Embed Size (px)
Citation preview

This content has been downloaded from IOPscience. Please scroll down to see the full text.
Download details:
IP Address: 141.219.152.23
This content was downloaded on 13/04/2016 at 20:04
Please note that terms and conditions apply.
Degradation of phosphorene in air: understanding at atomic level
View the table of contents for this issue, or go to the journal homepage for more
2016 2D Mater. 3 025011
(http://iopscience.iop.org/2053-1583/3/2/025011)
Home Search Collections Journals About Contact us My IOPscience

2DMater. 3 (2016) 025011 doi:10.1088/2053-1583/3/2/025011
PAPER
Degradation of phosphorene in air: understanding at atomic level
GaoxueWang1,William J Slough1, Ravindra Pandey1 and Shashi PKarna2
1 Department of Physics,Michigan Technological University, Houghton,MI 49931,USA2 USArmyResearch Laboratory,Weapons andMaterials ResearchDirectorate, ATTN: RDRL-WM,Aberdeen ProvingGround,MD
21005-5069, USA
E-mail: [email protected]
Keywords: phosphorene, degradation, density functional theory,first principlesmolecular dynamics
Supplementarymaterial for this article is available online
AbstractPhosphorene is a promising two-dimensional (2D)material with a direct band gap, high carriermobility, and anisotropic electronic properties. Phosphorene-based electronic devices, however, arefound to degrade upon exposure to air. In this paper, we provide an atomic level understanding of thestability of phosphorene in terms of its interactionwithO2 andH2O. The results based on densityfunctional theory together with first principlesmolecular dynamics calculations show thatO2 couldthe spontaneously dissociate on phosphorene at room temperature. H2Owill not strongly interactwith pristine phosphorene, however, an exothermic reaction could occur if phosphorene isfirstoxidized. The pathway of oxidationfirst, followed by exothermic reactionwithwater is themost likelyroute for the chemical degradation of phosphorene-based devices in air.
1. Introduction
Phosphorene is one of the group V elemental mono-layers [1–4] with a direct band gap, high carriermobility, and anisotropic electronic properties, mak-ing it a promising candidate for applications inelectronics and optoelectronics [5, 6]. The chemicaldegradation of phosphorene upon exposure to ambi-ent conditions, however, is a challenge to the stabilityand performance of phosphorene-based devices [7–11]. The presence of oxygen and humidity is suggestedto be the main cause of the degradation process [12–14]. Recent experiments have also demonstratedphoto-assisted degradation of phosphorene [15],which is predicted to be related to intrinsic defects[16]. Theoretically, it was reported that H2O adsorbedon phosphorene will induce a significant distortion toits structure [8]. Contradictory results were obtainedsuggesting that phosphorene is stable in the presenceof H2O [17]. Despite these experimental and theor-etical efforts, there are still some unanswered ques-tions regarding the degradation of phosphorene,including (i) atomic level of understanding on thedegradation process of phosphorene; (ii) the role ofH2O in the degradation process; and (3) the
environmental stability of other theoretically pre-dicted phosphorene allotropes (e.g., blue phosphorene[18]which has not been realized in experiments).
In order to address these questions, we have per-formed density functional theory (DFT) calculationscombined with first principles molecular dynamics(MD) simulations to investigate the interactions of O2
and H2O with phosphorene. In section 3.1, we focuson the interaction of O2 with phosphorene. Since sur-face reactionwithO2 has been reported to be crucial inthe degradation process of black phosphorene [14], wewill extend the discussion to blue phosphorene. Insection 3.2, the adsorption of H2O on phosphoreneallotropes is investigated in terms of adsorption con-figuration, binding energy, and bonding character-istics. In section 3.3, we discuss the degradation ofphosphorene by calculating the relative energies alonga likely interaction pathway. Our calculated resultsshow that O2 can spontaneously dissociate on phos-phorene at room temperature. H2O will not stronglyinteract with pristine phosphorene, however, an exo-thermic reaction could occur if phosphorene is firstoxidized. Other allotropes of phosphorene, e.g. bluephosphorene are also expected to deteriorate in air.
