Embed Size (px)
Citation preview

An in vitro assay reveals a role for the diaphragmprotein PV-1 in endothelial fenestra morphogenesisSofia Ioannidou*†, Katrin Deinhardt†‡, Jadwiga Miotla†, John Bradley*, Eunice Cheung*, Steven Samuelsson*,Yin-Shan Ng*, and David T. Shima*†§
*Eyetech Research Center, OSI Eyetech, 35 Hartwell Avenue, Lexington, MA 02420; and †Endothelial Cell Biology Laboratory, Cancer Research UK,44 Lincoln’s Inn Fields, London WC2A 3PX, United Kingdom
Edited by Judah Folkman, Harvard Medical School, Boston, MA, and approved September 25, 2006 (received for review May 15, 2006)
Fenestrae are small pores in the endothelium of renal glomerular,gastrointestinal, and endocrine gland capillaries and are involvedin the bidirectional exchange of molecules between blood andtissues. Although decades of studies have characterized fenestraeat the ultrastructural level, little is known on the mechanisms bywhich fenestrae form. We present the development of an in vitroassay in which rapid and abundant fenestra induction enables adetailed study of their biogenesis. Through the use of agents thatstabilize or disassemble actin microfilaments, we show that actinmicrofilament remodeling is part of fenestra biogenesis in thismodel. Furthermore, by using a loss-of-function approach, weshow that the diaphragm protein PV-1 is necessary for fenestralpore architecture and the ordered arrangement of fenestrae insieve plates. Together, these data provide insight into the cellbiology of fenestra formation and open up the future study of thefenestra to a combined morphological and biochemical analysis.
actin filaments � sieve plates � VEGF � vascular permeability
Regulated vascular permeability is essential for normal circula-tory function and tissue homeostasis. Throughout the vascular
network, endothelial cells employ a number of mechanisms tocontrol permeability. One such mechanism is the formation ofpore-like structures called fenestrae, which are implicated in thepermeability of water, solutes, and small macromolecules.Fenestrae occur at sites of high blood–tissue exchange, such as thekidney glomerulus, the gastrointestinal tract, endocrine glands, liversinusoids, and the choriocapillaris of the eye (1–3). In addition,fenestrae have been documented in vessels during cancer anddiabetic retinopathy (4–6), pathologies associated with uncon-trolled permeability and edema.
Ultrastructural studies have determined that fenestrae traversethe entire thickness of endothelial cells in attenuated regions as thinas 40 nm, forming a pore �60–70 nm in diameter (7, 8). In mostfenestrated vascular beds, the pore of fenestrae is dissected into 5-to 6-nm openings by a diaphragm composed of radially arrangedfibrils that converge in a central knob (9). Within an endothelial cell,fenestrae are organized in clusters referred to as ‘‘sieve plates,’’being arranged with equidistant spacing, often forming linear ortwo-dimensional arrays (8, 10). Despite the extensive morpholog-ical characterization of fenestrae, their composition and biogenesisremain poorly understood. The small size of fenestrae, togetherwith a lack of specific markers for these structures, has renderedtheir study dependent on ultrastructural methods, which are tech-nically demanding and provide only limited descriptive information.Attempts at analyzing fenestrae in vitro, an approach that facilitatesexperimental manipulation and observation, have been limited bythe rapid dedifferentiation and loss of the fenestrated phenotype inculture. Moreover, attempts at de novo induction of the fenestratedphenotype in cultured endothelial cells have resulted in low yieldsthat have limited the application of cell biological analyses (11–15).
VEGF is a prime candidate for induction of fenestrae in vivo.VEGF is a potent angiogenic and permeability mediator that isexpressed in epithelia adjacent to fenestrated vascular beds (16, 17)and fenestrated neovasculature (6, 18). Ectopic administration of
VEGF to certain tissues in vivo results in fenestra induction (19),and genetic ablation of components in the VEGF signaling pathway(20–22) or antagonism of its receptor (23, 24) lead to a loss of thefenestrated phenotype. However, the low levels of fenestra induc-tion in vitro obtained with VEGF [mean of �1 fenestra per cell(15)], have precluded in depth mechanistic studies.
The only known component of fenestrae is PV-1, a type IImembrane glycoprotein first discovered in caveolae (25) and laterrecognized to be a component of the diaphragm of endothelial cellcaveolae, fenestrae, and transendothelial channels (26). PV-1 ispresent in vascular beds containing diaphragmed fenestrae but isabsent from fenestrated endothelia of the adult liver sinusoids andkidney glomerulus, which are devoid of diaphragms (2, 26, 27).PV-1 has been proposed to associate in multiple coiled-coil ho-modimers to form the fibrils of the diaphragm (28) and was recentlyshown to be necessary and sufficient for the formation of dia-phragms (29). However, a role for PV-1 in fenestra formation andfunction has not been addressed.
To gain insight into the cellular and molecular events required forfenestra formation we developed an in vitro assay in which fenestraecan be induced at densities that approach those seen in vivo, therebyfacilitating cell biological studies. By establishing and applyingquantitative ultrastructural and light microscopy (LM) methods, wediscovered that actin microfilament depolymerization is a prereq-uisite for fenestra formation in our model. Furthermore, through aloss-of-function study using siRNA, we demonstrated that thediaphragm protein PV-1 is required for normal fenestra biogenesis.In particular, we found that PV-1 defines the dimensions ofindividual fenestrae and specifies their precise arrangement withina sieve plate.
