Embed Size (px)
Citation preview

Ps
AIAAa
Cb
dc
C
h
•••••
a
ARRAA
KDSAK
GU
h0
Behavioural Brain Research 326 (2017) 154–164
Contents lists available at ScienceDirect
Behavioural Brain Research
jou rn al hom epage: www.elsev ier .com/ locate /bbr
re-clinical investigation of Diabetes Mellitus as a risk factor forchizophrenia
lexandra S. Almeida Heylmanna, Lara Canevera, Katia Gressa, Sarah T. Gomesa,sadora Fachima, Carolina Michelsa, Geórgia C. Stopassoli a, Gustavo A. Mastellaa,manda V. Steckerta, Adriani P. Damianib, Vanessa M. de Andradeb, João Quevedoa,c,lexandra I. Zugnoa,∗
Laboratório de Neurociências and Instituto Nacional de Ciência e Tecnologia Translacional em Medicina (INCT-TM), Programa de Pós-Graduac ão emiências da Saúde, Unidade Acadêmica de Ciências da Saúde, Universidade do Extremo Sul Catarinense, 88806-000, Criciúma, SC, BrazilLaboratório de Biologia Celular e Molecular, Programa de Pós-graduac ão em Ciências da Saúde, Unidade Acadêmica de Ciências da Saúde, Universidadeo Extremo Sul Catarinense, Criciúma, SC, BrazilCenter for Experimental Models in Psychiatry, Department of Psychiatry and Behavioral Sciences, Medical School, University of Texas Health Scienceenter at Houston, Houston, TX 77054, USA
i g h l i g h t s
Type 1 Diabetes Mellitus can be considered a predisposing factor for schizophrenia.Association alloxan + ketamine induced deficit in social interaction between animals.Alloxan + ketamine group showed a PPI deficit at all three intensities evaluated.Alloxan + ketamine seems to have an exacerbated effect within the cholinergic system.Combination alloxan + ketamine intensified oxidative damage in the brain structures.
r t i c l e i n f o
rticle history:eceived 11 January 2017eceived in revised form 21 February 2017ccepted 24 February 2017vailable online 8 March 2017
eywords:iabetes mellituschizophrenialloxanetamine
a b s t r a c t
This study investigated the behavioral and biochemical parameters of DM1 as a risk factor in an ani-mal model of schizophrenia (SZ). All groups: 1 Control (saline + saline); 2 Alloxan (alloxan + saline); 3Ketamine (saline + ketamine); 4 (Alloxan + Ketamine) were fasted for a period of 18 h before the sub-sequent induction of DM via a single intraperitoneal (i.p) injection of alloxan (150 mg/kg). From the4th to the 10th days, the animals were injected i.p with ketamine (25 mg/kg) or saline, once a day, toinduce a model of SZ and 30 min after the last administration were subjected to behavioral testing. After,the animals were decapitated and the brain structures were removed. Ketamine induced hyperactiv-ity and in the social interaction, ketamine, alloxan and the association of alloxan + ketamine increasedthe latency and decreased the number of contacts between animals. The animals from the ketamine,alloxan and alloxan + ketamine groups showed a prepulse startle reflex (PPI) deficit at the three intensi-ties (65, 70 and 75 dB). Ketamine was shown to be capable of increasing the activity of acetylcholinesterase
(AChE) in the brain structures. Combination of alloxan + ketamine seems to have an exacerbated effectwithin the cholinergic system. For lipid peroxidation and protein carbonyls, alloxan + ketamine appearto have intensified lipid and protein damage in the three structures. Ketamine and the combination ofketamine + alloxan induced DNA damage in both frequency and damage index. This research found a1 and
relationship between DM∗ Corresponding author at: Laboratório de Neurociências, Programa de Pós-raduac ão em Ciências da Saúde, Unidade Acadêmica de Ciências da Saúde,niversidade do Extremo Sul Catarinense, 88806-000 Criciúma, SC, Brazil.
E-mail address: [email protected] (A.I. Zugno).
ttp://dx.doi.org/10.1016/j.bbr.2017.02.043166-4328/© 2017 Elsevier B.V. All rights reserved.
SZ.© 2017 Elsevier B.V. All rights reserved.
1. Introduction
Type 1 Diabetes Mellitus (DM1) is a metabolic disorder which is
characterized by an insulin deficiency, determined by the destruc-tion of pancreatic cells, which results in about 10% of the total casesof DM (ADA, 2013). Mediated by the immune system, this process
al Bra
laBdDei
plsslg[
dafiwGaassttf
cieacapo
twaadtioe
etiicfitaiirpi
ispT
A.S.A. Heylmann et al. / Behaviour
eads to a permanent state of hyperglycemia which is a main char-cteristic of this disease [1]. According to Diretrizes da Sociedaderasileira de Diabetes [2], this is one of the most aggressive types ofiabetes, occurring in both childhood and adolescence. People withM1 have a ten times greater risk of cardiovascular events Orchardt al., 2006, while the risk of developing mental disorders is equaln both DM1 and DM2 [3].
Additionally, schizophrenia (SZ) is one of the most devastatingsychiatric disorders, and affects about 0,5–1% of the global popu-
ation [4]. SZ is diagnosed based on the description of the patients’ymptoms – commonly seen as an abrupt change in the patients’ocial and emotional profile – which starts between the end of ado-escence and the beginning of adult life [5]. These symptoms arerouped into three classes: positive, negative and cognitive deficits6].
Despite a growing consensus regarding SZ as a mental disor-er, its physiopathology is still unknown [7]. There are no doubtsbout the existence of cerebral anatomic and biochemical modi-cations in the genesis of the disease [8], especially dysfunctionsithin the glutamatergic and dopaminergic systems, beyond theABAérgic and cholinergic systems [9]. In the same way that therere glutamatergic dysfunctions in schizophrenia, data in the liter-ture suggests there is an involvement of glutamate in DM [10],ince alterations in these neurotransmitters can lead to metabolicyndrome (obesity and hyperglycemia) [11]. A study has shownhat in B pancreatic cells, there is an expression of glutamate gliaransporters, which are responsible for protecting pancreatic cellsrom glutamatergic excitotoxicity [12].
Furthermore, evidence shows there is an increase in glucoseoncentrations within astrocytes, which can in turn lead to a risen glutamate that contributes to the development of glutamatergicxcitotoxicity. This is related to an increase of neuronal excitabilitynd glutamatergic receptor hyperactivity via an excessive influx ofalcium (Ca2+). The increase in the influx of Ca2+ combined with thectivation of several proteins (proteases, phosphatases and phos-holipase) induces a rise in the production of reactive species ofxygen (ROS) and nitrogen (RNS).
It is known that the Central Nervous System (CNS) is susceptibleo the deleterious effects of hipoinsulinmia and hiperinsulinmia,hich induces oxidative stress. This condition is able to initi-
te comorbidities in diabetic patients, such as depression, SZ andtherosclerosis [13]. The brain has relatively low levels of antioxi-ant defenses, but high levels of lipid and catecholamine, causing ito have a susceptibility to attack by reactive species [14]. Moreover,t is considered that isolated hyperglycemia, or that associated withbesity, can induce an increase in the production of ROS and RNS,specially within the brain [15].
Therefore, biochemical mechanisms have been proposed toxplain the structural and functional abnormalities associated withissues that are exposed to hyperglycemia, with evidence suggest-ng that the capacity of endogenous antioxidants are prejudicedn diabetics [16]. Additionally, hypoglycemia causes neurologicalonsequences and glial cell alterations that are not yet fully clari-ed, but which can influence brain metabolism and contribute tohe physiopathology of CNS disorders [17]. Cellular componentsre more susceptible to the actions of ROS and RNS, but because ofts membrane, the lipid bilayer is one of the most affected, caus-ng changes to its structure and permeability [18]. Thus, as in DM,esearch has shown that there is an increase in the levels of lipideroxidation and protein carbonylation in an animal model of SZ
nduced via different doses of ketamine [19].In animal models of DM, alterations have been observed includ-
ng; increases in the levels of hippocampal astrocyte reactivity,ynaptic plasticity, vascular alterations, decreases in dendritic com-lexity, prejudiced neurotransmission and memory losses [13].hese varying forms of damage are also all related to schizophre-
in Research 326 (2017) 154–164 155
nia, which strengthens the association between hyperglycemia,oxidative stress and mental disorders [20]. However, the correla-tion between SZ and DM is normally associated with weight gainand DM2 [21], with few studies showing the relationship betweenSZ and DM1.
Animal models of DM depict similar physiological character-istics to the ones found in diabetic humans, such as chronichyperglycemia, hyper or hypoinsulinemia, beyond the clinicalsymptoms of DM (polydipsia and polyuria) [22]. In this presentstudy, alloxan was used to induce DM1, which generates welldefined clinical and laboratorial alterations, such as increased lev-els of hydric ingest and diuresis, as well as glycemic values above300 mg/dL [23]. To induce the animal model of SZ, we emphasizethe administration of non-competitive antagonists of glutamater-gic receptors (NMDA), such as ketamine [19]. The use of thisanesthetic is already well described in the literature for its abil-ity to block the NMDA channel, preventing an influx of Ca2+ and soinducting a transitory and reversible psychotic state, which causesboth positive and negative symptoms as well the cognitive deficitsobserved in SZ in otherwise healthy people [24].
