Embed Size (px)
Citation preview

IDIOPATHIC/IMMUNE THROMBOCYTOPENIC
PURPURA
(ITP)
“Everybody bleeds sometimes,
I just bleed more………..”
Dr. Mohib Ali

• A 17 yr old adolescent is brought to the ER by her mother who is
concerned that she has bruised easily over the past few days and
has had a nose bleed this morning. There are no symptoms of other
illness and she is otherwise well. On examination there are
generalized petechiae and some bruising mainly affecting the
lower limbs. Blood testing reveals a platelet count of 35 * 109, but
the platelets are normal size. Hemoglobin is 11.1 g/dl and her white
blood cell count is normal. What diagnosis fits best with this clinical
picture ?
i. Immune thrombocytopenic purpura (ITP)
ii. Henoch Schonlein purpura
iii. Thrombotic thrombocytopenic purpura (TTP)
iv. Hemolytic Uremic Syndrome (HUS)
v. Generalized Bone Marrow Suppression

Platelets
• Platelets are released from the megakaryocyte.
• The normal blood platelet count is 150,000–450,000/L.
• The major regulator of platelet production is the hormone
thrombopoietin (TPO), which is synthesized in the liver.
• Synthesis is increased with inflammation and specifically
by interleukin 6.
• A reduction in platelet and megakaryocyte mass
increases the level of TPO, which then stimulates
platelet production.
• Platelets circulate with an average life span of 7 to 10
days.

• Thrombocytopenia can be broadly classified into five
categories based on the mechanism behind reduce
platelet count ;
– Pseudo or Spurious Thrombocytopenia
– Dilutional Thrombocytopenia
– Decreased Platelet Production
– Increased Platelet Destruction
– Altered Distribution of Platelets (Increased
Sequestraion)

Decreased
production of
platelets
Increased
destruction of
platelets
Increased
sequestration of
platelets
Other conditions
causing
thrombocytopenia
Congenital Bone
marrow failure(e.g.
fanconi anemia,
Wiskott-Aldrich
syndrome)
Immune
Thrombocytpenia
(including Hep C
& HIV related &
drug induced)
Hypersplenism
(e.g. related to
cirrhosis,
myeloproliferative
disorders,
lymphoma)
Gestational
thrombocytopenia
Acquired Bone
marrow failure (e.g.
Aplastic anemia,
myelodysplasia)
HIT , TMA , DIC ,
Post transfusion
purpura ,
Hemophagocytosi
s
Bernard-Soulier
syndrome , grey
platelet syndrome,
May-hegglin anomaly
Exposure to
chemotherapy,
irradiation
Neonatal
alloimmune
thrombocytopenia
Psudothrombocytope
nia
Marrow infiltration
(neoplastic,
infectious)
Von Willebrand
disease type2
Nutritional (def. of
vit.B12, folate, iron;
Mechanical (aortic
valvular

DRUGS THAT ARE STRONGLY ASSOCIATED
WITH THROMBOCYTOPENIA

ANTIBIOTICS AND
ANTIVIRALS
• Quinine/quinidine
• Penicillins
• Cephalosporins
• Vancomycin
• Trimethoprim/sulfamethoxazol
e
• Sulfonamides/sulfonylureas
• Linezolid
• Valacyclovir
• Ganciclovir
• Indinavir
CARDIOVASCULAR
MEDICATIONS
• Abciximab
• Tirofiban
• Eptifibatide
• Salicylates
• Digoxin
• Furosemide
MISCELLANEOUS
• Cimetidine
• Ranitidine
• Famotidine
• Valproate
• Interferon

Approach to the Thrombocytopenia Patient
• History
– Is the patient bleeding ?
– Do the sites of bleeding suggest a platelet defect ?
– Duration – Is thrombocytopenia acute or chronic ?
– Is there a history of medications, alcohol use, or recent
transfusion (post transfusion purpura) ?
– Are there any symptoms of a secondary illness ?
– (neoplasm , infection, autoimmune disease)
– Is there a family history of thrmobocytopenia ?
– Heparin exposure – recent or within past three months (HIT) ?
– Are there any risk factors for HIV infection ?
– History of liver disease ?

