorro Pereira de.
Preparo e caracterização de membranas de quitosana modificadas
com poli (ácido acrílico) / Maria do Socorro Pereira de Lima. – Natal
[RN], 2007.
89 f.
Orientador: Márcia Rodrigues Pereira.
Dissertação (Mestrado) – Universidade Federal do Rio Grande do
Norte. Programa de Pós-Graduação em Química.
1. Quitosona - Dissertação. 2. Poli(ácido acrílico) - Dissertação. 3.
Permeabilidade da membrana – Dissertação. 4. Complexos
polieletrolíticos - Dissertação. I. Pereira, Márcia Rodrigues. II.
Universidade Federal do Rio Grande do Norte. III. Título.
RN/UF/BCZM CDU 547.995(043.3)


"A ciência não é uma ilusão, ilusão seria buscar em outro lugar o que ela
nos proporciona"
S. Freud

Agradecimentos
A Deus.
À Profa. Dra. Márcia Rodrigues Pereira e ao Prof. Dr. José Luís Cardozo Fonseca pela
orientação, dedicação, interesse e por compartilharem seus conhecimentos profissionais.
Ao Prof. Dr. Francisco Gurgel de Azevedo, in memorian.
Aos amigos, cuja colaboração e convívio possibilitaram a realização deste trabalho
A todos que fazem parte do Laboratório de Membranas e Colóides.
E, em especial, à minha amada família, João e Mateus, que representa a minha força, dedico
este trabalho.

Resumo
O propósito deste estudo foi produzir uma membrana assimétrica biocompatível e biode-
gradável de quitosana modificada pelo contato com uma solução de poli(ácido acrílico) em
uma de suas superfícies à temperatura ambiente e a 60◦C. As membranas de quitosana pura,
quitosana com poli(ácido acrílico) a 25◦C e quitosana com poli(ácido acrílico) a 60◦C foram
caracterizadas por: espectroscopia no infravermelho (FTIR-ATR) em ângulos de incidência
de 39◦, 45◦ e 60◦, ganho de massa em água, análise térmica (TG/ DTG), microscopia ele-
trônica de varredura (MEV) e, ainda, através dos ensaios de permeação in vitro utilizando
como fármaco modelo o metronidazol em solução aquosa nas concentrações de 0,1 e 0,2%.
Os resultados obtidos comprovaram a existência de interações iônicas entre os dois políme-
ros, através da formação dos chamados complexos polieletrolíticos. Também mostraram que
a reticulação foi mais efetiva a 60◦C , uma vez que essa temperatura está acima da tempera-
tura de transição vítrea da quitosana, o que facilita a difusão do poli(ácido acrílico) e que as
membranas resultantes são assimétricas, mais estáveis termicamente e menos permeáveis ao
metronidazol do que a membrana de quitosana pura.
Palavras-chave: membranas, quitosana, poli(ácido acrílico), complexos polieletrolíticos e
permeabilidade.

Abstract
The aim of this study was to generate an asymmetric biocompactible and biodegradable
chitosan membrane modified by the contact with a poly(acrylic acid) solution at one of its
sides at room temperature and 60◦C. The pure chitosan membrane, as well as the ones treated
with poly(acrylic acid) were characterized by infrared spectroscopy (FTIR-ATR) at angles of
39◦, 45◦ and 60◦ , swelling capacity in water, thermal analysis (TG/DTG), scanning electronic
microscopy (SEM) and permeation experiments using metronidazole at 0,1% and 0,2% as a
model drug. The results confirmed the presence of ionic interaction between chitosan and
poly(acrylic acid) by means of a polyelectrolyte complex (PEC) formation. They also showed
that such interactions were more effective at 60◦C since this temperature is above the chitosan
glass transition temperature wich makes the diffusion of poly(acrylic acid) easier, and that
the two treated membranes were asymmetrics, more thermically stable and less permeable in
relation to metronidazole than the pure chitosan membrane.
Key words: membrane, chitosan, poly(acrylic acid), polyelectrolyte complex, permeability.
7

Símbolos e abreviaturas
DL50 dose letal capaz de causar o óbito de 50 % das cobaias;
mL mililitro;
g grama;
◦C graus Celsius;
D coeficiente de difusão do soluto em água (cm2s-1);
t tempo;
V volume;
α coeficiente angular;
ε absortividade;
mu massa da membrana úmida;
ms massa da membrana seca;
λ comprimento de onda;
A absorbância;
c concentração;
b caminho óptico;
PAA Poli(ácido acrílico);
pH Potencial hidrogeniônico;
S área da superfície da membrana;
l espessura da membrana;
8

K coeficiente de partição;
P constante de permeabilidade;
Q quantidade de substância difundida;
F fluxo de substância através da membrana;
c1 e c2 concentrações nas duas superfícies da membrana;
C1 e C2 concentrações das soluções nos dois lados da membrana;
QUI membrana de quitosana pura;
QUIPAA25 membrana de quitosana tratada com poli(ácido acrílico) a 25 ◦C;
QUIPAA60 membrana de quitosana tratada com poli(ácido acrílico) a 60 ◦C.
9

Lista de Figuras
1.1 Estrutura química da quitina. . . . . . . . . . . . . . . . . . . . . . . . . . . 16
1.2 Estrutura química da quitosana, com x representando o grau de acetilação. . 16
1.3 Estrutura química do poli(ácido acrílico). . . . . . . . . . . . . . . . . . . . 21
1.4 Processo de policomplexação entre a quitosana e o poli(ácido acrílico). . . . 24
1.5 Estrutura química do metronidazol. . . . . . . . . . . . . . . . . . . . . . . 31
1.6 Célula de reflectância total atenuada76. . . . . . . . . . . . . . . . . . . . . . 33
2.1 Sistema para tratamento superficial das membranas de quitosana. . . . . . . 42
2.2 Sistema de permeação in vitro. . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.1 Espectro no UV para uma solução aquosa de metronidazol. . . . . . . . . . 49
3.2 Curva de calibração do metronidazol. . . . . . . . . . . . . . . . . . . . . . 50
3.3 Espectros de FTIR-ATR para ambas as faces da membrana de quitosana pura
obtido a um ângulo de incidência de 45◦. . . . . . . . . . . . . . . . . . . . 52
3.4 Espectros de FTIR-ATR para ambas as faces da membrana tratada com poli(ácido
acrílico) à temperatura ambiente, obtidos a ângulo de incidência de 39◦, 45◦ e
60◦, respectivamente a, b e c. . . . . . . . . . . . . . . . . . . . . . . . . . 55
3.5 Espectros FTIR-ATR para ambas as faces da membrana tratada com poli(ácido
acrílico) a 60◦C, obtidos a ângulo de incidência de 39◦, 45◦ e 60◦ , respectiva-
mente a,b e c. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
10

Lista de Figuras
3.6 Espectros de FTIR-ATR para a membrana de quitosana pura, tratada com
poli(ácido acrílico) à temperatura ambiente e a 60◦C, obtidas a um ângulo
de incidência de 60◦. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
3.7 Porcentagem de ganho de massa para membranas QUI, QUIPAA25 e QUI-
PAA60 em função do tempo. . . . . . . . . . . . . . . . . . . . . . . . . . . 59
3.8 Curvas termogravimétricas (TG) das membranas QUI, QUIPAA25 e QUI-
PAA60 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
3.9 Curvas termogravimétricas derivadas (DTG) das membranas QUI, QUIPAA25
e QUIPAA60 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
3.10 Microscopia eletrônica da superfície da membrana QUI em contato com o ar
e em contato com a placa de Petri . . . . . . . . . . . . . . . . . . . . . . . 66
3.11 Microscopia eletrônica da superfície não tratada e tratada da membrana QUI-
PAA25 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
3.12 Microscopia eletrônica da superfície não tratada e tratada da membrana QUI-
PAA60 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
3.13 Microscopia eletrônica da superfície tratada das membranas QUIPAA25 e
QUIPAA60 na magnitude de 5000x . . . . . . . . . . . . . . . . . . . . . . 69
3.14 Curvas de absorbância em função de tempo para a permeação de metronidazol
em membranas QUI. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
3.15 Curvas de absorbância em função do tempo para a permeação do metronidazol
em membranas QUIPAA25. . . . . . . . . . . . . . . . . . . . . . . . . . . 73
3.16 Curvas de absorbância em função do tempo para a permeação de metronidazol
em membranas QUIPAA60. . . . . . . . . . . . . . . . . . . . . . . . . . . 74
11

Sumário
1 Introdução 141.1 Quitosana . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
1.2 Membranas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
1.2.1 Membranas de quitosana . . . . . . . . . . . . . . . . . . . . . . . . 19
1.3 Poli(ácido acrílico) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
1.4 Formação de complexos polieletrolíticos de quitosana e poli(ácido acrílico) . 21
1.5 Difusão . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
1.5.1 As leis de Fick . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
1.5.1.1 Primeira lei de Fick . . . . . . . . . . . . . . . . . . . . . 26
1.5.1.2 Segunda lei de Fick . . . . . . . . . . . . . . . . . . . . . 27
1.5.2 Difusão através de membranas . . . . . . . . . . . . . . . . . . . . . 27
1.5.2.1 Constante de permeabilidade . . . . . . . . . . . . . . . . 28
1.5.3 A permeabilidade da membrana . . . . . . . . . . . . . . . . . . . . 29
1.6 Metronidazol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
1.7 Espectroscopia na região do infravermelho . . . . . . . . . . . . . . . . . . 32
1.8 Análise térmica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
1.8.1 Análise termogravimétrica (TGA) . . . . . . . . . . . . . . . . . . . 37
1.9 Microscopia eletrônica de varredura (MEV) . . . . . . . . . . . . . . . . . . 38
2 Metodologia experimental 402.1 Materiais . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
2.2 Preparação das membranas . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
2.2.1 Membranas de quitosana . . . . . . . . . . . . . . . . . . . . . . . . 40
2.2.2 Membranas de quitosana/poli(ácido acrílico) . . . . . . . . . . . . . 41
2.3 Caracterização das membranas . . . . . . . . . . . . . . . . . . . . . . . . . 42
2.3.1 Espectroscopia de absorção na região do ultravioleta . . . . . . . . . 42
2.3.2 Espectroscopia de absorção na região do infravermelho . . . . . . . . 43
2.3.3 Ensaio de ganho de massa . . . . . . . . . . . . . . . . . . . . . . . 43
12

Sumário
2.3.4 Análise térmica . . . . . . . . . . . . . . . . . . . . . . . . . . . . 432.3.4.1 Análise termogravimétrica . . . . . . . . . . . . . . . . . 43
2.3.5 Microscopia eletrônica de varredura (MEV) . . . . . . . . . . . . . . 442.3.6 Ensaio de permeação . . . . . . . . . . . . . . . . . . . . . . . . . . 442.3.7 Cálculo da permeabilidade das membranas . . . . . . . . . . . . . . 45
3 Resultados e discussão 483.1 Espectroscopia na região do ultravioleta . . . . . . . . . . . . . . . . . . . . 48
3.1.1 Curva de calibração do metronidazol . . . . . . . . . . . . . . . . . . 483.2 Espectroscopia na região do infravermelho . . . . . . . . . . . . . . . . . . 513.3 Ensaio de ganho de massa . . . . . . . . . . . . . . . . . . . . . . . . . . . 573.4 Análise térmica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
3.4.1 Termogravimetria . . . . . . . . . . . . . . . . . . . . . . . . . . . . 603.5 Microscopia eletrônica de varredura (MEV) . . . . . . . . . . . . . . . . . . 653.6 Ensaio de permeação . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
4 Conclusões 77
5 Sugestões para futuras pesquisas 79
Referências Bibliográficas 80
13

1 Introdução
A tecnologia associada à modificação de preparações farmacêuticas para a liberação de
fármacos, tais como antibióticos, contraceptivos, enzimas e drogas contra o câncer, bem como
de moléculas de alta massa molar, como peptídeos e proteínas, sofreu um avanço notório nas
últimas décadas1.
Atualmente, além das formas farmacêuticas tradicionais, sólidas, pastosas e líquidas, pode-
mos destacar ainda cinco novas formas, que são: os comprimidos revestidos com membranas
semipermeáveis, em que se obtém um sistema de liberação de fármaco de forma controlada,
promovendo, assim, um aumento no tempo de permanência desse no organismo2; os adesivos
transdérmicos de liberação controlada utilizando membrana semi-permeável, que são fixados
sobre a pele sadia, funcionando como um sistema reservatório que possibilita ajustar e unifor-
mizar a liberação do fármaco, garantindo o fornecimento constante por um prolongado período
de tempo3; os implantes subcutâneos de liberação controlada, conhecidos como pellets, e que
são introduzidos na região subcutânea, possibilitando a liberação do fármaco lentamente, evi-
tando, desse modo, o uso das injeções que são usadas diariamente por pacientes de doenças
crônicas; as microesferas inaláveis, que consistem na combinação de um polímero degradável
com um fármaco, formando microesferas de 8 a 20µm de diâmetro, o que possibilita seu uso
através de inalação e reduz o uso das injeções diárias; e os lipossomas que são microvesículas
esféricas com dimensões que variam de 0,1 a 2,0µm constituídas de material lipossolúvel de
alta absortividade4. Essa inovação permite que fármacos que são adsorvidos com dificuldade
através de formas farmacêuticas convencionais tenham uma absorção completa.
Em todas estas formulações farmacêuticas, observa-se a necessidade de um biomaterial
14

1 Introdução
que controle a liberação do fármaco. Dentre os biomateriais, os hidrógeis biodegradáveis
têm emergido como uma importante classe devido a suas características biológicas, como
a biocompatibilidade, e pelo fato de seus produtos de degradação não serem imunogênicos,
carcinogênicos ou tóxicos ao ser humano5. Vários polímeros sintéticos e naturais têm sido in-
vestigados; dentre eles pode-se destacar o poli(ácido acrílico), o ácido hialurônico, o colágeno
e a quitosana. A taxa de liberação da droga nesses sistemas pode ser regulada por proprieda-
des poliméricas, tais como o inchamento, a degradabilidade, a porosidade e a permeabilidade
dos hidrógeis. O tipo de mecanismo que controla a liberação do fármaco também determina a
classificação dos sistemas, podendo ser: sistemas de difusão controlada, sistemas controlados
quimicamente, sistemas controlados por embebição e sistemas de liberação prolongada6.
Nesse trabalho foram obtidas membranas de quitosana pura e tratadas superficialmente com
uma solução aquosa de poli(ácido acrílico) a 25 e 60◦C com o objetivo de se estudar o seu po-
tencial uso em sistemas de liberação controlada. Para tanto, as interações ocorridas entre os
dois polímeros foram analisadas por diversas técnicas e também por ensaios de permeabili-
dade in vitro utilizando uma droga modelo. Os aspectos teóricos referentes aos materiais e às
técnicas utilizadas na pesquisa são relatados ainda na introdução desse trabalho. No segundo
capítulo, encontra-se descrita a metodologia experimental empregada na preparação, caracte-
rização e medidas de permeabilidade das membranas. No terceiro capítulo, são apresentados
e discutidos os resultados experimentais obtidos, no quarto capítulo, são apresentadas as con-
clusões alcançadas no trabalho e, por fim, no último capítulo, são descritas algumas sugestões
para futuras pesquisas.
1.1 Quitosana
A quitosana é um polímero natural, biodegradável, biocompatível e proveniente da reação
termoquímica alcalina de desacetilação parcial da quitina, um dos mais abundantes polissa-
carídeos de cadeia linear, constituído pela repetição de unidades β(1-4)-2-acetamida-2-deoxi-
15

