Embed Size (px)
Citation preview

bioexcel.eu
QM/MM approaches in CPMD
Presenters: Emiliano IppolitiHost: Adam Carter
BioExcel Educational Webinar Series #5
30 June, 2016
bioexcel.eu
This webinar is being recorded

bioexcel.eu
BioExcel Overview• Excellence in Biomolecular Software
- Improve the performance, efficiency and scalability of key codes
• Excellence in Usability- Devise efficient workflow environments
with associated data integration
• Excellence in Consultancy and Training- Promote best practices and train end users
DMI Monitor
DMI Enactor
DMI Executor
DMI Enactor
Data Delivery Point
Data Source
Monitoring flow
Data flow
Service Invocation
DMI Optimiser
DMI Planner
DMIValidator
DMI Gateway
DMI Gateway
DMI Gateway
DMI Enactor
Portal / Workbench
DMI Request
DADC Engineer
DMI Expert
Repository
Registry
DMI Expert
Domain Expert

bioexcel.eu
Interest Groups
• Integrative Modeling IG• Free Energy Calculations IG• Best practices for performance tuning IG• Hybrid methods for biomolecular systems IG• Biomolecular simulations entry level users IG• Practical applications for industry IG
Support platformshttp://bioexcel.eu/contact
Forums Code Repositories Chat channel Video Channel

bioexcel.eu
Today’s Presenter
Emiliano Ippoliti is a research associate in the Institute of Computational Biomedicine of ForschungszentrumJülich (Germany). He took his PhD in Theoretical Physics at the University of Trieste in 2005 with a thesis about the theoretical foundations of quantum mechanics. From 2006 to 2009 he was a postdoctoral fellow at the International School for Advanced Studies in Trieste where he became expert on predicting optical properties for biologically relevant systems. Then, he moved to the German Research School for Simulation Sciences in Aachen/Jülichas a research associate until 2014 where he applied diverse enhanced sampling techniques to biological systems. His current interests focus on HPC approaches and methodologies for systems of biological relevance, in particular classical and ab initio molecular dynamics.
5

bioexcel.eu6
QM/MM approaches in CPMD
BioExcel webinar 30/06/2016
Emiliano IppolitiForschungszentrum Jülich (Germany)

bioexcel.eu
Quantum DescriptionThere are cases where the dynamical behavior of the electrons of your systems cannot be neglected and a quantum mechanical description is required:
1. Chemical reactions (e.g. enzymatic reactions): electron transfer, bond breaking and bond formation
2. Systems with metal atoms (e.g. metallo proteins): no universal parametrization for metal atoms
3. Proton transport (e.g. aerobic generation of ATP and oxygen):Grotthuss mechanism (charge/topological diffusion vs mass diffusion)
4. Spectroscopic analysis and prediction (e.g. absorption and fluorescence spectra)
7

bioexcel.eu
CPMD
CPMD is an highly efficient parallel ab initioelectronic structure and molecular dynamics
program
Plane wave/pseudopotential implementation of
Density Functional Theory (DFT)
• Car-‐Parrinello MD• Born-‐Oppenheimer MD• EhrenfestMD
8
www.cpmd.org

bioexcel.eu
Additional Features
9
• Works with norm conserving or ultrasoft pseudopotentials• LDA, LSD and the most popular gradient correction schemes; free
energy density functional implementation• Isolated systems and system with periodic boundary conditions• Molecular and crystal symmetry• Wavefunction optimization: direct minimization and diagonalization• Geometry optimization: local optimization and simulated annealing• Molecular dynamics ensembles: NVE, NVT, NPT• Response functions• Excited states• Many electronic properties• Time-‐dependent DFT (excitations, molecular dynamics in excited states)• Coarse-‐grained non-‐Markovianmetadynamics• Path integral MD
bioexcel.eu
Scalability
10
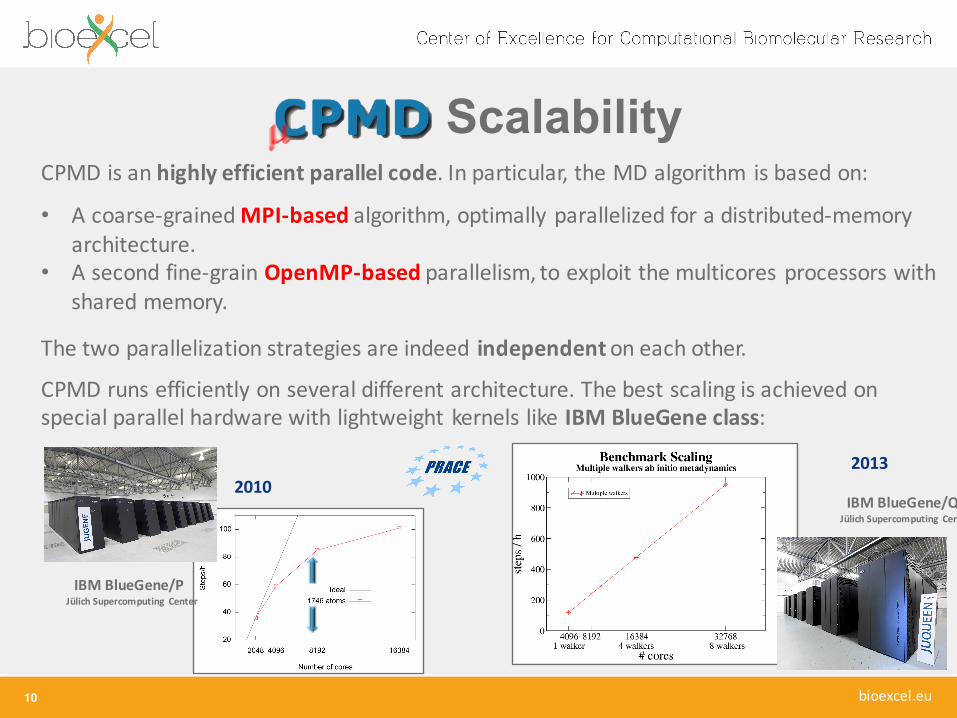
CPMD is an highly efficient parallel code. In particular, the MD algorithm is based on:
• A coarse-‐grained MPI-‐based algorithm, optimally parallelized for a distributed-‐memory architecture.
• A second fine-‐grain OpenMP-‐based parallelism, to exploit the multicores processors with shared memory.
The two parallelization strategies are indeed independenton each other.
CPMD runs efficiently on several different architecture. The best scaling is achieved on special parallel hardware with lightweight kernels like IBM BlueGene class:
IBM BlueGene/P Jülich Supercomputing Center
20102013
IBM BlueGene/Q Jülich Supercomputing Center

