Embed Size (px)
Citation preview

516
Alterations in Left Ventricular Mechanics,Energetics, and Contractile Reserve in
Experimental Heart FailureMatthew R. Wolff, Pieter P. de Tombe, Yasuhiko Harasawa, Daniel Burkhoff, Seth Bier,
William C. Hunter, Gary Gerstenblith, and David A. Kass
The contributions of changes in primary systolic and diastolic properties, limitations of contractilereserve, and alterations in energy efficiency to the left ventricular dysfunction seen with chronic pacingtachycardia were investigated. Seven dogs (heart failure group) were ventricularly paced at 250 beats perminute for 26.3 ±2.9 days and compared with a separate control group (n =8). Studies were performed withisolated, metabolically supported hearts coupled to a computer-controlled loading system. Pressure-volume relations and myocardial oxygen consumption (MVo2) were measured to assess chamber systolicand diastolic properties and efficiency (relation between MVo2 and pressure-volume area [PVA]).Systolic function was reduced in failure hearts versus controls as assessed by the slope of the end-systolicpressure-volume relation (1.29±0.94 versus 2.71±0.98 mm Hg/ml, p<0.01) and lowered end-systolicstiffness at a matched stress (956.1±123.5 versus 1,401.7±431.7 g/cm2, p<O.05). Diastolic chamber andmyocardial stiffness were unaltered in failure hearts, but the unstressed diastolic-arrested volume wassignificantly larger (33.3±3.9 versus 21.9±7.6 ml,p<0.01). Inotropic response to increased heart rate andexogenous ,-adrenergic stimulation (dobutamine HCI) was significantly impaired in failure comparedwith control hearts. Most interestingly, failure hearts had a lowered slope of the MVo2-PVA relation(2.1±1.1 versus 2.9±1.4 ml 02 mm Hg-' * ml-l * 100 g left ventricle-', p<0.001), indicating increasedeffliciency of chemomechanical energy conversion. The y intercept of the MVo2-PVA relation, whichreflects oxygen costs of basal metabolism and excitation-contraction coupling, was unchanged in the twogroups despite decreased contractility of the heart failure hearts. These results demonstrate reducedchamber and myocardial contractility, dilatation without alteration of passive myocardial properties,impaired contractile reserve, and novel alterations in cardiac efficiency in this model of heart failure.(Circulation Research 1992;70:516-529)KEY WORDs * pressure-volume relations * cardiomyopathy * force-frequency * ,-adrenergic
receptors * pacing tachycardia * diastole contractility canine heart failure model
R apid pacing over a period of several weeks leadsto ventricular dilation, decreased fractionalshortening, elevated filling pressures, and con-
gestive heart failure in several animal species.1-5 Priorstudies have revealed neurohumoral changes in thismodel similar to those observed in human congestiveheart failure.67 Evidence of myocardial dysfunction inpatients with persistent tachyarrhythmias suggests thatthis model may share some characteristics with humandilated cardiomyopathies.8 Although prior animal stud-ies have clearly demonstrated reduced ventricular pump
From the Division of Cardiology, Department of Medicine(M.R.W., D.B., S.B., G.G., D.A.K.) and Department of Biomed-ical Engineering (P.P. de T., Y.H., W.C.H.), The Johns HopkinsMedical Institutions, Baltimore, Md.
Supported by National Heart, Lung, and Blood Institute(NHLBI) Ischemic Heart Disease SCOR HL-17655, NHLBIHL-18912, NHLBI Physician Scientist Award HL-01820 (D.A.K.),and American Heart Association (Maryland Affiliate) traininggrants (P.P. de T. and D.B.). D.A.K. is an Established Investigatorof the American Heart Association.
Address for correspondence: David A. Kass, MD, Division ofCardiology, Carnegie 538, Johns Hopkins Hospital, 601 NorthWolfe Street, Baltimore, MD 21205.
Received April 12, 1991; accepted October 31, 1991.
performance from chronic rapid pacing, the mecha-nisms underlying this dysfunction remain poorly char-acterized. There are several mechanisms definable at achamber level that could contribute to reduced netpump function. These include 1) primary reductions inchamber and/or myocardial contractility, 2) abnormaldiastolic chamber and/or myocardial stiffness, 3) re-duced inotropic reserve caused by blunting of thepositive force-frequency relation or decreased responseto 83-adrenergic stimulation, and 4) a reduced ability toconvert consumed oxygen to mechanical work (de-creased metabolic efficiency). The purpose of the pres-ent study was to determine the contribution of each ofthese factors to the cardiac failure induced by chronicrapid pacing.To elucidate the role of each of these mechanisms,
studies were performed using isolated, metabolicallysupported canine hearts coupled to a computer-simu-lated vascular loading system. This preparation offersthe advantages of rigid control of chamber load, heartrate, and coronary perfusion and precise measurementsof ventricular volume and myocardial oxygen consump-tion (MVo2). The results demonstrate reductions inchamber and myocardial contractility, ventricular dila-
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from

Wolff et al Mechanics and Energetics of Heart Failure 517
tation without alterations in passive myocardial stiff-ness, and impairment of contractile reserve via theforce-frequency relation and ,B-adrenergic stimulation.In addition, evidence for alterations in metabolic effi-ciency in this form of heart failure is provided.
Materials and MethodsCongestive heart failure was produced in seven
adult mongrel dogs (mean weight, 25.3±+1.9 kg) bychronic rapid right ventricular pacing for 4 weeks oruntil signs of congestive heart failure were clinicallyapparent (mean, 26.3±2.9 days). An additional eighthealthy adult mongrel dogs (mean weight, 24.7±1.1kg) were used as controls.
Surgical PreparationDogs were anesthetized with pentobarbital sodium (30
mg/kg i.v.). A 7F balloon-tipped catheter was introducedinto the left jugular vein and advanced into the mainpulmonary artery. Right atrial, right ventricular, pulmo-nary artery, and capillary wedge pressures were ob-tained. The catheter was removed, and a bipolar screw-inendocardial pacing electrode was advanced to the rightventricular apex under fluoroscopic guidance. The leadwas tunneled to a subcutaneous pocket in the left lateralneck and connected to a pulse generator (model SX 5984or 5940, Medtronics, Inc., Minneapolis, Minn.), modifiedto pace rapidly by gluing a magnet to the canister, whichdisabled the rate limit. Animals were allowed to recover,and 1 or 2 days after pacemaker implantation a two-dimensional short-axis echocardiogram was obtained atthe level of the papillary muscle heads. The pacemakerwas then programmed to 250 beats per minute, andanimals were examined daily for evidence of pulmonaryand right heart congestion (ascites). Repeat echocardi-ography was performed after 2 weeks of pacing and 1-2days before the isolated heart study. All echocardiogramswere obtained with pacing temporarily suspended. En-docardial short-axis contours from at least three beatswere averaged to determine end-diastolic (maximal) andend-systolic (minimal) areas and area ejection fraction.
Isolated Heart PreparationDetails of the blood-perfused isolated canine heart
preparation have been previously reported.9 Briefly, asupport dog (weight, 25-30 kg) was anesthetized withpentobarbital sodium (30 mg/kg i.v.), heparinized(5,000 units i.v.), and mechanically ventilated. Thefemoral arteries and veins were cannulated and con-nected to a perfusion system used to supply oxygenatedblood to the isolated heart. The temperature of theperfusate was maintained at approximately 37°C with aheat exchanger. A donor animal from either the heartfailure or control group was anesthetized with fentanylcitrate (10 ,g/kg i.v.) and pentobarbital sodium (10-20mg/kg i.v.). Blood pressure was frequently supportedwith epinephrine (0.1-10 ug/kg/min) during initial in-duction in the heart failure dogs. A 7F balloon-tippedcatheter was advanced into the pulmonary artery, andright heart pressures were measured. In all but twocases, heart failure dogs were weaned from epinephrinebefore these hemodynamic measurements were obtained.
After right heart catheterization, the dog was me-chanically ventilated and the chest opened via midline
sternotomy. The left subclavian artery was cannulatedwith the arterial perfusion line from the support dogand the heart isolated and removed. The left atrium wasincised, the chordae tendineae cut from the mitralleaflets, and a metal adapter ring sutured onto themitral annulus. A water-filled latex balloon, connectedto a computer-controlled servo-pump system, wasplaced through the adapter into the left ventricularcavity. A small cannula inserted through the left ven-tricular apex was connected to negative pressure todrain thebesian blood flow and ensure close approxima-tion between balloon and endocardium. Left ventricular(LV) pressure was measured by a micromanometer-tipped catheter (PC-380, Millar Instruments, Houston,Tex.) placed inside the latex balloon. Coronary sinusblood was drained from the right ventricle through anin-line ultrasonic flowmeter (Transonics, Ithaca, N.Y.)to measure coronary blood flow. The difference be-tween arterial and coronary venous blood oxygen con-tent was measured continuously by absorption spec-trometry (A-VOX System) calibrated to a Lex-O2-Conoxygen analyzer. Electrodes were sutured to the ven-tricular surface and atrium for an electrocardiogramand pacing, respectively. Hearts were paced at 171 beatsper minute (cycle length, 350 msec) during data collec-tion, a rate chosen to ensure overdriving the atrialrhythm even during dobutamine infusion.
Several measures were taken to stabilize the prepa-ration and reduce fluctuations in sympathetic outflowfrom the support dog. First, pentobarbital sodium (2-3mg/kg/hr) was continuously infused to avoid fluctua-tions in the level of anesthesia. The support dog waspremedicated with sodium hydrocortisone 500 mg i.m.,diphenhydramine 25 mg i.m., and indomethacin 50 mgp.r. (likely minimizing release of platelet and whiteblood cell mediators during the extracorporeal circula-tion). Arterial and central venous pressures were con-tinuously monitored and arterial pH, Po2, and Pco2determined every 30 minutes. Adjustments of ventila-tion rate, supplemental oxygen, correction of base def-icits, and volume support were used when appropriateto maintain these parameters within normal rangesthroughout the experiment. Finally, a two-pump perfu-sion system was used to maintain a constant hemody-namic load on the support dog. Arterial blood waspumped from the support dog at a constant rate (400-500 ml/min) irrespective of coronary flow in the isolatedheart. A second pump shunted blood from the primarypump circuit back into the support dog, bypassing theisolated heart. This pump was servo-controlled to main-tain a constant perfusion pressure (90 mm Hg) to theisolated heart.LV volume was controlled by a servo-pump respond-
ing to a command signal generated by the computersolution of the cardiac interaction with a model vascularloading system (three-element Windkessel) as previ-ously described in detail.9"10 Windkessel parameterswere arterial resistance (2.0 mm Hg * second * ml-1),characteristic impedance (0.3 mm Hg* second * ml-l),and compliance (0.4 ml . mm Hg-1), values similar tothose derived from normal canine aortic impedancespectrum." This loading system could also be changedto allow isovolumic contraction.
