Embed Size (px)
Citation preview

Research Article
Antitumor Effects of Epidrug/IFNa CombinationDriven by Modulated Gene Signatures in BothColorectal Cancer and Dendritic CellsAlessandra Fragale1, Giulia Romagnoli1, Valerio Licursi2, Maria Buoncervello1,Giorgia Del Vecchio4, Caterina Giuliani1, Stefania Parlato1, Celeste Leone1,Marta De Angelis1, Irene Canini1, Elena Toschi1, Filippo Belardelli3, Rodolfo Negri4,5,Imerio Capone1, Carlo Presutti4, and Lucia Gabriele1
Abstract
Colorectal cancer results from the progressive accumulation ofgenetic and epigenetic alterations. IFN signaling defects play animportant role in the carcinogenesis process, in which the inabil-ity of IFN transcription regulatory factors (IRF) to access regula-tory sequences in IFN-stimulated genes (ISG) in tumors and inimmune cells may be pivotal. We reported that low-dose com-bination of two FDA-approved epidrugs, azacytidine (A) andromidepsin (R), with IFNa2 (ARI) hampers the aggressivenessof both colorectal cancer metastatic and stem cells in vivo andtriggers immunogenic cell death signals that stimulate dendriticcell (DC) function. Here, we investigated the molecular signalsinduced by ARI treatment and found that this drug combination
increased the accessibility to regulatory sequences of ISGs andIRFs that were epigenetically silenced in both colorectal cancercells andDCs. Likewise, specific ARI-induced histonemethylationand acetylation changes marked epigenetically affected ISG pro-moters in bothmetastatic cancer cells and DCs. Analysis by ChIP-seq confirmed such ARI-induced epigenetically regulated IFNsignature. The activation of this signal endowed DCs with amarked migratory capability. Our results establish a direct corre-lation between reexpression of silenced ISGs by epigenetic controland ARI anticancer activity and provide new knowledge for thedevelopment of innovative combined therapeutic strategies forcolorectal cancer. Cancer Immunol Res; 5(7); 604–16. �2017 AACR.
IntroductionColorectal cancer is a multistep process characterized by the
accumulation of genetic and epigenetic abnormalities that givesrise to genomic instability andmutations of tumor suppressor andoncogenic genes (1). Although immunologic parameters haveprognostic value, most colorectal cancer lesions are not sensitive(2, 3) to immune checkpoint inhibitor–based therapies (4).
Epigenetic reprogramming (the changing methylation/acety-lation status of DNA and histones) contributes to colorectalcancer development, progression, and acquisition of drug resis-tance to conventional agents, through the dysregulated expressionof key genes involved in tumor suppression, cell-cycle regulation,and DNA repair (1). Hence, epidrugs, that is, drugs that target
proteins responsible for making epigenetic changes, such asthe FDA-approved DNA methyltransferase inhibitor (iDNMT)5-azacytidine (A) and histone deacetylase inhibitor (iHDAC)romidepsin (R), are rational andpromising therapeutic approaches(5–7). Both iDNMTs and iHDACs are immunomodulatory andcan induce signals in cancer cells that in turn boost antitumorimmune response (8). These drugs control epigenetic alterationsdriving the expressionof genes, often transcriptionally repressed, atmultiple levels including transcription factor activity and signaltransduction pathways. These modifications also determine theproduction of factors modulating the immune system by targetingdirectly epigenetic mechanisms of immune cells (9–11). Thus,iHDACs have been shown to enhance the immunogenicity ofcancer cells and iHDAC therapeutic efficacy relies on a functionalintact immune system (12). The induction of immune signals andapoptosis in cancer cells by iDNMTs has been linked to theactivation of IFN signaling, triggered by cytosolic sensing of dsRNA(13). Similarly, the antitumor efficacy of the same iDNMT againstcolorectal cancer stem cells has been associated with the inductionof viralmimicry, and specifically to the activationof the type III IFN(IFN-III) pathway (14).
The IFN-IIIs, also known as IL29 (IFNl1) and IL28A, B(IFNl2, 3), besides stimulating the expression of antiviral IFN-stimulated genes (ISG), modulate both innate and adaptiveimmunity and exert antitumor activities (15). Although epithelialcells constitutively produce IFNl, myeloid-lineage cells such asdendritic cells (DCs) are major sources in response to syntheticdsRNA or viral infections (16). The expression of the IFNlreceptor (encoded by the IFNLR1 gene) is restricted to epithelial
1Department of Oncology and Molecular Medicine, Istituto Superiore di Sanit�a,Rome, Italy. 2Institute for System Analysis and Computer Science "AntonioRuberti", Consiglio Nazionale delle Ricerche, Rome, Italy. 3Institute of Transla-tional Pharmacology, CNR, Rome, Italy. 4Department of Biology andBiotechnol-ogies "C. Darwin," Sapienza University, Rome, Italy. 5Institute of MolecularBiology and Pathology, Consiglio Nazionale delle Ricerche, Rome, Italy.
Note: Supplementary data for this article are available at Cancer ImmunologyResearch Online (http://cancerimmunolres.aacrjournals.org/).
Corresponding Authors: Alessandra Fragale and Lucia Gabriele, Istituto Super-iore di Sanit�a, Viale Regina Elena 299, Rome 00161, Italy. Phone: 3906-4990-3291; Fax: 3906-4990-2140; E-mail: [email protected];[email protected]
doi: 10.1158/2326-6066.CIR-17-0080
�2017 American Association for Cancer Research.
CancerImmunologyResearch
Cancer Immunol Res; 5(7) July 2017604
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

cells, hepatocytes, and most immune cell types, and is epigenet-ically regulated (17). IFNl has been reported to induce apoptosisin colorectal cancer cells andhas potential for clinical applicationsas a less toxic alternative of IFNa, or in combinationwith it, on thebasis of shared signaling between these two cytokines (18).
Crucial regulators of both IFN-I and IFN-III signaling areinterferon regulatory factors (IRF; ref. 19). Among these, IRF8directs the developmental program of DCs (19). IRF8 is silencedin a large collection of primary colorectal cancer lines and itsepigenetic inactivation is associated with apoptotic-resistant andmetastatic phenotype of several tumor cells (20–23). A keymechanism by which IRF8 controls melanoma metastatic pro-cesses is through regulation of the cross-talk between cancer andimmune cells (24). Similarly, IRF7, themajor regulator of the IFNpathway, is silenced in many primary tumor cells and metastasesand implicated in epigenetically modulated colorectal cancerprogression (13).
The tumor inflammatory milieu plays a pivotal role in colo-rectal cancer development and progression (25), inducing mul-tifactorialmechanisms of colorectal cancer immune evasion, suchas functional defects of immune cells such as DCs (26, 27).Soluble factors derived from tumor microenvironment interferewith DC differentiation and migration, thereby contributing toloss of DC stimulatory activity. Similarly, quantitative and func-tional impairments of circulating DCs may significantly limitantitumor immune response contributing to tumor immuneescape (27). We and others demonstrated that IFN-I exerts mul-tiple effects on human DCs, affecting their differentiation, mat-uration, and antigen presentation (28, 29). A key cytokine, IFNa2can enhance the chemoattractant and stimulatory capacity of DCstoward antitumor CD8þ T lymphocytes, by stimulating produc-tion of high amounts of chemokines and DC cross-priming at thetumor microenvironment (28).
IFNa2b (IFN, hereafter) can cooperate with romidepsin and5-azacytidine (AR to IFN, ARI) to induce direct and immune-mediated antitumor effects on colorectal cancer cells and colo-rectal cancer stem cells in vitro and in vivo (30). Here, we examinethe molecular mechanisms of the ARI combination and find thatit concomitantly targets cell death signals and immune cell func-tions in RAS-mutated colorectal cancer cell lines and in DCs,respectively. We show that the ARI combination causes reactiva-tion of IRF8, IRF7, iNOS, IFNl1, and IFNLR1 by targeting specificepigenetic modifications. The resultant remodeling activates apeculiar IFN-III signature in both tumor and immune cells,leading to the pronounced migration of DCs toward ARI-treatedcolorectal cancer cells.
