Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY,0021-9193/00/$04.0010
July 2000, p. 3832–3838 Vol. 182, No. 13
Copyright © 2000, American Society for Microbiology. All Rights Reserved.
An Essential Two-Component Signal TransductionSystem in Mycobacterium tuberculosis
THOMAS C. ZAHRT AND VOJO DERETIC*
Department of Microbiology and Immunology, University of MichiganMedical School, Ann Arbor, Michigan 48109-0620
Received 4 January 2000/Accepted 30 March 2000
The bacterial two-component signal transduction systems regulate adaptation processes and are likely toplay a role in Mycobacterium tuberculosis physiology and pathogenesis. The previous initial characterization of anM. tuberculosis response regulator from one of these systems, mtrA-mtrB, suggested its transcriptional activa-tion during infection of phagocytic cells. In this work, we further characterized the mtrA response regulatorfrom M. tuberculosis H37Rv. Inactivation of mtrA on the chromosome of M. tuberculosis H37Rv was possible onlyin the presence of plasmid-borne functional mtrA, suggesting that this response regulator is essential for M. tu-berculosis viability. In keeping with these findings, expression of mtrA in M. tuberculosis H37Rv was detectableduring in vitro growth, as determined by S1 nuclease protection and primer extension analyses of mRNA levelsand mapping of transcript 5* ends. The mtrA gene was expressed differently in virulent M. tuberculosis and thevaccine strain M. tuberculosis var. bovis BCG during infection of macrophages, as determined by monitoring ofmtrA-gfp fusion activity. In M. bovis BCG, mtrA was induced upon entry into macrophages. In M. tuberculosisH37Rv, its expression was constitutive and unchanged upon infection of murine or human monocyte-derivedmacrophages. In conclusion, these results identify mtrA as an essential response regulator gene in M. tubercu-losis which is differentially expressed in virulent and avirulent strains during growth in macrophages.
Tuberculosis remains the leading cause of death in the worldfrom a single infectious agent (2). The capacity of Mycobacte-rium tuberculosis to establish infection within an individual andefficiently disseminate within the human population is medi-ated in part by its ability to survive within professional phago-cytic cells, remain dormant over long periods of latent infec-tion, and resume growth upon disease reactivation (27). Thephysiological and environmental signals during periods of ac-tive disease, dormancy, or disease reactivation are likely tocontribute to M. tuberculosis adaptation during various stagesof infection. One well-recognized class of ubiquitous bacterialregulatory elements associated with signal recognition andadaptive responses is that of the two-component signal trans-duction systems. Bacterial two-component systems regulatevarious functions, including transient adaptations, develop-mental phenomena, and production of secondary metabolites(reviewed in reference 16). In pathogenic organisms, two-com-ponent systems can also regulate expression of virulence de-terminants or factors that contribute to disease pathogenesis(reviewed in references 16 and 30). In addition to the majorityof two-component systems, which modulate nonvital albeit im-portant cellular functions, a limited number of essential two-component systems have also been described. Such systems,although rare, have been shown to regulate genes involved incell cycle control (29) and membrane permeability (23).
M. tuberculosis encodes a number of two-component sig-nal transduction systems. The MtrA-MtrB system was the firstsuch system to be characterized in the tubercle bacillus (5, 7,35). Since then, an additional 11 complete and 8 unlinked sen-sor kinase and response regulator homologs have been iden-tified in the M. tuberculosis H37Rv genome (4, 12, 14, 21, 22,33). Some of these two-component systems appear to be
differentially regulated during growth within cultured mac-rophages in vitro. For example, expression of the mtrA re-sponse regulator (Rv3246c), which has been studied in M. bovisBCG, is induced in infected murine macrophages (7, 35). Inaddition, cDNAs corresponding to transcripts encoding theprrA response regulator (Rv0903c) and the sensor kinase prrB(Rv0902c) have been recovered from M. tuberculosis grown inhuman peripheral blood monocyte-derived macrophages butnot from bacteria grown in standard laboratory medium (12).These limited examples reflect the preliminary nature of theinitial analyses of M. tuberculosis two-component systems. Incontinuation of our characterization of the mtrA-mtrB system,we attempted to disrupt the mtrA gene in M. tuberculosis. Herewe present data suggesting that mtrA is an essential gene inM. tuberculosis H37Rv. We also report the mapping of the 59end of the mtrA mRNA and its in vivo expression profiles inM. tuberculosis H37Rv and M. bovis BCG.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and electrotransformation. M. tubercu-losis H37Rv (ATCC 27294) and M. bovis BCG Pasteur (ATCC 27291) were used.All transformations done with Escherichia coli were performed with strainDH5a. Mycobacteria were grown under standard conditions in Middlebrook7H9 broth or on Middlebrook 7H10 agar (Difco Laboratories) supplementedwith 0.5% glycerol, 10% ADC or OADC (oleic acid-albumin-dextrose-catalase)(Difco) and 0.05% Tween 80 (Sigma) at 37°C in the presence of 5% CO2. E. coliwas grown in LB medium (Difco) and incubated at 37°C. When required,Middlebrook or LB medium was supplemented with 25 or 50 mg of kanamycinsulfate (Sigma) per ml, 50 or 200 mg of hygromycin B (Boehringer Mannheim)per ml, 25 or 100 mg of streptomycin sulfate (Sigma) per ml, and 2 or 10%sucrose, respectively. Preparation of electrocompetent cells and transformationof M. tuberculosis were performed as previously described (18).
Construction of plasmid vectors. Plasmid pmtrA-gfp has been described pre-viously (7). Plasmid pTZ113 was used for the disruption of mtrA and was con-structed as follows. A 2.7-kb SalI fragment containing the entire mtrA gene andthe 59 end of mtrB was filled in by treatment with Klenow enzyme and ligated intothe SmaI site of pSM243, a mycobacterial suicide vector carrying the sacB gene.Next, a 1.2-kb NheI-SpeI fragment carrying the Kmr gene from pMV206 (31) wasfilled in and cloned into a blunt-ended BglII site in mtrA. Finally, a 2.0-kbNheI-XbaI fragment encoding xylE from pHSX-1 (5) was ligated into the XbaIsite of the pSM243-derived polylinker. Temperature-sensitive (ts), mtrA1-com-
* Corresponding author. Mailing address: Department of Microbi-ology and Immunology, University of Michigan Medical School, Med-ical Science Bldg. II, Ann Arbor, MI 48109-0620. Phone: (734) 763-1580. Fax: (734) 647-6243. E-mail: [email protected].