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from

518 Circulation Research Vol 70, No 3 March 1992
Experimental ProtocolsSystolic and diastolic pressure-volume relations and ener-
getics. Steady-state data from ejecting beats were collectedat five to six end-diastolic volumes, with end-diastolicpressure ranging from 0 to 20 mm Hg, to determineend-systolic pressure-volume relations (ESPVRs), end-diastolic pressure-volume relations (EDPVRs), andMVo2. In each heart the volume intercept (VO) of theESPVR was directly measured by reducing intraventricu-lar volume until peak systolic pressure was zero. V0measurements were reproducible to within 1 ml withmultiple determinations.
Force-frequency relation. Heart rate was varied from125 to 225 beats per minute. Because of a limitedfrequency response of the impedance loading system,these data were obtained isovolumically. Peak systolicpressure was determined at each heart rate.Dobutamine dose-response. Dobutamine dose-
response curves were obtained under ejecting condi-tions. Steady-state data were collected at baseline andwith infusion of dobutamine into the isolated heartperfusion circuit at incremental rates (1, 5, 10, 20, 40,80, 160, and 320 ,g/min) up to the maximal toleratedrate, limited by arrhythmias and heart rate greater than171 beats per minute. Because of practical limitations inobtaining data at multiple infusion rates, a single end-diastolic volume was used (chosen for each heart toproduce an end-diastolic pressure of 8-12 mm Hg atbaseline). Contractile response was indexed by steady-state stroke work. At the maximal dobutamine infusionrates, pressure-volume and energetic relations werealso determined in the failure hearts by obtaining dataat five or six randomly chosen end-diastolic volumes.Postmortem examination. At the conclusion of the study
the isolated heart was arrested in diastole by slow intra-coronary infusion of saturated KCl solution. The heartwas removed from the volume servo-system, atrial tissueresected, and the apical vent in the left ventricle closed bya purse-string suture. The left ventricle was then filled tothe level of the mitral annulus with saline and submergedto that level in a saline bath. The intraventricular cavitywas carefully aspirated. At least three such measurementswere averaged to determine diastolic-arrested chambervolume at zero transmural pressure (Vref). Left ventricular(septum and free wall) and right ventricular free wallweights were also measured.
Data AnalysisData were recorded continuously on a strip-chart
recorder (model 2800, Gould, Cleveland, Ohio) andalso digitized at a rate of 500 Hz and stored on magneticdisk for off-line analysis. Data from five to 10 beats ateach steady-state condition were signal averaged foranalysis.
Systolic pressure-volume relations. Data obtained atmultiple preload volumes were used to derive severalsystolic chamber function indexes. The ESPVR wasderived from the points of maximal P/(V-VJ). End-systolic points were fit to a linear elastance (E,s) model
Pes=Ees (Ves -Vo) (1)where Pes is LV pressure at end systole and Ves is LVvolume at end systole. Because in many cases the
ESPVR was obviously nonlinear, the end-systolic pointswere also fit to a parabolic relation
Pes=a * (Ves-Vo)+b * (Ves-V0)2 (2)
where a is the slope of the ESPVR at V., and b is thecoefficient of nonlinearity. The statistical significance ofnonlinearity was determined by a Student's t test usingthe estimate and standard error of the estimate of the bcoefficient. The null hypothesis (b=0, thus ESPVR islinear) was rejected if p<0.05 for the b term.
Because nonlinearity rendered statistical compari-sons between ESPVRs somewhat difficult, two alterna-tives were used. One used the regression fits (linear ornonlinear) to estimate Ve, at a common Pes (50 mm Hg),a larger Ves being consistent with decreased ventricularcontractility. The second approach examined the rela-tion between stroke work and end-diastolic volume. Theslope of this linear relation is a load-insensitive measureof chamber contractility.12'13
Diastolic pressure-volume relations. End-diastolicpoints were fit to a monoexponential equation
Ped-PPo+c. (edVed-1 ) (3)where Ped and Ved are end-diastolic pressure and vol-ume, respectively, PO is the pressure at V., and PO, c, andd are nonlinear fit parameters. Diastolic chamber stiff-ness (dPed/dVed) was calculated from the first derivativeof Equation 3. For comparisons between groups, dia-stolic chamber stiffness was determined at a commonend-diastolic pressure (15 mm Hg).
Stress-strain relations. Systolic and diastolic pressure-volume relations have limitations in quantifying myo-cardial properties because of their dependence onchamber size and ventricular mass. Therefore a stress-strain analysis with a thick-walled spherical ventricularmodel was used ("Appendix").
Both end-systolic and end-diastolic stress-strain rela-tions were nonlinear and were fit well by monoexponen-tial equations. In the case of the end-systolic relation,the equation was
oes=A (eB(es-E)_1) (4)where cres is end-systolic stress, Ce, is end-systolic strain,,0 is the strain at VO, and A and B are fit parameters.End-diastolic stress-strain relations were fit to theequation
O'ed=C (eDIed-1) (5)where Led is end-diastolic stress, Ced iS end-diastolicstrain, and C and D are fit parameters. First derivativesof both stress equations with respect to strain were usedto determine myocardial end-systolic and end-diastolicstiffness (da/de). For comparisons between heartgroups, stiffness was determined at matched stresses (75and 15 g/cm2 for systole and diastole, respectively).Dobutamine dose-response. The contractile response
to 8-adrenergic stimulation (dobutamine HCl) was as-sessed by change in stroke work with hearts ejectingagainst a fixed afterload impedance at a constant Vedand heart rate. Intracoronary dobutamine concentra-tion was calculated by dividing infusion rate by mea-sured coronary blood flow. Because the relation be-tween dobutamine concentration and stroke work did
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from
Wolff et al Mechanics and Energetics of Heart Failure 519
not plateau in all hearts (particularly in the controlgroup in which increased heart rate and arrhythmiaslimited the dobutamine infusion rate), data could not bemeaningfully fit to the Hill equation. Instead, the rela-tions were linearized using the logarithm of the dobu-tamine concentration, and multivariate linear regres-
sion analysis was used (see below) to compare theresponses between animal groups.
Myocardial energetics. Ventricular energetics werecharacterized using the pressure-volume area (PVA)-MVo2 relation framework described by Suga.1415 PVAis a measure of the total mechanical work performed bythe left ventricle during a cardiac cycle and is defined as
the area bound by the ESPVR, the EDPVR, and thesystolic trajectory of the pressure-volume loop. Therelation between MVo2 and PVA is linear in the normalisolated and in situ dog heart.15 This framework allowspartitioning of the MVo2 between oxygen consumptionassociated with ventricular work and non-work-relatedMVo2. Hence, the efficiencies of these two aspects ofmyocardial energetics can be examined separately.MVo2 (per beat) was obtained as the difference
between arterial and coronary venous blood oxygencontent x coronary blood flow/heart rate, neglecting thesmall amount of LV thebesian flow. LV MVo2 wascalculated as suggested by Suga et al,15 by subtractingestimated unloaded right ventricular oxygen consump-tion from the total MVo2. LV MVo2 was expressed perbeat and was normalized to 100 g LV.PVA was determined by digitally integrating under
the pressure-volume loop from end diastole to endsystole and adding the area under the ESPVR betweenend systole and V, (obtained by analytic integration ofEquation 1 or 2). The area under the diastolic pressure-volume curve (EDPVR) between Ved and the zeropressure intercept (obtained by analytic integration ofEquation 3) was subtracted from the sum of the twoprevious areas. The small portion of PVA below thevolume axis was not included in the calculated PVA.PVA was also normalized to 100 g LV weight.
Statistical MethodsWe used a multivariate linear regression model to
compare MVo2-PVA relations as well as other linearrelations by using a dummy variable to represent thepresence or absence of heart failure and additionaldummy variables for each dog to adjust for interanimalvariation.16 Changes in echocardiographic parameterswere tested with analysis of variance (ANOVA). Com-parisons of other variables were made by two-tailed ttests. Differences were accepted as statistically significantfor p<0.05. All data are expressed as mean+±SD unlessotherwise specified. Statistical tests and nonlinear curvefitting were performed with commercially available soft-ware (SYSTAT, Inc., Evanston, Ill.).
ResultsThirteen dogs were instrumented and underwent
rapid ventricular pacing. Three animals died suddenlyduring the pacing period and two died during inductionof anesthesia for the isolated heart study. A sixth dogwas excluded because of pacer lead migration andextended interruption of pacing. The seven remainingdogs made up the heart failure group and were paced
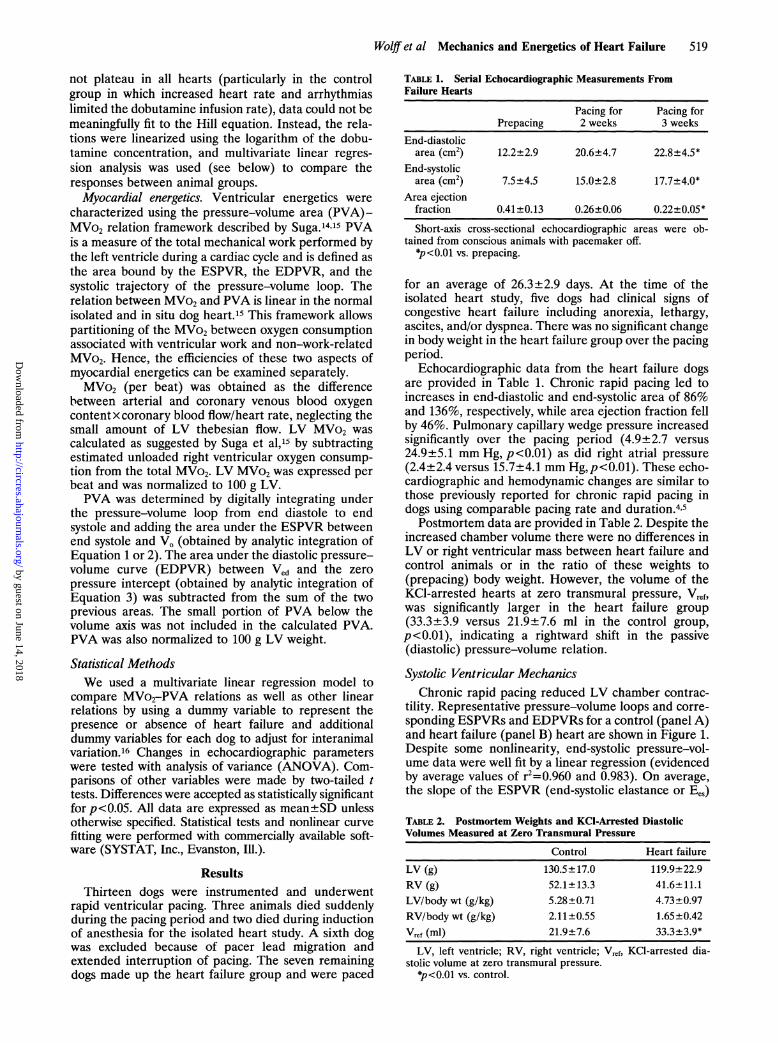
TABLE 1. Serial Echocardiographic Measurements FromFailure Hearts
Pacing for Pacing forPrepacing 2 weeks 3 weeks
End-diastolicarea (cm2) 12.2±2.9 20.6±4.7 22.8±4.5*
End-systolicarea (cm2) 7.5±4.5 15.0±2.8 17.7±4.0*
Area ejectionfraction 0.41±0.13 0.26±0.06 0.22±0.05*
Short-axis cross-sectional echocardiographic areas were ob-tained from conscious animals with pacemaker off.