bioexcel.eu
QM/MM Approach
11
The system is separated into two parts:
A small QM part comprises the chemically/photophysically active region treated by computationally demanding electronic structure methods.
The remainder MM part is described efficiently at a lower level of theory by classical force fields.

bioexcel.eu
Extends the range of applicability to the
much larger biologically relevant systems
12
QM/MM interfaces in/for CPMD developed during the years
2005: Gromacs 3.3/CPMD interface
M. Eichinger, P. Tavan, J. Hutter, and M. Parrinello, J. Chem. Phys. 110, 10452 (1999)
2002: CPMD/Gromos96 interface
2013: IPHIGENIE/CPMD interface
2002: CPMD/Gromos96 interface
1999: EGO/CPMD interface
A. Laio, J. VandeVondele, and U. Röthlisberger J. Chem. Phys. 116, 6941 (2002)
P. K. Biswas, V. Gogonea, J. Chem. Phys. 123,164114 (2005)
M. Schwörer, B. Breitenfeld, P. Tröster, S. Bauer, K. Lorenzen, P. Tavan, and G.Mathias, J. Chem. Phys., 138, 244103 (2013)
bioexcel.eu
QM/MM Coupling
13
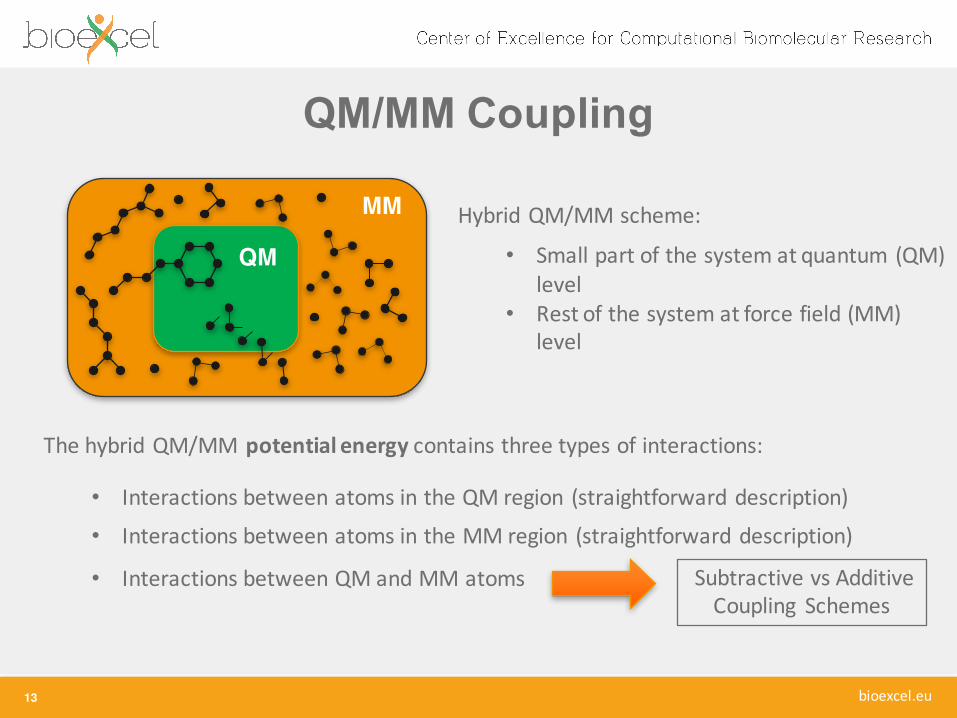
MM
QMHybrid QM/MM scheme:
• Small part of the system at quantum (QM) level
• Rest of the system at force field (MM) level
The hybrid QM/MM potential energy contains three types of interactions:
• Interactions between atoms in the QM region (straightforward description)
• Interactions between atoms in the MM region (straightforward description)
• Interactions between QM and MM atoms Subtractive vs AdditiveCoupling Schemes
bioexcel.eu
Subtractive Scheme
14
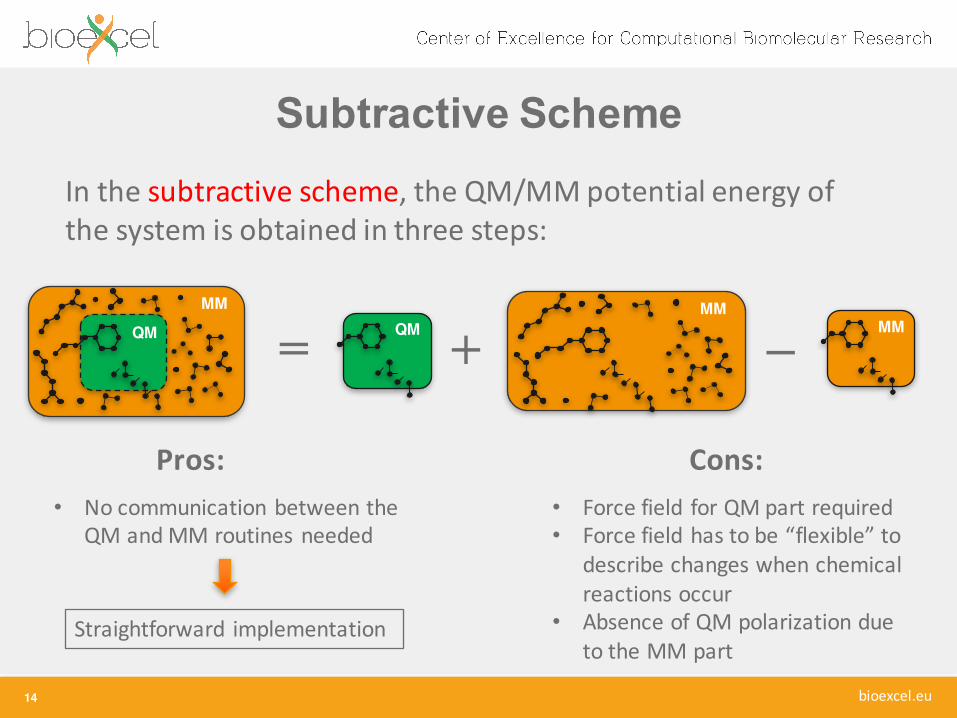
= + −MM
QM QMMM
MM
In the subtractive scheme, the QM/MM potential energy of the system is obtained in three steps:
Pros: Cons:• No communication between the
QM and MM routines needed
Straightforward implementation
• Force field for QM part required• Force field has to be “flexible” to
describe changes when chemical reactions occur
• Absence of QM polarization due to the MM part