It is understood that the oxidative stress that occurs becauseof chronic hyperglycemia plays a central role in the complicationsthat accompany DM1, and that ROS and RNS can injure cellularmacromolecules, such as proteins, lipids and the DNA, which thenact as pro-apoptosis agents. Based on this, the purpose of this paperwas to investigate the behavior and biochemical parameters of DM1as a risk factor in an animal model of SZ.
2. Methods
2.1. Experimental procedures
The animals included in this study were handled according tothe NIH Guide for the Care and Usage of Laboratory Animals, andalso in accordance with the rules of the Brazilian Society for Neuro-science and Behavior (SBNeC). The experiments were performed atthe Universidade do Extremo Sul Catarinense (UNESC) Brazil, at theLaboratory of Neurosciences, in partnership with the Pathophysi-ology Laboratory. All experimental procedures were performed inaccordance with the international recommendations for the careand use of laboratory animals, and additionally in compliance withthe recommendations for the use of animals as set out by the Brazil-ian Society of Neuroscience and Behavior (SBNeC).
2.2. Animals
Twenty-three day old male Wistar rats weighing around70–100 g were obtained from our breeding colony. Animals at theage of 23 days were chosen to mimic DM1 because it is often trig-gered in childhood and adolescence within humans [2]. The animalswere kept in cages (41 × 34 cm and 16 cm high) in groups of five,with free access to food and water, where they were maintainedon a 12-h light–dark cycle (lights on at 7:00 am), at a tempera-ture of 23 ± 1 ◦C. The only exception to this protocol was on theday that the animals received alloxan, when they were fasted for18 h prior to the administration of the drug. Fasting allows for bet-ter absorption of the drug, as determined by the protocol for DM1introduction in animals. These conditions were maintained con-stantly throughout the experiments. All experimental procedureswere performed in accordance with, and with the approval of, the
local Ethics Committee for the Use of Animals (Protocol 006/2015-2). All efforts were made to minimize animal suffering and to reducethe number of animals used in this study, utilizing alternatives toin vivo techniques where possible.
1 ral Bra
2
tssrTiotgac
2
21vaa
2
wot(fo4mtoastrmttbt
2
2
sablrtdm(Sosp
56 A.S.A. Heylmann et al. / Behaviou
.3. Induction of diabetes by alloxan
DM1 was induced in the animals through a single intraperi-oneal (i.p.) injection of alloxan (150 mg/kg), diluted in salineolution (0.9% NaCl), with the control group receiving the samealine solution. Both experimental groups (alloxan and saline)eceived an injection of alloxan or saline after fasting for 18 h [25].he content was quantitated using a commercial kit 48 h afternduction and also on the 10th day of the experiment. This studynly used animals that presented with both the clinical and labora-ory findings compatible with DM, as represented by a postprandiallucose level above 300 mg/dl, as well as weight loss, polydipsiand polyuria. Animals that did not meet these criteria were notonsidered in this study.
.4. Animal model of schizophrenia induced by ketamine
ketamine was administrated intraperitoneally (i.p.) at a dose of5 mg/kg, and was prepared in a saline solution at a volume of
mL/100 mg [26]. The use of a ketamine based animal model isiable because these animals have a low cost, are easy to work withnd have a physiology that is similar to that of humans in severalspects [27].
.5. Experimental draw
At the start of the study (DM induction), the animals wereeighed and this procedure was repeated until the final day (10th)
f the experiment. The rats were divided in 4 groups: 1) Con-rol (saline + saline), 2) Ketamine (ketamine + saline), 3) Alloxanalloxan + saline) and 4) Alloxan + ketamine. The animals were thenasted for 18 h prior to DM1 induction via an injection (i.p) of alloxanr saline (150 mg/kg). The animals’ glucose levels were evaluated8 h after the injection of alloxan to prove the induction of theodel. From the 4th to the 10th days, the animals received injec-
ions of ketamine (25 mg/kg) or saline over a period of 7 days inrder to mimic an animal model of SZ. On the 10th day, 30 minfter the last administration of ketamine or saline, the animals wereubmitted to the behavioral tests which evaluated their locomo-or activity, social interaction and prepulse inhibition of the startleeflex (PPI). Before guillotine decapitation, the animals were sub-itted to a glycemic test to verify that they were still diabetic at
he end of the experiment. The animals were then decapitated, andheir blood was collected for later analysis of DNA damage. Therain structures (frontal cortex, hippocampus and striatum) werehen dissected prior to biochemical analysis (Fig. 1).
.6. Behavioral evaluation
.6.1. Locomotor activityThirty minutes after the last injections of either ketamine or
aline, the animals’ locomotor activity was measured using anctivity monitor (40 × 60 cm). The activity monitor is surroundedy 50 cm high acrylic walls, with the test area containing 6 paral-
el bars, each bar containing 16 infrared sensors that can detect aat’s exact position and movement, making it possible to under-ake a detailed analysis of the animal’s behavior. The informationetected by the sensors over the 15 min test period was then trans-itted to a computer by the Open Source software Interbase 6.01
Activity Monitor – Insight Laboratory Equipment, Ribeirão Preto,
P). The distance covered by an animal was considered as the sumf the changes in position monitored in the activity arena, with theoftware calculating the distance between two locations, plus thereviously traveled distances [19].in Research 326 (2017) 154–164
2.6.2. Social interactionAfter the last injection of ketamine, the animals were socially
isolated for 6 h prior to the experiment. This test consisted of plac-ing two animals from different cages, but from within the samegroup, in the open field arena (50 × 25 × 50 cm) for a period of15 min [28]. During this period, three criteria were evaluated: thelatency to the first contact between animals, the number of contactsbetween animals and the total interaction time that the animalsremained together [29].
2.6.3. Prepulse inhibition of the startle reflex (PPI)The test session commenced by placing a subject in the sta-
bilimeter cage for a 5-min exposure to the background noise. Afterthis acclimatization period, the rats were presented with a seriesof 10 stimuli (pulse alone – 120 dB, 50 ms duration), with an inter-trial interval of 20 s. The purpose of this phase was to allow forwithin-session habituation to the startle stimulus. Thereafter, thePPI modulation of the acoustic startle was tested. The protocol con-sisted of 74 trials which were pseudo-randomly divided into sevendifferent categories. The trails were presented with an inter-trialinterval of 20 s: 20 presentations of just the pulse (120 dB, 50 msduration), eight presentations of each prepulse intensity (65, 70 and75 dB, 3000 Hz frequency, 20 ms duration) and 10 presentations ofeach prepulse intensity + pulse (with 50 ms interval) [30].
2.7. Biochemical evaluation
2.7.1. Glycemic mensurationThe levels of blood glucose were measured using a number 3
commercial kit on the 10th day of the experiment. To realize thisprocedure, it was necessary to puncture the rats tails with a needle(13 × 0,45 mm) allowing for blood to be collected and placed ontothe glucose measuring strip. To confirm DM1, the levels of glucoseafter fasting should be higher or equal to 300 mg/kg [23].
2.7.2. Measurement of AChE activityAChE activity in the brain structures of the animals was deter-
mined according to [31] Ellman’s method (1961). The hydrolysisrates were measured in 1 mL of a 0.8 mM solution containing100 mM of phosphate buffer (pH 7.5) and 1.0 mM of 5,5-dithiobis-2-nitrobenzoic acid (DTNB). A sample (50 mL) was then added tothe solution, and pre-incubated for a period of 3 min. Hydrolysiswas monitored spectrophotometrically at 412 nm for thiolate dian-ion formation, at 25 ◦C for 2–3 min in 30 s intervals. Samples wereevaluated in duplicates. Data were expressed in mmol ACSCh/h mgof protein.
2.7.3. Oxidative damage to lipids and proteinsThe formation of thiobarbituric acid reactive species (MDA
equivalents) was measured in the animal’s cerebral structuresto verify oxidative damage. As described by Draper and Hadley[66], samples of striatum, pre-frontal cortex and hippocampuswere mixed in 1 mL of 10% trichloroacetic acid and 1 mL of0.67% thiobarbituric acid, and then heated in a bath of boilingwater for a period of 30 min. The equivalents of malondialdehyde(MDA) were determined spectrophotometrically using absorbanceat 532 nm. The determination of carbonyl groups content was basedon the reaction with dinitrophenylhidrazine (DNPH), which wasundertaken to assess the damage to proteins. This protocol was
previously described by [32]. Briefly, proteins were precipitated bythe addition of 20% trichloroacetic acid, and were then re-dissolvedin DNPH. Absorbance was monitored spectrophotometrically at370 nm.
