Embed Size (px)
Citation preview

Joaquín Dopazo
Computational Genomics Department,
Centro de Investigación Príncipe Felipe (CIPF),
Functional Genomics Node, (INB),
Bioinformatics in Rare Diseases (BiER-CIBERER),
Valencia, Spain.
Digging into thousands of variants to
find disease genes in Mendelian and
complex diseases
http://bioinfo.cipf.es http://www.babelomics.org
@xdopazo
IV DCEX symposium, Barcelona, November 17th, 2015
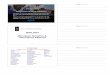
Precision medicine (P4*) is based on a better knowledge of phenotype-
genotype relationships
Requires of a better way of defining diseases by introducing genomic
technologies in the diagnostic procedures and treatment decisions
*P4: Predictive, Preventive, Personalized, Participative
Setting the problem in context: The transition to precision medicine
Intuitive Based on trial
and error
Identification of probabilistic
patterns
Decisions and actions based on knowledge
Intuitive Medicine Empirical Medicine Precision Medicine (P4)
Today Tomorrow
Degree of personalization
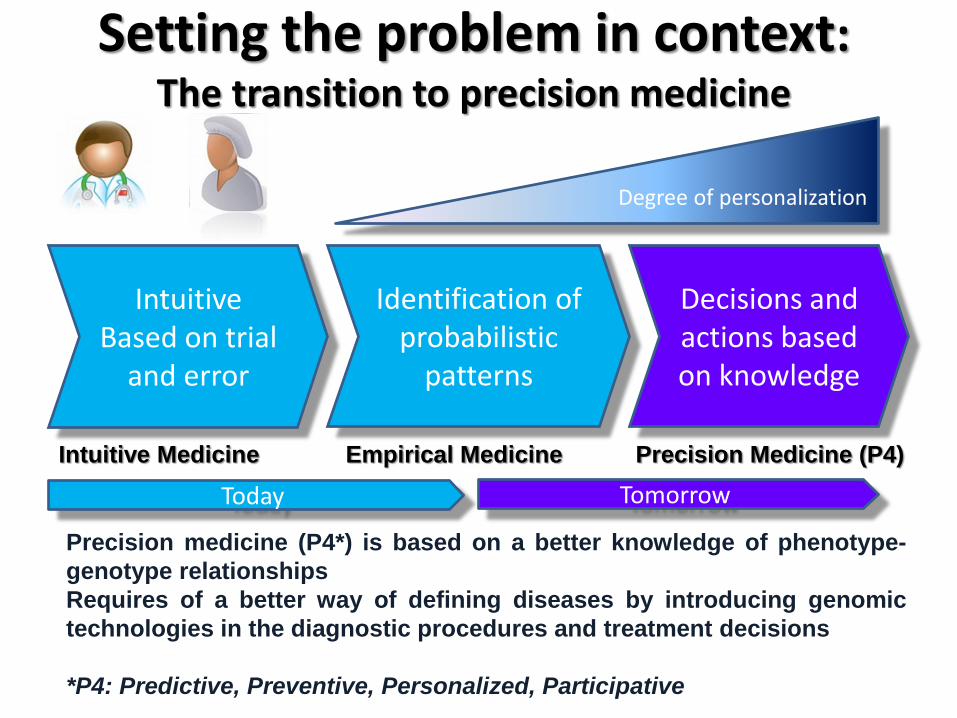
Empirical Medicine First Phase I: generation of the knowledge database
-------------------------------------------------------
sequencing
Patient Variants
Database. Query
Therapy outcome
System feedback
Genomic variants (biomarkers) can be quickly associated to precise diagnostics and therapy outcomes
Initially the system will need much feedback: Knowledge generation phase.
Empirical medicine
Knowledge database
Genome projects enable knowledge generation

Rare and familiar diseases
exome sequencing initiative
• Metabolic (86 samples)
• Optiz
• Atypical fracture
• coQ10 deficiency
• Congenital disorder of glycosylation types I and II
• Maple syrup urine disease
• Pelizaeus-like
• 4 unknown syndroms
• Genetic (24 samples)
• Charcot-Marie-Tooth
• Rett Syndrome
• Neurosensorial (35 samples)
• Usher
• AD non-syndromic hearing loss
• AR non-syndromic hearing loss
• RP
• Mitochondrial (28 samples)
• Progressive External Oftalmoplegy
• Multi-enzymatic deficiency in mitochondrial
respiratory complexes
• CoQ disease
• Other
• APL (10 samples)
Autism (37 samples)
Mental retardation (autosomal recessive) (24)
Immunodeficiency (18)
Leber's congenital amaurosis (9)
Cataract (2)
RP(AR) (60)
RP(AD) (46)
Deafness (24)
CLAPO (4)
Skeletal Dysplasia (3)
Cantú syndrome (1)
Dubowitz syndrome (2)
Gorham-Stout syndrome (1)
Malpuech syndrome (4)
Hirschprung’s disease (81)
Hereditary macrothrombocytopenia (3)
MTC (41)
Controls (301)
1044 samples = 183 samples + 200 controls + 360 samples + 301 controls
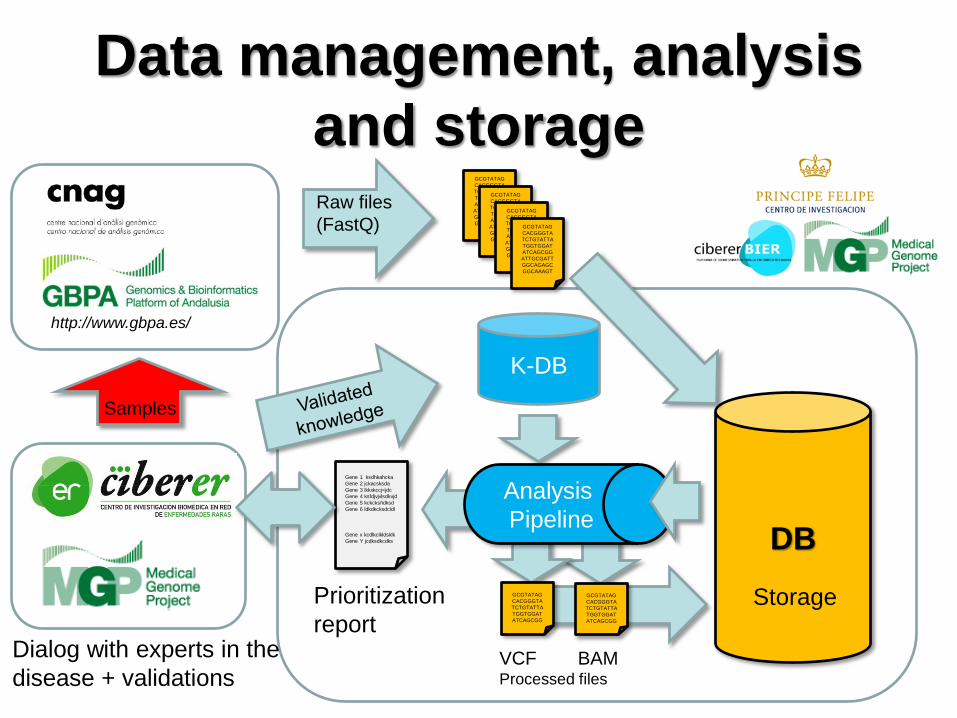
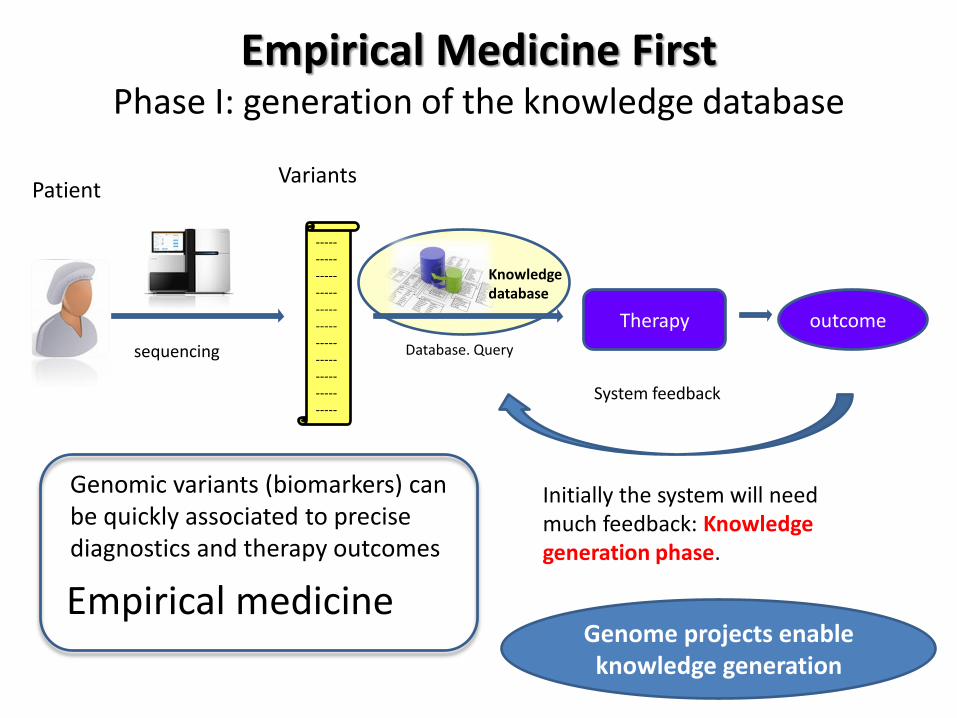
Data management, analysis
and storage
http://www.gbpa.es/
GCGTATAG
CACGGGTA
TCTGTATTA
TGGTGGAT
ATCAGCGG
ATTGCGATT
GGCAGAGC
GGCAAAGT
GCGTATAG
CACGGGTA
TCTGTATTA
TGGTGGAT
ATCAGCGG
ATTGCGATT
GGCAGAGC
GGCAAAGT
GCGTATAG
CACGGGTA
TCTGTATTA
TGGTGGAT
ATCAGCGG
ATTGCGATT
GGCAGAGC
GGCAAAGT
GCGTATAG
CACGGGTA
TCTGTATTA
TGGTGGAT
ATCAGCGG
ATTGCGATT
GGCAGAGC
GGCAAAGT
Raw files
(FastQ)
DB
Analysis
Pipeline
Storage
K-DB
Gene 1 ksdhkahcka
Gene 2 jckacsksda
Gene 3 lkkxkccj<jdc
Gene 4 ksfdjvjvlsdkvjd
Gene 5 kckcksñdksd
Gene 6 ldkdkcksdcldl
Gene x kcdlkclkldsklk
Gene Y jcdksdkcdks
Prioritization
report Dialog with experts in the
disease + validations
Samples
GCGTATAG
CACGGGTA
TCTGTATTA
TGGTGGAT
ATCAGCGG
GCGTATAG
CACGGGTA
TCTGTATTA
TGGTGGAT
ATCAGCGG
VCF BAM Processed files

Casablanca: Round up all suspicious characters and search them for stolen documents.
The prioritization process Typically, an exome renders between 40 and 60K variants (and a
genome about 1 million). Only one or a few among all of them are
expected to be the causative factors of the disease.
The prioritization process is like a police investigation in which
suspected are discarded by their alibies
Thus, through sequential heuristic filtering steps, unlikely candidates
are discarded and a final, reduced list with one or a few candidate
genes is (hopefully) produced
Agatha Christie
40-60K suspects?