3832
on March 6, 2020 by guest
http://jb.asm.org/
Dow
nloaded from

plementing plasmid pTZ178 was constructed by first ligating the SalI fragmentcarrying mtrA into the SalI site of pUC12 to create pTZ100. An SpeI-NotIfragment encoding Hygr from pOLYG, a derivative of p16R1 (11), was filled inand ligated into the SmaI site of pTZ100 to create pTZ175. Finally, a filled-inEcoRV-KpnI fragment encoding the ts origin of replication from pCG63 (13)was ligated into the ScaI site of pTZ175 to create pTZ178. pTZ195 is a derivativeof pTZ178 that lacks the SalI fragment encoding mtrA. Plasmid pTZ199 wasconstructed by ligating a 3.4-kb SpeI fragment encoding Strr from pSM240 intoan SpeI-NheI fragment carrying xylE1 from pHSX-1 (5).
Genetic scheme for mtrA::Kmr gene replacement. Plasmid pTZ113 (mtrA::Kmr
sacB1 xylE1) was used for allelic exchange of mtrA1 with mtrA::Kmr in M. tu-berculosis H37Rv via a two-step recombination process (Fig. 1). In the first step(integration), pTZ113 was transformed into M. tuberculosis H37Rv and recom-binants were selected on 7H10 agar containing kanamycin. Merodiploid trans-formants were distinguished from spontaneous Kmr mutants by spraying colonieswith 100 mM catechol (Fisher Scientific) (5). Transformants expressing xylE(detected as yellow colonies upon spraying with catechol) were screened forlegitimate single-crossover homologous recombination by PCR using primersmtrAupstm2 (59-CTGACCAAGCTGACCAAGGA-39), which is a primer up-stream of mtrA (and not carried on pTZ113), and KmUP2 (59-GTAAGCAGACAGTTTTATTGTTCATGA-39), which is a primer specific to and amplifyingout of the Kmr cassette. mtrA::Kmr-mtrA1 merodiploids resulting from single-crossover homologous recombination were subsequently resolved of pTZ113 toleave mtrA::Kmr or mtrA1 in the chromosome by growth on 7H10 agar (with orwithout kanamycin) and sucrose, respectively (28). Colonies resistant to sucroseand white upon spraying with catechol (loss of sacB and xylE markers) werefurther screened by PCR using primers that flank the Kmr disruption site in mtrA(RC4 [59-ACGTACCGGCGCGCACAAGGT-39] and RC13 [59-TCACGGAGGTCCGGCC-39]) and primers that amplify an internal portion of xylE (xylEstart[59-ATGAACAAAGGTGTAATGCG-39] and xylEend [59-GCGGTCGTGGTAAAAGATCG-39]).
Addition of mtrA1 to mtrA::Kmr-mtrA1 merodiploid strains. mtrA::Kmr-mtrA1 merodiploid recombinants resulting from single-crossover homologousrecombination of pTZ113 into the M. tuberculosis H37Rv chromosome weretransformed with pTZ178 [mtrA1 oriM (ts) Hygr], a conditionally replicatingplasmid carrying mtrA1. Resolution of pTZ113 in these strains to leave mtrA::Kmr or mtrA1 in the chromosome was achieved by plating on 7H10 agar (with orwithout kanamycin) containing sucrose and hygromycin and growth at 30°C(permissive temperature for pTZ178 replication). The resulting recombinantswere subjected to the screens previously described. Because pTZ178 contained ats origin of replication, loss of the mtrA1 complementing plasmid in mtrA::Kmr
mutants was attempted by growing strains at the nonpermissive temperature of39°C. In addition, loss of pTZ178 was also attempted by introduction of a secondplasmid, pTZ199 (pMV261 oriM xylE1 Strr), carrying the same origin of repli-cation as pTZ178.
DNA extraction and Southern analysis. Mycobacterial genomic DNA wasprepared as previously described (18). A 4-mg sample of genomic DNA wasdigested overnight with EcoRI (Gibco BRL), separated by electrophoresis on a0.8% agarose gel, transferred onto a Duralon-UV membrane (Stratagene), andused in subsequent high-stringency hybridization and washing steps (26). AnmtrA-specific probe was generated by random-primed labeling (Gibco) with[a-32P]dCTP (3,000 Ci mmol21; NEN Dupont) using PCR products generatedwith oligonucleotides RC4 and RC10 (59-CCCATCACCCGGCACC-39).
S1 nuclease protection and primer extension. Total RNA from M. tuberculosisH37Rv was isolated as previously described (8). To generate a uniformly labeledsingle-stranded DNA (ssDNA) probe for S1 nuclease protection, a 1.7-kb SalI-PstI fragment carrying the M. tuberculosis H37Rv mtrA gene and upstream se-quences was directionally cloned into an M13-based phagemid vector (24). 32P-radiolabeled ssDNA probes were prepared (25) using primers mtrAS5 (59-TCGCCGATGACCGCGGTGTC-39) and mtrAS6 (59-AGCGGCTACTCCGCGGTGTCGAAGCCTTCC-39). Probes generated from primer mtrAS6 contain a10-bp overhang (underlined nucleotides) at the 59 end that is not homologous tomtrA mRNA. Radiolabeled ssDNA polymerization products were digested withAgeI, heat denatured in formamide, and gel purified. Hybridization reactionswere performed using 75 mg of total RNA from M. tuberculosis H37Rv, and S1nuclease protection was carried out as previously described (25). Products of S1digestion were analyzed on sequencing gels and compared with the correspond-ing sequencing ladders to locate mRNA 59 ends. For primer extension, primermtrAS8 (59-TCCCCCCGCAGCACGATGGTGAGCATCTCA-39) was endlabeled with [g-32P]ATP (6,000 Ci mmol21; NEN Dupont) and purified on aSephadex G-25 spin column (Boehringer Mannheim). Radiolabeled primer wasadded to 10 mg of total M. tuberculosis H37Rv RNA in hybridization buffer (0.5M KCl, 0.25 M Tris-HCl, pH 8.3), and aliquots were denatured, annealed, andextended by the addition of 0.1 M dithiothreitol, 2.5 mM deoxynucleosidetriphosphates, reverse transcription buffer, and Superscript II reverse transcrip-tase (Gibco). Extension reactions were carried out at 44°C for 45 min, andsamples were loaded on a sequencing gel alongside the corresponding sequenceladder.