ResultsDevelopment of an in Vitro Assay for the Study of Fenestrae. Wescreened a large panel of endothelial cells for their response tosingle factors and combinations of factors previously reported toinduce fenestrae in vitro. The factors tested included extracellularmatrix, growth factors such as VEGF, actin microfilament depo-lymerizing agents, and phorbol 12-myristate 13-acetate. Most celllines and primary cell cultures examined demonstrated very fewfenestrae or no fenestrae at all under basal or induced conditions(data not shown). However, the greatest and most reproduciblefenestra induction occurred with the application of the actindepolymerizing agent latrunculin A to the bEND5 mouse endo-
Author contributions: S.I., K.D., J.M., Y.-S.N., and D.T.S. designed research; S.I., K.D., J.M.,and E.C. performed research; S.I., K.D., J.M., J.B., E.C., S.S., and D.T.S. analyzed data; and S.I.,J.B., and D.T.S. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS direct submission.
Abbreviations: TEM, transmission electron microscopy; LM, light microscopy; PECAM,platelet endothelial cell adhesion molecule; PFA, paraformaldehyde.
‡Present address: Molecular Neuropathobiology Laboratory, Cancer Research UK, 44 Lin-coln’s Inn Fields, London WC2A 3PX, United Kingdom.
§To whom correspondence should be addressed. E-mail: [email protected].
© 2006 by The National Academy of Sciences of the USA
16770–16775 � PNAS � November 7, 2006 � vol. 103 � no. 45 www.pnas.org�cgi�doi�10.1073�pnas.0603501103

thelioma cell line. Whole-mount transmission electron microscopy(TEM) and scanning electron microscopy (SEM) showed thatuntreated bEND5 cells had few fenestrae on their plasma mem-brane (Fig. 1A). Upon application of latrunculin A, most of theperipheral plasma membrane of bEND5 cells became perforatedwith hundreds of fenestrae that were organized in sieve plates andwere highly ordered (Fig. 1 A and B). Examination of thin sectionsof embedded monolayers and cell pellets confirmed that thestructures observed upon induction were surrounded by electron-dense material (Fig. 1C) and were indeed pores that traversed theentire thickness of endothelial cells (Fig. 1D). The fenestrae dis-played consistent pore diameters of �60 nm, diaphragms, lineararrangement, and equidistant spacing of 100–120 nm from onefenestral center to the next (Fig. 1 A–D).
To quantify the extent of fenestra formation, we applied stere-ology to randomly sampled SEM and TEM images. Quantificationconfirmed that fenestra formation was rapid, with large numbersoccurring as early as 20 min after induction (Fig. 1E). Maximumfenestra induction was �100-fold, reaching levels of 3.5–5.0fenestrae per squared micrometer after 3 h of latrunculin A
treatment (Fig. 1E). Because VEGF is a potent inducer offenestrae, we wondered whether the effects of latrunculin A weremediated by VEGF. However, a VEGF function-blocking antibody(recognizing all VEGF isoforms) did not affect latrunculin Ainduction of fenestrae, indicating that fenestrae form as a directresult of actin depolymerization (data not shown). Furthermore,cytochalasin B, an actin-depolymerizing agent with a mode ofaction different than latrunculin A, also produced large numbers offenestrae (Fig. 1E). Together, these data reveal a close relationshipbetween actin depolymerization and fenestra induction.
A LM Assay for the Study of Fenestrae in Vitro. We next developedan alternative approach for the characterization of fenestrae thatdid not require technically demanding and laborious ultrastructuralanalysis. Because the diaphragm protein PV-1 is the only knowncomponent of fenestrae and because almost all fenestrae in oursystem were spanned by a diaphragm (Fig. 1D, magnification), wereasoned that PV-1 would be enriched in fenestrated areas in vitro.Furthermore, we hypothesized that fenestrated areas in our modelwould not stain for cytoskeletal elements and organelles, becausesieve plates are only 40 nm thick and exclude such structures.