We need…
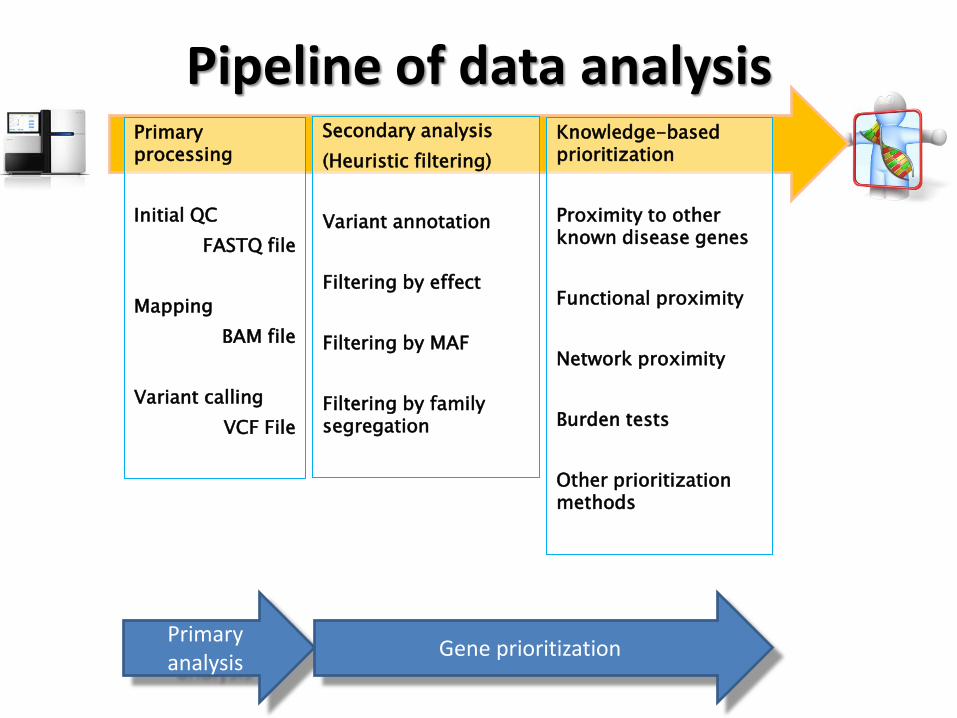
Pipeline of data analysis Primary processing
Initial QC
FASTQ file
Mapping
BAM file
Variant calling
VCF File
Knowledge-based prioritization
Proximity to other known disease genes
Functional proximity
Network proximity
Burden tests
Other prioritization methods
Secondary analysis
(Heuristic filtering)
Variant annotation
Filtering by effect
Filtering by MAF
Filtering by family segregation
Primary analysis
Gene prioritization
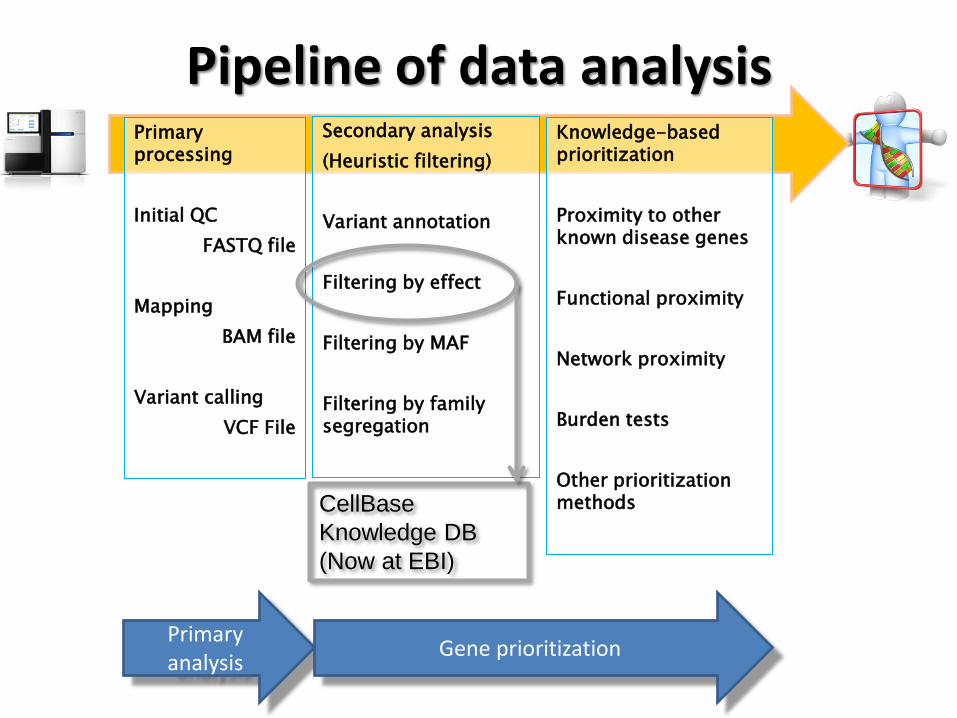
Pipeline of data analysis Primary processing
Initial QC
FASTQ file
Mapping
BAM file
Variant calling
VCF File
Knowledge-based prioritization
Proximity to other known disease genes
Functional proximity
Network proximity
Burden tests
Other prioritization methods
Secondary analysis
(Heuristic filtering)
Variant annotation
Filtering by effect
Filtering by MAF
Filtering by family segregation
Primary analysis
Gene prioritization
CellBase
Knowledge DB
(Now at EBI)

The knowledge database for filtering by effect
CellBase (Bleda, 2012, NAR), a comprehensive integrative database and RESTful Web Services API:
● Core features: genes, transcripts, exons, cytobands, proteins (UniProt),...
● Variation: dbSNP and Ensembl SNPs, HapMap, 1000Genomes, Cosmic, ClinVar, ...
● Functional: 40 OBO ontologies (Gene Ontology, HPO, etc.), Interpro, etc.
● Regulatory: TFBS, miRNA targets, conserved regions, tissue expression etc.
● System biology: Interactome (IntAct), Reactome database, co-expressed genes.
NoSQL (Mongo) and scales to TB
Wiki: http://docs.bioinfo.cipf.es/projects/cellbase/wiki
Project: http://bioinfo.cipf.es/compbio/cellbase
Now available at the EBI: http://www.ebi.ac.uk/cellbase/webservices/rest/v3/ and the
University of Cambridge: http://bioinfo.hpc.cam.ac.uk/cellbase/webservices/rest
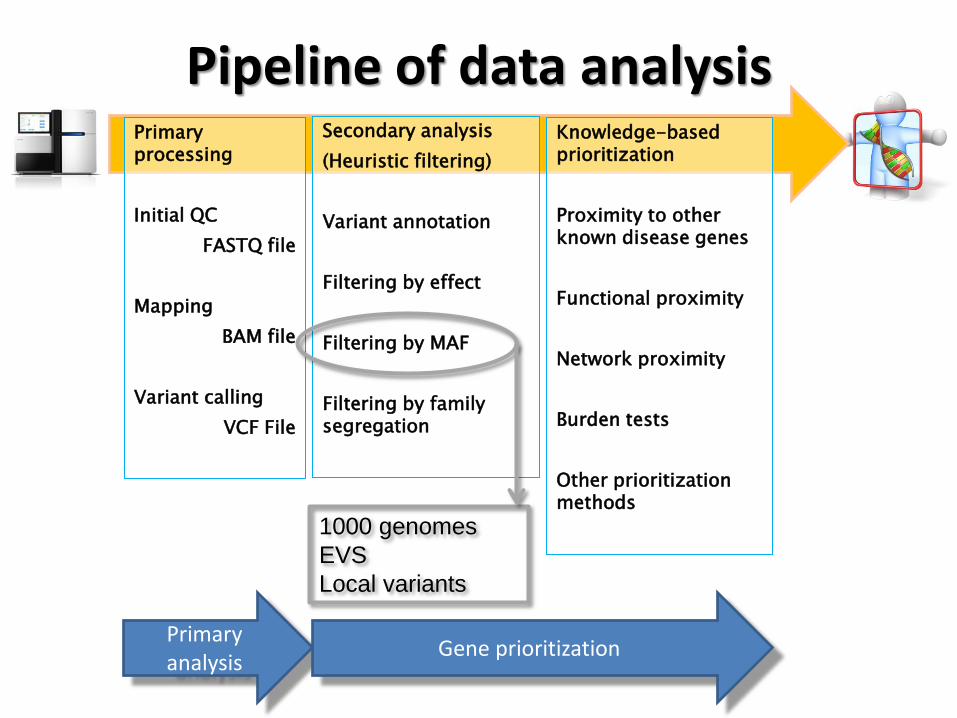
Pipeline of data analysis Primary processing
Initial QC
FASTQ file
Mapping
BAM file
Variant calling
VCF File
Knowledge-based prioritization
Proximity to other known disease genes
Functional proximity
Network proximity
Burden tests
Other prioritization methods
Secondary analysis
(Heuristic filtering)
Variant annotation
Filtering by effect
Filtering by MAF
Filtering by family segregation
Primary analysis
Gene prioritization
1000 genomes
EVS
Local variants
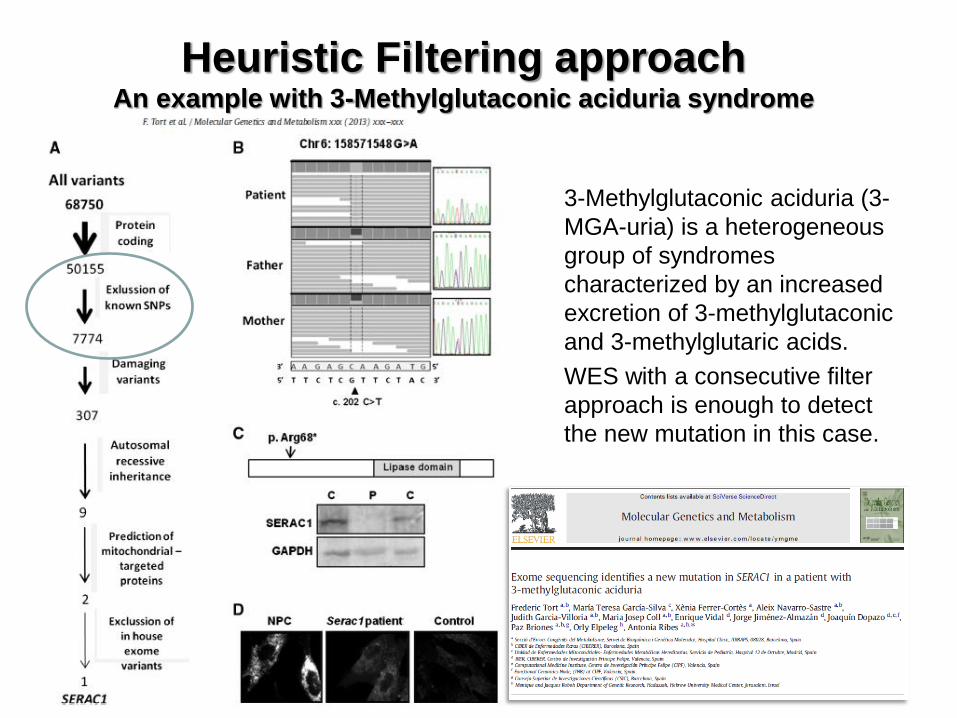
3-Methylglutaconic aciduria (3-
MGA-uria) is a heterogeneous
group of syndromes
characterized by an increased
excretion of 3-methylglutaconic
and 3-methylglutaric acids.