bioexcel.eu
CPMD/Gromos96 Coupling
15
In the more advanced additive scheme, the QM/MM potential energy of the whole system is a sum of the MM energy terms, QM energy terms and QM/MM coupling terms:
EQM / MM = EQM Rα{ }( )+ E MM RI{ }( )+ EQM−MM Rα{ }, RI{ }( )
From the DFT Kohn-‐Sham Hamiltonian
H eKS From a Gromos96 or
an Amber-‐like force field
EQM−MM = Ebonded
QM−MM + Enon−bondedQM−MM !=
EQM−MM = Ebonded
QM−MM + Enon−bondedQM−MM
bioexcel.eu
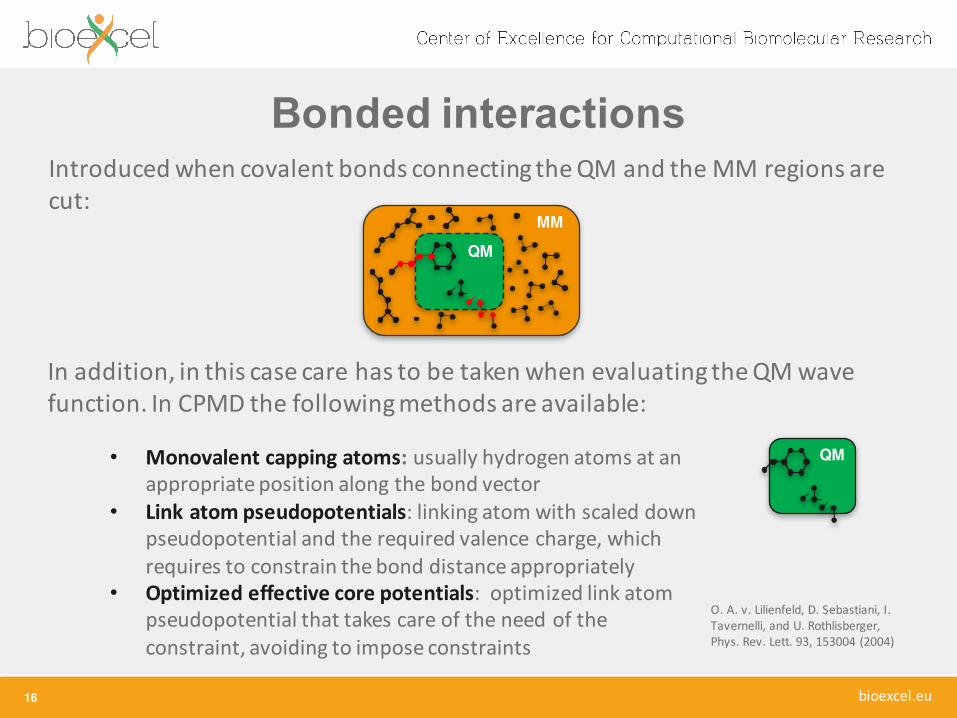
Bonded interactions
16
Introduced when covalent bonds connecting theQM and the MM regions are cut:
MM
QM
In addition, in this case care has to be taken when evaluating the QM wave function. In CPMD the following methods are available:
QM• Monovalent capping atoms: usually hydrogen atoms at an appropriate position along the bond vector
• Link atom pseudopotentials: linking atom with scaled down pseudopotential and the required valence charge, which requires to constrain the bond distance appropriately
• Optimized effective core potentials: optimized link atom pseudopotential that takes care of the need of the constraint, avoiding to impose constraints
O. A. v. Lilienfeld, D. Sebastiani, I. Tavernelli, and U. Rothlisberger, Phys. Rev. Lett. 93, 153004 (2004)