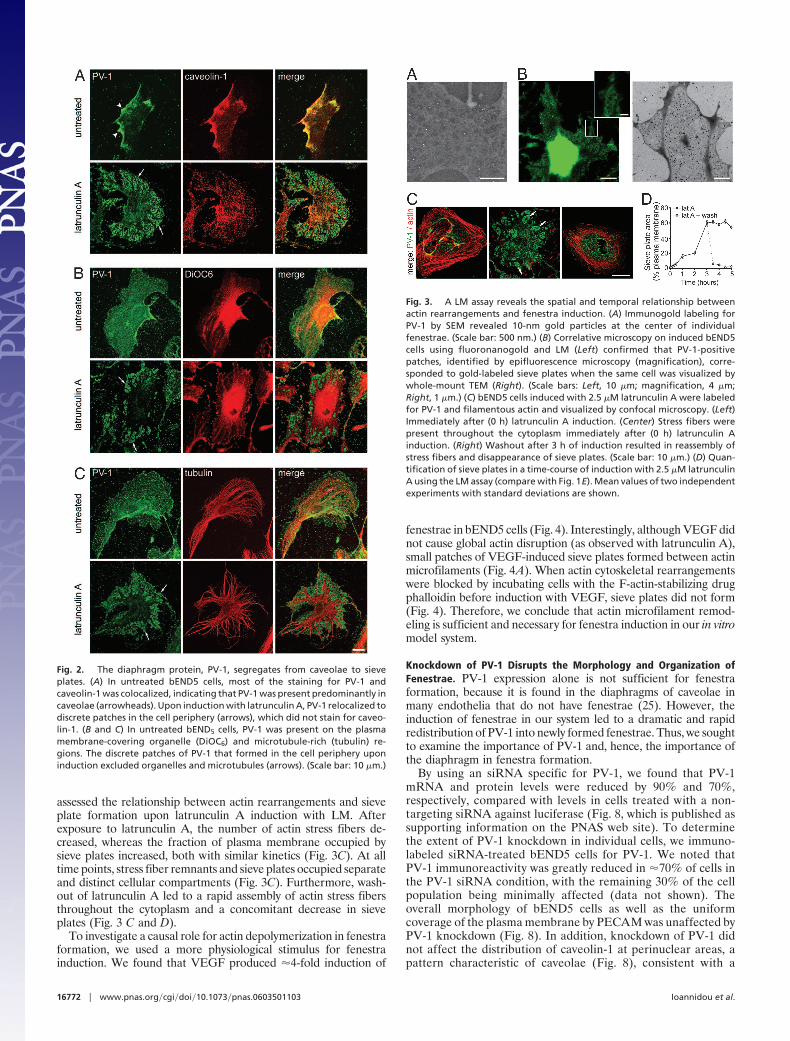
Double immunolabeling revealed that, in untreated bEND5 cells,PV-1 colocalized with caveolin-1 on the plasma membrane in apattern that is characteristic for caveolae (Fig. 2A) (29). DiOC6 andtubulin, markers for membrane-bound organelles and microtu-bules, respectively, were distributed widely throughout the cell andsignificantly overlapped with the localization of PV-1 and caveo-lin-1 in untreated bEND5 cells (Fig. 2 B and C). In contrast, doubleimmunolabeling of induced bEND5 cells for PV-1 and caveolin-1showed PV-1 concentrated in discrete patches at the cell periphery(Fig. 2A; see also Fig. 6, which is published as supporting infor-mation on the PNAS web site). Furthermore, labeling for PV-1, incombination with either the dye DiOC6 or an antibody againsttubulin, showed that PV-1 patches excluded organelles and micro-tubules in induced bEND5 cells (Figs. 2 B and C and 6). The PV-1patches appeared to be surrounded by a microtubule border, similarto that reported in fenestrated liver endothelial cells (30).
To determine whether the PV-1 patches observed with LM wereindeed sieve plates, we performed immunoelectron microscopy andcorrelative light–electron microscopy studies. First, direct immu-nogold labeling of induced cells examined by SEM showed mostPV-1 labeling in induced bEND5 cells to localize to the center ofeach fenestra (Fig. 3A). In contrast, the general plasma membranemarker platelet endothelial cell adhesion molecule (PECAM) wasdistributed across the entire plasma membrane (data not shown).Additionally, by using fluoronanogold to perform LM and electronmicroscopy on the same samples, we confirmed that PV-1 patches,which were identified by fluorescence LM, corresponded to gold-labeled fenestrae when the same cell was examined by TEM(Fig. 3B).
Finally, we used image analysis to quantify sieve plate area afterinduction. We defined sieve plates as areas positive for PV-1 andnegative for microtubules. Next, we defined the total area of cellsby using the cell-surface marker PECAM. By measuring the sum ofareas that were positive for PV-1 and negative for microtubules andby expressing this value as a fraction of the sum of areas that werepositive for PECAM we obtained an estimate of the percentage ofplasma membrane that was occupied by sieve plates (Fig. 7, whichis published as supporting information on the PNAS web site).
Cytoskeletal Remodeling Is Essential for Fenestra Formation. Micro-filament disruption agents have previously been shown to increasefenestra abundance by 3- to 4-fold in cultured fenestrated liverendothelium, leading researchers to suggest a role for the actincytoskeleton in modulating fenestra density (31, 32). However, ourobservations of a potent de novo induction of the fenestratedphenotype in the bEND5 cell model suggest that cytoskeletaldisassembly may be a basic prerequisite for fenestra formation. We
Fig. 1. In vitro system of fenestra formation. (A) bEND5 cells untreated (Left)or induced with 2.5 �M latrunculin A for 3 h (Right) were examined bywhole-mount TEM. (B) Numerous fenestrae in the plasma membrane ofinduced bEND5 cells were confirmed by SEM analysis. (C) Thin section cutalong the plane of a monolayer of induced bEND5 cells revealed that fenestraeare organized in a sieve plate (arrow). A section plane within the cytoplasmrevealed electron-dense material surrounding the pore (arrowheads). (D) TEMof scraped monolayers revealed fenestrae both en face (arrowheads) and incross-sectional view (arrows). Arrows in the magnification point to fenestraldiaphragms. (E) Time-course of fenestra formation after induction with twodifferent microfilament disruption agents. Mean values of two independentexperiments with standard deviations are shown. (Scale bars: 500 nm; mag-nification in D, 100 nm.)