WES with a consecutive filter
approach is enough to detect
the new mutation in this case.
Heuristic Filtering approach An example with 3-Methylglutaconic aciduria syndrome

Use known variants and their population frequencies to filter out.
• Typically dbSNP, 1000 genomes and
the 6515 exomes from the ESP are
used as sources of population
frequencies.
• We sequenced 300 healthy controls
(rigorously phenotyped) to add and
extra filtering step to the analysis
pipeline
Novembre et al., 2008. Genes mirror
geography within Europe. Nature Comparison of MGP controls to 1000g
How important do you
think local information is
to detect disease genes?
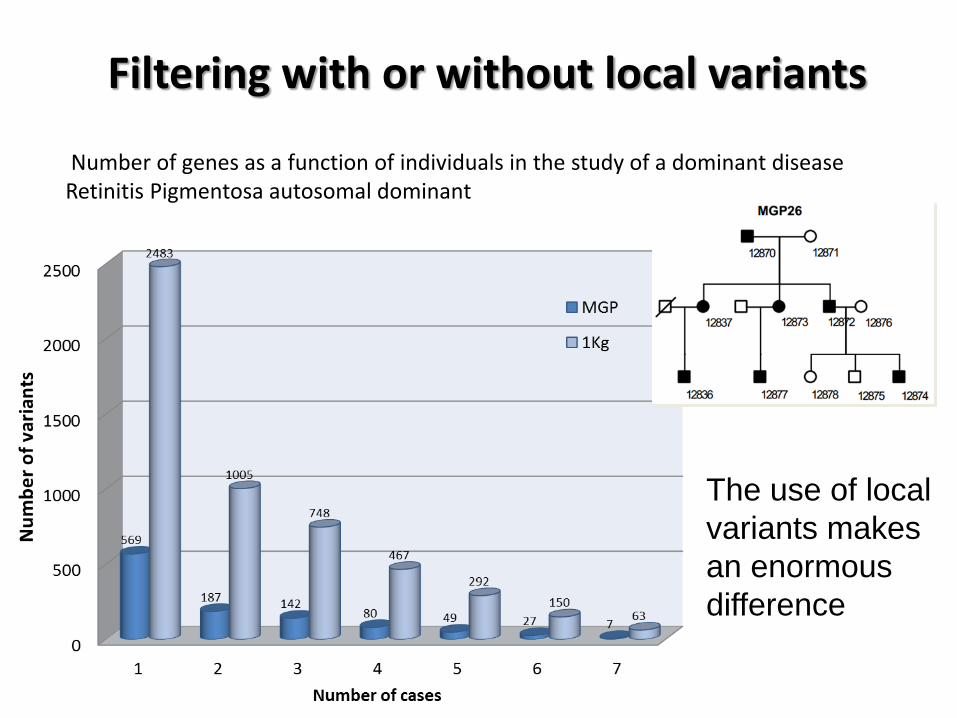
Filtering with or without local variants
Number of genes as a function of individuals in the study of a dominant disease Retinitis Pigmentosa autosomal dominant
The use of local
variants makes
an enormous
difference

The CIBERER Spanish Variant Server (CSVS): the first repository of variability of the Spanish population
Only another similar initiative
exists: the GoNL
http://www.nlgenome.nl/ http://ciberer.es/bier/exome-server/
And more recently
the Finnish
and the Icelandic
populations
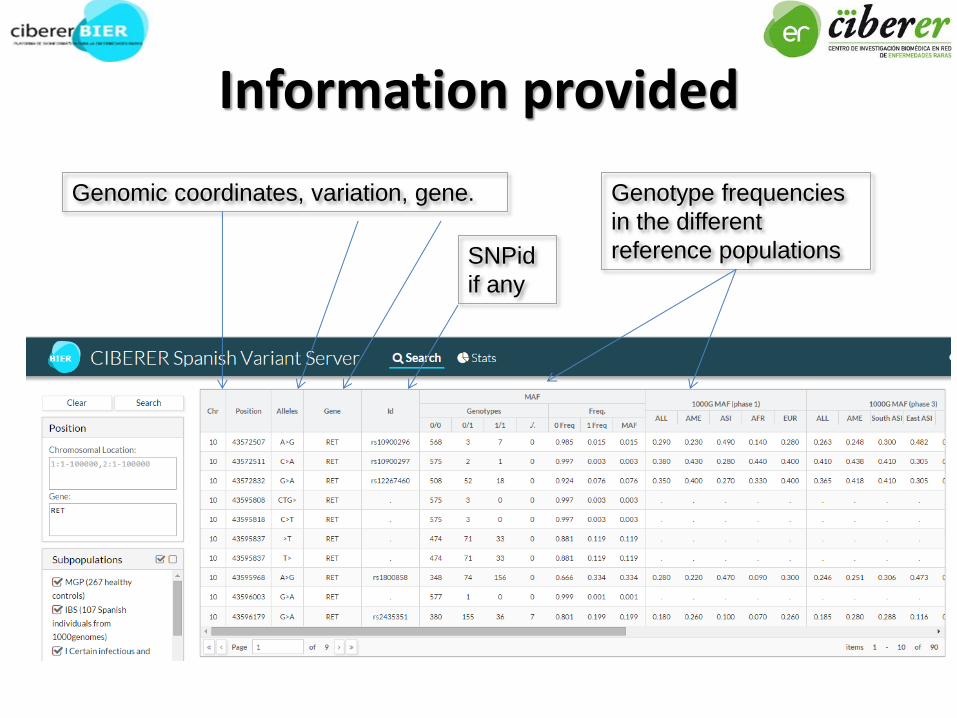
Information provided
Genotype frequencies
in the different
reference populations
Genomic coordinates, variation, gene.
SNPid
if any
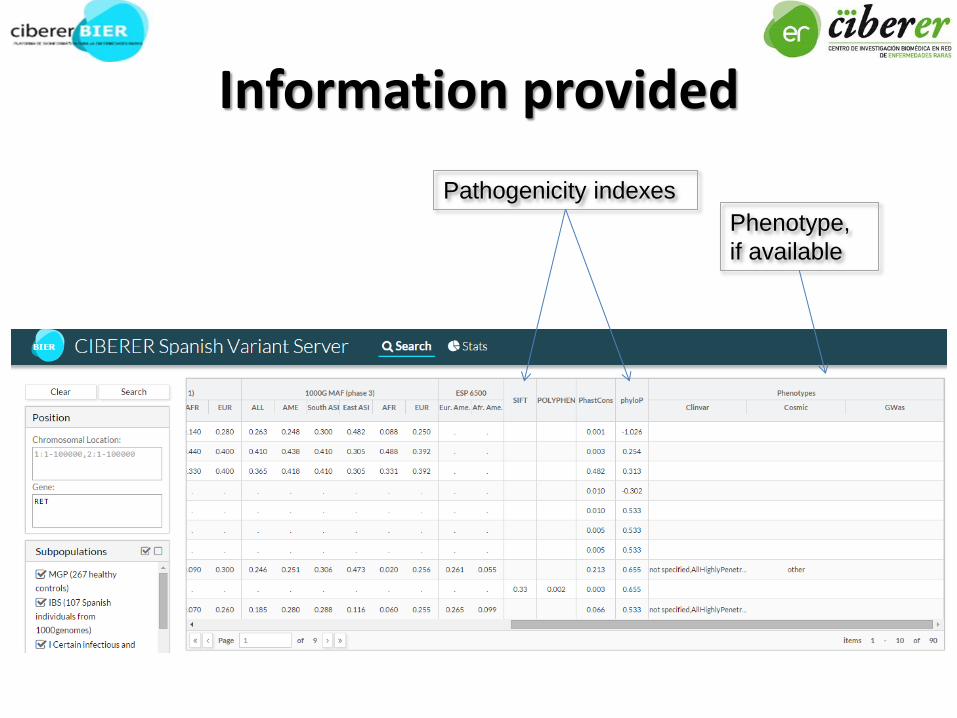
Information provided
Pathogenicity indexes
Phenotype,
if available
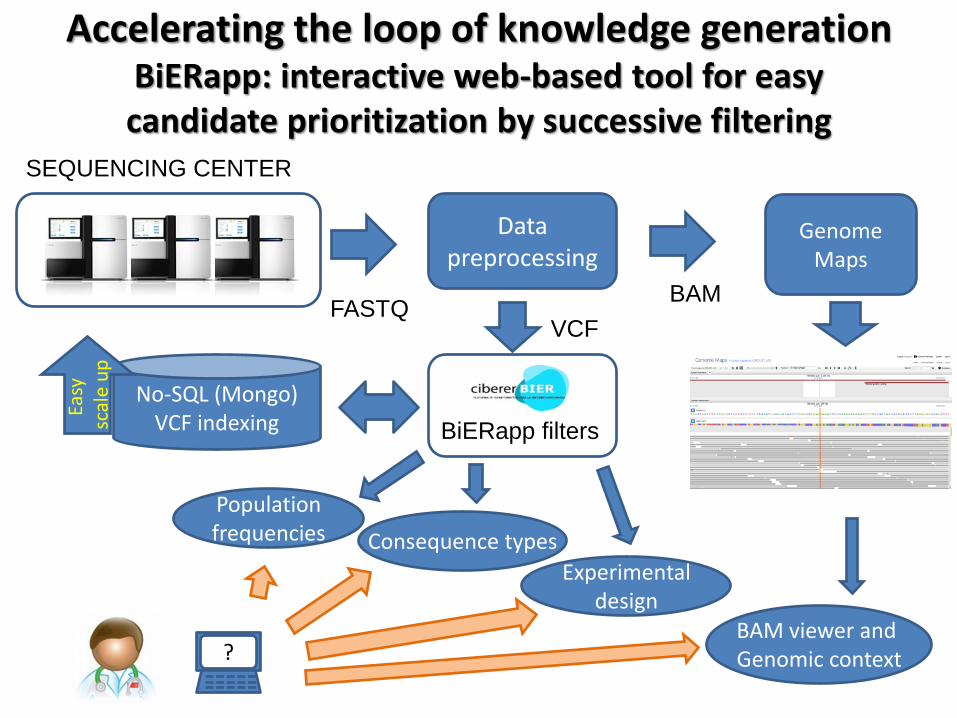
Accelerating the loop of knowledge generation BiERapp: interactive web-based tool for easy candidate prioritization by successive filtering
SEQUENCING CENTER
Data preprocessing
VCF FASTQ
Genome Maps
BAM
BiERapp filters
No-SQL (Mongo) VCF indexing
Population frequencies Consequence types
Experimental design
BAM viewer and Genomic context ?