A.S.A. Heylmann et al. / Behavioural Brain Research 326 (2017) 154–164 157
rimen
2
wa(itdc0dbgvImitanc
2
m
2
iauta(rfc(ocsasP%fa
Fig. 1. Expe
.7.4. Comet assayAn alkaline comet assay was performed as described by [33]
ith the modifications as suggested by [34]. The extent of the dam-ge to DNA was assessed using Collins’ visual classification method2004). Images of 100 randomly selected cells were analyzed perndividual. Cells were scored visually into five classes, accordingo the tail size and shape (from undamaged – 0, to maximallyamage – 4), and a value (Damage Index) was assigned to eachomet according to its class. Thus the damage index ranged from
(completely undamaged: 100 cells × 0) to 400 (with maximumamage: 100 cells × 4). The Damage Frequency (%) was calculatedased on the percentage of damaged cells (0–100%). Internationaluidelines and recommendations for the comet assay consider thatisual scoring of comets is a well-validated method of evaluation.t has a high correlation with computer-based image analysis. DNA
igration was determined visually by the categorization of cometsnto different ‘classes’ of migration Collins et al., 2004. In addi-ion, according to [34], research shows that the cells damaged bypoptosis/necrosis cannot be assessed using the Comet Assay tech-ique, thus, any cells that did not present the format required forlassification by the Comet Assay were excluded.
.7.5. Protein assayThe levels of proteins were determined using the [35] Lowry
ethod (1951), with bovine serum albumin used as a standard
.8. Statistical analysis
On the 3rd day, glucose analysis results were evaluated accord-ng to the Student’s T Test for independent samples. The glucosend weight measurements taken on the 10th day were analyzedsing the one way analysis of variance (ANOVA) test, followed byhe post hoc Tukey test. The interactions of ketamine, alloxan andlloxan + ketamine were described by two-way analysis of varianceANOVA) tests. Biochemical analysis of oxidative damage, as well asesults from the behavioral tests were obtained by two way ANOVA,ollowed by the Tukey post hoc test, when F values were signifi-ant. All data are presented as mean and standard error of meanSEM). The statistical significance was considered to have valuesf p < 0.05. The IPP test was analyzed after the P post sessions toalculate the average amplitude of shock, as well as the after PP + Pessions to calculate the average amplitude shock result for eachnimal. The PPI level for each animal was determined by the PP + Phock amplitude expression with a percentage reduction from the
shock amplitude, and was calculated according to the equation: IPP = 100 − [(PP/P) × 100], so, 0% corresponding to a lack of dif-
erence between shock amplitude after the P and PP + P sessionsnd consequently, the lack of shock inhibition. Statistical analyses
tal design.
were undertaken using the Statistica 8.0 software, and the Graph-Pad Prism 5 software package was used for graphic realization ofthe data shown in this study.
3. Results
3.1. Diabetes induction by alloxan
Glucose was measured on the 3rd and 10th days of the experi-ment. According to the T Test, which was undertaken on the 3rdday (Fig. 2A), an increase in the levels of glucose was observedin the alloxan animal group when compared to the control group(p < 0.01). When evaluating the levels of glucose on the 10th day(Fig. 2B), the two way variance test (ANOVA) did not show anyinteraction between alloxan and ketamine [F(1,56) = 0.27, p = 0.60,],but the one way ANOVA test did show a significant differencebetween groups [F(3,32) = 1555.76, p < 0.01]. On the 10th day therewas an increase in glycemia within the ketamine (p < 0.01), alloxan(p < 0.01) and alloxan + ketamine (p < 0.01) groups when comparedto the control group (p < 0.01). It is noteworthy that the alloxanand alloxan + ketamine groups had the most significant levels ofhyperglycemia (400–600 mg/dL). However, when comparing thealloxan + ketamine to the alloxan group, the data also showed asignificant difference in the levels of glycemia. It is yet to be shownthat the isolated administration of ketamine was able to increasethe levels of glycemia found in the animals, and this confirmsthat ketamine or ketamine associated with alloxan induced theDM1 model, which is characterized by a glycemic increase above300 mg/dL. The rats were weighed on the 10th day (Fig. 2C), andthe two way ANOVA test failed to reveal any interaction betweenthe alloxan and ketamine variables: [F(1,56) = 0.94, p = 0.33], how-ever, the one way ANOVA test did show a significant differencebetween the groups [F(3,56) = 6.17, p < 0.01]. These results show aconsiderable weight reduction in the groups that received alloxanand alloxan + ketamine, that is, the ones that developed hyper-glycemia when compared to the control (p < 0.01) and ketaminegroups (p < 0.01).
3.2. Behaviour assessment
3.2.1. Locomotor activityFig. 3 shows the distance traveled by the animals. The admin-
istration of Ketamine induced hyperlocomotion in the subject rats(p < 0.01). However, the alloxan + ketamine group showed a signif-
icant decrease in the distance traveled in relation to the ketamine(p < 0.01) and alloxan groups (p < 0.01). Two way ANOVA testrevealed a significant interaction between the variables: alloxanand ketamine [F(1,31) = 25.48, p < 0.01]. Besides this, the isolated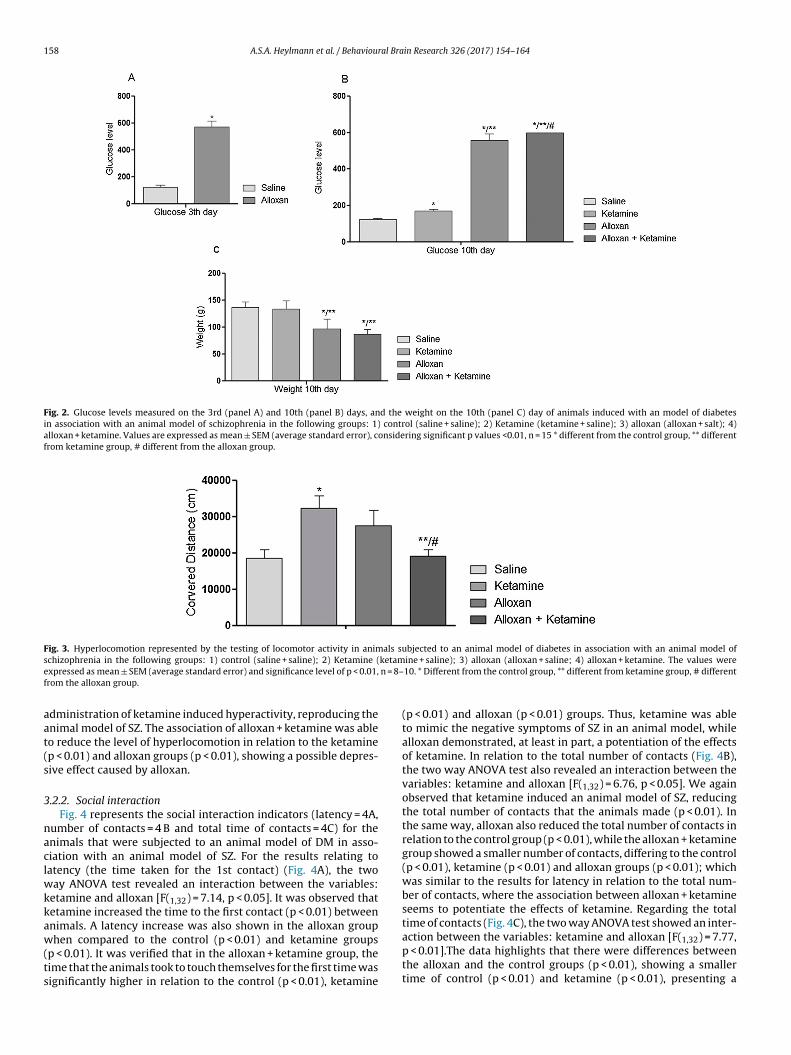
158 A.S.A. Heylmann et al. / Behavioural Brain Research 326 (2017) 154–164
Fig. 2. Glucose levels measured on the 3rd (panel A) and 10th (panel B) days, and the weight on the 10th (panel C) day of animals induced with an model of diabetesin association with an animal model of schizophrenia in the following groups: 1) control (saline + saline); 2) Ketamine (ketamine + saline); 3) alloxan (alloxan + salt); 4)alloxan + ketamine. Values are expressed as mean ± SEM (average standard error), considering significant p values <0.01, n = 15 * different from the control group, ** differentfrom ketamine group, # different from the alloxan group.
Fig. 3. Hyperlocomotion represented by the testing of locomotor activity in animals subjected to an animal model of diabetes in association with an animal model ofs ketame n = 8–f
aat(s
3
naclwkkaw(ts
chizophrenia in the following groups: 1) control (saline + saline); 2) Ketamine (xpressed as mean ± SEM (average standard error) and significance level of p < 0.01,rom the alloxan group.
dministration of ketamine induced hyperactivity, reproducing thenimal model of SZ. The association of alloxan + ketamine was ableo reduce the level of hyperlocomotion in relation to the ketaminep < 0.01) and alloxan groups (p < 0.01), showing a possible depres-ive effect caused by alloxan.