Materials and MethodsCells
The two RAS-mutated colorectal cancer cell lines, SW480 ade-nocarcinoma and the metastatic SW620, derived from a lymphnode metastasis of same patient, were grown in RPMI1640(Euroclone), colorectal adenocarcinoma HT-29 cells, and thecolorectal carcinoma HCT116 cells were grown in McCoy 5A(Modified) Medium (Thermo Fisher), with 10% FBS. Cell lineswere a gift of Dr. Michele Signore, (Istituto Superiore di Sanit�a,Rome, Italy) who purchased all the colorectal cancer cell linesfromATCC, froze cells for few passages after the first thawing, andidentified them by STR. Cells were passaged within 6 months.Monocytes were isolated from PBMCs from healthy donor by
using CD14 microbeads (Miltenyi Biotec) yielding a populationat least a 98%pure, as assessed by FACS analysis (31). The CD14þ
monocyteswere cultured inRPMI1640 containing 10%FBS in thepresence of GM-CSF (50 ng/mL; PeproTech) and IL4 (500 U/mL;R&DSystems; ref. 31). After 5 days, DCswere treatedwith ARI andPolyI:C (pI:C; Sigma Aldrich) at 25 mg/mL for 24 hours.
Drugs5-Azacytidine was purchased from Sigma; romidepsin (or
FK228, or depsipeptide, or istodax) from Selleckchem; IFNa2b(IntronA) from MSD, hrIFNg was from PeproTech. Concentra-tions were azacytidine, 0.01 mmol/L; romidepsin, 2 nmol/L;IFNa2b, 10,000 IU/mL; IFNg , 20 ng/mL for colorectal cancercells; romidepsin, 1 nmol/L for DC treatment.
Western blottingExpression of specific proteins in total extracts was analyzed as
described (31). Nitrocellulose was then incubated with mAbs toIRF8 (sc-365041), IRF7 (sc-74472), and b-actin (sc-47778; SantaCruz Biotechnology), and anti-CXCR4 (ab2074; Abcam). Chemi-luminescent signals were detected by autoradiograpy or by Bio-spectrum (UVP, Ultra-VioletProducts Ltd).
Real-time PCRTotal RNA was extracted from cells using the RNeasy Mini kit
(Qiagen). RNA was DNase-I digested (Roche) and reverse tran-scribed as described (31). Quantitative PCR (qPCR) was per-formed in duplicate by the real-time fluorescence detectionmeth-od with the fluorescent DNA binding dye SYBR green (PowerSYBR Green PCR master kit; Applied Biosystems) using an ABIPRISM 7900 (Applied Biosystems). Values were normalizedrespect to the HPRT gene. Primers used for real-time PCR arereported in Supplementary Table S1.
Ectopic expression of IRF8 and IRF7 and IRF8 silencingCells were transfected with TransIT-2020 Transfection Reagent
(MirusBio) with pCDNA3.1 empty vector (EV) or containinghuman IRF8 or an IRF8-inactive mutant in which all 12 tyrosineresidues were mutated to phenylalanine (IRF8 12Y), provided byDr. E. Eklund (Comprehensive Cancer Center of NorthwesternUniversity, Chicago, IL; ref. 31) or with CMV-IRF7 (32). Eighthours later, cells were treated with ARI and grown for further48 hours. Then, part of them were counted with trypan blue todetermine percentage of cell viability (live total cell count/totalcell count), while aliquots were either analyzed for RNA expres-sion or stained for apoptosis detection as described below.Transfections were performed in duplicate and repeated fivetimes. SW620 cellswere transfectedwith themixof 3 IRF8SilencersiRNA Pre-designed (siRNA-ID# s7098, s7099, S7100) or thesame amount of siRNA # 1 Silencer Select Negative Control(siRNA catalog no. 4390843; Ambion) byDharmaFECT (ThermoFisher Scientific).
Apoptosis assayTo assess the level of apoptosis, cells were incubated with
Annexin V and PI (AnnexinV ApoAlert Assay, Clontech). Cellswere analyzed by flow cytometry (FACSCalibur, BD Biosciences).Flow cytometry experiments were performed on a FACSCanto II(BD Biosciences) and analyzed with FlowJo vX (TreeStar Inc.).
IRF8 Drives Immune-Derived Antitumor Effect of Epidrugs/IFN
www.aacrjournals.org Cancer Immunol Res; 5(7) July 2017 605
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

Chromatin immunoprecipitationChromatin immunoprecipitation (ChIP) assays were per-
formed as described previously (31). Briefly, cells were cross-linked with 1% formaldehyde and quenched in 0.125 mol/Lglycine. Cell lysates were sonicated and immunoprecipitatedwith normal rabbit serum (BD Biosciences) or anti-H3K9me2(ab1220, Abcam), anti-H3K4me3 (C42D8, Cell Signaling Tech-nology), or anti-H3acetylK9 (ab4441, Abcam). The immuno-precipitated DNA was eluted and amplified by qPCR. Valueswere normalized to corresponding input controls andexpressed as fold enrichment relative to normal rabbit serumfor each experiment. Primer sequences are reported in Supple-mentary Table S2.
ChIP-seq data analysisThe reagents for library preparation were used according to the
manufacturer's protocol (Illumina Inc). DNA sequencing wascarried out by using an Illumina HiSeq2000/2500 sequencer as50-bp single-end reads, following the manufacturer's protocols.Fastq files (about 20 million reads/sample) were then mappedand aligned to Ensembl human reference, build GRCh37, usingbowtie (version 1.1.1; ref. 33) with the following settings: twomismatch was allowed, maximum one location in the genomewas allowed, and the highest quality match was reported.H3K9me2-enriched regions in ARI-treated and untreated cellswere identified using MACS (version 2.1.0) with q-value cutoffof 0.05 by comparing the ChIP-seq data to its matching input
Figure 1.
ARI combination modulatesexpression of IRF8 in SW620 andSW480 colorectal cancer cells. IRF8(A) and IRF7 expression (C) wasevaluated by qPCR at 24, 48, 72, and96 hours after treatment withazacytidine (A), romidepsin (R), andIFN, individually or in combination andwith azacytidine þ IFNg . Data areexpressed as ratios of the expressionof individual genes and HPRT.Experimental data are expressed asmean � SD of three independentexperiments, each conditionperformed in triplicates. Analysiswith parametric two-way ANOVAfollowed by multiple comparisonTukey test allowed ARI conditioncomparison with indicated doubletreatment groups. ��� , P < 0.001;��, P < 0.01; � , P < 0.05. Western blotanalysis of IRF8 (B), and IRF7 (D), fromprotein lysates of SW620 or SW480cells after 96 and 24 hours oftreatment. Representative blots out ofthree are shown. Intensity of bandswas measured by densitometry andnormalized values on the b-actinvalues as loading control.
Fragale et al.
Cancer Immunol Res; 5(7) July 2017 Cancer Immunology Research606
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

control. To identify sites that are differentially enriched inH3K9me2 histone mark between ARI-treated and untreated cells,R Bioconductor (34; version 3.2.2; R Core Team, 2015) packageDiffBind (version 1.14.5; ref. 35) was applied to the peak data toexamine the correlation of ARI-treated and untreated samples andto identify statistically significant differentially bound sites usingthe DESeqmethod (36). The resulting filtered (P < 0.05) peak setswere assigned to a genomic region and associated with nearestgenes and genomic features using "annotatePeak" function ofBioconductor R package ChIPseeker considering a transcriptionstart site (TSS) region ranging from3,000bpupstream to1,000bpdownstream. To understand biological meaning of the selectedenriched regions, all the peaks that are located inside promoterregion were filtered and the genes associated were significantly(q value < 0.05) clustered by enrichment pathway analysis usingBioconductor R package ClusterProfiler (37) with annotation ofKyoto Encyclopedia of Genes and Genomes (KEGG) and Reac-tome Pathway Database.