1 Introdução
D-glicopiranose e que constitui a maior fração dos exoesqueletos de insetos e crustáceos,
tais como caranguejo, camarão e lagosta7. A estrutura da quitosana é muito similar à sua
precursora quitina (figura1.1), exceto pela substituição do grupo acetamido (NHCOCH3) na
posição 2 do anel glicopiranosídeo por grupos amino (NH2). Estruturalmente, é um polissa-
carídeo linear com um número variável (em função do grau residual de acetilação) de grupos
N-acetil-glucosamina, aleatoriamente localizados, podendo ser definida como um copolímero
de 2-amino-2-deoxi-D-glicopiranose e 2-acetamido-2-deoxi-D-glicopiranose, cujas unidades
são unidas por ligações β(1-4), (figura 1.2). O termo quitosana abraça uma série de polímeros,
que variam em massa molar (em torno de 10.000 a 1 milhão de Daltons), dependendo da fonte
e procedimentos de preparação, e em grau de desacetilação de 50 a 95%8.
Figura 1.1: Estrutura química da quitina.
Figura 1.2: Estrutura química da quitosana, com x representando o grau de acetilação.
A quitosana é insolúvel em valores de pH neutro e alcalino, mas forma sais solúveis em
água com ácidos inorgânicos e orgânicos fracos, incluindo ácido glutâmico, clorídrico, láctico
16

1 Introdução
e acético, resultando em soluções viscosas9. Em meio ácido, é um polieletrólito catiônico que
tem seus grupos aminos protonados (NH+3 ), conferindo à molécula cargas positivas10. Quanto
maior a quantidade destes grupos protonados na molécula, maior será a repulsão eletrostá-
tica entre as cadeias e também maior a solvatação em água. A quitosana é um biopolímero
susceptível à mudanças estruturais, devido à grande quantidade de grupos reativos como as
hidroxilas e, principalmente, pela presença de grupos aminos, que caracterizam a sua forte
afinidade por moléculas polares11. Como um policátion, a quitosana pode formar complexos
eletrostáticos com espécies carregadas negativamente, incluindo proteínas, certos polímeros
aniônicos, fármacos e outros ânions de baixa massa molar12. A quitosana exibe excelente
propriedade de retenção de água, ação bactericida e uma dose letal, DL50 encontrada na faixa
de 16g×kg−1 de peso corporal, quando administrada oralmente em ratos, caracterizando uma
baixa toxicidade. Essas propriedades são características importantes que podem influenciar
no desempenho da quitosana em muitas das suas aplicações13.
Atualmente, a quitosana tem despertado muito interesse para as aplicações médicas e far-
macêuticas, por ter propriedades farmacológicas, tais como, ação hipolipêmica, propriedades
de cicatrização de feridas e em virtude dos seus produtos de degradação, os oligômeros de
N-acetil-D-glucosamina, terem atividade antiúlcera, antiácida, antimicrobiana e de serem to-
talmente absorvíveis pelo organismo, sendo assim, considerada um material biodegradável14.
Além dessas propriedades mencionadas anteriormente, a quitosana e seus derivados têm se
destacado pela propriedade da bio/mucoadesividade, uma vez que seus grupos podem sofrer
protonação em certos pHs fisiológicos e, assim, interagir eletrostaticamente com as cargas
negativas na mucosa ou superfícies celulares, caracterizando a sua propriedade de bioadesivi-
dade. O potencial da quitosana para a aplicação bio/mucoadesiva foi reforçado pelo primeiro
relato de L. Illum et al.15 sobre a capacidade que a quitosana possui de promover a absor-
ção transmucosa de pequenas moléculas polares, bem como de drogas peptídicas e protéicas
para o epitélio nasal. Posteriormente, P. Artursson et al.16 relatou que a quitosana pode au-
17

1 Introdução
mentar a permeabilidade de drogas peptídicas via mucosa epitelial intestinal e se mostrar um
promissor material bioadesivo em pHs fisiológicos, por possuir grupos OH e NH2 que podem
promover pontes de hidrogênio, propriedade considerada essencial para a mucoadesão; termo
esse usado, porque a interação é restrita à camada mucosa17, 18. Assim, quando a quitosana
está exercendo a função de sistema carreador de fármaco, essa interação pode resultar em
um aumento no tempo de permanência desse sistema nos sítios específicos de absorção do
fármaco, liberando-o controladamente e melhorando a sua biodisponibilidade19. Esta caracte-
rística bio/mucoadesiva tem potencializado seu uso como carreador para a liberação contro-
lada e sustentada de agentes farmacêuticos, incluindo prednisolona20, albumina21, cisplatina22,
diclofenaco sódico23 e melantonina24 e, ainda, para a liberação sustentada de agentes quimi-
oterápicos25. Por tudo isso, a quitosana vem sendo considerada um promissor material para a
preparação das mais diferentes formas físicas de liberação controlada, tais como, micropartí-
culas26, microesferas27, nanoesferas28, géis e membranas29.
1.2 Membranas
De modo geral, uma membrana pode ser definida como uma barreira que separa duas fases
e restringe o transporte de várias espécies químicas de maneira específica. Uma membrana
pode também ser definida como uma película polimérica ou inorgânica que funciona como
uma barreira semi-permeável para uma filtração em escala molecular, separando duas fases e
restringindo, total ou parcialmente, o transporte de uma ou várias espécies químicas (solutos)
presentes na solução. Uma membrana pode ser homogênea ou heterogênea, simétrica ou as-
simétrica na estrutura, pode ser sólida ou líquida, pode ser neutra, carregar cargas positivas
ou negativas, ou pode ser bipolar. Quanto à configuração, pode ser densa ou porosa. Sua
espessura pode variar entre menos que 100nm e mais do que 1cm30. O termo "membrana" in-
clui uma grande variedade de materiais e estruturas, embora todos esses materiais apresentem
uma característica comum, que é restringir a passagem de várias espécies químicas de maneira
18

1 Introdução
muito específica30. A seletividade à passagem de solutos presentes em soluções homogêneas
está relacionada com as dimensões da molécula ou partícula, o tamanho dos poros da mem-
brana, a difusividade do soluto no material que constitui a membrana e as cargas elétricas
associadas.
Em relação ao transporte de massa através da membrana, esse pode ser causado por con-
vecção ou por difusão de moléculas individuais, induzido por campo elétrico, concentração,
pressão ou gradiente de temperatura. A principal força motriz responsável pelo transporte
de uma espécie em processos com membrana é o seu gradiente de potencial químico entre
os dois lados da membrana que se traduz em seu gradiente de concentração, pressão e ou
temperatura30.
1.2.1 Membranas de quitosana
As membranas de quitosana foram descritas e caracterizadas pela primeira vez por Muzza-
relli et al31. Membranas de quitosana são comumente usadas como barreira, imitando a pele,
em sistemas de liberação de droga transdérmica, em rotina de controle de qualidade de testes
de sistemas de liberação de fármacos32, na osmose reversa33, na pervaporação34 e na ultrafiltra-
ção35. A técnica mais simples para a preparação das membranas de quitosana é a evaporação
de solvente em uma solução de quitosana sobre uma placa de vidro que, geralmente, produz
membranas resistentes e transparentes36, com elevada absortividade à água e lenta degradação
enzimática pela lisoenzima, presente em tecidos e fluidos corporais de mamíferos37.
Diferentes formas de membranas de quitosana29, incluindo as reticuladas utilizando rea-
gentes bifuncionais e as obtidas por quelação com íons ou por complexação com polímeros
e proteínas, assim como redes interpenetradas, têm sido investigadas38. Membranas reticu-
ladas podem ser obtidas através da reticulação da quitosana com os mais utilizados agentes
reticulantes que são os dialdeídos, tais como o glioxal, o formaldeído e, principalmente, o
glutaraldeído39. Outros agentes que reticulam a quitosana têm sido investigados, dentre eles
19

1 Introdução
encontra-se o poli(ácido acrílico), o qual é uma alternativa para a obtenção de membrana po-
licomplexada através de interações iônicas e intermoleculares, que ocorrem entre as cargas
positivas presentes ao longo da cadeia polimérica da quitosana e as cargas negativas presentes
no poli(ácido acrílico). Membranas de quitosana reticuladas foram estudadas utilizando como
fármaco modelo a vitamina B1240. A reticulação é um processo em que as cadeias poliméricas
são unidas por ligações físicas fortes (reticulação iônica) ou químicas (reticulação química),
através das ligações covalentes, entre o agente reticulante e o polímero, ou entre dois polí-
meros distintos, objetivando modificar as propriedades moleculares do polímero, como, por
exemplo, aumentando a estabilidade química e térmica, alterando a permeabilidade, entre ou-
tras41. Deste modo, a permeação através das membranas de quitosana pode ser controlada
variando-se a natureza e/ou a concentração de comonômeros, ligações cruzadas e plastifican-
tes29, e, ainda, o grau de desacetilação e a massa molar da quitosana usada na preparação das
membranas42.
1.3 Poli(ácido acrílico)
O poli(ácido acrílico)(PAA) (figura 1.3) é um polímero sintético, solúvel em água, dioxano,
etanol, metanol e álcool isopropílico (bons solventes polares), e insolúvel em benzeno, acetona
e outros43, obtido pela polimerização via radicais livres do ácido acrílico. Possui em sua
estrutura grupos carboxílicos terminais que conferem um caráter polianiônico à sua molécula,
rendendo-lhe habilidade de trocas iônicas e complexação com íons de carga positiva44.
O poli(ácido acrílico) é caracterizado por uma compatibilidade com biomateriais, tendo
também extensas aplicações como agente espessante em tintas, no campo de adesivos, nas
formulações de produtos farmacêuticos, em cosméticos e na agricultura45. Desse modo, um
grande interesse tem sido mostrado na sua utilização em diversas aplicações e, dentre os usos,
os mais interessantes são a fabricação de olhos artificiais, lentes de contato e dentaduras arti-
ficiais, uma vez que não causa irritação nem sensibilização à pele e aos olhos46.
20

1 Introdução
Figura 1.3: Estrutura química do poli(ácido acrílico).
O caráter aniônico polimérico do poli(ácido acrílico) em valores de pH alto (pH ≥ 6) torna
este polímero adequado para a cobertura de sistemas de liberação oral16, tendo sido inves-
tigado como agente que promove absorção para a liberação peroral de drogas peptídicas47,
prolonga o tempo de residência e aumenta o tempo de contato com a mucosa no sítio de ab-
sorção da droga. Sua alta solubilidade, porém, limita seu uso como sistema de liberação de
droga transmucosa, pois pode ser dissolvido antes do tempo desejado de permeação da droga
através da membrana48. Já seu caráter aniônico favorece a sua capacidade de se complexar
com álcool polivinílico(PVA)49, poli(vinil pirrolidona)(PVP)45, poli(óxido de etileno)(PEO)45
e quitosana50, entre outros51, através de pontes de hidrogênio e interações eletrostáticas, origi-
nando, assim, misturas de aspectos homogêneos que possuem baixa solubilidade em água.
1.4 Formação de complexos polieletrolíticos de
quitosana e poli(ácido acrílico)
Novas tecnologias requerem materiais com combinação de propriedades que não são en-
contradas nos materiais convencionais. A escolha adequada dos constituintes de um material,
com propriedades físicas complementares, tem levado ao desenvolvimento de complexos po-
lieletrolíticos de materiais poliméricos que apresentam características de interesse científico e
21

1 Introdução
tecnológico.
Os complexos poliméricos são formados pela associação de dois ou mais polímeros comple-
mentares, surgindo de atrações eletrostáticas, interações hidrofóbicas, pontes de hidrogênio,
forças de van der Waals ou combinações dessas interações52. A formação destes complexos
depende diretamente do grau de ionização do polímero catiônico e aniônico (pH e força iônica
das soluções), densidades de suas cargas (conformações da cadeia) e suas concentrações53. Al-
guns estudos têm confirmado que polímeros com propriedades complexantes têm alcançado
importância em termos de suas aplicações potenciais na ciência dos materiais e são extensi-
vamente usados nas mais diversas formas54. Usualmente, complexos polieletrolíticos (PEC)
podem ser aplicados na forma de hidrogéis, filmes e membranas, e são formados quando
cargas opostas de polieletrólitos carregados de grupos catiônico ou aniônico são misturados,
interagindo via interações eletrostáticas e causando precipitação em meio aquoso55, de modo
que o precipitado compacto obtido na forma de filmes contínuos é aplicado para membranas
de processo de separação como diálise, ultrafiltração e osmose reversa56; tais membranas têm,
também sido usadas na desidratação de álcoois55.
Na literatura, são descritos vários trabalhos sobre as propriedades peculiares de comple-
xos polieletrolíticos (PEC) adequados para aplicações biomédicas57, dentre estes complexos,
destacam-se os de quitosana/poli(estireno sulfonado) e quitosana/PAA que tiveram suas sínte-
ses e características físico-químicas estudadas58 e cujas estruturas têm levado a um novo ma-
terial polimérico com propriedades especiais, potencialmente proveitoso em vários campos,
incluindo liberação de droga57. Estudos também relatam formas de complexos polieletrolíti-
cos entre a quitosana, o alginato, a pectina e ainda interações entre membranas polieletrolíticas
e várias soluções de NaCl e proteínas plasmáticas59.
Os complexos de quitosana e poli(ácido acrílico)(PAA) são formados com o objetivo de
reduzir a solubilidade do PAA em água. Estudos mostraram que a solubilidade do PAA pode
ser reduzida, enquanto sua propriedade mucoadesiva pode ser melhorada ou mantida, quando
22

1 Introdução
ocorre sua complexação com a quitosana60. A insolubilidade é gerada pelas fortes intera-
ções eletrostáticas entre os polieletrólitos, poli(ácido acrílico) e quitosana61. De acordo com
a literatura55, 62, os complexos polieletrolíticos entre a quitosana e o poli(ácido acrílico) po-
dem ser obtidos pela mistura física entre os dois polímeros sob fusão, pela dissolução dos
polímeros em um solvente com posterior evaporação deste ou através da polimerização. A li-
teratura61 também relata a formação de membranas de complexos polieletrolíticos (PEC) entre
quitosana-PAA construída pela difusão do poli(ácido acrílico), na superfície de membranas de
quitosana pura, variando-se temperatura, tempo de difusão e a massa molar do PAA, com o
objetivo de serem usadas em sistemas de pervaporação61.
A quitosana, quando dissolvida em meio aquoso ácido pH ≤ 6,5, exibe uma alta densidade
de carga positiva no grupo NH+3 , permitindo que esta se agregue a compostos polianiônicos
como o PAA. Observa-se ainda a presença dos grupos hidroxilas nos carbonos 3 e 6 que
também podem participar das interações através das pontes de hidrogênio permitindo obter-se
membranas com excelentes propriedades que são exploradas pela indústria farmacêutica63.
Estudos mostraram a ocorrência de interações iônicas na mistura de quitosana e PAA que
incluem reação iônica entre íons NH+3 da quitosana e íons carboxílicos (COO−) do PAA,
pontes de hidrogênio entre H da quitosana e OH do grupo carboxílico do PAA ou entre OH
da quitosana e o H do PAA (figura1.4). Essas interações iônicas são geralmente confirmadas
por FTIR64 e todas as três ligações têm mostrado afinidade por água e repulsão por molé-
culas orgânicas. As ligações iônicas entre o íon amônio e carboxilato são das mais fortes e
induzem entrecruzamentos, prevenindo a dissolução e o excessivo inchamento da matriz po-
limérica na presença de água64. Em alguns casos de hidrogéis polieletrolíticos, o equilíbrio
de inchamento e tamanho das formas obtidas foram principalmente governadas pela percenta-
gem dos componentes iônicos no polímero e grau de ionização da droga65. Desta forma, um
completo entendimento da complexação interpolimérica pode facilitar a aplicação industrial
dos complexos com propriedades funcionais específicas.
23