bioexcel.eu
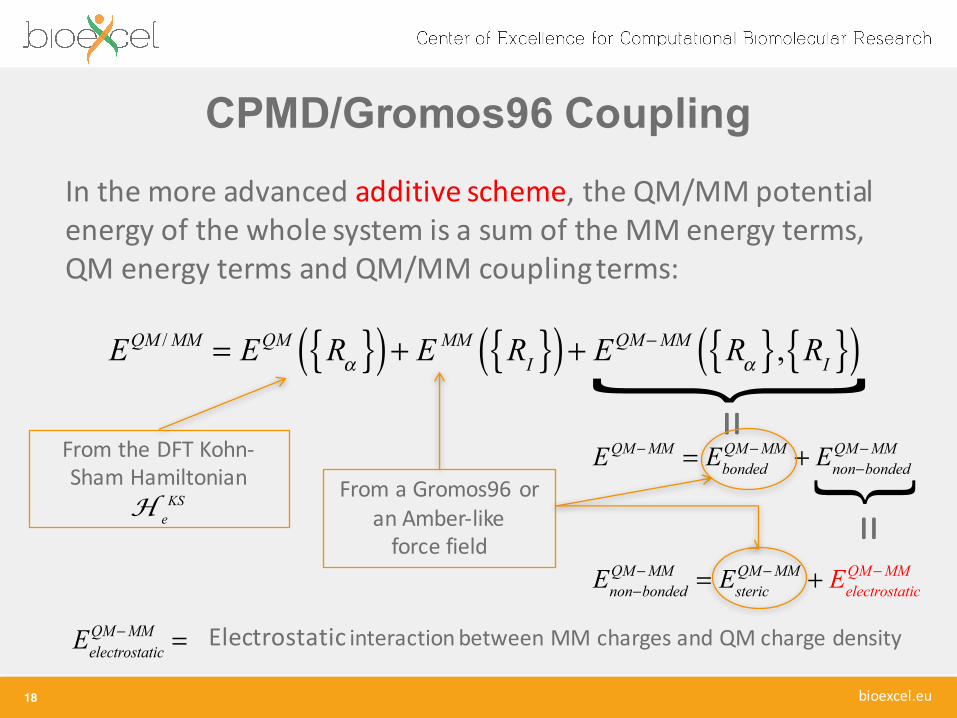
CPMD/Gromos96 Coupling
17
In the more advanced additive scheme, the QM/MM potential energy of the whole system is a sum of the MM energy terms, QM energy terms and QM/MM coupling terms:
EQM / MM = EQM Rα{ }( )+ E MM RI{ }( )+ EQM−MM Rα{ }, RI{ }( )
From the DFT Kohn-‐Sham Hamiltonian
H eKS From a Gromos96 or
an Amber-‐like force field
EQM−MM = Ebonded
QM−MM + Enon−bondedQM−MM
=
Enon−bondedQM−MM = Esteric
QM−MM + EelectrostaticQM−MM ! !=
bioexcel.eu
CPMD/Gromos96 Coupling
18
In the more advanced additive scheme, the QM/MM potential energy of the whole system is a sum of the MM energy terms, QM energy terms and QM/MM coupling terms:
EQM / MM = EQM Rα{ }( )+ E MM RI{ }( )+ EQM−MM Rα{ }, RI{ }( )
From the DFT Kohn-‐Sham Hamiltonian
H eKS From a Gromos96 or
an Amber-‐like force field
EQM−MM = Ebonded
QM−MM + Enon−bondedQM−MM
=
Enon−bondedQM−MM = Esteric
QM−MM + EelectrostaticQM−MM ! !=
Electrostatic interaction between MM charges and QM charge density EelectrostaticQM−MM =