Cell migrationIL4-DCs were left untreated or stimulated with ARI or pI:C and
24 hours later, washed, and seeded into the top compartment ofpre-equilibrated Transwell Permeable Inserts with polycarbonatemembrane with 8-mm pore size (Corning) that separated the topand bottom compartments. The bottom compartment was filledwith migration medium with 200 ng/mL CXCL12 (R&D Sys-
tems). Alternatively, the bottom compartments were filled withconditioned medium derived from SW620 cells treated or not for48 hours with ARI. After 3 hours, cells that had migrated to thebottom surface were collected and counted in ten different fields.Migration assays were performed in duplicate and repeated fourtimes (30).
Statistical analysisWhere appropriate, Student t tests, or parametric two-way
ANOVA followed by multiple comparison Tukey test was per-formed. All statistical analysis was done using native functions ofR language version 3.3.2. Differences in P values �0.05 wereconsidered statistically significant.
ResultsARI stimulated reexpressionof IFN-relatedmolecules and iNOS
IFN enhances the growth-inhibitory activity of 5-azacytidineand romidepsin on both metastatic cells and colorectal cancerstem cells (30). ARI treatment efficacy had been assayed in vivo, inNOD-SCID mice bearing colorectal cancer tumors (30). Thesingle drug dose used here was chosen on the basis of thoseprevious studies from our and other laboratories, a dose at whichin vivo toxicity of the triple treatment was totally absent. Tounderstand the molecular mechanisms underlying ARI-inducedeffects on colorectal cancer cells, we investigated whether specific
Figure 2.
ARI combination increases theexpression of IFN signature genes incolorectal cancer cells. mRNAs of iNOS(A), IFNLR1 (B), IFNl1 (C) wereanalyzed by qPCR at 42, 48, 72, and 96hours after treatment with azacytidine(A), romidepsin (R), and IFN,individually or in combination and withazacytidineþIFNg . Data are expressedas ratios of the expression of individualgenes and HPRT. Experimental dataare expressed as mean � SD of threeindependent experiments, eachcondition performed in triplicates.Analysis with parametric two-wayANOVA followed by multiplecomparison Tukey test allowed ARIcondition comparison withindicated double treatment groups.��� , P < 0.001; �� , P < 0.01; � , P < 0.05.
IRF8 Drives Immune-Derived Antitumor Effect of Epidrugs/IFN
www.aacrjournals.org Cancer Immunol Res; 5(7) July 2017 607
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

molecular signals characterized the selective antitumor effects ofARI treatment against the highly metastatic SW620 cells, withrespect to the poorly invasive SW480 cells. The epigenetic silenc-ing of the tumor suppressor gene IRF8 has been described as keyevent in colorectal cancer (20–23). Although IFN, as singletreatment, did not stimulate any IRF8 expression, it stronglycooperated with 5-azacytidine and romidepsin to elevate thistranscription factor specifically in highly metastatic SW620 cells,with respect to untreated cells, over 96 hours of treatment(Fig. 1A), increasing IRF8 protein expression to the level observed
in poorly invasive SW480 cells (Fig. 1B). IRF8 induction by ARItreatment was significant (P < 0.001) as compared with the 5-azacytidine þ IFNg-positive control, considered strong inducersfor IRF8 restoration (Fig. 1A; ref. 21). Similar resultswereobtainedin HT-29 adenocarcinoma cells and HCT-116 colorectal cancercells (Supplementary Fig. S1A). In contrast, IFN alone was suffi-cient to increase transient IRF7 expression, particularly in SW620cells (Fig. 1C). However, in this experimental condition, IRF7mRNA declined over time, whereas the addition of AR to IFN(ARI) was the only treatment able to sustain the expression of this
Figure 3.
IRF8 and IRF7 overexpression increases apoptosis in ARI-treated metastatic SW620 cells. Cells were transfected with human WT IRF8 or 12Y IRF8 (A) orwith EV or IRF7 vectors (B) and treated with ARI for 48 hours. Early apoptotic cells (Annexin Vþ/PI�), late apoptotic cells (Annexin Vþ/PIþ), necrotic cells(Annexin V�/PIþ), and vital cells (Annexin V�/PI�) can be distinguished. One representative experiment out of five independent transfection experiments isshown. Part of the cells as above were analyzed for expression of iNOS (C) and IFNL1R (D) by qPCR. Data are expressed as ratios between the expressionof individual genes and HPRT, as mean � SD of two independent experiments performed in triplicates.
Fragale et al.
Cancer Immunol Res; 5(7) July 2017 Cancer Immunology Research608
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080
transcription factor up to 96hours (Fig. 1C).Western blot analysisconfirmed the elevated expression of IRF7 after long exposure toARI in SW620 cells (Fig. 1D). Next, we investigated the modula-tion of ISGs. Inducible nitric oxide synthase (iNOS) expressionwas increased more in metastatic SW620, with respect to poorlyinvasive SW480 colorectal cancer cells, mainly following ARIcombined treatment. This confirmed the double control of iNOSby IFN and epigenetics, which is likely to occur through thereactivation of transcription factors such as IRF8 (Fig. 2A). Theexpression of IFN signal–relatedmolecules was further evaluated.Although a predominant role of histone deacetylation has beenreported for the IFNb promoter (38), IFNb was only transientlyincreased in SW620 and SW480 cells (Supplementary Fig. S1D).The IFNAR1, IFNAR2 receptors, as well as IFNa, were not signif-icantlymodulated at any experimental condition (SupplementaryFig. S1B, S1C, and S1E). Conversely, although high IFNl1 wasinduced in SW480 cells only by the combination of IFN andepidrugs, strongupmodulationof IFNlR1, andmoderate increaseof IFNl1, were observed in metastatic SW620 cells in the sameexperimental conditions (Fig. 2B and C). The expression of IFNl1in both cell lines, as well as of IFNlR1 and iNOS in SW620 cells,was retained up to 96 hours only after ARI treatment (Fig. 2).Therefore, IFN and epigenetics cooperate in preserving the expres-sion of key molecular factors.
Because IRF8 and IRF7, two key members of IRF family, werefound to be specifically modulated by ARI in metastatic colo-rectal cancer cells, we further investigated their function in thiscontext by carrying out overexpression assays of these factors inboth SW620 and SW480 cells. Although ectopic expression ofIRF8 per se increased the rate of basal apoptosis in bothcolorectal cancer cell types, treatment of IRF8-overexpressingcells with ARI stimulated significantly more apoptotic celldeath of the metastatic colorectal cancer cells than of the poorlyinvasive cells (Fig. 3A). Similarly, although the basal apoptosisrate of both colorectal cancer cell lines did not change uponIRF7 overexpression, treatment of cells with ARI generated 5- to10-fold more apoptosis only in SW620 cells (Fig. 3B). Bothcolorectal cancer cell lines transfected with functionally inactive12Y-IRF8 mutant or EV, untreated, or ARI-treated, showedlimited apoptosis (Fig. 3A and B); these cells were as viableas untransfected cells, independent of ARI treatment (Supple-mentary Fig. S2A). IRF8 overexpression slightly decreased cellviability in both untreated cell lines, whereas ARI treatmentreduced viability of SW620 and SW480 cells (P < 0.05; Sup-plementary Fig. S2A). Unlike IRF8, the overexpression of IRF7decreased cell viability only in both ARI-treated colorectalcancer cell lines (Supplementary Fig. S2B).