RECEIVED
2 September 2015
REVISED
18 February 2016
ACCEPTED FOR PUBLICATION
14March 2016
PUBLISHED
13April 2016
© 2016 IOPPublishing Ltd

2. Computational details
The electronic structure calculations were performedusing the Vienna ab initio simulation package [19, 20].The exchange-correlation was treated within thegeneralized gradient approximation (GGA) usingPerdew−Burke−Ernzerhof (PBE) [21] functional forthe calculations. We also employed the DFT-D2method of Grimme [22] to include contributions fromthe van der Waals (vdW) interactions. The energy ofconvergence was set to 1×10−6 eV and the residualforce on each atom was smaller than 0.01 eV Å−1
during the structural optimization. The cutoff energyfor the plane-wave basiswas set to 500 eV. The vacuumdistance normal to the plane was larger than 30 Å toeliminate interaction between the periodic replicas. Arectangular supercell of (3×4)was used for the blackphosphorene, and a parallelogram supercell of (4×4)was used for the blue phosphorene. The reciprocalspace was sampled by a grid of (2×2×1) k points inthe Brillouin Zone.
First principles MD simulations were also per-formed to simulate the interaction processes con-sidered. The MD simulation was based on the norm-conserving Troullier-Martins pseudopotential toge-ther with Nose ́ thermostat [23] as implemented in theSIESTA program package [24]. In order to minimizethe constraints induced by periodicity in the slabmodel for MD simulations, (5×6) and (5×5)supercells were used for black and blue phosphorene,respectively. The time step was set to 1 fs, and thesimulation temperature was set to 300 K. It is to benoted that most of the experiments on degradation ofphosphorene were done in the air. In our MD simula-tions, the number density of gas molecules was about65.8×1025 m–3 considering 9 O2 molecules in asimulation cell of (22.9 Å×19.9 Å×30 Å). Suchhigh pressure conditions were also used for MD simu-lations to study oxidation of SiC [25] and gra-phene [26].
3. Results and discussion
Black phosphorene has a puckered surface with twosub-layers of phosphorus atoms which are arranged ina rectangular lattice. At GGA-PBE level of theory, thelattice constants along the armchair and the zigzagdirection are 4.57 Å and 3.31 Å, respectively. The bondlengths are 2.22 Å and 2.25 Å. Blue phosphorene has abuckled honeycomb structure with lattice constant of3.28 Å and bond length of 2.26 Å. Our results are inagreement with the reported lattice constants andbond lengths of black and blue phosphorene [2, 18],thereby, demonstrating the reliability of the modelingelements used in the calculations.
3.1.O2 interactingwith phosphoreneIt has been established by both calculations andexperiments that O2 can easily dissociate on blackphosphorene [14, 27] leading to the formation of theoxidized lattice [28]. The O2 molecule prefers aperpendicular configuration to approach the surfaceof phosphorene with a distance of 2.5 Å as seen fromthe binding energy profile (figure S1 in the supplemen-tary materials). Thereafter, O2 tends to dissociate onthe surface with exothermic energy (ΔQ) of−4.07 eV/O2 molecule on black phosphorene, and−4.02 eV/O2 molecule on blue phosphorene(figure 1). The details of calculations forΔQ are givenin table S1 in the supplementary materials. Note thatthe dissociation energy is calculated using the tripletstate ofO2.
The calculated dissociation barrier of O2 on phos-phorene is 0.54 eV [14]. Considering that chemicalreaction with an energy barrier less than 0.9 eV(≈21 kcal mol−1) from DFT calculations could occurat room temperature [29], oxidation of phosphoreneis expected to occur readily at room temperature.Other external sources such as photon radiation mayspeed up the dissociation process. After dissociation,atomic oxygen finds the dangling position to be the
Figure 1.O2dissociation on phosphorene: (a) black phosphorene, (b) blue phosphroene. P(2O) represents black or blue phosphorenewith twoOadatoms.
2
2DMater. 3 (2016) 025011 GWang et al

preferred site on black phosphorene which is con-sistent with previous theoretical studies [14, 28, 30].This is not the case with blue phosphorene where thepreferred site is the top site (see figure S2 in the supple-mentary materials). The P–O bond shows similarbonding character as seen from the bond length andBader charges [31] given in table 1. Overall, the natureof interaction of oxygenwith phosphorene stems fromthe sp3 bonds which leave a lone electron pair on eachphosphorous atom, and the preferred binding site fol-lows the direction of the lone electron pair on bothallotropes.