Algorithm for Thrombocytopenia
Evaluation

Platelet count < 150,000/mcl
Hb & WBC count
Normal Abnormal
Peripheral
Smear
Bone Marrow
Examination
Normal RBC
morphology;
platelets
normal or
increased in
size
Fragmented
red blood cells
Microangiopathic
hemolytic anemias
(e.g. DIC, TTP)
Consider:
Drug induced thrombocytopenia
Infection induced thrombocytopenia
Idiopathic immune thrombocytopenia
Congenital thrombocytopenia
Platelets clumped: Redraw in
Sodium citrate or Heparin

ITP
• Immune thrombocytopenic purpura (ITP; also
termed idiopathic thrombocytopenic purpura) is
an acquired disorder in which there is immune-
mediated destruction of platelets and possibly
inhibition of platelet release from the
megakaryocyte.

Clinical Presentation
• Excessive bleeding with minor injuries
• Spontaneous bleeding from mouth & nose
• Unexplainable or spontaneous bruising
• Ecchymoses and Petechiae
• Hematuria
• Menorrhagia
• Intracranial bleed
• Purpura
• Thrombocytopenia


• Which of the following conditions is not associated with
ITP ?
i. SLE
ii. HIV
iii. Tuberculosis
iv. CLL
v. Hep B & C

Classification
• Primary
• Secondary;
– Post-Infectious : HIV, HCV, CMV, H.Pylori
– Antiphospholipid syndrome
– SLE
– Autoimmune thrombocytopenia (e.g. Evan’s
syndrome)
– Lymphoproliferative Disorders : CLL , NHL , HD
– Common Variable Immune Deficiency
– Drug Induced ITP


Types of ITP
ITP can be divided into acute and chronic forms:
Acute ITP
• More commonly seen in children
• May follow an infection or vaccination
• Usually runs a self-limiting course over 1-2 weeks
Chronic ITP
• More common in young/middle-aged women
• Tends to run a relapsing-remitting course
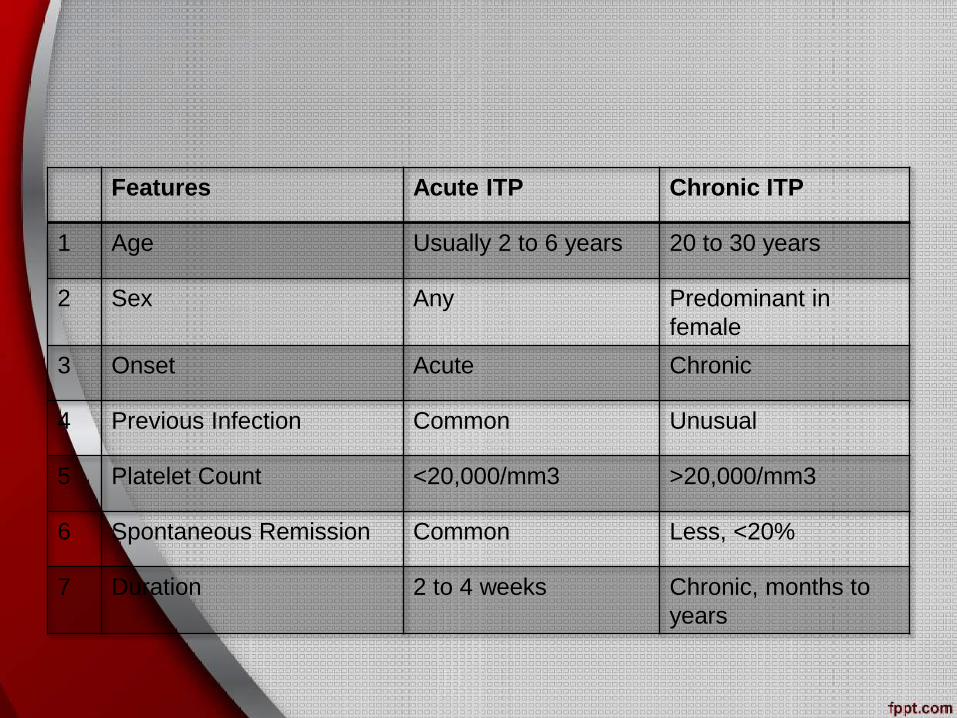
Features Acute ITP Chronic ITP
1 Age Usually 2 to 6 years 20 to 30 years
2 Sex Any Predominant in
female
3 Onset Acute Chronic
4 Previous Infection Common Unusual
5 Platelet Count <20,000/mm3 >20,000/mm3
6 Spontaneous Remission Common Less, <20%
7 Duration 2 to 4 weeks Chronic, months to
years