Easy
sc
ale
up
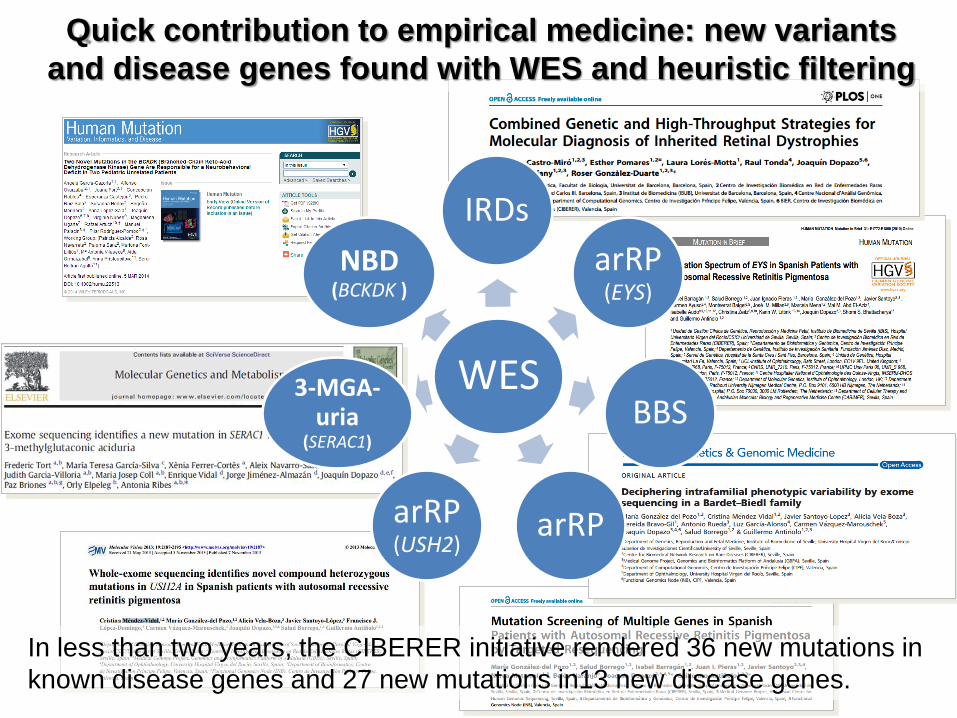
Quick contribution to empirical medicine: new variants
and disease genes found with WES and heuristic filtering
WES
IRDs
arRP (EYS)
BBS
arRP arRP (USH2)
3-MGA-uria
(SERAC1)
NBD (BCKDK )
In less than two years, the CIBERER initiative rendered 36 new mutations in
known disease genes and 27 new mutations in13 new disease genes.

Limitations of the single-gene
approach One might or might not be aware of it, but the single-gene
approach implies the following assumptions:
• Only one gene (or at least one main gene) is the
causative agent of the disease.
• The disease has complete (or very high) penetrance.
• Deleterious variation with no phenotype is not expected
(or expected to be marginal)
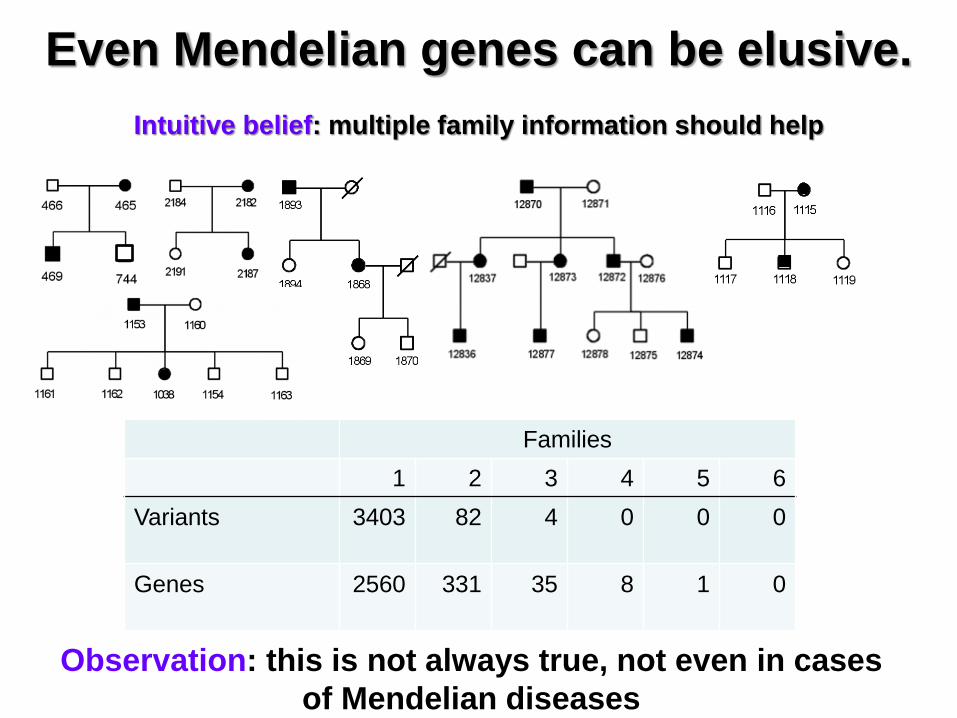
Even Mendelian genes can be elusive.
Intuitive belief: multiple family information should help
Families
1 2 3 4 5 6
Variants 3403 82 4 0 0 0
Genes 2560 331 35 8 1 0
Observation: this is not always true, not even in cases
of Mendelian diseases

Limitations of the single-gene
approach One might or might not be aware of it, but the single-gene
approach implies the following assumptions:
• Only one gene (or at least one main gene) is the
causative agent of the disease.
• The disease has complete (or very high) penetrance.
• Deleterious variation with no phenotype is not expected
(or expected to be marginal)

However, a high level of deleterious
variability exists in the human genome
Variants predicted to severely affect the function
of human protein coding genes (known as loss-of-
function -LOF- variants) were thought:
To have a potential deleterious effect
To be associated to severe Mendelian
disease
An unexpectedly large number of LOF variants
have been found in the genomes of apparently
healthy individuals: 281-515 missense
substitutions per individual, 40-85 of them in
homozygous state and predicted to be highly
damaging.
A similar proportion was observed in miRNAs
and possibly affect to any functional element in
the genome
Such apparently deleterious mutation must be first detected and
then distinguished from real pathological mutations

So far so good… but is the single-gene
approach realistic? Can we detect such
disease-related variants so easily?
There are several problems:
a) There is a non-negligible amount of apparently deleterious variants
that seem to have no pathologic effect (effect filter fails)
b) Sometimes we are not targeting rare but common variants (which
occur in normal population) (MAF filter fails)
c) Interrogating 60Mb sites produces too many variants. Many of these
segregating with our experimental design (segregation filter fails)
d) In many cases only one variant does not explain the disease but rather
a combination of them (single-gene concept fails)
That results in a low ratio of discovery of variants associated to the
disease, that usually account for a small portion of the trait heritability
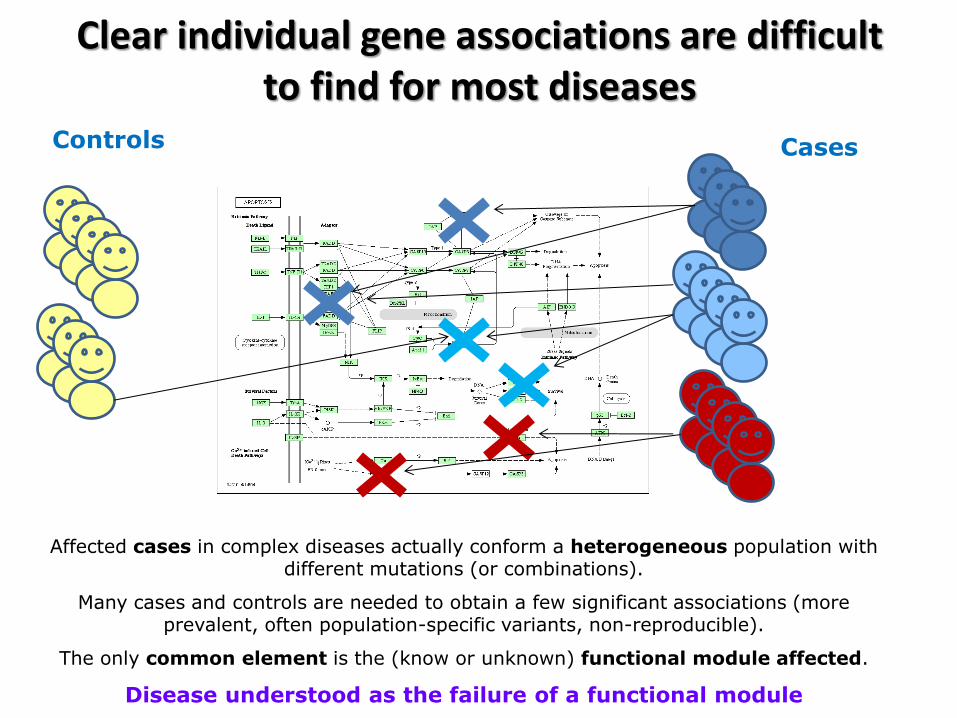
Clear individual gene associations are difficult to find for most diseases
Affected cases in complex diseases actually conform a heterogeneous population with different mutations (or combinations).
Many cases and controls are needed to obtain a few significant associations (more prevalent, often population-specific variants, non-reproducible).
The only common element is the (know or unknown) functional module affected.
Disease understood as the failure of a functional module
Cases Controls
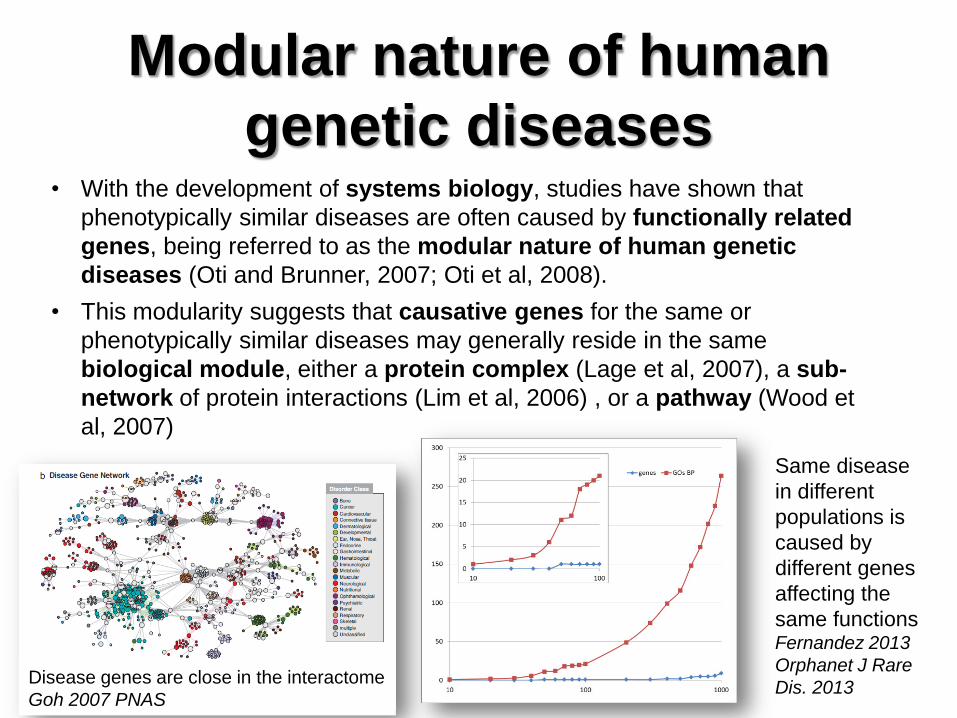
Modular nature of human
genetic diseases • With the development of systems biology, studies have shown that
phenotypically similar diseases are often caused by functionally related
genes, being referred to as the modular nature of human genetic
diseases (Oti and Brunner, 2007; Oti et al, 2008).
