
PH
PTahcPbowcomb
snemeo
F
A
9
rotein Aggregate Myopathiesans H. Goebel, MD, and Harald D. Müller, MD
Protein aggregate myopathies (PAMs) based on the morphologic phenomenon of aggre-gation of proteins within muscle fibers may occur in children (selenoproteinopathies,actinopathies, and myosinopathies) or adults (certain myofibrillar myopathies and myosi-nopathies). They may be mutation related, which includes virtually all childhood forms butcertain other forms as well, or sporadic, which are largely seen in adults. Their classifica-tion as myofibrillar or desmin-related myopathies, actinopathies, or myosinopathies isbased on the identification of respective mutant proteins, most of them components of thesarcomeres. Recognition of PAM requires muscle biopsy and an extensive immunohisto-chemical and electron microscopic workup of the biopsied muscle tissue after whichmolecular analysis of morphologically ascertained proteins should ensue to permit recog-nition of individual entities and genetic counseling of patients and families. Becausepathogenetic principles in PAMs are still incompletely known, causative therapy, at thistime, is not available.Semin Pediatr Neurol 13:96-103 © 2006 Elsevier Inc. All rights reserved.
KEYWORDS protein aggregation, myopathies, actinopathy, desminopathy, myosinopathy
Atppmdasets
oditmaqatmtmtpfit
rotein aggregate disorders are conditions morphologi-cally marked by the aggregation of proteins in tissues.
hey are classified among the slowly progressive degener-tive diseases. They might be hereditary/familial or non-ereditary/sporadic. Certain conditions may fall into bothategories (eg, Alzheimer disease and Parkinson disease).rotein aggregation may affect multiple organs representedy the various forms of amyloidoses or restricted to particularrgans, largely the brain or muscle. Proteins may aggregateithin cells or in the extracellular space, the latter examples
ontaining primary forms such as amyloidoses or secondarynes in which primary intracellular protein aggregates re-ain in the extracellular space after cell death (eg, neurofi-
rillary ghost tangles in Alzheimer disease).Protein aggregation within cells occurs outside of the lyso-
omal compartment; hence, protein aggregate disorders areot lysosomal diseases, which are marked by deficiencies ofnzymes, mostly with a genetic basis resulting in intralysoso-al accretion of noncatabolizable substrates. Lysosomal dis-
ases belong to metabolic diseases. Cause and pathogenesisf protein aggregation are still incompletely understood.
rom the Department of Neuropathology, Johannes Gutenberg University,Mainz, Germany.
ddress reprint requests to Hans H. Goebel, MD, Department of Neuropa-thology, Johannes Gutenberg University Mainz, Medical Center, Langen-beckstrasse 1, Bldg. 706, 55131 Mainz, Germany. E-mail: goebel@
tneuropatho.klinik.uni-mainz.de
6 1071-9091/06/$-see front matter © 2006 Elsevier Inc. All rights reserved.doi:10.1016/j.spen.2006.06.005
mong the hereditary forms, mutations result in the forma-ion of mutant proteins. These mutant proteins may defyroper extralysosomal proteolytic degradation followed byrotein aggregation. Such a process may affect more than theutant protein, resulting in accumulation of a multitude ofiverse proteins, for instance aggregates marked by desminnd other proteins. Involvement of the ubiquitin-proteasomeystem in protein aggregation in muscle fibers1 attests to thisxtralysosomal protein degradation. On the other hand, mu-ant proteins may resist proper integration into physiologicaltructures during development.
An important nosological feature of many diseases is thenset of clinical symptoms. Among the protein aggregateisorders, there are some commencing in childhood, others
n adulthood, and few show a wide spectrum, encompassinghose of infantile and adult onset. Although brain and skeletaluscle are the most frequently affected organs in protein
ggregate disorders, it is the skeletal muscle that is not infre-uently involved in those forms of protein aggregate myop-thies, which commence in childhood. It is this group amonghe protein aggregate disorders, childhood protein aggregateyopathies, formerly also termed protein surplus myopa-
hies,2 which are the topic of this article. Protein aggregateyopathies are a very telling example of forward genetics in
hat immunohistochemical identification of proteins withinrotein aggregates had led to respective genes and to identi-cation of mutations. Concerning desmin-related myopa-hies, desminopathies, actinopathies, and myosinopathies,
hese principles and pathways have gainfully been pursued.
PAAscdrifittsprhsetpdmg
sfttrbdpkeidb
MMamsodttrppdacupi
sposv
baEbewibpttcohaadwntMobn
ahotto
thEd
gtsgcctre
AAA
Protein aggregate myopathies 97
roteinggregation in Muscle Fibers
ggregation of proteins within muscle fibers may be a non-pecific feature when it is observed in ragged red fibers oraps,3 in neurogenic “targets”4 and cores, both of central coreisease4 and multiminicore disease.5 These lesions (ie, raggeded fibers, caps, and core-targetoid features) have a lack inntact regular sarcomeres in common. Because ragged redbers are not developmental defects but rather occur overime in mitochondrial myopathies and core-targetoid fea-ures may indicate damage to muscle fiber innervation, theyhow disturbance of structural integrity and destruction ofrimarily normal sarcomeres. It appears that proteins are notegularly catabolized when intact structures of sarcomeresave been compromised. Progressive failure to maintaintructural integrity of abnormal sarcomeres seems to impairxtralysosomal proteolytic capacities in such sarcomeres ando result in extralysosomal aggregation of noncatabolizedroteins. It is conceivable, although to our knowledge not yetocumented, that other such sarcomere-defective areas withinuscle fibers, such as sarcoplasmic masses, may also show ag-
regation of proteins.Morphologically, aggregates of proteins may be ill defined
uch as in ragged red fibers and core-targetoid lesions or mayorm distinct and well-described inclusion bodies, some-imes patches or plaques. In the context of protein aggrega-ion, certain inclusion bodies within muscle fibers have al-eady been shown to contain proteins such as nemalineodies, actin filament aggregates, cytoplasmic, and otheresmin-containing bodies. Even if the entire spectrum ofroteins composing individual inclusion bodies may not benown, certain inclusions within muscle fibers contain sev-ral proteins, but not a single individual protein, only. Othernclusions have not successfully been analyzed such as re-ucing bodies, cylindrical spirals, and others encountered iniopsied muscle tissues of respective congenital myopathies.