1 Introdução
Figura 1.4: Processo de policomplexação entre a quitosana e o poli(ácido acrílico).
24

1 Introdução
O mecanismo de formação do complexo entre a quitosana e o poli(ácido acrílico) irá de-
pender do pH do meio. De acordo com a literatura66 em valores de pH de 3, 4 e 5, o grau de
ionização da quitosana é alto (1,0; 0,95 e 0,85, respectivamente). Nas mesmas condições, o
grau de ionização do PAA é em torno de 0,1; 0,2 e 0,5, respectivamente. Em outras palavras,
na faixa de pH entre 3 e 5 a maior parte dos grupos NH2 da quitosana está sob a forma pro-
tonada (NH+3 ) enquanto que os grupos carboxílicos estão na forma de COOH. Nesse caso, o
mecanismo de formação do complexo sugerido é:
Q − NH+3 + P − COOH → Q − NH+
3−OOC − P + H+
Por outro lado, em pH = 6, o grau de ionização da quitosana é reduzido a cerca de 0,6
enquanto do PAA cerca de 0,8 ou seja, a maioria dos grupos amina está na forma de NH2
enquanto os grupos carboxila estão sob a forma de COO− . Isso sugere o seguinte mecanismo
de formação de complexo:
Q − NH2 + −OOC − P +H+−−→ Q − NH+
3−OOC − P
1.5 Difusão
De modo geral, entende-se por difusão o transporte de massa de moléculas individuais por
uma barreira ou espaço livre, que ocorre segundo um processo aleatório e depende de um gra-
diente de concentração ou pressão67. A difusão livre ou transporte passivo de uma substância
através de um líquido, de um sólido ou de membranas, é um processo de considerável impor-
tância na ciência farmacêutica. Exemplos de tópicos de fenômenos de transporte de massa
aplicados à farmácia são: dissolução de fármacos em tabletes, pós, grânulos, liofilização, ul-
trafiltração, liberação de fármacos de ungüentos e de bases de supositórios, passagem de vapor
de água, gases, aditivos ou fármacos atráves de filmes de revestimentos, cápsulas e paredes de
25

1 Introdução
embalagens3. É possível citar, ainda, a própria absorção passiva de fármacos pelo organismo
ou a distribuição de substâncias nos diferentes compartimentos fisiológicos do nosso corpo3.
A difusão é o principal meio de liberação de fármacos através de estruturas poliméricas,
tais como filmes e microesferas, nos quais a difusão acontece gradativamente, impedindo
a ocorrência de picos elevados na concentração plasmática e, com isso, os efeitos adversos
relacionados a muitos fármacos3.
A difusão também é conhecida como a tendência que as moléculas apresentam de migrar de
uma região de concentração elevada para outra região de concentração baixa em consequên-
cia direta do movimento browniano. O processo fundamenta-se em aspectos relacionados
com soluto e solvente, temperatura, pressão, potencial químico, entre outros68. O movimento
browniano das moléculas garante que o sistema passe de um estado inicial, sem equilíbrio,
para um estado final de energia livre mínima e entropia máxima e, portanto, em equilíbrio69.
1.5.1 As leis de Fick
1.5.1.1 Primeira lei de Fick
A difusão passiva rege-se pela primeira lei de Fick, a qual relaciona o fluxo com o gradiente
de concentração, sob condições de estado estacionário (Fin = Fex) , onde o fluxo interno é
igual ao fluxo externo, ou seja, o processo de difusão em que o gradiente de concentração(∂c∂t
)não varia com o tempo. O mecanismo de liberação é governado por essa lei, que pode
ser expressa pela equação 1.170, e que estabelece que o fluxo de matéria é proporcional à
variação da concentração (∂c) e inversamente proporcional à distância (∂x).
Fx = −D
(∂c
∂x
)(1.1)
em que Fx representa a quantidade de substância que se difunde no intervalo de tempo através
de uma área plana, e D é um coeficiente de difusão que se pode definir como sendo a quanti-
26

1 Introdução
dade de substância difundida por unidade de tempo através da unidade de superfície, quando
o gradiente de concentração(
∂c∂x
)é também unitário. O sinal negativo da equação significa
que a difusão ocorre na direção de diminuição da concentração do difusante. A maioria dos
métodos experimentais utilizados para estudar a difusão tem interesse na variação da concen-
tração com o tempo e a distância. Nesse caso, a primeira lei de Fick pode ser convertida em
uma equação diferencial parcial de segunda ordem, a segunda lei de Fick70.
1.5.1.2 Segunda lei de Fick
A segunda lei de Fick (equação 1.2) representa a velocidade de alteração da concentração
de soluto em função do tempo e do deslocamento, dois importantes fatores na determinação
do coeficiente de difusão de qualquer soluto em diferentes sistemas. A segunda lei de Fick
é usada no estado não estacionário, isto é, quando a concentração muda com o tempo, sendo
mais conhecida como equação da difusão:
∂c
∂t= D
∂2c
∂x2(1.2)
em que c é a concentração de substância difusora, t é o tempo, D é o coeficiente de difusão
e x é uma das direções em que o fluxo pode ocorrer em um corpo. Para qualquer situação, o
problema é encontrar uma solução apropriada para a equação 1.2. Diversos métodos para a
obtenção de soluções são encontrados no livro "The Mathematics of Difusion"70.
1.5.2 Difusão através de membranas
O estudo da difusão de uma dada substância pode ser desenvolvido usando uma membrana
de espessura l e coeficiente de difusão D, que separa dois compartimentos e cujas superfícies x
= 0 e x = l são mantidas à concentrações constantes c1 e c2, respectivamente. Depois de algum
tempo, o estado estacionário é alcançado e as concentrações permanecem constantes em todos
os pontos da membrana. Então, a partir da equação da difusão (equação 1.1), mantendo o
27

1 Introdução
coeficiente de difusão constante e integrando em relação a x, tem-se:
dc
dx= constante (1.3)
e, através de integração posterior, introduzindo as condições em que x = 0 e x = l:
c − c1
c2 − c1
=x
l(1.4)
As equações 1.3 e 1.4 mostram que as concentrações variam linearmente de c1 a c2 através
da membrana. Além disso, o fluxo de substância difusora é o mesmo através de todas as
seções da membrana e dado por:
F = −Ddc
dx= −D
(c2 − c1)
l(1.5)
Se a espessura l e as concentrações de superfícies c1 e c2 são conhecidas, D pode ser obtido
a partir de um valor observado de F usando a equação 1.5. No entanto, experimentalmente,
nem sempre os valores das concentrações c1 e c2 podem ser determinadas71.
1.5.2.1 Constante de permeabilidade
Em alguns sistemas, quando as concentrações das superfícies c1 e c2 não podem ser ob-
tidas, mas somente as concentrações C1 e C2 das soluções nos dois lados da membrana são
conhecidas, o fluxo de transferência no estado estacionário é então descrito como72:
F = −P (C2 − C1)
l(1.6)
e a constante P é denominada de constante de permeabilidade do sistema. Sendo o coeficiente
de difusão D constante e existindo uma relação linear entre a concentração das soluções e
a concentração de equilíbrio na superfície da membrana, então as equações 1.5 e 1.6 são
28

1 Introdução
equivalentes, resultando em:
−D (c2 − c1)
l= −P (C2 − C1)
l(1.7)
assim tem-se:
P = D(c2 − c1)
(C2 − C1)(1.8)
Sabendo-se que o coeficiente de partição é dado por:
K =(c2 − c1)
(C2 − C1)(1.9)
Substituindo a equação 1.9 na equação 1.8, tem-se:
P = DK (1.10)
1.5.3 A permeabilidade da membrana
O cálculo da permeabilidade da membrana pode ser desenvolvido assumindo uma mem-
brana de espessura l e área S entre dois compartimentos com soluções em diferentes concen-
trações C1 e C2. Se inicialmente o sistema estiver livre de diferenças de concentração entre as
faces da membrana, ou seja, c1=c2, pode-se dizer que a concentração inicial de permeante na
membrana é zero. Após adicionar uma solução de concentração C1 em um dos compartimen-
tos da célula de permeabilidade, a quantidade de matéria Q que difunde deste compartimento
para um outro, considerando um comportamento linear quando t → ∞, é dada por70:
Qt =Dc1
l
(t − l2
6D
)(1.11)
em que D é o coeficiente de difusão e l é a espessura da membrana hidratada.
No estado estacionário a equação 1.1 pode ser aplicada, então, a quantidade de substância
29

1 Introdução
que atravessa a membrana de área S no tempo t é70:
Q = −SD(c2 − c1)
lt (1.12)
sabendo que o coeficiente de partição K dado na equação 1.9 se aplica a ambas as superfícies
da membrana, a equação 1.12 se torna:
Q = −SDK(C2 − C1)
lt (1.13)
como P = DK, tem-se:
Q = −SP (C2 − C1)
lt (1.14)
desse modo, o coeficiente de permeabilidade P pode ser determinado através da reta Q × t
no estado estacionário. Em muitos experimentos tem-se uma situação em que C1� C2, nesse
caso, a equação 1.14 pode ser simplificada para:
Q =SPC1
lt (1.15)
1.6 Metronidazol
O metronidazol (2-metil-5-nitroimidazol) (figura 1.5) é um pó, cristalino, branco, muito so-
lúvel em água a 25◦C (10,5 mg×mL−1) e ativo contra um amplo espectro de infecções para-
sitárias, protozoários e bactérias anaeróbicas (B. fragilis, B. melaninogênicos e Clostridium),
possuindo atividade particularmente elevada in vitro contra T. vaginalis (tricomoníase), E.
histolítica (amebíase) e Giardia lamblia (giardíase)73, sendo a molécula de DNA celular dos
microorganismos o principal alvo de sua ação biólogica. O metronidazol tem uma estrutura
heterocíclica, consistindo de núcleos baseados em imidazol com um grupo nitro na posição 5.
No perfil farmacocinético, esse fármaco tem sua absorção completa após administração oral,
30

1 Introdução
atingindo concentrações plasmáticas de cerca de 10 mg×mL−1 após 1h de uma dose de 500
mg73.
Figura 1.5: Estrutura química do metronidazol.
A meia-vida do metronidazol é de cerca de 8h, e sua ligação às proteínas plasmáticas é de
10%. O metronidazol é encontrado inalterado em vários tecidos e na urina, após administração
oral. Apesar de ser geralmente bem absorvido após administração oral, alguns pacientes não
respondem bem ao tratamento em virtude da baixa concentração sistêmica, sendo esse caso
uma questão aberta. O fígado é o principal local do seu metabolismo, sendo responsável por
mais de 50% da depuração sistêmica do metronidazol. O metronidazol pode causar desconfor-
tos gastrointestinais e, ainda, reação eritematosa com prurido e urticária. A sua administração
em altas doses pode causar distúrbios neurológicos. Sob aspecto clínico, os efeitos colaterais
apenas raramente foram suficientemente graves para causar interrupção da medicação. Toda-
via, pesquisas relataram que a droga metronidazol com anéis nitroimidazol em sua estrutura
é suspeita de ser carcinogênica e mutagênica74. O metronidazol foi escolhido para ser usado
nesse trabalho por ser um fármaco solúvel em água que possui absorção na região do UV e
por produzir solução aquosa saturada com pH = 5,8.
31

1 Introdução
1.7 Espectroscopia na região do infravermelho
A espectroscopia na região do infravermelho é freqüentemente aplicada como um dos mais
importantes métodos analíticos usados na determinação de moléculas orgânicas no estado lí-
quido, sólido e gasoso. A espectroscopia vibracional tem sido aplicada em determinações
qualitativas e quantitativas de materiais poliméricos, onde vários parâmetros podem ser in-
vestigados, incluindo grupos terminais, ramificações de cadeias, configuração e conformação,
assim como, isomerismo geométrico e estérico.
A espectroscopia de infravermelho é baseada nas vibrações dos átomos nas moléculas e
pode ser obtida passando radiação através da amostra e determinando que fração de radiação
incidente é absorvida em uma energia particular; a energia em que algum pico em um espectro
de absorção aparece correspondente à freqüência de vibração de uma parte da amostra da
molécula.
Diversos métodos podem ser utilizados na espectroscopia de infravermelho, dentre eles está
a espectroscopia de transmissão, a espectroscopia de reflectância externa, a espectroscopia de
reflectância difusa (DRIFT), a espectroscopia fotoacústica (PAS), a espectroscopia por elip-
sometria e a espectroscopia de reflectância total atenuada (ATR), sendo este último o mais
comumente utilizado pelos cientista depois da transmitância75.
A espectroscopia de reflexão total atenuada (ATR) utiliza o fenômeno de reflexão total
interna, figura 1.6 que permite a obtenção de espectros qualitativos de sólidos ou líquidos.
Esta técnica baseia-se no fato de que um feixe de luz refletido internamente pela superfície
de um meio transmissor penetra uma pequena distância além da superfície refletora e retorna
ao meio transmissor durante o processo de reflexão. Os cristais usados na célula de ATR são
feitos de materiais que têm baixa solubilidade em água e altíssimo índice de refração, como,
por exemplo, o seleneto de zinco (ZnSe), que é insolúvel em água, em solventes orgânicos e
em ácidos diluídos e bases, o que permite uma análise de amostras líquidas e sólidas76.
Quando a luz passa através de dois meios com diferentes índices de refração n1 e n2, (n1>
32

1 Introdução
n2) e que estão em contato entre si, sua trajetória é refratada. A magnitude dessa distorção
depende do ângulo de incidência, θ, e das densidades óticas de ambos os meios. A um ângulo
de 90◦ a luz é refletida e transmitida, a um ângulo θ < θcritico ela será refletida e refratada e
a um ângulo θ > θcritico ela será totalmente refletida. Esta é a base para a espectroscopia de
reflectância interna. O ângulo crítico é função do índice de refração dos materiais em contato,
sendo definido por77:
θc = sin−1 n21 (1.16)
em que:
n21 =n2
n1
(1.17)
n1 é o índice de refração do cristal de ATR e o n2 é o índice de refração da amostra.
Figura 1.6: Célula de reflectância total atenuada 76.
O fenômeno da reflexão interna total é familiar. Pode ser observado em um copo de água,
por exemplo. Se a parte do vidro, localizado abaixo da água é observada obliquamente através
da superfície da água, ela parecerá espelhada e não podemos ver nenhum objeto atrás deste
vidro. Isto ocorre porque a luz que incide na superfície do vidro é totalmente refletida e, por-
tanto, não atravessa esta superfície para iluminar o objeto. No entanto, tocando-se o vidro com
33

1 Introdução
um dedo, por exemplo, os padrões da pele tornam-se evidentes, o que indica que a reflexão
total foi destruída onde o contato foi feito. Isto pode ser explicado pela penetração do campo
eletromagnético no meio de menor densidade um pouco além da superfície refletora e, en-
tão, quando um objeto é colocado suficientemente próximo desta superfície, interage com este
campo eletromagnético destruindo a reflexão total. Este fenômeno é explicado pela formação
de uma onda estacionária no meio denso, perpendicular à superfície refletora e de um campo
evanescente no meio de menor densidade que decai exponencialmente com a distância a partir
da superfície. Portanto, no caso da espectroscopia de reflexão interna, a reflectividade é uma
medida de interação de uma onda evanescente com a amostra, resultando em seu espectro77.
A exploração do fenômeno da reflectância interna ou reflectância total atenuada para pro-
dução de espectros foi, inicialmente, desenvolvida por N. J. Harrick77 e J. Fahrenfort78. Após
isto, uma enorme quantidade de publicações com diferentes aplicações desta técnica tornaram-
na a segunda técnica de infravermelho mais utilizada, depois da transmissão.
Em um ensaio de ATR a amostra é colocada em contato com o elemento de reflexão in-
terna, conhecido como cristal de ATR, com alto índice de refração. A radiação infravermelha
é focalizada na extremidade do cristal, refletida através dele e depois direcionada para o de-
tector (figura 1.6). Como citado anteriormente, embora ocorra reflexão interna na interface
cristal/amostra, a radiação (onda evanescente) penetra a uma curta distância na amostra, onde
pode ser absorvida. É obtido então um espectro de absorção. O grau de penetração na amostra,
dp, foi definido por N. J. Harrick77 como sendo a distância necessária para que a amplitude da
onda evanescente decaia a um valor e−1 do seu valor inicial na interface.
A profundidade de penetração em ATR é uma função do comprimento de onda (λ), do
índice de refração do cristal e do ângulo de incidência de radiação (θ). A profundidade de
34

