Embed Size (px)
Citation preview

American Journal of Medical Genetics 51:32-34 (1994)
Atypical Acrofacial Dysostosis Syndrome
Arnold L. Christianson, Hester Kruger, and Laurette Dini Departments of Human Genetics and Developmental Biology (A.L.C.), Obstetrics and Gynaecology (H.K.), and Pathology (L.D.), University of Pretoria, Pretoria, Republic of South Africa
We describe a male fetus with a combination of defects, including severe mandibulofacial dysostosis, holoprosencephaly, upper limb deficiency, and microgastria. These abnor- malities indicate a severe defect of gastrula- tiodblastogenesis predominantly affecting cephalad structures. This combination of anomalies has to our knowledge not previ- ously been described.
We propose that the anomaly pattern repre- sents either a blastogenesis-related associa- tion, or a microgastria-limb deficiency poly- topic field defect. o 1994 wiley-~iss, Inc.
KEY WORDS: acrofacial dysostosis, holopros- encephaly, limb deficiency, mi- crogastria
INTRODUCTION The lethal acrofacial dysostoses (AFD) can be sub-
divided, more or less confidently, into Nager, POADS (postaxial AFD type Genee-Wiedemann or Miller), Rodriguez, and other forms, some with autosomal domi- nant, others with autosomal recessive inheritance, but most being sporadic cases [Opitz et al., 19931. We report on a male fetus with a severe, lethal AFD syndrome and discuss cause and nosology.
CLINICAL REPORT The propositus, a 26-week male fetus, was examined
after termination of pregnancy because ultrasono- graphic findings consisting of microcephaly, absent arms, and no stomach bubble were observed. This was the fourth pregnancy of a 20-year-old mother and her 23-year-old non consanguineous husband. The first and third pregnancies terminated in spontaneous abortions at 7 and 8 weeks, respectively. No detailed data exist on these fetuses. The second pregnancy ended in the deliv- ery at term of a normal male infant. The family history was otherwise unremarkable.
Received for publication July 28,1993; revision received Novem- ber 29, 1993.
Address reprint requests to Dr. A.L. Christianson, Department of Human Genetics, Faculty of Medicine, University of Pretoria, P.O. Box 2034, Pretoria 0001, South Africa.
0 1994 Wiley-Liss, Inc.
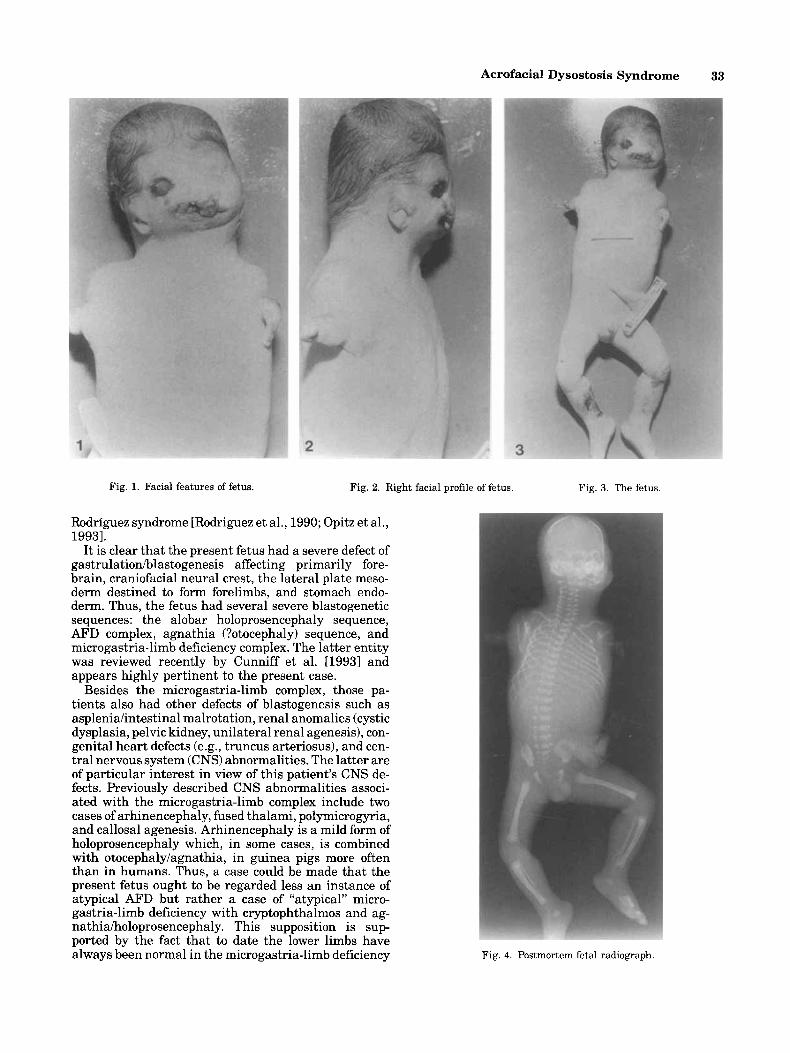
The fetus had a weight of 720 g, length 33.3 cm, and head circumference 17 cm. Three distinct patches of cutis aplasia were present on the scalp. The hair was pale and fine, descending low on the neck posteriorly, coming forward and parting in the midline over the forehead. Severe mandibulofacial dysostosis was pres- ent, with an agnathia-like appearance, macrostomia, cleft palate, single thick oral frenulum, hypoplastic tongue, flat broad nasal tip, with a large single nasal orifice opening directly into the mouth, and a broad nasal bridge. Malar hypoplasia, cryptophthalmos of the left eye, a right downslanting palpebral fissure, with ectropion and a coloboma of the lower eyelid were pres- ent. The ears were low set, dysplastic and the auditory canals were atretic (Figs. 1, 2).
