Embed Size (px)
Citation preview

3069
G. Keglevich et al. Short ReviewSyn thesis
SYNTHESIS0 0 3 9 - 7 8 8 1 1 4 3 7 - 2 1 0 X© Georg Thieme Verlag Stuttgart · New York2017, 49, 3069–3083short reviewen
d m
ater
ial.
Advantages of the Microwave Tool in Organophosphorus SynthesesGyörgy Keglevich* Nóra Zsuzsa Kiss Alajos Grün Erika Bálint Tamara Kovács
Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, [email protected]
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hte
Received: 14.03.2017Accepted after revision: 11.04.2017Published online: 20.06.2017DOI: 10.1055/s-0036-1589031; Art ID: ss-2017-z0162-sr
Abstract The microwave (MW) technique has become an importanttool also in the organophosphorus field of organic chemistry. On theone hand, otherwise reluctant reactions, such as the esterification of P-acids, may be enhanced by the effect of MW, while on the other hand,catalysts may be omitted, or catalyst systems may be simplified on MWirradiation. This later group includes the Kabachnik–Fields reactions, al-kylation of active methylene-containing compounds, O-alkylations, de-oxygenations, as well as the Hirao reaction. It is also the purpose of thisreview to elucidate the scope and limitations of the MW tool, to inter-pret the MW effects, and to model the distribution of local overheatingsand their beneficial effect.1 Introduction2 The Esterification of Phosphinic Acids2.1 Synthetic Results2.2 Scope and Limitation of the Application of MWs2.3 Modelling the Distribution of the Local Overheatings and
Predicting Their Effect3 The Simplification of Catalytic Systems3.1 Replacement of the Catalyst by MW Irradiation3.1.1 The Kabachnik–Fields Reaction3.1.2 Solid–Liquid Phase Alkylation of Active Methylene-Containing
Compounds3.1.3 Solid–Liquid Phase Alkylation of Phosphinic Acids3.1.4 The Deoxygenation of Phosphine Oxides3.2 The Hirao Reaction without Added P-Ligands4 Conclusions
Key words organophosphorus chemistry, microwave irradiation,phosphinates, Kabachnik–Fields reaction, alkylation, deoxygenation,Hirao reaction
1 Introduction
The spread of the microwave (MW) technique has had agreat impact not only in organic chemistry in general,1–4
but also on organophosphorus chemistry. Guenin summa-rized the initial results.5 The Keglevich group elaborated the
synthesis of P-heterocycles and other phosphinic, phos-phonic, and phosphine oxide derivatives utilizing the MWtechnique.6–12 The reactions investigated are shown inFigure 1.
Figure 1 MW-assisted organophosphorus reactions investigated by the Keglevich group
The role of MWs in organic syntheses induced vehe-ment disputes that led to different theories.13 Initially, non-thermal effects were assumed for polar transition states inapolar aprotic solvents, or under solvent-free conditions.14
Other non-thermal explanations include supposition ofchange in the thermodynamic parameters,15 or that of in-crease in the pre-exponential factor.15,16 Nowadays, non-thermal theories are mostly rejected,17,18 and rather ther-mal effects are considered.19 According to a major approach,local overheatings occurring statistically in the bulk of themixture are responsible for the beneficial effect of MWs.4,20
The finally distributed ‘nano-size’ overheatings cannot bemeasured individually. Modest thermal differences mayhave a significant impact on the reaction time.21 A newertheory suggests that MW-absorbing solutes in MW-trans-parent solvents may be responsible for the special rate en-hancing effect.22–25
In our work, we have emphasized green chemical as-pects.7,8 Besides elaborating efficient syntheses (short reac-tion times and good yields), it was possible to enhance re-
MW
Arbuzov reaction Diels–Alder reaction
Hirao reaction
deoxygenation
Pudovik reaction
Kabachnik–Fields reaction
transesterification
esterification
inverse Witttig reaction
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3070
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
luctant or slow reactions. Another advantage was to substi-tute catalysts by MW irradiation, or to simplify catalyticsystems. A new challenge was for us to evaluate the scopeand limitations of the use of the MW technique in organicchemistry, and to model the effect of MWs.2 The Esterification of Phosphinic Acids
2.1 Synthetic Results
Although phosphinic acids do not undergo direct esteri-fication under conventional conditions,26 we observed thatthis reaction may be successful under MW conditions.27 On
MW irradiation at 160–180 °C, phenylphosphinic acids 1reacted readily with alcohols used in excess (Scheme 1).Phenyl-H-phosphinates were obtained in high (73–90%)yields. The sterically hindered diphenylphosphinic acid (1,Y = Ph) showed lower reactivity; after heating at 220 °C for6 hours, the corresponding phosphinate 2 (Y = Ph, R = octyl)was isolated in a yield of 42%.
Scheme 1 Direct esterification of phenylphosphinic acids 1
Then the esterification of cyclic phosphinic acids, suchas hydroxyphospholene 1-oxides 3 and hydroxyphos-pholane 1-oxides 5 was also elaborated (Scheme 2 andScheme 3).28 The use of less volatile alcohols with longercarbon atom chains as the reagents proved to be more ad-vantageous.29 In a few cases, comparative thermal experi-ments (Δ) were also performed under similar conditions,and the beneficial effect of MWs was proved.
Scheme 2 Direct esterification of hydroxyphospholene 1-oxides 3
Due to the somewhat decreased reactivity of thehydroxyphospholane oxides 5 compared to that of thehydroxy-3-phospholene oxides 3, there was a need for ahigher temperature, and these reactions were less efficient.The monomethyl esters were formed as a ca. 1:1 mixture oftwo diastereomers, while the dimethyl compoundscomprised three isomers, two optically inactive species anda racemate.
The obvious disadvantage of our MW-assisted esterifi-cation is the relatively high temperature (optimally 200–220 °C) and the long reaction times required. When alco-hols with low boiling point were used, the yields of thephosphinates were low. We then developed a more efficientesterification method utilizing ionic liquids (ILs) in a cata-
György Keglevich graduated from the Technical University of Buda-pest in 1981 as a chemical engineer. He obtained D.Sc. in 1994, and be-came a professor in 1996. He has been the Head of the Department of Organic Chemistry and Technology, Budapest University of Technology and Economics since 1999. He deals with organophosphorus, P-hetero-cyclic, and environmentally friendly chemistry. He is the author or co-author of ca. 450 papers. He is a member of the Editorial Board of Heteroatom Chem., Phosphorus, Sulfur, Silicon Relat. Elem., and Curr. Microwave Chem. He holds different editor responsibilities with Curr. Org. Chem., Curr. Org. Synth., Curr. Green Chem., Lett. Org. Chem., Lett. Drug Des. Discovery, and Curr. Catal.Nóra Zsuzsa Kiss graduated from Budapest University of Technology and Economics in 2011 as a chemical engineer. She obtained her Ph.D. in 2014 on the subject of microwave-assisted organophosphorus chemistry. Since 2016, she has worked as an assistant professor at the same university. Her interest lies in synthetic organophosphorus chemistry with a focus on MW-assisted and green chemistry.Alajos Grün graduated in 1992 as chemical engineer and obtained his Ph.D. in 2000 at the Budapest University of Technology and Economics. He holds an Associate Professor status. His main research interests embrace calixarenes and organophosphorus chemistry including the synthesis of dronic acid derivatives.Erika Bálint graduated from the Budapest University of Technology (BUTE) and Economics in 2009 as a chemical engineer. She received her Ph.D. in 2013. She became a Research Associate at the Research Group of the Hungarian Academy of Sciences at the Department of Organic Chemistry and Technology at the BUTE in 2013. Her main research topics include the synthesis of aminophosphonate derivatives under MW conditions. She is the co-author of approximately 50 papers and book chapters.Tamara Kovács graduated from the Budapest University of Technology and Economics in 2013 as a chemical engineer. She has been a member of the Organophosphorus Research Group since 2011. Since 2013, she has been a Ph.D. student at the same university, and her research topics include the deoxygenation of phosphine oxides.
PO
OHY+ ROH
MW
160–220 °C, 2–10 bar0.5–6 h
PO
ORY
PO
OEtH80%
PO
OBuH90%
PO
OnOctH84%
PO
OnOct
42%
(15 equiv)1 2
73–90% (Y = H)
+ R2OHMW (Δ)
180–235 °C, 1–8 bar, 2–4 h(15 equiv)P
Me
O OH
R1
P
Me
O OR2
R1
P
Me
O OBu
58% (<9%)
P
Me
O OiOct
76% (<25%)
P
Me
O OBu
60% (<11%)
P
Me
O OiOct
82% (<20%)
Me Me
3 458–95%
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3071
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
lytic amount. The esterifications were carried out asdescribed above, but in the presence of catalytic amounts ofimidazolium salts, out of which [bmim][PF6] was found tobe the best.30 Applying [bmim][PF6] as an additive in the es-terification of several cyclic phosphinic acids 7 with a seriesof simple alcohols at 160–210 °C under MW irradiation,higher yields (up to 95%) could be obtained (Scheme 4).
Scheme 4 Direct esterification of cyclic phosphinic acids 7 in the presence of 10% of an ionic liquid
Thus, we have developed an environmentally friendlymethod for the preparation of phosphinates that may evenbe quantitative.
The MW-assisted direct thioesterification of phosphinicacids using thiols was also investigated.31 However, thesynthesis of thiophosphinates was not efficient enough,and took place in lower conversions and yields of 36–40%(Scheme 5).
