Embed Size (px)
Citation preview

Deletion of the NESP55differentially methylatedregion causes loss of maternalGNAS imprints andpseudohypoparathyroidismtype IbMurat Bastepe1, Leopold F Frohlich1, Agnes Linglart1,Hilal S Abu-Zahra1, Katsuyoshi Tojo2, Leanne M Ward3 &Harald Juppner1,4
Epigenetic defects in the imprinted GNAS cluster areassociated with pseudohypoparathyroidism type Ib. In twokindreds with this disorder, we now report deletions thatremove the differentially methylated region encompassing exonNESP55 and exons 3 and 4 of the antisense transcript. When
inherited from a female, either deletion abolishes all maternalGNAS imprints and derepresses maternally silenced transcripts,suggesting that the deleted region contains a cis-acting elementthat controls imprinting of the maternal GNAS allele.
The GNAS locus (and its mouse homolog) encodes the alphasubunit of the heterotrimeric stimulatory G protein (Gsa), as wellas NESP55, a chromogranin-like neuroendocrine secretory protein,and XLas, a large Gsa variant (Fig. 1a). The Gsa transcript isbiallelically expressed in most tissues, whereas the NESP55 andXLas transcripts are expressed from the maternal and the paternalGNAS allele, respectively1–3. In addition, the GNAS locus yields twopaternally derived, noncoding RNAs: the A/B transcript (also called1A; ref. 4), and the antisense (AS) transcript5 (called Nespas inmice6). The promoter of the Gsa transcript is not differentiallymethylated, although transcription of paternal Gsa is silenced insome tissues (e.g., renal proximal tubules)7. In contrast, promotersof the imprinted GNAS transcripts are located in individual differen-tially methylated regions (DMRs). Ablation of the exon A/B DMR inmice identified a cis-acting element that controls tissue-specific
c
a
b
XL
f
NESP55A/B
2–13
7.318.3
Gsα-1MatPat
DMR
g
3.2 10.7
Kindred Y2P1
P2425
1,561
1,191
EcoRV
15.3
3.2
15.310.7 10.7
EcoRV
PCR
Kindred C
1,191
539
1,0871,561
BamHI
7.3
5.0
7.718.3 7.0
P1
P2
BamHI
BBB B B
PCR
SNP AL121917-21574P1 P2
WT (a/b)WT (c/d)
Mut (a/d)
WT (a/b)
WT (c/d)
Mut (a/e)
Mut (f/d)
5.0 7.018.3
a c
3.2 15.3
E E
WT
Mut
P1 P2
WT
BB B
Mut
P1 P2 P1 P2
AS-1
~5 kb
2345
XL
XL XL
XL
DMR DMRDMR
+–
–+
–+
–+
7.7
E
E EE
B
b d a
a d a e f d
cb d
U A U A U A U A U A U A
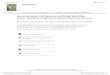
Figure 1 GNAS and deletion of the NESP55
DMR in AD-PHP-Ib. (a) Exons and introns are
depicted by boxes (black, sense; gray, antisense)
and connecting lines, respectively. mat, maternal;
pat, paternal; +, methylated; –, nonmethylated.Arrows indicate direction (rightward, sense;
leftward, antisense) and allelic origin (upward,
maternal; downward, paternal) of transcription.
Primers f and g (arrowheads) were used to
genotype for SNP AL121917-21574 near the
end of the duplicated region (white rectangle;
nucleotides 6,703–21,662 of AL121917;
double arrows indicate orientation). (b) The
deletion in kindred Y2 located between
nucleotides 105,010 and 109,696 of
AL132655 (between vertical lines) was detected
by Southern-blot analysis using EcoRV (E) and
probes P1 and P2 (horizontal bars) and by PCR
using primers a, b, c and d (arrowheads). (c) The
deletion in kindred C located between
nucleotides 105,410 and 109,440 of
AL132655 (between vertical lines) was
detected by Southern-blot analysis using BamHI
(B), and the rearrangement detected by PCRusing primers a, b, c, d, e and f (arrowheads).
U, unaffected; A, affected; WT, wild-type;
Mut, mutant.
Published online 12 December 2004; doi:10.1038/ng1487
1Endocrine Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA. 2Division of Diabetesand Endocrinology, Department of Internal Medicine, Jikei University School of Medicine, Tokyo, Japan. 3Division of Endocrinology and Metabolism,Children’s Hospital of Eastern Ontario, Ottawa, Ontario, Canada. 4Pediatric Nephrology Unit, MassGeneral Hospital for Children, Massachusetts GeneralHospital and Harvard Medical School, Boston, Massachusetts, USA. Correspondence should be addressed to M.B. ([email protected])or H.J. ([email protected]).