We investigated whether IRF8 and IRF7 overexpression couldaffect that of iNOS and IFNl1. iNOS expression was upregulatedabout 400-fold by transfecting selectively IRF8 in metastatic cells(Fig. 3C). In contrast, IFNl1was upregulated byoverexpression of
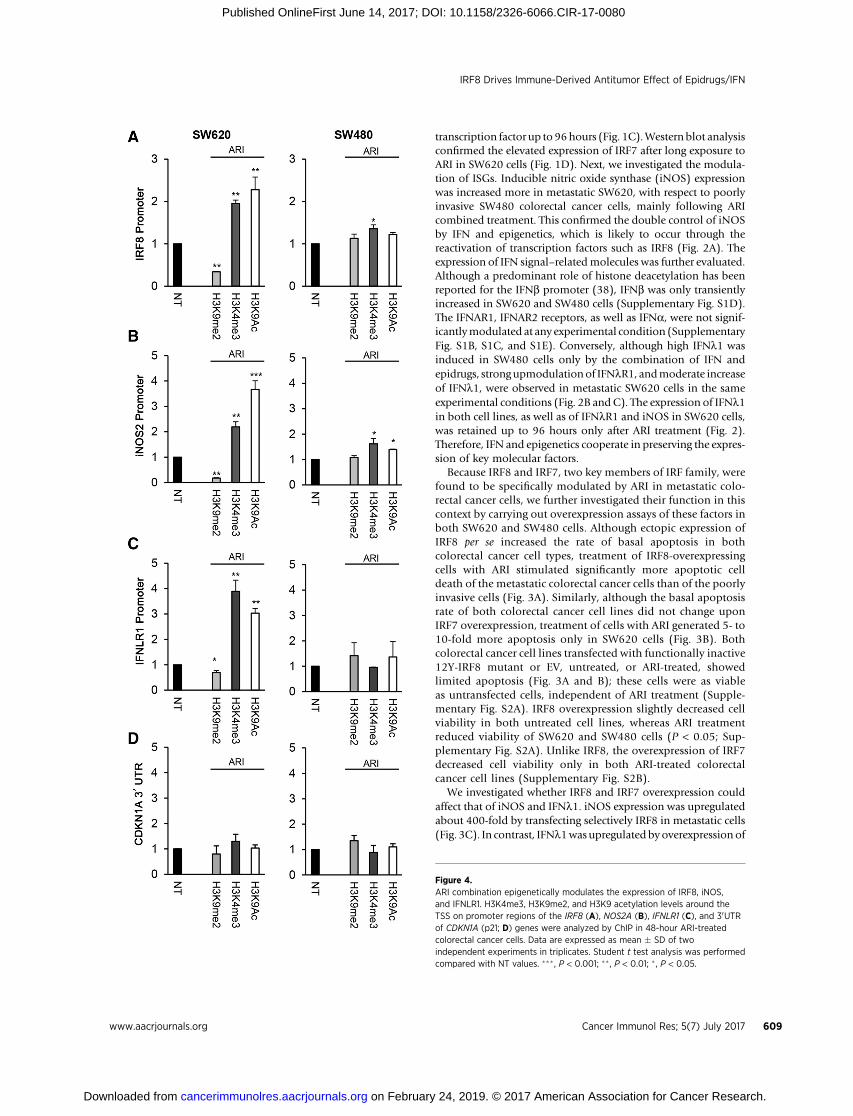
Figure 4.ARI combination epigenetically modulates the expression of IRF8, iNOS,and IFNLR1. H3K4me3, H3K9me2, and H3K9 acetylation levels around theTSS on promoter regions of the IRF8 (A), NOS2A (B), IFNLR1 (C), and 30UTRof CDKN1A (p21; D) genes were analyzed by ChIP in 48-hour ARI-treatedcolorectal cancer cells. Data are expressed as mean � SD of twoindependent experiments in triplicates. Student t test analysis was performedcompared with NT values. ��� , P < 0.001; �� , P < 0.01; � , P < 0.05.
IRF8 Drives Immune-Derived Antitumor Effect of Epidrugs/IFN
www.aacrjournals.org Cancer Immunol Res; 5(7) July 2017 609
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080
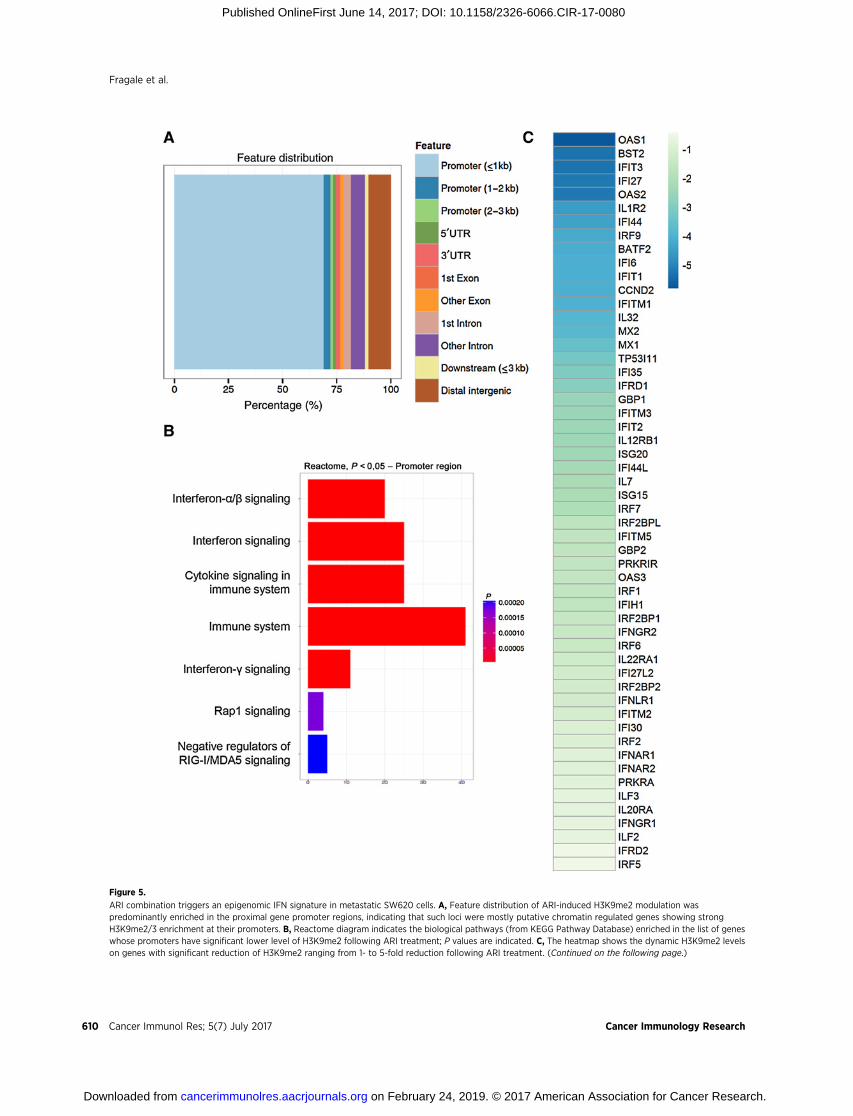
Figure 5.
ARI combination triggers an epigenomic IFN signature in metastatic SW620 cells. A, Feature distribution of ARI-induced H3K9me2 modulation waspredominantly enriched in the proximal gene promoter regions, indicating that such loci were mostly putative chromatin regulated genes showing strongH3K9me2/3 enrichment at their promoters. B, Reactome diagram indicates the biological pathways (from KEGG Pathway Database) enriched in the list of geneswhose promoters have significant lower level of H3K9me2 following ARI treatment; P values are indicated. C, The heatmap shows the dynamic H3K9me2 levelson genes with significant reduction of H3K9me2 ranging from 1- to 5-fold reduction following ARI treatment. (Continued on the following page.)