The direct amidation of 1-hydroxy-3-phospholene 1-oxide 10 was also attempted, and it was found that thisderivatization took place with a ca. 33% conversion on MWirradiation (Scheme 6). The 1-alkylamino-3-phospholeneoxides 11 were obtained in low yields (25–29%).32
Scheme 6 Direct amidation of hydroxyphospholene 1-oxide 10
A better choice for the preparation of the phosphinicamides is the traditional reaction of the correspondingphosphinic chlorides (e.g., 12) with amines. It was observedthat the reaction with primary amines resulted in a mix-ture of the expected 1-amino-3-phospholene 1-oxide 11and its N-phosphinoyl derivative 13. Depending on the mo-lar ratio of the reactants and on the order of mixing, theproduct composition could be fine-tuned, and either 11 or13 could be obtained almost exclusively in high yields(Scheme 7).33
Scheme 3 Direct esterification of hydroxyphospholane 1-oxides 5
+ R2OHMW
210–235 °C, 1–8 bar, 2–4 h(15 equiv)P
Me
O OH
R1
P
Me
O OR2
R1
P
Me
O OBu
54%
P
Me
O OiOct
86%
P
Me
O OBu
45%
P
Me
O OiOct
50%
Me Me
5 645–86%
+ ROH
MW
160–210 °C, 1–18 bar, 0.5–3 h
10% [bmim][PF6]
PO OH
P
Me
O OMe38%
P
Me
O OPent
72%
P
Me
O OPent
89%
P
Me
O OPent
84%
MeMe
P
Me
O OPent
P
Me
O OBu83%
P
Me
O OnPent94%
P
Me
O OnDodecyl94%
P
Me
O OnOct85%
42%
PO OR
7 838–95%
(15 equiv)
Scheme 5 Thioesterification of hydroxyphospholene 1-oxides 3
PO SBu
Me
38%
PO OH
MeR1
MW
200 °CR2SH
PO SR2
MeR1
PO SPent
Me
40%
PO SPent
Me
36%
Me
3 936–40%
+
(15 equiv)
+ RNH2
MW
220 °C, ~7 bar, 2 hP
Me
O OHP
Me
O NHR
P
Me
O NHnHexP
Me
O NHcHexP
Me
O NHBn26% 25% 29%
10 1125–29%
(15 equiv)
Scheme 7 Synthesis of 1-amino-2,5-dihydro-1H-phosphole 1-oxides 11 and their N-phosphinoyl derivatives 13
PO NHR2
Me R2NH2
Et3N
PhMePO Cl
R1 Me
PO
R1Me
PO
MeR1
NR2
R2NH2
Et3N
PhMe
PO NHnBu
Me
85%
PO NHnHex
Me
82%
PO NHcHex
Me
85%
PO
Me
PO
Me
NnBu95%
PO
Me
PO
Me
NBn
95%
PO
Me
PO
Me
NPh72%
PO
Me
PO
Me
NnBu94%
MeMe
11 12 1353–95%
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083
3072
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
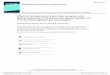
Phosphonic ester-acid derivatives 14 were also subjectedto MW-assisted direct esterification, and the dialkyl phenyl-phosphonates 15 were obtained in variable yields(Scheme 8).34 This is the first example of the MW-assisteddirect esterification of phosphonic acid derivatives.
Scheme 8 Direct esterification of phosphonic acid derivatives 14
2.2 Scope and Limitation of the Application of MWs
Each reaction may be characterized by its thermody-namic and kinetic parameters. According to the enthalpychange (ΔH0), a reaction may be exothermic, thermo-neutral, and endothermic. The other important factor is theenthalpy of activation (ΔH#) that determines the rate of thereaction. It is possible that a higher ΔH# prevents the reac-tion. In order to be able to predict the usefulness of MW ir-radiation for a particular reaction, let us consider a fewcases with typical energetics as shown in Figure 2. For thecases, where ΔH# is not too high, and there is a significantenthalpy gain (Figure 2, (I)/1), or the reaction is practicallywithout an enthalpy change (Figure 2, (II)/1), commonheating should be applied to perform the chemical reac-tions. For the variations with a higher ΔH# (Figure 2, (I)/2and Figure 2, (II)/2), it can be expected that the reactionsmay be promoted by MW irradiation. It can be said thatthermoneutral transformations with a higher ΔH# (Figure 2,(II)/2) may be the ideal subjects for MW activation. Thismay be exemplified by the esterification of phosphinicacids (vide infra). At the same time, endothermic transfor-mations shown in Figure 2, (III)/1 and (III)/2 will remain in-
complete even under MW irradiation. Such reactions arethe thioesterification and amidation of phosphinic acids(vide infra).
It was mentioned above that phosphinic acids cannot beesterified directly, and it was also shown that on MW irra-diation above 200 °C and using the alcohols in a 15-fold ex-cess, the esterifications of phosphinic acids took place.27–29
After a 3 hour irradiation at 200 °C, the reaction of 1-hydroxy-3-methyl-3-phospholene 1-oxide (10) with butanol pro-ceeded with a conversion of 58%. In a comparative thermalexperiment at 200 °C for 2 hours, the conversion was only17%. The energetics of this esterification were calculated bythe B3LYP/6-31G(d,p) method utilizing the explicit–implicitsolvent model. A relatively high ΔH# of 135.0 kJ mol–1 wasobtained (Figure 3).35
Figure 3 Enthalpy profile for the esterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide (10)
As a comparison, the esterification of acetic acid wasalso calculated. This reaction is practically also thermo-neutral, but the ΔH# is significantly lower (75.0 kJ mol–1).36
It was shown that the MW-assisted esterifications maybe further promoted in the presence of 10% of an IL.30
MW+ ROHP
OH
O ORP
OR
O OR220–235 °C, 2–15 bar, 6 h
POBu
O OBuP
OnPent
O OnPentP
OnOct
O OnOct
31% 51% 62%
14 1525–62%
(15 equiv)
Figure 2 Typical enthalpy diagrams for organic chemical transformations
reaction coordinate
ΔE2#
ΔE0
0
(I)
ΔH (kJ mol–1)
ΔE2#
0
ΔH (kJ mol–1)
ΔE0
ΔE2#
0
ΔH (kJ mol–1)
ΔE0
reaction coordinate(II)
reaction coordinate(III)
1 1 1
2 2 2
ΔE1# ΔE1
# ΔE1#
ΔH (kJ mol–1)
ΔH0 = 3.3
P
Me
O OH
+ BuOHP
Me
O OBu
+ H2O
reaction coordinate
ΔH# = 135.0
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3073
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
At the same time the MW-assisted thioesterification of1-hydroxy-3-methyl-3-phospholene oxide (10) withthiobutanol remained incomplete at 200 °C, the final con-version being ca. 30%. The outcome is in accord with thesignificant endothermicity (ΔH0 = 48.5 kJ mol–1) suggestedby the B3LYP/6-31++G(d,p) calculations. At the same time,the high ΔH# of 145.4 kJ mol–1 was comparable with that ofthe esterification with butanol (Figure 4).31
Figure 4 Enthalpy profile for the thioesterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide (10)
The amidation of 1-hydroxy-3-methyl-3-phospholeneoxide (10) with cyclohexylamine under MW irradiation at220 °C also remained incomplete at a conversion of 33%.Theoretical calculations suggested again endothermicity(ΔH0 = 32.6 kJ mol–1), but the ΔH# was rather low (79.4kJ mol–1) (Figure 5).32
Figure 5 Enthalpy profile for the amidation of 1-hydroxy-3-methyl-3-phospholene 1-oxide (10)
The examples shown in Figures 3–5 demonstrated wellthat MW irradiation may enhance thermoneutral reactionswith a relatively high ΔH#, but endothermic transforma-
tions cannot be performed in a complete conversion. Thetheory outlined above was also applied for other reactionsby de la Hoz et al.37–40
2.3 Modelling the Distribution of the Local Over-heatings and Predicting Their Effect
The potential of MW heating stems in the beneficial ef-fect of local overheatings occurring statistically in the bulkof the mixture. Even a small extent of the overheatings mayhave a significant impact on the rate, but higher over-heatings are also possible. The sum of the randomly occur-ring overheatings may overcome the barriers due to thehigher enthalpy of activation.
When modelling the thermal effect of local over-heatings, the key is the Arrhenius equation (Equation 1)that allows the calculation of the rate constants in the bulkof the mixture, and also in the overheated segments.
Equation 1
The different temperature (T) and volume (V) parame-ters are defined in Figure 6.
Figure 6 Definition of the volume (V) and temperature (T) parameters necessary for the modelling
The overall rate constant (koverall) is the weighted sum ofkbulk and the individual rate constants (ki-s) of the overheatedsegments as shown by Equation 2), where kbulk and kOH
i maybe obtained by Equation 1. Equation 3 shows the rate accel-eration.
Equation 2
ΔH# = 145.4
ΔH0 = 48.5
reaction coordinate
ΔH (kJ mol–1)
PO OH
Me
PO SnBu
Me
– H2O
MW200 °CnBuSH
reaction coordinate
ΔH (kJ mol–1)
ΔH# = 79.4
ΔH0 = 32.6
– H2OP
Me
O OH
+ NH2C6H13P
Me
O NHC6H13
Equation 3
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083
3074
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
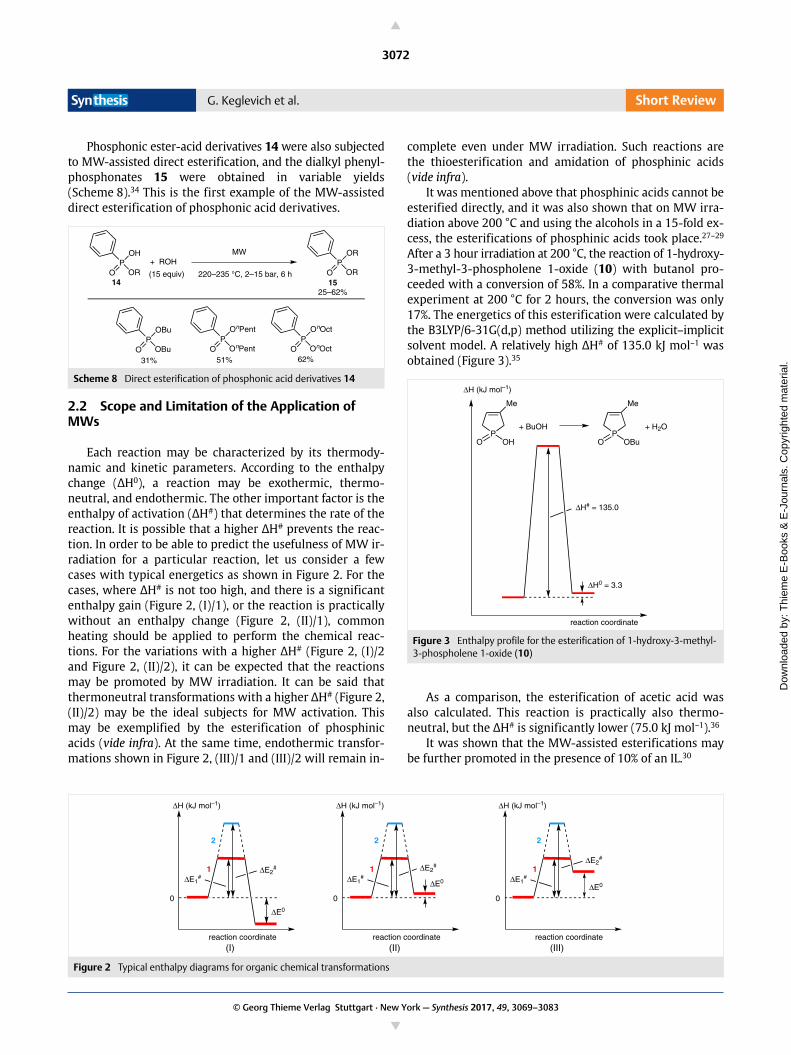
It is possible to model the thermal effect of MWs ac-cording to a simplified approach assuming a percentagesegment (VOH/VO) with a distinct overheating (TOH – Tbulk).41
In this case, Equation 2 takes a simpler form (Equation 4).