bioexcel.eu
Electrostatic Coupling Schemes
19
• Mechanical embedding: no influence of MM charges on the QM part, i.e QM calculation is gas-‐phase-‐like without additional potential due to the MM atoms.
• Electrostatic embedding: QM polarization due to the MM part included.
• Polarized embedding: MM polarization due to the QM part included as well(non-‐self consistently or fully self-‐consistently).
EelectrostaticQM−MM
EelectrostaticQM−MM
• Neglected• Established by assigning a fixed set of effective charges to the
QM region (for example, those given by the force field) • QM partial charges re-‐compued at every integration step of the
simulation (least-‐squares fitting procedure to reproduce
Estimated through an additional term to the QM Hamiltonian, where it is taken into account in terms of an external charge distribution.

bioexcel.eu
Electrostatic Embedding
20
• Electrostatic interactions between QM and MM subsystems are handled during the computation of the electronic wave function
• The charged MM atoms enter the QM Hamiltonian as one-‐electron operators:
Eelectrostatic
QM−MM = −qIe
ri − R II∈MM∑
i∈QM∑ ⎯→⎯ qI
ρ(r)r − R I
d∫I∈MM∑ r
• Thus, the electrons “see” the MM atoms as special nuclei with non-‐integer and possibly negative charge
Is the polarization induced by qI realistic?
Electron spill-‐out problem at the bondaries

bioexcel.eu
Is the polarization induced by qI realistic?
21
• qI have been parametrized to provide a realistic description of an MM systemrather than of a physically correct charge distribution.
• In principle, one would need to re-‐derive the charges for use in QM/MM frameworks
• In practice, most researchers use default force field parameters.
• In force fields, only the combination of atomic charges and Lennard-‐Jones parametersprovides a reasonable description of all the relevant effects (including polarization, dispersion, Pauli repulsion, etc) taken together: part of the interaction due to polarization of the QM region is thus already accounted for by the Lennard-‐Jones potential.
Ideally not only qI, but also the Lennard-‐Jones parameters would need to be reparametrized for usage in electrostatic
embedding QM/MM simulations

bioexcel.eu
Electron Spill-out
22
A further problem that may arise when using standard MM atomic chargesto describe the charge distribution in the MM system, is the risk of over-‐polarization near the boundary:
The point charges on the MM side of the interface may attract (or repel) the electrons too strongly
Electron density spilling out into the MM region
QMMM
QM/MM boundary
+
Such artefacts can become serious if large flexible basis set (e.g., withpolarization and diffuse functions), or as in our case plane waves are usedin theQM calculations

bioexcel.eu
Electron Spill-out
23
Eelectrostatic
QM−MM = qI
ρ(r)υ Ismear r − R I( )r − R I
d∫I∈MM∑ r
The electron spill-‐out can be avoided by using smeared-‐out charges insteadof the traditional point charges:
where the smearing function can be a Gaussian distribution or anothersuitable function centered at the MM atom. In the CPMD/Gromos96scheme:
υ I
smear r( ) = rc,I4 − r 4
rc,I5 − r5 r rc,I = covalent radius of the atom I
In contrast to the point charge model, the Coulomb interaction between theQM electrons and the smeared charge distributions does not diverge if theelectrons approach theMM atoms.
bioexcel.eu
Flavors of QM/MM Schemes
24
QM/MM methods
Subtractive schemeAdditive scheme
Mechanicalembedding
Electrostatic embedding
Polarizedembedding
Fixed partial charges
Non self consistent
Full selfconsistent
Recalculated partial charges
EelectrostaticQM−MM = 0
CPMD/Gromos96
Point charge model
Smeared-‐outcharge model