*p<0.01 vs. prepacing.
for an average of 26.3+2.9 days. At the time of theisolated heart study, five dogs had clinical signs ofcongestive heart failure including anorexia, lethargy,ascites, and/or dyspnea. There was no significant changein body weight in the heart failure group over the pacingperiod.
Echocardiographic data from the heart failure dogsare provided in Table 1. Chronic rapid pacing led toincreases in end-diastolic and end-systolic area of 86%and 136%, respectively, while area ejection fraction fellby 46%. Pulmonary capillary wedge pressure increasedsignificantly over the pacing period (4.9+ 2.7 versus24.9+5.1 mm Hg, p<0.01) as did right atrial pressure(2.4 +2.4 versus 15.7+ 4.1 mm Hg,p <0.01). These echo-cardiographic and hemodynamic changes are similar tothose previously reported for chronic rapid pacing indogs using comparable pacing rate and duration.4,5Postmortem data are provided in Table 2. Despite the
increased chamber volume there were no differences inLV or right ventricular mass between heart failure andcontrol animals or in the ratio of these weights to(prepacing) body weight. However, the volume of theKCl-arrested hearts at zero transmural pressure, Vref,was significantly larger in the heart failure group(33.3+3.9 versus 21.9+7.6 ml in the control group,p<0.01), indicating a rightward shift in the passive(diastolic) pressure-volume relation.
Systolic Ventricular MechanicsChronic rapid pacing reduced LV chamber contrac-
tility. Representative pressure-volume loops and corre-sponding ESPVRs and EDPVRs for a control (panel A)and heart failure (panel B) heart are shown in Figure 1.Despite some nonlinearity, end-systolic pressure-vol-ume data were well fit by a linear regression (evidencedby average values of r2=0.960 and 0.983). On average,the slope of the ESPVR (end-systolic elastance or Ees)
TABLE 2. Postmortem Weights and KCI-Arrested DiastolicVolumes Measured at Zero Transmural Pressure
Control Heart failure
LV (g) 130.5±17.0 119.9±22.9RV (g) 52.1±13.3 41.6±11.1LV/body wt (g/kg) 5.28±0.71 4.73±0.97RV/body wt (g/kg) 2.11±0.55 1.65±0.42Vref (ml) 21.9±7.6 33.3±3.9*
LV, left ventricle; RV, right ventricle; Vref, KCl-arrested dia-stolic volume at zero transmural pressure.
$p<0.01 vs. control.
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from
520 Circulation Research Vol 70, No 3 March 1992
Pressure-Volume
A
125 -
100 -
NE 7
5
0
g 50-(n
25 -
-1 0 -0.100 -0.2
Volume (ml)
125
100B
NEu1-Ci
(0
75
Stress-Strain
C
-0.1 0.0Strain
80 100 -0.2 -0.1 0.0
Stroin
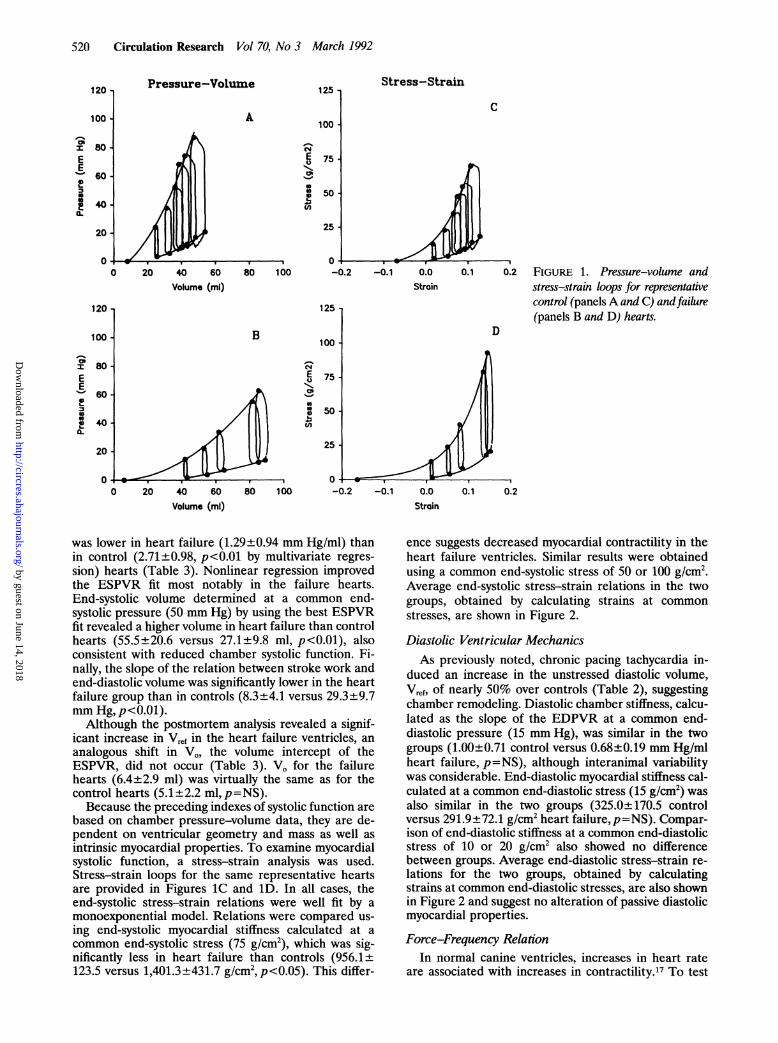
0.1 0.2 FIGURE 1. Pressure-volume andstress-strain loops for representativecontrol (panelsA and C) andfailure(panels B and D) hearts.
D
0.1 0.2
was lower in heart failure (1.29+±0.94 mm Hg/mi) thanin control (2.71±0.98, p<0.01 by multivariate regres-sion) hearts (Table 3). Nonlinear regression improvedthe ESPVR fit most notably in the failure hearts.End-systolic volume determined at a common end-systolic pressure (50 mm Hg) by using the best ESPVRfit revealed a higher volume in heart failure than controlhearts (55.5±20.6 versus 27.1±9.8 ml, p<0.01), alsoconsistent with reduced chamber systolic function. Fi-nally, the slope of the relation between stroke work andend-diastolic volume was significantly lower in the heartfailure group than in controls (8.3+4.1 versus 29.3±9.7mm Hg, p<0.01).
Although the postmortem analysis revealed a signif-icant increase in Vref in the heart failure ventricles, ananalogous shift in V,, the volume intercept of theESPVR, did not occur (Table 3). VO for the failurehearts (6.4±2.9 ml) was virtually the same as for thecontrol hearts (5.1+2.2 ml, p=NS).
Because the preceding indexes of systolic function arebased on chamber pressure-volume data, they are de-pendent on ventricular geometry and mass as well asintrinsic myocardial properties. To examine myocardialsystolic function, a stress-strain analysis was used.Stress-strain loops for the same representative heartsare provided in Figures 1C and 1D. In all cases, theend-systolic stress-strain relations were well fit by amonoexponential model. Relations were compared us-ing end-systolic myocardial stiffness calculated at acommon end-systolic stress (75 g/cm'), which was sig-nificantly less in heart failure than controls (956.1+±123.5 versus 1,401.3+±431.7 g/cm2,p<0.05). This differ-
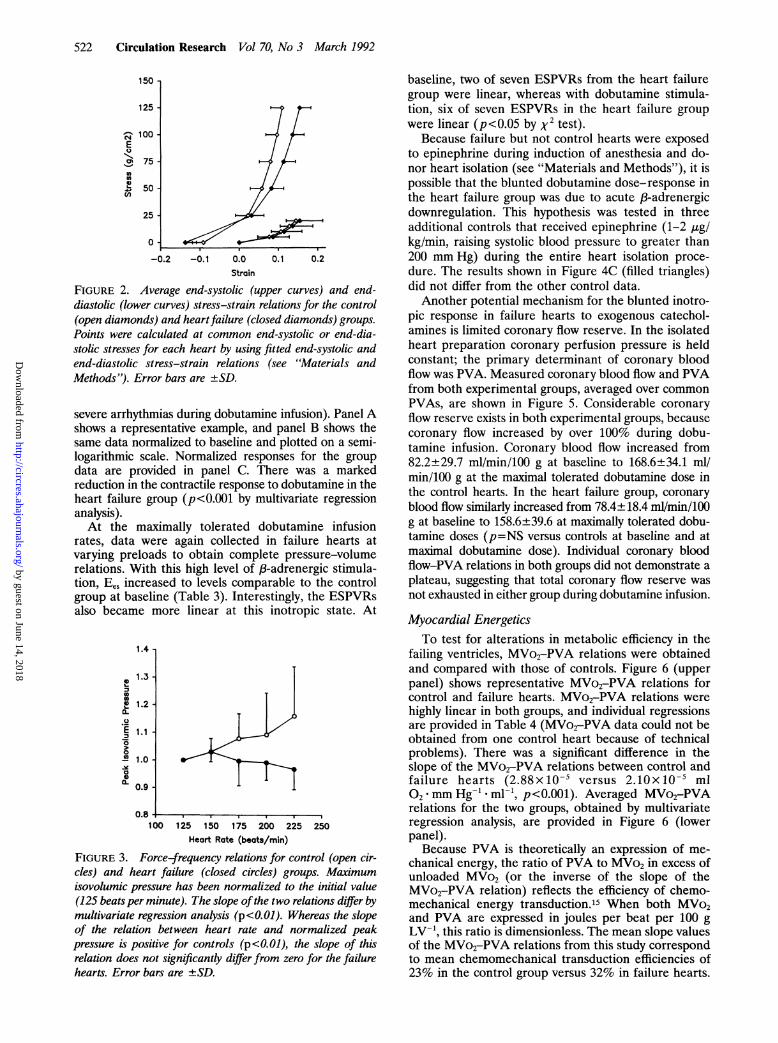
ence suggests decreased myocardial contractility in theheart failure ventricles. Similar results were obtainedusing a common end-systolic stress of 50 or 100 g/cm'.Average end-systolic stress-strain relations in the twogroups, obtained by calculating strains at common
stresses, are shown in Figure 2.
Diastolic Ventricular MechanicsAs previously noted, chronic pacing tachycardia in-
duced an increase in the unstressed diastolic volume,Vref, of nearly 50% over controls (Table 2), suggestingchamber remodeling. Diastolic chamber stiffness, calcu-lated as the slope of the EDPVR at a common end-diastolic pressure (15 mm Hg), was similar in the twogroups (1.00±0.71 control versus 0.68±0.19 mm Hg/mlheart failure, p=NS), although interanimal variabilitywas considerable. End-diastolic myocardial stiffness cal-culated at a common end-diastolic stress (15 g/cm2) wasalso similar in the two groups (325.0+170.5 controlversus 291.9+72.1 g/cm2 heart failure, p=NS). Compar-ison of end-diastolic stiffness at a common end-diastolicstress of 10 or 20 g/cm2 also showed no differencebetween groups. Average end-diastolic stress-strain re-lations for the two groups, obtained by calculatingstrains at common end-diastolic stresses, are also shownin Figure 2 and suggest no alteration of passive diastolicmyocardial properties.