Except for two small appendages at the shoulders, the upper limbs were absent (Fig. 3).
A postmortem roentgenogram (Fig. 4) indicated the presence of two rudimentary bones in the right arm appendage and three similar bones in the left append- age, The scapulae appeared hypoplastic and no mandi- ble was visible. The ribs and lower limbs appeared nor- mal and no other skeletal abnormalities were noted. Chromosomes from blood and skin were normal (46, XY). Autopsy demonstrated alobar holoprosencephaly and a small stomach.
DISCUSSION The fetus reported here had severe mandibulofacial
dysostosis (MFD) with malar hypoplasia, malformed ears, atresia of auditory canals, cleft palate, coloboma of lower lid, macrostomia, and most unusual additional anomalies of unilateral cryptophthalmos, virtual ag- nathia with alobar holoprosencephaly and single nos- tril, microgastria, and virtual upper limb amelia save for a tiny limb rudiment at each shoulder. The clavicles appeared to be of normal size on the radiograph, but the scapulae were clearly hypoplastic.
This fetus appeared to have some type of acrofacial dysostosis, but could also be described as a severe MFD with virtual upper limb amelia. Since the lower limbs were unaffected, it was not possible to determine pre- dominantly pre- or postaxial limb involvement. Thus, it was impossible to state whether this fetus had a severe Nager or POADS anomaly, the Rodriguez syndrome, or another, previously undescribed entity. No known Nager or POADS patient has had this combination of anomalies, and the condition of this fetus does not fit the
Acrofacial Dysostosis Syndrome 33
Fig. 1. Facial features of fetus. Fig. 2. Right facial profile of fetus.
Rodriguez syndrome [Rodriguez et al., 1990; Opitz et al., 19931.
It is clear that the present fetus had a severe defect of gastrulatiodblastogenesis affecting primarily fore- brain, craniofacial neural crest, the lateral plate meso- derm destined to form forelimbs, and stomach endo- derm. Thus, the fetus had several severe blastogenetic sequences: the alobar holoprosencephaly sequence, AFD complex, agnathia (?otocephaly) sequence, and microgastria-limb deficiency complex. The latter entity was reviewed recently by Cunniff et al. U9931 and appears highly pertinent to the present case.
Besides the microgastria-limb complex, those pa- tients also had other defects of blastogenesis such as aspleniahntestinal malrotation, renal anomalies (cystic dysplasia, pelvic kidney, unilateral renal agenesis), con- genital heart defects (e.g., truncus arteriosus), and cen- tral nervous system (CNS) abnormalities. The latter are of particular interest in view of this patient’s CNS de- fects. Previously described CNS abnormalities associ- ated with the microgastria-limb complex include two cases of arhinencephaly, fused thalami, polymicrogyria, and callosal agenesis. Arhinencephaly is a mild form of holoprosencephaly which, in some cases, is combined with otocephalylagnathia, in guinea pigs more often than in humans. Thus, a case could be made that the present fetus ought to be regarded less an instance of atypical AFD but rather a case of “atypical” micro- gastria-limb deficiency with cryptophthalmos and ag- nathiaiholoprosencephaly. This supposition is sup- ported by the fact that to date the lower limbs have always been normal in the microgastria-limb deficiency
Fig. 3. The fetus.
Fig. 4. Postmortem fetal radiograph.

34 Christianson et al.
complex; i.e., this complex seems to represent a defect of gastrulation involving primarily the head end of the fetus.
No family observations of the microgastria-limb defi- ciency defect have been observed. Thus, it may repre- sent a single but polytopic field defect, or, as in this case, an association, i.e., an idiopathic pattern of multiple anomalies of blastogenesis.
ACKNOWLEDGMENTS We would like to thank Dr. J. Opitz and Prof. G.S.
Gericke for their help in reviewing the text and Mrs. S. Swarts for her patience and care in preparing the manu- script.
REFERENCES Cunniff C, Williamson-Kruse L, Olney AH (1993): Congenital micro-
gastria and limb reduction defects. Pediatrics 91:1192-1194. Opitz JM, Mollica F, Sorge G, Milana G, Cimino G, Caltabiano M
(1993): The acrofacial dysostoses--Review and report of a previ- ously undescrihed condition-the autosomal or X-linked dominant Catania form of acrofacial dysostosis. Am J Med Genet 47560-678.
Rodriquez JI, Palacios J, Urioste M (1990): New acrofacial dysostosis syndrome in three sibs. Am J Med Genet 35:484-489.





