• In ITP , the autoantibodies are most commonly directed
at ?
A. Platelet activating factor
B. Glycoprotein IIb/IIIa complex
C. ATP receptor
D. Anti-thrombin III receptor
E. ADP Receptor

Pathophysiology
• Increased platelet destruction caused by antiplatelet
antibodies Antibodies directed against platelet
membrane antigens such as glycoprotein IIb/IIIa
The platelets coated with immune complexes bind to Fc
portion of macrophages in spleen and other RES and
are removed
• Lack of compensatory response by megakaryocytes due
to suppressive effect of antiplatelet antibodies
• So a combination of increased platelet destruction +
ineffective megakaryopoiesis

Diagnosis
• Careful and detailed history, clinical examination
• CBC
• Peripheral Smear
• PT , apTT
• LFTs , RFTs
• Hep B & C Serology
• ANA
• Antiphospholipid antibodies
• IgG levels to rule out CVID
• HIV , EBV , CMV Serology
• Bone Marrow Examination
• Nutrient workup (Iron, B12, folate )

When to do Bone Marrow Examination
• Bone marrow examination can be reserved for older
adults (usually >60 years) or
• those who have other signs or laboratory abnormalities
not explained by ITP, or
• in patients who do not respond to initial therapy.
• Megakaryocyte abnormalities and hypocellularity or
hypercellularity are not characteristic of ITP.

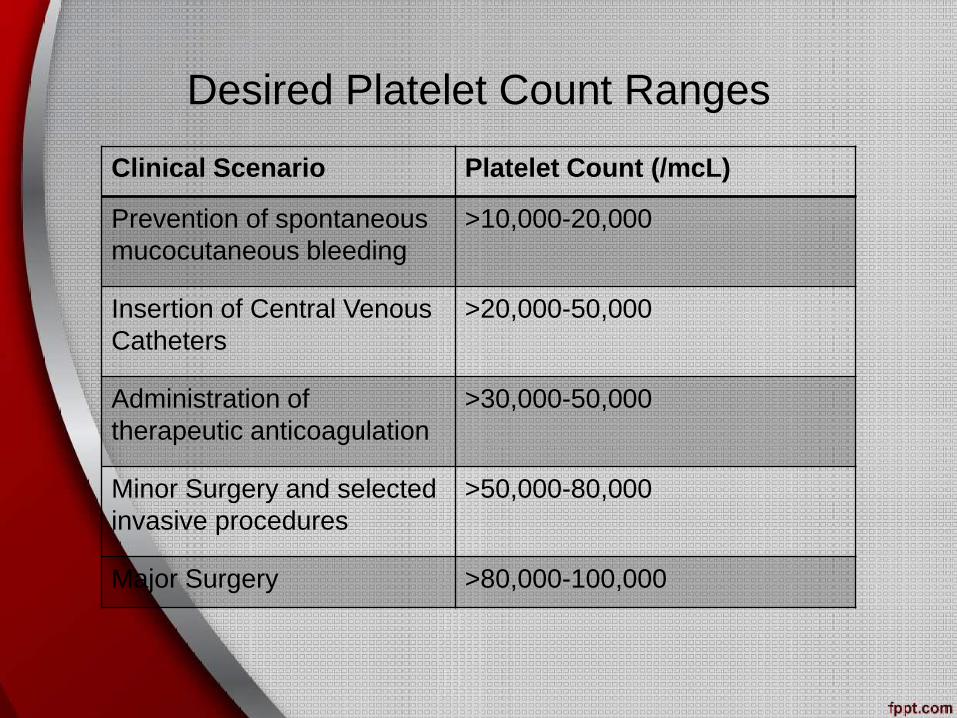
Desired Platelet Count Ranges
Clinical Scenario Platelet Count (/mcL)
Prevention of spontaneous
mucocutaneous bleeding
>10,000-20,000
Insertion of Central Venous
Catheters
>20,000-50,000
Administration of
therapeutic anticoagulation
>30,000-50,000
Minor Surgery and selected
invasive procedures
>50,000-80,000
Major Surgery >80,000-100,000