Fragale et al.
Cancer Immunol Res; 5(7) July 2017 Cancer Immunology Research610
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

both IRF7 and IRF8 in both colorectal cancer cell lines (Fig. 3D),consistent with expression analysis of IFNl1 in SW480 cells (Fig.2C). To further elucidate the IRF8 antitumor role in SW620 cells,we silenced IRF8 in ARI-treated cells. Expression of iNOS and theproapoptotic BAX genes (both direct targets of IRF8) were signif-icantly reduced (Supplementary Fig. S2C).
ARI reprogrammed metastatic colorectal cancer cells viaepigenetic control
Because dimethylation of lysine 9 on histone 3 (H3K9me2) is ahallmark event regulating the expression IFN-I–inducible genes(39), we investigated whether this modification, as well as otherrelevant altered epigenetic changes implicated in transcriptionalregulation, was involved in the reexpression of key genes by ARI.To this end, we evaluated the amount of H3K9me2, H3K4me3,and H3K9Ac at IRF8, NOS2A (iNOS), and IFNLR1 gene promo-ters in colorectal cancer cell lines byChIP assay. The 30-UTR regionof the CDKN1A gene (p21) was a negative control (40). ARIdecreased H3K9me2 and increased both H3K4me3 andH3K9Ac,all markers of active gene transcription, specifically on the pro-moter of IRF8, NOS2A, and IFNLR1 genes inmetastatic cells, withonly limited changes in SW480 cells (Fig. 4). As expected, nosignificant methylation or acetylation modification of histone
residues was observed in 30UTR of the CDKN1A gene by ARI (Fig.4D). We examined H3K9me2 to determine the mechanismunderlying the antitumor effects exerted by ARI in metastaticcells, because acetylation and methylation are mutually exclusiveat K9, andHDAC activity is crucial in determining H3K9 genome-wide distribution (39). To get a more extensive view of theinduced epigenomic changes, we performed genome-wideChIP-seq analysis for this histone modification. The extensivelist of genes involved are listed in Supplementary Table S3.The distribution of H3K9me2 peaks identified in the ChIP-seqexperiments was grouped into 11 classes that included both genicand intergenic regions. The distribution of H3K9me2 histonemodification peaks that were associated with the differentgenome categories was predominantly enriched in the geneproximal promoter regions (around 70%), likely correspondingto chromatin-regulated genes (Fig. 5A). Promoters of genesinvolved in IFN signaling, immune response, and cytokine sig-naling showed an overall decrease in H3K9me2 occupancy fol-lowing ARI treatment, indicating their potential transcriptionalactivation (Fig. 5B). The heatmap shows that H3K9me2 changedon many ISG and cytokine promoters (Fig. 5C). The enrichmentin promoters belonging to particular KEGG (Kyoto Encyclopediaof Genes and Genomes) pathways highlighted the IFN signature
Figure 5.
(Continued. ) D, IFN signaling (modified from the HCV KEGG pathways) highlighting the related genes, which show reduced H3K9me2 following ARI treatment.
IRF8 Drives Immune-Derived Antitumor Effect of Epidrugs/IFN
www.aacrjournals.org Cancer Immunol Res; 5(7) July 2017 611
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

induced by ARI treatment. H3K9me2 occupancy was uniformlydecreased in IFN-controlled genes that appear in key positions inpathways of response to viral infections and genes related tosurvival and apoptosis (Fig. 5D). Besides the evident IFN signa-ture, the list of gene promoters with decreased H3K9me2 wasenriched for some other biological processes (SupplementaryFig. S3).
ARI-treated DCs acquired marked epigenetic IFN signaturesand migratory ability
Given the modulation of immune-related genes by ARI, weinvestigated the effects of this combination treatment on DCs,immune cells that counter cancer cells by stimulating an antitu-mor immune response. We first evaluated whether ARI couldmodulate the expression of IRF8, a key regulator of DC differen-tiation. We generated DCs from peripheral monocytes by incu-bationwith GM-CSF and IL4 for 5 days (IL4-DC) and then treatedthem for a further 24 hourswith single or combined agents or pI:C(a positive inducer of IFN andDCmigration andmaturation). Notoxicity to DCs was observed by ARI treatment, as evaluated bytrypan blue exclusion (Supplementary Fig. S4A), or by stainingwith SYTOX Green (Supplementary Fig. S4B). Not only was theviability of these cells maintained, but ARI treatment enhancedthe expression of the DC activation markers CD83 and CD86(Supplementary Fig. S4C). Upon romidepsin and IFN singletreatments, IRF8 transcripts were increased, but romidepsin andIFN (RI) or ARI combined treatments enhanced this induction upto 100- and 150-fold, respectively (Fig. 6A). Next, we assessed theexpression of the other major factors modulated by ARI incolorectal cancermetastatic cells. Besides the significant upregula-tion of IRF7, iNOS, IFNl1, and IFNlR1, we also found that ARIinduced increased TLR3 and TLR7mRNA, suggesting the skewingof treatedDCs toward an activated phenotype (Fig. 6B). A detailedanalysis of IFN genes control by single and combined drugsrevealed that romidepsin and I, as single treatments, stimulatedthe expression of IFNl1, IFNLR1, and IFNb, whereas the RIcombination was sufficient to generate additive effects on all IFNgenes; however, ARI was the most effective treatment (Supple-mentary Fig. S4). The strong induction of IRF8 and IRF7 by ARItreatment in DC was further confirmed by Western blot analysis,which revealed increased CXCR4 proteins in the same experimen-tal condition (Fig. 6C). The additional analysis of the IRF8promoter by ChIP for H3K9me2, H3K4me3, and H3K9Acrevealed that ARI can reprogram the expression of this gene inDCs by epigenetic modulation to the same extent as in metastaticSW620 cells (Fig. 6D). The functional effects of the overall ARI-induced DC transcriptional reprogramming were assessed. Giventhe elevated amount of ARI-induced CXCR4, untreated or treatedDC with either ARI or pI:C were assayed for migration towardCXCL12. ARI-treated IL4-DCs acquired the specific competence tomigrate toward CXCL12; in contrast, pI:C-treated IL4-DCs exhib-ited per se a strong ability to migrate, independent of the presenceof CXCL12 (Fig. 6E). Finally, we evaluated whether simultaneoustreatment of SW620 cells and IL4-DCs with ARI could impact thedialog between these cells. DC migration assays were performedin the presence of conditioned medium of untreated or ARI-treated (CM-ARI) SW620 cells, for 48 hours. CM-ARI was theonly treatment that conferred migratory competence to IL4-DCsto the same extent as pI:C–treatedDCs (Fig. 6F), suggesting that inthe course of ARI treatment, the maximum DC-mediated antitu-
mor benefit can be achieved in the context of cancer–immunesystem cross-talk.
DiscussionEpigenetic drugs have the attractive ability to prime cancer cells
to be more responsive to the subsequent therapies (5–8). Pre-clinical and translational research is under way to test whetherepidrugs in combination with other antitumor agents might offeran alternative approach to render targeted immunotherapy and/or conventional protocols for solid tumors more effective (5–8).Low dose treatment with two epidrugs, 5-azacytidine and romi-depsin, can potentiate the antitumor effect exerted by IFN, whichunderscores the potential of the low-dose ARI combination as atreatment for controlling the metastatic behavior of colorectalcancer (30).Here, we established themechanism triggered byARI,highlighting a crucial role for IRF8, whose expression is underepigenetic control, in both metastatic colorectal cancer cells andDC. Consistent with the findings that both iHDACs and iDNMTsenhance IRF8 expression (20–23), our data establish that thecombination of romidepsin and azacytidine restores IRF8 expres-sion in an additive fashion and that this effect is synergistic incombination with IFN. Selective epigenetic-regulated expressionof IRF8 in highly metastatic colorectal cancer cells is associatedwith the reexpression of IRF8 target genes with antitumor activ-ities and induction of apoptosis.