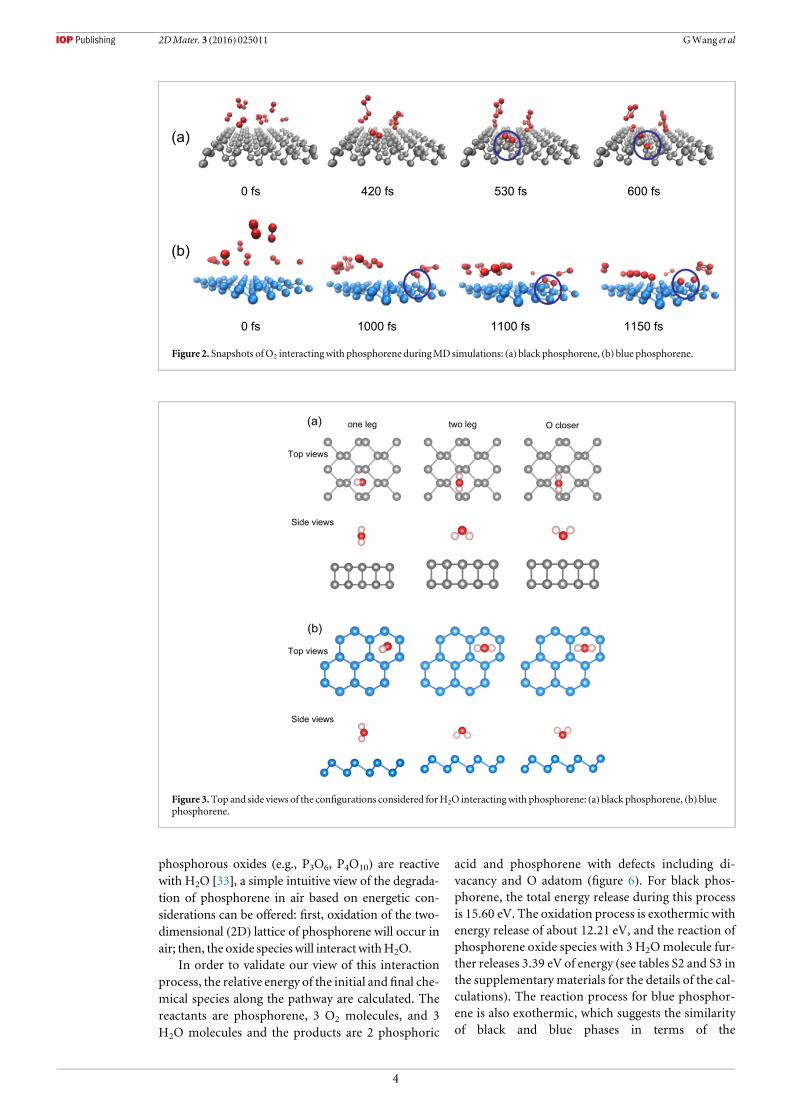
The calculated results based on first principlesMDsimulations further affirm the dissociation of O2 onphosphorene. Figure 2 shows time-dependent snap-shots of the configurations showing interaction ofoxygen with phosphorene during MD simulations.These configurations were obtained by placing a fewO2 molecules 4 Å initially above the surface at a con-stant temperature of 300 K. For the case of black phos-phorene, some O2 molecules will first move closer tothe native phosphorus atoms, then dissociate intoatomic oxygen atoms after 600 fs (see video V1 in thesupplementary materials). A similar O2 dissociationprocess is seen on the blue phosphorene after 1150 fs(see videoV2 in the supplementarymaterials).
Considering the relatively high pressure condi-tions in ourMD simulations, we have performed addi-tional MD calculations with only one O2 molecule inthe simulation box of (22.9 Å× 19.9 Å× 85 Å)whichappears to mimic number density of gas molecules of2.5×1025 m–3 under standard atmospheric condi-tions. The results shown in figure S3, video V3 andvideo V4 (supplementary materials) find that dissocia-tion of O2 on the surface does occur under the rela-tively reduced pressure conditions. We may thereforeconclude that both allotropes of phosphorene will gothrough the spontaneous oxidation process uponexposure toO2 at room temperature due to the affinityof P and O atoms forming P=O bonds with a largeexothermic energy (seefigure 1).