yofibrillar Myopathiesyofibrillar myopathies (MFMs)6 are the longest-known
nd largest group of PAM, also designated as desmin-relatedyopathies,7 because desmin, the intermediate filament of
triated and smooth muscle cells, is a consistent componentf their protein aggregates. Hence, MFMs can be further sub-ivided according to mutations in proteins (ie, desminopa-hies,8 �-B crystallinopathies,9 selenoproteinopathy,10 myo-ilinopathies,11 ZASPopathies,12 filaminopathy,13 and mostecently also a form of laminopathy14 in which the majority ofatients are adults) (Table 1). Because MFMs indicate a mor-hologically defined muscle lesion, namely derangement andestruction of sarcomeres, together with aggregation of proteinsmong them desmin and other proteins, electron microscopi-ally either as inclusion bodies [eg, cytoplasmic bodies (Fig-re 1)], or granulofilamentous material, genetically not com-letely identified forms seem to exist among MFM, for
nstance when related to gene loci 2q21,15 10q23,16 chromo- m
ome 12,17 or, recently, 15q22.18 Although the majority ofatients with MFM have an adult onset of muscle weakness,ften more distally than proximally and not infrequently as-ociated with cardiomyopathy, some MFMs commenced atarious stages of childhood in several patients (Table 2).2
Selenoproteinopathy has often been observed in childrenecause it originated from observations earlier designated asparticular form of congenital muscular dystrophy, the
ichsfeld type, by Goebel and coworkers19 or later as Malloryody-like inclusion myopathy by Fidzianska and cowork-rs.20 These children have proximal or generalized muscleeakness, scoliosis, and often severe respiratory failure lead-
ng to premature death. Selenoproteinopathies are markedy mutations in the selenoprotein N1 gene10 and may mor-hologically show accumulation of desmin and other pro-eins,20,21 thus, qualifying as a myofibrillar myopathy, al-hough the aggregation of selenoprotein has not yet beenlearly documented, perhaps because of lacking availabilityf a suitable antibody. Other conditions occurring in child-ood, originating from mutations in the selenoprotein N1,re multiminicore disease, multiminicores also marked byccumulation of diverse proteins,5,22 and a rigid spine syn-rome.23 At another instance, Mallory body-like inclusionsere found in biopsied muscle tissue of 2 children, althoughot related to a mutation in the SEPN1 gene.24 This suggestedhat heterogeneity of the descriptively labeled myopathy with
allory body-like inclusions was further confirmed by an-ther study on 3 adolescents/young adults who had Malloryody-like inclusions in their biopsied muscle specimens buto mutations in the SEPN1 gene.25
Spheroid body myopathy, known for many years to affectlarge kinship,26,27 has a wide clinical spectrum with child-ood onset of muscle weakness in several patients. Presencef aggregated proteins, among them desmin in and aroundhe spheroid bodies27 as well as recently myotilin,28 promp-ed final recognition of a missense mutation, S39F, in exon 2f the myotilin gene (MYOT/TTID).28
A particular mutation in the desmin gene, R406W, in the Cerminus vicinity of the 2B helix (exon 6) of the desmin geneas repeatedly and independently been observed in Westernurope, associated with onset in adolescence of severe car-iomyopathy and subsequent skeletal muscle weakness.29
One patient with a heterozygous mutation in the desminene had a rather early (ie, childhood onset of clinical symp-oms) and a severe course because, in addition, he had aecond heterozygous mutation in the lamin A/C (LAMNA)ene.30 This observation not only indicated aggravation oflinical symptoms and, thus, perhaps, of clinical severity be-ause beside the desmin mutation recently a LAMNA muta-ion14 has been found independently causing MFM, but itepresents also one of the still rare although increasinglymerging digenic conditions.