Force-Frequency RelationIn normal canine ventricles, increases in heart rate
are associated with increases in contractility.17 To test
120
100_
z: 80EE'~60
240a.
20
0
120,
100
OhX 80EE
60o0.4020
00 20 40 60
Volume (ml)
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from
Wolff et al Mechanics and Energetics of Heart Failure 521
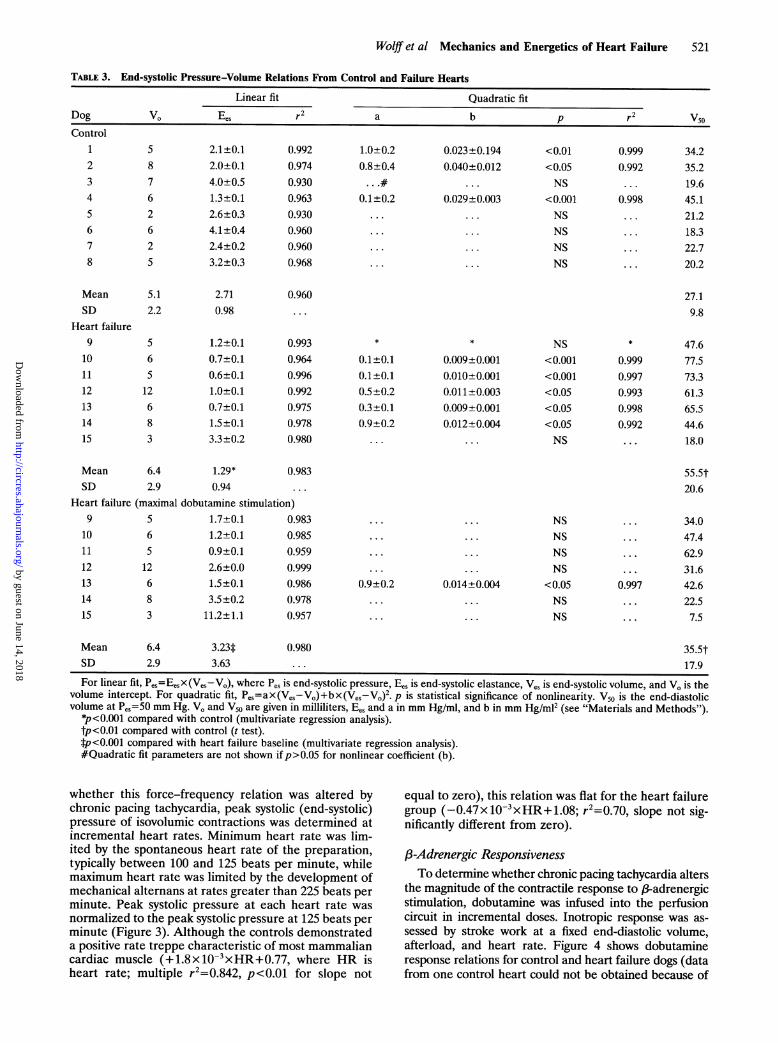
TABLE 3. End-systolic Pressure-Volume Relations From Control and Failure Hearts
Linear fit Quadratic fitDog VO Ees r2 a b p r2 V5oControl
12345678
MeanSD
Heart failure9
101112131415
58762625
5.12.2
2.1±0.12.0±0.14.0±0.51.3±0.12.6±0.34.1±0.42.4±0.23.2±0.3
2.710.98
S6S12683
1.2±0.10.7±0.10.6±0.11.0±0.10.7±0.11.5±0.13.3±0.2
0.9920.9740.9300.9630.9300.9600.9600.968
0.960. . .
0.9930.9640.9960.9920.9750.9780.980
1.0+0.20.8+0.4
0.1±0.2
.
0.023±0.1940.040±0.012
. .
0.029±0.003. ..
. . .
..
<0.01<0.05NS
<0.001NSNSNSNS
0.9990.992
0.998
. . .
. . .
34.235.219.645.121.218.322.720.2
27.19.8
0.1+0.10.1±0.10.5±0.20.3±0.10.9±0.2
0.009±0.0010.010±0.0010.011±0.0030.009±0.0010.012±0.004
. ..
NS<0.001<0.001<0.05<0.05<0.05NS
0.9990.9970.9930.9980.992
47.677.573.361.365.544.618.0
6.4 1.29*2.9 0.94
Heart failure (maximal dobutamine stimulation)665
12 1213 614 815
Mean
SD
1.7+0.11.2±0.10.9±0.12.6±0.01.5±0.13.5±0.2
3 11.2±1.1
6.42.9
3.23*3.63
0.980 35.5t17.9
For linear fit, Pes=EesX(Ves-Vo), where Pes is end-systolic pressure, Ees is end-systolic elastance, Ves is end-systolic volume, and V. is thevolume intercept. For quadratic fit, Pes=ax(Ves-Vo)+bx(Ves-Vo)2. p is statistical significance of nonlinearity. V50 is the end-diastolicvolume at PeS=50 mm Hg. VO and V50 are given in milliliters, Ees and a in mm Hg/ml, and b in mm Hg/m12 (see "Materials and Methods").
*p<0.001 compared with control (multivariate regression analysis).tp<0.01 compared with control (t test).*p <0.001 compared with heart failure baseline (multivariate regression analysis).#Quadratic fit parameters are not shown ifp>0.05 for nonlinear coefficient (b).
whether this force-frequency relation was altered bychronic pacing tachycardia, peak systolic (end-systolic)pressure of isovolumic contractions was determined atincremental heart rates. Minimum heart rate was lim-ited by the spontaneous heart rate of the preparation,typically between 100 and 125 beats per minute, whilemaximum heart rate was limited by the development ofmechanical alternans at rates greater than 225 beats perminute. Peak systolic pressure at each heart rate wasnormalized to the peak systolic pressure at 125 beats perminute (Figure 3). Although the controls demonstrateda positive rate treppe characteristic of most mammaliancardiac muscle (+1.8x1O-3xHR+0.77, where HR isheart rate; multiple r2=0.842, p<O.01 for slope not
equal to zero), this relation was flat for the heart failuregroup (-0.47x10'3xHR+1.08; r2=0.70, slope not sig-nificantly different from zero).
13-Adrenergic ResponsivenessTo determine whether chronic pacing tachycardia alters
the magnitude of the contractile response to 3-adrenergicstimulation, dobutamine was infused into the perfusioncircuit in incremental doses. Inotropic response was as-sessed by stroke work at a fixed end-diastolic volume,afterload, and heart rate. Figure 4 shows dobutamineresponse relations for control and heart failure dogs (datafrom one control heart could not be obtained because of
MeanSD
0.983
91011
0.9830.9850.9590.9990.9860.9780.957
. . .
. . .
0.9+0O.2
. . .
. ..
0.014±0.004
NSNSNSNS
<0.05NSNS
55.5t20.6
34.047.462.931.642.622.57.5
. . .
0.997
. . .
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from
522 Circulation Research Vol 70, No 3 March 1992
150
125 -
c- 100-E
\ 75-000
~e 50- 7-C')
25-
0--0.2 -0. 1 0.0 0.1
Strain
0.2
FIGURE 2. Average end-systolic (upper curves) and end-diastolic (lower curves) stress-strain relations for the control(open diamonds) and heart failure (closed diamonds) groups.
Points were calculated at common end-systolic or end-dia-stolic stresses for each heart by using fitted end-systolic andend-diastolic stress-strain relations (see "Materials andMethods"). Error bars are ±SD.
severe arrhythmias during dobutamine infusion). Panel Ashows a representative example, and panel B shows thesame data normalized to baseline and plotted on a semi-logarithmic scale. Normalized responses for the groupdata are provided in panel C. There was a markedreduction in the contractile response to dobutamine in theheart failure group (p<O.OO1 by multivariate regressionanalysis).At the maximally tolerated dobutamine infusion
rates, data were again collected in failure hearts atvarying preloads to obtain complete pressure-volumerelations. With this high level of ,B-adrenergic stimula-tion, Ees increased to levels comparable to the controlgroup at baseline (Table 3). Interestingly, the ESPVRsalso became more linear at this inotropic state. At
1.4 -
1.3
00
E 1.2-CL
E 1.10
0
1.0
X 0.9'
0.8100 125 150 175 200 225 250
Heart Rate (beats/min)
FIGURE 3. Force-frequency relations for control (open cir-cles) and heart failure (closed circles) groups. Maximumisovolumic pressure has been normalized to the initial value(125 beats per minute). The slope ofthe two relations differ bymultivariate regression analysis (p <0.01). Whereas the slopeof the relation between heart rate and normalized peakpressure is positive for controls (p<O.Ol), the slope of thisrelation does not significantly differ from zero for the failurehearts. Error bars are ±SD.
baseline, two of seven ESPVRs from the heart failuregroup were linear, whereas with dobutamine stimula-tion, six of seven ESPVRs in the heart failure groupwere linear (p<0.05 by x2 test).
Because failure but not control hearts were exposedto epinephrine during induction of anesthesia and do-nor heart isolation (see "Materials and Methods"), it ispossible that the blunted dobutamine dose-response inthe heart failure group was due to acute ,-adrenergicdownregulation. This hypothesis was tested in threeadditional controls that received epinephrine (1-2 ,ug/kg/min, raising systolic blood pressure to greater than200 mm Hg) during the entire heart isolation proce-dure. The results shown in Figure 4C (filled triangles)did not differ from the other control data.Another potential mechanism for the blunted inotro-
pic response in failure hearts to exogenous catechol-amines is limited coronary flow reserve. In the isolatedheart preparation coronary perfusion pressure is heldconstant; the primary determinant of coronary bloodflow was PVA. Measured coronary blood flow and PVAfrom both experimental groups, averaged over commonPVAs, are shown in Figure 5. Considerable coronaryflow reserve exists in both experimental groups, becausecoronary flow increased by over 100% during dobu-tamine infusion. Coronary blood flow increased from82.2±29.7 ml/min/100 g at baseline to 168.6±34.1 ml!min/100 g at the maximal tolerated dobutamine dose inthe control hearts. In the heart failure group, coronaryblood flow similarly increased from 78.4±18.4 ml/min/100g at baseline to 158.6+39.6 at maximally tolerated dobu-tamine doses (p=NS versus controls at baseline and atmaximal dobutamine dose). Individual coronary bloodflow-PVA relations in both groups did not demonstrate aplateau, suggesting that total coronary flow reserve wasnot exhausted in either group during dobutamine infusion.