• This modularity suggests that causative genes for the same or
phenotypically similar diseases may generally reside in the same
biological module, either a protein complex (Lage et al, 2007), a sub-
network of protein interactions (Lim et al, 2006) , or a pathway (Wood et
al, 2007)
Disease genes are close in the interactome
Goh 2007 PNAS
Same disease
in different
populations is
caused by
different genes
affecting the
same functions Fernandez 2013
Orphanet J Rare
Dis. 2013
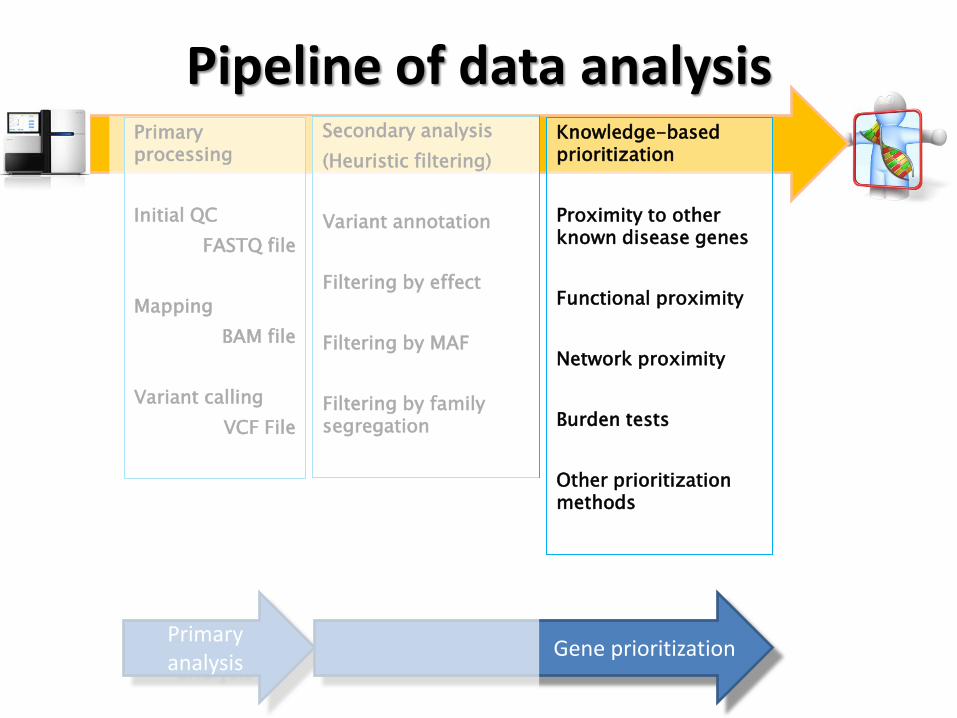
Pipeline of data analysis Primary processing
Initial QC
FASTQ file
Mapping
BAM file
Variant calling
VCF File
Knowledge-based prioritization
Proximity to other known disease genes
Functional proximity
Network proximity
Burden tests
Other prioritization methods
Secondary analysis
(Heuristic filtering)
Variant annotation
Filtering by effect
Filtering by MAF
Filtering by family segregation
Primary analysis
Gene prioritization
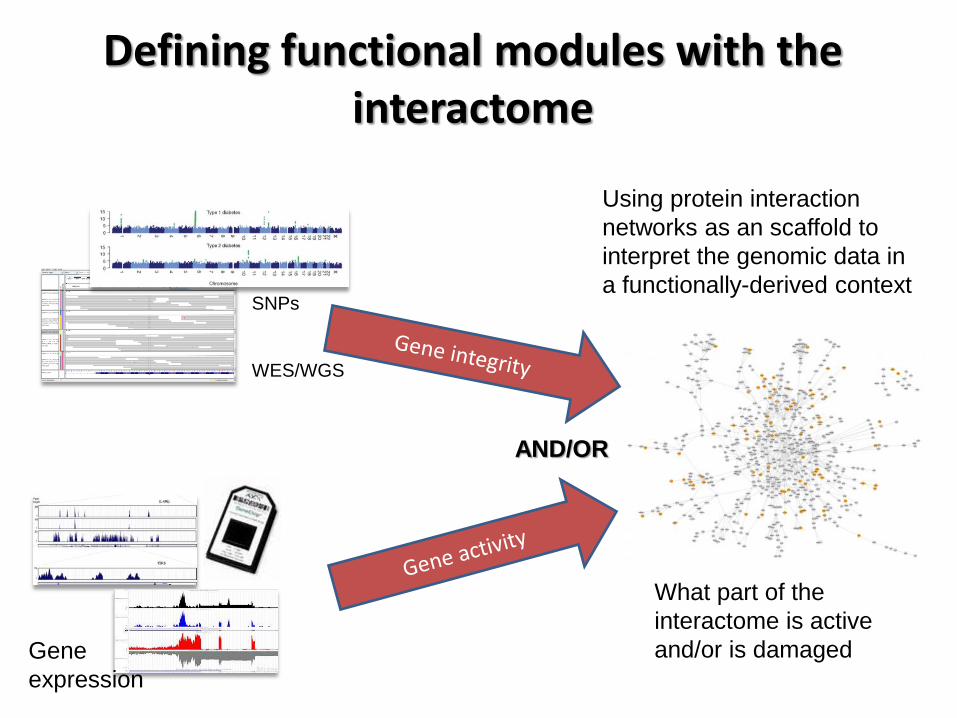
Defining functional modules with the interactome
SNPs
WES/WGS
Gene
expression
Using protein interaction
networks as an scaffold to
interpret the genomic data in
a functionally-derived context
AND/OR
What part of the
interactome is active
and/or is damaged

CHRNA7 (rs2175886 p = 0.000607)
IQGAP2 (rs950643 p = 0.0003585)
DLC1 (rs1454947 p = 0.007526)
SNPs validated in independent cohorts
Network analysis helps to find disease genes
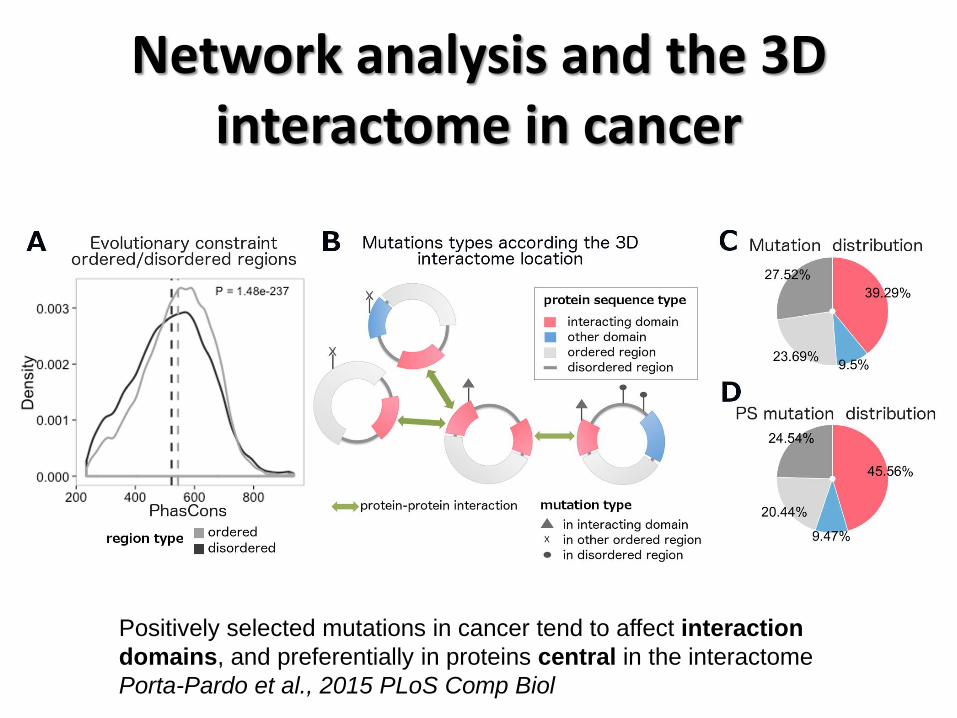
Network analysis and the 3D interactome in cancer
Positively selected mutations in cancer tend to affect interaction
domains, and preferentially in proteins central in the interactome
Porta-Pardo et al., 2015 PLoS Comp Biol
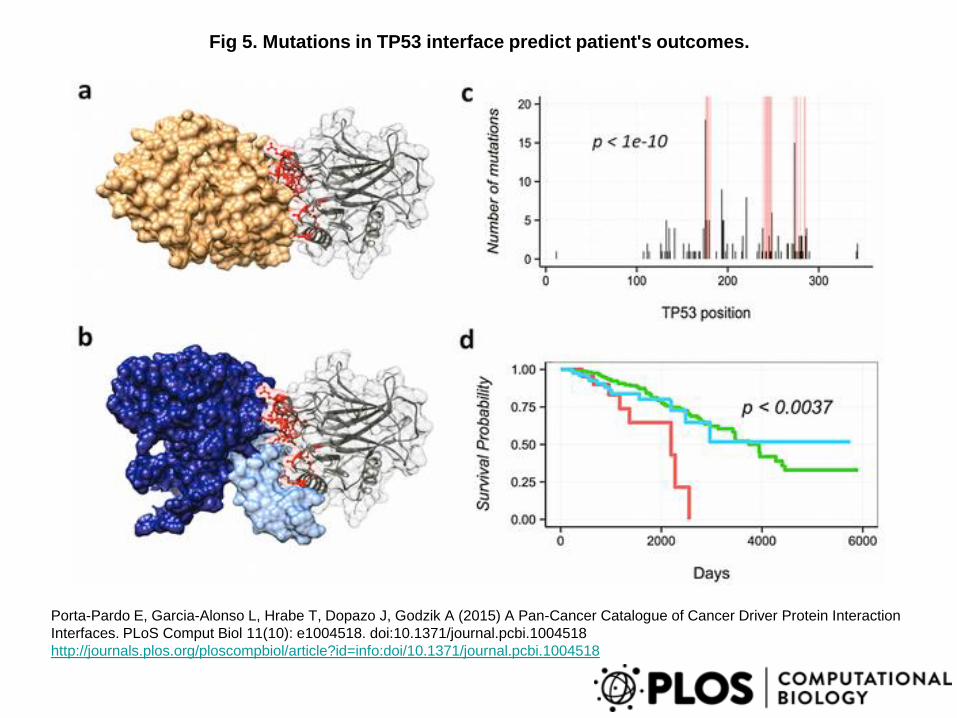
Fig 5. Mutations in TP53 interface predict patient's outcomes.