ctin Filamentggregate Myopathy
ctin filament aggregate myopathy (AFAM) is a congenital
yopathy morphologically marked by aggregation of actin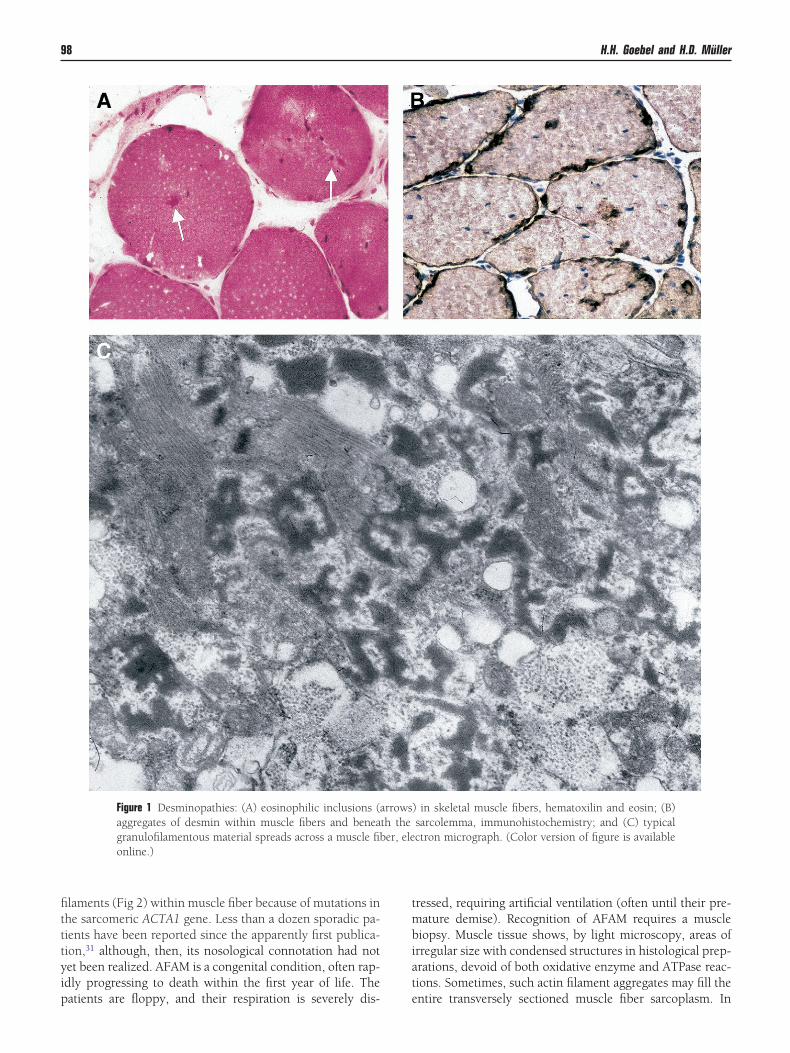
fitttyip
tmbiat
98 H.H. Goebel and H.D. Müller
laments (Fig 2) within muscle fiber because of mutations inhe sarcomeric ACTA1 gene. Less than a dozen sporadic pa-ients have been reported since the apparently first publica-ion,31 although, then, its nosological connotation had notet been realized. AFAM is a congenital condition, often rap-dly progressing to death within the first year of life. The
Figure 1 Desminopathies: (A) eosinophilic inclusions (aaggregates of desmin within muscle fibers and beneagranulofilamentous material spreads across a muscle fibonline.)
atients are floppy, and their respiration is severely dis- e
ressed, requiring artificial ventilation (often until their pre-ature demise). Recognition of AFAM requires a muscle
iopsy. Muscle tissue shows, by light microscopy, areas ofrregular size with condensed structures in histological prep-rations, devoid of both oxidative enzyme and ATPase reac-ions. Sometimes, such actin filament aggregates may fill the
) in skeletal muscle fibers, hematoxilin and eosin; (B)sarcolemma, immunohistochemistry; and (C) typicalctron micrograph. (Color version of figure is available
rrowsth theer, ele
ntire transversely sectioned muscle fiber sarcoplasm. In
obpoimieshIn
ipenn1pmpAtpAciw
mm
pAtbpruaatio
T
A
DAMZSFLV
Protein aggregate myopathies 99
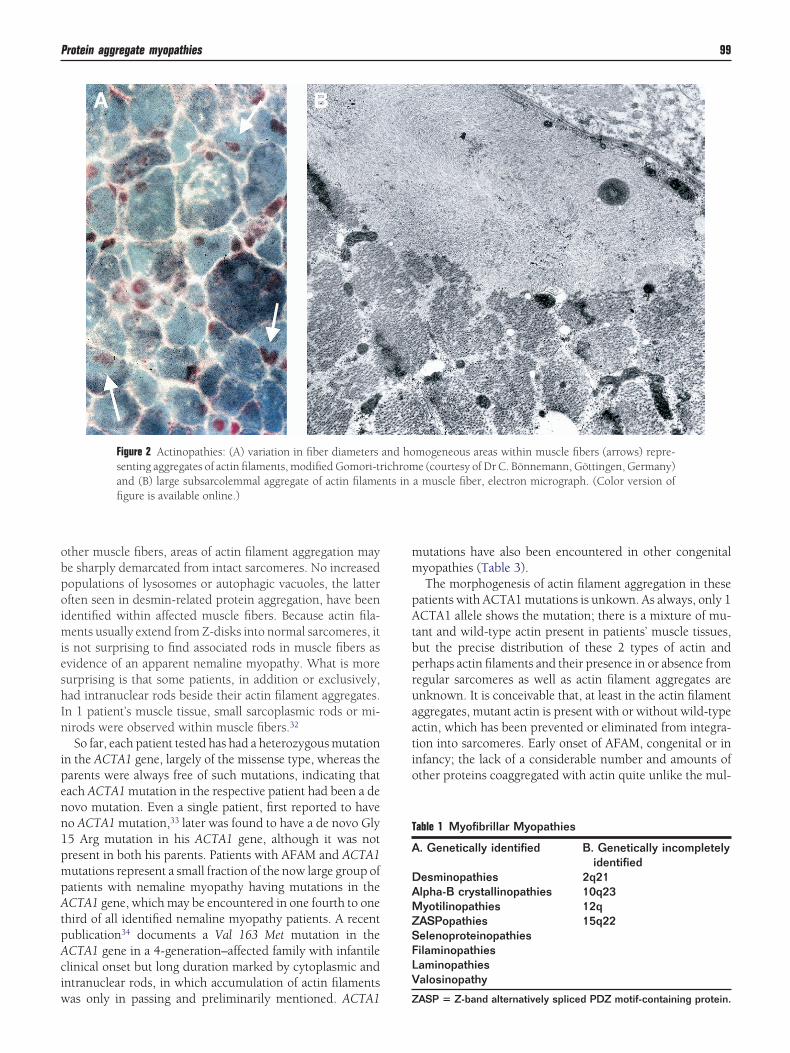
ther muscle fibers, areas of actin filament aggregation maye sharply demarcated from intact sarcomeres. No increasedopulations of lysosomes or autophagic vacuoles, the latterften seen in desmin-related protein aggregation, have beendentified within affected muscle fibers. Because actin fila-
ents usually extend from Z-disks into normal sarcomeres, its not surprising to find associated rods in muscle fibers asvidence of an apparent nemaline myopathy. What is moreurprising is that some patients, in addition or exclusively,ad intranuclear rods beside their actin filament aggregates.n 1 patient’s muscle tissue, small sarcoplasmic rods or mi-irods were observed within muscle fibers.32
So far, each patient tested has had a heterozygous mutationn the ACTA1 gene, largely of the missense type, whereas thearents were always free of such mutations, indicating thatach ACTA1 mutation in the respective patient had been a deovo mutation. Even a single patient, first reported to haveo ACTA1 mutation,33 later was found to have a de novo Gly5 Arg mutation in his ACTA1 gene, although it was notresent in both his parents. Patients with AFAM and ACTA1utations represent a small fraction of the now large group ofatients with nemaline myopathy having mutations in theCTA1 gene, which may be encountered in one fourth to one