Myocardial EnergeticsTo test for alterations in metabolic efficiency in the
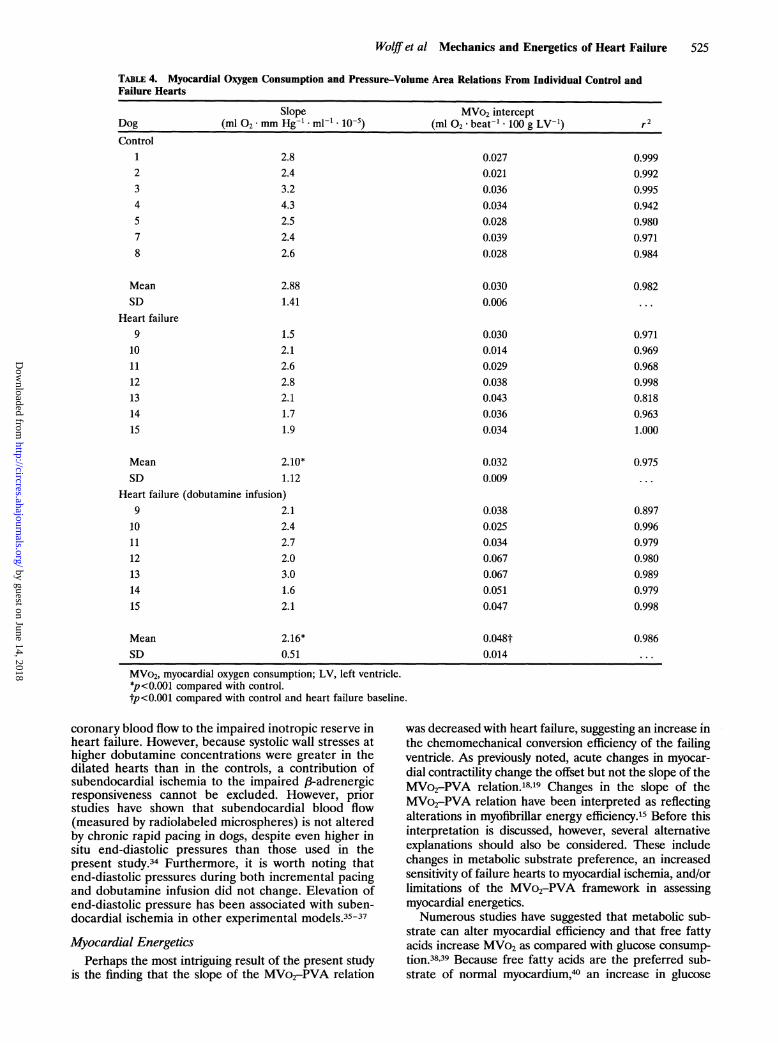
failing ventricles, MVo2-PVA relations were obtainedand compared with those of controls. Figure 6 (upperpanel) shows representative MVo2-PVA relations forcontrol and failure hearts. MVo2-PVA relations werehighly linear in both groups, and individual regressionsare provided in Table 4 (MVo2-PVA data could not beobtained from one control heart because of technicalproblems). There was a significant difference in theslope of the MVo2-PVA relations between control andfailure hearts (2.88x10` versus 2.10x10 ml02*mm Hg-1 * ml-1, p<0.001). Averaged MVo2-PVArelations for the two groups, obtained by multivariateregression analysis, are provided in Figure 6 (lowerpanel).
Because PVA is theoretically an expression of me-chanical energy, the ratio of PVA to MVo2 in excess ofunloaded MVo2 (or the inverse of the slope of theMVo2-PVA relation) reflects the efficiency of chemo-mechanical energy transduction.15 When both MVo2and PVA are expressed in joules per beat per 100 gLV- , this ratio is dimensionless. The mean slope valuesof the MVo2-PVA relations from this study correspondto mean chemomechanical transduction efficiencies of23% in the control group versus 32% in failure hearts.
4
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from
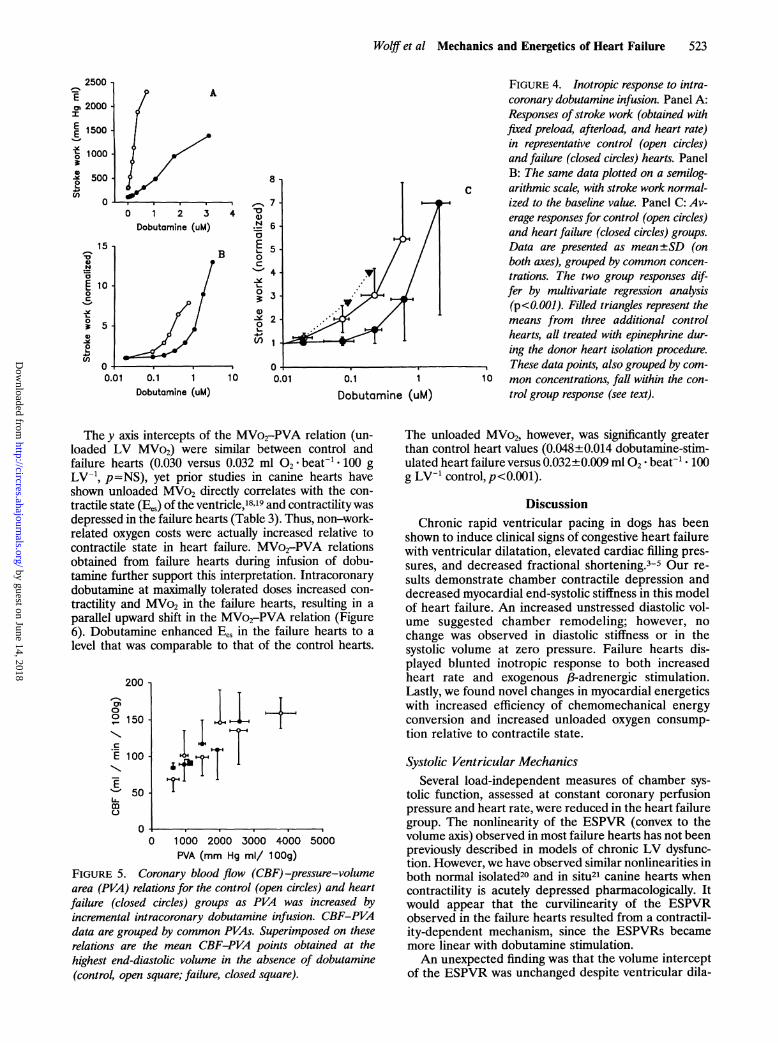
Wolff et al Mechanics and Energetics of Heart Failure 523
8
0 1 2 3Dobutomine (uM)
41 n-14 o
0N
_RaE
00c
04-,(bi)_.
.01 0.1 1 10Dobutomine (uM)
0.01 0.1 1
Dobutamine (uM)
FIGURE 4. Inotropic response to intra-coronary dobutamine infusion. Panel A:Responses ofstroke work (obtained withfixed preload, afterload, and heart rate)in representative control (open circles)and failure (closed circles) hearts. PanelB: The same data plotted on a semilog-arithmic scale, with stroke work normal-ized to the baseline value. Panel C: Av-erage responses for control (open circles)and heart failure (closed circles) groups.Data are presented as mean±SD (onboth axes), grouped by common concen-trations. The two group responses dif-fer by multivariate regression analysis(p<0.001). Filled triangles represent themeans from three additional controlhearts, all treated with epinephrine dur-ing the donor heart isolation procedure.These data points, also grouped by com-
10 mon concentrations, fall within the con-trol group response (see text).
The y axis intercepts of the MVo2-PVA relation (un-loaded LV MVo2) were similar between control andfailure hearts (0.030 versus 0.032 ml °2* beat- 100 gLV-1 ,p=NS), yet prior studies in canine hearts haveshown unloaded MVo2 directly correlates with the con-tractile state (Bes) of the ventricle,'8 9 and contractility wasdepressed in the failure hearts (Table 3). Thus, non-work-related oxygen costs were actually increased relative tocontractile state in heart failure. MVo2-PVA relationsobtained from failure hearts during infusion of dobu-tamine further support this interpretation. Intracoronarydobutamine at maximally tolerated doses increased con-
tractility and MVo2 in the failure hearts, resulting in a
parallel upward shift in the MVo2-PVA relation (Figure6). Dobutamine enhanced Bes in the failure hearts to a
level that was comparable to that of the control hearts.
200
1`._
E'0
50
C
.E 100
mc0
0-0 1000 2000 3000 4000 5000
PVA (mm Hg mI/ 1 00g)
FIGURE 5. Coronary blood flow (CBF)-pressure-volumearea (PVA) relations for the control (open circles) and heartfailure (closed circles) groups as PVA was increased byincremental intracoronary dobutamine infusion. CBF-PVAdata are grouped by common PVAs. Superimposed on theserelations are the mean CBF-PVA points obtained at thehighest end-diastolic volume in the absence of dobutamine(control, open square; failure, closed square).
The unloaded MVo2, however, was significantly greaterthan control heart values (0.048±0.014 dobutamine-stim-ulated heart failure versus 0.032+0.009 ml 02 beat' * 100g LV`1 control, p<0.001).
DiscussionChronic rapid ventricular pacing in dogs has been
shown to induce clinical signs of congestive heart failurewith ventricular dilatation, elevated cardiac filling pres-
sures, and decreased fractional shortening.3-5 Our re-
sults demonstrate chamber contractile depression anddecreased myocardial end-systolic stiffness in this modelof heart failure. An increased unstressed diastolic vol-ume suggested chamber remodeling; however, no
change was observed in diastolic stiffness or in thesystolic volume at zero pressure. Failure hearts dis-played blunted inotropic response to both increasedheart rate and exogenous ,B-adrenergic stimulation.Lastly, we found novel changes in myocardial energeticswith increased efficiency of chemomechanical energyconversion and increased unloaded oxygen consump-tion relative to contractile state.
Systolic Ventricular MechanicsSeveral load-independent measures of chamber sys-
tolic function, assessed at constant coronary perfusionpressure and heart rate, were reduced in the heart failuregroup. The nonlinearity of the ESPVR (convex to thevolume axis) observed in most failure hearts has not beenpreviously described in models of chronic LV dysfunc-tion. However, we have observed similar nonlinearities inboth normal isolated20 and in situ2' canine hearts whencontractility is acutely depressed pharmacologically. Itwould appear that the curvilinearity of the ESPVRobserved in the failure hearts resulted from a contractil-ity-dependent mechanism, since the ESPVRs becamemore linear with dobutamine stimulation.An unexpected finding was that the volume intercept
of the ESPVR was unchanged despite ventricular dila-
2500-C>Eo 2000I
E 1500
-t 1000
.O 500~0
0
150N
E 100c
-E03 5.0
(n -0*0.