1 Introdução
penetração, dp, para uma absorbância nominal média é dada pela equação 1.18:
dp =
(λn1
)
2π
[sin2θ −
(n2
n1
)2] 1
2
(1.18)
O dp dá, portanto, uma idéia da faixa de espessura da amostra que está sendo analisada. De
acordo com a equação 1.18, uma das formas de se alterar o valor da dp é através da variação
no ângulo de incidência θ. O grau de penetração aumenta à medida que nos aproximamos do
ângulo crítico, de modo que várias profundidades podem ser atingidas, ajustando-se o ângulo
de incidência. No entanto, o número de reflexões, e, também, o dp irão diminuir à medida que
θ se aproxima de uma incidência rasa. O número de reflexões no cristal dá uma medida da
intensidade do espectro resultante e pode ser calculado através da seguinte equação77:
N =l × cotθ
t(1.19)
em que l é o comprimento do cristal e t sua espessura. Estes pontos foram enfatizados porque
frequentemente se deseja observar camadas finas da superfície através da variação destes pa-
râmetros (θ e n1), no entanto, isto possui sempre a desvantagem de diminuir a intensidade do
sinal.
Os espectrômetros modernos geralmente possuem acessório que permite a variação no ân-
gulo de incidência, θ. Observando-se a figura 1.6, verifica-se que o cristal possui a forma de
um paralelepípedo cortado para produzir um ângulo de 45◦. Caso o ângulo de incidência seja
diferente de 45◦, a radiação não será mais normal à superfície e, desse modo, ocorrerão perdas
por refração. Nesse caso, o ângulo efetivo pode ser calculado usando a lei de Snell e algumas
relações trigonométricas75. A relação obtida é :
θef = 45 − sen−1
[(n1
n2
)sen (45 − θescala)
](1.20)
35

1 Introdução
em que θef é o ângulo efetivo e θescala é o ângulo indicado na escala do acessório.
1.8 Análise térmica
Talvez nenhum campo de caracterização de polímeros tenha se expandido tão rapidamente
como as análises térmicas. Desde a introdução de modernos instrumentos em 1962, a popu-
laridade da grande variedade de técnicas de análise térmica tem crescido satisfatoriamente.
Análise térmica é um termo que abrange um grupo de técnicas que medem as propriedades
físicas e/ou químicas de uma substância e seus produtos de reação em função da tempera-
tura, com métodos que envolvem mudanças da massa e energia, à medida que a substância é
submetida a um programa de temperatura controlada79.
Na análise térmica, a amostra é aquecida ou resfriada à taxa constante ou mantida à tem-
peratura constante. Em diversos experimentos, a atmosfera é igualmente importante, podendo
ter um efeito significativo nos estágios de decomposição de uma amostra. Particularmente, ela
diferencia entre o uso de gases inertes e oxidantes.
As vantagens da análise térmica podem ser resumidas como as seguintes:
• a amostra pode ser analisada em larga faixa de temperatura, usando-se vários programas
de temperatura;
• qualquer forma física de amostra (sólida, líquida ou gel), pode ser acomodada, usando
uma variedade de porta-amostras;
• é necessário somente uma pequena quantidade de amostra;
• a atmosfera do ensaio é padronizada (inerte, oxidante ou uma composição de gases);
• flexibilidade no tempo de ensaio, podendo levar desde alguns minutos até várias horas.
Existem várias técnicas em análise térmica e cada uma delas apresenta respostas específi-
cas do comportamento dos materiais diante da variação de temperatura, como é mostrado na
36

1 Introdução
tabela 1.1. Nesse trabalho, foi utilizada a análise termogravimétrica (TGA) para analisar a
estabilidade térmica das membranas de quitosana e quitosana/PAA.
Tabela 1.1: Técnicas de análise térmica.
Análise Sigla Propriedades
Análise termogravimétrica TGA Variação de massa em função da temperatura
Análise termogravimétrica diferencial DTG Velocidade de variação da massa em
função da temperatura
Análise térmica diferencial DTA Mudança na quantidade de calor liberado ou absorvido
Calorimetria diferencial de varredura DSC Medida quantitativa das mudanças de entalpia
em função da temperatura e do tempo
Análise dinâmico mecânica DMA Variação de módulo dinâmico e/ou amortecimento
de uma substância sob uma carga oscilatória em
em função da temperatura e frequência
Dilatometria ou análise TMA Variação da dimensão linear em função da temperatura
termomecânica e medida de coeficiente de expansão térmica sob carga
não oscilatória
1.8.1 Análise termogravimétrica (TGA)
O termo análise termogravimétrica (TGA) é comumente empregado, particularmente, em
polímeros, no lugar de TG, por ser seu precendente histórico e para minimizar a confusão
verbal com Tg, abreviação da temperatura de transição vítrea. A análise termogravimétrica é
usada para determinar alterações da massa de amostras em função da temperatura ou de tempo
a uma temperatura constante, usando uma microbalança. A aplicação típica desta técnica está
na avaliação da estabilidade térmica e temperatura de decomposição. Em uma TGA, há um
grande número de fatores que afetam a natureza e a precisão dos resultados experimentais,
37

1 Introdução
sendo de natureza instrumental (taxa de aquecimento do forno, atmosfera do forno, geome-
tria do porta-amostra e do forno, entre outras) ou dependentes das características da amostra
(quantidade, solubilidade dos gases envolvidos na amostra, tamanho da partícula, calor de
reação, empacotamento da amostra, condutividade térmica, entre outras). A análise termo-
gravimétrica também pode ser usada para se obter algumas informações sobre distribuição de
seqüência de copolímeros, da composição de polímeros carregados e para analisar a cinética
dos processos físico-químicos que ocorrem nas amostras80.
A análise termogravimétrica geralmente é realizada de três formas:
1. Termogravimetria isotérmica, em que as variações de massa da amostra são medidas em
função do tempo, a uma temperatura constante;
2. Termogravimetria quase-isotérmica, em que a variação de massa da amostra é medida
em função do tempo a várias temperaturas constantes;
3. Termogravimetria dinâmica, em que o registro de variação de massa da amostra é feito
em função da temperatura, a uma taxa de aquecimento pré-determinada e preferencial-
mente constante, sendo essa a técnica utilizada nesse trabalho.
A termogravimetria derivada (DTG) é uma técnica que fornece a derivada primeira da curva
termogravimétrica (TG), e esta é caracterizada por apresentar a variação de massa em função
da temperatura ou em relação ao tempo sob a forma de picos, sendo útil nos casos em que a
análise da TG apresenta sobreposições, permitindo distinguir com maior facilidade o ínicio e
o fim de um processo. A aréa sob a curva da DTG é proporcional à variação de massa, o que
permite análises quantitativas precisas81.
1.9 Microscopia eletrônica de varredura (MEV)
Em 1965, o equipamento de microscopia eletrônica de varredura (MEV) tornou-se comerci-
almente disponível e imprescindível nas mais diversas áreas: eletrônica, geologia, ciência dos
38

1 Introdução
materiais, ciência da vida, entre outras, uma vez que o desenvolvimento de novos materiais
tem exigido um número de informações bastante detalhado de características só possíveis de
serem observadas no MEV. Essa técnica é utilizada para analisar a morfologia de materiais
sólidos, pois permite uma caracterização rápida e precisa da superfície e de suas subestrutu-
ras. A área a ser analisada é bombardeada por um fino feixe de elétrons de alta voltagem,
que são direcionados na superfície da amostra coberta por um filme condutivo. Como resul-
tado da interação do feixe de elétrons com a superfície da amostra em análise ponto a ponto,
uma série de radiações é emitida, e, quando captada corretamente, fornece informações da
natureza topográfica da amostra, cujas imagens se apresentam com aparência natural em três
dimensões82.
Na microscopia eletrônica de varredura, as amostras em análise devem ser recobertas com
uma fina camada de material condutor (Au, Pd, C), para evitar que estas se alterem ou mesmo
queimem, devido a alta voltagem empregada para a aceleração dos elétrons e, também, pelo
fato de alguns materiais serem maus condutores de elétrons, como, por exemplo, as membra-
nas preparadas a partir de materiais poliméricos. Pode-se também usar uma baixa voltagem
de aceleração no feixe de elétrons. Para a maioria dos materiais, o uso de voltagem entre 1 e
3kV não produz alteração na amostra82.
39

2 Metodologia experimental
2.1 Materiais
A quitosana em pó (Polymar Ltda, Brasil) usada neste trabalho apresenta grau de desace-
tilação em torno de 90,47%, segundo o fabricante. Sua massa molar média (M v = 2,2×105
Daltons) foi determinada pelo método de viscometria, utilizando a equação de Mark-Howink-
Sakurada, cujos valores das constantes específicas a e K são 0,76 e 3,5×10−4, respectiva-
mente83. O fármaco metronidazol (MM = 171, 15g×mol−1, Aldrich, USA), o Poli(ácido
acrílico) (MM = 250.000 Da, Aldrich, USA), o ácido acético (P. A., Reagen Quimibrás
Ltda, Brasil) e o hidróxido de sódio (P.A., Vetec Ltda, Brasil) foram utilizados sem qualquer
purificação prévia.
2.2 Preparação das membranas
2.2.1 Membranas de quitosana
A quitosana foi dissolvida em solução aquosa de ácido acético 2 % v/v, e mantida sob
agitação constante durante 24h, em temperatura ambiente, de modo a se obter uma solução
1,5 % m/v do polímero. Após esse período, a solução foi filtrada, sendo esse procedimento
realizado em duas etapas. Na primeira, foi utilizado um filtro com tela de náilon a fim de
eliminar resíduos sólidos derivados do processo de obtenção da quitosana. Na segunda, foi
40

2 Metodologia experimental
utilizado um filtro Millex Millipore R© com diâmetro de poros de 41µm. Em seguida, o filtrado
foi deixado em repouso por cerca de 6h para permitir o desaparecimento de bolhas de ar. Um
volume de 25mL da solução de quitosana foi colocado em placas de Petri, que foram deixadas
em repouso por cerca de 1h para eliminar as bolhas e, em seguida, colocadas em estufa a
50◦C por 24h para evaporação do solvente. Depois de retiradas da estufa, as membranas
foram neutralizadas com uma solução de NaOH 5 % por 2h e lavadas com água destilada
repetidamente para a remoção dos resíduos da solução alcalina. Após o estiramento e secagem,
à temperatura ambiente, por 24h, foram obtidas membranas de quitosana denominadas (QUI)
com espessura na faixa de 32 a 42µm (micrômetro digital Check-line DCF 900).
2.2.2 Membranas de quitosana/poli(ácido acrílico)
As membranas de quitosana/poli(ácido acrílico) foram preparadas a partir de membranas de
quitosana pura obtidas como descrito na subseção 2.2.1, entretanto, antes do processo de se-
cagem em extensor à temperatura ambiente, as membranas foram fixadas em suporte ilustrado
na figura 2.1. Um volume de aproximadamente 50mL de uma solução aquosa de poli(ácido
acrílico) na concentração de 2,0g×L−1 e (pH 3,2) foi colocado em contato com a camada su-
perior das membranas fixadas ao suporte e mantidas em duas diferentes temperaturas: 25◦C
e a 60◦C. A temperatura de 60◦C foi obtida utilizando-se um banho termostático (Tecnal-TE
184), pré-aquecido. Em ambos os casos, o tempo de tratamento foi de 2h. Depois do tra-
tamento com o poli(ácido acrílico)(PAA), as membranas foram cuidadosamente retiradas do
suporte e lavadas em água destilada para a remoção dos resíduos de PAA. Após isso, foram
estiradas e secadas à temperatura ambiente por 24h, obtendo-se assim, membranas de quito-
sana/poli(ácido acrílico) denominadas QUIPAA25 e QUIPAA60, com espessura na faixa de
44 a 48µm, respectivamente (micrômetro digital Check-line DCF 900).
41

2 Metodologia experimental
Figura 2.1: Sistema para tratamento superficial das membranas de quitosana.
2.3 Caracterização das membranas
2.3.1 Espectroscopia de absorção na região do ultravioleta
A curva de calibração do metronidazol foi obtida a partir de soluções de metronidazol
na faixa de concentração de 0,006g×L−1 a 0,028g×L−1, em comprimento de onda (λ) de
320nm por espectrofotometria na região de UV, em espectrofotômetro Varian, modelo Cary
100UV/Visível. Todas as medidas foram feitas em duplicatas. A espectrofotometria é funda-
mentada na Lei de Lambert-Beer (equação 2.1), utilizada nesse experimento para a obtenção
do valor da absortividade do metronidazol, o qual é um dos parâmetros utilizados para o
cálculo da permeabilidade das membranas. Para medidas de absorção de radiação em deter-
minado comprimento de onda, tem-se a equação 2.1:
A = εbc (2.1)
em que A é a absorbância, ε é a absortividade molar, b é o caminho óptico da célula do
espectrofotômetro e c a concentração da solução absorvente.
42

2 Metodologia experimental
2.3.2 Espectroscopia de absorção na região do infravermelho
As medidas espectroscópicas na região do infravermelho das membranas obtidas, como
descrito na seção 2.2, foram realizadas com um espectrofotômetro FT-IR Thermo Nicolet
Nexus 470. As membranas QUI, QUIPAA25 e QUIPAA60 foram analisadas pela técnica de
reflexão total atenuada (ATR) em cristal de ZnSe, com comprimento de 50mm e a espessura
de 3mm, utilizando-se ângulos de incidência de 39◦, 45◦ e 60◦.
2.3.3 Ensaio de ganho de massa
A porcentagem de ganho de massa por intumescimento das membranas foi determinada pela
imersão destas individualmente em becker de 250mL contendo água destilada a temperatura
ambiente. O procedimento foi realizado no período de tempo de 15min a 48h, usando uma
balança analítica B-TEC-U210A Tecnal. A porcentagem de água absorvida foi calculada,
através da equação 2.2:
% =
(mu − ms
ms
)× 100 (2.2)
onde mu e ms representam, respectivamente, as massas das membranas úmidas e secas.
2.3.4 Análise térmica
2.3.4.1 Análise termogravimétrica
As membranas QUI, QUIPAA25 e QUIPAA60 foram analisadas em um analisador termo-
gravimétrico DTG-60 de marca SHIMADZU que consiste de uma balança eletrônica acoplada
a um forno tipo mufla de temperatura máxima de trabalho de 1500◦C e com um controle de
temperatura que permite a aplicação de taxas de aquecimento de 0,1 até 25◦C×min−1. As aná-
lises foram feitas com amostras de 5mg submetidas à uma razão de aquecimento de 10◦C×min−1 em uma atmosfera dinâmica de N2 (30mL× min−1) e em uma faixa de temperatura de
43

