Embed Size (px)
Citation preview

REVISING THE COMMON RULE: ANPRM to Final Rule and Beyond
HOLLY FERNANDEZ LYNCH JANUARY 23, 2017

FEDERAL POLICY FOR THE PROTECTION OF HUMAN SUBJECTS • Adopted in 1991 • The “Common Rule”
• Shared regulations governing human subjects research conducted/supported by 18 fed departments and agencies
• Codified in separate regulations for each • HHS: 45 CFR Part 46 • FDA rules are NOT part of the Common Rule
• But similar at 21 CFR Part 50 and Part 56 • Likely to become more harmonized after 21st Century Cures
(authority to waive consent in certain cases) • Covers IRBs, informed consent

ANPRM – JULY 2011 • “Enhancing Protections for
Research Subjects and Reducing Burden, Delay, and Ambiguity for Investigators”
• Motivation: Human subjects research landscape changed dramatically since 1980s • More research • More multi-site studies • New technologies – especially
genomics and informatics • New “scandals” • Major changes proposed re:
research with specimens, multisite studies, and “excluded” research

NPRM – SEPTEMBER 2015 • 1,142 comments received on ANPRM (but NPRM largely the same) • Intervening changes:
• NIH proposed single IRB review for its studies • SUPPORT controversy over standard of care research/risk • Newborn blood spots defined as HSR by statute
• Rationale: • HSR has grown in scale and diversity, especially use of repositories
(biospecimens and data), research in clinical care settings • Risks shifting from physical to informational
• Data privacy and security • Ability to reidentify data/specimens
• “[P]eople want to be asked for their permission”

NPRM – PRIMARY PROPOSALS • Revised definition of HSR to include all biospecimen research • New consent approaches to secondary research use of
biospecimens and identifiable private information • Standardized privacy safeguards for research w/ specimens and IPI • New research categories:
• Excluded = no regulatory requirements or review process • Exempt = determined through IRB review or investigator tool (but still
some privacy safeguards, documentation reqs) • Non-exempt = full regs + privacy safeguards
• Improved informed consent (organization and elements) • IRB accountability and streamlined review, including single IRB • Apply Common Rule to all clinical trials conducted at US institutions
receiving federal research funding

NPRM RESPONSE – PUBLIC COMMENTS • 2,186 received (not necessarily representative) • Concern about complexity and length of NPRM, lack of key
components for review and comment (e.g., broad consent template) • Most comments received re: biospecimen provisions
• Strong majority in opposition, across all subgroups (patients, public, research organizations) – but different reasons
• Mixed reaction on single IRB as mandate • Institutions generally opposed, individuals generally in favor
• Concern re: complexity of approaches to excluded/exempt research • Generally positive reactions on certain approaches to improving IC,
eliminating some continuing review, extending CR coverage, and improved privacy protections
• Several comments asked for a new NPRM, rather than finalization
Source: Lauren Hartsmith, OHRP, NPRM Update: Summary of Public Comments, May 2016

FINAL RULE – BREAKING NEWS! • Published FR Jan. 19, 2017 • Effective date: Jan. 19, 2018
(except single IRB – 1/20/20) • Major changes from what was
originally proposed (based on public comments)
• Major changes to status quo in several areas: • Research with data and
specimens • Informed consent • Exemption categories • IRB review
Or is it?