-1
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from
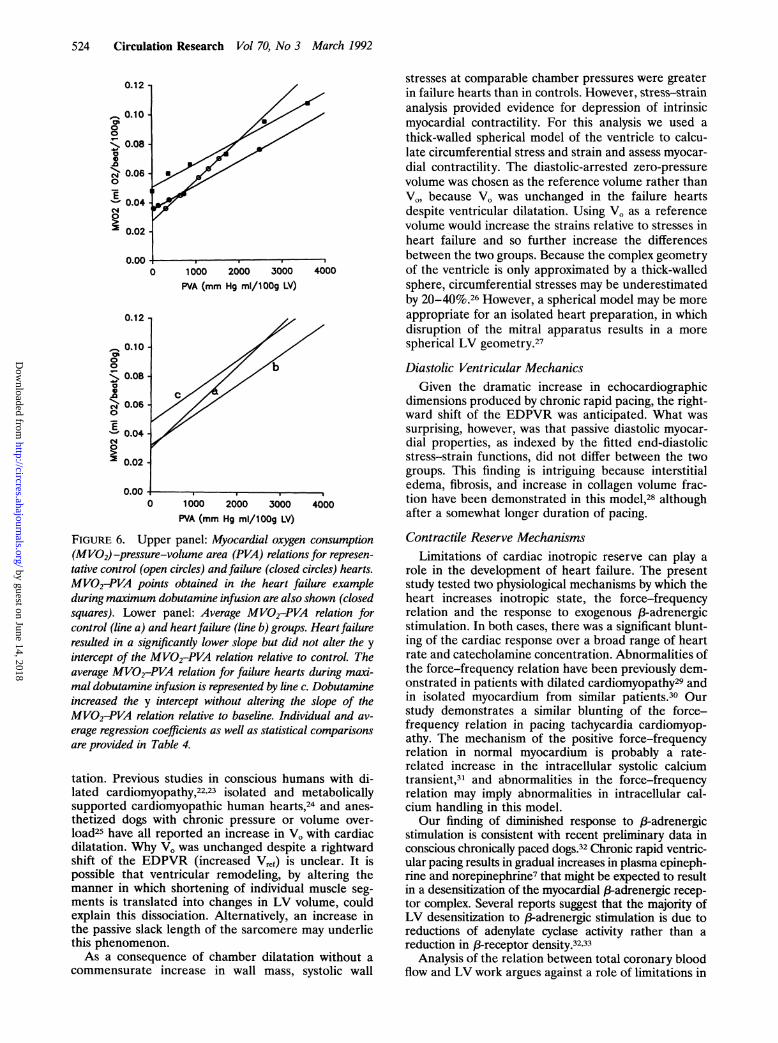
524 Circulation Research Vol 70, No 3 March 1992
0.08
0.06
0.04
fi nn13
0.00
%_.
ob00
0.080
.0
,- 0.060
O 0.04
9 0.02
0 1000 2000 3000 4000PVA (mm Hg mi/100g LV)
0 1000 2000 3000 4000
PVA (mm Hg mi/100g LV)
FIGURE 6. Upper panel: Myocardial oxygen consumption(MVO2)-pressure-volume area (PVA) relations for represen-
tative control (open circles) and failure (closed circles) hearts.MVO2-PVA points obtained in the heart failure exampleduring maximum dobutamine infusion are also shown (closedsquares). Lower panel: Average MVO2-PVA relation forcontrol (line a) and heartfailure (line b) groups. Heartfailureresulted in a significantly lower slope but did not alter the y
intercept of the MVO2-PVA relation relative to control. Theaverage MVO2-PVA relation for failure hearts during maxi-mal dobutamine infusion is represented by line c. Dobutamineincreased the y intercept without altering the slope of theMVO2-PVA relation relative to baseline. Individual and av-
erage regression coefficients as well as statistical comparisonsare provided in Table 4.
tation. Previous studies in conscious humans with di-lated cardiomyopathy,2223 isolated and metabolicallysupported cardiomyopathic human hearts,24 and anes-thetized dogs with chronic pressure or volume over-load25 have all reported an increase in VO with cardiacdilatation. Why VO was unchanged despite a rightwardshift of the EDPVR (increased Vref) is unclear. It ispossible that ventricular remodeling, by altering themanner in which shortening of individual muscle seg-ments is translated into changes in LV volume, couldexplain this dissociation. Alternatively, an increase inthe passive slack length of the sarcomere may underliethis phenomenon.As a consequence of chamber dilatation without a
commensurate increase in wall mass, systolic wall
stresses at comparable chamber pressures were greaterin failure hearts than in controls. However, stress-strainanalysis provided evidence for depression of intrinsicmyocardial contractility. For this analysis we used athick-walled spherical model of the ventricle to calcu-late circumferential stress and strain and assess myocar-dial contractility. The diastolic-arrested zero-pressurevolume was chosen as the reference volume rather thanVO, because VO was unchanged in the failure heartsdespite ventricular dilatation. Using VO as a referencevolume would increase the strains relative to stresses inheart failure and so further increase the differencesbetween the two groups. Because the complex geometryof the ventricle is only approximated by a thick-walledsphere, circumferential stresses may be underestimatedby 20-40%.26 However, a spherical model may be moreappropriate for an isolated heart preparation, in whichdisruption of the mitral apparatus results in a morespherical LV geometry.27
Diastolic Ventricular MechanicsGiven the dramatic increase in echocardiographic
dimensions produced by chronic rapid pacing, the right-ward shift of the EDPVR was anticipated. What wassurprising, however, was that passive diastolic myocar-dial properties, as indexed by the fitted end-diastolicstress-strain functions, did not differ between the twogroups. This finding is intriguing because interstitialedema, fibrosis, and increase in collagen volume frac-tion have been demonstrated in this model,28 althoughafter a somewhat longer duration of pacing.
Contractile Reserve MechanismsLimitations of cardiac inotropic reserve can play a
role in the development of heart failure. The presentstudy tested two physiological mechanisms by which theheart increases inotropic state, the force-frequencyrelation and the response to exogenous ,B-adrenergicstimulation. In both cases, there was a significant blunt-ing of the cardiac response over a broad range of heartrate and catecholamine concentration. Abnormalities ofthe force-frequency relation have been previously dem-onstrated in patients with dilated cardiomyopathy29 andin isolated myocardium from similar patients.30 Ourstudy demonstrates a similar blunting of the force-frequency relation in pacing tachycardia cardiomyop-athy. The mechanism of the positive force-frequencyrelation in normal myocardium is probably a rate-related increase in the intracellular systolic calciumtransient,31 and abnormalities in the force-frequencyrelation may imply abnormalities in intracellular cal-cium handling in this model.Our finding of diminished response to 3-adrenergic
stimulation is consistent with recent preliminary data inconscious chronically paced dogs.32 Chronic rapid ventric-ular pacing results in gradual increases in plasma epineph-rine and norepinephrine7 that might be expected to resultin a desensitization of the myocardial ,B-adrenergic recep-tor complex. Several reports suggest that the majority ofLV desensitization to j-adrenergic stimulation is due toreductions of adenylate cyclase activity rather than areduction in ,3-receptor density.3233
Analysis of the relation between total coronary bloodflow and LV work argues against a role of limitations in
U.UZ
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from
Wolff et al Mechanics and Energetics of Heart Failure 525
TABLE 4. Myocardial Oxygen Consumption and Pressure-Volume Area Relations From Individual Control andFailure Hearts
Slope MVo2 interceptDog (ml 02*mm Hg-l'ml-'1 10-5) (ml O2 *beat- -100 g LV-1) r2Control
1234578
2.82.43.24.32.52.42.6
MeanSD
Heart failure9
101112131415
Mean
0.0270.0210.0360.0340.0280.0390.028
2.881.41
1.52.12.62.82.11.71.9
0.9990.9920.9950.9420.9800.9710.984
0.0300.006
0.0300.0140.0290.0380.0430.0360.034
2.10*
0.982
0.9710.9690.9680.9980.8180.9631.000
0.0320.009SD 1.12
Heart failure (dobutamine infusion)9
101112131415
2.12.42.72.03.01.62.1
0.0380.0250.0340.0670.0670.0510.047
0.975
0.8970.9960.9790.9800.9890.9790.998
MeanSD
2.16*0.51
MVo2, myocardial oxygen consumption; LV, left ventricle.*p<0.001 compared with control.tp<0.001 compared with control and heart failure baseline.
coronary blood flow to the impaired inotropic reserve inheart failure. However, because systolic wall stresses athigher dobutamine concentrations were greater in thedilated hearts than in the controls, a contribution ofsubendocardial ischemia to the impaired 8-adrenergicresponsiveness cannot be excluded. However, priorstudies have shown that subendocardial blood flow(measured by radiolabeled microspheres) is not alteredby chronic rapid pacing in dogs, despite even higher insitu end-diastolic pressures than those used in thepresent study.34 Furthermore, it is worth noting thatend-diastolic pressures during both incremental pacingand dobutamine infusion did not change. Elevation ofend-diastolic pressure has been associated with suben-docardial ischemia in other experimental models.35-37
Myocardial EnergeticsPerhaps the most intriguing result of the present study
is the finding that the slope of the MVo2-PVA relation
was decreased with heart failure, suggesting an increase in
the chemomechanical conversion efficiency of the failingventricle. As previously noted, acute changes in myocar-dial contractility change the offset but not the slope of theMVo2-PVA relation.'819 Changes in the slope of theMVo2-PVA relation have been interpreted as reflectingalterations in myofibrillar energy efficiency.15 Before thisinterpretation is discussed, however, several alternativeexplanations should also be considered. These includechanges in metabolic substrate preference, an increasedsensitivity of failure hearts to myocardial ischemia, and/orlimitations of the MVo2-PVA framework in assessingmyocardial energetics.Numerous studies have suggested that metabolic sub-
strate can alter myocardial efficiency and that free fattyacids increase MVo2 as compared with glucose consump-tion.38,39 Because free fatty acids are the preferred sub-strate of normal myocardium,40 an increase in glucose
0.048t0.014
0.986
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from

526 Circulation Research Vol 70, No 3 March 1992
metabolism could favorably affect the efficiency of ATPsynthesis in failing hearts. Recent studies have demon-strated increased glucose and decreased free fatty aciduptake in pressure-overloaded rat hearts.4142 However,Burkhoff et al43 found that in crystalloid perfused rathearts, hexanoate (a short-chain fatty acid) alters theMVo2-PVA relation by increasing the y intercept but notthe slope when compared with a glucose-containing per-fusate. Also inconsistent with this hypothesis is our findingof an unchanged unloaded MVo2 in the failure hearts,which would be expected to decrease if the efficiency ofATP synthesis were improved.Marked reductions in coronary perfusion pressure
have been shown to decrease the slope of the MVo2-PVA relation in the normal isolated canine heart.19 Thisfinding was not attributed to increased efficiency of thecontractile apparatus but rather to load sensitivity ofboth basal metabolism and the oxygen costs of excita-tion-contraction coupling under conditions of myocar-dial ischemia. A similar mechanism could apply to thefailing ventricle. However, total coronary blood flow atthe highest end-diastolic volume in each heart wassimilar in the two groups (92.6±34.9 control versus90.7±21.9 ml/min/100 g heart failure, p=NS) and wasmuch less than measured coronary blood flow duringintracoronary dobutamine infusion (168.6±34.1 controlversus 158.8 ±39.6 ml/min/100 g heart failure at maximaldobutamine doses). Also, the slope of the MVo2-PVArelation was not altered by high doses of dobutamine, asituation that would likely exacerbate ischemia if al-ready present under baseline conditions.