.2.2. Social interactionFig. 4 represents the social interaction indicators (latency = 4A,
umber of contacts = 4 B and total time of contacts = 4C) for thenimals that were subjected to an animal model of DM in asso-iation with an animal model of SZ. For the results relating toatency (the time taken for the 1st contact) (Fig. 4A), the two
ay ANOVA test revealed an interaction between the variables:etamine and alloxan [F(1,32) = 7.14, p < 0.05]. It was observed thatetamine increased the time to the first contact (p < 0.01) betweennimals. A latency increase was also shown in the alloxan group
hen compared to the control (p < 0.01) and ketamine groupsp < 0.01). It was verified that in the alloxan + ketamine group, theime that the animals took to touch themselves for the first time wasignificantly higher in relation to the control (p < 0.01), ketamine
ine + saline); 3) alloxan (alloxan + saline; 4) alloxan + ketamine. The values were10. * Different from the control group, ** different from ketamine group, # different
(p < 0.01) and alloxan (p < 0.01) groups. Thus, ketamine was ableto mimic the negative symptoms of SZ in an animal model, whilealloxan demonstrated, at least in part, a potentiation of the effectsof ketamine. In relation to the total number of contacts (Fig. 4B),the two way ANOVA test also revealed an interaction between thevariables: ketamine and alloxan [F(1,32) = 6.76, p < 0.05]. We againobserved that ketamine induced an animal model of SZ, reducingthe total number of contacts that the animals made (p < 0.01). Inthe same way, alloxan also reduced the total number of contacts inrelation to the control group (p < 0.01), while the alloxan + ketaminegroup showed a smaller number of contacts, differing to the control(p < 0.01), ketamine (p < 0.01) and alloxan groups (p < 0.01); whichwas similar to the results for latency in relation to the total num-ber of contacts, where the association between alloxan + ketamineseems to potentiate the effects of ketamine. Regarding the totaltime of contacts (Fig. 4C), the two way ANOVA test showed an inter-
action between the variables: ketamine and alloxan [F(1,32) = 7.77,p < 0.01].The data highlights that there were differences betweenthe alloxan and the control groups (p < 0.01), showing a smallertime of control (p < 0.01) and ketamine (p < 0.01), presenting a
A.S.A. Heylmann et al. / Behavioural Brain Research 326 (2017) 154–164 159
Fig. 4. Representation of social interaction (panel A = latency, panel B = number of contacts and panel C = total time of contact) of the animals subjected to an animal model ofd s: 1) c4 d errod
sgcttw
3
atkppitapwsgaTmesaatkt
3
3
cto
iabetes in association with an animal model of schizophrenia in the following group) alloxan + ketamine. The values were expressed as mean ± SEM (average standarifferent from ketamine group, # different from the alloxan group.
maller time of contact between animals. In the alloxan + ketamineroup, the total contact time also was significant, but only whenompared to the ketamine group (p < 0.01). It must be emphasizedhat ketamine was not able to induce an animal model of SZ inhis parameter. However, alloxan and its association with ketamineere shown to reduce the total time of contacts between animals.
.2.3. Prepulse inhibition of the startle reflex (PPI)The effects of the motor-sensory deficits of DM associated with
n animal model of SZ were evaluated using the PPI test (Fig. 5). Thewo way ANOVA test revealed an interaction between alloxan andetamine at the three available intensities – 65 dB [F(1,42) = 5.79,
< 0.05]; 70 dB [F(1,42) = 160.27, p < 0.01] and 75 dB [F(1,40) = 60.072, < 0.01]. The data shows that the control group P (pulse) was
nhibited by the PP (Pre pulse) because they showed much bet-er IPP values that were a higher% of IPP. The data belonging to thenimals from the ketamine group showed an IPP deficit when com-ared to the control group at all three intensities studied (p < 0.01),hich indicates a possible effect for ketamine in starting a motor
ensory profile deficit in animals. In the same way, the alloxanroup demonstrated a significant IPP deficit when compared to thenimals from the control group at the same intensities (p < 0.01).his result reveals the prejudicial effect of alloxan on the ani-als’ cognitive profiles, showing that its action is similar to the
ffect of ketamine. Furthermore, at 65 dB, the results showed aignificant motor sensory deficit in the animals belonging to thelloxan + ketamine group when compared to the control (p < 0.01)nd alloxan groups (p < 0.01). It also revealed a decrease in IPP inhe alloxan group at both 70 dB and 75 dB when compared to theetamine group (p < 0.01). These results show an important inhibi-ion of IPP in association with both pathologies.
.3. Biochemical analysis
.3.1. Activity of acetylcholinesterase (AChE)
Fig. 6 depicts the activity of the AChE enzyme in the frontalortex, hippocampus and striatum of animals that were subjectedo an animal model of DM in association with an animal modelf SZ. The two way ANOVA test revealed interactions between
ontrol (saline + saline); 2) Ketamine (ketamine + saline); 3) alloxan (alloxan + saline;r) and significance level of p < 0.01, n = 8–10. * Different from the control group, **
the variables: alloxan and ketamine within the frontal cortex:[F(1,20) = 49.89, p < 0.01]; hippocampus: [F(1,20) = 50.40, p < 0.01]and striatum: [F(1,21) = 10.23, p < 0.01]. Ketamine increased theactivity of AChE in all three brain structures that were evaluatedwhen compared to the control group (p < 0.01), which indicates thepossibility that the action of ketamine plays a part in the inductionof the cholinergic system. The activity of AChE increased signif-icantly within the frontal cortex, hippocampus and striatum ofanimals from the alloxan group when compared to the controlgroup (p < 0.01). The association of alloxan + ketamine induced aconsiderable increase in the activity of the AChE when comparedto the control (p < 0.01), ketamine (p < 0.01) and alloxan (p < 0.01)groups. The group treated solely with alloxan also showed anincrease in the levels of AChE activity when compared to the con-trol group (p < 0.01), but this increase was not as exacerbated asthe association of alloxan + ketamine. The data observed regardingthe activity of AChE indicates that the effect of ketamine in animalsis to induce biochemical changes that are similar to those seen inschizophrenic patients, which suggests that there is an increasedrisk in the association of DM with SZ (alloxan + ketamine). Ketaminewas capable of increasing the amount of AChE activity in all of thecerebral structures evaluated in this study, reproducing the animalmodel of SZ, and highlighting the effect that this anesthetic haswithin the cholinergic system.
3.3.2. Oxidative damage parametersFigs. 7 and 8, respectively, show the results of lipid peroxida-
tion (MDA equivalents) and protein carbonylation in animals thatwere subjected to a model of DM in association with a model ofSZ. The two way ANOVA test revealed that there were interac-tions in lipid peroxidation (Fig. 7) between the variables: alloxanand ketamine in the frontal cortex [F = (1,16) = 39.03,p < 0.01]; hip-pocampus [F = (1,16) = 7.71, p 0.01] and striatum [F = (1,16) = 7.49,p < 0.05]. Fig. 8 shows that the two way ANOVA test revealed inter-actions in protein carbonylation between the variables: alloxan and
ketamine, within the frontal cortex [F = (1,16) = 36.22, p < 0.01]; hip-pocampus [F = (1,16) = 8.54, p < 0.01] and striatum: [F = (1,20) = 35.89,p < 0.01]. For both parameters of oxidative damage, ketamine wasonly able to increase the levels of TBARS and protein carbonylation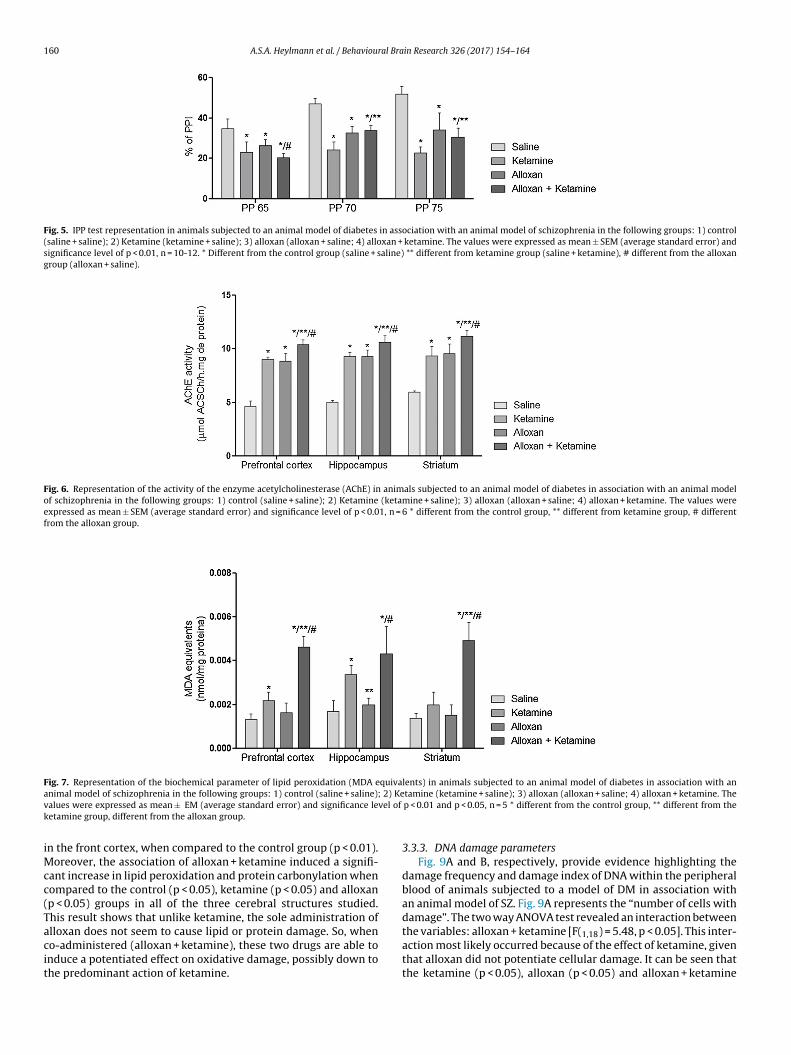
160 A.S.A. Heylmann et al. / Behavioural Brain Research 326 (2017) 154–164
Fig. 5. IPP test representation in animals subjected to an animal model of diabetes in association with an animal model of schizophrenia in the following groups: 1) control(saline + saline); 2) Ketamine (ketamine + saline); 3) alloxan (alloxan + saline; 4) alloxan + ketamine. The values were expressed as mean ± SEM (average standard error) andsignificance level of p < 0.01, n = 10-12. * Different from the control group (saline + saline) ** different from ketamine group (saline + ketamine), # different from the alloxangroup (alloxan + saline).