NATURE GENETICS VOLUME 37 [ NUMBER 1 [ JANUARY 2005 2 5
B R I E F C O M M U N I C AT I O N S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
egen
etic
s

imprinting of Gsa without effects on the imprinting of other tran-scripts8. Individual roles of the DMRs for NESP55, AS and XLas inregulation of GNAS imprinting have not yet been determined inhumans and mice.
Most individuals with the autosomal dominant form of pseudo-hypoparathyroidism type Ib (AD-PHP-Ib; OMIM 603233), a disorderof renal parathyroid hormone (PTH) resistance9, have loss ofGNAS methylation at the exon A/B DMR alone10,11. Nearly all suchindividuals carry an identical maternally inherited 3-kb deletion at aclosely linked, apparently nonimprinted gene, STX16, thus identifyingthe location of a putative cis-acting element required for establishmentor maintenance of methylation at exon A/B12. In contrast, manysporadic cases of pseudohypoparathyroidism type Ib and a few casesof AD-PHP-Ib lack evidence for STX16 mutations or deletions.They have, however, epigenetic abnormalities at all GNAS DMRs12,suggesting that the disease-causing mutation in these individualsinvolves another cis-acting element that is required for imprinting ofthe entire GNAS locus.
To locate this element, we searched for GNAS mutations intwo unrelated kindreds with AD-PHP-Ib (Y2 and C) who lackedSTX16 mutations or deletions and in whom the disease waslinked to 20q13.3 (Supplementary Fig. 1 online). Fine-mappingof the GNAS locus showed, in affected individuals and unaffectedcarriers of both kindreds, allelic loss of several single-nucleotidepolymorphisms (SNPs) in the exon NESP55 region. Southern-blotanalyses identified for kindred Y2, a heterozygous 4.7-kb deletionremoving the entire NESP55 DMR including AS exons 3 and 4(Fig. 1b). Analyses in kindred C indicated a heterozygous 4-kbdeletion at a similar location, which was complicated by genomicduplication (Fig. 1c). Genome-walking and Southern-blot analysesidentified, in place of the 4-kb deleted region, an insertion of B15 kbduplicated from the region between exons XL and A/B. In its
native location, the latter region appeared intact, as determined bySouthern blotting (data not shown) and analysis of SNP AL121917-21574 (Fig. 1a), which was heterozygous in several carriers whengenotyped by PCR using an outside reverse primer (SupplementaryFig. 1 online).
DNA from affected individuals, who inherited the deletions mater-nally, showed loss of methylation at all neighboring DMRs that arenormally methylated on the maternal allele. In contrast, DNA fromunaffected carriers, who inherited the deletions paternally, or fromhealthy family members showed no epigenetic alterations of theseregions (Fig. 2a and Supplementary Fig. 1 online). Affected indivi-duals and unaffected carriers seemed to have either gained or lostmethylation at the NESP55 DMR, respectively, owing to the deletion.RT-PCR using total RNA from lymphoblastoid cells of an affectedindividual (individual C/II-1) amplified both paternally derived wild-type and maternally derived mutant AS transcripts (Fig. 2b). Similarto the finding in an individual with paternal uniparental isodisomy of20q (patUPD20q; ref. 13), real-time RT-PCR detected roughly twotimes more A/B mRNA in these cells than in cells from a normalcontrol (Fig. 2c). Differential methylation of NNAT, a distinctimprinted gene centromeric of GNAS, was unaltered in DNA fromindividuals carrying the deletion, unlike in patUPD20q (Fig. 2d).Therefore, maternal inheritance of the NESP55 DMR deletion causedloss of imprinting in cis at the entire GNAS locus without affectingdistant imprinted sites.
The identified deletion, when inherited maternally, is predicted toresult in a complete lack of NESP55 protein and, due to derepressionof the maternal XLas promoter, two times more XLas14. Thesechanges, however, do not seem to contribute to the development ofPTH resistance. Instead, PTH resistance probably results from a lackor reduction of Gsa in renal proximal tubules, which seems to bedue to loss of exon A/B methylation8,9. The latter can occur either
~0.5 kb
db
AS Exon 5 Exon 4
Mutant
Wild-type
C-II/1 Y2-II/1
NormalpatUPD20q
a2–13
XL
NESP55
DMR
5 4 3 2
DMR
A/BGsα-1
DMR
AS-1
ASpromoter
A/B
3NNAT
(bp)
200
100
Norm
al
C-II/1
500
No deletion (C-III/11) Maternal (C-II/1) Maternal (Y2-II/1)Paternal (C-III/13)
c
XL
~1 kb
0
2
4
6
8
10
12
14
**
A/B
mR
NA
(%
of G
sα)
Norm
al
patU
PD20q
Affecte
d (C
-II/1
)
Mar
ker
DMR
*AS Exon 5 Exon 2
– + – + RT
1 2
Figure 2 Imprinting analysis of GNAS and NNAT.
(a) The GNAS locus and its methylation analysis.