3.2.H2O interactingwith phosphoreneFigure 3 shows the configurations of H2O interactingwith phosphorene considered for the calculations: oneleg, two leg, and O closer. The configuration referredto as ‘one leg’ is the configuration in which one of theH atoms is closer to the surface, ‘two leg’ means both
H atoms are closer to the surface, and ‘O closer’meanstheO atom is closer to the surface.
The calculated binding energy profiles with vdWcorrection using DFT-D2 method of Grimme [22] areshown infigure 4. Some of the results deduced are:
(i) The ‘two leg’ configuration is the most stableconfiguration suggesting that H atoms prefer tomove towards the surface. This is due to the well-known polar nature of the H2O molecule inwhich H atoms tend to attract the lone electronpairs of phosphorene.
(ii) The calculated binding energy which includesvdW correction term is about 180 and 125 meVfor H2O on the black and blue phosphorene,respectively. It is larger than that of H2O ongraphene at the same level of theory (in the rangefrom 60 to 120 meV [32]), mainly because of thepresence of the lone electron pairs on phosphor-ene. The attraction betweenH atoms and the loneelectron pairs of phosphorene is clearly demon-strated by the deformation charge density plots(see figure S4 in the supplementary materials)showing the increase in the electron density inthe region betweenH and P atoms.
In order to further examine the interaction of H2Owith phosphorene, we considered the initial config-uration to consist of a ‘forced’ H2O molecule at theinterstitial site of the phosphorene lattice. If H2O pre-fers to interact strongly with phosphorene, then theoptimized configuration should show that H and Oatoms remain in the lattice. This is not the case as H2Omoves out of the lattice to a surface site (figure S5 inthe supplementary materials) without distorting thesurface for both allotropes. Our first principles MDsimulations up to 10 ps also find that H2O moleculesstay near the phosphorene surface without any chemi-cal interaction within 10 ps (see video V5 in the sup-plementary materials). Therefore, H2O prefers to bindto the surface through hydrogen bonds.
3.3. Stability of phosphorene in air : exposure toO2
andH2OBased on the calculated results given in sections 3.1and 3.2, we can state that the O2 molecule couldspontaneously dissociate on phosphorene at roomtemperature, and the H2O molecule will not interactstrongly with the pristine phosphorene.
The dissociation of H2O on pristine black and bluephosphorene is endothermic with an energy increaseof 1.24 eV and 1.37 eV, respectively (figure 5). This isnot the case with the oxidized phosphorene mono-layers for which the endothermic energy significantlydecreases to 0.26 eV and 0.48 eV, respectively. There-fore, oxidation of phosphorene may enable dissocia-tion of H2O on the surface. Also considering that the