Porta-Pardo E, Garcia-Alonso L, Hrabe T, Dopazo J, Godzik A (2015) A Pan-Cancer Catalogue of Cancer Driver Protein Interaction
Interfaces. PLoS Comput Biol 11(10): e1004518. doi:10.1371/journal.pcbi.1004518
http://journals.plos.org/ploscompbiol/article?id=info:doi/10.1371/journal.pcbi.1004518
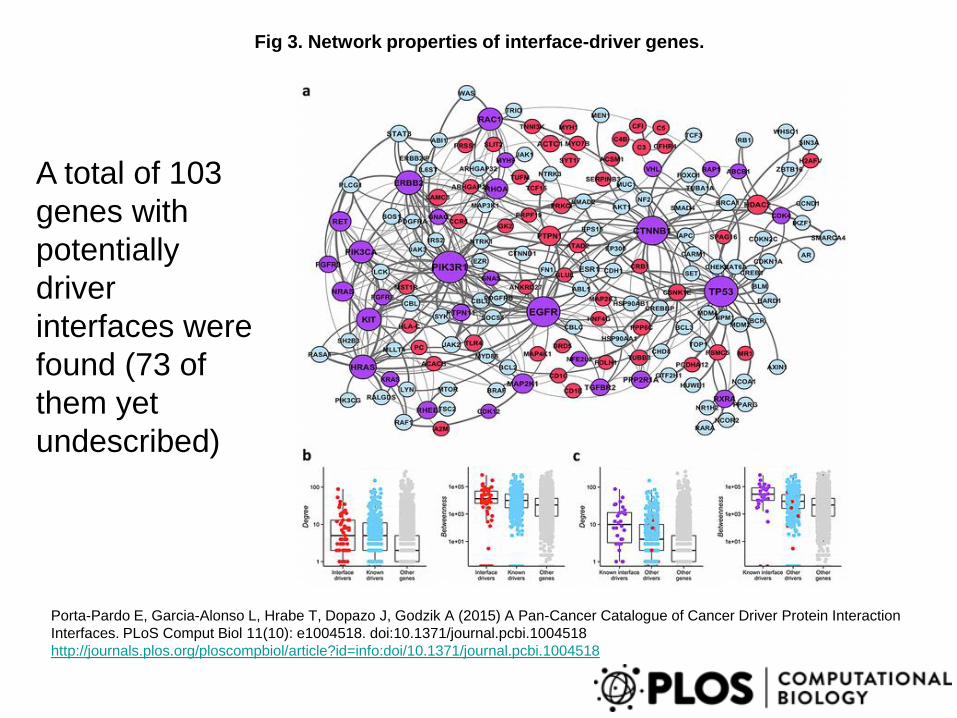
Fig 3. Network properties of interface-driver genes.
Porta-Pardo E, Garcia-Alonso L, Hrabe T, Dopazo J, Godzik A (2015) A Pan-Cancer Catalogue of Cancer Driver Protein Interaction
Interfaces. PLoS Comput Biol 11(10): e1004518. doi:10.1371/journal.pcbi.1004518
http://journals.plos.org/ploscompbiol/article?id=info:doi/10.1371/journal.pcbi.1004518
A total of 103
genes with
potentially
driver
interfaces were
found (73 of
them yet
undescribed)
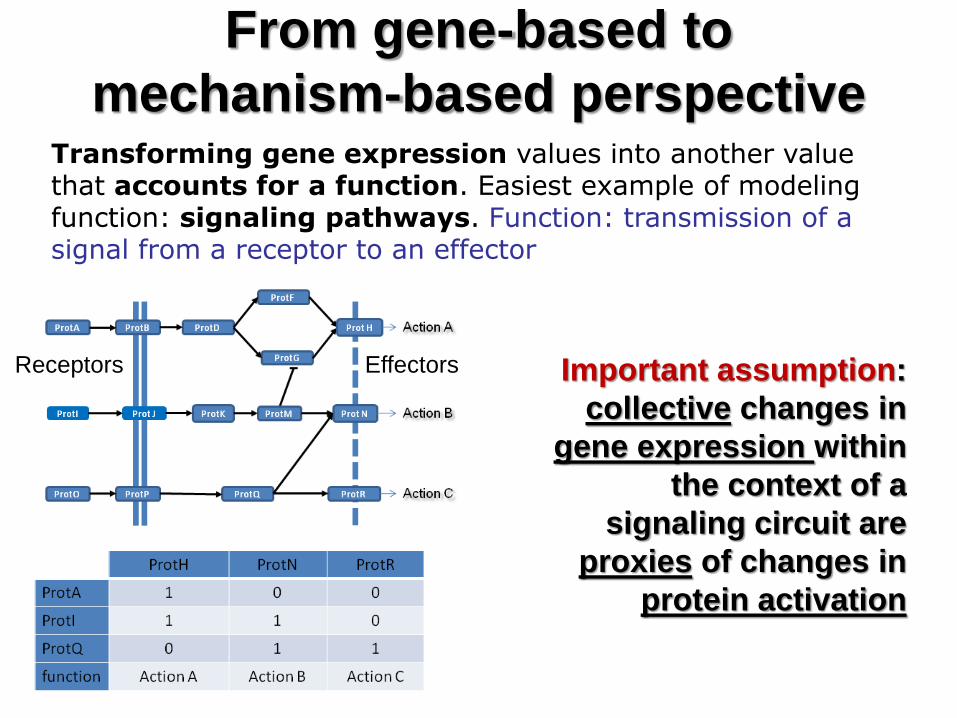
From gene-based to
mechanism-based perspective Transforming gene expression values into another value that accounts for a function. Easiest example of modeling function: signaling pathways. Function: transmission of a signal from a receptor to an effector
Receptors Effectors Important assumption:
collective changes in
gene expression within
the context of a
signaling circuit are
proxies of changes in
protein activation

What would you
predict about the
consequences of
gene activity changes
in the apoptosis
pathway in a case
control experiment of
colorectal cancer?
The figure shows the
gene up-regulations
(red) and down-
regulations (blue)
The effects of changes in gene
activity are not obvious
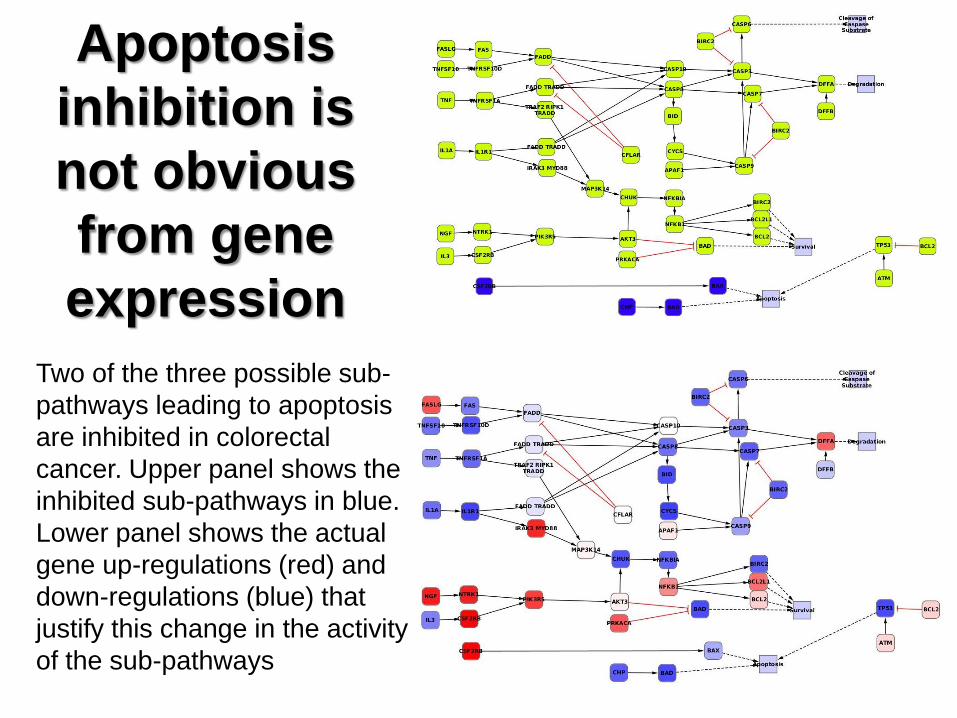
Apoptosis
inhibition is
not obvious
from gene
expression
Two of the three possible sub-
pathways leading to apoptosis
are inhibited in colorectal
cancer. Upper panel shows the
inhibited sub-pathways in blue.
Lower panel shows the actual
gene up-regulations (red) and
down-regulations (blue) that
justify this change in the activity
of the sub-pathways
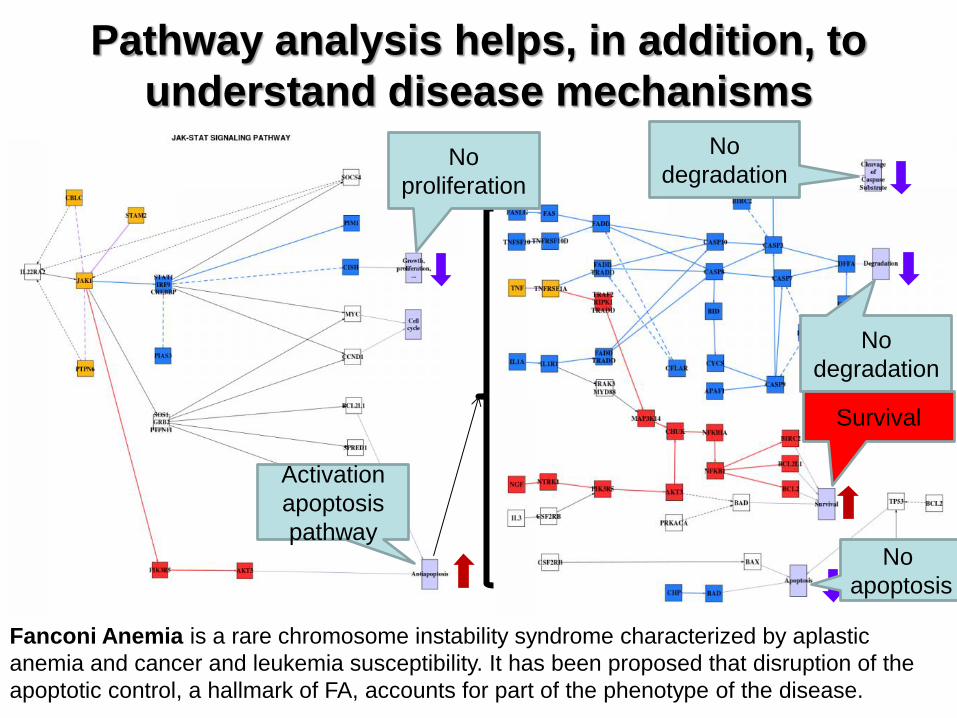
Pathway analysis helps, in addition, to
understand disease mechanisms
Fanconi Anemia is a rare chromosome instability syndrome characterized by aplastic
anemia and cancer and leukemia susceptibility. It has been proposed that disruption of the
apoptotic control, a hallmark of FA, accounts for part of the phenotype of the disease.