Preparation of mycobacteria, infection of macrophage monolayers, and fluo-rescence microscopy. M. tuberculosis H37Rv or M. bovis BCG Pasteur was grownin static cultures until cells reached mid-exponential phase (optical density at 600nm of 0.5). Bacterial cells were prepared for macrophage infection by washing in
phosphate-buffered saline (PBS; pH 7.2) and resuspension either in Dulbecco’smodified Eagle’s medium (Bio Whittaker) supplemented with 10% fetal bovineserum (Hyclone) and 4 mM L-glutamine (Bio Whittaker) or in RPMI 1640medium (Bio Whittaker) supplemented with 5% human AB serum (Sigma) and2 mM L-glutamine. Single-cell bacterial suspensions were obtained by vortexingbacteria with 3-mm glass beads (Fisher), low-speed centrifugation, and passageof the resulting supernatant through a 5-mm-pore-size filter (Micron SeparationsInc.). The number of organisms was determined by staining with Bac-Light(Molecular Probes) and counting in a hemocytometer. Mycobacteria were usedto infect murine BALB/c macrophage cell line J774A (ATCC TIB-67) or humanmacrophages derived from peripheral blood monocytes (3) obtained from theAmerican Red Cross. J774 cells and human monocyte-derived macrophageswere cultured before infection and maintained during infection in supplementedDulbecco’s modified Eagle’s medium and RPMI medium, respectively. Bothmurine and human macrophage monolayers were maintained at 37°C in humid-ified air containing 5% CO2. For infections, macrophage monolayers were es-tablished by plating 105 cells per well in 12-well tissue culture plates (Corning)containing no. 1 thickness, 18-mm-diameter glass coverslips (Fisher). Macro-phages were infected with mycobacteria at a multiplicity of infection of 10 bacilliper macrophage. Macrophages were allowed to take up bacteria for 2 h beforeextracellular bacteria were removed by washing in PBS. Macrophages wereincubated for 2 h, 3 days, or 5 days before harvest with no apparent damage. Atharvest, macrophage monolayers were washed in PBS, fixed in 3.8% paraformal-dehyde, and mounted on glass slides with Permafluor (Lipshaw Immunon).Epifluorescence images were captured using a Kodak Kaf 1400-2 Olympix cam-era connected to an Olympus BX60 microscope. Images were captured with ashutter speed of 500 ms and analyzed using Espirit software (Life SciencesResources). NIH Image (version 1.62; National Institutes of Health) was used toquantitate mean pixel density from individual bacilli present within monolayers.At the settings used, macrophage autofluorescence was not observed. Statisticalanalysis (analysis of variance [ANOVA] and Fisher’s protected least significantdifference) was performed with ANOVA (version 1.11; Abicus Software).
RESULTS AND DISCUSSION
Gene replacements with mtrA::Kmr in M. tuberculosis H37Rv.To further examine the role of mtrA in M. tuberculosis, we setout to disrupt mtrA in strain H37Rv. We constructed a myco-bacterial suicide plasmid, pTZ113 (see Materials and Meth-ods), that carried a copy of mtrA disrupted by a Kmr cassette,the counterselectable marker sacB, and the xylE gene as a con-venient scorable marker for subsequent recombination steps.Following electroporation of pTZ113 into M. tuberculosis H37Rv,we obtained recombinants expressing xylE (detected as yellowcolonies upon spraying with catechol), of which 2.6% hadundergone legitimate single-crossover homologous recom-bination into the chromosome, resulting in a tandem mtrA::Kmr-mtrA1 merodiploid (Fig. 1A and Table 1, row A). Thelow frequency of legitimate single-crossover homologous re-combination was most likely a result of illegitimate integrationof pTZ113 into the M. tuberculosis genome (1, 19). Subsequentattempts to produce the desired mtrA::Kmr gene replacementsby resolving the mtrA::Kmr-mtrA1 merodiploids via a secondcrossover after selection against sacB failed (Fig. 1B and Table1, row B). None of the colonies recovered as resistant to su-crose (loss of the sacB marker) and not expressing xylE (whiteupon spraying with catechol) were true double-crossover re-combinants and most likely harbored other types of mutationseliminating or precluding sacB and xylE activity (data notshown). The inability to obtain an mtrA::Kmr gene replace-ment was not simply the result of inefficient resolution by ho-mologous recombination, because double-crossover recombi-nants which had lost the plasmid moiety along with mtrA::Kmr,leaving the wild-type copy of mtrA in the chromosome, oc-curred efficiently (Fig. 1C and Table 1, row C).