Equation 4
For the esterification of 1-hydroxy-3-methyl-3-phos-pholene oxide (10) with butanol at 200 °C, and applying acalculated ΔH# of 135 kJ mol–1 (Scheme 9),35 rate enhance-ments up to 3.2 were obtained as a consequence of theoverheating (up to 30 °C) and the proportion of this seg-ment (up to 30%).41
Scheme 9 The model esterification selected
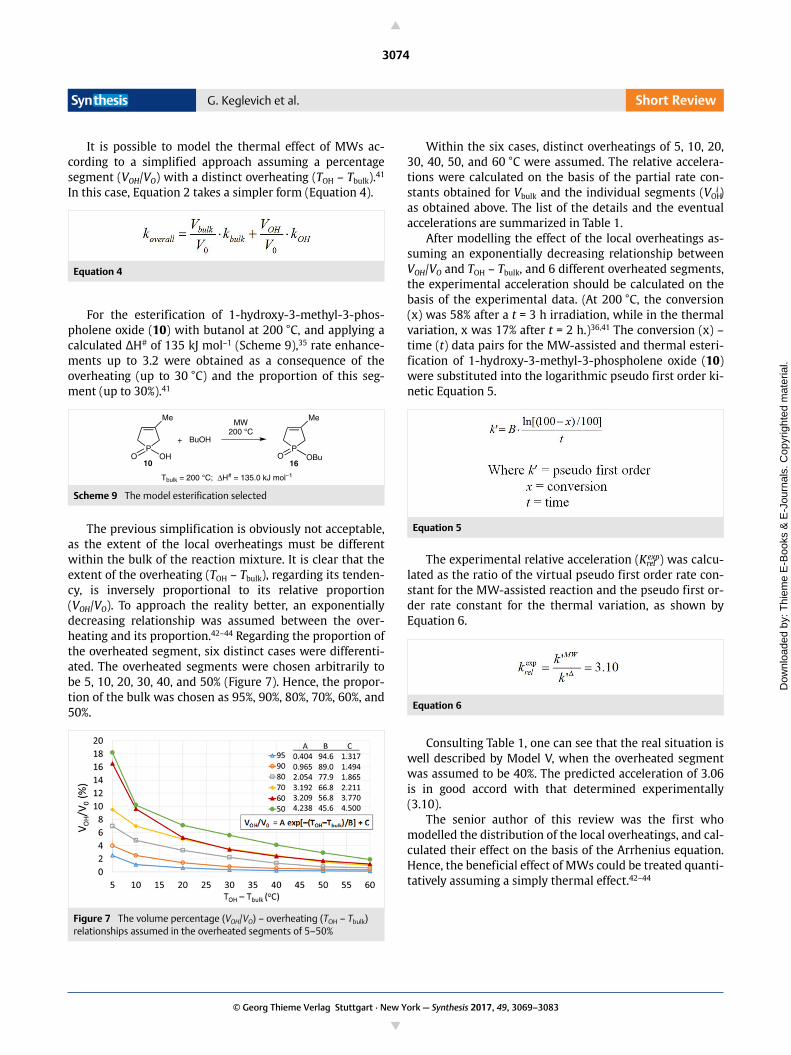
The previous simplification is obviously not acceptable,as the extent of the local overheatings must be differentwithin the bulk of the reaction mixture. It is clear that theextent of the overheating (TOH – Tbulk), regarding its tenden-cy, is inversely proportional to its relative proportion(VOH/VO). To approach the reality better, an exponentiallydecreasing relationship was assumed between the over-heating and its proportion.42–44 Regarding the proportion ofthe overheated segment, six distinct cases were differenti-ated. The overheated segments were chosen arbitrarily tobe 5, 10, 20, 30, 40, and 50% (Figure 7). Hence, the propor-tion of the bulk was chosen as 95%, 90%, 80%, 70%, 60%, and50%.
Figure 7 The volume percentage (VOH/VO) – overheating (TOH – Tbulk) relationships assumed in the overheated segments of 5–50%
Within the six cases, distinct overheatings of 5, 10, 20,30, 40, 50, and 60 °C were assumed. The relative accelera-tions were calculated on the basis of the partial rate con-stants obtained for Vbulk and the individual segments (VOH
i )as obtained above. The list of the details and the eventualaccelerations are summarized in Table 1.
After modelling the effect of the local overheatings as-suming an exponentially decreasing relationship betweenVOH/VO and TOH – Tbulk, and 6 different overheated segments,the experimental acceleration should be calculated on thebasis of the experimental data. (At 200 °C, the conversion(x) was 58% after a t = 3 h irradiation, while in the thermalvariation, x was 17% after t = 2 h.)36,41 The conversion (x) –time (t) data pairs for the MW-assisted and thermal esteri-fication of 1-hydroxy-3-methyl-3-phospholene oxide (10)were substituted into the logarithmic pseudo first order ki-netic Equation 5.
Equation 5
The experimental relative acceleration (Krelexp) was calcu-
lated as the ratio of the virtual pseudo first order rate con-stant for the MW-assisted reaction and the pseudo first or-der rate constant for the thermal variation, as shown byEquation 6.
Equation 6
Consulting Table 1, one can see that the real situation iswell described by Model V, when the overheated segmentwas assumed to be 40%. The predicted acceleration of 3.06is in good accord with that determined experimentally(3.10).
The senior author of this review was the first whomodelled the distribution of the local overheatings, and cal-culated their effect on the basis of the Arrhenius equation.Hence, the beneficial effect of MWs could be treated quanti-tatively assuming a simply thermal effect.42–44
PO OH
Me
PO
BuOH+
OBu
MeMW
200 °C
Tbulk = 200 °C; ΔH# = 135.0 kJ mol−1
10 16
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083
3075
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
3 The Simplification of Catalytic Systems
3.1 Replacement of the Catalyst by MW Irradiation
3.1.1 The Kabachnik–Fields Reaction
The Kabachnik–Fields (phospha-Mannich) reaction isthe three-component condensation of primary or secondaryamines, oxo compounds (aldehydes or ketones), and >P(O)Hreagents, such as dialkyl phosphites or secondary phos-phine oxides, resulting in the formation of α-aminophos-phonates or α-aminophosphine oxides.45–47 α-Aminophos-phonates are considered as potential bioisosteres of α-ami-no acids. Due to their versatile bioactivity, they areimportant targets in biochemistry,48,49 medicinal chemis-try,50–53 and pesticide chemistry.54–56 The three-componentcondensation may be accomplished in many ways. A num-ber of papers have been published, suggesting the use ofspecial catalysts,57–66 such as a phthalocyanine–AlCl3 com-plex,57 metal triflates,58 lanthanide triflates,59,60 gallium(III)iodide,61 bismuth(I) nitrate,62 samarium(II) iodide,63 indi-um(III) chloride,64 and magnesium perchlorate,65,66 etc. insolvents, or without the use of any solvent. The Keglevichgroup have found that under MW irradiation, there was noneed for any catalyst and solvent.67 Green chemicalaccomplishments are of special interest.68,69
A series of α-aminophosphonates 17 were synthesizedby solvent- and catalyst-free MW-assisted Kabachnik–
Fields reactions (Scheme 10).67,70–72 The condensation ofaniline or benzylamine, simple oxo compounds, such asformaldehyde, benzaldehyde, acetophenone, or cyclohexa-none, and dimethyl or diethyl phosphite gives the corre-sponding α-aminophosphonates in yields of 80–93%.67 Thismethod was extended to the synthesis of N-heterocycliccontaining α-aminophosphonates applying heterocyclicamines.70 3-Amino-6-methyl-2H-pyran-2-ones were alsosuitable starting materials in the Kabachnik–Fields reac-tion, and a series of N-(2-oxo-2H-pyranyl)-α-aminophos-phonates were obtained in yields of 61–91% (Scheme 10).71
Ethyl octyl phosphite, a dialkyl phosphite with mixed alkylgroups was prepared, and utilized as P-reagent in the three-component condensation with primary or secondaryamines and paraformaldehyde (Scheme 10).72 Several ethyloctyl alkylaminomethylphosphonates were prepared inhigh yields (72–92%) under MW irradiation in the absenceof catalyst and solvent.