No
proliferation
No
degradation
Survival
No
degradation
No
apoptosis
Activation
apoptosis
pathway
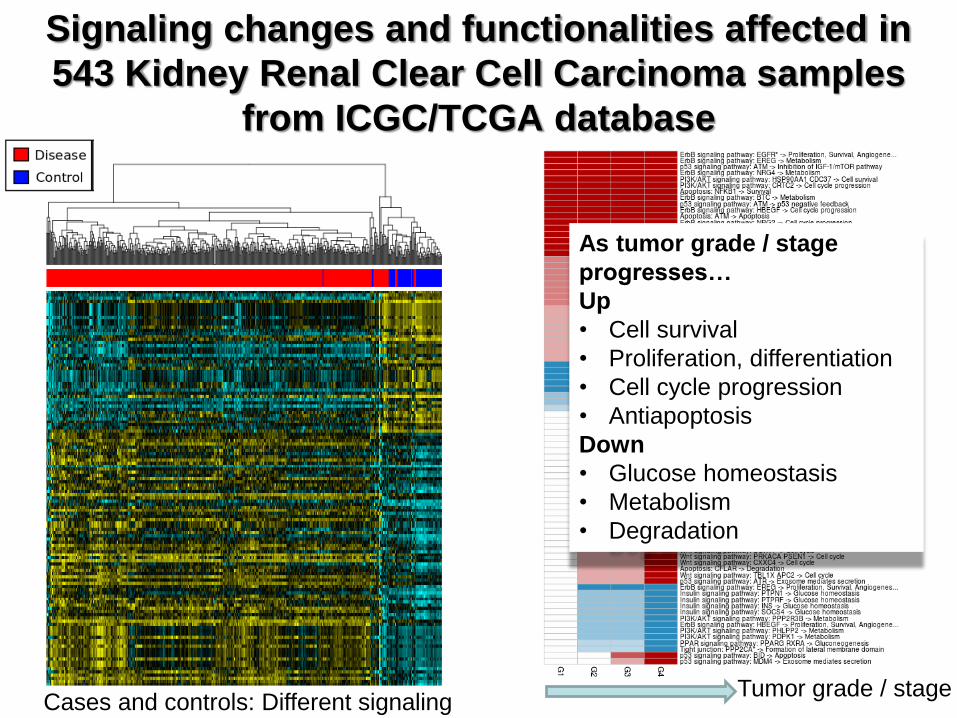
Cases and controls: Different signaling
As tumor grade / stage
progresses…
Up
• Cell survival
• Proliferation, differentiation
• Cell cycle progression
• Antiapoptosis
Down
• Glucose homeostasis
• Metabolism
• Degradation
Tumor grade / stage
Signaling changes and functionalities affected in
543 Kidney Renal Clear Cell Carcinoma samples
from ICGC/TCGA database
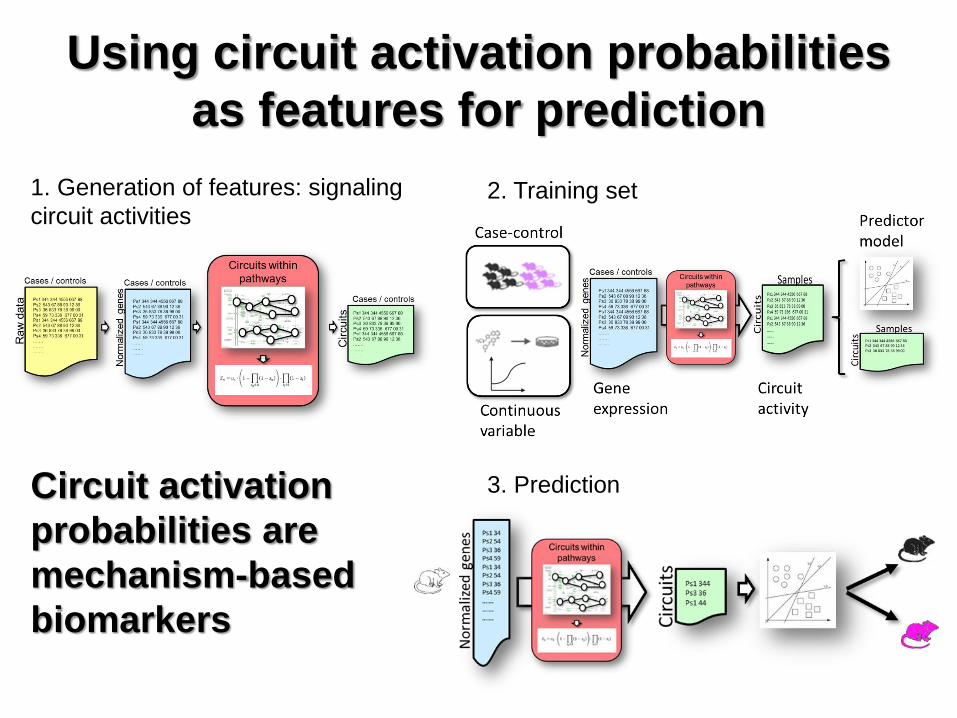
Using circuit activation probabilities
as features for prediction
Circuit activation
probabilities are
mechanism-based
biomarkers
1. Generation of features: signaling
circuit activities 2. Training set
3. Prediction
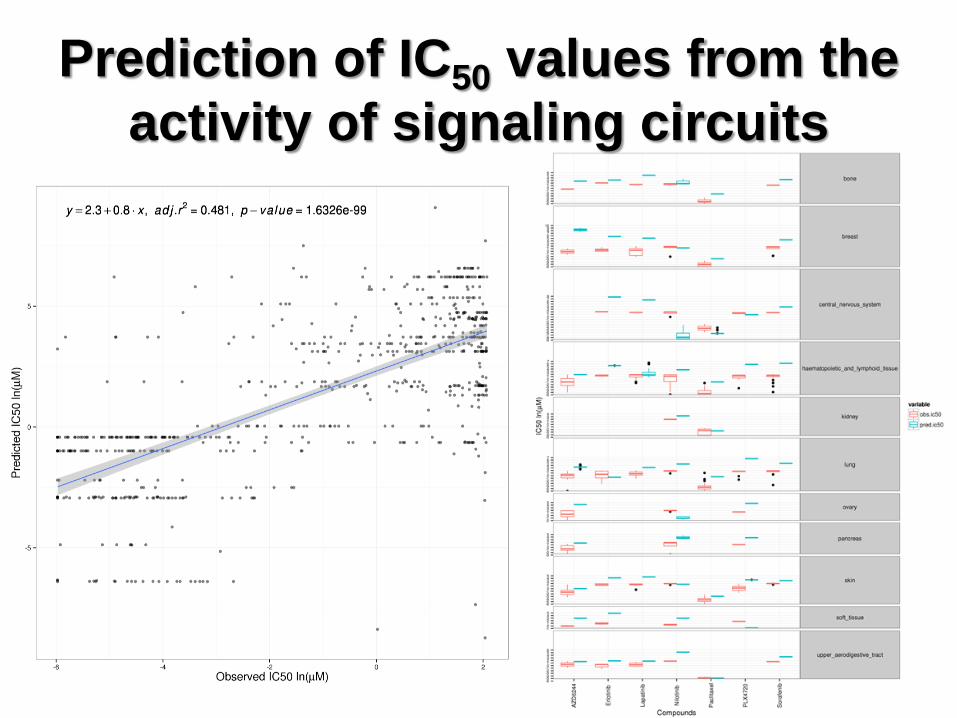
Prediction of IC50 values from the
activity of signaling circuits
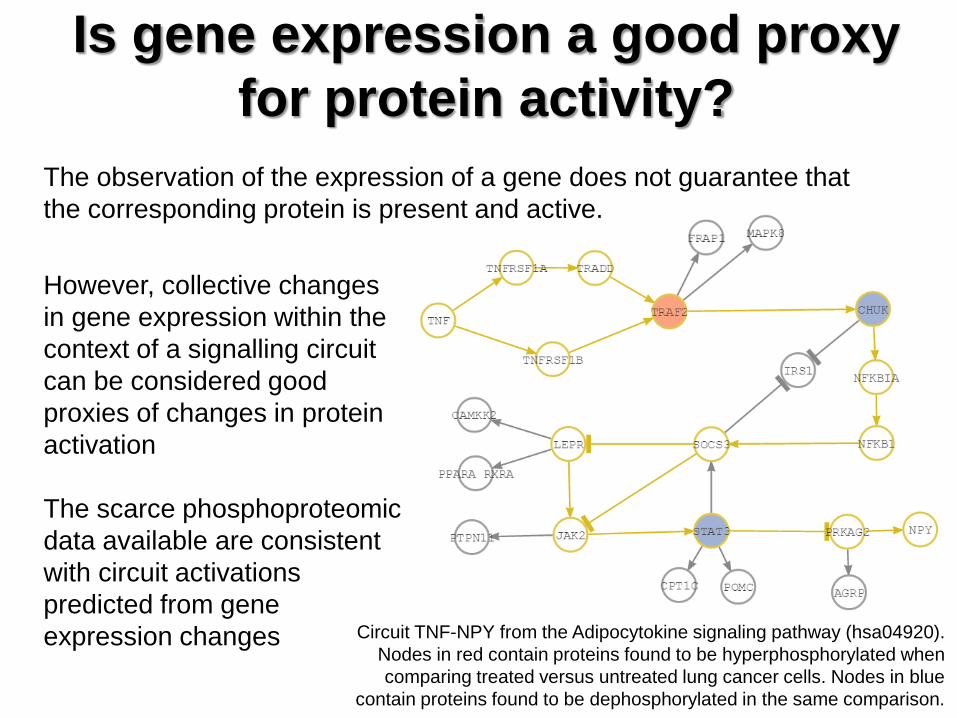
Is gene expression a good proxy
for protein activity?
However, collective changes
in gene expression within the
context of a signalling circuit
can be considered good
proxies of changes in protein
activation
The scarce phosphoproteomic
data available are consistent
with circuit activations
predicted from gene
expression changes
The observation of the expression of a gene does not guarantee that
the corresponding protein is present and active.
Circuit TNF-NPY from the Adipocytokine signaling pathway (hsa04920).
Nodes in red contain proteins found to be hyperphosphorylated when
comparing treated versus untreated lung cancer cells. Nodes in blue
contain proteins found to be dephosphorylated in the same comparison.
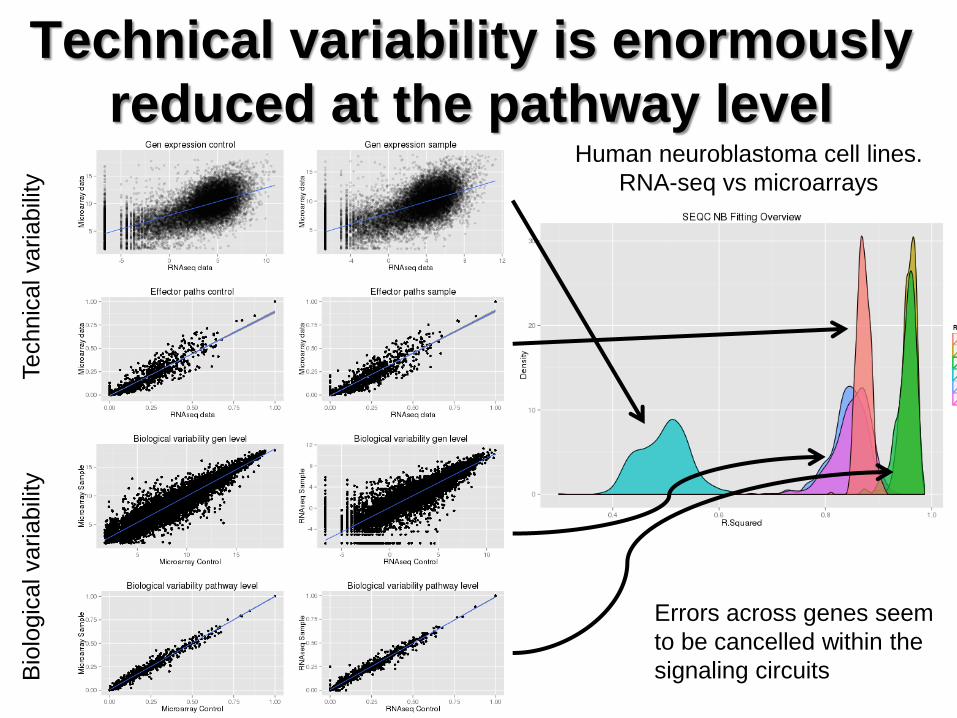
Bio
logic
al variabili
ty
Technic
al variabili
ty
Technical variability is enormously
reduced at the pathway level Human neuroblastoma cell lines.