Among the genes targeted by IRF8 that are strongly inducedby ARI in an epigenetic-dependent manner is iNOS, which hasboth anti- and protumoral effects (41). Its function is depen-dent upon its concentration, the spatiotemporality of theaction, and the cellular redox state of its metabolite NO, aswell as the timing of the apoptotic stimulus (41). In fact,modest concentrations of NO have protumoral action, whereashigh quickly inducible concentrations act by promoting apo-ptosis and inhibiting angiogenesis, thus driving antitumorresponses. In our study, treatment of metastatic colorectalcancer cells, but not poorly invasive ones, with the ARI com-bination strongly induced iNOS expression, suggesting iNOS asan initiator of the antitumor response, potentially as result ofepigenetic activation of the IRF8–iNOS–NO axis. Indeed, IRF8overexpression in metastatic colorectal cancer cells was foundto selectively induce large amount of iNOS. In agreement withour findings, the upstream portion of the NOS2A gene pro-moter contains IFN-stimulated response elements (ISREs)where the IRF8/IRF1 complex was reported to play a role(42). The cooperation between epidrugs and IFN in inducingmolecular and functional proapoptotic changes in metastaticSW620 cells lasted over 96 hours, supporting the importance ofthis treatment in maintaining active cellular signals that met-astatic cells tend to shut down by acquiring drug resistance. Thistight connection between IRF8 and iNOS also occurred in DCs,suggesting ARI as a strong stimulator of DC maturation andactivation (19).
Distinct from the need for the triple combination to stimulatestrong persistent IRF8 expression, IFN alone was sufficient tostrongly induce IRF7, whereas its combination with epigeneticswas essential for maintaining high concentrations of this tran-scription factor in metastatic colorectal cancer cells. Ectopicexpression of IRF7 in these cells resulted in an increased apoptoticrate, in agreement with a previous report showing that theactivation of the extrinsic pathway of apoptosis in response to
Fragale et al.
Cancer Immunol Res; 5(7) July 2017 Cancer Immunology Research612
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

Figure 6.
ARI treatment induces IRF and IFN-related genes and confer migration capability to IL4-DC. A, IRF8 expression by qPCR after 24 hours of romidepsin (R),I, RI, ARI, and pI:C treatment. B, Indicated gene expression by qPCR of IL4-DC treated with ARI. Data are expressed as ratios of the expression of individualgenes and HPRT. Experimental data are expressed as mean � SD of three independent experiments performed in duplicates. C, Western blot analysis ofIRF8, IRF7, and CXCR4 expression in IL4-DC treated with ARI treatment or pI:C. b-Actin as a loading control. D, ChIP analysis shows the levels of H3K9me2,H3K4me3, and H3K9acetylated on the promoter of IRF8. Migration ability of ARI-DC or pI:C-DC was assayed with CXCL12 (E), or with conditioned mediumfrom untreated (CM) or treated with ARI (CM-ARI) SW620 cells (F). Experimental data are expressed as mean � SD of two independent experimentsperformed in triplicates. Student t test analysis was performed compared with NT values. ��� , P < 0.001; �� , P < 0.01; � , P < 0.05.
IRF8 Drives Immune-Derived Antitumor Effect of Epidrugs/IFN
www.aacrjournals.org Cancer Immunol Res; 5(7) July 2017 613
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

IFNa is also IRF-7–dependent (43). Treatment of DCs with ARIalso led to the induction of elevated IRF7, whoseMYC-dependentexpression is critical in pDCs for IFN-I production (44). Therefore,the induction and the maintenance of IRF7 expression in bothcancer cells and DCs may contribute to the activation of pairedantitumor properties by ARI.
We also found that the ARI combination induced a clear-cutstimulation of the IFN-III axis, particularly in metastatic cells. TheIFN-III system is potently antiproliferative andperhaps a powerfultherapeutic option (15). Its antitumor role has been described forcolorectal cancer cells treated with low doses of 5-azacytidine(14). Here we report that ARI combined treatment could induceboth IFNl1 and IFNLR1 specifically in metastatic SW620 cells,whereas significant production of only IFNl1was found in poorlyinvasive SW480 cells, suggesting the occurrence of IFNl-mediatedantitumor effects only in the former cells. Both IRF8 and IRF7regulated the expression of this cytokine, highlighting an IRF-dependent control of the expression of this factor also in cancer(45). Findings showing that low-dose of iDNMTs targets colo-rectal cancer cells by inducing IFN response also via activation ofIRF7 are consistent with this (13, 14). IFNLR1 expression ispositively regulated by iDNMTs and iHDACs in a cell type–specific fashion (17), confirming epigeneticmechanisms as essen-tial in regulating the IFN-III axis (14). We envisage that ARItreatment may increase accessibility of IFNLR1 to transcriptionactivators, promoting its elevated expression in highly nonre-sponsive SW620 cells and rendering them sensitive to the tumorsuppressor activities of IFNl (14, 17). Conversely, these events donot occur in poorly invasive colorectal cancer cells. From amechanistic point of view, as stated above and similarly to IRF8
and iNOS, the expression of IFNLR1 was tightly tied to themodulation of H3K9me2, H3K9me3, and H3K9Ac epigeneticalterations. Although the presence of H3K9me2 around the TSSgene promoter is associated with transcriptional repression, theextent of this epigenetic alteration at individual gene promoters,including IFN-related sequences, is a crucial factor in determiningtheir transcription activity (39). Some evidence indicates "natu-ral" H3K9me2 depletion as an epigenetic hallmark of activeproinflammatory genes (39). High levels of H3K9me2 at IFNand ISG promoters inversely correlate with the amplitude of theexpression of these genes in fibroblasts and DCs, highlighting therole of this epigenetic modification in counteracting IFN stimu-lation. In contrast, H3K4me3 on TSS of gene promoters is gen-erally associated with transcriptional activation, as H3K9Ac (46).Here, we report the simultaneous downmodulation of H3K9me2and upmodulation of H3K4me3 and H3K9Ac on the promotersof IRF8, NOS2A, and IFNLR1 by ARI treatment, specifically inSW620 cells, suggesting the activation of a coordinate epigeneticregulation of these genes ultimately potentiating complementaryand interconnected antitumor signals. We envisage that in a firstphase ARI treatment may primarily favor the expression of reg-ulators of IFN signaling, such as IRF8 and IRF7, that in turn maytake advantage ofmultiple epigenetic alterations on the promoterof target genes. These coordinate events ultimately lead to theexpression of molecules mediating antitumor effects, such asIFNl, IFNLR1, and iNOS. The cooperation between the twodiverse epigenetic drugs in this study is supported by severalstudies on promoters of numerous genes, including p16, SALL3,andGATA4, in colorectal cancer cell lines, suggesting the ability ofromidepsin to decrease the recruitment of DNMT1 to these sites,
Figure 7.
ARI treatment reactivates IFNsignaling in metastatic colorectalcancer cells and in DC. In colorectalcancer cells, ARI therapy decreasesH3K9me2 and increases H3K4me3andH3K9Acetylated levels; stimulatesreexpression of silenced genes such asIRF8, IFNLR1, and iNOS; and inducesapoptosis and immunogenic celldeath. Likewise, in immature DC (iDC)ARI therapy reactivates genes such asIRF8, iNOS, IFNLR1, and IFNl1 andconcomitantly stimulates CXCR4expression and ARI-DC migrationcapability selectively towardapoptotic/dying ARI-treatedcolorectal cancer cells.