The Kabachnik–Fields condensation was extended usingsecondary phosphine oxides, such as diphenylphosphineoxide, dibenzylphosphine oxide, and bis(4-methylphe-nyl)phosphine oxide, as the P-reactants in reaction with al-dehydes or ketones and different types of amines resultingα-aminophosphine oxides 18 (Scheme 11).67,70,71,73 Only afew examples were known earlier for the synthesis of α-aminophosphine oxides. The condensations leading tothese products were carried out applying a large excess of
Table 1 Dependence of the Relative Rate Enhancement (krel) on the Overheated Segment (VOH/VO) Assuming an Exponential Distribution of the TOH – Tbulk
Model (Vbulk/V0)
Reference A (bulk)
B C D E F G H Overall effect (krel)
I (95%) TOH – Tbulk (°C) 0 0 5 10 20 30 40 50 60
VOH/VO (%) 0 0 2.5 1.1 0.6 0.32 0.2 0.155 0.125
krel 1.00 0.95 0.04 0.02 0.03 0.03 0.03 0.05 0.07 Σ = 1.20
II (90%) TOH – Tbulk (°C) 0 0 5 10 20 30 40 50 60
VOH/VO (%) 0 0 4 2.5 1.4 0.8 0.55 0.4 0.35
krel 1.00 0.90 0.06 0.05 0.06 0.07 0.09 0.12 0.19 Σ = 1.53
III (80%) TOH – Tbulk (°C) 0 0 5 10 20 30 40 50 60
VOH/VO (%) 0 0 7 4.8 3.3 2.2 1.3 0.8 0.6
krel 1.00 0.80 0.10 0.10 0.14 0.18 0.20 0.24 0.32 Σ = 2.08
IV (70%) TOH – Tbulk (°C) 0 0 5 10 20 30 40 50 60
VOH/VO (%) 0 0 9.5 7 5 3.5 2.5 1.5 1
krel 1.00 0.70 0.14 0.15 0.21 0.29 0.39 0.44 0.54 Σ = 2.85
V (60%) TOH – Tbulk (°C) 0 0 5 10 20 30 40 50 60
VOH/VO (%) 0 0 16.5 9.6 5.2 3.4 2.4 1.70 1.2
krel 1.00 0.60 0.24 0.20 0.22 0.28 0.38 0.50 0.64 Σ = 3.06
VI (50%) TOH – Tbulk (°C) 0 0 5 10 20 30 40 50 60
VOH/VO (%) 0 0 18.2 10.2 7.1 5.6 4.1 2.9 1.9
krel 1.00 0.50 0.26 0.21 0.30 0.46 0.65 0.85 1.02 Σ = 4.26
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3076
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
the amine components, high temperatures and a long reac-tion time, or using benzene as the solvent.74–76 The Keglevichgroup accomplished the three-component condensationsunder catalyst-free, and in most cases solvent-free MWconditions at 80–100 °C for 30–60 minutes, and the corre-sponding α-aminophosphine oxides 18 were obtained inhigh yields (80–98%).67,70,71,73
Scheme 11 Synthesis of α-aminophosphine oxides 18
As a further development, double Kabachnik–Fields re-actions were also carried out using two equivalents of thealdehyde, and two equivalents of the P-reagent in reactionwith the amine component to make available bis(phospho-nylmethyl)amines 19 (Scheme 12).72,77–81 The condensationof primary amines with two equivalents of paraformalde-hyde and dialkyl phosphites were accomplished under MWirradiation in the absence of catalysts and solvents.77–79 Thebest result (98% yield) was obtained starting from benzyl-amine. The reaction was also carried out using ethyl octylphosphite as the P-reagent.72 α-, β-, and γ-Amino acid de-
rivatives were novel starting materials in the doubleKabachnik–Fields reactions.80,81 A few bis(dialkoxyphos-phonylmethyl)amino acid derivatives were synthesized inyields of 60–97%.
Scheme 12 Synthesis of bisphosphonates 19
The beneficial effect of the MW technique was demon-strated by comparative thermal experiments.
The double Kabachnik–Fields condensations were alsoperformed with secondary phosphine oxides as the P-re-agents without any catalyst in acetonitrile to overcome theheterogeneity of the reaction mixture (Scheme 13).73,77–81
The corresponding bis(phosphinoylmethyl)amines 20 wereobtained in high yields (75–98%).
Most of the bis(phosphine oxides) were utilized as pre-cursors of bidentate P-ligands. Several bis(phosphinoyl-methyl)amines 20 prepared were subjected to doubledeoxygenation using phenylsilane to afford bisphosphines21, which were converted into cyclic platinum complexes22 on reaction with dichlorodibenzonitrileplatinum(II)(Scheme 14).73,77–79 These cis platinum complexes 22showed good activity and chemoselectivity, along with anunexpected regioselectivity as catalyst in the hydroformy-lation of styrene.73,79
In summary, new mono- and bis(aminophosphonates),as well as aminophosphine oxides were synthesized byMW-assisted three-component Kabachnik–Fields reactions.It was proved that there is no need for any catalyst and, in
Scheme 10 Synthesis of α-aminophosphonates 17
1761-93%
O
R3 R4P
O
CN
R3
R4
OR5
OR6P
OR6
O
H+
OR5
+solvent-free
80–120 °C, 15–150 min
MW R1
R2
93%
P
O
CHNHOEt
OEt
91%
CH2 P
OOEt
OEt
88% 87%
P
O
CH2NOEt
OOct
Bu
Me
O O
NH CH2 P
OMe
OBu
OBu
NH N
73%
P
O
CH2
OEt
OEt
NH
R1
R2
N
91%
P
O
CH2
Ph
Ph
1880–98%
R1 NH2
O
R2 R3P
O
CNH
R2
R3
Y
YP
Y
O
H+
Y+
solvent-free or MeCN*80–100 °C, 30–60 min
MWR1
Y = Ph, 94%Y = Bn, 94%*Y = 4-MeC6H4, 94%*
P
O
CH2NHY
Y
87%
P
O
CHNHPh
Ph
80%
CH2 P
OPh
Ph
98%
O O
NH CH2 P
OMe
Ph
Ph
NH
2 (HCHO)n
1965–98%
89%
P
O
CH2
N
OR2
OR3
POR3
O
H+
OR2
+solvent-free
100 °C, 1–1,5 h
MWR1R1 NH2 2
P
O
CH2
OR2
OR3
P
O
CH2
N
OMe
OMePr
P
O
CH2
OMe
OMe86%
P
O
CH2
N
OEt
OEt
P
O
CH2
OEt
OEt
CH2
89%
P
O
CH2
N
OEt
OOct
P
O
CH2
OEt
OOct
P
O
CH2
N
OBn
OBn
P
O
CH2
OBn
OBn
CH2
98%
P
O
CH2
N
OBu
OBu
P
O
CH2
OBu
OBu
EtOOC (CH2)n
n = 1, 94%n = 2, 91%n = 3, 97%
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3077
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
most cases, for any solvent in the condensations. The bis-phosphines obtained by the deoxygenation of bisphosphineoxides were utilized as bidentate P-ligands in platinumcomplexes.
3.1.2 Solid–Liquid Phase Alkylation of Active Methy-lene-Containing Compounds
Alkylation of compounds with active methylene groupunder MW conditions is a less studied field.82 Diethylmalonate,83 acetoacetic ester,84 and other derivatives85 wereeasily substituted by alkyl halides in the presence of alkalicarbonates or other bases under solvent-free and MW-assisted phase-transfer catalytic conditions.
The Keglevich group found that there was no need for aphase-transfer catalyst in the solid–liquid-phase reactionon MW irradiation.86,87
There are only a few examples for the MW-assisted al-kylation of the CH-acidic compounds with P-function.88 TheKeglevich group found that tetraethyl methylenebisphos-phonate (23, Y = P(O)(OEt)2) could be alkylated by alkali ha-lides in the presence of Cs2CO3 at 120–140 °C on MW irradi-ation (Scheme 15).89 The alkylation of bis(diphenylphosphi-noyl)methane (23, Y = P(O)Ph2) required more forcingconditions (mostly 180 °C and TEBAC), and the use of aceto-nitrile as the solvent due to the heterogeneity (Scheme15).90
Scheme 15 MW-assisted alkylation of P-functionalized CH acidic com-pounds
In the alkylation of diethyl ethoxycarbonylmethylene-phosphonate (25, Z = CO2Et)91 and diethyl cyanomethyl-phosphonate (25, Z = CN)90 Cs2CO3 and K2CO3, respectively,were the suitable bases (Scheme 16).