Table 1. Structural properties of atomicO adsorbed onphosphorene.
Phosphorene Black Blue
Bond lengthRP–O (Å) 1.50 Å 1.50 Å
Bond angle∠P–P–O (°) 112°, 117° 123°Bader charge, oxygen −1.31e −1.32e
3
2DMater. 3 (2016) 025011 GWang et al
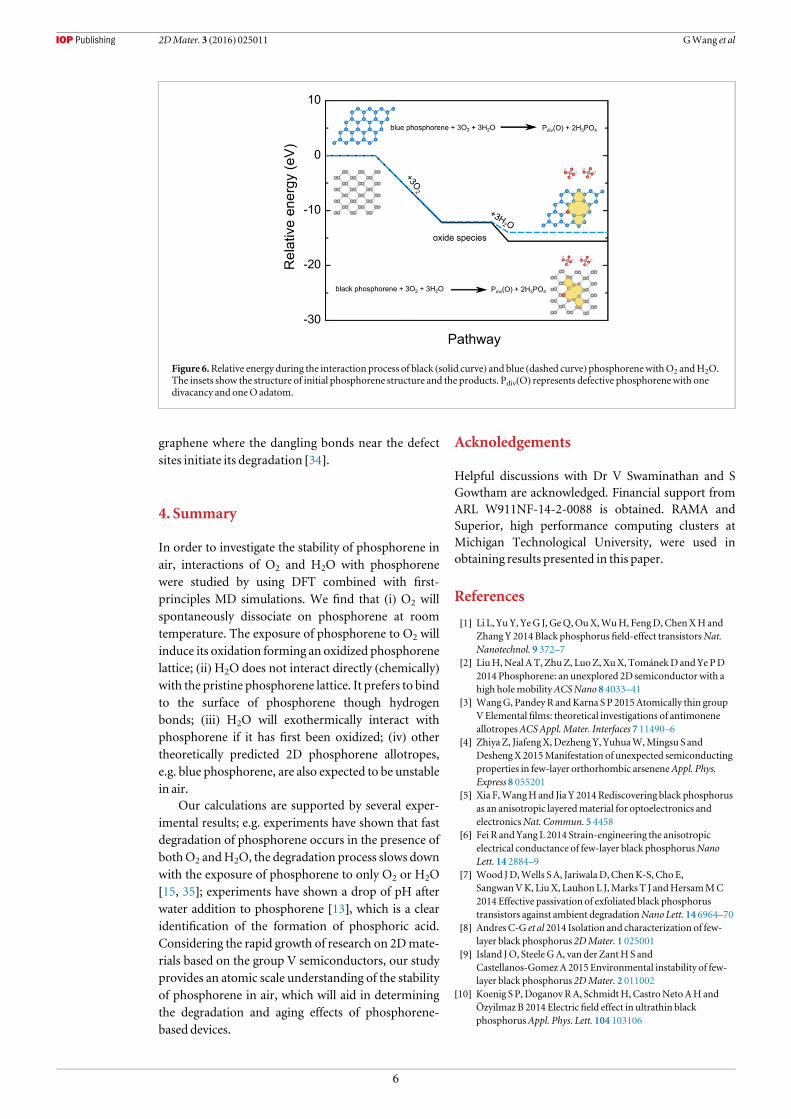
phosphorous oxides (e.g., P3O6, P4O10) are reactivewith H2O [33], a simple intuitive view of the degrada-tion of phosphorene in air based on energetic con-siderations can be offered: first, oxidation of the two-dimensional (2D) lattice of phosphorene will occur inair; then, the oxide species will interact withH2O.
In order to validate our view of this interactionprocess, the relative energy of the initial and final che-mical species along the pathway are calculated. Thereactants are phosphorene, 3 O2 molecules, and 3H2O molecules and the products are 2 phosphoric
acid and phosphorene with defects including di-vacancy and O adatom (figure 6). For black phos-phorene, the total energy release during this processis 15.60 eV. The oxidation process is exothermic withenergy release of about 12.21 eV, and the reaction ofphosphorene oxide species with 3 H2Omolecule fur-ther releases 3.39 eV of energy (see tables S2 and S3 inthe supplementary materials for the details of the cal-culations). The reaction process for blue phosphor-ene is also exothermic, which suggests the similarityof black and blue phases in terms of the
Figure 2. Snapshots ofO2 interactingwith phosphorene duringMD simulations: (a) black phosphorene, (b) blue phosphorene.
Figure 3.Top and side views of the configurations considered forH2O interactingwith phosphorene: (a) black phosphorene, (b) bluephosphorene.
4
2DMater. 3 (2016) 025011 GWang et al

environmental stability. Overall, the exothermic pro-cess implies that H2O will react with phosphorene ifit is oxidized on the surface. The proposed pathwaywill lead to the formation of phosphoric acid anddefective phosphorene. The defective phosphorene
could further be photo-oxidized [16], and then theoxide species will further react with H2O. This reac-tion circle results in the fast degradation of phos-phorene in air. The predicted mechanism forphosphorene is therefore different from the case of
Figure 4.The calculated binding energy profiles of aH2Omolecule approaching phosphorene: (a) black phosphorene, (b) bluephosphorene.
Figure 5.H2Odissocation on pristine and oxide phosphorene: (a) black phosphorene, (b) blue phosphorene. P(OH,O) representsblack or blue phosphorene withOHgroup andOadatom.
5
2DMater. 3 (2016) 025011 GWang et al
graphene where the dangling bonds near the defectsites initiate its degradation [34].