Fig. 6. Representation of the activity of the enzyme acetylcholinesterase (AChE) in animals subjected to an animal model of diabetes in association with an animal modelof schizophrenia in the following groups: 1) control (saline + saline); 2) Ketamine (ketamine + saline); 3) alloxan (alloxan + saline; 4) alloxan + ketamine. The values wereexpressed as mean ± SEM (average standard error) and significance level of p < 0.01, n = 6 * different from the control group, ** different from ketamine group, # differentfrom the alloxan group.
Fig. 7. Representation of the biochemical parameter of lipid peroxidation (MDA equivalents) in animals subjected to an animal model of diabetes in association with ana 2) Kev vel of
k
iMcc(Tacit
nimal model of schizophrenia in the following groups: 1) control (saline + saline);alues were expressed as mean ± EM (average standard error) and significance leetamine group, different from the alloxan group.
n the front cortex, when compared to the control group (p < 0.01).oreover, the association of alloxan + ketamine induced a signifi-
ant increase in lipid peroxidation and protein carbonylation whenompared to the control (p < 0.05), ketamine (p < 0.05) and alloxanp < 0.05) groups in all of the three cerebral structures studied.his result shows that unlike ketamine, the sole administration of
lloxan does not seem to cause lipid or protein damage. So, wheno-administered (alloxan + ketamine), these two drugs are able tonduce a potentiated effect on oxidative damage, possibly down tohe predominant action of ketamine.tamine (ketamine + saline); 3) alloxan (alloxan + saline; 4) alloxan + ketamine. Thep < 0.01 and p < 0.05, n = 5 * different from the control group, ** different from the
3.3.3. DNA damage parametersFig. 9A and B, respectively, provide evidence highlighting the
damage frequency and damage index of DNA within the peripheralblood of animals subjected to a model of DM in association withan animal model of SZ. Fig. 9A represents the “number of cells withdamage”. The two way ANOVA test revealed an interaction between
the variables: alloxan + ketamine [F(1,18) = 5.48, p < 0.05]. This inter-action most likely occurred because of the effect of ketamine, giventhat alloxan did not potentiate cellular damage. It can be seen thatthe ketamine (p < 0.05), alloxan (p < 0.05) and alloxan + ketamine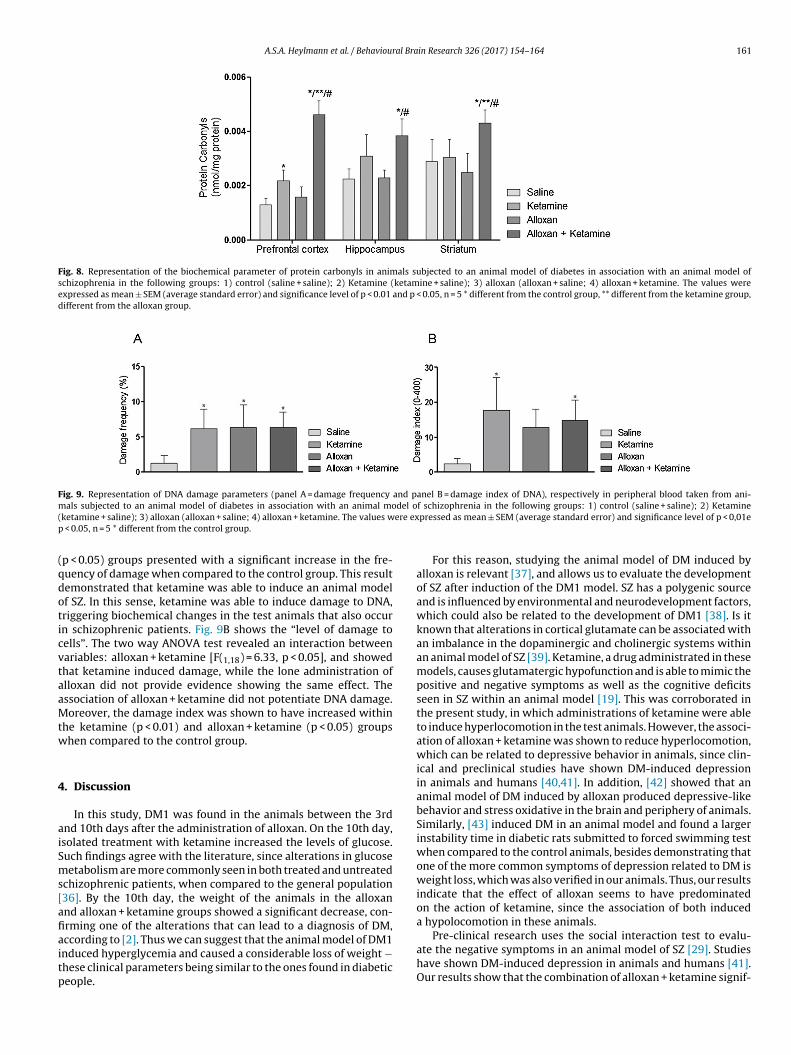
A.S.A. Heylmann et al. / Behavioural Brain Research 326 (2017) 154–164 161
Fig. 8. Representation of the biochemical parameter of protein carbonyls in animals subjected to an animal model of diabetes in association with an animal model ofschizophrenia in the following groups: 1) control (saline + saline); 2) Ketamine (ketamine + saline); 3) alloxan (alloxan + saline; 4) alloxan + ketamine. The values wereexpressed as mean ± SEM (average standard error) and significance level of p < 0.01 and p < 0.05, n = 5 * different from the control group, ** different from the ketamine group,different from the alloxan group.
Fig. 9. Representation of DNA damage parameters (panel A = damage frequency and panel B = damage index of DNA), respectively in peripheral blood taken from ani-m del o( ere exp
(qdoticvtaaMtw
4
aiSms[afiaitp
als subjected to an animal model of diabetes in association with an animal moketamine + saline); 3) alloxan (alloxan + saline; 4) alloxan + ketamine. The values w
< 0.05, n = 5 * different from the control group.
p < 0.05) groups presented with a significant increase in the fre-uency of damage when compared to the control group. This resultemonstrated that ketamine was able to induce an animal modelf SZ. In this sense, ketamine was able to induce damage to DNA,riggering biochemical changes in the test animals that also occurn schizophrenic patients. Fig. 9B shows the “level of damage toells”. The two way ANOVA test revealed an interaction betweenariables: alloxan + ketamine [F(1,18) = 6.33, p < 0.05], and showedhat ketamine induced damage, while the lone administration oflloxan did not provide evidence showing the same effect. Thessociation of alloxan + ketamine did not potentiate DNA damage.oreover, the damage index was shown to have increased within
he ketamine (p < 0.01) and alloxan + ketamine (p < 0.05) groupshen compared to the control group.