The mtrA gene is an essential response regulator in M. tu-berculosis. To test further whether the encountered difficul-ties in inactivating mtrA could be explained by its potentiallyessential function, we introduced into the mtrA::Kmr-mtrA1
merodiploids plasmid pTZ178 containing an mtrA1 copy on aconditionally replicating (ts) mycobacterial shuttle vector andrepeated the selection procedure for mtrA::Kmr gene replace-
VOL. 182, 2000 mtrA IS ESSENTIAL IN M. TUBERCULOSIS 3833
on March 6, 2020 by guest
http://jb.asm.org/
Dow
nloaded from
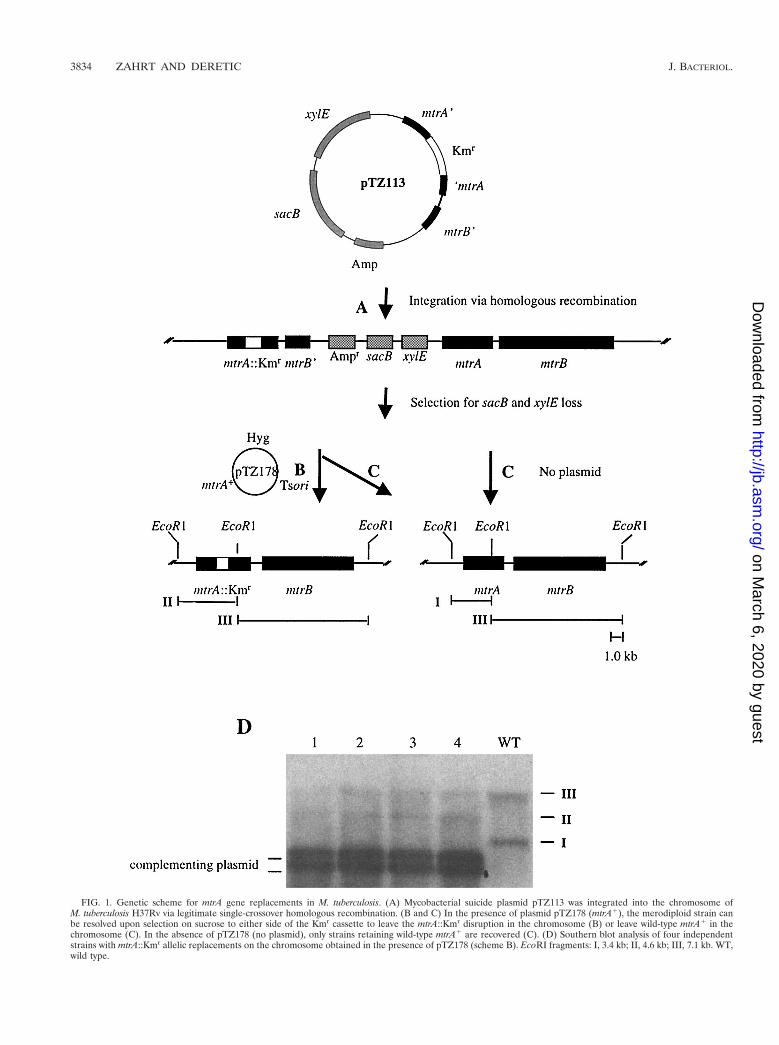
FIG. 1. Genetic scheme for mtrA gene replacements in M. tuberculosis. (A) Mycobacterial suicide plasmid pTZ113 was integrated into the chromosome ofM. tuberculosis H37Rv via legitimate single-crossover homologous recombination. (B and C) In the presence of plasmid pTZ178 (mtrA1), the merodiploid strain canbe resolved upon selection on sucrose to either side of the Kmr cassette to leave the mtrA::Kmr disruption in the chromosome (B) or leave wild-type mtrA1 in thechromosome (C). In the absence of pTZ178 (no plasmid), only strains retaining wild-type mtrA1 are recovered (C). (D) Southern blot analysis of four independentstrains with mtrA::Kmr allelic replacements on the chromosome obtained in the presence of pTZ178 (scheme B). EcoRI fragments: I, 3.4 kb; II, 4.6 kb; III, 7.1 kb. WT,wild type.
3834 ZAHRT AND DERETIC J. BACTERIOL.
on March 6, 2020 by guest
http://jb.asm.org/
Dow
nloaded from

ments. This time, in the presence of plasmid-borne mtrA1, truemtrA::Kmr gene replacements were obtained via a secondcrossover on the chromosome (Fig. 1B and Table 1, row D).Southern hybridization analysis performed on four randomlyselected recombinants confirmed the replacement of mtrA1
with the mtrA::Kmr allele on the M. tuberculosis chromosome(Fig. 1D). Two EcoRI chromosomal fragments in wild-typeM. tuberculosis H37Rv hybridized with the mtrA probe (3.4-and 7.1-kb fragments I and III, respectively). In mtrA::Kmr
recombinants, fragment I was lost but, instead, a new fragment(fragment II) of 4.6 kb (corresponding to 3.4-kb EcoRI frag-ment I carrying the 1.2-kb Kmr insert) was detected in each ofthe four recombinants tested. These strains, however, also har-bored a plasmid (pTZ178) borne wild-type mtrA gene.
Because pTZ178 carried a mycobacterial ts origin of repli-cation, we next tried to eliminate the plasmid by growing themtrA::Kmr (pTZ178) strains at the nonpermissive temperatureof 39°C. However, none of the colonies arising followinggrowth at the nonpermissive temperature lost the complement-ing plasmid (Table 1, row E). In contrast, loss of pTZ193 (aderivative of pTZ178 lacking the mtrA gene) from the mtrA::Kmr-mtrA1 merodiploid parental strain was observed at highfrequency (100%) upon growth at 39°C (Table 1, row F). Be-cause two plasmids containing the same origin of replicationcannot be maintained simultaneously in the same cell, we alsotried to eliminate pTZ178 from mtrA::Kmr mutants by intro-duction of plasmid pTZ199. This approach also failed to curethe mtrA1 plasmid from mtrA::Kmr mutants despite the factthat the merodiploid parental strain carrying pTZ193 could becured of this plasmid (Table 1, rows G and H). Based on theseexperiments, we conclude that mtrA encodes a response regu-lator that is essential for M. tuberculosis viability in vitro. Theviability-associated function resided within the mtrA gene andwas not due to polar effects on mtrB, as it was possible toknockout mtrA on the chromosome in the presence of themtrA1 complementing plasmid, which did not contain a com-plete mtrB gene (Fig. 1D).