In the MVo2-PVA framework, MVo2 is partitionedinto unloaded MVo2 (the sum of the oxygen consump-tions associated with basal metabolism and excitation-contraction coupling) and the MVo2 associated withmechanical work. This construction depends on theassumption that oxygen consumption associated withbasal metabolism and excitation-contraction coupling isindependent of LV volume. As mentioned above, thismay not be true in the case of reduced coronaryperfusion pressure. In addition, a length dependence ofboth the systolic calcium transient and basal metabolismhave been demonstrated in isolated cardiac muscle.44,45Despite these potential problems, the assumption seemsreasonable in normal blood-perfused canine hearts, inwhich the MVo2 of ejecting contractions from a highpreload against a negligible afterload (resulting in avery small PVA) is nearly the same as the MVo2 ofunloaded contractions.46
It is also possible that PVA does not provide anadequate measure of total mechanical energy producedby crossbridge cycling because it does not incorporatethe energetic consequences of an internal resistance toejection or the positive and negative effects of shorten-ing on contractility.47 These effects, however, likelyaccount for only a small fraction of work-related oxygenconsumption,15 and the magnitude of the efficiencychange seen in this study is probably too large to beaccounted for by these factors.
Alterations in the slope of the MVo2-PVA relationmay reflect changes in myofibrillar efficiency.15 Onlyhyperthyroidism in rabbits has been shown to chroni-cally alter the slope of the MVo2-PVA relation,48 whilemany acute interventions alter the MVo2 interceptwithout altering the slope of the relation. Thyroid
hormone causes an increase in the myosin isoformV1/V3 ratio in rabbit ventricles, and the increase in theslope of the MVo2-PVA relation was attributed to thechange in the myosin ATPase accompanying this shift.Although the V3 myosin isoform predominates in thecanine heart and is not altered with chronic rapidpacing, myofibrillar Ca2-ATPase activity is decreasedin vitro by 26%.49 It is possible that chronic posttrans-lational modification of the myosin ATPase or alter-ation of another protein constituent of the myofilamentcould alter the efficiency of mechanical energy produc-tion and that this change could be adaptive to condi-tions of increased stress and limited energy supply.
It is unclear whether our finding of increased chemo-mechanical efficiency of the contractile apparatus can beextended to other models of congestive heart failure or tohuman cardiomyopathies. Both reduced50,51 and nor-mal52,53 myosin ATPase activity are reported in humancardiomyopathic hearts, the discrepancy in part relating todifferent techniques used to assay enzyme activity. Alpertand Mulieri54 reported increases in myothermal economy(the relation between the tension-dependent heat liber-ated with isometric contractions and the force-time inte-gral of those contractions) in isolated papillary musclesfrom rabbits with pressure-overload hypertrophy. A simi-lar increase in myothermal economy has recently beenreported in myocardium obtained from patients with heartfailure.55 Myothermal economy is not equivalent to theefficiency of the contractile apparatus reported in thisstudy because it has units of velocity, in contrast to thedimensionless efficiency of the contractile apparatus ob-tained from MVo2-PVA relations. However, these find-ings raise the possibility that alterations of the myofibrilexist in the failing human heart, affecting its chemome-chanical efficiency.The mean efficiency of chemomechanical energy
transduction of the control hearts in this study is lessthan the 35-45% that has been frequently reported forthe normal canine heart.15 However, previous studiesfrom our56 and other57 laboratories with an isolated,blood-perfused preparation have reported efficienciesof less than 30% for normal canine hearts. A similarlylow efficiency has also been reported for in situ caninehearts.58 Although there is no obvious explanation forthe differences in efficiencies of normal hearts betweenthese studies, it should be noted that the presentexperiments were performed using the identical instru-mentation under the same conditions for the controland failure groups. Furthermore, the order in whichcontrol or failure animals were studied was random.Nevertheless, the marked variability in efficiency mustbe kept in mind for comparison of data among differentstudies.The second major observation made regarding the
MVo2-PVA relation was that unloaded MVO2 (represent-ing the oxygen costs of basal metabolism and excitation-contraction coupling) was similar in the heart failure andcontrol groups. Given the positive relation between con-tractile state and the unloaded MVo2 previously demon-strated in the normal isolated canine heart,'8"19 MVo2 isthus increased relative to ventricular or myocardial con-tractility in heart failure. Consistent with this interpreta-tion is the finding that increasing contractility with dobu-tamine to levels comparable to controls led to significantlyhigher unloaded MVo2 in the failing hearts. Increased
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from

Wolff et al Mechanics and Energetics of Heart Failure 527
unloaded MVo2 at a given contractile state could resultfrom greater oxygen costs of basal metabolism or excita-tion-contraction coupling in heart failure. The lattercould result from altered stoichiometry of the sarcoplas-mic reticulum Ca2+-ATPase and/or a decreased sensitivityof the myofilaments to calcium, although these issuesremain speculative.
It should be noted that the overall efficiency of theventricle, the ratio of external (stroke) work to MVo2,depends on contractile state and loading conditions aswell as the MVo2-PVA relation. Because the contrac-tility of the isolated heart preparation is certainly lessthan in situ and because the loading conditions used inthis study were somewhat arbitrary, our data cannot beused to predict the effects of rapid-pacing-inducedheart failure on the overall cardiac efficiency (ratio ofstroke work to MVo2) in conscious animals.
LimitationsA potential limitation of this study is that the loading
conditions at which the two groups were compared donot reflect in situ loading conditions. In particular, therange of end-diastolic pressures used did not overlapthe measured in situ pulmonary wedge pressures in theanesthetized failure dogs. High end-diastolic pressureswere avoided because some normal canine hearts dem-onstrate altered systolic behavior and more rapid dete-rioration of function when operating at end-diastolicpressures of 25 mm Hg or greater. DPVRs and stress-strain relations were determined up to pressures of17.7+ 6.4 mm Hg in control and 18.6 ±6.7 mm Hg infailure hearts, a range that should be sufficient to allowmeaningful comparisons of the two groups. Also, theheart rate at which the study was performed (171 beatsper minute) may have magnified differences betweenthe two groups, because the force-frequency relationclearly differs at higher heart rates. This rate was chosento allow data collection at a constant heart rate, evenduring dobutamine stimulation, and the effects of dif-ferences in the force-frequency relation were relativelysmall. Finally, afterload impedance for ejecting beatswas identical for the two groups although aortic inputimpedance likely differs in situ. Although we did notattempt to duplicate in situ vascular physiology, anidentical afterload allowed more direct comparisons ofventricular properties.
SummaryThe present study demonstrates several important fea-
tures of chronic pacing tachycardia-induced heart failure.Reduction of systolic chamber function was confirmed byseveral load-independent indexes and was found to be aconsequence of depressed myocardial contractility as wellas increased wall stress caused by chamber dilatationwithout hypertrophy. Unstressed diastolic volume in-creased without alteration of passive myocardial stiffness.The observation that the zero-pressure intercept of theESPVR also remained unchanged cautions against the useof extrapolated ESPVRs in this as well as other forms ofventricular dilatation. Contractile reserve to inotropicstimulation is impaired in this form of heart failure as it isin others. Finally, this is the first study to examine theMVo2-PVA relation in a dilated cardiomyopathy model.The alterations demonstrated in this relation imply both
increased efficiency of chemomechanical energy transduc-tion and increased oxygen costs of basal metabolismand/or excitation-contraction coupling.
AppendixInstantaneous natural strain (e) for the midwall fiber layer
was determined from instantaneous LV volume (LVV) andLV mass
E=0.33 xln(LVV+fxVw)/(Vref+cxVw) (A1)where Vw is the ventricular wall volume (1.04 mlIg LV mass) andVref is the KCl-arrested diastolic volume (reference volume). Thefactor f is the fraction of the ventricular wall volume enclosed bythe midwall circumferential fibers at the reference (i.e., diastolic-arrested) state
f=ax (rmid3-ri3)/Vw (A2)
where rmid=(ri+ro)/2, ri=(Vref/a)l , r0=[(Vref+ Vj)/aI1', anda=41r/3.A balanced-force equation for a thick-walled spherical
geometry was used to determine instantaneous circumferen-tial stress (u-) from the LV pressure (LVP) and LVV signals
cr (g/cm2)=1.36xLVPx/3xXLVV2/3/A, (A3)
where A. is the cross-sectional area of the ventricular wall inthe equatorial plane and ,3=wrx[3/(4wr)]2'3. Rather than useinstantaneous LV cross-sectional wall area, the area deter-mined from the diastolic-arrested reference state (Vref) wasused for all stress calculations
AO=pX[(Vref+Vw)213-Vref2/3] (A4)
The rationale for this approach is that the force is distributedthroughout the same number of muscle fibers at different vol-umes regardless of the instantaneous cross-sectional wall area.59
AcknowledgmentsWe gratefully acknowledge the excellent technical assis-
tance of Kenneth Rent and Richard Tunin.