Ioannidou et al. PNAS � November 7, 2006 � vol. 103 � no. 45 � 16771
CELL
BIO
LOG
Y
assessed the relationship between actin rearrangements and sieveplate formation upon latrunculin A induction with LM. Afterexposure to latrunculin A, the number of actin stress fibers de-creased, whereas the fraction of plasma membrane occupied bysieve plates increased, both with similar kinetics (Fig. 3C). At alltime points, stress fiber remnants and sieve plates occupied separateand distinct cellular compartments (Fig. 3C). Furthermore, wash-out of latrunculin A led to a rapid assembly of actin stress fibersthroughout the cytoplasm and a concomitant decrease in sieveplates (Fig. 3 C and D).
To investigate a causal role for actin depolymerization in fenestraformation, we used a more physiological stimulus for fenestrainduction. We found that VEGF produced �4-fold induction of
fenestrae in bEND5 cells (Fig. 4). Interestingly, although VEGF didnot cause global actin disruption (as observed with latrunculin A),small patches of VEGF-induced sieve plates formed between actinmicrofilaments (Fig. 4A). When actin cytoskeletal rearrangementswere blocked by incubating cells with the F-actin-stabilizing drugphalloidin before induction with VEGF, sieve plates did not form(Fig. 4). Therefore, we conclude that actin microfilament remod-eling is sufficient and necessary for fenestra induction in our in vitromodel system.
Knockdown of PV-1 Disrupts the Morphology and Organization ofFenestrae. PV-1 expression alone is not sufficient for fenestraformation, because it is found in the diaphragms of caveolae inmany endothelia that do not have fenestrae (25). However, theinduction of fenestrae in our system led to a dramatic and rapidredistribution of PV-1 into newly formed fenestrae. Thus, we soughtto examine the importance of PV-1 and, hence, the importance ofthe diaphragm in fenestra formation.
By using an siRNA specific for PV-1, we found that PV-1mRNA and protein levels were reduced by 90% and 70%,respectively, compared with levels in cells treated with a non-targeting siRNA against luciferase (Fig. 8, which is published assupporting information on the PNAS web site). To determinethe extent of PV-1 knockdown in individual cells, we immuno-labeled siRNA-treated bEND5 cells for PV-1. We noted thatPV-1 immunoreactivity was greatly reduced in �70% of cells inthe PV-1 siRNA condition, with the remaining 30% of the cellpopulation being minimally affected (data not shown). Theoverall morphology of bEND5 cells as well as the uniformcoverage of the plasma membrane by PECAM was unaffected byPV-1 knockdown (Fig. 8). In addition, knockdown of PV-1 didnot affect the distribution of caveolin-1 at perinuclear areas, apattern characteristic of caveolae (Fig. 8), consistent with a
Fig. 2. The diaphragm protein, PV-1, segregates from caveolae to sieveplates. (A) In untreated bEND5 cells, most of the staining for PV-1 andcaveolin-1 was colocalized, indicating that PV-1 was present predominantly incaveolae (arrowheads). Upon induction with latrunculin A, PV-1 relocalized todiscrete patches in the cell periphery (arrows), which did not stain for caveo-lin-1. (B and C) In untreated bEND5 cells, PV-1 was present on the plasmamembrane-covering organelle (DiOC6) and microtubule-rich (tubulin) re-gions. The discrete patches of PV-1 that formed in the cell periphery uponinduction excluded organelles and microtubules (arrows). (Scale bar: 10 �m.)
Fig. 3. A LM assay reveals the spatial and temporal relationship betweenactin rearrangements and fenestra induction. (A) Immunogold labeling forPV-1 by SEM revealed 10-nm gold particles at the center of individualfenestrae. (Scale bar: 500 nm.) (B) Correlative microscopy on induced bEND5cells using fluoronanogold and LM (Left) confirmed that PV-1-positivepatches, identified by epifluorescence microscopy (magnification), corre-sponded to gold-labeled sieve plates when the same cell was visualized bywhole-mount TEM (Right). (Scale bars: Left, 10 �m; magnification, 4 �m;Right, 1 �m.) (C) bEND5 cells induced with 2.5 �M latrunculin A were labeledfor PV-1 and filamentous actin and visualized by confocal microscopy. (Left)Immediately after (0 h) latrunculin A induction. (Center) Stress fibers werepresent throughout the cytoplasm immediately after (0 h) latrunculin Ainduction. (Right) Washout after 3 h of induction resulted in reassembly ofstress fibers and disappearance of sieve plates. (Scale bar: 10 �m.) (D) Quan-tification of sieve plates in a time-course of induction with 2.5 �M latrunculinA using the LM assay (compare with Fig. 1E). Mean values of two independentexperiments with standard deviations are shown.
16772 � www.pnas.org�cgi�doi�10.1073�pnas.0603501103 Ioannidou et al.

previous study that showed intact caveola morphology afterPV-1 siRNA treatment (29).
We used whole-mount TEM to assess the formation of fenestraein cells upon PV-1 knockdown (Fig. 5A). Interestingly, we found nodifference in total sieve plate areas between the luciferase and PV-1siRNA conditions. Furthermore, quantitative analysis of fenestradensity within sieve plates revealed no significant difference be-tween the two conditions, although a trend toward a lower overalldensity was observed in the PV-1 siRNA condition (Fig. 5B).However, the appearance and order of fenestrae within the sieveplates of cells in the PV-1 siRNA condition was significantlydifferent from that of controls (Fig. 5 C–E). The spacing betweenfenestrae in the PV-1 siRNA condition was highly variable, beingboth shorter and longer than the average distance of 100–120 nmin the luciferase siRNA condition (Fig. 5C). The diameters of 75%of fenestrae in the PV-1 siRNA condition fell in the range of 60–80nm, similar to all fenestrae in the luciferase siRNA condition.However, 25% of fenestrae in the PV-1 siRNA condition showeddiameters as small as 20 nm and as large as 400 nm (Fig. 5D).