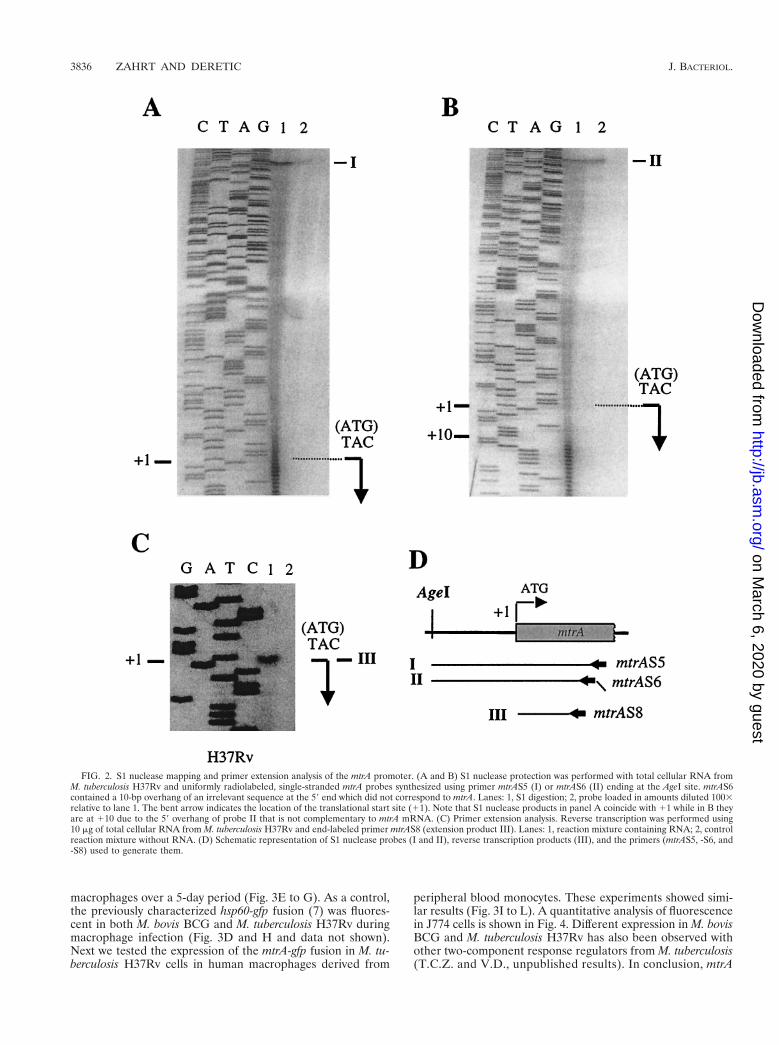
Expression of mtrA in M. tuberculosis grown outside the host.The inability to recover viable mtrA mutants would require thatmtrA be expressed in vitro. Expression of the mtrA gene isinducible in M. bovis BCG during growth in cultured J774macrophages (35). However, this does not preclude the possi-bility that mtrA is expressed at baseline levels in vitro outsidethe host. To test this possibility, we analyzed mtrA transcriptionand mapped mtrA mRNA 59 ends in M. tuberculosis H37Rv byS1 nuclease protection and primer extension analyses. Total
cellular RNA was isolated from bacteria grown in 7H9 mediumand hybridized with a uniformly labeled ssDNA probe, and theproducts of the hybridization reaction were digested with S1nuclease. In keeping with our prediction that mtrA was ex-pressed during in vitro growth, a band of protection corre-sponding to mtrA transcripts was observed (Fig. 2A). The 59end of the protected fragments corresponded to the mtrA ini-tiation codon. Similar results were obtained using a probe (Fig.2D) generated with a primer that contained a 59 (10-bp) over-hang that did not correspond to the mtrA sequence. The ad-dition of the heterologous 10-bp overhang sequence resulted inthe corresponding reduction in the size of the protected frag-ment obtained upon S1 nuclease treatment (Fig. 2B), confirm-ing that the assigned transcript was mtrA specific and indicatingthat its 59 end coincided with the translational initiation site.Due to the intrinsic heterogeneity of the products of uniformlylabeled S1 nuclease probes, which introduced some uncer-tainty regarding the exact position of the mRNA 59 end, it wasimportant to test whether the mtrA mRNA 59 end included themtrA translational start. With this aim, we performed primerextension analysis. The results of these experiments indicatedthat the translation and transcriptional initiation start sites ofmtrA overlap (Fig. 2C). Thus, in M. tuberculosis H37Rv, mtrAis expressed in vitro from a transcript with the 59 mRNA endoverlapping the translational start site. Overlapping transcrip-tional and translational start sites have been identified forseveral other mycobacterial genes, including the major oxyRtranscript of M. leprae (6) and the furA promoter from M. tu-berculosis (unpublished data). In addition, a large number ofgenes from actinomycetes, a phylogenetic group closely relatedto mycobacteria, also contain overlapping transcriptional andtranslational sites (32). In conclusion, the in vitro expression ofmtrA in M. tuberculosis H37Rv was in keeping with our findingthat mtrA is essential for the viability of this organism.
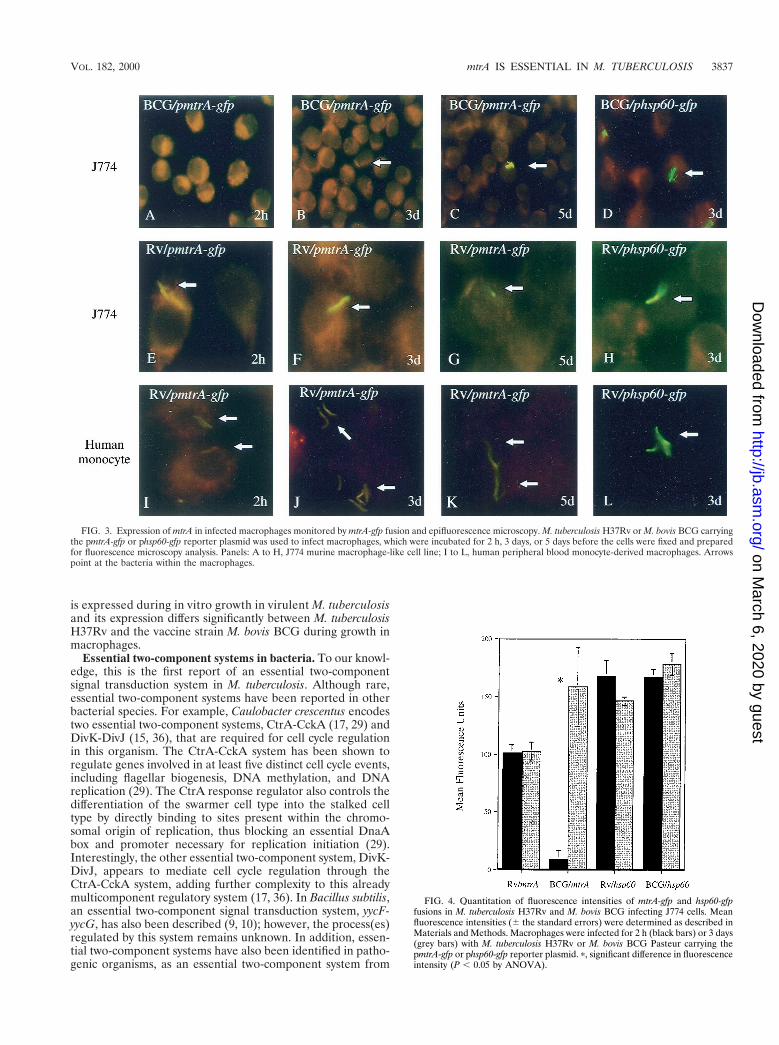
The mtrA promoter is induced in M. bovis BCG but is con-stitutively active in M. tuberculosis H37Rv within macrophages.To examine mtrA expression in M. tuberculosis during intracel-lular growth, we tested whether the mtrA induction previouslydetected in M. bovis BCG could also be observed in M. tuber-culosis H37Rv during infection of J774 murine macrophages.As reported previously, green fluorescent protein fluorescencein M. bovis BCG carrying pmtrA-gfp became detectable after 3days of incubation in J774 cells (Fig. 3A to C) (35). However,green fluorescent protein fluorescence from pmtrA-gfp wasbright in M. tuberculosis H37Rv even prior to infection and nofurther induction was observed during growth in J774 murine
TABLE 1. Screening for mtrA::Kmr gene replacements in M. tuberculosis H37Rv
Row Stepa Parental strainplasmid(s)b
Selection/screen
Expectedgenotype
Resulting strainplasmidc
No. of coloniesscreened