. Discussion
In this study, DM1 was found in the animals between the 3rdnd 10th days after the administration of alloxan. On the 10th day,solated treatment with ketamine increased the levels of glucose.uch findings agree with the literature, since alterations in glucoseetabolism are more commonly seen in both treated and untreated
chizophrenic patients, when compared to the general population36]. By the 10th day, the weight of the animals in the alloxannd alloxan + ketamine groups showed a significant decrease, con-rming one of the alterations that can lead to a diagnosis of DM,
ccording to [2]. Thus we can suggest that the animal model of DM1nduced hyperglycemia and caused a considerable loss of weight −hese clinical parameters being similar to the ones found in diabeticeople.f schizophrenia in the following groups: 1) control (saline + saline); 2) Ketaminepressed as mean ± SEM (average standard error) and significance level of p < 0,01e
For this reason, studying the animal model of DM induced byalloxan is relevant [37], and allows us to evaluate the developmentof SZ after induction of the DM1 model. SZ has a polygenic sourceand is influenced by environmental and neurodevelopment factors,which could also be related to the development of DM1 [38]. Is itknown that alterations in cortical glutamate can be associated withan imbalance in the dopaminergic and cholinergic systems withinan animal model of SZ [39]. Ketamine, a drug administrated in thesemodels, causes glutamatergic hypofunction and is able to mimic thepositive and negative symptoms as well as the cognitive deficitsseen in SZ within an animal model [19]. This was corroborated inthe present study, in which administrations of ketamine were ableto induce hyperlocomotion in the test animals. However, the associ-ation of alloxan + ketamine was shown to reduce hyperlocomotion,which can be related to depressive behavior in animals, since clin-ical and preclinical studies have shown DM-induced depressionin animals and humans [40,41]. In addition, [42] showed that ananimal model of DM induced by alloxan produced depressive-likebehavior and stress oxidative in the brain and periphery of animals.Similarly, [43] induced DM in an animal model and found a largerinstability time in diabetic rats submitted to forced swimming testwhen compared to the control animals, besides demonstrating thatone of the more common symptoms of depression related to DM isweight loss, which was also verified in our animals. Thus, our resultsindicate that the effect of alloxan seems to have predominatedon the action of ketamine, since the association of both induceda hypolocomotion in these animals.
Pre-clinical research uses the social interaction test to evalu-ate the negative symptoms in an animal model of SZ [29]. Studieshave shown DM-induced depression in animals and humans [41].Our results show that the combination of alloxan + ketamine signif-

1 ral Bra
ingtfk
diFcfa
DtdpOtiatsso[nd
iActaiibh
icsiaitac
cmtpinnes
h[taos
62 A.S.A. Heylmann et al. / Behaviou
cantly increased the level of latency in animals, and reduced theumber of contacts in relation to the control, ketamine and alloxanroups. These findings emphasize the negative symptoms relatedo behavior, and suggest that alloxan may be partially responsibleor potentiating the depressive-like behavior which is an effect ofetamine.
It is known that mental disorders are associated with chroniciseases, particularly DM1 which is related to the depression, anx-
ety and cognitive impairments seen in diabetic patients [44].urthermore, research has shown that patients with DM1 haveognitive deficits associated with worsening performance in dif-erent areas of cognitive function [45], and according to [46], suchlterations are generally related to NMDA receptors.
Thus, in this study, the effects of the motor-sensory deficits ofM associated with an animal model of SZ were evaluated using
he PPI test. The results from this study show that there is a PPIeficit in the animals treated with ketamine and alloxan at the pre-ulse intensities analyzed in this research (65 dB, 70 dB e 75 dB).ur findings confirm the results of previous studies, which showed
hat NMDA receptor antagonists, such as ketamine and MK-801,nduced prejudice towards PPI [47]. Generally, our results indicate
loss in the cognitive profile of the diabetic animals that is similaro the damage caused by ketamine at the three evaluated inten-ities. Thus, it is emphasized that children’s brains could be moreusceptible to the effects of DM than the brains of adults. This canccur due the difficulties in maintaining glycemic levels at this age48]. The cognitive dysfunctions seen in DM1 emerge at the begin-ing of the disease, and continue for a period of two years after theiagnosis [49].
Regarding the biochemical analysis, the results from this studyndicate that ketamine significantly increased the activity levels ofChE in the frontal cortex, hippocampus and striatum. This dataonfirms the findings of previous studies in which antagonists ofhe NMDA receptors were also able to increase the levels of enzymectivity [47]. This effect can be explained by the fact that ketamines considered an antagonist of nicotinic receptors (nAChR), whichncrease the amount of ACh in the synaptic slit, due to an exacer-ated activation of AChE, compromising memory formation in theippocampus [50].
Taken as a whole, this study confirms that the alterations seenn the levels of AChE activity support the behavioral results of theognitive profile (PPI) shown in animals. The prejudice of motor-ensory filtering induced by ketamine, with a consequent loss ofnhibition by pre-pulse, accompanies the increased levels of AChEctivity in the brain structures studied. In the same way, alloxanncreased the levels of AChE activity in all three cerebral struc-ures in relation to the control group, while the association oflloxan + ketamine potentiated the increase in this enzyme whenompared to the control, ketamine and alloxan groups.
Studies have shown that altered levels of AChE are also asso-iated with delusions and hallucinations, which proves that theetabolism of this neurotransmitter is directly linked to cogni-
ive functions [51]. Cognitive deficits were observed in diabeticatients and experimental models of DM, being related to changes
n AChE activity, which may indicate alterations in cholinergiceurotransmission [52]. Finally, SZ is also linked to a series of cog-itive dysfunctions, so the cholinergic system has been consideredssential in modulating the neuronal mechanisms underlying theseymptoms [53].
It has been postulated that oxidative stress caused by chronicyperglycemia plays a central role in the complications of DM54]. It is known that lipid peroxidation and protein carbonyla-
ion alter cell membrane permeability and therefore impair thectivity of enzymes and receptors [55]. In addition, high levelsf oxidative damage markers have been observed in DM andchizophrenic patients, which reinforces the relationship betweenin Research 326 (2017) 154–164
DM and psychiatric disorders, as oxidative stress may be involvedin the pathophysiology of both diseases [55].
Thus, the present data indicates an imbalance in the oxidativedamage induced by the chronic administration of ketamine, whichincreased the levels of TBARS and protein carbonylation in thefrontal cortex. These findings corroborate previous studies from ourgroup [19]. It is known that a reduction in the positive symptomsof SZ is involved in a decrease in lipid peroxidation, consideringthat dopaminergic hyperactivity triggers damage in lipids due tothe formation of free radicals during the metabolism of dopamine[56]. Moreover, surveys have shown increased levels of TBARS inthe plasma, erythrocytes and cerebrospinal fluid collected fromschizophrenic patients, as well as an accumulation of protein car-bonyls in these patients [55].
Our results also demonstrate that the association ofalloxan + ketamine increased lipid peroxidation and proteincarbonylation when compared to the control, ketamine andalloxan groups within the frontal cortex, hippocampus and stria-tum. These findings indicate that unlike ketamine, alloxan was notable to induce lipid and protein damage. In DM, oxidative stressis associated with hyperglycemia and in particular with cardio-vascular complications and damage to the CNS [57]. Furthermore,hyperglycemia, insulin resistance and dysfunction of pancreatic� cells seem to play an important role in the redox imbalance,creating cytotoxic effects in the phospholipids of cell membranes[57,15]. In general, study showed that oxidative stress secondaryto hyperglycemia occurs before the late complications of diabetesmanifest as mental disorders. Therefore, it is believed that thiscondition plays an important role in the pathogenesis of DM, andhas proven to be involved in the pathophysiology of SZ [15].
Based in this, studies justify that hyperglycemia is the majorcause of disruptions seen in behavioral [43] and biochemical[58,59,60] parameters, which might trigger SZ. It is kwon that DMmay modify CNS functions and is associated with cognitive deficitsand changes in the brain, including biochemical alterations. In addi-tion, the prevalence of depression and SZ in diabetic patients ishigher than in the general population [43]. The metabolic syn-drome as well as cognitive dysfunctions are common, particulary inpatients with SZ, yet there is no general consensus concerning theeffects of the components of the metabolic syndrome on severalcognitive domains [61]. Additionally, oxidative stress may play arole in the development of diabetes complications, being a commoncondition for psychiatric disorders, in particular SZ.
Many studies have shown that hyperglycemia is associated withincreased production of ROS and RNS in the brain, liver, pancreasand endothelial cells of diabetic patients, inducing oxidative stress[58]. Thus, biochemical mechanisms have been proposed to explainthe structural and functional abnormalities associated with pro-longed tissue exposure to hyperglycemia with indications thatendogenous antioxidant capacity is impaired in diabetic individ-uals, making it difficult to remove ERO [16].
In addition, hyperglycemia may trigger neurological conse-quences, which, although not yet fully understood, can influencebrain metabolism. An environment with high glucose concen-tration may cause changes in glia cells, contributing to thepathophysiology of CNS disorders observed in hyperglycemia andmay be due to a direct effect of glucose, not necessarily involvingan insulin deficit [18,17].
Studies, including the present one, have shown that ketamineis associated with increased levels of oxidative stress. In this con-text, of all the cellular targets, DNA is characterized as the mostvulnerable, since it cannot simply be replaced as can proteins and
lipids [62]. Thus, this study evaluated DNA damage parameters,and the results showed that ketamine, alloxan and the associa-tion of alloxan + ketamine induced an increase in the frequency ofdamage when compared to the control group. Within the index of
al Bra
da
goeatbs(at
5
tira
C
A
Ntofa
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
A.S.A. Heylmann et al. / Behaviour
amage, the groups treated with ketamine and alloxan + ketaminessociation increased in this parameter.