hird of all identified nemaline myopathy patients. A recentublication34 documents a Val 163 Met mutation in theCTA1 gene in a 4-generation–affected family with infantilelinical onset but long duration marked by cytoplasmic andntranuclear rods, in which accumulation of actin filaments
Figure 2 Actinopathies: (A) variation in fiber diameterssenting aggregates of actin filaments, modified Gomori-trand (B) large subsarcolemmal aggregate of actin filamefigure is available online.)
as only in passing and preliminarily mentioned. ACTA1 Z
utations have also been encountered in other congenitalyopathies (Table 3).The morphogenesis of actin filament aggregation in these
atients with ACTA1 mutations is unkown. As always, only 1CTA1 allele shows the mutation; there is a mixture of mu-
ant and wild-type actin present in patients’ muscle tissues,ut the precise distribution of these 2 types of actin anderhaps actin filaments and their presence in or absence fromegular sarcomeres as well as actin filament aggregates arenknown. It is conceivable that, at least in the actin filamentggregates, mutant actin is present with or without wild-typectin, which has been prevented or eliminated from integra-ion into sarcomeres. Early onset of AFAM, congenital or innfancy; the lack of a considerable number and amounts ofther proteins coaggregated with actin quite unlike the mul-
mogeneous areas within muscle fibers (arrows) repre-e (courtesy of Dr C. Bönnemann, Göttingen, Germany)
a muscle fiber, electron micrograph. (Color version of
able 1 Myofibrillar Myopathies
. Genetically identified B. Genetically incompletelyidentified
esminopathies 2q21lpha-B crystallinopathies 10q23yotilinopathies 12qASPopathies 15q22elenoproteinopathiesilaminopathiesaminopathiesalosinopathy
and hoichromnts in
ASP � Z-band alternatively spliced PDZ motif-containing protein.
tmfimdd
MMMh
mMmah2hrnha
T
W
W
N
N
N
F ardio)r
100 H.H. Goebel and H.D. Müller
itude of additional proteins seen together with desmin accu-ulation; and the absence of autophagy close to the actinlament aggregates or within muscle fibers with actin fila-ent aggregates suggest anabolic or synthetic/developmentalefects rather than a degradative/catabolic one as seen inesmin-related protein aggregation.