The dialkylation of diethyl ethoxycarbonylmethylene-phosphonate (27) was also elaborated in four steps. Intro-duction of the second alkyl group required three consecu-tive alkylations to obtain satisfactory yields (Scheme 17).92
Somewhat analogous compounds, hydroxy-methylenebisphosphonates 30 were prepared by the addi-
Scheme 13 Synthesis of bisphosphine oxides 20
2 (HCHO)n
2075–98%
78%*
P
O
CH2
N
Y
YP
Y
O
H
+Y
+ RR NH2 2
P
O
CH2
Y
Y
P
O
CH2
N
Ph
PhPr
P
O
CH2
Ph
Ph90%
P
O
CH2
N
Ph
Ph
P
O
CH2
Ph
Ph
CH2
95%
P
O
CH2
N
Ph
Ph
P
O
CH2
Ph
Ph
solvent-free* or MeCN80–100 °C, 30–90 min
MW
P
O
CH2
N
Y
YBu
P
O
CH2
Y
Y
Y = Bn, 96%Y = 4-MeC6H4, 98%
P
O
CH2
N
Ph
Ph
P
O
CH2
Ph
Ph
MeOOC (CH2)n
n = 1, 92%n = 2, 89%n = 3, 94%
Scheme 14 Synthesis of platinum complexes 22
2229–75%
29%A
P
O
CH2
N
Y
YR
P
O
CH2
Y
Y
solvent-free120 °C, 2.5 h
MW
PCH2
N
Y
YR
PCH2
Y
Y25 °C, 12 h
PCH2
N
Y Y
R
PCH2
YY
PtCl2
PCH2
N
Ph Ph
PCH2
PhPh
PtCl2
38%A
PCH2
N
Ph Ph
Pr
PCH2
PhPh
PtCl2
60%A
PCH2
N
Ph Ph
PCH2
PhPh
PtCl2CH2
PCH2
N
Y Y
Bu
PCH2
YY
PtCl2
Y = Bn, 75%B
Y = 4-MeC6H4, 74%B
PhH
(PhCN)2PtCl2
PhSiH3
80 °C, 3 dPhSiH3
PhH
A)
B)
21
Y2P CH2 PY2
O OY2P CH PY2
O O
R
MW
Cs2CO3
RX
120–180 °C, 1.5–4 h(TEBAC, MeCN)*
EtOP
EtOCH P
OEt
OEt
O O
Et80%
EtOP
EtOCH P
OEt
OEt
O O
Pr~45%
PhP
PhCH P
Ph
Ph
O O
Pr40%*
2440–80%
PhP
PhCH P
Ph
Ph
O O
Bu46%*
PhP
PhCH P
Ph
Ph
O O
Bn45%*
23
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3078
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
tion of dialkyl phosphites to α-oxophosphonates 29 underMW conditions (Scheme 18).93,94
3.1.3 Solid–Liquid Phase Alkylation of Phosphinic Acids
A few examples are known of the alkylation of phos-phinic acids using diazomethane as the alkylating agent;however, only methyl esters can be synthesized in thisway.95,96 The solid–liquid-phase alkylation of phosphinicacids is a less studied field,97 and the alkylation of cyclicphosphinic acids has not been dealt with before. A solvent-free MW-assisted method was developed for the solid–liquid-phase alkylation of cyclic phosphinic acids 7 with al-kyl halides (Scheme 19). The corresponding phosphinates 8were obtained in yields of 56–98%.28,98 The reactions werecarried out in the presence of K2CO3, and the effect of aphase-transfer catalyst (TEBAC) was also studied. It wasfound, that applying alkylating agents of increased reactivi-ty (ethyl iodide and benzyl bromide), the MW irradiationwas beneficial, and there was no need to add TEBAC. The
use of the catalyst is advantageous, when alkyl halides ofnormal reactivity (propyl and butyl bromide) were applied.In this case, the effect of onium salt was synergetic with theMW irradiation, and the reactions were complete. The al-kylation with isopropyl bromide was quite reluctant, theyield was 56%, and 65% in the presence of TEBAC.
3.1.4 The Deoxygenation of Phosphine Oxides
The deoxygenation of tertiary phosphine oxides is animportant reaction, as it provides useful intermediates andreagents.99,100 The resulting phosphines may be useful as P-ligands in transition metal complexes. Phosphines are alsoused as reagents in various organic reactions, such as theWittig and the Mitsunobu reactions. In these transforma-tions, the phosphine oxides applied in a catalytic quantityare regenerated by in situ silane reduction.101–104
Scheme 16 MW-assisted alkylation of CH acidic compounds with mixed functionalities
EtOP
EtOCH2 Z
O EtOP
EtOCH Z
O
R
MW
M2CO3
RX
100–120 °C, 2 h
EtOP
EtOCH C OEt
O O
Et
M = Cs, K
EtOP
EtOCH C OEt
O O
Bu
EtOP
EtOCH CN
O
Pr
EtOP
EtOCH CN
O
Bu
70% 70%
64% 59%
2664–70%
25
Scheme 17 Dialkylation of ethoxycarbonylmethylphosphonate under MW conditions
1) MW
Cs2CO3
120 °C, 2 h EtOP
EtOC C OEt
O O
R1
EtOP
EtOCH2 C OEt
O O
EtOP
EtOC C OEt
O O
Pr39%
36–64%
R2
R1X
Cs2CO3
120 °C, 2 h
2–4) MW
Et
EtOP
EtOC C OEt
O O
Bn
Et
40%
EtOP
EtOC C OEt
O O
Et
Et
64%
EtOP
EtOC C OEt
O O
Bu
Bu
48%
R2I
(X = I, Br)
27 28Scheme 18 Synthesis of 1-hydroxy-methylenebisphosphonates
C P(OR2)2
O
(R2O)2P C P(OR3)2
O O
R1
MW
60–120 °C, 20–180 min
EtOP
EtOC P
OEt
OEt
O O
Me81%
MeOP
MeOC P
OMe
OMe
O O
Me85%
3034–85%
O
R1
EtOP
EtOC P
OBn
OBn
O O
Me55%
EtOP
EtOC P
OMe
OMe
O O
Me70%
MeOP
MeOC P
OMe
OMe
O O
Bn51%
OH
OH OH
OH OH
OH
(R3O)2PH
(R2O)2PHC P(OR3)2
O O
R1
O
29
31
Et2NH
Scheme 19 Solid–liquid phase alkylation of cyclic phosphinic acids 7
+ 1.2 RX
MW
100–120 °C, 1–1.5 h
K2CO3, TEBAC
PO OH
PO OR
7 856–98%
solvent-free
P
Me
O ORP
Me
O OnBu
95%
P
Me
O OnBu
98%
Me
P
Me
O OnBuP
Me
O OR
81%
Me
R = Et, 90%R = iPr, 65%R = nBu, 96%
R = Et, 95%R = iPr, 56%R = nBu, 90%
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3079
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
The most common means for the deoxygenation ofphosphine oxides involves silanes as the reagents.99,100
Many silanes have been described as reducing agents. It is areal challenge to accomplish the deoxygenation of phos-phine oxides by cheap and user-friendly silanes, and toelaborate green chemical protocols. Within silanes, trichlo-rosilane is the most widespread reagent,105,106 but it is vola-tile (bp 32 °C) and corrosive, implying a significant disad-vantage. Phenylsilane (PhSiH3) may be a good choice as thereagent for the deoxygenation; however, it is rather expen-sive.107,108 Tetramethyldisiloxane (TMDS) and polymethyl-hydrosiloxane (PMHS) are cheap, but not too reactive re-ducing agents (Figure 8). To promote their reactivity, thedeoxygenations by TMDS and PMHS were performed withcopper, titanium, and indium catalysis,109–113 or with aphosphoric acid diester as the catalyst.114 It was reportedthat these catalysts were quite helpful at 100–110 °C intoluene.
Figure 8 The preferred silanes applied in our work
It was a challenge for us, within the Keglevich group, totry to substitute the catalysts suggested for the TMDS- andPMHS-promoted deoxygenations by MW irradiation.115–117
In our work, 1-phenyl- and 1-alkyl-3-methyl-3-phospho-lene oxides 32 were subjected to deoxygenation by TMDSand PMHS under solvent-free conditions. It was found thatthe MW-assisted accomplishments were significantly faster(2–5 h). The comparative thermal experiment requiredheating for 3–9 hours. Selected results obtained with PMHSare shown in Scheme 20.115–118
Scheme 20 MW-assisted deoxygenation of 1-substituted 3-methyl-3-phospholene 1-oxides 32 by PMHS
With TMDS, there was a need for a longer reaction timeof 3–5 hours under MW irradiation, and the yield of phos-phine 33 was 82–92%. Under MW conditions, the expensivePhSiH3 could be replaced well by PMHS (and TMDS), al-though somewhat higher temperatures, and longer reactiontimes were necessary.
Other 5-ring phosphine oxides, such as a 3,4-dimethyl-3-phospholene 1-oxide, 2-phospholene 1-oxides, and aphospholane 1-oxide (in general 34) were deoxygenated byPMHS under similar conditions, shown above for 1-substi-tuted 3-methyl-3-phospholene 1-oxides 32 in Scheme 20(Scheme 21).116,119 The phosphines (in general 35) wereobtained in 87–92% yields. On traditional heating, thecompletion required 4 hours, and the yields were similar(86–90%).
Scheme 21 MW-assisted reduction of a series of phospholene 1-ox-ides and a phospholane 1-oxide by PMHS
The PMHS- or TMDS-promoted deoxygenations wereextended to the reduction of acyclic tertiary phosphine ox-ides 36. The deoxygenation of sterically hindered modelsrequire a higher temperature (Scheme 22).115,120
Scheme 22 Deoxygenation of acyclic tertiary phosphine oxides 36 by PMHS or TMDS under MW conditions
In summary, PMHS and TMDS allow a user-friendly andcheap deoxygenation of a series of tertiary phosphine ox-ides under MW conditions.
3.2 The Hirao Reaction without Added P-Ligands
The Hirao reaction is the P–C coupling reaction of arylhalides and dialkyl phosphites in the presence of Pd(PPh3)4and triethylamine to afford arylphosphonates 38. Manyvariations including in situ formed Pd, Ni, and Cu catalystsincorporating mono- and bidentate P ligands have beendescribed over the last two decades (Scheme 23).121–124
Green chemical approaches involving phase-transfercatalytic and MW-assisted protocols were also described.125–133
Arylphosphonates may also be obtained by the MW-as-sisted Arbuzov reaction of aryl bromides and trialkyl phos-phites.134
MW
110 °C, 2 hP
Me
O RP
Me
R
P
Me
Ph
92%
P
Me
Et
93%
P
Me
Pr
93%
P
Me
Bu93%
32 33
no solvent+
(2 equiv)PMHS
MW
110 °C, 2 h
P
Me
Ph
87%
P
Me
Ph
88%
PPh
90%
P
Me
Ph
92%
34 35
no solvent+
(2 equiv)P
PhO
PPh
Me
PMHS
MW
150–175 °C, 4–7.5 h
90%93% 88%
36 37
no solvent+
PMHSor
TMDS(5 equiv)
PR R
Ph
OPR R
Ph
PPh Ph
Ph
PMe Me
Ph
P
Ph
MeMe
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3080
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
The first ‘P-ligand-free’ Hirao reaction was reported bythe Keglevich group.135,136 In homogenous media, thePd(OAc)2 was found to be a suitable catalyst using triethyl-amine under solvent-free MW conditions (Scheme 24). At150–200 °C dialkyl phosphites reacted easily with substi-tuted bromobenzenes, and the arylphosphonates 39 wereformed, in most cases, in good yields (60–93%). 3-Methyl-benzene and fluorobenzene were the most reactive arenesafter bromobenzene. These compounds took part in a quan-titative reaction with diethyl phosphite in the presence of 5mol% of Pd(OAc)2 at 175 °C after a reaction time of 5–10minutes. It was observed that both electron-donating andelectron-withdrawing substituents in the aryl ring at posi-tion 4 and 3 decreased the reactivity of aryl bromides.