bioexcel.eu
Electrostatic Embedding in theCPMD/Gromos96 Interface
25
• Hierarchical approach to deal with electrostatic interactions in combination with an empirical modification of the short-‐range terms that does not require a refit of the MM force field used and that overcomes the computational bottleneck in computing the integral term of the interacting Hamiltonian within a plane-‐wave scheme.
• Electrostatic embedding tailored to study dynamics of complex biomolecular systems and chemical reactions
• “Fully” Hamiltonian formulation from which the additional external electrostatic potential to be included in the Kohn-‐Sham Hamiltonian and the one acting onto the MM atoms as results of the QM charge distribution can be obtained consistently.
bioexcel.eu
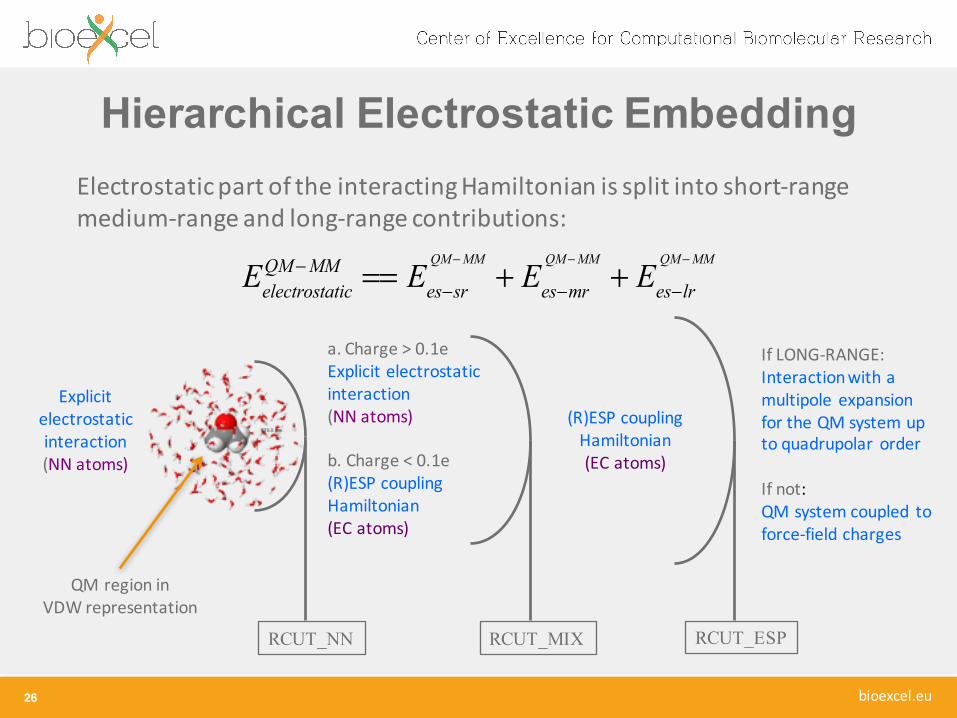
Hierarchical Electrostatic Embedding
26
Electrostatic part of the interacting Hamiltonian is split into short-‐range medium-‐range and long-‐range contributions:
EelectrostaticQM−MM == Ees−sr
QM−MM
+ Ees−mr
QM−MM
+ Ees−lr
QM−MM
Explicit electrostatic interaction(NN atoms)
RCUT_NN RCUT_MIX RCUT_ESP
a. Charge > 0.1eExplicit electrostatic interaction(NN atoms)
b. Charge < 0.1e(R)ESP coupling Hamiltonian(EC atoms)
(R)ESP coupling Hamiltonian(EC atoms)
If LONG-‐RANGE:Interaction with a multipole expansion for the QM system up to quadrupolar order
If not:QM system coupled to force-‐field charges
QM region in VDW representation

bioexcel.eu
Short-range term
27
Ees−sr
QM−MM
== qI ρ(r)υ Ieff r − RI( )dr∫
I∈NN∑
υ I
eff r( ) = rc,I4 − r 4
rc,I5 − r5
• Only involving the „nearest-‐neighbour’’ (NN) atoms:
1. MM atoms within a certain cutoff range (RCUT_NN) around QM atoms
2. MM atoms with a charge larger than 0.1e and at a distance r between RCUT_NN and RCUT_MIX from any QM atom
• Evaluated exactly by summing all grid points/MM atom contributions in the NN class explicitly:
Modified Coulomb interaction
(smeared charges)RCUT_NN RCUT_MIX
Charge > 0.1e
bioexcel.eu
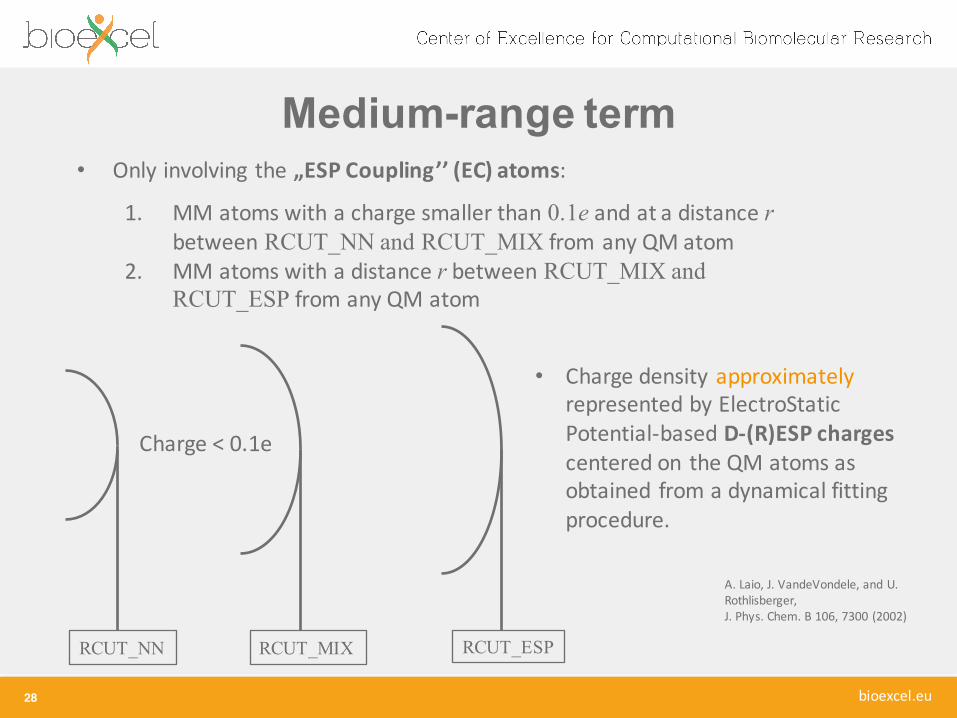
Medium-range term
28
RCUT_NN RCUT_MIX
Charge < 0.1e
RCUT_ESP
• Only involving the „ESP Coupling’’ (EC) atoms:
1. MM atoms with a charge smaller than 0.1e and at a distance r between RCUT_NN and RCUT_MIX from any QM atom
2. MM atoms with a distance r between RCUT_MIX and RCUT_ESP from any QM atom
• Charge density approximatelyrepresented by ElectroStaticPotential-‐based D-‐(R)ESP chargescentered on the QM atoms as obtained from a dynamical fitting procedure.
A. Laio, J. VandeVondele, and U. Rothlisberger, J. Phys. Chem. B 106, 7300 (2002)