yosin Storageyopathy or Myosinopathy
yosin storage myopathy, in the premolecular era known as
able 2 Myofibrillar Myopathies in Children and Adolescents
Onset y/gender Presentation We
ith desmin mutationChildhood Cardiomyopathy
Conduction defectYoung adult:
prog. to prmuscles in
15 y/F Cardiac conductiondefect
Distal, legs (
18 y/M Atrial tachycardia Anterior tibia
ith other mutations3 y/F Difficulty running
No cardiac signsLimb girdle
Childhood Muscle weaknessNo cardiac signs
Proximal
Early childhood HypotoniaRespiratory distress
Proximal & fa
o known mutationAge 10 years Tiptoe walking Head flexion
weaknessPeripheral ne
Age 12 years Syncope Distal weaknCardiac dysrhythmia Bulbar musc
Age 14 years Dyspnoea Proximal & dChest pain Hoarse voice
o known mutationInfancy Delayed milestones Proximal andInfancy Weak suck Never able tInfancy Severe dysphagia Hypotonia
Ophthalmoplegia Persisting mEarly childhood Exercise intolerance None
Abnormal heart rateChildhood Waddling gait Axial and proChildhood Exercise intolerance
TirednessTeens: Proxi
muscle weAge 8 years Exercise intolerance
DyspnoeaLimb-girdle-lPolyneuropa
o known mutationAge 8 years Exercise intolerance
Heart murmurGeneral mus
(marked diperipheral
Age 9 years Frequent fallsWaddling gait
Limb-girdle-l
� female; M � male; prog. to prox. � progressive to proximal; (c
yaline body myopathy35 or originally as “myofibrillar lysis a
yopathy,”36 is now, after identification of mutations in theYH7 gene,38,39 considered a myosinopathy. Although theajority of patients are adults, some were only diagnosed as
dults but, retrospectively, had been affected since child-ood.40,41 As a matter of fact, the original report36 concernedvery young siblings affected by muscle weakness and whatad been termed “myofibrillar lysis” suggesting autosomal-ecessive inheritance. Another family with autosomal-domi-ant heredity has also been described.42 Affected childrenad mild proximal weakness, somewhat enlarged calves,36
nd slight elevation of creatine kinase. The electromyogram
rted Before and After 2002)2
sCardioresp. and
cardiac
Ref. no. andgene
mutation
myopathylbar
Cardiomyopathy 47
.) Cardiomyopathy 29, Pat. 2R406W
scles Cardiomyopathy 29, Pat. 4R406W
No cardiomyopathy 14 W498CLMNA
No cardiomyopathyRespiratory insufficiency
28 S39FTTID/MYOT
Respiratory insufficiency 10 SEPN1 del
neal Cardiomyopathy 49
thyECHO normal 56
olvedeakness Cardiomyopathy 46
l 51Death: resp. failure 52
53al weakn.
Cardiomegaly 54CardiomyopathyECG normal 21
istals
Cardiomyopathy 55
Restrictive cardiomyopathy 45
rophy. Mildpathy
CardiomyopathyReduced vital capacity
48
Restrictive respiratoryfunction
50
esp. � (cardio)respiratory.
(Repo
aknes
distalox. Buvolved22 yrs
lis mu
cial
, pero
uropaessles invistal w
distao walk
ild axi
ximalmal>daknesikethy
cle atstally)neuroike
ppears myopathic. Although lifespan usually is not re-

saCvpcwfiwi[amwpspa
1acmfmith
mqA
CAmpgtpdsb(o
T
Protein aggregate myopathies 101
tricted, one of the orginally described siblings36 has died asyoung adult and had a novel MYH7 Leal793Pro mutation.37
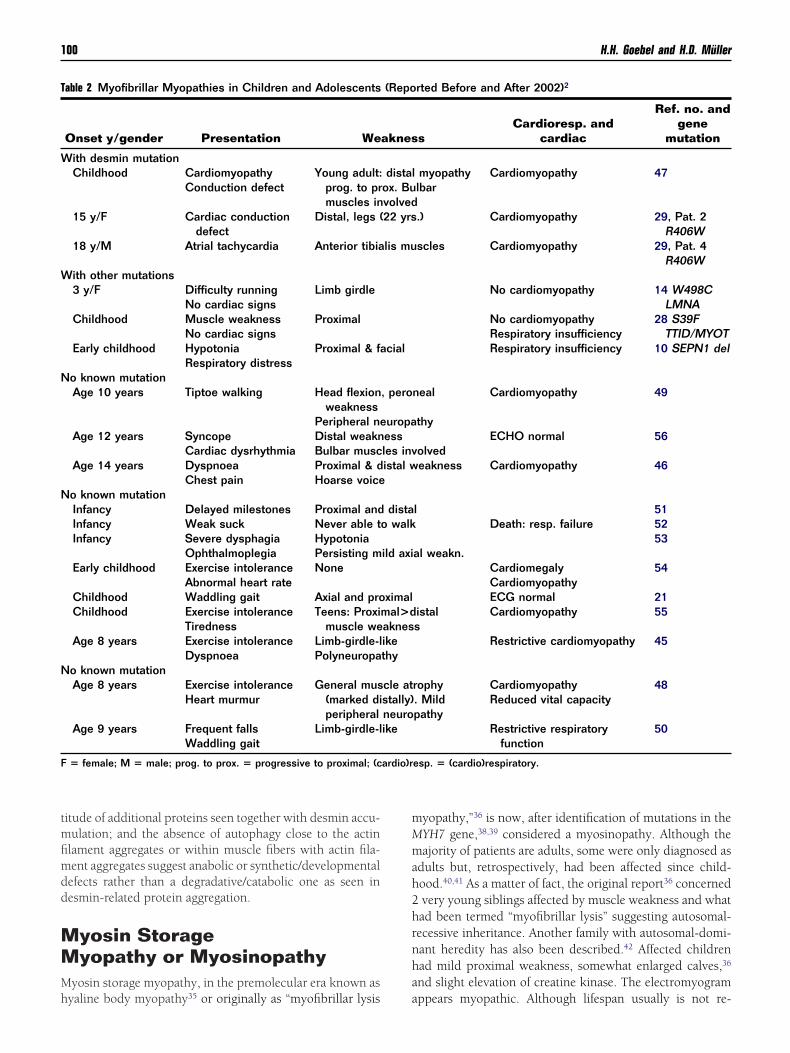
ardiomyopathy is not a consistent feature, although a mildentricular hypertrophy was recorded by echocardiogra-hy.40,41 However, mutations in the MYH7 gene may causeardiomyopathy. Diagnosis is established by muscle biopsy,hich shows, largely subsarcolemmally, hyaline bodies (ie,nely granular, homogeneous, sharply demarcated pale areasithin muscle fibers [Fig 3] that lack oxidative enzyme activ-
ties but show fibrillar/myosin adenosine triphosphataseATPase] activity, especially in type I fibers, where these hy-line bodies prevail). Immunohistochemically, slow myosinay be encountered in these hyaline bodies, which, togetherith the histochemical myofibrillar ATPase activity, actuallyrompted mutational studies of the MYH7 gene. Two mis-ense mutations have separately been identified in differentatients: an Arg 1845 Trp mutation in European patients38,41
nd a His 1904 Leu mutation in a large Saudi family.43 The Arg
Figure 3 Myosinopathy: large homogeneous, opaque(arrows), modified trichrome preparation (courtesy of
able 3 Congenital Myopathies With ACTA1 Mutations
Nemaline/Rod myopathy57
With or without intranuclear rodsWith intranuclear rods only
Actin filament aggregate myopathy57
Core myopathy58
Congenital fiber type disproportion59
available online.)