Scheme 24 The ‘P-ligand-free’ Hirao reaction with dialkyl phosphites
The alkyl phenyl-H-phosphinates were also found to besuitable reagents in the MW-assisted ‘P-ligand-free’ Hiraoreaction (Scheme 25),135,136 and the corresponding products40 were formed in high yields (68–92%).
Scheme 25 The ‘P-ligand-free’ Hirao reaction with alkyl phenyl-H-phosphinates
A series of phosphine oxides 41 were synthesized by theP–C coupling reaction of secondary phosphine oxides witharyl bromides (Scheme 26).136 The coupling of diphenyl-phosphine oxide and aryl bromides afforded the triaryl-phosphine oxides 41 (Y = Ar) in yields of 83–89% using 5mol% Pd(OAc)2 at 150 °C after a reaction time of 5 minutes.Dialkylphosphine oxides were found to be less reactive, andthe transformation required a longer reaction time of 1.5hours at 175 °C. Furthermore, in the coupling of dialkyl-phosphine oxides, there was need for acetonitrile as thesolvent to avoid side reactions. It was found that diphenyl-phosphine oxide had a higher reactivity than other >P(O)Hcompounds including alkyl-phenyl-H-phosphinates and di-alkyl phosphites.
Scheme 26 The ‘P-ligand-free’ Hirao reaction with secondary phos-phine oxides
The reaction of 2-nitro-5-bromoanisole with diethylphosphite was carried out using Pd(OAc)2 as the catalystand K2CO3 as the base in xylene, and the corresponding arylphosphonate was obtained in a yield of 69%.137
Cu salts may also be suitable catalysts in ligand-free P–Ccouplings. A variety of Cu salts, bases and solvents weretested in the reaction of aryl halides with dialkyl phos-phites, H-phosphinates, and secondary phosphine ox-ides.138 The best catalyst was found to be CuI with Cs2CO3 asthe base in DMF or toluene.
It was found that the P–C coupling can also be per-formed in the presence of NiCl2 and K2CO3 under ‘P-ligand-free’ conditions (Scheme 27).139 Bromobenzene undergoesefficient P–C coupling with dialkyl phosphites using 5 mol%Pd(OAc)2 at 150 °C for 45 minutes. More forcing conditions(10% Pd(OAc)2 and ≥1.5 h) were necessary in the reaction of4-alkoxy- and 4-alkyl-substituted bromobenzene to attainacceptable yields. The 3-methyl-, 3-chloro-, and fluoro-substituted bromobenzenes were more reactive startingmaterials, and the couplings were almost complete after45–60 minutes. It can be concluded that in the ‘P-ligand-free’ Ni-catalyzed P–C coupling, the substituted aryl bro-mides have a lower reactivity than bromobenzene. Themethod under discussion was successfully applied to thesynthesis of alkyl diphenylphosphinates and tertiary phos-phine oxides (Scheme 27).139 The alkyl phenyl-H-phosphi-nates were found to be more reactive than dialkyl phos-phites.
Scheme 23 The classical Hirao reaction
+
Br
Y
Y = a wide range of substituents
solvent(RO)2P
O
H
R = alkyl
P
Y
Pd(PPh3)4Et3N
Δ
O
OR
OR
38
ArBr +
MWPd(OAc)2 (5–10 mol%)
Et3N
solvent-freeP
O
H
RO
ROP
O
Ar
RO
RO39
60–93%
(EtO)2P(O)Ph, 93%(EtO)2P(O)ArAr = 3-MeC6H4, 91%Ar = 4-FC6H4, 91%Ar = 3-FC6H4, 88%Ar = 4-EtO2CC6H4, 89%Ar = 3-MeC(O)C6H4, 92%
(BuO)2P(O)Ph, 77%
150–200 °C, 2–10 min
PO
H
Ph
ROPhBr +
MWPd(OAc)2 (5 mol%)
Et3N
solvent-freeP
O
Ph
Ph
RO40
68–92%
PO
Ph
Ph
EtO85%
PO
Ph
Ph
PrO91%
PO
Ph
Ph
Bu87%
PO
Ph
Ph
PentO92%
150 °C, 5 min
ArBrH +
MWPd(OAc)2 (5 mol%)
Et3N
solvent-free or MeCNP
O
H
Y
YP
O
Ar
Y
Y
Pr2P(O)Ph, 80%Bu2P(O)Ph, 88%Bn2P(O)Ph, 95%
Ph2P(O)ArAr = Ph, 88%Ar = 4-MeC6H4, 83%Ar = 4-ClC6H4, 89%Ar = 4-FC6H4, 87%
4180–95%
150–175 °C, 5–90 min
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3081
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
Scheme 27 Ni-catalyzed ‘P-ligand-free’ Hirao reactions
This is the first instance that the Hirao reaction was per-formed without directly added P-ligands. In the casesstudied, the trivalent tautomeric form (Y1Y2POH) of the P-reagent may have served as the P-ligand in the in situformed Pd complex. For this, the Y1Y2P(O)H reagent had tobe used in a 1.05–1.10 equivalents quantity.
In special cases, the P–C coupling reactions could alsobe carried out without any catalyst (Scheme 28).140 More-over, secondary diarylphosphine oxides could be reactedwith halobenzoic acids in water, in the presence of K2CO3under MW conditions. The phosphinylated products 43were formed in variable yields.
Scheme 28 The ‘greenest’ Hirao reaction in water without any catalyst
In summary, the ‘P-ligand-free’ Pd- and Ni-catalyzedcoupling reactions proved to be suitable in the arylation ofdifferent >P(O)H reagents under MW conditions. SimplePd(OAc)2 and NiCl2 could be used as catalysts instead of ex-pensive and air sensitive Pd(PPh3)4, or other variations.
4 Conclusions
In summary our recent results in MW-assisted organo-phosphorus syntheses were surveyed. Beside the usual ad-vantages of the application of the MW technique in synthe-ses, either thermally reluctant reactions, such as the esteri-fication of phosphinic and phosphonic acids werepromoted, or catalytic systems were simplified. The lattercase was illustrated via the Kabachnik–Fields reaction, thealkylation of CH acidic compounds, O-alkylations, deoxy-genations, and the Hirao reaction. It was found that, ingeneral, MWs may be useful to enhance thermoneutralorganic reactions with a higher enthalpy of activation. Itwas possible to model the distribution of local overheatingsand their effect utilizing the Arrhenius equation. Hence, wewere successful in selecting the best model giving the samerate enhancement obtained experimentally for the esterifi-cation of a phosphinic acid.
Funding Information
This project was supported by the Hungarian Research Developmentand Innovation Fund (K119202).Hungarian Research Development and Innovation Fund (K119202)
References
(1) Microwave-Assisted Organic Synthesis; Tierney, J. P.; Lidsröm, P.,Eds.; Blackwell: Oxford, 2005.
(2) Microwaves in Organic Synthesis; de la Hoz, A.; Loupy, A., Eds.;Wiley-VCH: Weinheim, 2012.
(3) Milestones in Microwave Chemistry (SpringerBriefs in MolecularScience); Keglevich, G., Ed.; Springer: Switzerland, 2016.
(4) Kranjc, K.; Kočevar, M. Curr. Org. Chem. 2010, 14, 1050.(5) Guenin, E.; Meziane, D. Curr. Org. Chem. 2011, 15, 3465.(6) Keglevich, G.; Grün, A.; Bálint, E.; Kiss, N. Z.; Jablonkai, E. Curr.
Org. Chem. 2013, 17, 545.(7) Keglevich, G.; Greiner, I. Curr. Green Chem. 2014, 1, 2.(8) Keglevich, G.; Kiss, N.; Mucsi, Z.; Jablonkai, E.; Bálint, E. Green
Process Synth. 2014, 3, 103.(9) Keglevich, G. Phosphorus, Sulfur, Silicon Relat. Elem. 2014, 189,
1266.(10) Keglevich, G. In Green Synthetic Approaches for Biologically Rele-
vant Heterocycles; Brahmachari, G., Ed.; Elsevier: Amsterdam,2015, 559.
(11) Keglevich, G.; Grün, A.; Bagi, P.; Bálint, E.; Kiss, N. Z.; Kovács, R.;Jablonkai, E.; Kovács, T.; Fogassy, E.; Greiner, I. Period. Polytech.,Chem. Eng. 2015, 59, 82.
(12) Kiss, N. Z.; Bálint, E.; Keglevich, G. In Milestones in MicrowaveChemistry (SpringerBriefs in Molecular Science); Keglevich, G.,Ed.; Springer: Switzerland, 2016, Chap. 2, 11.
(13) Bana, P.; Greiner, I. In Milestones in Microwave Chemistry (Sprin-gerBriefs in Molecular Science), Keglevich G; Springer: Switzer-land, 2016, Chap. 4 77.
(14) Perreux, L.; Loupy, A. Tetrahedron 2001, 57, 9199.(15) Miklavc, A. ChemPhysChem 2001, 2, 552.(16) Binner, J. G. P.; Hassine, N. A.; Cross, T. E. J. Mater. Sci. 1995, 30,
5389.(17) Yadav, G. D.; Bisht, P. M. J. Mol. Catal. A: Chem. 2005, 236, 54.