There is positive evidence that hyperglycemia can also causelycation of antioxidant enzymes, which causes the inactivationf these enzymes and decreased antioxidant defense [63]. Merkelt al. [64] showed that diabetic patients who do not produce orre sensitive to insulin, may show greater genomic instability dueo an inefficient repair system. For this reason, this disease maye related to the accumulation of damage seen in different tis-ues over time, and the dysfunction and failure of various organseyes, heart, kidneys, blood vessels, etc) [65]. Thus, oxidative dam-ge has been considered as one of the main factors responsible forhe complications presented by diabetics [63].
. Conclusion
Overall, our findings suggest that DM1 can trigger SZ, sincehe constant hyperglycemia found in DM1 may initiate changesn behavioral and biochemical parameters, as evidenced in thisesearch. However, more studies are needed to unravel the mech-nisms involved and to allow implementation of the findings.
onflict of interest
There are no conflict of interest.
cknowledgments
Laboratory of Neurosciences (Brazil) is one of the centers of theational Institute for Translational Medicine (INCT-TM) and one of
he members of the Center of Excellence in Applied Neurosciencesf Santa Catarina (NENASC). This research was supported by grantsrom CNPq (A.I.Z and J.Q.), FAPESC (A.I.Z.), Instituto Cérebro e Mentend UNESC. A.I.Z. and J.Q. are CNPq Fellows.
eferences
[1] C.A. Balda, A. Pacheco-Silva, Aspectos imunológicos do diabetes mellitos tipo1, Rev. Ass. Med. Bras. 45 (2) (1999) 175–180.
[2] Sociedade Brasileira de Diabetes. Diretrizes da Sociedade Brasileira deDiabetes, 2014.
[3] R. Fraguas, D.V. Iosifescu, B. Bankier, R. Perlis, N. Clementi-Craven, J. Alpert, M.Fava, Major depressive disorder with anger attacks and cardiovascular riskfactors, Int. J. Psychiatry Med. 37 (1) (2007) 99–111.
[4] J. McGrath, S. Saha, T. Chant, J. Welham, Schizophrenia: a concise overview ofincidence, prevalance, and mortality, Epidemiol. Rev. 30 (2008) 67–76.
[5] A. Marsman, M.P. Van Den Heuvel, D.W. Klomp, R.S. Kahn, P.R. Luijten, H.E.Hulshoff Pol, Glutamate in schizophrenia: a focused review and meta-analysisof (1)H-MRS studies, Schizophr. Bull. 39 (1) (2013) 120–129.
[6] S. Shamsi, A. Lau, T. Lencz, K.E. Burdick, P. DeRosse, R. Brenner, J.P.Lindenmayer, A.K. Malhotra, Cognitive and symptomatic pre¬dictors offunctional disability in schizophrenia, Schizophr. Res. 126 (2011) 257–264.
[7] U. Meyer, J. Feldon, Epidemiology-driven neurodevelopmental animal modelsof schizophrenia, Prog. Neurobiol. 90 (3) (2010) 285–326.
[8] M.S. Keshavan, H.A. Nasrallah, R. Tandon, Schizophrenia: just the facts 6.Moving ahead with the schizophrenia concept: from the elephant to themouse, Schizophr. Res. 127 (1-3) (2011) 3–13.
[9] P.J. Harrison, D.R. Weinberger, Schizophrenia genes, gene expression, andneuropathology: on the matter of their convergence, Mol. Psychiatry 10 (1)(2005) 40–68.
10] A. Nawa, W. Fujita-Hamabe, S. Tokyyama, Altered instestinal Pgycoproteinexpressionmlevels a monosodium glutamateinduced obese mouse model,Life Sci. 89 (23) (2011) 834–838.
11] J.F. Morrison, S. Shehab, R. Sheen, S. Dhanasekaran, M. Shaffullah, E.Mensah-Brown, Sensory and autonomic nerve changes in the monosodiumglutamate-treated rat: a model of type II diabetes, Exp. Physiol. 93 (2) (2008)213–222.
12] E.S. Di Cairano, A.M. Davalli, L. Perego, S. Sala, V.F. Sacchi, S. La Rosa, G. Finzi, C.Placidi, C. Capella, P. Conti, V.E. Centonze, F. Casiraghi, F. Bertuzzi, F. Folli, C.Perego, The glial glutamate transporter 1 (GLT1) is expressed by pancreaticbeta-cells and prevents glutamate-inducedbeta-cell death, J. Biol. Chem. 286(16) (2011) 14007–14018.
[
in Research 326 (2017) 154–164 163
13] S.W. Jung, O.K. Han, S.J. Kim, Increased expression of � amyloid precursor genein the hippocampus of streptozotocin-induced diabetic mice with memorydeficit and anxiety induction, J. Neural Transm. 117 (12) (2010) 1411–1418.
14] A.S. Haeser, Oxidative stress parameters in diabetic rats submitted to forcedswimming test: the clonazepan effect, Brain Res. 1154 (2007) 137–143.
15] F.D. Rocha, V.L. Teixeira, R.C. Pereira, M.A. Kaplan, Diabetes mellitus e estresseoxidativo: produtos naturais como alvo de novos modelos terapêuticos, Rev.Bras. Farm. 87 (2) (2006) 49–54.
16] S.A. Santini, G. Marra, B. Giardina, P. Cotroneo, A. Mordente, G.E. Martorana, A.Manto, G. Ghirlanda, Defective plasma antioxidant defenses and enhancedsusceptibility to lipid peroxidation in uncomplicated IDDM, Diabetes 46 (11)(1997) 1853–1858.
17] T. Valente, A. Gella, M. Solé, N. Durany, M. Unzeta, Immunohistochemicalstudy of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1in the hippocampal vasculature: pathological synergy of Alzheimer’s diseaseand diabetes mellitus, J. Neurosci. Res. 90 (2012) 1989–1996.
18] A.S. Mello, A. Quincozes-Santos, C. Funchal, Correlac ão entre hiperglicemia ecélulas do SNC, com enfoque na atividade glial, Rev. Neurosci. 20 (2) (2012)294–301.
19] L. De Oliveira, D.B. Fraga, R.D. De Luca, L. Canever, F.V. Ghedim, M.P. Matos,E.L. Streck, J. Quevedo, A.I. Zugno, Behavioral changes and mitochondrialdysfunction in a rat model of schizophrenia induced by ketamine, Metab.Brain Dis. 26 (1) (2011) 69–77.
20] M. Boarolli, N.C. Ferreira, D.V. Bavaresco, D.F. Felipe, G. Amboni,Manifestac ões psiquiátricas e possíveis danos cognitivos em pacientesdiabéticos tipo II, Revista de Iniciac ão Científica 5 (2) (2014) 134–143.
21] A.P. Azevedo, M. Papelbaum, F. D’Elia, Diabetes and eating disorders: a highrisk association, Rev. Bras. Psiquiatric 24 (3) (2002) 77–80.
22] K. Srinivasan, P. Ramarao, Animal models in type 2 diabetes research: anoverview, Indian J. Med. Res. 125 (3) (2007) 451–472.
23] F. Spiller, D. Carlos, F.O. Souto, A. De Freitas, F.S. Soares, S.M. Viera, F.J. Paula,J.C. Alves-Filho, F.Q. Cunha, A1-Acid Glycoprotein decreases neutrophilmigration a dincreases susceptibility to sepsis in diabetic mice, Diabetes 61(2012) 1584–1591.
24] A. Mechri, M. Saoud, G. Khiari, T. D’amato, J. Dalery, L. Gaha, Glutaminergichypothesis of schizophrenia: clinical research studies with ketamine,Encephale 27 (1) (2010) 53–59.
25] M.M. Lerco, C.T. Spadella, J.L.M. Machado, A.S. Schellini, C.R. Padovani,Caracterizac ão de um modelo experimental de Diabetes Mellitus, induzidopor aloxano em ratos. Estudo clínico e laboratorial, Acta Cir. Bras. 18 (2003)132–142.
26] L. Canever, L. Oliveira, R. D’altoe De Luca, P.T. Correa, B.F.D. De, M.P. Matos, G.Scaini, J. Quevedo, E.L. Streck, A.I. Zugno, A rodent model of schizophreniareveals increase in creatine kinase activity with associated behavior changes,Oxid. Med. Cell Longev. 3 (6) (2010) 421–427.
27] F.C. Silva, R.T. Dantas, C.O. Citó, I.G. Silva, M.M. Vasconcelos, M.F. Fonteles, S.B.Viana, C.F. Sousa, Cetamina, da anestesia ao uso abusivo: artigo de revisão,Rev. Neurosci. 18 (2) (2010) 227–237.
28] T. Schneider, R. Przewlocki, Behavioral alterations in rats prenatally exposedto valproic acid: animal model of autism, Neuropsychopharmacology 30 (1)(2005) 80–89.
29] C.S. Gama, M. Salvador, A.C. Anndreazza, F. Kapczinski, P. Belmonte-de Abreu,Elevated serum superoxide dismutase and thiobarbituric acid reactivesubstances in schizophrenia: a study of patients treated with haloperidol orclozapine, Prog. Neuropsychopharmacol. Biol. Psychiatry 30 (3) (2006)512–515.