RNA-seq vs microarrays
Errors across genes seem
to be cancelled within the
signaling circuits
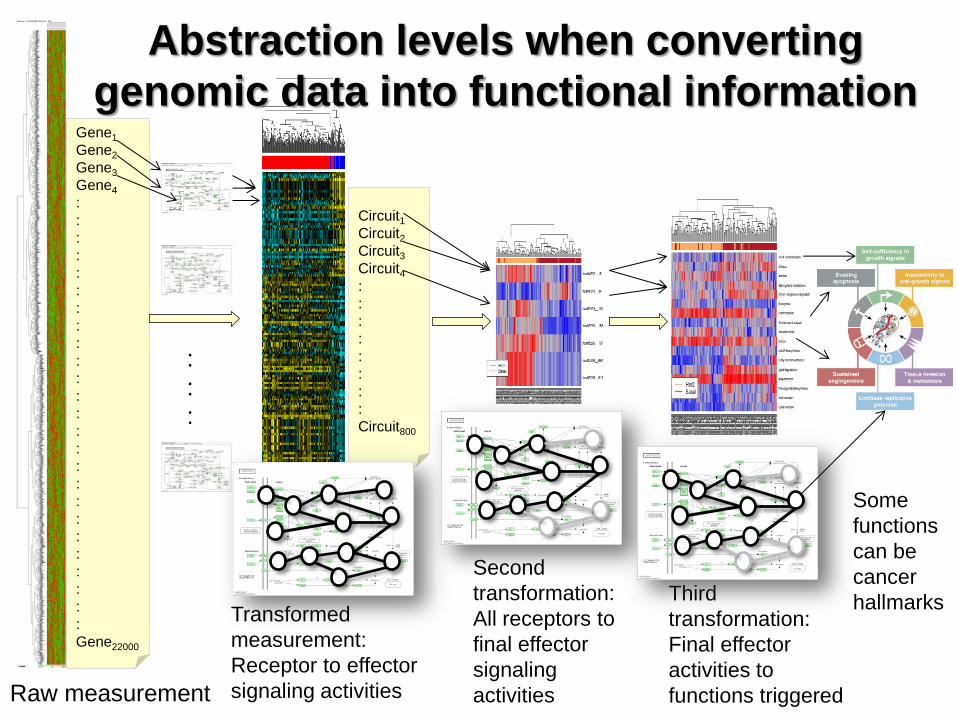
Gene1
Gene2
Gene3
Gene4
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
:
Gene22000
Raw measurement
Transformed
measurement:
Receptor to effector
signaling activities
Circuit1
Circuit2
Circuit3
Circuit4
:
:
:
:
:
:
:
:
Circuit800
:
:
:
Abstraction levels when converting
genomic data into functional information
Second
transformation:
All receptors to
final effector
signaling
activities
Third
transformation:
Final effector
activities to
functions triggered
Some
functions
can be
cancer
hallmarks
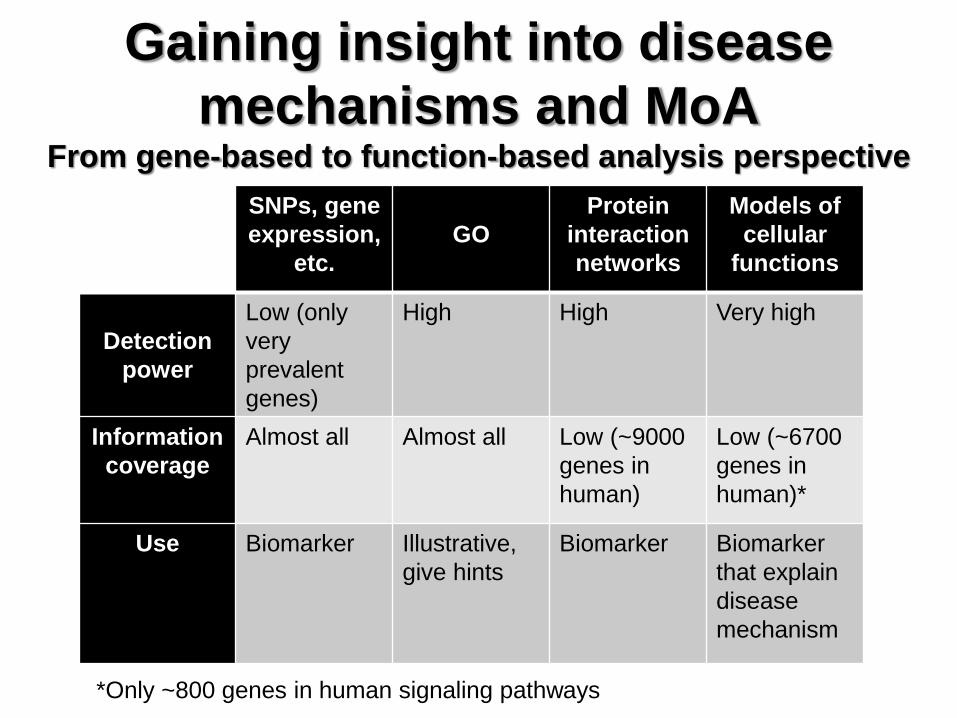
Gaining insight into disease
mechanisms and MoA From gene-based to function-based analysis perspective
SNPs, gene
expression,
etc.
GO
Protein
interaction
networks
Models of
cellular
functions
Detection
power
Low (only
very
prevalent
genes)
High High Very high
Information
coverage
Almost all Almost all Low (~9000
genes in
human)
Low (~6700
genes in
human)*
Use Biomarker Illustrative,
give hints
Biomarker Biomarker
that explain
disease
mechanism
*Only ~800 genes in human signaling pathways
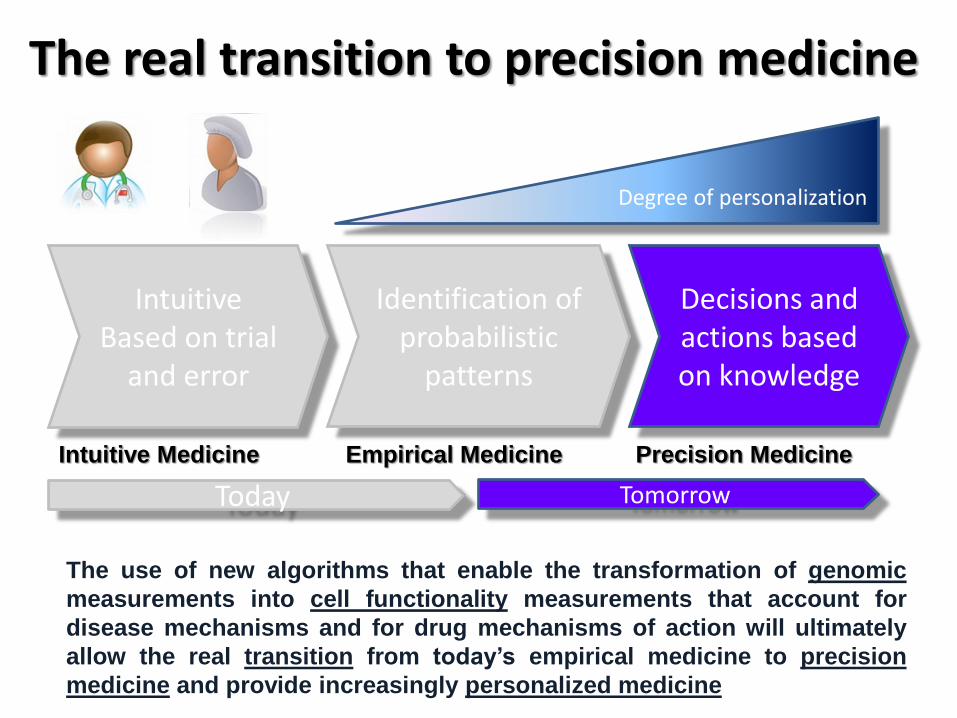
The use of new algorithms that enable the transformation of genomic
measurements into cell functionality measurements that account for
disease mechanisms and for drug mechanisms of action will ultimately
allow the real transition from today’s empirical medicine to precision
medicine and provide increasingly personalized medicine
The real transition to precision medicine
Intuitive Based on trial
and error
Identification of probabilistic
patterns
Decisions and actions based on knowledge
Intuitive Medicine Empirical Medicine Precision Medicine
Today Tomorrow
Degree of personalization
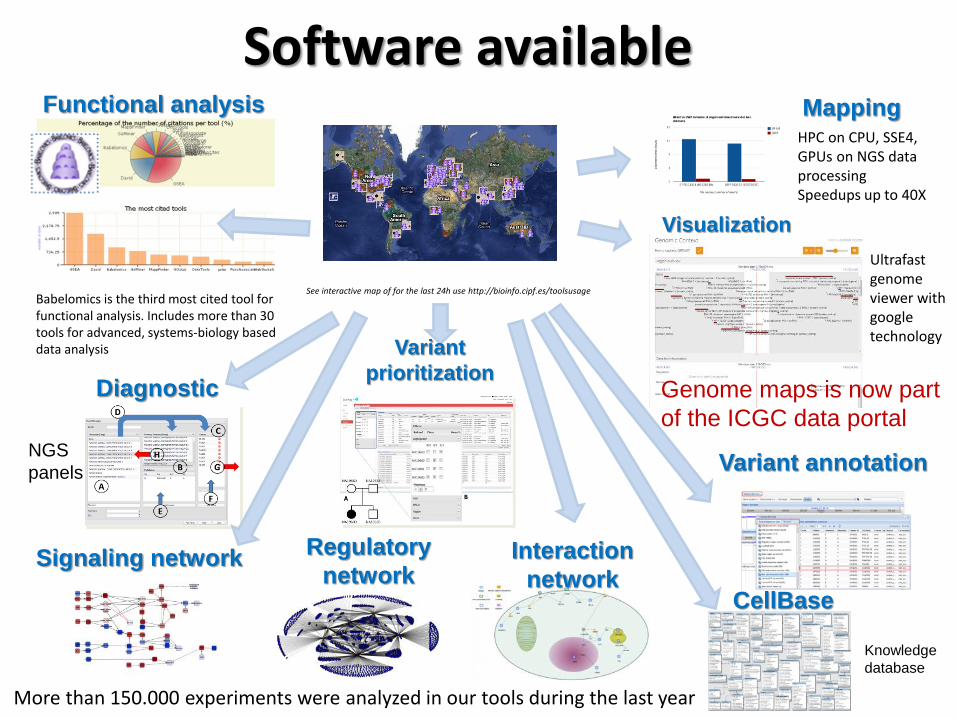
Software available
See interactive map of for the last 24h use http://bioinfo.cipf.es/toolsusage Babelomics is the third most cited tool for functional analysis. Includes more than 30 tools for advanced, systems-biology based data analysis
More than 150.000 experiments were analyzed in our tools during the last year
HPC on CPU, SSE4, GPUs on NGS data processing Speedups up to 40X
Genome maps is now part
of the ICGC data portal
Ultrafast genome viewer with google technology
Mapping
Visualization
Functional analysis
Variant annotation
CellBase
Knowledge
database
Variant
prioritization
NGS
panels
Signaling network Regulatory
network Interaction
network
Diagnostic

The Computational Genomics Department at the
Centro de Investigación Príncipe Felipe (CIPF),
Valencia, Spain, and… ...the INB, National
Institute of Bioinformatics
(Functional Genomics Node)
and the BiER (CIBERER Network of
Centers for Rare Diseases)
@xdopazo
@bioinfocipf
Follow us on twitter