% Legitimateeventsd
A Integration pTZ113 (mtrA::Kmr xylE1 sacB1) Kmr/XylE1 Sucs mtrA::Kmr-mtrA1 Cointegrate 191 2.6B Resolution Cointegrate Sucr/XylE2 Kmr mtrA::Kmr — 500 0C Resolution Cointegrate Sucr/XylE2 Kms mtrA1 — 600 100D Resolution pTZ178 (mtrA1 Hygr) Sucr/XylE2 Kmr mtrA::Kmr pTZ178 (mtrA1 Hygr) 200 96E pTZ178 loss pTZ178 (mtrA1 Hygr) 39°C/Hygs mtrA::Kmr — 100 0F pTZ193 loss pTZ193 (Hygr) 39°C/Hygs mtrA::Kmr-mtrA1 — 200 100G pTZ178 loss pTZ178 (mtrA1 Hygr), pTZ199 (Strr) Strr/Hygs mtrA::Kmr pTZ199 (Strr) 100 0H pTZ193 loss pTZ193 (Hygr), pTZ199 (Strr) Strr/Hygs mtrA::Kmr-mtrA1 pTZ199 (Strr) 100 100
a Steps involved in the construction or resolution of the mtrA disruption. Parental strains were M. tuberculosis H37Rv (row A), M. tuberculosis H37Rv mtrA::Kmr-mtrA1 sacB1 xylE1 (rows B and C), M. tuberculosis H37Rv mtrA::Kmr-mtrA1(pTZ178) (rows D, E, and G), and M. tuberculosis H37Rv mtrA::Kmr-mtrA1(pTZ193)(rows F and H).
b Plasmids were pTZ113 (pUC18 mtrA::Kmr sacB1 xylE1 [Fig. 1]), pTZ178 [pUC12 oriM (ts) Hygr mtrA1], pTZ193 [pUC12 oriM (ts) Hygr], and pTZ199 (pMV261oriM xylE1 Strr). Cointegrate refers to pTZ113 integrated on the chromosome of H37Rv via single-crossover homologous recombination with the mtrA gene.
c —, no plasmid.d A legitimate event is achievement of the expected genotype.
VOL. 182, 2000 mtrA IS ESSENTIAL IN M. TUBERCULOSIS 3835
on March 6, 2020 by guest
http://jb.asm.org/
Dow
nloaded from
macrophages over a 5-day period (Fig. 3E to G). As a control,the previously characterized hsp60-gfp fusion (7) was fluores-cent in both M. bovis BCG and M. tuberculosis H37Rv duringmacrophage infection (Fig. 3D and H and data not shown).Next we tested the expression of the mtrA-gfp fusion in M. tu-berculosis H37Rv cells in human macrophages derived from
peripheral blood monocytes. These experiments showed simi-lar results (Fig. 3I to L). A quantitative analysis of fluorescencein J774 cells is shown in Fig. 4. Different expression in M. bovisBCG and M. tuberculosis H37Rv has also been observed withother two-component response regulators from M. tuberculosis(T.C.Z. and V.D., unpublished results). In conclusion, mtrA
FIG. 2. S1 nuclease mapping and primer extension analysis of the mtrA promoter. (A and B) S1 nuclease protection was performed with total cellular RNA fromM. tuberculosis H37Rv and uniformly radiolabeled, single-stranded mtrA probes synthesized using primer mtrAS5 (I) or mtrAS6 (II) ending at the AgeI site. mtrAS6contained a 10-bp overhang of an irrelevant sequence at the 59 end which did not correspond to mtrA. Lanes: 1, S1 digestion; 2, probe loaded in amounts diluted 1003relative to lane 1. The bent arrow indicates the location of the translational start site (11). Note that S1 nuclease products in panel A coincide with 11 while in B theyare at 110 due to the 59 overhang of probe II that is not complementary to mtrA mRNA. (C) Primer extension analysis. Reverse transcription was performed using10 mg of total cellular RNA from M. tuberculosis H37Rv and end-labeled primer mtrAS8 (extension product III). Lanes: 1, reaction mixture containing RNA; 2, controlreaction mixture without RNA. (D) Schematic representation of S1 nuclease probes (I and II), reverse transcription products (III), and the primers (mtrAS5, -S6, and-S8) used to generate them.
3836 ZAHRT AND DERETIC J. BACTERIOL.
on March 6, 2020 by guest
http://jb.asm.org/
Dow
nloaded from
is expressed during in vitro growth in virulent M. tuberculosisand its expression differs significantly between M. tuberculosisH37Rv and the vaccine strain M. bovis BCG during growth inmacrophages.