ArBr +
MWNiCl2
K2CO3P
O
H
Y1
Y2P
O
Ar
Y1
Y2
4231–92%
(EtO)2P(O)ArAr = Ph, 92%Ar = 4-MeOC6H4, 40%Ar = 4-MeC6H4, 81%Ar = 3-MeC6H4, 87%Ar = 3-ClC6H4, 75%Ar = 4-FC6H4, 91%Ar = 3-FC6H4, 92%
(BuO)2P(O)Ph, 94% Ar2P(O)PhAr = Ph, 91%Ar = 4-MeOC6H4, 86%Ar = 4-MeC6H4, 85%Ar = 4-FC6H4, 87%Ar = 4-ClC6H4, 88%
R = Et, 85%R = Pr, 87%R = Bu, 90%
R = iPent, 84%
PO
Ph
Ph
RO
150 °C, 0.5–2.5 minMeCN
CO2H
+
MWK2CO3
H2O
CO2H
PO
H
Ar
ArP
O
Ar
Ar
X
43~ 59–82%
PO
CO2H
Ar = Ph, 82%Ar = 4-MeC6H4, 80%
PO
Ar = Ph, 70%Ar = 4-MeC6H4, 59%
CO2H
Ar Ar
ArAr
180 °C, 1–6 h
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3082
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
(18) Kappe, C. O.; Pieber, B.; Dallinger, D. Angew. Chem. Int. Ed. 2013,52, 1088.
(19) de la Hoz, A.; Díaz-Ortiz, A.; Moreno, A. Chem. Soc. Rev. 2005, 34,164.
(20) Kuhnert, N. Angew. Chem. Int. Ed. 2002, 41, 1863.(21) Hayes, B. L. Microwave Synthesis: Chemistry at the Speed of Light;
CEM Publishing: Matthews NC, 2002, 23.(22) Yeboah, K. A.; Boyd, J. D.; Kyeremateng, K. A.; Shepherd, C. C.;
Ingersoll, I. M.; Jackson, D. L. Jr.; Holland, A. W. React. Kinet.,Mech. Catal. 2014, 112, 295.
(23) Rosana, M. R.; Tao, Y.; Stiegman, A. E.; Dudley, G. B. Chem. Sci.2012, 3, 1240.
(24) Rosana, M. R.; Hunt, J.; Ferrari, A.; Southworth, T. A.; Tao, Y.;Stiegman, A. E.; Dudley, G. B. J. Org. Chem. 2014, 79, 7437.
(25) Chen, P.-K.; Rosana, M. R.; Dudley, G. B.; Stiegman, A. E. J. Org.Chem. 2014, 79, 7425.
(26) Kiss, N. Z.; Keglevich, G. Curr. Org. Chem. 2014, 18, 2673.(27) Kiss, N. Z.; Ludányi, K.; Drahos, L.; Keglevich, G. Synth. Commun.
2009, 39, 2392.(28) Keglevich, G.; Bálint, E.; Kiss, N. Z.; Jablonkai, E.; Hegedűs, L.;
Grün, A.; Greiner, I. Curr. Org. Chem. 2011, 15, 1802.(29) Kiss, N. Z.; Böttger, É.; Drahos, L.; Keglevich, G. Heteroat. Chem.
2013, 24, 283.(30) Kiss, N. Z.; Keglevich, G. Tetrahedron Lett. 2016, 57, 971.(31) Keglevich, G.; Kiss, N. Z.; Drahos, L.; Körtvélyesi, T. Tetrahedron
Lett. 2013, 54, 466.(32) Keglevich, G.; Kiss, N. Z.; Körtvélyesi, T. Heteroat. Chem. 2013,
24, 91.(33) Kiss, N. Z.; Simon, A.; Drahos, L.; Huben, K.; Jankowski, S.;
Keglevich, G. Synthesis 2013, 45, 199.(34) Kiss, N. Z.; Mucsi, Z.; Böttger, É.; Drahos, L. Curr. Org. Synth.
2014, 11, 767.(35) Mucsi, Z.; Kiss, N. Z.; Keglevich, G. RSC Adv. 2014, 4, 11948.(36) Keglevich, G.; Kiss, N. Z.; Mucsi, Z.; Körtvélyesi, T. Org. Biomol.
Chem. 2012, 10, 2011.(37) de Cózar, A.; Millán, M. C.; Cebrián, C.; Prieto, P.; Díaz-Ortiz, A.;
de la Hoz, A.; Cossío, F. P. Org. Biomol. Chem. 2010, 8, 1000.(38) Rodríguez, A. M.; Prieto, P.; de la Hoz, A.; Díaz-Ortiz, A.; García,
J. I. Org. Biomol. Chem. 2014, 12, 2436.(39) Rodríguez, A. M.; Prieto, P.; de la Hoz, A.; Díaz-Ortiz, Á.; Martín,
D. R.; García, J. I. ChemistryOpen 2015, 4, 308–1363;http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2191.
(40) Prieto, P.; de la Hoz, A.; Diaz-Ortiz, A.; Rodriguez, A. M. Chem.Soc. Rev. 2017, 46, 431.
(41) Keglevich, G.; Kiss, N. Z.; Jablonkai, E.; Bálint, E.; Mucsi, Z. Phos-phorus, Sulfur, Silicon Relat. Elem. 2015, 190, 647.
(42) Keglevich, G.; Greiner, I.; Mucsi, Z. Curr. Org. Chem. 2015, 19,1436.
(43) Keglevich, G.; Kiss, N. Z.; Mucsi, Z. Pure Appl. Chem. 2016, 88,931.
(44) Keglevich, G.; Kiss, N. Z.; Mucsi, Z. Curr. Phys. Chem. 2017, DOI:10.2174/1877946806666161103142955.
(45) Kabachnik, M. I.; Medved, T. Y. Dokl. Akad. Nauk SSSR 1952, 83,689.
(46) Fields, E. K. J. Am. Chem. Soc. 1952, 74, 1528.(47) Cherkasov, R. A.; Galkin, V. I. Russ. Chem. Rev. 1998, 67, 857.(48) Kukhar, V. P.; Hudson, H. R. Aminophosphonic and Aminophos-
phinic Acids: Chemistry and Biological Activity; Wiley: Chiches-ter, 2000.
(49) Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955.(50) Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassall, C. H.; Holmes, S.
W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature (London)1978, 272, 56.
(51) Lavielle, G.; Hautefaye, P.; Schaeffer, C.; Boutin, J. A.; Cudennec,C. A.; Pierre, A. J. Med. Chem. 1991, 34, 1998.
(52) Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. J. Med. Chem.2003, 46, 2641.
(53) Sieńczyk, M.; Oleksyszyn, J. Curr. Med. Chem. 2009, 16, 1673.(54) Forlani, G.; Berlicki, L.; Duo, M.; Dziedziola, G.; Giberti, S.;
Bertazzini, M.; Kafarski, P. J. Agric. Food Chem. 2013, 61, 6792.(55) Forlani, G.; Occhipinti, A.; Berlicki, Ł.; Dziędzioła, G.; Wieczorek,
A.; Kafarski, P. J. Agric. Food Chem. 2008, 56, 3193.(56) Long, N.; Cai, X. J.; Song, B. A.; Yang, S.; Chen, Z.; Bhadury, P. S.;
Hu, D. Y.; Jin, L. H.; Xue, W. J. Agric. Food Chem. 2008, 56, 5242.(57) Matveeva, E. D.; Podrugina, T. A.; Tishkovskaya, E. V.; Tomilova,
L. G.; Zefirov, N. S. Synlett 2003, 2321.(58) Firouzabadi, H.; Iranpoor, N.; Sobhani, S. Synthesis 2004, 2692.(59) Lee, S.; Park, J. H.; Kang, J.; Lee, J. K. Chem. Commun. 2001, 1698.(60) Lee, S.; Lee, J. K.; Song, C. E.; Kim, D.-C. Bull. Korean Chem. Soc.
2002, 23, 667.(61) Sun, P.; Hu, Z.; Huang, Z. Synth. Commun. 2004, 34, 4293.(62) Bhattacharya, A. K.; Kaur, T. Synlett 2007, 745.(63) Xu, F.; Luo, Y.; Deng, M.; Shen, Q. Eur. J. Org. Chem. 2003, 4728.(64) Ranu, B. C.; Hajra, A.; Jana, U. Org. Lett. 1999, 1, 1141.(65) Bhagat, S.; Chakraborti, A. K. J. Org. Chem. 2007, 72, 1263.(66) Wu, J.; Sun, W.; Xia, H.-G.; Sun, X. Org. Biomol. Chem. 2006, 4,
1663.(67) Keglevich, G.; Szekrényi, A. Lett. Org. Chem. 2008, 5, 616.(68) Keglevich, G.; Bálint, E. Molecules 2012, 17, 12821.(69) Kafarski, P.; Górniak, M. G.; Andrasiak, I. Curr. Green Chem.
2015, 2, 218.(70) Prauda, I.; Greiner, I.; Ludányi, K.; Keglevich, G. Synth. Commun.
2007, 37, 317.(71) Bálint, E.; Takács, J.; Drahos, L.; Juranovic, A.; Kocevar, M.;
Keglevich, G. Heteroat. Chem. 2013, 24, 221.(72) Tajti, Á.; Bálint, E.; Keglevich, G. Curr. Org. Synth. 2016, 13, 638.(73) Bálint, E.; Tripolszky, A.; Jablonkai, E.; Karaghiosoff, K.; Czugler,
M.; Mucsi, Z.; Kollár, L.; Pongrácz, P.; Keglevich, G. J. Organomet.Chem. 2016, 801, 111.
(74) Petrov, K. A.; Chauzov, V. A.; Erokhina, T. S.; Chernobrovkina, L.P. Zh. Obsh. Khim. 1976, 46, 493.
(75) Petrov, K. A.; Chauzov, V. A.; Erokhina, T. S. Khim. Elementoorg.Soedin. 1976, 200.
(76) Garifzyanov, A. R.; Vasil’ev, R. I.; Cherkasov, R. A. Russ. J. Gen.Chem. 2005, 75, 217.
(77) Keglevich, G.; Szekrényi, A.; Szöllősy, Á.; Drahos, L. Synth.Commun. 2011, 41, 2265.
(78) Bálint, E.; Fazekas, E.; Pintér, G.; Szöllősy, Á.; Holczbauer, T.;Czugler, M.; Drahos, L.; Körtvélyesi, T.; Keglevich, G. Curr. Org.Chem. 2012, 16, 547.