2 Metodologia experimental
25 a 800◦C.
2.3.5 Microscopia eletrônica de varredura (MEV)
Para a realização das análises, foram retirados fragmentos de 0,5cm do centro de cada mem-
brana. As fotomicrografias das amostras das membranas de QUI, QUIPAA25 e QUIPAA60
foram obtidas de sua superfície, de modo a se observar a morfologia externa. As membranas
foram fixadas com fita adesiva em suportes de alumínio próprios para o MEV, sendo pos-
teriormente recobertas com uma fina camada de ouro de 20nm de espessura. Usou-se um
microscópio eletrônico de varredura da marca Phillips-XL30, operando com feixe de elétrons
secundários com aceleração 2kV e que permite visualização morfológica da amostra em dife-
rentes magnitudes.
2.3.6 Ensaio de permeação
A medida de permeabilidade do fármaco é outro método importante na caracterização de
membranas. A determinação é simples e não destrutiva para a membrana. O experimento
foi realizado com as membranas de QUI, QUIPAA25 e QUIPAA60, previamente imersas em
água destilada, à temperatura ambiente, por aproximadamente 24h.
As membranas foram colocadas entre dois compartimentos A e B de uma célula de perme-
abilidade em formato de ”U”, feita de PVC. A superfície tratada ficou sempre voltada para o
lado A da célula. No compartimento B foi colocado um volume de 230mL de solvente puro
(água destilada) e no compartimento A igual volume de uma solução aquosa de metronidazol
nas concentrações de 0,1 e 0,2%. O solvente e a solução foram submetidos à agitação mecâ-
nica constante sincronizada. Todo o sistema foi mantido em banho termostático à temperatura
constante de 30 ± 0,1 ◦C (figura 2.2).
Uma bomba peristáltica (Micronal B 332 II), ligada através de duas mangueiras ao compar-
timento B e à cubeta do espectrofotômetro (Varian Cary 100 UV/visível), coletou amostras de
44

2 Metodologia experimental
metronidazol que difundiram do compartimento A para o compartimento B em intervalos de
tempo regulares de 5min e a absorbância foi medida no comprimento de onda (λ) de 320nm;
a alíquota retirada foi devolvida automaticamente ao compartimeno de origem, após análise.
Todos os experimentos foram feitos em duplicata e duraram em torno de 3h.
Figura 2.2: Sistema de permeação in vitro.
2.3.7 Cálculo da permeabilidade das membranas
Os valores de permeabilidade foram determinados utilizando o modelo descrito por Crank70
para o fluxo através de membrana. Assumindo que a membrana possui inicialmente concen-
tração (c0 = 0) e que a concentração em uma das faces da membrana é muito maior que a
outra, isto é, c1 � c2, tem-se que c1 − c2 ≈ c1. Nesse caso, a quantidade total de substância
Q que difunde através da membrana de área S no tempo t é dada pela equação 2.3 mostrada,
abaixo:
Q =Dc1
l
(t − l2
6D
)(2.3)
45

2 Metodologia experimental
em que D é o coeficiente de difusão e l é a espessura da membrana hidratada. Como a quan-
tidade de fármaco que passa através da membrana foi determinada espectroscopicamente, Q
pode ser dado por:
Q =V c
S(2.4)
pela equação 2.1, lei de Lambert-Beer, a concentração é dada por:
c =A
εb(2.5)
portanto, substituindo a equação 2.5 na equação 2.4, tem-se:
Q =V A
εbS(2.6)
em que V é o volume da célula de difusão, A é a absorbância, ε é a absortividade calculada
a partir do coeficiente angular da curva de calibração, b é o caminho ótico da célula e S é a
área de superfície da membrana. Igualando a equação 2.6 com a equação 2.3 e rearranjando,
obtém-se:
A =Dc1εbS
V lt − c1lεbS
6V(2.7)
em que c1 é a concentração inicial da substância difusora na superfície da membrana, D é o co-
eficiente de difusão e l a espessura da membrana. Entretanto, como as membranas em análise
são relativamente finas, a determinação da concentração do fármaco na superfície da mem-
brana, necessária para a determinação do valor de D, é muito difícil. Dessa forma, substitui-se
o cálculo do coeficiente de difusão D pelo cálculo da permeabilidade, utilizando-se para isto
a equação 1.10 que mostra a relação existente entre estes dois fatores. Substituindo a equação
46

2 Metodologia experimental
1.10 na equação 2.7, obtém-se:
A =PC1εbS
V lt − c1lεbS
6V(2.8)
Desse modo, o coeficiente de permeabilidade pode ser determinado através da inclinação
da curva gerada pelo gráfico de absorbância versus tempo no estado estacionário, em que:
α =PC1εbS
V l(2.9)
47

3 Resultados e discussão
Neste capítulo são apresentados os resultados experimentais referentes à preparação das
membranas poliméricas, sua caracterização morfológica, térmica e espectroscópica, assim
como o comportamento dessas membranas em relação à permeação do fármaco metronida-
zol.
3.1 Espectroscopia na região do ultravioleta
3.1.1 Curva de calibração do metronidazol
A figura 3.1 mostra o espectro de ultravioleta obtido para a solução aquosa de metronida-
zol a 0,022g×L−1. De acordo com a figura 3.1, observa-se a existência de três bandas de
absorção. A banda com máximo localizado em λ= 320nm foi escolhida para ser usada na ela-
boração da curva de calibração por apresentar maior intensidade. De acordo com a literatura84
o metronidazol apresenta tal absorção na faixa de 200 a 350nm.
A figura 3.2 apresenta o gráfico com os valores de absorbância versus concentração usados
na curva de calibração. Considerando a relação existente entre a absorbância e a concentração
da solução dada pela lei de Lambert-Beer, equação 2.1, tem-se que o coeficiente angular da
reta gerada fornece o valor de absortividade (α=εb). O valor obtido para a absortividade do
metronidazol foi ε= 54806g−1×cm2.
Com o valor da absortividade e da variação da absorbância da solução com o tempo no com-
48

3 Resultados e discussão
partimento B da célula de permeação, os valores de permeabilidade das diferentes membranas
puderam ser calculados em relação ao metronidazol.
Figura 3.1: Espectro no UV para uma solução aquosa de metronidazol.
49

3 Resultados e discussão
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0.005 0.01 0.015 0.02 0.025 0.03
Ab
sorb
ânci
a
C (g/m3)
Figura 3.2: Curva de calibração do metronidazol.
50

3 Resultados e discussão
3.2 Espectroscopia na região do infravermelho
A técnica FTIR-ATR com ângulo variável foi utillizada nesse trabalho com o objetivo de,
através da variação no ângulo de incidência, obter-se espectros mais representativos da su-
perfície das membranas. Para se determinar a faixa de ângulo a ser utilizada, a primeira
providência foi determinar o ângulo crítico de acordo com a equação 1.16. Utilizando um
valor de 1,50 para o índice de refração da quitosana85, 86 e 2,40 para o cristal de seleneto de
zinco (ZnSe)76, foi obtido um valor de ângulo crítico de 38,7◦. A partir desse valor, os ângulos
escolhidos foram 39◦, 45◦e 60◦.
A tabela 3.1 fornece algumas informações características de cada ângulo utilizado, como
valores de profundidade de penetração (dp), número de reflexões e ângulo efetivo, que foram
calculados utilizando-se as respectivas equações 1.18, 1.19 e 1.20, já descritas na seção 1.7.
Tabela 3.1: Características de cada ângulo utilizado na técnica FTIR-ATR.
θescala θefetivo n◦ de reflexões dp(µm) p/ ν = 1.600 cm−1
39◦ 41,2◦ 19,0 5,3245◦ 45,0◦ 16,6 1,2560◦ 54,0◦ 12,1 0,69
Observa-se que, quanto maior o ângulo de incidência, menor o número de reflexões e o
grau de penetração na amostra; e conclui-se que, utilizando-se um ângulo de incidência de
60◦com grau de penetração de 0,69µm, é possível se obter um espectro mais representativo da
superfície das membranas hidratadas, em relação a um ângulo de incidência de 39◦, com grau
de penetração de 5,32µm. Deve-se citar que as membranas hidratadas possuem espessura na
faixa de 69 e 72µm.
A figura 3.3 apresenta os espectros da quitosana pura obtidos usando os dois lados da mem-
brana, a um ângulo de incidência de 45◦. O lado A refere-se à face inferior da membrana, que
secou em contato com a placa de Petri, enquanto o lado B refere-se à face superior que secou
em contato com o ar. O objetivo desse experimento foi demonstrar a semelhança inicial das
51

3 Resultados e discussão
duas faces. A primeira observação que pode ser feita está relacionada à qualidade dos espec-
tros obtidos. Observa-se que a face A produz espectros de melhor qualidade e com nível de
ruído consideravelmente pequeno, já a face B produz um espectro com bandas menos intensas
e com um ruído maior. Com esse experimento, demonstra-se a semelhança espectral das duas
faces. Em relação à posição e/ou surgimento de novas bandas, pode-se afirmar que as super-
fícies são praticamente idênticas e que a diferença observada na qualidade dos espectros está
relacionada, provavelmente, à diferença de rugosidade nas faces. Nesse caso, a face B (em
contato com o ar) deve ser um pouco mais rugosa do que a face A, o que prejudica o contato
entre a membrana e o cristal e, assim, a qualidade do espectro. Resultados semelhantes foram
obtidos para os ângulos de 39◦ e 60◦ para a membrana QUI.
Figura 3.3: Espectros de FTIR-ATR para ambas as faces da membrana de quitosana pura ob-tido a um ângulo de incidência de 45◦.
52

3 Resultados e discussão
Outra observação importante a ser feita é que, inicialmente, tentou-se obter os espectros a
partir das membranas secas. Os espectros obtidos, todavia, não apresentaram boa qualidade,
provavelmente devido à dificuldade de se obter um bom contato da membrana-cristal, causada
pela rigidez da membrana seca. Por esse motivo, optou-se pelo uso de membranas hidratadas.
A literatura55 tem nos mostrado que a água possui efeito plastificante nas membranas de qui-
tosana, tornando-as mais macias, o que facilita o contato membrana-cristal. Além disso, as
informações sobre as membranas hidratadas são também muito valiosas, visto que durante o
seu uso nas medidas de permeabilidade estas estão sempre hidratadas.
A partir dos espectros da figura 3.3, as principais absorções relativas à quitosana foram
caracterizadas e encontram-se descritas na tabela 3.2. Comparando os espectros da figura 3.3
com os espectros encontrados na literatura87, observa-se que as principais diferenças surgem
na região entre 3000-3600 cm−1 e a 1640 cm−1, devido à presença de água nas membranas.
Na primeira região tem-se as deformações axiais simétricas (3490 cm−1) e assimétricas (3280
cm−1) das ligações O-H da água; em 1640 cm−1, tem-se a deformação angular da mesma
ligação, tornando estas bandas mais intensas do que no espectro da quitosana seca.
Tabela 3.2: Bandas de absorção no FTIR-ATR para membrana QUI.
Número de onda (cm−1) Atribuição3300 deformação axial de O-H e N-H do grupo NH2
2920 e 2880 deformação axial de C-H de grupos CH2 e CH3
1640 deformação axial de C= O (amida I)1593 deformação angular N-H do grupo NH2 (amida II)1420 e 1323 deformação angular de O-H e deformação angular
de C-H do anel glicosídico1380 deformação angular do C-H de CH3
1150,1074 e 1031 deformação axial do C-O da ligação éter
A partir da diferença de qualidade dos espectros obtidos em lados diferentes da membrana,
optou-se por realizar o tratamento com o PAA sempre na face A (inferior) da membrana.
A figura 3.4 apresenta os espectros de ambas as faces de uma membrana tratada com PAA à
temperatura ambiente, obtidos a partir de ângulos de 39◦, 45◦ e 60◦, (a,b e c) respectivamente.
53

3 Resultados e discussão
A faixa de número de onda mostrada é, de acordo com a literatura,59 a região onde se observa
as bandas referentes à formação do complexo entre o poli(ácido acrílico) e a quitosana. O
restante do espectro não apresentou nenhuma variação para os três ângulos utilizados.
A análise da figura 3.4 (a) mostra que, para o ângulo de 39◦, ambos os lados da membrana
produzem praticamente o mesmo espectro. Já os espectros obtidos em ângulos de 45◦e 60◦,
mostrados nas figuras 3.4 (b e c), evidenciam o surgimento de uma nova banda, próximo a
1560 cm−1 que, de acordo com a literatura62, corresponde à absorções características dos gru-
pos NH+3 e COO−, evidenciando, assim, a formação do complexo61, 62. O não surgimento
dessa banda no espectro obtido a 39◦ pode ser explicado pelo fato de que, nesse ângulo, o
grau de penetração do campo evanescente na amostra é bem maior, como mostra a tabela 3.1,
e, portanto, o espectro obtido é característico do volume da amostra e não de sua superfície.
Já os espectros obtidos nos ângulos de 45◦ e 60◦ são os que possuem a menor profundidade
de penetração, sendo os mais característicos para análise da superfície da membrana. A partir
desses resultados, confirma-se que o tratamento com o PAA foi apenas superficial e que a pro-
fundidade de penetração do PAA foi de apenas alguns micrômetros e, portanto, as membranas
obtidas são assimétricas.
A figura 3.5 mostra a mesma sequência de espectros da figura 3.4, só que, agora, usando
membranas tratadas com PAA a 60◦C. Nesse caso, percebe-se que, mesmo a um ângulo de
39◦ na figura 3.5, já é possível se observar o surgimento da banda de absorção referente à
formação do complexo e, à medida que o ângulo de incidência aumenta, essa absorção torna-
se mais evidente. Esses resultados indicam que, com o aumento da temperatura, o processo
de formação do complexo ocorre não apenas próximo à superfície, mas também à camadas
mais internas da membrana. Uma provável explicação para isto seria o fato de que, a 60◦C,
a quitosana já passou pela sua temperatura de transição vítrea (em torno de 50◦C)88 estando,
agora, no estado borrachoso, em que existe uma maior mobilidade das cadeias, o que facilitaria
a penetração das moléculas de poli(ácido acrílico).
54

3 Resultados e discussão
Figura 3.4: Espectros de FTIR-ATR para ambas as faces da membrana tratada com poli(ácidoacrílico) à temperatura ambiente, obtidos a ângulo de incidência de 39◦, 45◦ e 60◦,respectivamente a, b e c.
55

3 Resultados e discussão
Figura 3.5: Espectros FTIR-ATR para ambas as faces da membrana tratada com poli(ácidoacrílico) a 60◦C, obtidos a ângulo de incidência de 39◦, 45◦ e 60◦ , respectivamentea,b e c.
56

3 Resultados e discussão
Figura 3.6: Espectros de FTIR-ATR para a membrana de quitosana pura, tratada compoli(ácido acrílico) à temperatura ambiente e a 60◦C, obtidas a um ângulo de inci-dência de 60◦.
A figura 3.6, por sua vez, apresenta os espectros de quitosana pura (60Q_A), tratada à
temperatura ambiente (60QPAA_25_T) e a 60◦C (60QPAA_60_T), obtidos a um ângulo de
incidência de 60◦. Observa-se, aqui, que a 60◦C, a banda de absorção referente à formação do
complexo torna-se ainda mais evidente do que à temperatura ambiente, confirmando que com
o aumento da temperatura ocorre também aumento no grau de complexação. Os resultados
obtidos a 39◦ e 45◦, embora menos evidentes, também apresentaram a mesma tendência.
3.3 Ensaio de ganho de massa
O ganho de massa devido à sorção de água pelas membranas de QUI, QUIPAA25 e QUI-
PAA60 foi analisado através da imersão dessas em água destilada por diferentes intervalos de
tempo. A figura 3.7 e a tabela 3.3 apresentam os resultados obtidos nesses experimentos.
A análise da figura 3.7 permite observar que as três membranas analisadas apresentam ga-
57