RESEARCH WITH DATA AND SPECIMENS

RESEARCH WITH DATA AND SPECIMENS Final result: • Maintain status quo keeping secondary research with
nonidentified data and specimens outside HSR definition • Proposal had been based on apparent public sentiment,
but public comment strongly opposed • Treat research with data and specimens consistently • Introduce option for broad consent for secondary
research use of identified private information/identified biospecimens • And retain realistic possibility of waiving consent
• Provide more information about secondary research uses as part of consent process
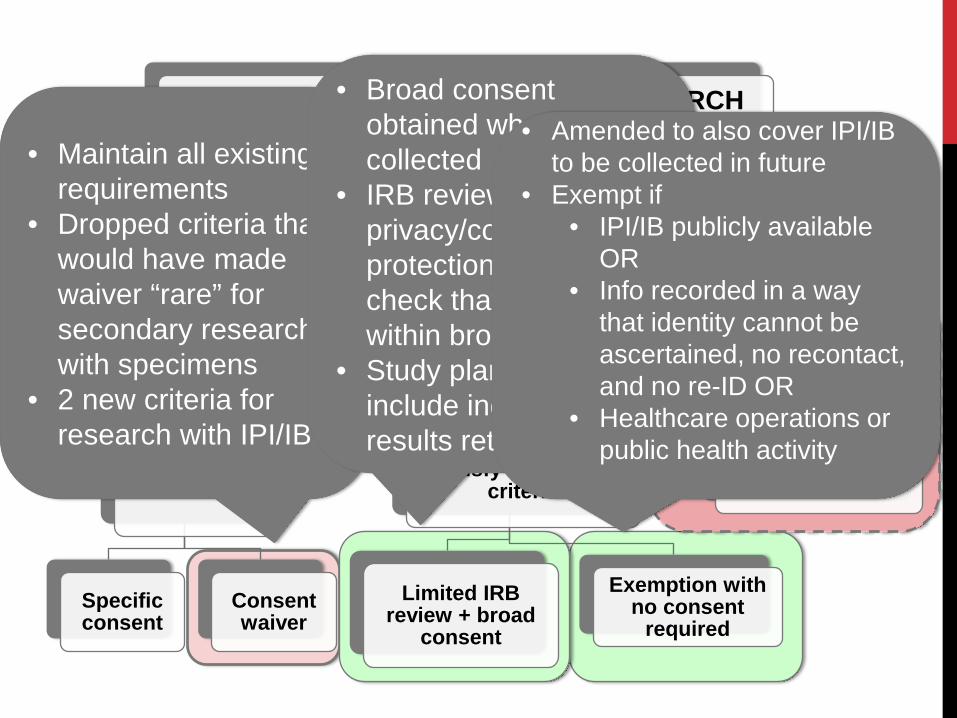
SCOPE OF POSSIBILITIES FOR RESEARCH WITH DATA AND SPECIMENS
Collected for THIS research
IRB review/consen
t (standard)
Secondary research
Identified/identifiable
IRB review
Specific consent
Consent waiver
Satisfy exemption criteria
Limited IRB review + broad
consent
Exemption with no consent
required
Nonidentified
NOT HSR (no IRB review/consent)
• Maintain all existing requirements
• Dropped criteria that would have made waiver “rare” for secondary research with specimens
• 2 new criteria for research with IPI/IB
• Broad consent obtained when IPI/IB collected
• IRB reviews for privacy/confidentiality protection and to check that research is within broad consent
• Study plan does not include individual results return
• Amended to also cover IPI/IB to be collected in future
• Exempt if • IPI/IB publicly available
OR • Info recorded in a way
that identity cannot be ascertained, no recontact, and no re-ID OR
• Healthcare operations or public health activity

RESEARCH WITH DATA AND SPECIMENS • Definition of identifiable continues to matter – BUT UNSTABLE
• Regulation requires reexamination of meaning of “identifiable biospecimen” and “identifiable private information” w/I 1 year and every 4 years after to account for technological developments
• “If appropriate and permitted by law, such Federal departments and agencies may alter the interpretation of these terms, including though the use of guidance.”
• Guidance is not subject to notice and comment • Similar process to assess whether there are new
technologies/techniques that should be considered to generate IPI/IB when applied to otherwise nonidentified information/specimens
• Create a list via notice and comment • UPSHOT: more activities could be deemed identifiable later –
could become more difficult to do research with data/specimens • Should we care about identifiability per se OR risk?

RESEARCH WITH DATA AND SPECIMENS People were surprised to learn what was happening with their specimens/data, so: • (1) New research consent elements – part of standard consent
• Identifiers might be removed and info/specimens used/shared for future studies without new consent (OR research info/specimens will not be used/shared for future research, with or w/o identifiers)
• Specimens may be used for commercial profit (and if subject will share in profit)
• Whether clinically relevant research results will be returned, and under what conditions
• Whether research will or might include whole genome sequencing