Cancer Immunol Res; 5(7) July 2017 Cancer Immunology Research614
Fragale et al.
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

thus inducing demethylation and reactivation (47). A further levelof interaction between epigenetic drugs occurs through the sup-pressionof histonemethyltransferasesG9Aand SUV39H1expres-sion by romidepsin, which in turn results in a decrease ofH3K9me2/me3 around the promoters mentioned above (47).Inactivation of G9A, which is essential for the deposition ofH3K9me2, can convert fibroblasts into highly potent IFN-pro-ducing cells (39), confirmed by our ChIP-seq data showing thetight correlation between a decrease in H3K9me2 and an IFNsignature in metastatic cells upon ARI treatment. This IFN signa-ture is evident in the gene ontology classification of affectedpromoters and in the pathways enriched among demethylatedgene promoters.
Epigenetic alterations of immune cells, including DCs, arepivotal in reprogramming the milieu of tumor microenviron-ments to restore immune surveillance programs (11, 48). Wereport that ARI-DCs gain the capability to upmodulate the expres-sion of critical mediators of their effector function such as IRF7,IRF8, IFNl1, IFNLR1, iNOS, TLR3, and TLR7. We can speculatethat upon ARI exposure, different immune signals may convergein DCs to induce both IFN-I and IFN-III. These data have partic-ular relevance because IFN-I can act in an autocrine/paracrinemanner to activate DCs (19). All of these signals converge in thecharacterization of DCs in their ability to respond to inflamma-tory stimuli with effector functions. Indeed, the ARI-induced DCactivated phenotype correlated well with the enhanced capabilityof these cells to specifically migrate toward ARI-treated colorectalcancer cells. One of the most important factors regulating DC–cancer cell interactions is IRF8 (49), which acts through epigeneticregulation (50). We previously reported that IRF8 expression isprogressively downmodulated during melanoma growth in miceand in humanmetastatic melanoma cells with respect to primarytumors. Moreover, we demonstrated that treatment of tumorswith azacytidine stimulates IRF8 reexpression, which establishesappropriate intratumoral chemokines and chemokine receptorprofiles thatwould in turn control immune infiltration and tumorgrowth (24). On the basis of these insights, we reported that theARI combination downregulated the chemokine receptor CXCR4in metastatic colorectal cancer cells controlling their migrationability (30). Here, we report that ARI treatment stimulates IRF8expression in DCs through the modulation of specific epigenetic
modifications in the promoter. This event is associated withincreased CXCR4, which increases the ability of DCs to migratetoward its ligand CXCL12 and specifically toward ARI-treatedcolorectal cancer cells. Therefore, we envisage that ARI treatmenthas double potential to reprogram the transcription of metastaticcells and to dictate DC maturation and function, directly or viafactors released bydrug-treated cancer cells (Fig. 7). Thesefindingssuggest the strong therapeutic potential of this approach toregulate the cross-talk between cancer and immune cells in thetumor microenvironment and modulating key signals that regu-late cell trafficking.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: A. Fragale, E. Toschi, C. Presutti, L. GabrieleDevelopment of methodology: A. Fragale, G. Romagnoli, M. Buoncervello,C. Giuliani, S. Parlato, C. Leone, M. De Angelis, I. Canini, E. ToschiAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): M. Buoncervello, C. Leone, M. De AngelisAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis):A. Fragale, G. Romagnoli, V. Licursi,M. Buoncervello,G. Del Vecchio, S. Parlato, C. Leone, M. De Angelis, I. Capone, C. Presutti,L. GabrieleWriting, review, and/or revision of the manuscript: A. Fragale, G. Romagnoli,F. Belardelli, R. Negri, I. Capone, C. Presutti, L. GabrieleStudy supervision: A. Fragale, E. Toschi, L. Gabriele
AcknowledgmentsWe are grateful to Dr. Edvige Perrotti for excellent technical assistance.
Grant SupportThe research was supported by grants from the Italian Association for
Research against Cancer (AIRC) grant #11610 (to L. Gabriele) and AIRC grant#16891 (to F. Belardelli).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received February 8, 2017; revised April 12, 2017; accepted June 6, 2017;published OnlineFirst June 14, 2017.
References1. Okugawa Y, GradyWM, Goel A. Epigenetic alterations in colorectal cancer:
emerging biomarkers. Gastroenterology 2015;149:1204–25.2. XiaoY, FreemanGJ. Themicrosatellite instable subset of colorectal cancer is
a particularly good candidate for checkpoint blockade immunotherapy.Cancer Discov 2015;5:16–8.
3. Le DT, Uram JN,WangH, Bartlett BR, KemberlingH, Eyring AD, et al. PD-1blockade in tumors with mismatch-repair deficiency. N Engl J Med2015;372:2509–20.
4. Kroemer G, Galluzzi L, Zitvogel L, FridmanWH. Colorectal cancer: the firstneoplasia found to be under immunosurveillance and the last one torespond to immunotherapy? Oncoimmunology 2015;4:e1058597.
5. Juo YY, Gong XJ, Mishra A, Cui X, Baylin SB, Azad NS, et al. Epigenetictherapy for solid tumors: from bench science to clinical trials. Epigenomics2015;7:215–35.
6. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to ther-apy. Cell 2012;150:12–27.
7. Vaiopoulos AG, Athanasoula K, Papavassiliou AG. Epigenetic modifica-tions in colorectal cancer: molecular insights and therapeutic challenges.Biochim Biophys Acta 2014;1842:971–80.
8. Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining epigeneticand immunotherapy to combat cancer. Cancer Res 2016;76:1683–9.
9. Li X, Mei Q, Nie J, Fu X, Han W. Decitabine: a promising epi-immu-notherapeutic agent in solid tumors. Expert Rev Clin Immunol 2015;11:363–75.
10. Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transientlow doses of DNA-demethylating agents exert durable antitumoreffects on hematological and epithelial tumor cells. Cancer Cell 2012;21:430–46.
11. Logie C, Stunnenberg HG. Epigenetic memory: a macrophage perspective.Semin Immunol 2016;28:359–67.
12. West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDACinhibitors require the immune system. Oncoimmunology 2014;3:e27414.
13. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al.Inhibiting DNA methylation causes an interferon response in cancer viadsRNA including endogenous retroviruses. Cell 2015;162:974–86.
14. Roulois D, LooYauH, Singhania R,WangY,DaneshA, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mim-icry by endogenous transcripts. Cell 2015;162:961–73.
www.aacrjournals.org Cancer Immunol Res; 5(7) July 2017 615
IRF8 Drives Immune-Derived Antitumor Effect of Epidrugs/IFN
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

15. Lasfar A, Zloza A, Cohen-Solal KA. IFN-lambda therapy: current status andfuture perspectives. Drug Discov Today 2016;21:167–71.
16. LauterbachH,Bathke B,Gilles S, Traidl-HoffmannC, Luber CA, FejerG, et al.Mouse CD8alphaþ DCs and human BDCA3þ DCs are major producers ofIFN-lambda in response to poly IC. J Exp Med 2010;207:2703–17.
17. Ding S, Khoury-Hanold W, Iwasaki A, Robek MD. Epigenetic reprogram-ming of the type III interferon response potentiates antiviral activity andsuppresses tumor growth. PLoS Biol 2014;12:e1001758.
18. Li W, Lewis-Antes A, Huang J, Balan M, Kotenko SV. Regulation ofapoptosis by type III interferons. Cell Prolif 2008;41:960–79.
19. Gabriele L,OzatoK. The role of the interferon regulatory factor (IRF) familyin dendritic cell development and function. Cytokine Growth Factor Rev2007;18:503–10.
