
TEXTAL - Automated Crystallographic Protein Structure Determination Using Pattern Recognition
Principal Investigators: Thomas Ioerger (Dept. Computer Science)
James Sacchettini (Dept. Biochem/Biophys)
Other contributors: Tod D. Romo, Kreshna Gopal, Erik McKee,
Lalji Kanbi, Reetal Pai & Jacob Smith
Funding: National Institutes of Health
Texas A&M University

X-ray crystallography• Most widely used method for
protein modeling
• Steps: – Grow crystal
– Collect diffraction data
– Generate electron density map (Fourier transform)
– Interpret map i.e. infer atomic coordinates
– Refine structure
• Model-building– Currently: crystallographers
– Challenges: noise, resolution
– Goal: automation

X-ray crystallography• Most widely used method for
protein modeling
• Steps: – Grow crystal
– Collect diffraction data
– Generate electron density map (Fourier transform)
– Interpret map i.e. infer atomic coordinates
– Refine structure
• Model-building– Currently: crystallographers
– Challenges: noise, resolution
– Goal: automation

• Automated model-building program
• Can we automate the kind of visual processing of patterns that crystallographers use?– Intelligent methods to interpret density, despite noise– Exploit knowledge about typical protein structure
• Focus on medium-resolution maps– optimized for 2.8A (actually, 2.6-3.2A is fine)
– typical for MAD data (useful for high-throughput)
– other programs exist for higher-res data (ARP/wARP)
Overview of TEXTAL
Electron density map(or structure factors) TEXTAL Protein model
(may need refinement)

SCALE MAP
TRACE MAP
CALCULATE FEATURES
PREDICT Cα’s
BUILD CHAINS
PATCH & STITCH CHAINS
REFINE CHAINS
LOOKUP: model side chains CAPRA: models backbone
POST-PROCESSING
SEQUENCE ALIGNMENT
REAL SPACE REFINEMENT
Crystal Collect data Diffraction data Electron density map
Model of backbone
Model of backbone & side chains
Corrected & refined model

CAPRA: C-Alpha Pattern-Recognition Algorithm
tracing
linking
Neural network:estimates whichpseudo-atoms areclosest to true C’s
Best-first search with heuristicscoring function based on: • neural net scores• density• connectivity• secondary structure

Example of C-chains fit by CAPRA
% built: 84%# chains: 2lengths: 47, 88RMSD: 0.82A
Rat 2 urinary protein (P. Adams)data: 2.5A MRmap generated at 2.8A

Stage 2: LOOKUP
• LOOKUP is based on Pattern Recognition – Given a local (5A-spherical) region of density, have we seen a
pattern like this before (in another map)?
– If so, use similar atomic coordinates.
• Use a database of maps with known structures– 200 proteins from PDB-Select (non-redundant)
– back-transformed (calculated) maps at 2.8A (no noise)
– regions centered on 50,000 C’s
• Use feature extraction to match regions efficiently– feature (e.g. moments) represent local density patterns
– features must be rotation-invariant (independent of 3D orientation)
– use density correlation for more precise evaluation
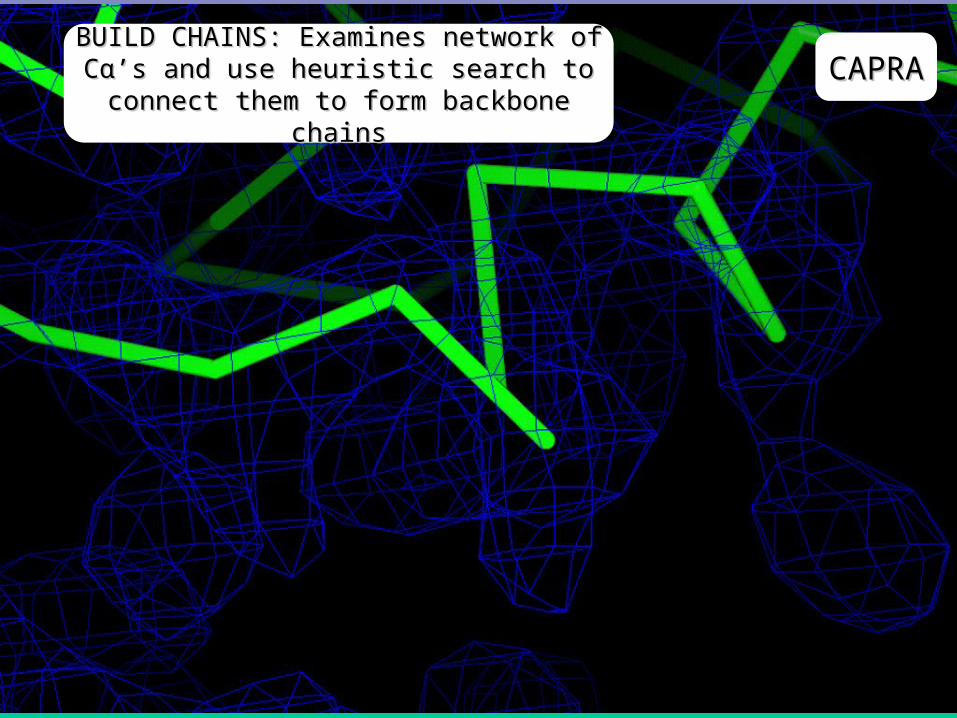
CAPRACAPRABUILD CHAINS: Examines network of BUILD CHAINS: Examines network of Cα’s and use heuristic search to Cα’s and use heuristic search to
connect them to form backbone chainsconnect them to form backbone chains