RESEARCH WITH DATA AND SPECIMENS • (2) Broad consent for future use of IPI/IB
• Obtained when data/specimens collected in clinical care or research • Optional – just needed for exemption
• Content: • Some standard elements • General description of types of research that may be conducted • General description of types of IPI/IB that might be used • Whether sharing of IPI/IB might occur • Types of institutions or researchers • Period of time for storage/use (could be indefinite) • Subject will not be provided details about specific future uses, and
could include some things they would have chosen not to consent to • Results will not be returned (unless they will be in all circumstances) • Who to contact with questions or if harmed
• No template provided

RESEARCH WITH DATA AND SPECIMENS • Doesn’t cover ALL surprises re: use of clinical specimens/data:
• Would not have had research consent – so no notice of deidentified use
• May not have had broad consent • Could still be deidentified and used (not HSR) • Could be used with identifiers under waiver of consent or
exemption • Does improve info for people whose specimens/data are used:
• After being collected in research • After being collected with broad consent
• Some disincentives to seeking broad consent • Infrastructure needed to track • If you ask and people decline, can’t get a consent waiver for them

NEW CONSENT CHANGES – BEYOND DATA/SPECIMENS • Goal: improved decisionmaking
• Provide information that a “reasonable person” would want to have to make a decision about whether to participate
• Begin with “concise and focused” presentation of key information
• Do not just present list of isolated facts – organization to facilitate understanding of reasons why one might or might not participate
• Will require guidance from OHRP as to how to do this

NEW CONSENT CHANGES • Must post consent document on public government
website for federally funded “clinical trials” • After trial is closed to recruitment and within 60 days after
last study visit • Agency supporting or conducting trial can make
exceptions if certain information should not be made publicly available • Can permit or require redactions, e.g., of confidential
commercial information

EXEMPT RESEARCH • No category of “excluded” research, as proposed • Either not HSR, exempt, exempt with limited IRB review for
privacy/confidentiality protection, or non-exempt • Important changes for social/behavioral research (among others):
• Surveys/interviews and benign behavioral interventions in adults with prospective agreement to participate exempt if:
• Recorded in a way identity of subject cannot be readily ascertained • Disclosure of responses would not place subject at risk • Recorded in a way identity CAN be ascertained + IRB conducts
limited review • Scholarly and journalistic activities that focus directly on specific
individuals about whom the info is collected is deemed not HSR • Also exemptions re: secondary research with IPI/IB • May develop exemption determination tool in the future

IRB REVIEW • Single IRB review required for multi-site studies in the US
• Basically the same as new NIH policy • More flexible than what had been proposed because fed agency
supporting research can determine that single IRB is not appropriate for particular context (broad groups of studies rather than case by case determination)
• Single IRB selected by supporting agency or proposed by lead institution
• Reviewing and relying institutions must document respective responsibilities
• New authority to enforce regulations against stand-alone IRBs • Eliminated need for continuing review of some research

UPSHOTS, IMPLICATIONS, AND OPEN QUESTIONS • Status quo on nonidentified
specimens/info • For now…
• Additional options for identified specimens/info • Will broad consent option be useful?
• More information for public about how specimens/data are used for research • Will it be enough?
• Improved consent • Will it be more than platitude?
• Public posting of consent materials • What can we learn?
• Fewer restrictions on SBER • Single IRB review
• Will it really be better/more efficient?
What is still missing? (NAS 2016 Report) • Big data research • Cluster randomized
trials • Standard of care
research • Clinical innovation,
QA/QI
AND
Penalty for reidentification

NOW WHAT? • Once a rule is finalized, it takes a new rulemaking to change
• New notice-and-comment process • Identification of defensible reasons for repeal
• BUT…

NOW WHAT? • Congressional Review Act
• Congress always has the power to stop any rule it disapproves of • CRA provides streamlined process and bars agencies from
imposing a similar rule afterward • 60 legislative day period in which Congress can pass a resolution of
disapproval – and then president can sign or veto • Midnight Rules Relief Act – passed House 1/4/17
• Amendment to CRA to allow Congress to disapprove multiple rules at once
• REINS Act – passed House 1/5/17 • “Major” rules would need joint resolution of Congressional approval
within 70 session days to take effect • On list of rules House Freedom Caucus is looking to overturn in
first 100 days of Trump administration (pre-finalization) • BUT intervention less likely with output of final rule on specimens

WE WAIT…





