3 Resultados e discussão
nhos de massa superiores a 90%, o que é uma indicação da forte afinidade desses polímeros,
quitosana e poli(ácido acrílico), pela água. No caso da quitosana, sabe-se que esta hidrofilici-
dade é consequência da presença de grupos amino e hidroxila em sua estrutura, enquanto que,
no caso do poli(ácido acrílico), é devido à presença dos grupos carboxílicos, que tornam esse
polímero solúvel em água.
Outra observação que pode ser feita é quanto à velocidade com que a água é retida pelas
membranas. No caso da membrana de quitosana pura, observa-se que o processo de intumes-
cimento ocorre muito rapidamente tendendo a um valor de equílibrio. De acordo com a tabela
3.3, a variação no percentual de ganho de massa entre o tempo inicial de 15 minutos e o final
de 48 horas é de apenas 3,0%. Já no caso das membranas tratadas com PAA, observa-se que o
mesmo processo ocorre de forma mais gradual e não parece ainda ter atingido seus valores de
equílibrio, isso pode ser consequência do tratamento na superfície das membranas. De acordo
com a literatura54, a interação entre os grupos NH+3 da quitosana e COO− do PAA induz à
reticulação iônica na camada superficial da membrana, fazendo com que haja uma redução na
velocidade de retenção de água pela membrana. De acordo com a tabela 3.3, a variação no
percentual de ganho de massa entre os tempos inicial e final nas membranas tratadas com o
PAA é de cerca de 6,4%.
Uma última observação refere-se à quantidade de água retida pelas membranas. Neste caso,
a figura 3.7 mostra que as membranas tratadas com poli(ácido acrílico) têm uma maior capaci-
dade de retenção de água do que a membrana de quitosana pura. Este resultado é inicialmente
supreendente, visto que, como já relatado, a reticulação deveria dificultar a absorção de água,
entretanto pode ser explicado considerando que os sítios iônicos formados pela reticulação exi-
bem grande afinidade pela água, assim como também o poli(ácido acrílico) adicionado para
o tratamento. Além disso, deve-se considerar que as membranas utilizadas são relativamente
finas, o que significa dizer que suas superfícies possuem grande influência em seu compor-
tamento. Mais adiante serão analisadas as imagens de microscopia eletrônica de varredura
58

3 Resultados e discussão
Figura 3.7: Porcentagem de ganho de massa para membranas QUI, QUIPAA25 e QUIPAA60em função do tempo.
59

3 Resultados e discussão
das membranas usadas neste experimento. Desde já, pode-se adiantar que as superfícies trata-
das com poli(ácido acrílico) são substancialmente diferentes da superfície da membrana não
tratada. Com isso, sugere-se que o tratamento produz uma modificação na superfície da mem-
brana, aumentando sua área superficial e, consequentemente, permitindo uma maior adsorção
de água nesta superfície. Estima-se que, no caso das membranas tratadas, o aumento na quan-
tidade de água retida na membrana foi de 8% para QUIPAA25 e 14% para QUIPAA60 e está
mais relacionado a um fenômeno de superfície, ou seja, as moléculas de água estariam distri-
buídas na superfície da membrana (adsorvidas) além de fixadas em seu interior (absorvidas).
Tabela 3.3: Porcentagem de ganho de massa pelas membranas QUI, QUIPAA25 e QUIPAA60.
Tempo (h) QUI (%) σn−1 QUIPAA25(%) σn−1 QUIPAA60 (%) σn−1
0,25 98 2 103 3 108 30,50 99 2 106 3 109 3
1 98 2 107 3 108 22 99 2 107 4 109 23 100 3 107 4 111 24 100 2 106 3 112 25 99 2 104 3 111 46 103 3 105 3 112 27 101 2 106 4 112 28 100 2 106 4 112 224 100 2 108 3 114 348 101 2 109 3 115 2
3.4 Análise térmica
3.4.1 Termogravimetria
As curvas termogravimétricas (TG) e as curvas termogravimétricas derivadas (DTG) das
membranas QUI, QUIPAA25 e QUIPAA60 são mostradas nas figuras 3.8 e 3.9, respectiva-
mente. O termograma da quitosana exibe um comportamento típico para esse polissacarídeo,
60

3 Resultados e discussão
apresentando dois estágios distintos de decomposição. O primeiro estágio inicia-se a 31◦C e
vai até cerca de 150◦C, com pico máximo em torno de 52,6◦C, correspondendo a uma perda
de massa de 10,3%. O segundo estágio tem início próximo a 200◦C indo até 400◦C, com
pico máximo em torno de 300◦C e perda de massa de 49,5%. As curvas TG e DTG das
membranas QUIPAA25 e QUIPAA60 também apresentam dois estágios de degradação. A
membrana QUIPAA25 apresenta o primeiro estágio na faixa entre 31 e 200◦C, com pico má-
ximo a 58,9◦C e perda de massa de 7,6%, e um segundo estágio na faixa entre 200 e 400◦C,
com pico máximo a 300◦C e perda de massa de 35,0%. Na membrana QUIPAA60, observa-se
que o primeiro estágio ocorre entre 31 e 200◦C , com pico máximo a 59,2◦C e perda de massa
de 6,8%, enquanto o segundo estágio ocorre entre 200 e 400◦C , com pico máximo a 300◦C e
perda de massa de 34,6%.
De acordo com a literatura57, 89, 90, a primeira perda de massa (1◦estágio) para as membra-
nas de quitosana está relacionada à perda de moléculas de água que estariam associadas aos
grupos aminos e hidroxilas deste polímero, através de pontes de hidrogênio. Portanto, varia-
ções nesse estágio estão relacionadas tanto com as intensidades das interações polímero-água
quanto com a capacidade de retenção de água pelo material. Comparando o termograma da
figura 3.9 para as membranas de quitosana pura e tratada, observa-se que a temperatura de
saída da água é praticamente a mesma para as três membranas, indicando, portanto, que as
interações polímero-água são semelhantes para os três materiais. No entanto, observa-se uma
redução na perda de massa para a membrana QUIPAA25 e outra ainda maior para a QUI-
PAA60. Estes resultados são uma forte indicação de que o tratamento superficial ocorreu
como esperado. Ou seja, as membranas foram ionicamente reticuladas a partir de uma super-
fície e, como consequência, tiveram sua capacidade de absorção de água reduzida, visto que
alguns grupos aminos, que funcionavam como sítios de absorção de água, estão agora proto-
nados, formando complexos polieletrolíticos com o poli(ácido acrílico). Observa-se também
que na membrana tratada a 60◦C a perda de massa é ainda menor, indicando que o processo
61

3 Resultados e discussão
20
30
40
50
60
70
80
90
100
0 100 200 300 400 500 600 700 800
Mas
sa (
%)
Temperatura (°C)
QUIQUIPAA25QUIPAA60
Figura 3.8: Curvas termogravimétricas (TG) das membranas QUI, QUIPAA25 e QUIPAA60
62

3 Resultados e discussão
−15
−10
−5
0
0 100 200 300 400 500 600 700 800
DT
G (
%/
mim
)
Temperatura (°C)
QUIQUIPAA25QUIPAA60
Figura 3.9: Curvas termogravimétricas derivadas (DTG) das membranas QUI, QUIPAA25 eQUIPAA60
63

3 Resultados e discussão
de reticulação, neste material ocorreu em maior extensão como já mostrado nos resultados do
infravermelho. Os resultados obtidos com a membrana ”seca” confirmam também a hipótese
levantada anteriormente de que o elevado ganho de massa apresentado pelas membranas tra-
tadas com o poli(ácido acrílico), quando imersas em água, estava relacionado apenas com um
efeito de superfície e não propriamente com o aumento de hidrofilicidade.
A segunda perda de massa para a quitosana, que ocorre a 300◦C, é atribuída a um complexo
processo de decomposição que tem início através da quebra aleatória das ligações glicosídi-
cas seguida de decomposição das unidades acetiladas e desacetiladas do polímero57, 89. Nesse
estágio, observa-se mais uma vez uma redução na perda de massa para a membrana QUI-
PAA25 e outra ainda maior para a QUIPAA60. Estes resultados confirmam mais uma vez
a reticulação iônica que ocorre através da formação dos complexos polieletrolíticos entre a
quitosana e o PAA e que este processo ocorre em maior extensão a 60◦C. A consequência da
formação destas atrações iônicas seria o aumento da estabilidade térmica desses materiais. A
pequena diferença observada entre as membranas QUIPAA25 e QUIPAA60 deve-se ao fato
do tratamento ter sido apenas na superfície da membrana.
A tabela 3.4 mostra a perda percentual de massa das membranas em cada estágio e a quan-
tidade de resíduo de massa após 500◦C. O resíduo de massa comprova que as membranas
tratadas possuem uma maior estabilidade térmica devido às interações iônicas ocorridas entre
a quitosana e o poli(ácido acrílico).
Tabela 3.4: Perda de massa das membranas QUI, QUIPAA25, QUIPAA60 em dois estágios
distintos e quantidade de resíduo após 500oC .
Membranas 1oestágio 2oestágio ResíduoT◦C Perda de massa % T◦C Perda de massa % após 500◦C
QUI 52,6◦C 10,3% 300◦C 49,5% 40,2%QUI/PAA25 58,9◦C 7,6% 300◦C 35,0% 57,3%QUI/PAA60 59,2◦C 6,8% 300◦C 34,6% 58,6%
64

3 Resultados e discussão
3.5 Microscopia eletrônica de varredura (MEV)
As micrografias das superfícies das membranas QUI, QUIPAA25 e QUIPAA60 são apre-
sentadas nas figuras 3.10, 3.11 e 3.12, respectivamente, e ainda na figura 3.13 onde pode ser
observada a superfície das membranas tratadas com o poli(ácido acrílico) na magnitude de
5000x. Nas três primeiras figuras são mostrados os resultados obtidos em ambos os lados,
na magnitude de 2000x, com o objetivo de verificar a existência ou não de diferenças signi-
ficativas entre os dois lados. Os resultados apresentados na figura 3.10 mostram que, para a
quitosana pura, não existem diferenças significativas entre o lado superior da membrana seca
em contato com o ar e o lado inferior, que estava em contato com a placa de Petri. Ambas
as superfícies apresentam-se homogêneas e, pelo menos na magnificação utilizada, não se
observa a presença de poros.
No caso das membranas tratadas, QUIPAA25 e QUIPAA60, os resultados obtidos são bas-
tante diferentes. A figura 3.11 mostra que a superfície tratada à temperatura ambiente é signi-
ficativamente diferente da superfície não tratada, apresentando agora um padrão mais rugoso.
Na figura 3.12 que representa a membrana tratada a 60◦C, estas diferenças são mais evidentes.
Esses resultados confirmam que o tratamento com o PAA foi realmente superficial, ou seja, o
polímero, mesmo a 60◦C, não se difundiu completamente pela membrana de quitosana, desta
forma não atingiu o outro lado da membrana, como pode ser visto na microscopia eletrônica
de varredura. Isto deve-se provavelmente a sua alta massa molar, que dificulta sua mobili-
dade. Como consequência, podemos afirmar que as membranas obtidas são assimétricas, ou
seja, possuem uma camada formada pelo complexo polimérico quitosana-poli(ácido acrílico)
e o restante do volume formado por quitosana pura. Os resultados mostram também que o
tratamento feito a 60◦C é mais efetivo, causando maiores modificações na superfície da mem-
brana. Estes resultados estão de acordo com os discutidos anteriormente para os espectros de
infravermelho e o ensaio de ganho de massa.
65

3 Resultados e discussão
Figura 3.10: Microscopia eletrônica da superfície da membrana QUI em contato com o ar eem contato com a placa de Petri
66

3 Resultados e discussão
Figura 3.11: Microscopia eletrônica da superfície não tratada e tratada da membrana QUI-PAA25
67

3 Resultados e discussão
Figura 3.12: Microscopia eletrônica da superfície não tratada e tratada da membrana QUI-PAA60
68

3 Resultados e discussão
Figura 3.13: Microscopia eletrônica da superfície tratada das membranas QUIPAA25 e QUI-PAA60 na magnitude de 5000x
69

3 Resultados e discussão
3.6 Ensaio de permeação
O fármaco modelo escolhido para ser permeado foi o metronidazol, em virtude de algumas
de suas características físico-químicas, como solubilidade em água e absorção na região do
ultravioleta e, ainda, por formar solução com pH igual a 5,8. A permeabilidade foi determi-
nada em membranas de quitosana pura e tratadas com poli(ácido acrílico). As concentrações
do metronidazol utilizadas foram de 0,1 e 0,2%, enquanto as espessuras das membranas secas
variaram entre 42 e 48µm.
Os resultados dos ensaios de permeação das membranas QUI, QUIPAA25 e QUIPAA60 em
relação ao metronidazol 0,1 e 0,2% podem ser vistos nas figuras 3.14, 3.15 e 3.16, respecti-
vamente. Todos os experimentos foram feitos em duplicata e apresentaram boa reprodutibili-
dade. O tempo de duração dos experimentos foi determinado a partir dos resultados obtidos
com a membrana de quitosana pura. Conforme visto na figura 3.14, a um tempo aproximado
de 200 minutos, o valor da absorbância medido no compartimento A do sistema de perme-
ação atinge um valor próximo a 2,0 para as concentrações de metronidazol de 0,1 e 0,2%.
Sabendo-se que, de acordo com a lei de Beer, valores de absorbância acima de 2,0 significam
menos de 1% de luz transmitida chegando ao detector, optou-se por limitar o tempo de todos
os experimentos a 200 minutos para efeito comparativo com a membrana de quitosana pura.
Observa-se, a partir das figuras 3.14, 3.15 e 3.16, que os experimentos apresentam um com-
portamento bastante linear para o período de tempo e concentrações utilizadas, indicando que
o estado estacionário é atingido rapidamente e que a permeabilidade pode ser obtida a partir
de valores de coeficiente angular das retas, como já descrito na subseção 2.3.7 da metodologia.
De acordo com a literatura70, 71, algumas vezes este estado estacionário leva algum tempo para
ser estabelecido, produzindo um comportamento inicial conhecido como tempo de retardo
(time lag), que pode ser definido como o tempo necessário para que o sistema alcance o seu
estado estacionário. No caso dos experimentos aqui relatados, a ausência do tempo de retardo
pode estar relacionada a pequena espessura das membranas usadas em relação aos seus valores
70