References1. Coleman HN, Taylor RR, Pool PE, Whipple GH, Covell JW, Ross
J Jr, Braunwald E: Congestive heart failure following chronictachycardia. Am Heart J 1971;81:790-798
2. Chow E, Woodard JC, Farrar DJ: Rapid ventricular pacing in pigs:An experimental model of congestive heart failure. Am J Physiol1990;258:H1603-H1605
3. Armstrong PW, Stopps TP, Ford SE, De Bold AJ: Rapid ventric-ular pacing in the dog: Pathophysiologic studies of heart failure.Circulation 1986;74:1075-1084
4. Wilson JR, Douglas P, Hickey WF, Lanoce V, Ferraro N, Muham-mad A, Reichek N: Experimental congestive heart failure pro-duced by rapid ventricular pacing in the dog: Cardiac effects.Circulation 1987;75:857-867
5. Howard RJ, Stopps TP, Moe GW, Gotlieb A, Armstrong PW:Recovery from heart failure: Structural and functional analysis ina canine model. Can J Physiol Pharmacol 1988;66:1505-1512
6. Riegger AJG, Liebau G: The renin-angiotension-aldosterone sys-tem, antidiuretic hormone and sympathetic nerve activity in anexperimental model of congestive heart failure in the dog. Clin Sci1982;62:465-469
7. Moe GW, Stopps TP, Angus C, Forster C, DeBold AJ, ArmstrongPW: Alterations in serum sodium in relation to atrial natriureticfactor and other neuroendocrine variables in experimental pacing-induced heart failure. JAm Coll Cardiol 1989;13:173-179
8. Schachnow N, Spellman S, Rugin I: Persistent supraventriculartachycardia: Case report with review of the literature. Circulation1954;10:232-236
9. Suga H, Sagawa H: Instantaneous pressure-volume relationshipand their ratio in the excised, supported canine left ventricle. CircRes 1974;35:117-126
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from

528 Circulation Research Vol 70, No 3 March 1992
10. Sunagawa K, Lim KO, Burkoff D, Sagawa K: Microprocessorcontrol of a ventricular volume servo-pump. Ann Biomed Eng1983;10:154-159
11. Burkhoff D, Alexander J, Schipke J: Assessment of Windkessel as
a model of aortic input impedance. Am J Physiol 1988;255:H742-H753
12. Glower DD, Spratt JA, Snow NK, Kabas JS, Davis JW, Olsen CO,Tyson GS, Sabiston DC Jr, Rankin JS: Linearity of the Frank-Starling relationship in the intact heart: The concept of preloadrecruitable stroke work. Circulation 1985;71:944-1009
13. Kass DA, Maughan WL, Guo ZM, Kono A, Sunagawa K, SagawaK: Comparative influence of load versus inotropic states on indexesof ventricular contractility: Experimental and theoretical analysisbased on pressure-volume relations. Circulation 1987;76:1422-1436
14. Suga H: Total mechanical energy of a ventricular model andcardiac oxygen consumption. Am J Physiol 1979;236:H498-H505
15. Suga H: Ventricular energetics. Physiol Rev 1990;70:247-27716. Slinker BK, Glantz SA: Multiple linear regression is a useful
alternative to traditional analyses of variance. Am J Physiol 1988;255:R353-R367
17. Maughan WL, Sunagawa K, Burkhoff D, Graves WL, Hunter WC,Sagawa K: Effect of heart rate on the canine end-systolic pressure-
volume relation. Circulation 1985;72:654-65918. Burkhoff D, Yue DT, Oikawa RY, Franz MR, Schaefer J, Sagawa
K: Influence of ventricular contractility on non-work-related myo-
cardial oxygen consumption. Heart Vessels 1987;3:66-7219. Suga H, Goto Y, Yasumura Y, Nozawa T, Futaki S, Tanaka N,
Uenishi M: 02 consumption of dog heart under decreased coro-
nary perfusion and propranolol. Am J Physiol 1988;254:H292-H303
20. Burkoff D, Sugiura S, Yue DT, Sagawa K: Contractility-dependentcurvilinearity of end-systolic pressure-volume relations. Am JPhysiol 1987;252:H1218-H1227
21. Kass DA, Beyer R, Lankford E, Heard M, Maughan WL, SagawaK: Influence of contractile state on curvilinearity of in situend-systolic pressure-volume relations. Circulation 1989;79:167-178
22. McKay RG, Aroesty JM, Heller GV, Royal HD, Warren SE,Grossman W: Assessment of the end-systolic pressure-volumerelationship in human beings with the use of a time-varyingelastance model. Circulation 1986;74:97-104
23. Grossman W, Braunwald E, Mann T, McLaurin LP, Green LH:Contractile state of the left ventricle in man as evaluated from theend-systolic pressure-volume relations. Circulation 1977;56:845-852
24. Burkoff D, Flaherty JT, Herskowitz A, Oikawa RY, Sugiura S,Franz MR, Baumgarter WA, Schaefer J, Reitz BA, Sagawa K: Invitro studies of isolated supported human hearts. Heart Vessels1988;4:185-196
25. Alyono D, Ring WS, Crumbley AJ, Schneider JR, O'Connor MJ,Parrish D, Bache RJ, Anderson RW: Global left ventricularcontractility in three models of hypertrophy evaluated with Em,,.JSurg Res 1984;37:48-54
26. Yin CPF: Ventricular wall stress. Circulation 1981;49:829-84127. Hansen DE, Cahill PD, DeCampli WM, Harrison DC, Derby GC,
Mitchell RS, Miller DC: Valvular-ventricular interaction: Impor-tance of the mitral apparatus in canine left ventricular systolicperformance. Circulation 1986;73:1310-1320
28. Weber KT, Pick R, Silver MA, Moe GW, Janicki JS, Zucker IH,Armstrong PW: Fibrillar collagen and remodeling of dilated canineleft ventricle. Circulation 1990;82:1387-1401
29. Feldman MD, Alderman JD, Aroesty JM, Royal HD, Ferguson JJ,Owen RM, Grossman W, McKay RG: Depression of systolic anddiastolic myocardial reserve during atrial pacing tachycardia inpatients with dilated cardiomyopathy. J Clin Invest 1988;82:1661-1668
30. Phillips PJ, Gwathmey JK, Feldman MD, Schoen FJ, Grossman W,Morgan JP: Post-extrasystolic potentiation and the force-frequencyrelationship: Differential augmentation of myocardial contractilityin working myocardium from patients with end-stage heart failure.JMol Cell Cardiol 1990;22:99-110
31. Morgan JP, Blinks JR: Intracellular calcium transients in the cat
papillary muscle. Can J Physiol Pharmacol 1982;60:524-52832. Shannon RP, Vatner DE, Komamura K, Homcy CJ, Stambler BS,
Vatner SF: Impaired 3-adrenergic responsiveness in consciousdogs with pacing cardiomyopathy (abstract). Circulation 1990;82(suppl III):III-160
33. Fray MJ, Manning D, Wilson JR, Molinoff PB: Alterations in thecatalyst of adenylate cyclase and the inhibitory guanine nucleotideregulatory protein in experimental heart failure (abstract). J AmCoil Cardiol 1989;13(suppl A):179A
34. lanussa CD, Montgomery C, Moe G, Armstrong P: Energy statusof canine myocardium with congestive heart failure induced byrapid ventricular pacing (abstract). FASEB J 1990;4:A281
35. Bache RJ, Arentzen CE, Simon AB, Vrobel TR: Abnormalities inmyocardial perfusion during tachycardia pacing in dogs with leftventricular hypertrophy: Metabolic evidence for myocardial isch-emia. Circulation 1984;69:409-417
36. Fujii AM, Gelpi RJ, Mirsky I, Vatner SF: Systolic and diastolicdysfunction during atrial pacing in conscious dogs with left ven-tricular hypertrophy. Circ Res 1988;62:462-470
37. Hittinger L, Shannon RP, Kohin S, Manders T, Kelley P, VatnerSF: Exercise-induced subendocardial dysfunction in dogs with leftventricular hypertrophy. Circ Res 1990;66:329-343
38. Mjos OD: Effect of free fatty acids on myocardial function andoxygen consumption in intact dogs. J Clin Invest 1971;50:1386-1389
39. Willebrands AF, van der Veen KJ: Influence of substrate onoxygen consumption of isolated perfused rat heart. Am J Physiol1967;212:1529-1535
40. Drake AJ: Substrate utilization in the myocardium. Basic ResCardiol 1982;77:1-11
41. Kagaya Y, Kanno Y, Takeyama D, Ishide N, Maruyama Y,Takahashi T, Ido T, Takishima T: Effects of long-term pressureoverload on regional myocardial glucose and free fatty acid uptakein rats. Circulation 1990;81:1353-1361
42. Yonekura Y, Brill AB, Som P, Yamamoto K, Srivastava SC, Iwai J,Elmaleh DR, Livni E, Strauss HW, Goodman MM, Knapp FF Jr:Regional myocardial substrate uptake in hypertensive rats: Aquantitative autoradiographic measurement. Science 1985;227:1494-1496
43. Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, WannenburgT, Gerstenblith G: Influence of metabolic substrate on myocardialcontractile state and metabolism at different coronary flows. Am JPhysiol 1991;261:H741-H750
44. Allen DG, Kurihara S: The effects of muscle length on intracellularcalcium transients in mammalian cardiac muscle. J Physiol (Lond)1982;327:79-94
45. Whalen WJ: Some factors influencing 02 consumption of isolatedheart muscle. Am J Physiol 1960;198:1153-1156
46. Yasumura Y, Nozawa T, Futaki S, Tanaka N, Suga H: Minorpreload dependence of 02 consumption of unloaded contraction indog heart. Am J Physiol 1989;256:H1289-H1294
47. Cooper G: Load and length regulation of cardiac energetics. AnnuRev Physiol 1990;52:505-522
48. Goto Y, Slinker BK, LeWinter MM: Decreased contractileefficiency and increased nonmechanical energy cost in hyperthy-roid rabbit heart: Relation between 02 consumption and systolicpressure-volume area or force-time integral. Circ Res 1990;66:999-1011
49. O'Brien PJ, Ianuzzu CD, Moe GW, Stopps TP, Armstrong PW:Rapid ventricular pacing of dogs to heart failure: Biochemical andphysiological studies. Can J Physiol Pharmacol 1989;68:34-39
50. LeClercq JF, Swynghedauw B: Myofibrillar ATPase, DNA, andhydroxyproline content of human hypertrophied heart. Eur J ClinInvest 1976;6:27-33
51. Alpert NR, Gordon MS: Myofibrillar adenosine triphosphataseactivity in congestive heart failure. Am J Physiol 1962;202:940-946
52. Mercadier JJ, Bouveret P, Gorza L, Schiaffino S, Clark WA, ZakR, Swynghedauw B, Schartz K: Myosin isoenzymes in normal andhypertrophied human ventricular myocardium. Circ Res 1983;53:52-62
53. Lauer B, Van Thiem N, Swynghedauw B: ATPase activity of thecross-linked complex between cardiac myosin subfragment 1 andactin in several models of chronic overloading: A new approachto the biochemistry of contractility. Circ Res 1989;64:1106-1115
54. Alpert NR, Mulieri LA: Increased myothermal economy of iso-metric force generation in compensated cardiac hypertrophyinduced by pulmonary artery constriction in the rabbit: A charac-terization of heat liberation in normal and hypertrophied rightventricular papillary muscles. Circ Res 1982;50:491-500
55. Hasenfuss G, Mulieri LA, Blanchard EM, Holubarsch C, LeavittBJ, Ittleman F, Alpert NR: Energetics of isometric force develop-
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from

Wolff et al Mechanics and Energetics of Heart Failure 529
ment in control and volume-overload human myocardium: Com-parison with animal species. Circ Res 1991;68:836-846
56. Burkhoff D, de Tombe PP, Hunter WC, Kass DA: Contractilestrength and mechanical efficiency of left ventricle are enhanced byphysiologic afterload. Am J Physiol 1991;260:H569-H578
57. Izzi G, Zile MR, Gaasch WH: Myocardial oxygen consumptionand the left ventricular pressure-volume area in normal andhypertrophic canine hearts. Circulation 1991;84:1384-1392
58. Nozawa T, Yasumura Y, Futaki S, Tanaka N, Igarashi Y,Goto Y, Suga H: Relation between oxygen consumption andpressure-volume area of in situ dog heart. Am J Physiol 1987;253:H31-H40
59. Hunter WC, Baan J: The role of wall thickness in the relationbetween sarcomere dynamics and ventricular dynamics, in Baan J,Arntzenius AC, Yellin EL (eds): Cardiac Dynamics. The Hague,Martinus Nijhoff Publishing, 1980, pp 123-134
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from

A KassM R Wolff, P P de Tombe, Y Harasawa, D Burkhoff, S Bier, W C Hunter, G Gerstenblith and D
experimental heart failure.Alterations in left ventricular mechanics, energetics, and contractile reserve in
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 1992 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.70.3.5161992;70:516-529Circ Res.
http://circres.ahajournals.org/content/70/3/516World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 14, 2018http://circres.ahajournals.org/
Dow
nloaded from





