(79) Bálint, E.; Fazekas, E.; Pongrácz, P.; Kollár, L.; Drahos, L.;Holczbauer, T.; Czugler, M.; Keglevich, G. J. Organomet. Chem.2012, 717, 75.
(80) Bálint, E.; Fazekas, E.; Drahos, L.; Keglevich, G. Heteroat. Chem.2013, 24, 510.
(81) Bálint, E.; Fazekas, E.; Kóti, J.; Keglevich, G. Heteroat. Chem.2015, 26, 106.
(82) Deshayes, S.; Liagre, M.; Loupy, A.; Luche, J.-L.; Petit, A. Tetrahe-dron 1999, 55, 10851.
(83) Wang, Y.; Deng, R.; Mi, A.; Jiang, Y. Synth. Commun. 1995, 25,1761.
(84) Deng, R.; Wang, Y.; Jiang, Y. Synth. Commun. 1994, 24, 111.(85) Deng, R.; Wang, Y.; Jiang, Y. Synth. Commun. 1994, 24, 1917.(86) Keglevich, G.; Novák, T.; Vida, L.; Greiner, I. Green Chem. 2006, 8,
1073.
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083

3083
G. Keglevich et al. Short ReviewSyn thesis
Dow
nloa
ded
by: T
hiem
e E
-Boo
ks &
E-J
ourn
als.
Cop
yrig
hted
mat
eria
l.
(87) Keglevich, G.; Majrik, K.; Vida, L.; Greiner, I. Lett. Org. Chem.2008, 5, 224.
(88) Grün, A.; Bálint, E.; Keglevich, G. Catalysts 2015, 5, 634.(89) Greiner, I.; Grün, A.; Ludányi, K.; Keglevich, G. Heteroat. Chem.
2011, 22, 11.(90) Keglevich, G.; Grün, A.; Blastik, Z.; Greiner, I. Heteroat. Chem.
2011, 22, 174.(91) Grün, A.; Blastik, Z.; Drahos, L.; Keglevich, G. Heteroat. Chem.
2012, 23, 241.(92) Grün, A.; Blastik, Z.; Drahos, L.; Keglevich, G. Heteroat. Chem.
2014, 25, 107.(93) Grün, A.; Molnár, I. G.; Bertók, B.; Greiner, I.; Keglevich, G.
Heteroat. Chem. 2009, 20, 350.(94) Keglevich, G.; Grün, A.; Molnár, I. G.; Greiner, I. Heteroat. Chem.
2011, 22, 640.(95) Tokutake, N.; Hiratake, J.; Katoh, M.; Irie, T.; Kato, H.; Oda, J.
Bioorg. Med. Chem. 1998, 6, 1935.(96) Martin, M. T.; Angeles, T. S.; Sugasawara, R.; Aman, N. I.; Napper,
A. D.; Darsley, M. J.; Sanchez, R. I.; Booth, P.; Titmas, R. C. J. Am.Chem. Soc. 1994, 116, 6508.
(97) Golubski, Z. E. Synthesis 1980, 632.(98) Bálint, E.; Jablonkai, E.; Bálint, M.; Keglevich, G. Heteroat. Chem.
2010, 21, 211.(99) Hérault, D.; Nguyen, D. H.; Nuel, D.; Buono, G. Chem. Soc. Rev.
2015, 44, 2508.(100) Kovács, T.; Keglevich, G. Curr. Org. Chem. 2017, 21, 569.(101) O'Brien, C. J.; Tellez, J. L.; Nixon, Z. S.; Kang, L. J.; Carter, A. L.;
Kunkel, S. R.; Przeworski, K. C.; Chass, G. A. Angew. Chem. Int. Ed.2009, 48, 6836.
(102) O’Brien, C. J.; Nixon, Z. S.; Holohan, A. J.; Kunkel, S. R.; Tellez, J.L.; Doonan, B. J.; Coyle, E. E.; Lavigne, F.; Kang, L. J.; Przeworski,K. C. Chem. Eur. J. 2013, 19, 15281.
(103) O’Brien, C. J.; Lavigne, F.; Coyle, E. E.; Holohan, A. J.; Doonan, B. J.Chem. Eur. J. 2013, 19, 5854.
(104) Coyle, E. E.; Doonan, B. J.; Holohan, A. J.; Walsh, K. A.; Lavigne,F.; Krenske, E. H.; O’Brien, C. J. Angew. Chem. Int. Ed. 2014, 53,12907.
(105) Fritzsche, H.; Hasserodt, U.; Korte, F. Chem. Ber. 1965, 98, 171.(106) Quin, L. D.; Caster, K. C.; Kisalus, J. C.; Mesch, K. A. J. Am. Chem.
Soc. 1984, 106, 7021.(107) Fritzsche, H.; Hasserodt, U.; Korte, F. Chem. Ber. 1964, 97, 1988.(108) Demchuk, O. M.; Jasiński, R.; Pietrusiewicz, K. M. Heteroat.
Chem. 2015, 26, 441.(109) Li, Y. H.; Das, S.; Zhou, S. L.; Junge, K.; Beller, M. J. Am. Chem. Soc.
2012, 134, 9727.(110) Berthod, M.; Favre-Réguillon, A.; Mohamad, J.; Mignani, G.;
Docherty, G.; Lemaire, M. Synlett 2007, 1545.(111) Petit, C.; Favre-Réguillon, A.; Albela, B.; Bonneviot, L.; Mignani,
G.; Lemaire, M. Organometallics 2009, 28, 6379.(112) Pehlivan, L.; Metay, E.; Delbrayelle, D.; Mignani, G.; Lemaire, M.
Tetrahedron 2012, 68, 3151.
(113) Coumbe, T.; Lawrence, N. J.; Muhammad, F. Tetrahedron Lett.1994, 35, 625.
(114) Li, Y. H.; Lu, L. Q.; Das, S.; Pisiewicz, S.; Junge, K.; Beller, M. J. Am.Chem. Soc. 2012, 134, 18325.
(115) Keglevich, G.; Kovács, T.; Csatlós, F. Heteroat. Chem. 2015, 26,199.
(116) Kovács, T.; Urbanics, A.; Csatlós, F.; Binder, J.; Falk, A.; Uhlig, F.;Keglevich, G. Curr. Org. Synth. 2016, 13, 148.
(117) Keglevich, G.; Kovács, T. Curr. Green Chem. 2014, 1, 182.(118) Kovács, T.; Urbanics, A.; Csatlós, F.; Keglevich, G. Heteroat. Chem.
2017, in press.(119) Kovács, T.; Keglevich, G. Phosphorus, Sulfur, Silicon Relat. Elem.
2016, 191, 359.(120) Kovács, T.; Csatlós, F.; Urbanics, A.; Uhlig, F.; Keglevich, G. Phos-
phorus, Sulfur, Silicon Relat. Elem. 2016, 191, 1597.(121) Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Tetrahedron Lett.
1980, 21, 3595.(122) Hirao, T.; Masunaga, T.; Yamada, N.; Ohshiro, Y.; Agawa, T. Bull.
Chem. Soc. Jpn. 1982, 55, 909.(123) Jablonkai, E.; Keglevich, G. Curr. Org. Synth. 2014, 11, 429.(124) Jablonkai, E.; Keglevich, G. Org. Prep. Proced. Int. 2014, 46, 281.(125) Kalek, M.; Ziadi, A.; Stawinski, J. Org. Lett. 2008, 10, 4637.(126) Andaloussi, M.; Lindh, J.; Sävmarker, J.; Sjöberg, P. J. R.; Larhed,
M. Chem. Eur. J. 2009, 15, 13069.(127) Villemin, D.; Jaffrès, P.-A.; Simèon, F. Phosphorus, Sulfur, Silicon
Relat. Elem. 1997, 130, 59.(128) Julienne, D.; Lohier, J.-F.; Delacroix, O.; Gaumont, A.-C. J. Org.
Chem. 2007, 72, 2247.(129) Rummelt, S. M.; Ranocchiari, M.; van Bokhoven, J. A. Org. Lett.
2012, 14, 2188.(130) Novikova, Z. S.; Demik, N. N.; Agarkov, A. Yu.; Beletskaya, I. P.
Russ. J. Org. Chem. 1995, 31, 129.(131) Kabachnik, M. M.; Solntseva, M. D.; Izmer, V. V.; Novikova, Z. S.;
Beletskaya, I. P. Russ. J. Org. Chem. 1998, 34, 93.(132) Beletskaya, I. P.; Neganova, E. G.; Veits, Yu. A. Russ. J. Org. Chem.
2004, 40, 1782.(133) Beletskaya, I. P.; Karlstedt, N. B.; Nifant’ev, E. E.; Khodarev, D. V.;
Kukhareva, T. S.; Nikolaev, A. V.; Ross, A. J. Russ. J. Org. Chem.2006, 42, 1780.
(134) Keglevich, G.; Grün, A.; Bölcskei, A.; Drahos, L.; Kraszni, M.;Balogh, G. T. Heteroat. Chem. 2012, 23, 574.
(135) Jablonkai, E.; Keglevich, G. Tetrahedron Lett. 2013, 54, 4185.(136) Keglevich, G.; Jablonkai, E.; Balázs, L. B. RSC Adv. 2014, 4, 22808.(137) Amaya, T.; Abe, Y.; Inada, Y.; Hirao, T. Tetrahedron Lett. 2014, 55,
3976.(138) Xiong, B.; Li, M.; Liu, Y.; Zhou, Y.; Zhao, C.; Goto, M.; Yin, S.-F.;
Han, L.-B. Adv. Synth Catal. 2014, 356, 781.(139) Jablonkai, E.; Balázs, L. B.; Keglevich, G. Curr. Org. Chem. 2015,
19, 197.(140) Jablonkai, E.; Keglevich, G. Tetrahedron Lett. 2015, 56, 1638.
© Georg Thieme Verlag Stuttgart · New York — Synthesis 2017, 49, 3069–3083