20. Zhang Q, Zhang L, Li L, Wang Z, Ying J, Fan Y, et al. Interferon regulatoryfactor 8 functions as a tumor suppressor in renal cell carcinoma and itspromoter methylation is associated with patient poor prognosis. CancerLett 2014;354:227–34.
21. Yang D, Thangaraju M, Greeneltch K, Browning DD, Schoenlein PV,Tamura T, et al. Repression of IFN regulatory factor 8 by DNAmethylationis a molecular determinant of apoptotic resistance and metastatic pheno-type in metastatic tumor cells. Cancer Res 2007;67:3301–9.
22. Lee KY, Geng H, Ng KM, Yu J, van Hasselt A, Cao Y, et al. Epigeneticdisruption of interferon-gamma response through silencing the tumorsuppressor interferon regulatory factor 8 in nasopharyngeal, esophagealand multiple other carcinomas. Oncogene 2008;27:5267–76.
23. Banik D, Khan AN, Walseng E, Segal BH, Abrams SI. Interferon regulatoryfactor-8 is important for histone deacetylase inhibitor-mediated antitumoractivity. PLoS One 2012;7:e45422.
24. Mattei F, Schiavoni G, Sestili P, Spadaro F, Fragale A, Sistigu A, et al. IRF-8controlsmelanoma progression by regulating the cross talk between cancerand immune cells within the tumor microenvironment. Neoplasia 2012;14:1223–35.
25. Wang K, Karin M. Tumor-elicited inflammation and colorectal cancer. AdvCancer Res 2015;128:173–96.
26. Mager LF, Wasmer MH, Rau TT, Krebs P. Cytokine-induced modulation ofcolorectal cancer. Front Oncol 2016;6:96.
27. LegitimoA,Consolini R, Failli A,OrsiniG, Spisni R.Dendritic cell defects inthe colorectal cancer. Hum Vaccin Immunother 2014;10:3224–35.
28. Schiavoni G, Mattei F, Gabriele L. Type I interferons as stimulators of DC-mediated cross-priming: impact on anti-tumor response. Front Immunol2013;4:483.
29. Parlato S, Romagnoli G, Spadaro F, Canini I, Sirabella P, Borghi P, et al.LOX-1 as a natural IFN-alpha-mediated signal for apoptotic cell uptakeand antigen presentation in dendritic cells. Blood 2010;115:1554–63.
30. Buoncervello M, Romagnoli G, Buccarelli M, Fragale A, Toschi E, Parlato S,et al. IFN-alpha potentiates the direct and immune-mediated antitumoreffects of epigenetic drugs on both metastatic and stem cells of colorectalcancer. Oncotarget 2016;7:26361–73.
31. Fragale A, Stellacci E, Ilari R, Remoli AL, Lanciotti A, Perrotti E, et al. Criticalrole of IRF-8 in negative regulation of TLR3 expression by Src homology 2domain-containing protein tyrosine phosphatase-2 activity in humanmyeloid dendritic cells. J Immunol 2011;186:1951–62.
32. SgarbantiM,Marsili G, Remoli AL, Orsatti R, Battistini A. IRF-7: new role inthe regulation of genes involved in adaptive immunity. Ann N Y Acad Sci2007;1095:325–33.
33. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.Genome Biol 2009;10:R25.
34. Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al.Orchestrating high-throughput genomic analysis with Bioconductor. NatMethods 2015;12:115–21.
35. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ,et al. Differential oestrogen receptor binding is associated with clinicaloutcome in breast cancer. Nature 2012;481:389–93.
36. Anders S, Huber W. Differential expression analysis for sequence countdata. Genome Biol 2010;11:R106.
37. Yu G,Wang LG, Han Y, HeQY. clusterProfiler: an R package for comparingbiological themes among gene clusters. Omics 2012;16:284–7.
38. Shestakova E, Bandu MT, Doly J, Bonnefoy E. Inhibition of histonedeacetylation induces constitutive derepression of the beta interferonpromoter and confers antiviral activity. J Virol 2001;75:3444–52.
39. Fang TC, Schaefer U,Mecklenbrauker I, StienenA,Dewell S, ChenMS, et al.Histone H3 lysine 9 di-methylation as an epigenetic signature of theinterferon response. J Exp Med 2012;209:661–9.
40. Choi WI, Jeon BN, Yoon JH, Koh DI, Kim MH, Yu MY, et al. The proto-oncoprotein FBI-1 interacts with MBD3 to recruit the Mi-2/NuRD-HDACcomplex and BCoR and to silence p21WAF/CDKN1A by DNA methyla-tion. Nucleic Acids Res 2013;41:6403–20.
41. SinghS,GuptaAK.Nitric oxide: role in tumourbiologyand iNOS/NO-basedanticancer therapies. Cancer Chemother Pharmacol 2011;67:1211–24.
42. Xiong H, Zhu C, Li H, Chen F, Mayer L, Ozato K, et al. Complex formationof the interferon (IFN) consensus sequence-binding protein with IRF-1 isessential for murine macrophage IFN-gamma-induced iNOS gene expres-sion. J Biol Chem 2003;278:2271–7.
43. PotuH, Sgorbissa A, Brancolini C. Identification of USP18 as an importantregulator of the susceptibility to IFN-alpha and drug-induced apoptosis.Cancer Res 2010;70:655–65.
44. Kim TW, Hong S, Lin Y, Murat E, Joo H, Kim T, et al. Transcriptionalrepression of IFN regulatory factor 7 by MYC is critical for type I IFNproduction in human plasmacytoid dendritic cells. J Immunol 2016;197:3348–59.
45. Ueki IF,Min-OoG,Kalinowski A, Ballon-Landa E, Lanier LL,Nadel JA, et al.Respiratory virus-induced EGFR activation suppresses IRF1-dependentinterferon lambda and antiviral defense in airway epithelium. J Exp Med2013;210:1929–36.
46. Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr OpinCell Biol 2003;15:172–83.
47. Wu LP, Wang X, Li L, Zhao Y, Lu S, Yu Y, et al. Histone deacetylaseinhibitor depsipeptide activates silenced genes through decreasing bothCpG and H3K9 methylation on the promoter. Mol Cell Biol 2008;28:3219–35.
48. PapaioannouNE, BeniataOV, Vitsos P, TsitsilonisO, Samara P.Harnessingthe immune system to improve cancer therapy. Ann Transl Med 2016;4:261.
49. Abrams SI, Netherby CS, TwumDY, Messmer MN. Relevance of interferonregulatory factor-8 expression in myeloid-tumor interactions. J InterferonCytokine Res 2016;36:442–53.
50. Kurotaki D, Tamura T. Transcriptional and epigenetic regulation of innateimmune cell development by the transcription factor, interferon regulatoryfactor-8. J Interferon Cytokine Res 2016;36:433–41.
Cancer Immunol Res; 5(7) July 2017 Cancer Immunology Research616
Fragale et al.
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080

2017;5:604-616. Published OnlineFirst June 14, 2017.Cancer Immunol Res Alessandra Fragale, Giulia Romagnoli, Valerio Licursi, et al. Dendritic CellsModulated Gene Signatures in Both Colorectal Cancer and
Combination Driven byαAntitumor Effects of Epidrug/IFN
Updated version
10.1158/2326-6066.CIR-17-0080doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerimmunolres.aacrjournals.org/content/suppl/2017/06/14/2326-6066.CIR-17-0080.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerimmunolres.aacrjournals.org/content/5/7/604.full#ref-list-1
This article cites 50 articles, 13 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerimmunolres.aacrjournals.org/content/5/7/604To request permission to re-use all or part of this article, use this link
on February 24, 2019. © 2017 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Published OnlineFirst June 14, 2017; DOI: 10.1158/2326-6066.CIR-17-0080





