LOOKUP: Uses case-based reasoning LOOKUP: Uses case-based reasoning to find, for each Cto find, for each Cαα, the best , the best
matching local region in a database matching local region in a database

Databaseof knownmaps
Region in map to be interpreted
The LOOKUP ProcessFind optimalrotation
i
iii RFRFwRRdist 22121 ))()((),(
“2-norm”: weighted Euclideandistance metric for retrieving matches:
Two-step filter: 1) by features 2) by density correlation

Examples of Numeric Density Features
•Distance from center-of-sphere to center-of-mass•Moments of inertia - relative dispersion along orthogonal axes•Geometric features like “Spoke angles” •Local variance and other statistics
Features are designed to be rotation-invariant, i.e. samevalues for region in any orientation/frame-of-reference.
TEXTAL uses 19 distinct numeric features to represent the pattern of density in a region, each calculated over 4 different radii, for a total of 76 features.

F=<1.72,-0.39,1.04,1.55...> F=<1.58,0.18,1.09,-0.25...>
F=<0.90,0.65,-1.40,0.87...> F=<1.79,-0.43,0.88,1.52...>

SLIDER: Feature-weighting algorithm• Euclidean distance metric used for retrieval: • importance of relevant features, avoid noisy features• Goal: find optimal weight vector w the generates highest
probability of hits (matches) in top K candidates from database• Concept of Slider:
• analyze distances between representative matches and mismatches• adjust features so the most matches are ranked higher than mismatches
i
iii RFRFwRRdist 22121 ))()((),(
Slider Algorithm(w,F,{Ri},matches,mismatches) choose feature fF at random for each <Ri,Rj,Rk>, Rjmatches(Ri),Rkmismatches(Ri) compute cross-over point i where: dist’(Ri,Rj)=dist’(Ri,Rk) dist’(X,Y)= (Xf-Yf)2+(1-)dist\f(X,Y) pick that is best compromise among i
ranks most matches above mismatches update weight vector: w’update(w,f,), wf’= repeat until convergence

SLIDER ResultsConvergence of feature selection/weighting
algorithms
60
70
80
90
100
0 50 100 150 200 250
Iterations
Acc
ura
cy o
f ra
nki
ng
SLIDER
SFS
SBS
DIET
Accuracy of case retrieval
012345678
SLIDER SBS DIET SFS Uniformweights
Nu
mb
er o
f m
atch
es r
etri
eved
Speed of convergence
0
500
1000
1500
2000
SLIDER SFS SBS DIET
Tim
e (s
eco
nd
s)
Effectiveness of retrieval using Euclidean (tolerance = .02)
0
1
2
3
4
5
6
7
0 1000 2000 3000 4000
k
Ave
rag
e n
o o
f m
atch
es
cau
gh
t in
to
p k
Uniform-weighted
Slider-weighted

Stage 3: Post-Processing

Quality of TEXTAL models
• Typically builds >80% of the protein atoms
• Accuracy of coordinates: ~1Å error (RMSD)– Depends on resolution and quality of map

PcaA• Mycolic acid cyclopropyl synthase (Smith&Sacchettini)
• original structure solved at 2.0A via MADR-value = 0.22, R-free = 0.27
• 287 residues, fold
Example of density quality (~1 contour with C trace)