4. Summary
In order to investigate the stability of phosphorene inair, interactions of O2 and H2O with phosphorenewere studied by using DFT combined with first-principles MD simulations. We find that (i) O2 willspontaneously dissociate on phosphorene at roomtemperature. The exposure of phosphorene to O2 willinduce its oxidation forming an oxidized phosphorenelattice; (ii)H2O does not interact directly (chemically)with the pristine phosphorene lattice. It prefers to bindto the surface of phosphorene though hydrogenbonds; (iii) H2O will exothermically interact withphosphorene if it has first been oxidized; (iv) othertheoretically predicted 2D phosphorene allotropes,e.g. blue phosphorene, are also expected to be unstablein air.
Our calculations are supported by several exper-imental results; e.g. experiments have shown that fastdegradation of phosphorene occurs in the presence ofbothO2 andH2O, the degradation process slows downwith the exposure of phosphorene to only O2 or H2O[15, 35]; experiments have shown a drop of pH afterwater addition to phosphorene [13], which is a clearidentification of the formation of phosphoric acid.Considering the rapid growth of research on 2Dmate-rials based on the group V semiconductors, our studyprovides an atomic scale understanding of the stabilityof phosphorene in air, which will aid in determiningthe degradation and aging effects of phosphorene-based devices.
Acknoledgements
Helpful discussions with Dr V Swaminathan and SGowtham are acknowledged. Financial support fromARL W911NF-14-2-0088 is obtained. RAMA andSuperior, high performance computing clusters atMichigan Technological University, were used inobtaining results presented in this paper.
References
[1] Li L, YuY, YeG J, GeQ,OuX,WuH, FengD,ChenXH andZhangY 2014 Black phosphorus field-effect transistorsNat.Nanotechnol. 9 372–7
[2] LiuH,Neal AT, ZhuZ, LuoZ, XuX, TománekD andYe PD2014 Phosphorene: an unexplored 2D semiconductor with ahigh holemobilityACSNano 8 4033–41
[3] WangG, Pandey R andKarna S P 2015Atomically thin groupVElemental films: theoretical investigations of antimoneneallotropesACSAppl.Mater. Interfaces 7 11490–6
[4] Zhiya Z, JiafengX,Dezheng Y, YuhuaW,Mingsu S andDeshengX 2015Manifestation of unexpected semiconductingproperties in few-layer orthorhombic arseneneAppl. Phys.Express 8 055201
[5] Xia F,WangHand Jia Y 2014Rediscovering black phosphorusas an anisotropic layeredmaterial for optoelectronics andelectronicsNat. Commun. 5 4458
[6] Fei R andYang L 2014 Strain-engineering the anisotropicelectrical conductance of few-layer black phosphorusNanoLett. 14 2884–9
[7] Wood JD,Wells S A, JariwalaD, ChenK-S, ChoE,SangwanVK, LiuX, Lauhon L J,Marks T J andHersamMC2014 Effective passivation of exfoliated black phosphorustransistors against ambient degradationNano Lett. 14 6964–70
[8] AndresC-G et al 2014 Isolation and characterization of few-layer black phosphorus 2DMater. 1 025001
[9] Island JO, Steele GA, van der ZantH S andCastellanos-GomezA 2015 Environmental instability of few-layer black phosphorus 2DMater. 2 011002
[10] Koenig S P,DoganovRA, SchmidtH, CastroNetoAH andÖzyilmaz B 2014 Electricfield effect in ultrathin blackphosphorusAppl. Phys. Lett. 104 103106
Figure 6.Relative energy during the interaction process of black (solid curve) and blue (dashed curve) phosphorenewithO2 andH2O.The insets show the structure of initial phosphorene structure and the products. Pdiv(O) represents defective phosphorenewith onedivacancy and oneO adatom.