Some Facts
• ITP was originally described in 1735 by a German
physician, Paul Gottlieb Werlhof, and was therefore
known as Werlhof’s disease.
• In 1916, Paul Kaznelson reported the first successful
treatment for ITP after a patient showed a response to
splenectomy
• Splenectomy was then used as the first-line therapy for
ITP until 1950.

Management

Initial Treatment
Prednisone 1 mg/kg/d for 7-10
days followed by rapid taper
Or
Dexamethasone 40mg/d orally
for 4 days monthly for 6 months
IVIG, 1g/kg/d i/v for 2 days
Or
Anti-D, 75mcg/kg i/v for 1 dose
Platelets, if bleeding
+/-
+/-

Relapsed or Persistent ITP
Prednisone 1 mg/kg/d for 7-10 days followed by rapid taper
Or
Dexamethasone 40mg/d orally for 4 days monthly for 6
months
Rituximab
375mg/m2 i/v
weekly for 4
weeks
Anti-D
75mcg/kg i/v
serially as
needed for
platelets <
30,000/mcl
IVIG, 1g/kg/d
i/v for 2 days
serially as
needed for
platelets <
30,000/mcl
Thrombopoeiti
n Receptor
Agonist
Romiplostim
(s/c)
Eltrombopag
(orally)
Splenectomy
(Laparoscopic)
or or or
and

Persistent or Worsening ITP
Mycophenolate mofetil – Azathioprine/Danazol – Cyclosporine -
Chemotherapy
Enrollment in Clinical trial – Autologous transplantation
Splenectomy
(Laparoscopic)
Trial of additional agent(s) above

THERAPY FOR THE INITIAL
MANAGEMENT OF
IMMUNE THROMBOCYTOPENIC
PURPURA

ORAL PREDNISONE
• Effect is dose dependent—approximately 80% of
patients respond to 1 mg/kg/day.
• Toxicity also increases with dose and duration of
treatment and includes glucose intolerance,
immunosuppression, osteoporosis, and cataracts.
Relapse is typical once therapy is discontinued.

• Steroids are presumed to reduce the risk of symptoms in
ITP patients by:
– Reducing antibody production
– Reducing reticuloendothelial system phagocytosis of
antibody-coated platelets
– Improving vascular integrity
– Improving platelet production

INTRAVENOUS IMMUNOGLOBULIN (IVIG)
• More rapid than daily prednisone. Administered at a
dose of 1 g/kg/day for 2 consecutive days or 0.4g/kg/day
for 5 consecutive days.
• Response rates are approximately 80%, and effects
typically last 2-4 wk.
• Toxicity includes headache, allergic reactions, and,
rarely, thrombosis.

ANTI-D IMMUNOGLOBULIN
• Administered at a dose of 50-75 μg/kg IV.
• Response rates are dose dependent but can approach
75-80%.
• Hemolysis is a common toxicity. Rarely, hemolysis can
be life threatening and can be associated with
disseminated intravascular coagulation, renal failure, and
end-organ infarction.

THERAPY FOR THE MANAGEMENT
OF REFRACTORY IMMUNE
THROMBOCYTOPENIC PURPURA

ORAL PREDNISONE
• Effect is dose dependent and rapidly dissipates after
discontinuation of the medication. Some patients can be
maintained on a very low and tolerable daily dose (e.g.,
5 mg).
• Long-term use is associated with infections, diabetes,
osteoporosis, avascular necrosis, weight gain, and
cataracts.

ORAL DEXAMETHASONE
• Administered at a dose of 40 mg/day for 4 consecutive
days. Repeated every 2-4 wk for several months.
• Sustained response rates of 29-42% are possible.
• Toxicity is similar to that of oral prednisone.