Electron density map (2.8A)
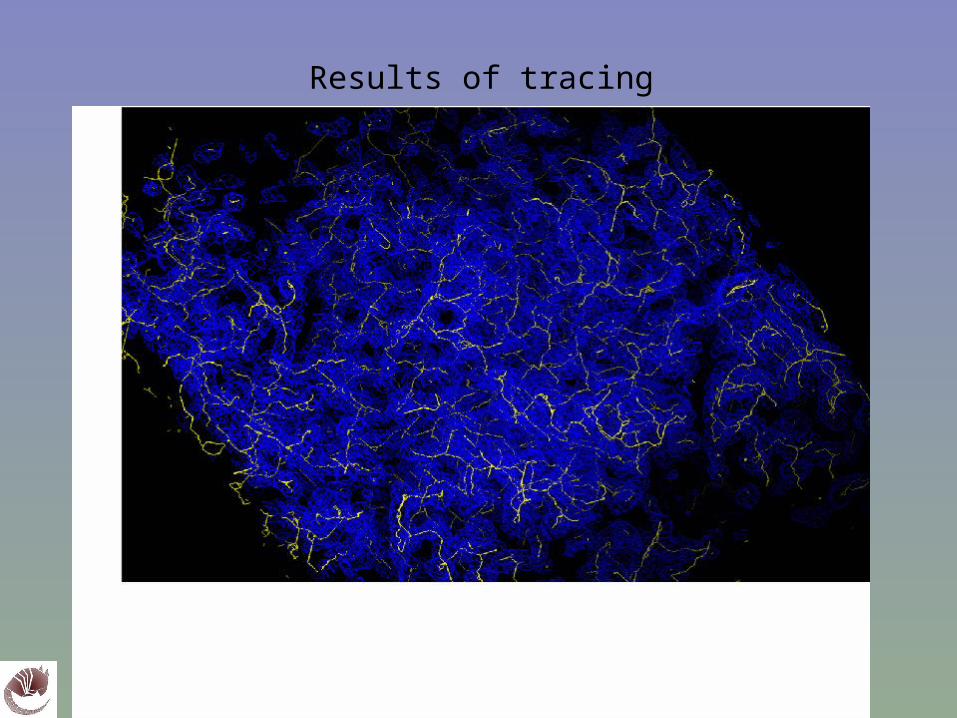
Results of tracing


Strip off branches of trace (linearize)

Linearized trace shows backbone connectivity

Pick C’s using neural net; link together

Results of CAPRA

Comparison to backbone of true structure (white)
Percent built = 89% (missing: 15-residue N-terminus, 17-residue disordered loop)4 single-atom insertions; 5 single-atom deletionsRMSD = 0.81A

CAPRA model consists of 3 chains
Chain lengths: 14, 96, 145 residues

Results of LOOKUP (modeling side-chains)
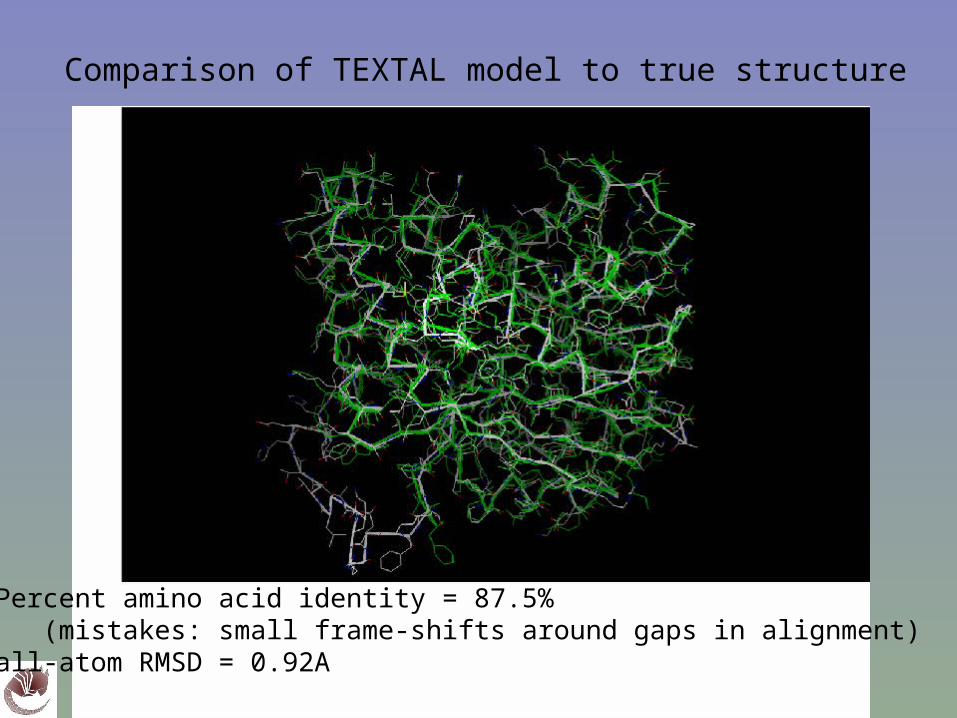
Comparison of TEXTAL model to true structure
Percent amino acid identity = 87.5% (mistakes: small frame-shifts around gaps in alignment)all-atom RMSD = 0.92A