6
2DMater. 3 (2016) 025011 GWang et al

[11] BuscemaM,GroenendijkD J, Blanter S I, Steele GA,van der ZantH S J andCastellanos-GomezA 2014 Fast andbroadband photoresponse of few-layer black phosphorusfield-effect transistorsNano Lett. 14 3347–52
[12] Yau S-L,Moffat T P, BardA J, Zhang Z and LernerMM1992STMof the (010) surface of orthorhombic phosphorusChem.Phys. Lett. 198 383–8
[13] HanlonD et al 2015 Liquid exfoliation of solvent-stabilizedfew-layer black phosphorus for applications beyondelectronicsNat. Commun. 6 8563
[14] Ziletti A, CarvalhoA,Campbell DK, CokerDF andCastroNetoAH2015Oxygen defects in phosphorene Phys.Rev. Lett. 114 046801
[15] FavronA,Gaufres E, Fossard F, Phaneuf-Lheureux A-L,TangNYW, Levesque P L, LoiseauA, Leonelli R,Francoeur S andMartel R 2015 Photooxidation and quantumconfinement effects in exfoliated black phosphorusNat.Mater. 14 826–32
[16] UttK L, Rivero P,MehboudiM,Harriss EO, BorundaMF,Pacheco SanJuanAA andBarraza-Lopez S 2015 Intrinsicdefects,fluctuations of the local shape, and the photo-oxidation of black phosphorusACSCent. Sci. 1 320–7
[17] Sa B, Li Y-L,Qi J, Ahuja R and SunZ 2014 Strain engineeringfor phosphorene: the potential application as a photocatalystJ. Phys. Chem.C 118 26560–8
[18] ZhuZ andTománekD 2014 Semiconducting layered bluephosphorus: a computational study Phys. Rev. Lett. 112 176802
[19] KresseG and Furthmüller J 1996 Efficiency of ab-initio totalenergy calculations formetals and semiconductors using aplane-wave basis setComput.Mater. Sci. 6 15–50
[20] KresseG andHafner J 1993Ab initiomolecular dynamics foropen-shell transitionmetals Phys. Rev.B 48 13115–8
[21] Perdew JP,BurkeK andErnzerhofM1996Generalized gradientapproximationmade simplePhys. Rev. Lett. 773865–8
[22] Grimme S 2006 Semiempirical GGA—type density functionalconstructedwith a long—range dispersion correctionJ. Comput. Chem. 27 1787–99
[23] Nosé S 1984Aunified formulation of the constant temperaturemolecular dynamicsmethods J. Chem. Phys. 81 511–9
[24] JoséMS, Emilio A, JulianDG, AlbertoG, Javier J, PabloO andDaniel S-P 2002The SIESTAmethod for ab initio order-Nmaterials simulation J. Phys.: Condens.Matter 14 2745
[25] NewsomeDA, SenguptaD, ForoutanH, RussoMF andvanDuinAC2012Oxidation of silicon carbide byO2 andH2O: a ReaxFF reactivemolecular dynamics study: I J. Phys.Chem.C 116 16111–21
[26] Zandiatashbar A, LeeG-H, An S J, Lee S,MathewN,TerronesM,Hayashi T, PicuCR,Hone J andKoratkarN 2014Effect of defects on the intrinsic strength and stiffness ofgrapheneNat. Commun. 5 3186
[27] Kang J,Wood JD,Wells S A, Lee J-H, LiuX, ChenK-S andHersamMC2015 Solvent exfoliation of electronic-grade, two-dimensional black phosphorusACSNano 9 3596–604
[28] WangG, Pandey R andKarna S P 2015 Phosphorene oxide:stability and electronic properties of a novel two-dimensionalmaterialNanoscale 7 524–31
[29] YoungD 2004Computational Chemistry: A Practical Guide forApplying Techniques to RealWorld Problems (NewYork:Wiley)
[30] WangG, Pandey R andKarna S P 2015 Effects of extrinsicpoint defects in phosphorene: B, C,N,O, and F adatomsAppl.Phys. Lett. 106 173104
[31] HenkelmanG, ArnaldssonA and JónssonH2006A fast androbust algorithm for Bader decomposition of charge densityComput.Mater. Sci. 36 354–60
[32] Ambrosetti A and Silvestrelli P L 2011Adsorption of rare-gasatoms andwater on graphite and graphene by van derWaals-corrected density functional theory J. Phys. Chem.C 1153695–702
[33] Twarowski A 1995Reduction of a phosphorus oxide and acidreaction setCombust. Flame 102 41–54
[34] Carlsson JM,Hanke F, Linic S and SchefflerM2009Two-stepmechanism for low-temperature oxidation of vacancies ingraphene Phys. Rev. Lett. 102 166104
[35] KuntzK L et al 2015Understanding the effect of oxygen andwater on 2Dblack phosphorus oxidation fromphotoelectronspectroscopyAbstract in Int. Phosphorene Symp. (Lansing,USA) (http://nanotube.pa.msu.edu/IPS15/IPS15abstracts.pdf)
7
2DMater. 3 (2016) 025011 GWang et al