845 Trp mutation is said to prevent myosin polymerizationnd, apparently, filament formation because, by electron mi-roscopy, the hyaline bodies consist of finely granular non-embrane-bound material, suggesting a developmental de-
ect, although incomplete extralysosomal degradation ofyosin proteins has not been ruled out. A second gene locus
n autosomal-recessive hyaline body myopathy has been es-ablished at 3p22.2 to 21.32,44 although a defective proteinas not yet been communicated from this gene locus.Hyaline body myopathy is a telling example how primaryorphologic studies of muscle tissue have prompted ade-
uate and successful molecular analysis (ie, from granularTPase-positive myosin to mutations in its MYH7 gene).
onclusionslthough aggregation of proteins is now a firmly recognizedorphologic phenomenon in muscle fibers, formerly encom-assing inclusion bodies of varying types (ie, filamentous orranular material), although now further characterized byheir protein composition having given rise to the group ofrotein aggregate myopathies among neuromuscular disor-ers, both in childhood and adulthood, progress in under-tanding these slowly or rapidly progressing disorders haseen accelerated by molecular identification of new formseg, selenoproteinopathies and myosinopathies) or additionf newly identified mutations in desminopathies and acti-
ccupy major parts of cross-sectioned muscle fiberszzarelli, Indianapolis, IN). (Color version of figure is
areas oDr B. A

npttifiatmcuqcTmfiufd
AWa
R
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
3
3
3
3
3
3
3
3
102 H.H. Goebel and H.D. Müller
opathies. Recent studies have brought some insight into theathogenetic mechanisms linking pathomorphologic fea-ures to molecular defects as well as clinical phenotypes. Athis time, however, our understanding of the basic principlesnvolved in the formation of protein aggregates in musclebers is still too scant to allow any promising therapeuticpproaches. Such basic principles entail extralysosomal pro-eolytic degradation as well as development, integration, andaturation of sarcomeric protein components, both pro-
esses resulting in faulty aggregation of proteins. The originalltrastructural description of aggregates and their subse-uent immunohistochemical characterization has led to suc-essful molecular analyses in many but possibly not all PAMs.his investigative path of morphologic features leading toolecular data ought to be further pursued in still-unidenti-ed individual PAMs and their patients. Recognition of still-nknown sarcomeric proteins may also add, enlarge, andurther clarify PAMs as a growing cohort of neuromuscularisorders with both early and late clinical onset.
cknowledgmentse are grateful to Walther Wagner for photographic support
nd to Astrid Wöber for editorial assistance.
eferences1. Ferrer I, Martín B, Castaño JG, et al: Proteasomal expression, induction
of immunoproteasome subunits, and local MHC class I presentation inmyofibrillar myopathy and inclusion body myositis. J Neuropathol ExpNeurol 63:484-498, 2004
2. Goebel HH, Borchert A: Protein surplus myopathies and other rarecongenital myopathies. Semin Pediatr Neurol 9:160-170, 2002
3. Ceuterick-de Groote CM, Lübke U, Durling HJ, et al: A congenitalmyopathy associated with subsarcolemmal ‘cap-like’ myofibrillar alter-ations. Myopathology and molecular genetic analysis. NeuromusculDisord 15:566, 2005 (abstr)
4. De Bleecker JL, Ertl BB, Engel AG: Patterns of abnormal protein expres-sion in target formations and unstructured cores. Neuromuscul Disord6:339-349, 1996
5. Bönnemann C, Thompson TG, van der Ven PFM, et al: Filamin Caccumulation is a strong but nonspecific immunohistochemical markerof core formation in muscle. J Neurol Sci 206:71-78, 2003
6. Selcen D, Ohno K, Engel AG: Myofibrillar myopathy: Clinical, morpho-logical and genetic studies in 63 patients. Brain 127:439-451, 2004
7. Goebel HH, Warlo IAP: Progress in desmin-related myopathies. J ChildNeurol 15:565-572, 2000
8. Goldfarb LG, Vicart P, Goebel HH, et al: Desmin myopathy. Brain127:723-734, 2004
9. Fardeau M, Vicart P, Caron A, et al: Myopathie familiale avec surchargeen desmine, sous forme de matériel granulo-filamentaire dense en mi-croscopie électronique, avec mutation dans le gène de l’alpha-B-cristal-line. Rev Neurol (Paris) 156:497-504, 2000
0. Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al: Desmin-relatedmyopathy with Mallory body-like inclusions is caused by mutations ofthe selenoprotein N gene. Ann Neurol 55:676-686, 2004
1. Selcen D, Engel AG: Mutations in myotilin cause myofibrillar myop-athy. Neurology 62:1363-1371, 2004
2. Selcen D, Engel AG: Mutations in ZASP define a novel form of musculardystrophy in humans. Ann Neurol 57:269-276, 2005
3. Vorgerd M, van der Ven PFM, Bruchertseifer V, et al: A mutation in thedimerization domain of filamin C causes a novel type of autosomaldominant myofibrillar myopathy. Am J Hum Genet 77:297-304, 2005
4. D’Amico A, Benedetti S, Petrini S, et al: Major myofibrillar changes inearly onset myopathy due to de novo heterozygous missense mutation
in lamin A/C gene. Neuromuscul Disord 15:847-850, 2005 35. Xiang F, Nicolao P, Chapon F, et al: A second locus for autosomaldominant myopathy with proximal muscle weakness and early respi-ratory muscle involvement: A likely chromosomal locus on 2q21. Neu-romuscul Disord 9:308-312, 1999
6. Melberg A, Oldfors A, Blomström-Lundqvist C, et al: Autosomal dom-inant myofibrillar myopathy with arrhythmogenic right ventricular car-diomyopathy linked to chromosome 10q. Ann Neurol 46:684-692,1999
7. Wilhelmsen KC, Blake DM, Lynch T, et al: Chromosome 12-linkedautosomal dominant scapuloperoneal muscular dystrophy. Ann Neu-rol 39:507-520, 1996
8. Lamont PJ, Davis MR, Mastaglia FL, et al: Adult-onset dominant my-opathy with nemaline rods, unstructured cores and desmin on musclebiopsy, linked to chromosome 15q22. J Neurol Sci 199:S104, 2002(abstr)
9. Goebel HH, Lenard H-G, Langenbeck U, et al: A form of congenitalmuscular dystrophy. Brain Dev 2:387-400, 1980
0. Fidzianska A, Goebel HH, Osborn M, et al: Mallory body-like inclu-sions in a hereditary congenital neuromuscular disease. Muscle Nerve6:195-200, 1983
1. Fidzianska A, Ryniewicz B, Barcikowska M, et al: A new familial con-genital myopathy in children with desmin and dystrophin reactingplaques. J Neurol Sci 131:88-95, 1995
2. Ferreiro A, Quijano-Roy S, Pichereau C, et al: Mutations of the seleno-protein N gene, implicated in rigid spine muscular dystrophy, cause theclassical phenotype of multi-minicore disease. Am J Hum Genet 71:739-749, 2002
3. Gonzales V, Quijano-Roy S, Parain K, et al: SEPN-related myopathy:an emerging entity phenotypical and molecular analysis of 80 cases.Neuromuscul Disord 15:715, 2005 (abstr)
4. Taratuto AL, Saccoliti M, Espada G, et al: Early-onset myopathy withcytoplasmic and/or Mallory body-like inclusions. Neuromuscul Disord15:693-694, 2005 (abstr)
5. Parain K, Milic Rasic V, Cabello A, et al: Genetic heterogeneity ofdesmin-related myopathy with Mallory body-like inclusions. Neuro-muscul Disord 15:694, 2005 (abstr)
6. Goebel HH, Muller J, Gillen HW, et al: Autosomal dominant “spheroidbody myopathy.” Muscle Nerve 1:14-26, 1978
7. Goebel HH, D’Agostino AN, Wilson J, et al: Spheroid body myop-athy—Revisited. Muscle Nerve 20:1127-1136, 1997
8. Foroud T, Pankratz N, Batchman AP, et al: A mutation in myotilincauses spheroid body myopathy. Neurology 65:1936-1940, 2005
9. Dagvadorj A, Olivé M, Urtizberea J-A, et al: A series of West Europeanpatients with severe cardiac and skeletal myopathy associated with a denovo R406W mutation in desmin. J Neurol 251:143-149, 2004
0. Muntoni F, Mercuri E, Bonne G, et al: Double lamin-emerin and lamin-desmin trouble. Neuromuscul Disord 15:677, 2005 (abstr)
1. Dubowitz V: Muscle Biopsy: A Practical Approach. London, BaillièreTindall, 1985, pp 664-670
2. Agrawal PB, Strickland CD, Midgett C, et al: Heterogeneity of nemalinemyopathy cases with skeletal muscle �-actin gene mutations. Ann Neu-rol 56:86-96, 2004
3. Goebel HH, Brockmann K, Bönnemann C, et al: Actin-related myop-athy without any missense mutation in the ACTA1 gene. J Child Neurol19:149-153, 2004
4. Hutchinson DO, Charlton A, Laing NG, et al: Autosomal dominantnemaline myopathy with intranuclear rods due to mutation of theskeletal muscle ACTA1 gene: Clinical and pathological variabilitywithin a kindred. Neuromuscul Disord 16:113-121, 2006
5. Barohn RJ, Brumback RA, Mendell JR: Hyaline body myopathy. Neu-romuscul Disord 4:257-262, 1994
6. Cancilla PA, Kalyanaraman K, Verity MA, et al: Familial myopathy withprobable lysis of myofibrils in type I fibres. Neurology 21:579-585,1971