Closeup of -strand (TEXTAL model in green)
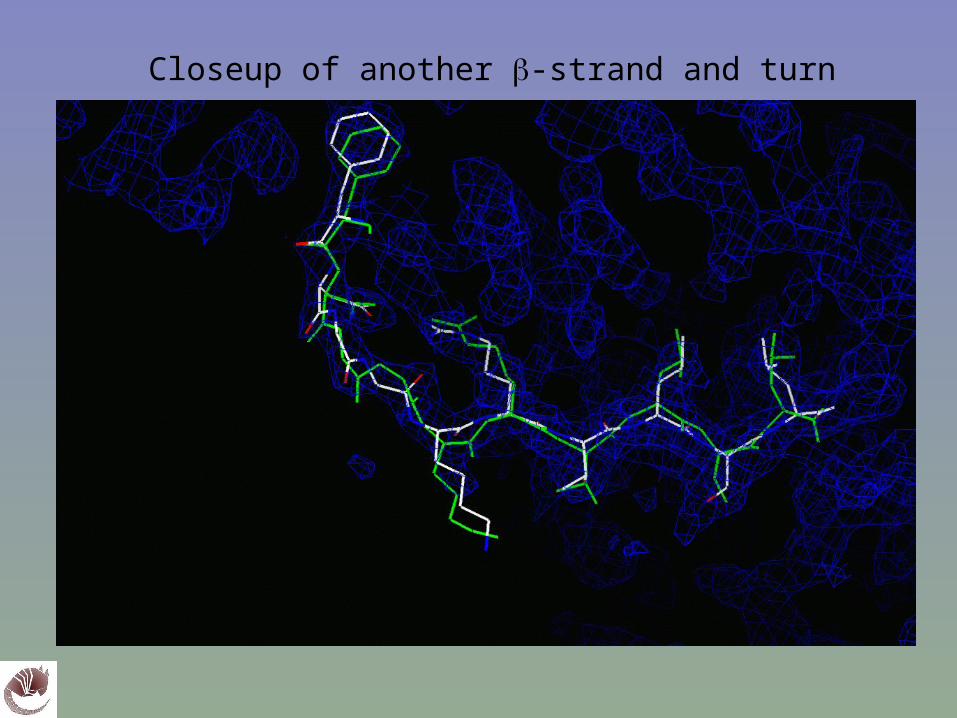
Closeup of another -strand and turn

Implementation
• Project started in 1998 – Collaboration between TAMU Computer Science & Biochemistry
departments
• 100,000 lines of C/C++, Perl, Python code• ~8 developers • CVS for version management• Platforms: Irix, Linux, OSX, Win32• Speed: 1-3 hours for medium-sized proteins

Deployment
• September 2004: Linux and OSX distributions– Can be downloaded from http://textal.tamu.edu:12321– 40 trial licenses granted so far
• June 2002: WebTex (http://textal.tamu.edu:12321)– Till May 2005: TB Structural Genomics Consortium members only– Recently open to the public– ~500 jobs successfully processed– 120 users from 70 institutions in 20 countries
• July 2003: Model building component of PHENIX– Python-based Hierarchical ENvironment for Integrated Xtallography– Consortium members:
• Lawrence Berkeley National Lab• University of Cambridge• Los Alamos National Lab• Texas A&M University
– April 2005: Alpha release - over 300 downloads so far

Python-based Hierarchical ENvironment for Integrated Xtallography
HYSS, CCTBX (Lawrence Berkeley Lab)
Crystallography toolbox, heavy atom search, refinement
PHASER (University of Cambridge)
Maximum likelihood phasing
SOLVE/RESOLVE (Los Alamos National Lab)
Statistical density modification, minimum bias phasing
TEXTAL™ (Texas A&M University)
Model building
PH
EN
IXdiffractiondata
refined molecular model

Conclusions• Pattern recognition is a successful technique for
macromolecular model-building• Future directions:
– recognizing disulfide bridges, metal ions, detergents...
– building ligands, co-factors, etc.
– using models built to iteratively improve phases
– building at higher or lower resolutions
– intelligent agent for guiding model-completion
– detecting and exploiting non-crystallographic symmetry
– building nucleic acids (RNA and DNA)
• Importance and challenges of interdisciplinary research

Acknowledgements• Funding:
– National Institutes of Health
• Our group:– Jacob Smith, Kreshna Gopal, Lalji Kanbi, Erik McKee,
Reetal Pai, Tod Romo • Our association with the PHENIX group:
– Paul Adams (Lawrence Berkeley National Lab)
– Randy Read (Cambridge University)
– Tom Terwilliger (Los Alamos National Lab)
Recommended

