bioexcel.eu
Long-range term
29
• Computed approximately by expanding the charge density of the QM system in terms of multipoles up to quadrupolar order:
Ees−lrQM−MM
= qIC
R I −R+ Di
R I −R3 RIi − Ri( )
i∑
⎧⎨⎪
⎩⎪I∉NN∑
+ 12
Qij
R I −R5 RIi − Ri( ) RIj − Rj( )
i, j∑
⎫⎬⎪
⎭⎪
• Coefficients C, Di, Qij are the usual multipole moments computed from the charge density ρ(r) of the QM part with respect to the geometric center of the QM system, , where i=1,2,3 are the cartesiancomponents.
R = (R1,R2,R3)
bioexcel.eu30
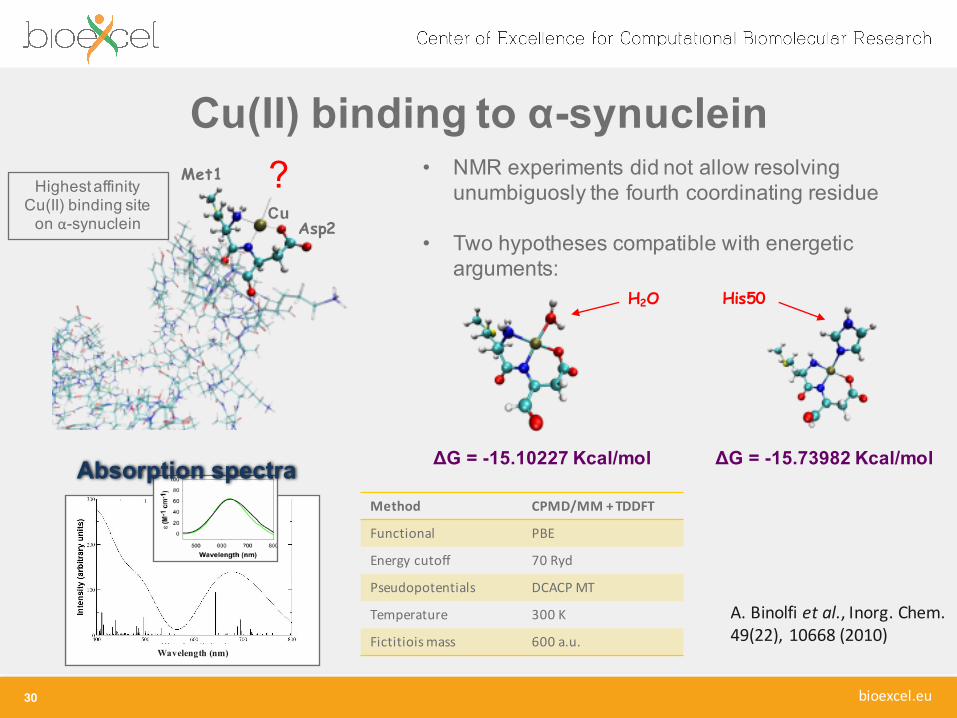
Cu(II) binding to α-synuclein
Asp2
Met1
Cu
?HighestaffinityCu(II) binding site on α-synuclein
• NMR experiments did not allow resolvingunumbiguosly the fourth coordinating residue
• Two hypotheses compatible with energeticarguments:
His50H2O
ΔG = -15.10227 Kcal/mol ΔG = -15.73982 Kcal/mol
A. Binolfi et al., Inorg. Chem. 49(22), 10668 (2010)
Method CPMD/MM + TDDFT
Functional PBE
Energy cutoff 70 Ryd
Pseudopotentials DCACP MT
Temperature 300 K
Fictitioismass 600 a.u.
Absorption spectra
Wavelength (nm)
bioexcel.eu
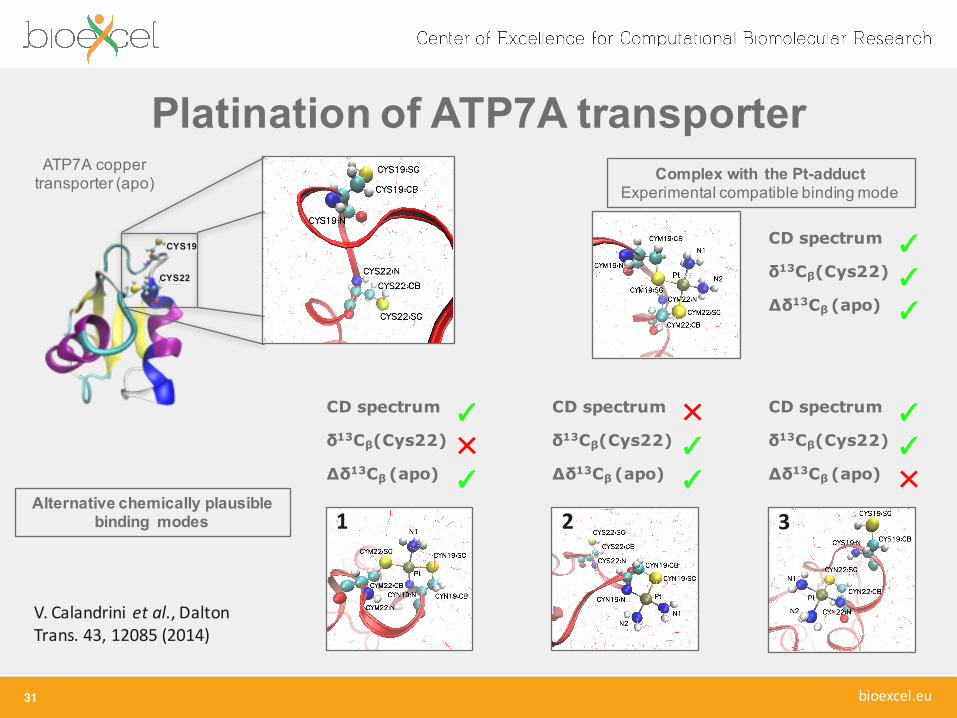
Platination of ATP7A transporter
31
V. Calandrini et al., Dalton Trans. 43, 12085 (2014)
ATP7A coppertransporter (apo)
CYS19
CYS22
Complex with the Pt-adductExperimental compatible bindingmode
1 2 3Alternative chemically plausible
binding modes
CD spectrum ✓δ13Cβ(Cys22) ✕Δδ13Cβ (apo) ✓
CD spectrum ✓δ13Cβ(Cys22) ✓Δδ13Cβ (apo) ✓
CD spectrum ✕δ13Cβ(Cys22) ✓Δδ13Cβ (apo) ✓
CD spectrum ✓δ13Cβ(Cys22) ✓Δδ13Cβ (apo) ✕