7. Dye DE, Azzarelli B, Goebel HH, et al: Novel slow-skeletal myosin(MYH7) mutation in the original myosin storage myopathy kindred.Neuromuscul Disord 16:357-360, 2006
8. Tajsharghi H, Thornell L-E, Lindberg C, et al: Myosin storage myop-

3
4
4
4
4
4
4
4
4
4
4
5
5
5
5
5
5
5
5
5
5
Protein aggregate myopathies 103
athy associated with a heterozygous missense mutation in MYH7. AnnNeurol 54:494-500, 2003
9. Bohlega S, Abu-Amero SN, Wakil SM, et al: Mutation of the slowmyosin heavy chain rod domain underlies hyaline body myopathy.Neurology 62:1518-1521, 2004
0. Ceuterick C, Martin J-J, Martens C: Hyaline bodies in skeletal muscle ofa patient with a mild chronic nonprogressive congenital myopathy.Clin Neuropathol 12:79-83, 1993
1. Laing NG, Ceuterick-de Groote C, Dye DE, et al: Myosin storage my-opathy: Slow skeletal myosin (MYH7) mutation in two isolated cases.Neurology 64:527-529, 2005
2. Masuzugawa S, Kuzuhara S, Narita Y, et al: Autosomal dominant hya-line body myopathy presenting as scapuloperoneal syndrome: Clinicalfeatures and muscle pathology. Neurology 48:253-257, 1997
3. Bohlega S, Lach B, Meyer BF, et al: Autosomal dominant hyaline bodymyopathy: Clinical variability and pathologic findings. Neurology 61:1519-1523, 2003
4. Önengüt S, Ugur SA, Karasoy H, et al: Identification of a locus for anautosomal recessive hyaline body myopathy at chromosome 3p22.2-p21.32. Neuromuscul Disord 14:4-9, 2004
5. Bertini E, Bosman C, Ricci E, et al: Neuromyopathy and restrictivecardiomyopathy with accumulation of intermediate filaments: A clini-cal, morphological and biochemical study. Acta Neuropathol (Berl)81:632-640, 1991
6. Amato AA, Kagan-Hallet K, Jackson CE, et al: The wide spectrum ofmyofibrillar myopathy suggests a multifactorial etiology and pathogen-esis. Neurology 51:1646-1655, 1998
7. Park K-Y, Dalakas MC, Goebel HH, et al: Desmin splice variants caus-ing cardiac and skeletal myopathy. J Med Genet 37:851-857, 2000
8. Liu HM, Gumbinas M: Axonal filamentous spheroids associated with car-
diomyopathy with “targetoid fibres.” Neurology 24:547-554, 19749. Sabatelli M, Bertini E, Ricci E, et al: Peripheral neuropathy withgiant axons and cardiomyopathy associated with desmin type inter-mediate filaments in skeletal muscle. J Neurol Sci 109:1-10, 1992
0. Bertini E, Salviati G, Apollo F, et al: Reducing body myopathy anddesmin storage in skeletal muscle: Morphological and biochemicalfindings. Acta Neuropathol (Berl) 87:106-112, 1994
1. Prelle A, Moggio M, Comi GP, et al: Congenital myopathy associatedwith abnormal accumulation of desmin and dystrophin. NeuromusculDisord 2:169-175, 1992
2. Bertini E, Ricci E, Boldrini R, et al: Involvement of repiratory muscles incytoplasmic body myopathy. Brain Dev 12:798-806, 1990
3. Sarnat HB: Myofibrillar myopathy in infancy and childhood: five casesin two families of an unclassified congenital myopathy. J Child Neurol12:132-133, 1997 (abstr)
4. Calderon A, Becker LE, Murphy EG: Subsarcolemmal vermiform de-posits in skeletal muscle, associated with familial cardiomyopathy: Re-port of two cases of a new entity. Pediatr Neurosci 13:108-112, 1987
5. Vajsar J, Becker LE, Freedom RM, et al: Familial desminopathy: Myop-athy with accumulation of desmin-type intermediate filaments. J Neu-rol Neurosurg Psychiatry 56:644-648, 1993
6. Telerman-Toppet N, Bauherz G, Noël S: Auriculo-ventricular blockand distal myopathy with rimmed vacuoles and desmin storage. ClinNeuropathol 10:61-64, 1991
7. Sparrow JC, Nowak KJ, Durling HJ, et al: Muscle disease caused bymutations in the skeletal muscle alpha-actin gene (ACTA1). Neuromus-cul Disord 13:519-531, 2003
8. Kaindl AM, Rüschendorf F, Krause S, et al: Missense mutations ofACTA1 cause dominant congenital myopathy with cores. J Med Genet41:842-848, 2004
9. Laing NG, Clarke NF, Dye DE, et al: Actin mutations are one cause of
congenital fibre type disproportion. Ann Neurol 56:689-694, 2004Recommended
![Version 2 Please don’t nore this - Blood · Inflammatory myopathies – inclusion body myositis (IBM) (formerly Inflammatory myopathies: polymyositis [PM], dermatomyositis [DM]](https://img.pdfslide.us/doc/110x75/602246494e545541c973d779/version-2-please-donat-nore-this-blood-inflammatory-myopathies-a-inclusion.jpg)
