3 Resultados e discussão
de permeabilidade. Outra observação importante a ser feita é que o comportamento linear está
geralmente relacionado à ausência de fortes interações entre o fármaco e a membrana63, 91.
A tabela 3.5 apresenta os valores de permeabilidade calculados para as três membranas
estudadas. Os valores podem ser analisados de duas formas: influência do tratamento feito na
membrana e influência da variação na concentração do fármaco. Analisando-se a influência
do tratamento feito nas membranas, verifica-se que para ambas as concentrações de fármacos
utilizadas (0,1% e 0,2%) ocorreu uma grande redução nos valores de permeabilidade para
as duas membranas. Para a membrana QUIPAA25, a redução no valor de permeabilidade foi
cerca de cinco vezes para ambas as concentrações, enquanto que para a membrana QUIPAA60
foi de 36 vezes para a concentração de 0,1% e 56 vezes para a concentração de 0,2%. Os
resultados mostram que o tratamento, embora apenas superficial, influenciou grandemente a
permeabilidade das membranas.
Para compreender estes resultados, deve-se recorrer aos modelos existentes na literatura
para a permeação em membranas92. Dentre os vários existentes, podem ser destacados o mo-
delo de poros e o modelo do volume livre. No primeiro, assume-se a presença de poros na
membrana, enquanto no segundo considera-se a membrana compacta ou homogênea. Como
mostrado nas imagens do MEV, as membranas preparadas nesse trabalho não possuem poros,
sendo membranas densas com alguma rugosidade na superfície. Portanto, o modelo aplicável
é o do volume livre. Nesse modelo, a membrana é considerada como sendo uma rede ho-
mogênea hidratada. A difusão ocorre por ”saltos” sucessivos por entre estes espaços vazios
(volume livre) que seriam maiores do que o soluto. A difusão é dependente da quantidade
desses espaços vazios. No caso das membranas tratadas, pode-se considerar que as interações
quitosana-poli(ácido acrílico) estariam produzindo uma estrutura mais compacta e densa com
redução do volume livre e, consequentemente, redução de permeabilidade. A maior redução
para a membrana tratada a 60◦C está coerente com os resultados mostrados anteriormente,
que indicaram que o aumento na temperatura aumenta a profundidade da camada reticulada,
71
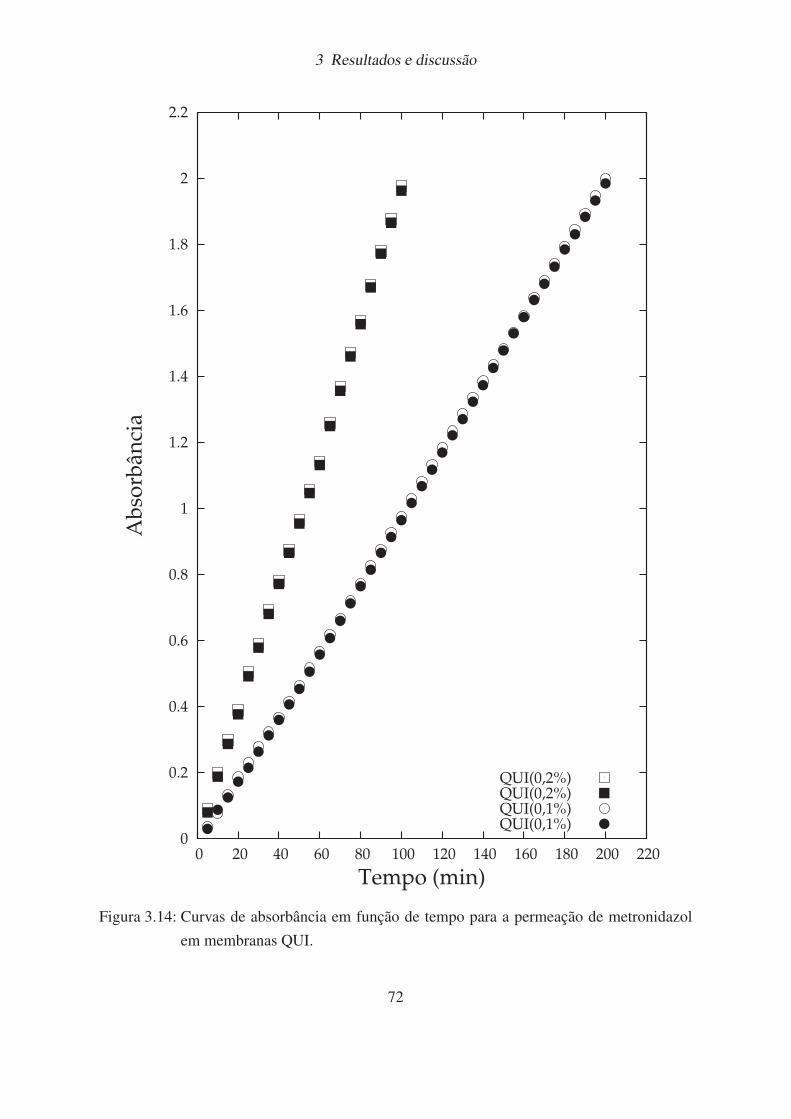
3 Resultados e discussão
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
2.2
0 20 40 60 80 100 120 140 160 180 200 220
Ab
sorb
ânci
a
Tempo (min)
QUI(0,2%)QUI(0,2%)QUI(0,1%)QUI(0,1%)
Figura 3.14: Curvas de absorbância em função de tempo para a permeação de metronidazolem membranas QUI.
72

3 Resultados e discussão
0
0.2
0.4
0.6
0.8
1
0 20 40 60 80 100 120 140 160 180 200 220
Ab
sorb
ânci
a
Tempo (min)
QUIPAA25(0,2%)QUIPAA25(0,2%)QUIPAA25(0,1%)QUIPAA25(0,1%)
Figura 3.15: Curvas de absorbância em função do tempo para a permeação do metronidazolem membranas QUIPAA25.
73
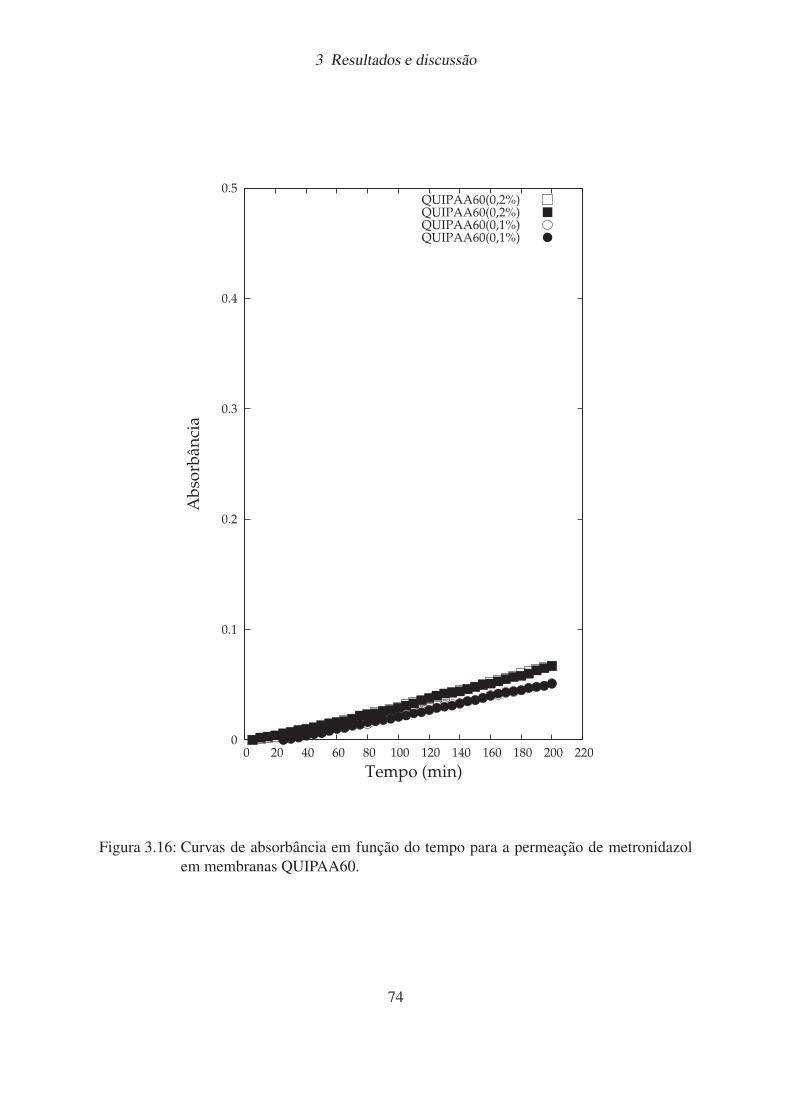
3 Resultados e discussão
0
0.1
0.2
0.3
0.4
0.5
0 20 40 60 80 100 120 140 160 180 200 220
Ab
sorb
ânci
a
Tempo (min)
QUIPAA60(0,2%)QUIPAA60(0,2%)QUIPAA60(0,1%)QUIPAA60(0,1%)
Figura 3.16: Curvas de absorbância em função do tempo para a permeação de metronidazolem membranas QUIPAA60.
74

3 Resultados e discussão
Tabela 3.5: Cálculo do coeficiente de permeabilidade para as diferentes membranas estudadas.
(P± σn−1) × 105cm2.min−1
MEMBRANA METZ (0,1%) METZ (0,2%)
QUI 3,5±0,1 3,4±0,2
QUIPAA25 0,695±0,003 0,652±0,005
QUIPAA60 0,098±0,003 0,061±0,003
causando, com isso, uma maior redução da permeabilidade. Os resultados estão também co-
erentes com a hipótese levantada na discussão dos resultados de ganho de massa, de que o
aumento na massa das membranas hidratadas deve estar relacionado à água adsorvida na su-
perfície das mesmas e não distribuídas em seu interior, visto que um aumento na quantidade
de água no interior da membrana deveria levar a um aumento de permeabilidade e não uma
redução, como foi observado experimentalmente.
Com relação à influência da concentração, observa-se uma redução nos valores de perme-
abilidade com o aumento na concentração do fármaco para as três membranas estudadas e,
mais uma vez, percebe-se que essa redução é mais acentuada na membrana tratada a 60◦C.
Aparentemente, esse não era o resultado esperado a partir das figuras 3.14, 3.15 e 3.16. Nes-
sas, observa-se que ocorre sempre um aumento no valor do coeficiente angular das retas com
o aumento da concentração. De acordo com a equação 2.9, o valor da permeabilidade tam-
bém deveria aumentar, mas a mesma equação 2.9 também mostra que a permeabilidade é
inversamente proporcional à concentração, ou seja, à medida que a concentração aumenta, a
permeabilidade deve diminuir. Observa-se, portanto, que nesses experimentos, as variações
de concentrações foram predominantes na determinação dos valores de P, visto que estas va-
riações foram provavelmente maiores do que as variações nos valores de α. Essa situação fica
bastante clara na figura 3.16 obtida para a membrana QUIPAA60 que mostra que os valores
75

3 Resultados e discussão
de α são muito próximos para as duas concentrações usadas, já os valores de permeabili-
dade são significantemente diferentes (causados pelo aumento na concentração). Do ponto de
vista químico, essa diminuição da permeabilidade deve estar relacionada com a saturação da
membrana pelo fármaco, dificultanto, assim, o transporte através desta. Conclui-se, portanto,
que o tratamento superficial apresenta-se como uma alternativa bastante simples e rápida,
mas que produz diferenças significativas nas características de permeação nas membranas de
quitosana, estabelecendo uma camada reticulada ionicamente e provocando uma resistência
adicional além daquelas já oferecidas pela membrana.
76

4 Conclusões
Para este estudo de caracterização de membranas policomplexadas, foram estudadas três
tipos de membrana: a primeira sem a presença do PAA (membrana de quitosana pura), que
serviu como parâmetro de comparação para as demais membranas, a segunda tratada com
PAA na temperatura ambiente e a terceira tratada com PAA a 60◦C . Com base nos resultados
obtidos para cada membrana estudada pode-se concluir que:
1. É possível promover uma reticulação gradual na membrana de quitosana controlando-se
a temperatura da solução de PAA a ser difundido.
2. O processo de difusão do PAA na superfície da membrana de quitosana pura levou a for-
mação de um complexo polieletrolítico como mostram os resultados da espectroscopia
de infravermelho utilizando o acessório de reflectância total atenuada (ATR) com vari-
ação do ângulo de incidência que evidencia o aparecimento de uma banda de absorção
na região de 1560 cm−1 correspondente à formação do complexo.
3. O aspecto macroscópico das membranas policomplexadas como coloração e formato foi
preservado, mas o aspecto morfológico superficial comparando os dois lados da mem-
brana na microscopia eletrônica de varredura foi alterado, obtendo-se uma membrana
assimétrica, isto também pode ser comprovado pela técnica de ATR, que mostrou banda
de absorção na região de 1560 cm−1 apenas no lado tratado da membrana.
4. Avaliando conjuntamente todos os ensaios estudados (FTIR-ATR, análise térmica (TG
77

4 Conclusões
e DTG), ganho de massa e ensaio de permeação para as três membranas estudadas,
conclui-se que a presença de PAA aumentou a capacidade de ganho de massa de água
nas membranas tratadas, e que esse aumento no ganho de massa está relaciondado com
a superfície da membrana, pois, possivelmente, ocorreu uma maior adsorção de água na
camada superficial tratada.
5. Os valores de permeabilidade nas membranas tratadas foram menores do que os da
membrana de quitosana pura, em virtude do aumento da quantidade de interações ocor-
ridas na superfície da membrana.
6. O aumento na concentração do fármaco leva a uma diminuição na permeabilidade das
membranas tratadas possivelmente devido a uma saturação destas.
7. O maior grau de reticulação iônica entre a quitosana e poli(ácido acrílico), ocorreu na
temperatura de 60◦C, uma vez que essa temperatura está acima da temperatura de tran-
sição vítrea da quitosana, o que facilita a difusão do poli(ácido acrílico).
8. As membranas tratadas são promissores materiais para a permeação de fármacos para
períodos longos de tratamento.
78

5 Sugestões para futuras pesquisas
O presente trabalho constitui-se em uma base para futuras pesquisas sobre membranas de
quitosana pura modificadas com poli(ácido acrílico). A investigação mais acurada das pro-
priedades dessa nova membrana leva à necessidade de se aprofundar os estudos relacionados
com a permeação in vitro utilizando-se novos fármacos modelos.
Novas pesquisas deverão ser orientadas no intuito de averiguar a eficiência de ensaio de
permeação in vitro com novos fármacos modelos, tendo em vista a tendência atual da procura
de material biocompatível e biodegradável de liberação controlada, que possa liberar fármacos
em níveis esperados e com reduzida toxicidade.
Deve-se averiguar, também, a influência da temperatura corporal, tempo de reação, massa
molar e concentração do PAA, procurando determinar a possível influência desses parâmetros
nos ensaios de permeação in vitro.
79

Referências Bibliográficas
[1] DAS, N.; DAS, S. Controlled release of oral dosage forms. Pharmaceutical Technology,
v. 27, p. 10–17, 2003.
[2] ANSEL, H.; POLOVICH, N.; JR, L. Farmacotécnica: Formas farmacêuticas e sistemas
de liberação de fármacos. 6. ed. São Paulo: Editorial Premier, 2000.
[3] PRISTA, L.; ALVES, C.; MORGADO, R. Tecnologia farmacêutica e farmácia galênica.
4 ed. Lisboa: Fundação Calouste Gulbenkian, 1996.
[4] SAHLE, A. et al. The stability of heparin-coated lipossomes in plasma and their effect on
its coagulation. Colloids and Surfaces Biointerfaces, v. 10, p. 205–215, 1998.
[5] MUZZARELLI, R. Biochemica significance of exogenous chitins, chitosan in animals
and patients. Carbohydrate Polymers, v. 20, p. 7–16, 1993.
[6] BAKER, R. Controlled release of biologically active agents. New York; Willey Intersci-
ence, p. 279, 1987.
[7] RATHKE, T.; HUDSON, S. Determination of the degree of N-deacetylation in chitin
and chitosan as well as their monomer sugar ratios by near infrared spectroscopy. Polymer
Chemical, v. 31, p. 749–753, 1993.
[8] NUNTHANID, J. et al. Physical properties and molecular behavior of chitosan films.
Drug Delivery Ind. Pharmaceutics, v. 27, n. 2, p. 143–157, 2001.
80

Referências Bibliográficas
[9] DYER, M. et al. Nasal delivery of insulin using novel chitosan based formulations: A
comparative study in two animal models between simple chitosan formulation and chitosan
nanoparticles. Pharm.Research, v. 19, n. 7, p. 50–95, 2002.
[10] MUZZARELLI, R.; JEUNIAUX, C.; GOODOY, G. Chitin in Nature and Technology.
Eds.;Plenum Press; New York, 1986.
[11] SANTOS, J. et al. Caracterização de quitosanas comerciais de diferentes origens. Polí-
meros; Ciência e Tecnologia, v. 13, p. 242–249, 2003.
[12] ZHANG, M. et al. Properties and biocompatibility of chitosan film modified by blending
with PEG. Biomaterials, v. 23, p. 2641–2648, 2002.
[13] CANELLA, K.; GARCIA, R. Caracterização de quitosana por cromatografia de perme-
ação em gel - influência do método de preparação e do solvente. Química Nova, v. 24, p.
13–17, 2001.
[14] MINANI, S. et al. Effects of chitosan on wound healing. Carbohydrate Polymers, v. 2,
p. 141–152, 1992.
[15] ILLUM, L.; FARRAJ, N.; DAVIS, S. Chitosan as a novel nasal delivery sistems for
peptide drugs. Pharm.Research, v. 11, p. 1186–1189, 1994.
[16] ARTURSSON, P. et al. Chitosan on the permeability of monolayers of intestinal epithe-
lial cells(caco-2). Pharm.Research, v. 11, p. 1358–1361, 1994.
[17] PEPPAS, N.; BURI, P. Surface, interfacial and molecular aspects of bioadhesion on soft
tissues. Journal of Controlled Release, v. 2, p. 257–275, 1985.
[18] ROBINSON, J.; MLYNEK, G. Bioadhesive phase-change polymers for ocular drug de-
livery. Advanced Drug Delivery Reviews, v. 16, p. 45–50, 1985.
81