Essential two-component systems in bacteria. To our knowl-edge, this is the first report of an essential two-componentsignal transduction system in M. tuberculosis. Although rare,essential two-component systems have been reported in otherbacterial species. For example, Caulobacter crescentus encodestwo essential two-component systems, CtrA-CckA (17, 29) andDivK-DivJ (15, 36), that are required for cell cycle regulationin this organism. The CtrA-CckA system has been shown toregulate genes involved in at least five distinct cell cycle events,including flagellar biogenesis, DNA methylation, and DNAreplication (29). The CtrA response regulator also controls thedifferentiation of the swarmer cell type into the stalked celltype by directly binding to sites present within the chromo-somal origin of replication, thus blocking an essential DnaAbox and promoter necessary for replication initiation (29).Interestingly, the other essential two-component system, DivK-DivJ, appears to mediate cell cycle regulation through theCtrA-CckA system, adding further complexity to this alreadymulticomponent regulatory system (17, 36). In Bacillus subtilis,an essential two-component signal transduction system, yycF-yycG, has also been described (9, 10); however, the process(es)regulated by this system remains unknown. In addition, essen-tial two-component systems have also been identified in patho-genic organisms, as an essential two-component system from
FIG. 4. Quantitation of fluorescence intensities of mtrA-gfp and hsp60-gfpfusions in M. tuberculosis H37Rv and M. bovis BCG infecting J774 cells. Meanfluorescence intensities (6 the standard errors) were determined as described inMaterials and Methods. Macrophages were infected for 2 h (black bars) or 3 days(grey bars) with M. tuberculosis H37Rv or M. bovis BCG Pasteur carrying thepmtrA-gfp or phsp60-gfp reporter plasmid. p, significant difference in fluorescenceintensity (P , 0.05 by ANOVA).
FIG. 3. Expression of mtrA in infected macrophages monitored by mtrA-gfp fusion and epifluorescence microscopy. M. tuberculosis H37Rv or M. bovis BCG carryingthe pmtrA-gfp or phsp60-gfp reporter plasmid was used to infect macrophages, which were incubated for 2 h, 3 days, or 5 days before the cells were fixed and preparedfor fluorescence microscopy analysis. Panels: A to H, J774 murine macrophage-like cell line; I to L, human peripheral blood monocyte-derived macrophages. Arrowspoint at the bacteria within the macrophages.
VOL. 182, 2000 mtrA IS ESSENTIAL IN M. TUBERCULOSIS 3837
on March 6, 2020 by guest
http://jb.asm.org/
Dow
nloaded from

Staphylococcus aureus showing high similarity to yycF-yycGfrom B. subtilis has recently been reported (23). Initial char-acterization of this system in S. aureus suggests that its roleincludes the proper regulation of bacterial cell wall or mem-brane composition (23). In addition, among the 13 two-com-ponent signal transduction systems present in Streptococcuspneumoniae, one two-component response regulator could notbe inactivated (20, 34), suggesting that this system is also re-quired for an essential cellular function.
Although mtrB is located immediately downstream of mtrAand probably encodes its cognate sensor histidine kinase (35),our results suggest that mtrB is not essential for the growth ofM. tuberculosis in vitro. The identification of a nonessentialhistidine kinase closely linked to an essential response regula-tor has also been observed in S. pneumoniae (34); however,other essential two-component systems appear to require botha response regulator and its cognate histidine kinase forgrowth in vitro (9, 10, 17, 23, 29). It is not known whether mtrAis also essential in M. bovis BCG. As low-level expression ofmtrA may be sufficient to exert its function, we anticipate thatmtrA could be essential for the growth of M. bovis BCG invitro. Regardless, the presence of an essential two-componentsignal transduction system in M. tuberculosis underscores theneed for further characterization of the functions controlled bythis and other regulators of this type in the tubercle bacillus.
ACKNOWLEDGMENTS
We thank V. Vishwanath for the preparation of human peripheralblood monocyte-derived macrophages. Plasmid pCG63 was kindly pro-vided by Brigitte Gicquel.
This work was supported by a National Research Service award(AI10278) to T.C.Z. and by grants (AI35217 and AI42999) from theNational Institute of Allergy and Infectious Diseases to V.D.
REFERENCES
1. Aldovini, A., R. N. Husson, and R. A. Young. 1993. The uraA locus andhomologous recombination in Mycobacterium bovis BCG. J. Bacteriol. 175:7282–7289.
2. Bloom, B. R., and C. J. Murray. 1992. Tuberculosis: commentary on areemergent killer. Science 257:1055–1064.
3. Boyum, A. 1968. Separation of leukocytes from blood and bone marrow.Introduction. Scand. J. Clin. Lab. Investig. Suppl. 97:7.
4. Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, et al.1998. Deciphering the biology of Mycobacterium tuberculosis from the com-plete genome sequence. Nature 393:537–544.
5. Curcic, R., S. Dhandayuthapani, and V. Deretic. 1994. Gene expression inmycobacteria: transcriptional fusions based on xylE and analysis of the pro-moter region of the response regulator mtrA from Mycobacterium tubercu-losis. Mol. Microbiol. 13:1057–1064.
6. Dhandayuthapani, S., M. Mudd, and V. Deretic. 1997. Interactions of OxyRwith the promoter region of the oxyR and ahpC genes from Mycobacteriumleprae and Mycobacterium tuberculosis. J. Bacteriol. 179:2401–2409.
7. Dhandayuthapani, S., L. E. Via, C. A. Thomas, P. M. Horowitz, D. Deretic,and V. Deretic. 1995. Green fluorescent protein as a marker for gene ex-pression and cell biology of mycobacterial interactions with macrophages.Mol. Microbiol. 17:901–912.
8. Dhandayuthapani, S., Y. Zhang, M. H. Mudd, and V. Deretic. 1996. Oxida-tive stress response and its role in sensitivity to isoniazid in mycobacteria:characterization and inducibility of ahpC by peroxides in Mycobacteriumsmegmatis and lack of expression in M. aurum and M. tuberculosis. J. Bacte-riol. 178:3641–3649.
9. Fabret, C., V. A. Feher, and J. A. Hoch. 1999. Two-component signal trans-duction in Bacillus subtilis: how one organism sees its world. J. Bacteriol. 181:1975–1983.
10. Fabret, C., and J. A. Hoch. 1998. A two-component signal transductionsystem essential for growth of Bacillus subtilis: implications for anti-infectivetherapy. J. Bacteriol. 180:6375–6383.