Importantly, diaphragms were still evident in a population offenestrae in the PV-1 siRNA condition, a partial phenotype that ispredictable because we observed a reduction rather than a com-plete elimination of PV-1 protein. To examine the correlationbetween the observed morphological irregularities and a reductionin PV-1 levels, we determined the pore size of fenestrae as afunction of the presence or absence of a diaphragm. Quantitativeassessment of whole-mount TEM images from the PV-1 siRNAcondition revealed that fenestrae that were devoid of diaphragmshad enlarged and variable diameters, whereas fenestrae that dis-played regular or smaller-than-usual diameters contained a dia-phragm (Fig. 5E). Taken together, our observations strongly sug-gest that PV-1 is required during fenestra biogenesis to define thedimensions of fenestrae as well as their organization within sieveplates.
DiscussionIn this study we have described an in vitro culture system thatproduces fenestrae similar to those observed in capillary beds in
vivo in terms of morphology, abundance, and higher-order orga-nization. Our model system confers a number of advantages overother approaches: (i) Fenestrae are formed in a transformedendothelial cell line that can be maintained for numerous passages;(ii) fenestra induction occurs within minutes rather than days; (iii)fenestrae are abundant; (iv) the fold-induction is large and issuitable for studies using comparative biochemical methods andLM; and (v) the model system is amenable to gain- and loss-of-function approaches.
In developing a system to study fenestrae, we found that numer-ous endothelial cell lines and primary cell cultures failed to inducefenestrae upon treatment with a variety of stimuli. At present, wedo not know what predisposes the bEND5 brain endothelioma cellline to respond so robustly to latrunculin A. Although our systemshould prove useful in gaining insight into fenestra morphology andbiogenesis, care must be taken when extrapolating these data tofenestra regulation in vivo, because we observed only a low responseto VEGF, a physiological stimulus for fenestra formation. Under-standing the predisposing factors behind the robust response to
Fig. 4. Actin rearrangements are necessary for fenestra induction by VEGF.(A) bEND5 cells untreated (Left), induced with 100 ng�ml VEGF 164 for 4 h(Center), or induced with VEGF in the presence of phalloidin (Right), wereanalyzed by epifluorescence microscopy. In VEGF-induced cells, small sieveplates (arrows) were apparent between rearranged stress fibers. In cellspretreated with labeled phalloidin, sieve plate formation was prevented.(Scale bars: Right, 10 �m; magnification in Center, 4 �m.) (B) Quantification ofsieve plates using the LM assay (P � 0.05; one-way ANOVA with post hocBonferroni test).
Fig. 5. Reduction of PV-1 disrupts the morphology of fenestrae. (A) Variablefenestra diameters and spacing after PV-1 knockdown. (Left) The luciferasesiRNA condition. (Right) The PV-1 siRNA condition. The magnifications showthe recordings of fenestra centers (yellow dots) that were used to determinefenestra spacing. (Scale bars: 500 nm; magnification, 250 nm.) (B) Fenestradensity within sieve plates was not significantly different between luciferaseand PV-1 siRNA conditions. (C) Distances between fenestrae in the PV-1condition were significantly smaller or greater than in the luciferase siRNAcondition (P � 0.01; see also Fig. 9, which is published as supporting informa-tion on the PNAS web site). (D) Fenestra diameters showed a wider distribu-tion in the PV-1 versus the luciferase siRNA condition but were not signifi-cantly different. The discontinuous y axis simplifies comparisons betweenconditions within one graph. (E) Fenestra diameters within the PV-1 siRNAcondition were significantly greater in fenestrae without diaphragms versusfenestrae with diaphragms (P � 0.01).
Ioannidou et al. PNAS � November 7, 2006 � vol. 103 � no. 45 � 16773
CELL
BIO
LOG
Y

latrunculin A and the low response to VEGF in bEND5 cells willhelp in the future characterization of the signaling cascades andsubcellular changes controlling fenestra biogenesis.
Absolute numbers of �5.0 fenestrae per squared micrometer anda relative induction of 100-fold favorably compare to previous invitro studies where adrenal cortex endothelial cells or humanumbilical vein endothelial cells induced with VEGF, phorbol esters,or retinoic acid attained maximal levels of only 0.187 fenestrae persquared micrometer, with peak levels requiring days of treatmentand yielding a relatively low fold-induction (11–15). Fenestra den-sities of �9.0 per squared micrometer were reported for liverendothelial cells treated with swinholide A (33), but these outcomesrepresented only a �3-fold induction and were restricted to alreadyfenestrated, freshly isolated, cells (34). Our estimated density offenestrae in sieve plate areas (30 fenestrae per squared micrometer)is in agreement with the value reported for kidney capillaries (8).