Referências Bibliográficas
[19] THANOU, M.; VERHOEF, J.; JUNGINGER, H. Oral drug absorption enhancement by
chitosan and its derivatives separation. Advanced Drug Delivery Reviews, v. 52, p. 117–126,
2001.
[20] KANKE, M. et al. Application of chitin and chitosan to pharmaceutical preparati-
ons.I.film preparation and in vitro evaluation. Chemical of Pharmaceutical Bulletin, v. 37,
p. 523–525, 1989.
[21] POLK, A. et al. Controlled release of albumina from chitosan-alginate microcapsules.
Journal of Pharmaceutical Science, v. 83, p. 178–185, 1994.
[22] WANG, M. et al. Optimization of the formulation design of chitosan microspheres con-
taining cisplatin. Journal of Pharmaceutical Science, v. 85, p. 1204–1210, 1996.
[23] AçIKGöZ, M. et al. Chitosan microspheres of diclofenac sodium: I. application of fac-
torial design and evaluation of release kinetics. Journal of Microencapsulation, v. 13, p.
141–160, 1996.
[24] LEE, B.; CHOE, J. S.; KIM, C. Preparation and characterization of melatonin-loaded
stearyl alcohol microspheres. Journal of Microencapsulation, v. 15, p. 775–787, 1998.
[25] FELT, O.; BURI, P.; GURNY, R. Chitosan: a unique polysaccharide for drug delivery.
Drug Delivery Ind. Pharmaceutics, v. 24, p. 979–993, 1998.
[26] VERHOEF, J.; LUBBEN, I. Chitosan microparticles for oral vaccinations.Preparation,
characterization and preliminary in vivo uptake studies in murine reyerts patches. Bioma-
terials, v. 22, p. 685–694, 2001.
[27] GENTA, I.; PERUGINI, R.; PAVANETTO, K. Different molecular weight chitosan mi-
crospheres.Influence on drug leading and drug release. Drug Delivery Ind. Pharmaceutics,
v. 24, p. 779–784, 1998.
82

Referências Bibliográficas
[28] OBYA, Y.; HARA, H. N. K.; OUCHI, T. Preparation of PEG-grafted chitosan nanopar-
ticles as peptide drug carriers. STP Pharma Science, v. 10, p. 77–82, 2000.
[29] ROCHA, A. et al. Permeation of drug in chitosan membranes. Journal of Applied Poly-
mer Science, v. 84, p. 44–49, 2002.
[30] MARK, C. Handbook of industrial membrane technology. Westwood; New Jessey, USA,
1990.
[31] MUZZARELLI, R.; PARISER, E. Application of chitin and chitosan in wound-healing
acceleration. Cambridge, Ed.MIT Press, 1978.
[32] DUREJA, H.; TIWARY, A.; GUPTA, S. Simulation of skin permeability in chitosan
membranes. Journal of Controlled Release, v. 213, p. 193–198, 2001.
[33] YANG, T.; ZALL, R. Chitosan membrane for reverse osmose application. Journal Food
Science, p. 49–91, 1984.
[34] ZENG, X. et al. Pervaporation separation of ethanol-water mixtures using cross-linked
chitosan/PAN membrane. Membranes Science Technology, p. 13–29, 1993.
[35] MUSALE, D.; KUMAR, A.; PLEIZIER, G. Formation and characterization of
poly(acrylonitrile)/chitosan composite ultrafiltration membranes. Journal of Membrane
Science, v. 154, p. 163–173, 1999.
[36] BEPPU, M.; SANTANA, C.; ARRUDA, E. Síntese e caracterização de estruturas densas
e porosas de quitosana. Polímeros; Ciência e Tecnologia, 1999.
[37] ALLAN, G. et al. Biomedical application of chitin and chitosan. In: Chitin,Chitosan and
Related Enzymes.Ed. Academic Prees., 1984.
83

Referências Bibliográficas
[38] THACHARODI, D.; RAO, K. Collagen-chitosan composite membranes controlled trans-
dermal delivery of nifedipen and propanolol hydrochloride. International Journal of Phar-
maceutics, v. 134, p. 239–241, 1996.
[39] PATEL, V.; AMIJI, M. Preparation and characterization of freeze-dried chitosan-
poly(ethylene oxide) hidrogels for site especific antibiotic delivery in the stomach.
Pharm.Research, v. 13, p. 588–593, 1996.
[40] MATSUYAMA, H.; SHIRAISHI, H.; KITAMURA, Y. Effect of membrane preparation
conditions on solute permeability in chitosan membrane. Journal of Applied Polymer Sci-
ence, v. 73, p. 2715–2725, 1999.
[41] NETO, C. et al. Permeability studies in chitosan membranes.Effects of crosslinking and
poly(ethylene oxide) addition. Carbohydrate Research, v. 340, p. 2630–2636, 2005.
[42] CHEN, R.; TSAIH, M. Effect of preparation method and characteristics of chitosan on
the mechanical and release properties of the prepared capsule. Journal of Applied Polymer
Science, v. 66, p. 161–169, 1997.
[43] J.W.NEMEC; BAUER, J. In Encyclopedia of Polymer Science and Engineering; Wiley
Interscience, New York, p. 211, 1985.
[44] ALPATOVA, A. et al. Ultrafiltration of water containing natural organic material: heavy
metal removing in the hybrid complexation-ultrafiltration process. Separation and Purifi-
cation Technology, v. 40, p. 155–162, 2004.
[45] KACZMAREK, H.; SZALLA, A. Photochemical transformation in poly(acrylic
acid)/poly (ethylene oxide) complexes. Journal of Photochemistry and Photobiology A.
Chemistry, v. 180, p. 46–53, 2006.
[46] DITTGEN, M.; DURRANI, M.; LEHMANN, K. Acrylic polymers: A review of phar-
maceutical application. STP Pharma Science, v. 7, p. 403–437, 1997.
84

Referências Bibliográficas
[47] LUESSEN, H. et al. Bioadhesive polymers for the peroral delivery of peptide drugs.
Carbohydrate Research, v. 29, p. 329–338, 1994.
[48] NEEDLEMAN, I.; SMALES, F. In vitro assessment of bioadhesion for periodontal and
bucal drug delivery. Biomaterials, v. 16, p. 617–624, 1995.
[49] WU, G.; LIN, S.; YANG, C. Alkaline Zn-air and Al-air cells based on novel solid
PVA/PAA polymer electrolyte membrane. Journal of Membrane Science, v. 280, p. 802–
808, 2006.
[50] TORRADO, S. et al. Chitosan-poly(acrylic acid) polyionic complex: in vivo study to
demonstrate prolonged gastric retention. Biomaterials, v. 25, p. 917–923, 2004.
[51] CHING-LING, P. et al. A pH-sensitive EVAL membrane by blending with PAA. Journal
of Membrane Science, v. 275, p. 89–96, 2006.
[52] SCRANTON A, B. J.; ARONSON C, L. ACS Symposium; In American Chemical So-
ciety, Washington, p. 171–189, 1992.
[53] LL’INA, V.; VARLAMOV, V. Chitosan-based polyelectrolyte complexes. Applied Bio-
chemistry and Microbiology, v. 41, p. 5–11, 2005.
[54] NGE, T. et al. Synthesis and characterization of chitosan/poly(acrylic acid) polyelec-
trolyte complex. Journal of Applied Polymer Science, v. 83, p. 1025–1035, 2002.
[55] DHANUJA, G.; SMITH, B.; SRIDHAR, S. Pervaporation of isopropanol-water mixtures
through polyion complex membranes. Separation and Purification Technology, v. 44, p.
130–138, 2005.
[56] JENG-JYH, S.; HUANG, R. Pervaporation with chitosan membranes II. blend mem-
branes of chitosan and polyacrylic acid and comparison of homogeneous and composite
membrane based on polyelectrolyte complexes of chitosan and polyacrylic acid for the
85

Referências Bibliográficas
separation of ethanol-water mixtures. Journal of Membrane Science, v. 127, p. 185–202,
1997.
[57] PENICHE, C. et al. Self-curing membranes of chitosan/PAA IPNs obtained by radical
polymerization: preparation, characterization and interpolymer complexation. Biomateri-
als, v. 20, p. 1869–1878, 1999.
[58] MALTINTI, S. et al. In vitro cytotoxicity testing of chitosan-containing polyelectrolyte
complexes. Journal Material Science, v. 9, p. 73–76, 1998.
[59] NAM, S. Y.; LEE, Y. M. Pervaporation and properties of chitosan-poly(acrylic acid)
complex membranes. Journal of Membrane Science, v. 135, p. 161–171, 1997.
[60] AHN JAE-SOON.;CHOI, H. K. et al. Release of triamcinolone acetomide from muco-
adhesive polymer composed of chitosan and poly(acrylic acid) in vitro. Biomaterials, v. 23,
p. 1411–1416, 2002.
[61] IWATSUBO, T.; KUSUMOCAHYO, S.; SHINBO, T. Water/ethanol pervaporation per-
formance of asymmetric polyelectrolyte complex membrane constructed by the diffusion
of poly(acrylic acid) in chitosan membrane. Journal of Applied Polymer Science, v. 86, p.
265–271, 2002.
[62] WANG, H. et al. Studies on chitosan and poly(acrylic acid) interpolymer complex. i.
preparation, structure, pH-sensitivity, and salt sensitivity of complex-forming poly(acrylic
acid): chitosan semi-interpenetrating polymer network. Journal of Applied Polymer Sci-
ence, v. 65, p. 1445–1450, 1997.
[63] KRAJEWSKA, B. Diffusional properties of chitosan hydrogel membranes. Journal of
Chemical Technology Biotechnology, v. 76, p. 636–642, 2001.
86

Referências Bibliográficas
[64] SMITHA, B.; SRIDHAR, S.; KHAN, A. A. Polyelectrolyte complexes of chitosan and
poly(acrylic acid) as proton exchange membranes for fuel cells. Macromolecules, v. 37, p.
2233–2239, 2004.
[65] BELL, C.; PEPPAS, N. Modulation of drug permeation through interpolymer complexed
hydrogels for drug delivery applications. Journal of Controlled Release, v. 39, p. 201–207,
1996.
[66] CHAVASIT, V.; KIENZLE-STERZER, C.; TORRES, J. A. Formation and characteriza-
tion of an insoluble polyelectrolyte complex: chitosan-polyacrylic acid. Polymer Bulletin,
v. 19, p. 223–230, 1988.
[67] SHOEMAKER et al. Experiment in physical chemistry. McGraw-Hill, New York, 1981.
[68] YAMASHITA, F.; HASHIDA, M. Mechanistic and empirical modeling of skin permea-
tion of drug. Advanced Drug Delivery Reviews, v. 55, p. 1185–1199, 2005.
[69] REMASCO, M. Fundamentos de transferência de massa. Editora da Unicamp, 2002.
[70] CRANK, J. The Mathematics of Difusion. London; Claredrom Press, 2 ed., 1975.
[71] COMYN, J. Polymer permeability. London: Elsevier Applied Science Publishers LTD,
1985.
[72] THEEUWES, F.; GALE, R.; BAKER, R. Transference: a compreensive parameter go-
verning permeation of solutes through membranes. Journal of Membrane Science, v. 1, p.
3–16, 1976.
[73] LAU, A. et al. Clinical pharmackinetics of metronidazole and other nitroimidazole anti-
infective agents. International Journal of Pharmaceutics, v. 23, p. 328–364, 2005.
[74] GOODMAN, L.; GILMAN, A. As bases farmacológicas da terapêutica. Guanabara Ko-
ogan, Rio de Janeiro, 4 edição, 1973.
87

Referências Bibliográficas
[75] PEREIRA, M. FTIR-ATR studies on polymer/polymer and polymer/liquid interfaces.
Tese (Doutorado)-University of Durham, 1994.
[76] STUART, B.; GEORGE, W.; MCINTYRE, P. Modern infrared spectroscopy. John Wil-
ley and Sons, Chichester, 1996.
[77] HARRICK, N. Internal reflection spectroscopy. New York; USA; John Willey Sons,
1979.
[78] FAHRENFORT, J. Attenuated total reflection-A new principle for the production of use-
ful infrared reflection spectra of organic compounds. Journal Spectrochimica acta, v. 17, p.
698–709, 1961.
[79] WENDLANDT, W. Thermal analysis. A.Wiley, New York, 1986.
[80] HATAKEYAMA, T.; QUINN F, X. Thermal analysis: Fundamentals and applications to
polymer science. John Wiley and Sons Ltd; Chichester, 1995.
[81] LUCAS, E.; SOARES, B.; MONTEIRO, E. Caracterização de polímeros-determinação
de peso molecular e análise térmica. E-papers serviços editoriais LTDA; Rio de Janeiro,
2001.
[82] KESTENBACH, H.; FILHO, W. Microscopia eletrônica:transmissão e varredura. Asso-
ciação Brasileira de Metalurgia e Materiais, 1994.
[83] RINAUDO, M.; MILAS, M.; DUNG, P. Characterization of chitosan. Influence of ionic
strength and degree of acetylation on chain expansion. International Journal of Biological
Macromolecules, v. 15, p. 281–285, 1993.
[84] PHARMACOPEIA, Portuguesa. Volume VII, 2002.
[85] ROBERTS, G. In Chitin Chemistry. Ed.Houndmills; Macmillan Press Ltd, New York, 1
edição, 1992.
88

Referências Bibliográficas
[86] NOSAL, W. et al. Infrared optical properties and AFM of spin-cast chitosan films che-
mically modified with 1,2 epoxy-3-phenoxy-propane. Colloids and Surfaces Biointerfaces,
v. 46, p. 26–31, 2005.
[87] SHIGEMASA, Y. et al. Evaluation of different absorbance ratios from infrared spectros-
copy for analyzing the degree of deacetylation in chitin. International Journal of Biological
Macromolecules, v. 18, p. 237–242, 1996.
[88] LAZARIDOU, A.; BILIADERIS, C. Thermophysical properties of chitosan, chitosan-
starch and chitosan pullulan films near the glass transition. Carbohydrate Polymers, v. 48,
p. 179–190, 2002.
[89] NETO, C. Obtenção de membranas de quitosana modificadas e estudo das suas proprie-
dades térmicas e de permeabilidade. Dissertação (Mestrado)-UFRN, Natal, 2004.
[90] MUKOMA, P.; JOOSTE, B.; VOSLOO, H. Synthesis and characterization of crosslinked
chitosan membranes for application as alternative proton exchange membrane materials in
fuel cells. Journal of Power Sources, v. 136, p. 16–23, 2004.
[91] TSIPOURAS, N.; RIX, C.; BRADY, P. Passage of silver ions through membrane-
mimetic materials, and its relevance to treatment of burn wounds with silver sulfadiazine
cream. Clinical Chemistry, v. 43, p. 290–301, 1997.
[92] WIJMANS, J.; BAKER, R. The solution-diffusion model:a review. Journal of Membrane
Science, v. 107, p. 1–21, 1995.
89





