bioexcel.eu
CPMD/Gromos96
32
Cons:• Bad scaling performance in comparison with “full-QM” CPMD• Non-free Gromos96 license required to use this interface
Pros:• Full Hamiltonian QM/MM coupling• Support of both Gromos96 and Amber force fileds• Extensively validated for biologically relevant systems• Most popular among CPMD community

bioexcel.eu
The New QM/MM Interface
33
in collaboration with:
U. Rothlisberger’s group (EPFL Lausanne)(developer of the CPMD/Gromos96 interface)
Zürich(owner of CPMD)
J. M. Haugaard Olsen (University of Southern Denmark)S. Meloni (University La Sapienza, Rome)
V. Bolnykh (RWTH Aachen and Cyprus Institute)
is developing a new QM/MM interface

bioexcel.eu
The New QM/MM Interface
34
• Implementation: Interface built in the CPMD code
• Physics: Full Hamiltonian coupling based on the well-validated CPMD/Gromos96 interface
• Abstraction: interfacing any classical MD code with minimal code intervention (Gromacs 5 will be first used as proof of concept)
• Flexible: managing two (QM and MM) or even more layers at the same time
• Running model: separate pools of processes running CPMD+Interface and the partner MD code(s)
Propagation of the equation of motion done by CPMD

bioexcel.eu
Advantages
35
The new QM/MM interface of CPMD will allow one to:
1. Improve the poor HPC scaling performance of the current most used QM/MM interface in CPMD (CPMD/Gromos96).
1. Provide a QM/MM interface with the same coupling of the CPMD/Gromos96 one but completely free (for academic users) as CPMD is.
1. Have additional features, including the possibility to work with: • different MD codes interfacing CPMD• more resolution layers at the same time• different (polarizable) force fields• more general simulation boxes (triclinic ones)
bioexcel.eu
Audience Q&A session
Please use the Questions function in GoToWebinar application

bioexcel.eu
www.bioexcel.eu/contact
NEXT BIOEXCEL WEBINAR
Mid July – To Be Confirmed
Find out more at www.bioexcel.eu/webinars
Also coming up:
BDE Webinar: The Open PHACTS pilotIncluding a talk by Stian Soiland-‐Reyes (BioExcel)6th July, 14:00 BST / 15:00 CESThttps://www.big-‐data-‐europe.eu/event/webinar-‐pilot/





