Although the development of a fenestra biogenesis model wasfocused on obtaining the maximum amount of fenestrae, our modelhas provided some insight into the cell biology of fenestra forma-tion. The potent effect of microfilament disassembly on fenestrainduction in bEND5 cells and the spatial and temporal correlationbetween stress fiber disassembly and sieve plate formation high-lighted the importance of actin microfilaments. Whereas previousstudies had addressed the role of actin disassembly in sustaining ormodulating the number of pores in fenestrated liver endothelialcells (31–33, 35) and isolated kidney glomeruli (36), we demon-strated a role for actin disassembly in de novo fenestra formation.Our observations of a rearrangement of large actin filaments duringfenestra induction by the physiological stimulus VEGF and theprevention of such rearrangements and fenestra induction by theF-actin-stabilizing drug phalloidin further supported a role for actinremodeling in fenestra formation. Actin remodeling in response toVEGF was not as dramatic as the complete stress fiber disassemblyseen with latrunculin A; however, in light of the reported lowstress-fiber content of capillaries (37), it is likely to reflect moreclosely the subcellular changes in vivo. Furthermore, a recent studyshowed that an antagonist of the actin regulator Rac, blockedVEGF-mediated induction of fenestrae in the corneal micropocketassay (38). Together with evidence for a modulation of actinpolymerization by VEGF signaling in other systems (39), these datasuggest that actin remodeling may be part of VEGF-driven fenestrabiogenesis in vivo.
We hypothesize that actin stress fibers are a structural barrier,the disruption or remodeling of which is required to bring theapical and basal plasma membranes of the cell in close proximityfor fusion. A parallel can be drawn with organelle membranefusion, where persistence of a surrounding actin coat acts as aphysical barrier that prevents the recruitment of tethering factorsand, in turn, the close apposition of membranes (40, 41).Furthermore, the absence of stress fibers from fenestrated areasappears to be required for their maintenance, given that washoutof latrunculin A was followed by a reassembly of actin stressfibers and the disappearance of fenestrae. The reversal in cellularattenuation suggested by the increase in actin stress fibers andthe redistribution of organelles to the cell periphery is likelysimilar to the increase in thickness observed in vivo duringfenestra disappearance after VEGF inhibition (42) or preven-tion of fenestra formation by a Rac antagonist (38).
The timing of fenestra appearance and disappearance in ourmodel and the dynamic and reversible nature of the relationshipbetween actin microfilaments and fenestrae are suggestive of aplasticity of endothelial cells. Although the low response to VEGFin our model precludes direct comparisons to the situation in vivo,a similar endothelial plasticity has been reported in studies usingVEGF and VEGF inhibitors to induce or remove fenestrae (19, 42).Furthermore, the gradual appearance of fenestrae in our model, incontrast to their rapid disappearance upon stimulus removal, likelyreflects the kinetics of actin filament polymerization. Actin filament
disassembly after latrunculin A treatment also was gradual, whereasactin filament reassembly after stimulus removal was more rapid,consistent with the mode of action of latrunculin A, an inhibitor ofactin polymerization (43).
Our data indicate that expression of PV-1, the only recognizedconstituent of fenestrae, is not sufficient for their formation,consistent with previous studies of PV-1 overexpression in endo-thelial cells (29). However, the dysmorphic appearance of fenestraeand disorganized sieve plates after PV-1 knockdown by siRNAsuggested that PV-1 is required for the morphology of fenestrae.Furthermore, we observed a reduction of diaphragms in fenestraeafter PV-1 knockdown, consistent with previous reports showingthat PV-1 is the major constituent of caveolar and fenestraldiaphragms (29). Analysis restricted to fenestrae without dia-phragms revealed an increase in fenestra diameter and in thevariability of these diameters, further supporting a role for PV-1 infenestra morphogenesis.
Observations on the fenestrated capillary beds of the kidneyglomerulus and liver sinusoids indicated a periodic presence ofdiaphragms during vascular development, despite the absence of adiaphragm in the adult tissues (refs. 27 and 44 and unpublishedobservation). Taken together, these observations and our siRNAdata are suggestive of a requirement for PV-1 during fenestraformation but not maintenance. PV-1 is unlikely to play a role in thefusion of the apical and basal plasma membranes, given thatfenestral pores, albeit devoid of diaphragms, were evident in thePV-1 knockdown condition. Plausible functions of PV-1 includeconstraining the fenestral opening by oligomerizing with otherPV-1 molecules at opposite sides of the pore or regulating thespacing between fenestrae through interactions with intracellularscaffolds.
The in vitro model for fenestra formation and the initial charac-terization described here should provide a platform and conceptualframework to enable further studies of fenestra biogenesis tocharacterize fenestra components and their function, to help probethe function of fenestrae in vivo, and to eventually modulate thebarrier properties of the endothelium for therapeutic purposes.
Materials and MethodsAntibodies and Reagents. The following primary antibodies wereused for immunocytochemistry: rat anti-PV-1 (MECA-32; Devel-opmental Studies Hybridoma Bank, Iowa City, IA), rabbit anti-PV-1 (prepared against the 12 C-terminal amino acids), mouseanti-caveolin 1 (BD Biosciences, Franklin Lakes, NJ), mouseanti-tubulin (Sigma, St. Louis, MO), rat anti-PECAM (MEC13.3;BD Biosciences), allophycocyanin-conjugated rat anti-PECAM(MEC13.3; BD Biosciences). Rhodamine phalloidin (MolecularProbes, Eugene, OR) and DiOC6 (Molecular Probes) were used tovisualize F-actin and intracellular organelles, respectively. All stan-dard chemicals were obtained from Sigma unless otherwiseindicated.