30] E.D. Levin, A. Petro, D.P. Caldwell, Nicotine and clozapine actions on pre-pulseinhibition deficits caused by N-methyl-D-aspartate (NMDA) glutamatergicreceptor blockade, Prog. Neuropsychopharmacol. Biol. Psychiatry 29 (4)(2005) 581–586.
31] G.L. Ellman, K.D. Courtney, V. Andres Jr., R.M. Feather-Stone, A new and rapidcolorimetric determination of acetylcholinesterase activity, Biochem.Pharmacol. 7 (1961) 88–95.
32] R.L. Levine, D. Garland, C.N. Oliver, Dettermination of carbonyl content inoxidatively modified proteins, Methods Enzymol. 186 (1994) 464–478.
33] N.P. Singh, M. McCoy, R.R. Tice, E.L. Schneider, A simple technique forquantitation of low levels of DNA damage in individual cells, Exp. Cell Res.175 (1) (1988) 184–191.
34] R.R. Tice, E. Agurell, D. Anderson, B. Burlinson, A. Hartmann, H. Kobayashi, Y.Miyama, E. Rojas, J.C. Ryu, Y.F. Sasaki, Single cell gel/comet assay: guidelinesfor in vitro and in vivo genetic toxicology testing, Environ. Mol. Mutagen. 35(3) (2000) 206–221.
35] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, Protein measurement withthe Folin phenol reagent, J. Biol. Chem. 193 (1) (1951) 265–275.
36] E.P. Sena, A.S. Sampaio, L.C. Quarantini, I.R. Oliveira, Diabetes mellitus andatypical antipsychotics, Rev. Bras. Psiquiatric 25 (4) (2003) 253–257.
37] T. Szkudelski, The mechanism of alloxan and streptozotocin action in B cellsof the rat pancreas, Physiol. Res. 50 (6) (2001) 537–546.
38] J. Van Os, G. Kenis, B.P. Rutten, The environment and schizophrenia, Nature468 (7321) (2010) 203–212.
39] J.T. Coyle, G. Tsai, D. Goff, Converging evidence of NMDA receptor
hypofunction in the pathophysiology of schizophrenia, Ann. N. Y. Acad. Sci.(2003) 318–327.40] L.B. Ceretta, G.Z. Réus, R.B. Stringari, K.F. Ribeiro, G. Zappellini, B.W. Aguiar, B.Pfaffenseller, C. Lersh, F. Kapczinski, J. Quevedo, Imipramine treatment

1 ral Bra
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[expression of the DNA repair enzyme XPD, Mol. Cell. Endocrinol. 2 (2003)75–85.
[65] S.I. Taylor, Deconstructing type 2 diabetes, Cell 97 (1) (1999) 9–12.
64 A.S.A. Heylmann et al. / Behaviou
reverses depressive-like behavior in alloxan-diabetic rats, Diabetes Metab.Res. Rev. 28 (2) (2012) 139–144.
41] H.S. Dhavale, V. Panikkar, B.S. Jadhav, M. Ghulghule, A.D. Agari, Depressionand diabetes: impact of antidepressant medications on glycaemic control, J.Assoc. Phys. India 61 (12) (2013) 896–899.
42] G.Z. Réus, M.A. Dos Santos, H.M. Abelaira, S.E. Titus, A.S. Carlessi, B.I. Matias, L.Bruchchen, D. Florentino, A. Vieira, F. Petronilho, L.B. Ceretta, A.I. Zugno, J.Quevedo, Antioxidant treatment ameliorates experimental diabetes-induceddepressive-like behaviour and reduces oxidative stress in brain and pancreas,Diabetes Metab. Res. Rev. 32 (3) (2016) 278–288.
43] C.A. Wayhs, V. Manfredini, A. Sitta, M. Deon, G. Ribas, C. Vanzin, G. Biancini, M.Ferri, M. Nin, H.M. Barros, C.R. Vargas, Protein and lipid oxidative damage instreptozotocin-induced diabetic rats submitted to forced swimming test: theinsulin and clonazepam effect, Metab. Brain Dis. 25 (3) (2010) 297–304.
44] B. Cholerton, L.D. Baker, S. Craft, Insulin, cognition, and dementia, Eur. J.Pharmacol. (2013) 170–179.
45] E.R. Mayeda, R.A. Whitmer, K. Yaffe, Diabetes and cognition, Clin. Geriatr.Med. 31 (2015) 101–115.
46] A.R. Mohn, R.R. Gainetdinov, M.G. Caron, B.H. Koller, Mice with reducedNMDA receptor expression display behaviors related to schizophrenia, Cell 98(4) (1999) 427–436.
47] A.I. Zugno, H.L. Chipindo, A.M. Volpato, J. Budni, A.V. Steckert, M.B. DeOliveira, A.S. Heylmann, S.F. Da Rosa, G.A. Mastella, S.G. Maravai, P.G. Wessler,A.R. Binatti, B. Panizzutti, P.F. Schuck, J. Quevedo, C.S. Gama, Omega-3prevents behavior response and brain oxidative damage in the ketaminemodel of schizophrenia, Neuroscience 259 (2014) 223–231.
48] C.M. Ryan, Diabetes and brain damage: more (or less) than meets the eye?Diabetologia (2006) 2229–2233.
49] M.A. Cato, N. Mauras, J. Ambrosino, A. Bondurant, A.L. Conrad, C. Kollman, P.Cheng, R.W. Beck, K.J. Ruedy, T. Aye, A.L. Reiss, N.H. White, T. Hershey,Cognitive functioning in young children with type 1 diabetes, J. Int.Neuropsychol. Soc. 20 (2014) 238–247.
50] M. Chatterjee, R. Verma, S. Ganguly, G. Palit, Neurochemical and molecularcharacterization of ketamine-induced experimental psychosis model in mice,
Neuropharmacology 63 (6) (2012) 1161–1171.51] M.E. Hasselmo, The role of acetylcholine in learning and memory, Curr. Opin.Neurobiol. 16 (6) (2006) 710–715.
52] G. Sánchez-Chávez, R. Salceda, Effect of streptozotocin-induced diabetes onactivities of cholinesterases in the rat retina, IUBMB Life 49 (2000) 283–287.
[
in Research 326 (2017) 154–164
53] M. Sarter, C. Lustig, S.F. Taylor, Cholinergic contributions to the cognitivesymptoms of schizophrenia and the viability of cholinergic treatments,Neuropharmacology 62 (3) (2012) 1544–1553.
54] F. Giacco, M. Brownlee, Oxidative stress and diabetic complications, Cir Res.107 (2010) 1058–1070.
55] I. Dalle-Donne, G. Aldini, M. Carini, R. Colombo, R. Rossi, A. Milzani, Proteincarbonylation, cellular dysfunction, and disease progression, J. Cell Mol. Med.10 (2) (2006) 389–406.
56] X.Y. Zhang, Y.l. Tan, L.Y. Cao, G.Y. Wu, Q. Xu, Y. Shen, D.F. Zhou, Antioxidantenzymes and lipid peroxidation in different forms of schizophrenia treatedwith typical and atypical antipsychotics, Schizophr. Res. 81 (2006) 291–300.
57] D. Pitocco, F. Zaccardi, E. Di Stasio, F. Romitelli, S.A. Santini, C. Zuppi, G.Ghirlanda, Oxidative stress, nitric oxide, and diabetes, Rev. Diabetic Stud. 7(2010) 15–25.
58] S. Furukawa, T. Fujita, M. Shimabukuro, M. Iwaki, Y. Yamada, Y. Nakajima, O.Nakayama, M. Makishima, M. Matsuda, I. Shimomura, Increased oxidativestress in obesity and its impact on metabolic syndrome, J. Clin. Invest. 114(12) (2004) 1752–1761.
59] A.P. Kudin, N.Y. Bimpong-Buta, S. Vielhaber, C.E. Elger, W.S. Kunz,Charactezation of superoxide producing sites in isolated brain mitochondria,J. Biol. Chem. 279 (6) (2004) 4129–4135.
60] B.L. Wajchenberg, B-cell failure in diabetes and preservation by clinicaltreatment, Endocr. Rev. 28 (2007) 187–218.
61] A.S. Goughari, S. Mazhari, A.M. Pourrahimi, M.M. Sadeghi, N. Nakhaee,Associations between components of metabolic syndrome and cognition inpatients with schizophrenia, J. Psychiatr. Pract. 21 (3) (2015) 190–197.
62] B. Hallwell, J.M.C. Gutteridge, Free Radicals in Biology and Medicine, 4. ed.,Oxford University Press, 2007.
63] N.F. Wiernsperger, Oxidative stress as a therapeutic target in diabetes:revisiting the controversy, Diabetes Metab. 296 (2003) 579–585.
64] P. Merkel, N. Khoury, C. Bertolotto, R. Perfetti, Insulin and glucose regulate the
66] H.H. Draper, M. Hadley, Malondialdehyde determination as index of lipidperoxidation, Methods Enzymol. 186 (1990) 421–431.





