Deleted region is in brackets. Dotted lines, splice
patterns of AS transcripts; arrowheads, primers
for AS RT-PCR. PCR products (white rectangles)
from bisulfite-treated DNA were cloned and
sequenced. Each row of circles represents a
clone (black circles, methylated CpG; white
circles, nonmethylated CpG). (b) RT-PCR showing
expression of both wild-type and mutant AS
transcripts in lymphoblastoid cells from C-II/1.
Asterisk, amplicon from a previously undescribed
splice variant (Supplementary Fig. 2 online).
Direct sequencing of the additional short
amplicon identified no sequences from exons 3and 4. (c) Real-time RT-PCR showing the level
of A/B mRNA relative to Gsa mRNA (mean 7s.e.m. of four independent experiments; asterisk,
P o 0.05 compared to normal). (d) The NNAT
locus, its CpG islands (gray rectangles) and
regions analyzed for differential methylation by
sequencing bisulfite-modified genomic DNA
(white rectangles). Sequence traces for
informative CpGs in the DMR comprising exon 1
are shown in comparison to those in an individual
with patUPD20q (ref. 13). Analysis of the region
further upstream, done by both direct sequencing
and digestion of the PCR product with FauI,
HhaII or BstUI (not shown), verified these results.
Primer information and conditions are available
on request.
2 6 VOLUME 37 [ NUMBER 1 [ JANUARY 2005 NATURE GENETICS
B R I E F C O M M U N I C AT I O N S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
egen
etic
s

alone, caused by the 3-kb STX16 deletion12, or in combinationwith methylation defects of other DMRs, caused by the deletionreported here.
Our findings indicate that the NESP55 DMR contains a cis-actingelement essential for imprinting of the maternal GNAS allele. TheNESP55 DMR, unlike the exon A/B DMR and the DMR encompass-ing the AS promoter4,15, is established during postimplantation inmice. The identified element may, therefore, be involved in main-tenance rather than establishment of imprinting at the maternal allele.It might bind a chromatin insulator or control imprinting by allowingthe methylation-sensitive binding of a trans-acting factor, which mayinteract, directly or indirectly, with DNA methyltransferases. Consis-tent with these hypotheses, several regions of strong DNase hypersen-sitivity have been identified in the mouse Nesp55 DMR15, and thereare at least two putative binding sites for the CTCF insulator proteinin the NESP55 and Nesp55 promoters. Additional investigations arerequired to understand the mechanisms governing the actions of thiscontrol element.
This study was approved by the Subcommittee on Human Studiesof Massachusetts General Hospital (No. 92-7338). Informed consentwas obtained from all human subjects.
Note: Supplementary information is available on the Nature Genetics website.
ACKNOWLEDGMENTSWe thank H.M. Kronenberg and J.T. Lee for critically reviewing themanuscript. This work was funded, in part, by separate grants fromNational Institute of Diabetes and Digestive and Kidney Diseases(to M.B. and H.J.).
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Received 26 October; accepted 19 November 2004
Published online at http://www.nature.com/naturegenetics/
1. Hayward, B. et al. Proc. Natl. Acad. Sci. USA 95, 10038–10043 (1998).2. Hayward, B.E., Moran, V., Strain, L. & Bonthron, D.T. Proc. Natl. Acad. Sci. USA 95,
15475–15480 (1998).3. Peters, J. et al. Proc. Natl. Acad. Sci. USA 96, 3830–3835 (1999).4. Liu, J., Yu, S., Litman, D., Chen, W. & Weinstein, L. Mol. Cell. Biol. 20, 5808–5817
(2000).5. Hayward, B. & Bonthron, D. Hum. Mol. Genet. 9, 835–841 (2000).6. Wroe, S.F. et al. Proc. Natl. Acad. Sci. USA 97, 3342–3346 (2000).7. Yu, S. et al. Proc. Natl. Acad. Sci. USA 95, 8715–8720 (1998).8. Williamson, C.M. et al. Nat Genet. 36, 894–899 (2004).9. Spiegel, A.M. & Weinstein, L.S. Annu. Rev. Med. 55, 27–39 (2004).10. Liu, J. et al. J Clin. Invest. 106, 1167–1174 (2000).11. Bastepe, M. et al. Hum. Mol. Genet. 10, 1231–1241 (2001).12. Bastepe, M. et al. J Clin. Invest. 112, 1255–1263 (2003).13. Bastepe, M., Lane, A.H. & Juppner, H. Am J. Hum. Genet. 68, 1283–1289 (2001).14. Plagge, A. et al. Nat. Genet. 36, 818–826 (2004).15. Coombes, C. et al. Mol Cell. Biol. 23, 5475–5488 (2003).
NATURE GENETICS VOLUME 37 [ NUMBER 1 [ JANUARY 2005 2 7
B R I E F C O M M U N I C AT I O N S©
2005
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
egen
etic
s