Cell Culture, siRNA Transfection, and Fenestra Induction. The bEND5endothelioma cell line (45) was kindly provided by Urban Deutschand Britta Engelhardt (University of Bern, Bern, Switzerland).bEND5 cells were maintained in high-glucose DMEM (Invitrogen,Carlsbad, CA) containing 10% FBS and antibiotics. Cells weretransfected with Oligofectamine (Invitrogen) in culture mediumlacking FBS and antibiotics. Predesigned siRNA against PV-1 (ID85339; Ambion, Austin, TX) and luciferase negative control siRNA(Ambion) were used at a final concentration of 5 nM. PV-1knockdown and cell phenotypes were assessed 72 h later.
Twenty-four hours before fenestra induction, glass coverslips,formvar grids, or culture dishes were coated with 1% gelatin in PBSand bEND5 cells were seeded at a density equivalent to 1.5 � 106
cells per 100-mm dish. Cells were induced with either 10 �Mcytochalasin B for 2 h (Sigma), 2.5 �M latrunculin A for 3 h(Molecular Probes), or 100 ng�ml murine VEGF 164 for 4 h
16774 � www.pnas.org�cgi�doi�10.1073�pnas.0603501103 Ioannidou et al.

(Peprotech, Rocky Hill, NJ). For actin stabilization studies, cellswere preincubated with 80 nM rhodamine phalloidin for 30 min(Molecular Probes).
Immunocytochemistry. Cells on coverslips were fixed with eithermethanol at �20°C for 8 min or 4% paraformaldehyde (PFA) for15 min. PFA-fixed cells were permeabilized in PBT (0.1% TritonX-100�PBS). Before incubation with primary and secondaryantibodies (30 min each at room temperature), cells wereblocked in 10% goat serum�0.2% fish skin gelatin in PBS for 15min. Fluorescent images were captured with an LSM510 laserscanning confocal microscope (Zeiss, Gottingen, Germany) anda DMRA2 epifluorescence microscope (Leica Microsystems,Wetzlar, Germany).
Immunolabeling for SEM was performed on live cells at 4°C,followed by fixation in 1% PFA�3% glutaraldehyde (ElectronMicroscopy Sciences, Hatfield, PA) in cacodylate buffer (0.1 Msodium cacodylate, pH 7.4). For whole-mount TEM immuno-labeling, cells were fixed in 4% PFA�0.5% glutaraldehyde in 0.1M phosphate buffer for 15 min, labeled, and postfixed with 2.5%glutaraldehyde.
Electron Microscopy. For SEM, cells on coverslips were fixed in2% PFA (EM grade; Electron Microscopy Sciences)�2.5% glu-taraldehyde in cacodylate buffer, dehydrated, dried in hexam-ethyldisalazane, gold-coated, and examined under a JSM-6700field emission SEM (JEOL, Tokyo, Japan) in Backscatter mode.Immunogold labeling was visualized in Backscatter mode. Cellsgrown on formvar grids were fixed in 1.25% glutaraldehyde and2.5% PFA in cacodylate buffer, postfixed, dehydrated, dried asfor SEM and examined under a JEOL 1010 TEM. For TEM thinsections, intact monolayers or pellets of monolayers that hadbeen scraped and centrifuged at 1,000 � g for 10 min were fixedin 2.5% glutaraldehyde�2% PFA in cacodylate buffer for 20 min.After osmium fixation and dehydration, specimens were infil-trated with resin and polymerized overnight at 60°C. Sections
(70–100 nm) were stained with uranyl acetate and Reynoldsstain for 2 min and dried.
Morphometrics. We randomly captured 20–30 SEM images pertime-point at a magnification of �10,000. A grid with 150 pointsof intersection was placed on each image, and the number of gridpoints falling on any cellular structure versus the number of gridpoints falling on sieve plates were counted. The density offenestrae was estimated by multiplying the fraction of total cellmembrane area that was occupied by sieve plates with theaverage density of fenestrae within 10 representative sieve plates(30 fenestrae per squared micrometer of sieve plate area).
For sieve plate quantification by LM, seven images wererandomly acquired per coverslip from preparations triple stainedfor PV-1, tubulin, and PECAM. The total cell area was calcu-lated by measuring the PECAM-positive area with Openlabsoftware (Improvision, Lexington, MA). The area occupied bysieve plates was calculated by measuring the PV-1-positive,tubulin-negative area. Sieve plate area per cell was estimated asthe percentage of the total PECAM-positive area that waspositive for PV-1 and negative for tubulin (Fig. 7).
For the siRNA phenotype quantification, 36 whole-mountTEM images per condition were randomly captured at a mag-nification of �25,000. Only images containing sieve plates(20–30 images per condition) were included in the analysis (seealso Supporting Materials and Methods, which is published assupporting information on the PNAS web site).
Statistical analysis was performed by using one-way ANOVAwith a post hoc Bonferroni test for which P � 0.05 wasconsidered a statistically significant difference.
We thank Amy Snodgrass, Peter Munro, Kirsty Roberts, Rose Watson,and Steve Gschmeissner for technical assistance; Vladimir Mastyugin forhelp in designing siRNA transfections; and members of the EndothelialCell Biology Laboratory and the Eyetech Research Center for usefuldiscussions. This work was supported by Cancer Research UK and OSIEyetech.