11. Garbe, T. R., J. Barathi, S. Barnini, Y. Zhang, C. Abou-Zeid, D. Tang, et al.1994. Transformation of mycobacterial species using hygromycin resistance
as selectable marker. Microbiology 140:133–138.12. Graham, J. E., and J. E. Clark-Curtiss. 1999. Identification of Mycobacte-
rium tuberculosis RNAs synthesized in response to phagocytosis by humanmacrophages by selective capture of transcribed sequences (SCOTS). Proc.Natl. Acad. Sci. USA 96:11554–11559.
13. Guilhot, C., B. Gicquel, and C. Martin. 1992. Temperature-sensitive mutantsof the Mycobacterium plasmid pAL5000. FEMS Microbiol. Lett. 77:181–186.
14. Haydel, S. E., N. E. Dunlap, and W. H. Benjamin, Jr. 1999. In vitro evidenceof two-component system phosphorylation between the Mycobacterium tu-berculosis TrcR/TrcS proteins. Microb. Pathog. 26:195–206.
15. Hecht, G. B., T. Lane, N. Ohta, J. M. Sommer, and A. Newton. 1995. Anessential single domain response regulator required for normal cell divisionand differentiation in Caulobacter crescentus. EMBO J. 14:3915–3924.
16. Hoch, J. A., and T. J. Silhavy (ed.). 1995. Two-component signal transduc-tion. American Society for Microbiology, Washington, D.C.
17. Jacobs, C., I. J. Domian, J. R. Maddock, and L. Shapiro. 1999. Cell cycle-dependent polar localization of an essential bacterial histidine kinase thatcontrols DNA replication and cell division. Cell 97:111–120.
18. Jacobs, W. R., Jr., G. V. Kalpana, J. D. Cirillo, L. Pascopella, S. B. Snapper,R. A. Udani, et al. 1991. Genetic systems for mycobacteria. Methods Enzy-mol. 204:537–555.
19. Kalpana, G. V., B. R. Bloom, and W. R. Jacobs, Jr. 1991. Insertional mu-tagenesis and illegitimate recombination in mycobacteria. Proc. Natl. Acad.Sci. USA 88:5433–5437.
20. Lange, R., C. Wagner, A. de Saizieu, N. Flint, J. Molnos, M. Stieger, et al.1999. Domain organization and molecular characterization of 13 two-com-ponent systems identified by genome sequencing of Streptococcus pneumo-niae. Gene 237:223–234.
21. Magdalena, J., P. Supply, and C. Locht. 1998. Specific differentiation be-tween Mycobacterium bovis BCG and virulent strains of the Mycobacteriumtuberculosis complex. J. Clin. Microbiol. 36:2471–2476.
22. Magdalena, J., A. Vachee, P. Supply, and C. Locht. 1998. Identification of anew DNA region specific for members of Mycobacterium tuberculosis com-plex. J. Clin. Microbiol. 36:937–943.
23. Martin, P. K., T. Li, D. Sun, D. P. Biek, and M. B. Schmid. 1999. Role in cellpermeability of an essential two-component system in Staphylococcus aureus.J. Bacteriol. 181:3666–3673.
24. Misra, T. K. 1987. DNA sequencing: a new strategy to create ordereddeletions, modified M13 vector, and improved reaction conditions for se-quencing by dideoxy chain termination method. Methods Enzymol. 155:119–139.
25. Mohr, C. D., D. W. Martin, W. M. Konyecsni, J. R. Govan, S. Lory, and V.Deretic. 1990. Role of the far-upstream sites of the algD promoter and thealgR and rpoN genes in environmental modulation of mucoidy in Pseudomo-nas aeruginosa. J. Bacteriol. 172:6576–6580.
26. Pagan-Ramos, E., J. Song, M. McFalone, M. H. Mudd, and V. Deretic. 1998.Oxidative stress response and characterization of the oxyR-ahpC and furA-katG loci in Mycobacterium marinum. J. Bacteriol. 180:4856–4864.
27. Parrish, N. M., J. D. Dick, and W. R. Bishai. 1998. Mechanisms of latency inMycobacterium tuberculosis. Trends Microbiol. 6:107–112.
28. Pelicic, V., J. M. Reyrat, and B. Gicquel. 1996. Expression of the Bacillussubtilis sacB gene confers sucrose sensitivity on mycobacteria. J. Bacteriol.178:1197–1199.
29. Quon, K. C., G. T. Marczynski, and L. Shapiro. 1996. Cell cycle control byan essential bacterial two-component signal transduction protein. Cell 84:83–93.
30. Rappuoli, R., V. Scarlato, and B. Arico (ed.). 1995. Signal transduction andbacterial virulence. R. G. Landes Company, Austin, Tex.
31. Stover, C. K., V. F. de la Cruz, T. R. Fuerst, J. E. Burlein, L. A. Benson, L. T.Bennett, et al. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460.
32. Strohl, W. R. 1992. Compilation and analysis of DNA sequences associatedwith apparent streptomycete promoters. Nucleic Acids Res. 20:961–974.
33. Supply, P., J. Magdalena, S. Himpens, and C. Locht. 1997. Identification ofnovel intergenic repetitive units in a mycobacterial two-component systemoperon. Mol. Microbiol. 26:991–1003.
34. Throup, J. P., K. K. Koretke, A. P. Bryant, K. A. Ingraham, A. F. Chalker,Y. Ge, et al. 2000. A genomic analysis of two-component signal transductionin Streptococcus pneumoniae. Mol. Microbiol. 35:566–576.
35. Via, L. E., R. Curcic, M. H. Mudd, S. Dhandayuthapani, R. J. Ulmer, and V.Deretic. 1996. Elements of signal transduction in Mycobacterium tuberculosis:in vitro phosphorylation and in vivo expression of the response regulatorMtrA. J. Bacteriol. 178:3314–3321.
36. Wu, J., N. Ohta, and A. Newton. 1998. An essential, multicomponent signaltransduction pathway required for cell cycle regulation in Caulobacter. Proc.Natl. Acad. Sci. USA 95:1443–1448.
3838 ZAHRT AND DERETIC J. BACTERIOL.
on March 6, 2020 by guest
http://jb.asm.org/
Dow
nloaded from





![[VII]. Regulation of Gene Expression Via Signal Transduction Reading List VII: Signal transduction Signal transduction in biological systems](https://img.pdfslide.us/doc/110x75/56649e385503460f94b28319/vii-regulation-of-gene-expression-via-signal-transduction-reading-list-vii.jpg)