SPLENECTOMY
• Durable (often lifelong), significant responses are seen in
65-70% of patients who undergo this procedure.
• Produces lifelong immunosuppression to encapsulated
and gram-positive organisms.
• American Society of Hematology guidelines recommend
waiting until at least 12 months after diagnosis, if
possible.
• When possible, surgery should be performed using
laparoscopic techniques.

When to vaccinate the patient before & after
splenectomy ?

• Patients should receive pneumococcal, Haemophilus
influenza type b, and meningococcal vaccination at least
2 weeks before the procedure.
• Post Splenectomy;
– Pneumococcal 5 yearly
– Meningococcal 3 yearly
– Hemophilus Influenza type b 1 yearly

• RITUXIMAB
• Given at a dose of 375 mg/m2/wk IV for a total of 4 wk.
Significant responses are seen in 28-44% of patients
and typically last for months.
• Toxicity includes reactivation of hepatitis B,
immunosuppression, and, rarely, progressive multifocal
leukoencephalopathy.

• For selected refractory patients, other
immunosuppressives, such as cyclophosphamide,
azathioprine, cyclosporine A, mycophenolate mofetil,
dapsone, interferon, and etanercept, can be used.

• THROMBOPOIETIN RECEPTOR AGONISTS
• Administered daily (eltrombopag) or weekly
(romiplostim).
• An effect is typically seen in 2-3 weeks and disappears
a few weeks after discontinuation of the medication.
• Toxicity from long-term use is not well known but may
include excessive thrombosis and bone marrow fibrosis.


ITP in Pregnancy
• The goal of management of pregnancy-associated ITP is
a platelet count of 10,000–30,000/mcL in the first
trimester,
• > 30,000/mcL during the second or third trimester, and
• > 50,000/mcL prior to cesarean section or vaginal
delivery.
• Moderate-dose oral prednisone or intermittent infusions
of IVIG
are standard.
• Splenectomy is reserved for failure to respond to these

ITP with HIV & HCV
• For thrombocytopenia associated with HIV or hepatitis C
virus, treatment of either infection leads to an
amelioration in the platelet count in most cases;
• Refractory thrombocytopenia may be treated with
infusion of IVIG or anti-D (HIV and hepatitis C virus),
splenectomy (HIV), or interferon-alpha or eltrombopag
(hepatitis C virus, including eradication).
• Treatment with corticosteroids is not recommended in
hepatitis C virus infection.

Response after Initial Treatment
(Steroids)
• Complete Responders
– Platelet count > 100,000
• Partial Responders
– Platelet count 50,000-100,000
• Non Responders
– Platelet count < 50,000

New Terminologies in ITP
i. Complete Response
ii. Partial response
iii. No response
iv. Loss of complete response
v. Loss of response

• Complete response
– A platelet count of >/= 100 * 109/L measured on two occasions
more than 7 days apart and the absence of bleeding.
• Partial response
– A platelet count of >/= 30 * 109/L and a > 2fold increase in
platelet count from baseline measured on two occasions more
than 7 days apart and the absence of bleeding.
• No response
– A platelet count of < 30 * 109/L or a < 2 fold increase in platelet
count from baseline or the presence of bleeding. Platelet count
must be measured on two occasions more than a day apart.

• Loss of complete response
– A platelet count of < 100 * 109/L measured on two occasions
more than a day apart and/or the presence of bleeding.
• Loss of response
– A platelet count of < 30 * 109/L or < 2 fold increase in platelet
count from baseline or the presence of bleeding. Platelet count
must be measured on two occasions more than a day apart.

• A 40-year old lady , 3 years post splenectomy for chronic ITP
presents with a petechial rash and gum bleeding . Her blood count
shows;
– WBC of 6.3 *109 /L
– Hb of 11.5 g/dL
– platlet count of 3*109 /L
• What would be the most appropriate next treatment modality
provided ?
A. Thrombopoietin agonist ( eltrombopag or romiplostim)
B. Rituximab
C. Cyclosporine
D. Mycophenolate mofetil
E. All of the above are acceptable, treatment should be patient-
centered as regard to choice of the immunosuppressive agent

Thank You





