1. Roberts WG, Palade GE (2000) in Morphogenesis of Endothelium, eds Risau W,Rubanyi GM (Hardwood, Amsterdam), pp 23–41.
2. Braet F, Wisse E (2002) Comp Hepatol 1:1.3. Pino RM, Essner E (1980) Cell Tissue Res 208:21–27.4. Jin E, Ghazizadeh M, Fujiwara M, Nagashima M, Shimizu H, Ohaki Y, Arai
S, Gomibuchi M, Takemura T, Kawanami O (2001) Pathol Int 51:691–700.5. Madigan MC, Penfold PL (1997) Ultrastruct Pathol 21:95–107.6. Wallow IH, Geldner PS (1980) Invest Ophthalmol Vis Sci 19:1176–1183.7. Bennett HS, Luft JH, Hampton JC (1959) Am J Physiol 196:381–390.8. Rhodin JA (1962) J Ultrastruct Res 6:171–185.9. Bearer EL, Orci L (1985) J Cell Biol 100:418–428.
10. Simionescu M, Simionescu N, Palade GE (1974) J Cell Biol 60:128–152.11. Lombardi T, Montesano R, Furie MB, Silverstein SC, Orci L (1986) J Cell Biol
102:1965–1970.12. Lombardi T, Montesano R, Orci L (1987) Eur J Cell Biol 44:86–89.13. Lombardi T, Montesano R, Furie MB, Silverstein SC, Orci L (1988) J Cell Sci
91:313–318.14. Milici AJ, Furie MB, Carley WW (1985) Proc Natl Acad Sci USA 82:6181–6185.15. Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W (1998) J Cell
Biol 140:947–959.16. Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM (1999) Curr Top
Microbiol Immunol 237:97–132.17. Breier G, Albrecht U, Sterrer S, Risau W (1992) Development (Cambridge, UK)
114:521–532.18. Dvorak AM, Feng D (2001) J Histochem Cytochem 49:419–432.19. Roberts WG, Palade GE (1995) J Cell Sci 108:2369–2379.20. Carpenter B, Lin Y, Stoll S, Raffai RL, McCuskey R, Wang R (2005)
Development (Cambridge, UK) 132:3293–3303.21. Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP,
Kikkawa Y, Miner JH, Quaggin SE (2003) J Clin Invest 111:707–716.22. Lammert E, Gu G, McLaughlin M, Brown D, Brekken R, Murtaugh LC,
Gerber HP, Ferrara N, Melton DA (2003) Curr Biol 13:1070–1074.23. Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, Hu-Lowe
DD, Shalinsky DR, Thurston G, Yancopoulos GD, McDonald DM (2004) Am JPathol 165:35–52.
24. Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR,Norberg SM, O’Brien SM, Davis RB, Gowen LC, et al. (2006) Am J Physiol290:H560–H576.
25. Stan RV, Ghitescu L, Jacobson BS, Palade GE (1999) J Cell Biol 145:1189–1198.
26. Stan RV, Kubitza M, Palade GE (1999) Proc Natl Acad Sci USA 96:13203–13207.
27. Reeves WH, Kanwar YS, Farquhar MG (1980) J Cell Biol 85:735–753.28. Stan RV (2004) Am J Physiol 286:H1347–H1353.29. Stan RV, Tkachenko E, Niesman IR (2004) Mol Biol Cell 15:3615–3630.30. Braet F, De Zanger R, Baekeland M, Crabbe E, Van Der Smissen P, Wisse E
(1995) Hepatology 21:180–189.31. Steffan AM, Gendrault JL, Kirn A (1987) Hepatology 7:1230–1238.32. Braet F, De Zanger R, Jans D, Spector I, Wisse E (1996) Hepatology
24:627–635.33. Braet F, Spector I, De Zanger R, Wisse E (1998) Proc Natl Acad Sci USA
95:13635–13640.34. Braet F, Wisse E, Probst I (2005) Eur J Cell Biol 84:745–748.35. Braet F, De Zanger R, Kalle W, Raap A, Tanke H, Wisse E (1996) Scanning
Microsc Suppl 10:225–235, and discussion (1996) 10:235–236.36. Andrews PM (1981) Kidney Int 20:549–562.37. Thurston G, Baldwin AL (1994) Am J Physiol 266:H1896–H1909.38. Eriksson A, Cao R, Roy J, Tritsaris K, Wahlestedt C, Dissing S, Thyberg J, Cao
Y (2003) Circulation 107:1532–1538.39. Gong C, Stoletov KV, Terman BI (2004) Angiogenesis 7:313–321.40. Orci L, Gabbay KH, Malaisse WJ (1972) Science 175:1128–1130.41. Muallem S, Kwiatkowska K, Xu X, Yin HL (1995) J Cell Biol 128:589–598.42. Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR,
Norberg SM, O’Brien SM, Davis RB, Gowen LC, et al. (2005) Am J Physiol290:H509–H511.
43. Morton WM, Ayscough KR, McLaughlin PJ (2000) Nat Cell Biol 2:376–378.
44. Bankston PW, Pino RM (1980) Am J Anat 159:1–15.45. Wagner EF, Risau W (1994) Semin Cancer Biol 5:137–145.
Ioannidou et al. PNAS � November 7, 2006 � vol. 103 � no. 45 � 16775
CELL
BIO
LOG
Y





























