Embed Size (px)
Citation preview

Whole-genome sequencing improves MS-based proteotyping of clinically-relevant bacteria
Francisco Salvà Serra
7th Congress of European Microbiologists
Valencia, Spain
12th July 2017
Department of Infectious DiseasesSahlgrenska AcademyUniversity of Gothenburg
MicrobiologyDepartment of Biology
University of the Balearic Islands

Faculty Disclosure
Company NameHonoraria/
Expenses
Consulting/
Advisory Board
Funded
Research
Royalties/
Patent
Stock
Options
Ownership/
Equity
Position
EmployeeOther
(please specify)
Example: company XYZ x x x
x No, nothing to disclose
Yes, please specify:
Era of antibiotics

Overuse & missuse of antibiotics:

Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. 2014
Annual deaths attributable to antibiotic resistance
Economic impact: 100 trillions (100 · 1012) USD

Rapid and better diagnostics of infectious diseases
Less antibiotic resistance
Reduce overuse & misuse
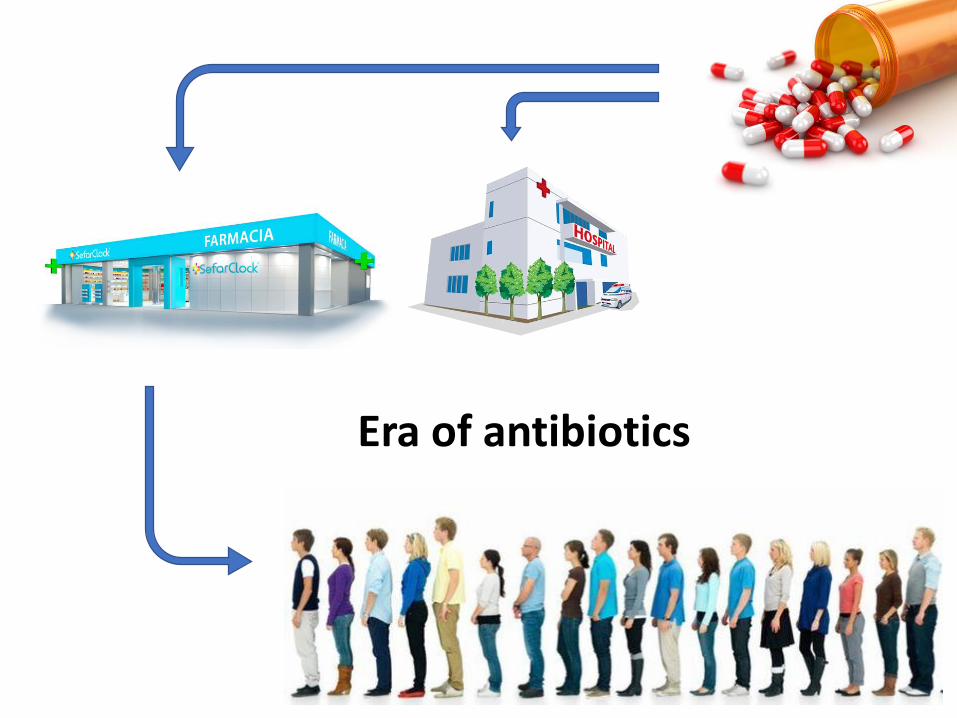
CRITICAL POINT
Database must have:• Species diversity (comprehensive)• Correct classifications
‘Proteotyping’: characterization, identification and diagnostics of microorganisms with MS-based proteomics.
Peptides
Genomes DB
All complete genomes from the NCBI
Reference Sequence Database (RefSeq)
Taxonomic
assignment
Expressed
proteins
LC-MS/MS

PROBLEM 1 - Insufficient and biased number of genomes
S. mitis
S. pneumoniae (major human pathogen)
Members of theStreptococcus mitis group

A single strain does not cover the whole repertoire of genes of a species!
PROBLEM 1 - Insufficient and biased number of genomes
Figure source: Soucy et al., 2015
We need several strains per species!
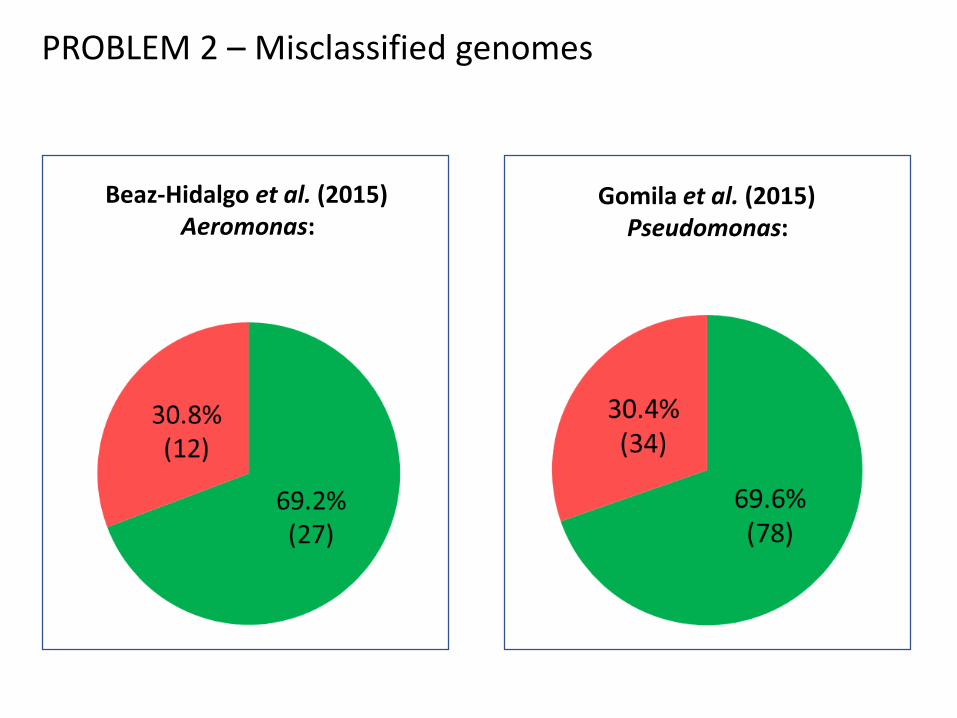
PROBLEM 2 – Misclassified genomes
Beaz-Hidalgo et al. (2015)Aeromonas:
Gomila et al. (2015)Pseudomonas:

PROBLEM 2 – Misclassified genomes
< 93% 93 - 96% ≥ 96%
Streptococcus pneumoniae 323 0 0 323
Streptococcus mitis 40 18 22 0
Streptococcus australis 0 0 0 0
Streptococcus cristatus 2 0 1 1
Streptococcus infantis 4 3 1 0
Streptococcus oligofermentans 7 7 0 0
Streptococcus oralis 11 7 4 0
Streptococcus parasanguinis 9 1 6 2
Streptococcus peroris 0 0 0 0
Streptococcus pseudopneumoniae 6 0 0 6
Streptococcus sanguinis 23 2 16 5
Streptococcus sinensis 0 0 0 0
Total 425 38 50 337
100% 8.94% 11.76% 79.29%
Total number of
genome sequences
ANIb
Organism

PROBLEM 2 – Misclassified genomes
< 93% 93 - 96% ≥ 96%
Streptococcus pneumoniae
Streptococcus mitis 40 18 22 0
Streptococcus australis 0 0 0 0
Streptococcus cristatus 2 0 1 1
Streptococcus infantis 4 3 1 0
Streptococcus oligofermentans 7 7 0 0
Streptococcus oralis 11 7 4 0
Streptococcus parasanguinis 9 1 6 2
Streptococcus peroris 0 0 0 0
Streptococcus pseudopneumoniae 6 0 0 6
Streptococcus sanguinis 23 2 16 5
Streptococcus sinensis 0 0 0 0
Total 102 38 50 14
100% 37.25% 49.02% 13.73%
Organism
Total number of
genome sequences
ANIb

Consequences PROBLEM 1 Insufficient and biased number of genomes
S. mitisS. pseudopneumoniae
S. pneumoniae
REALITY
S. mitisS. pseudopneumoniae
S. pneumoniae
DATABASE
Species poorly represented or not present: Loss of peptide hits
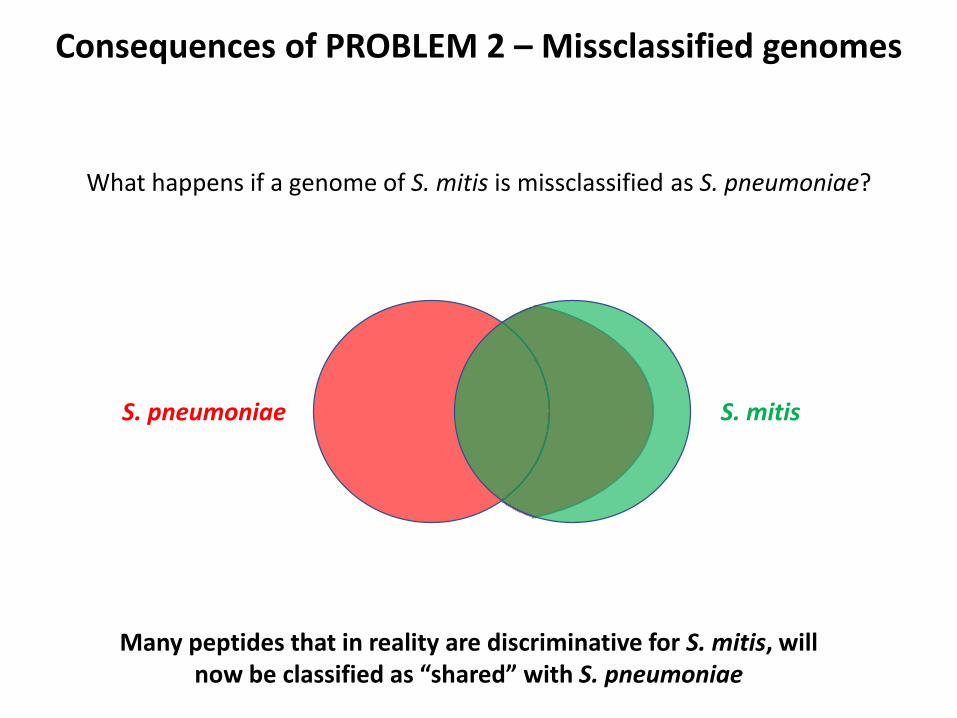
Consequences of PROBLEM 2 – Missclassified genomes
S. mitisS. pneumoniae
What happens if a genome of S. mitis is missclassified as S. pneumoniae?
Many peptides that in reality are discriminative for S. mitis, willnow be classified as “shared” with S. pneumoniae

Solution
Genome DB
Additional genomes from
GenBank
In-house whole-genome
sequencing
Taxonomical verification
(ANIb, MLSA, core-genome)

Improvement
2015-02 2016-09
Organism Before addition of reference genomes After addition of reference genomes
Streptococcus pneumoniae 27 (0) 31(1)
Streptococcus pseudopneumoniae 1 (0) 6 (1)Streptococcus mitis 1 (0) 30 (1)
Type strains included
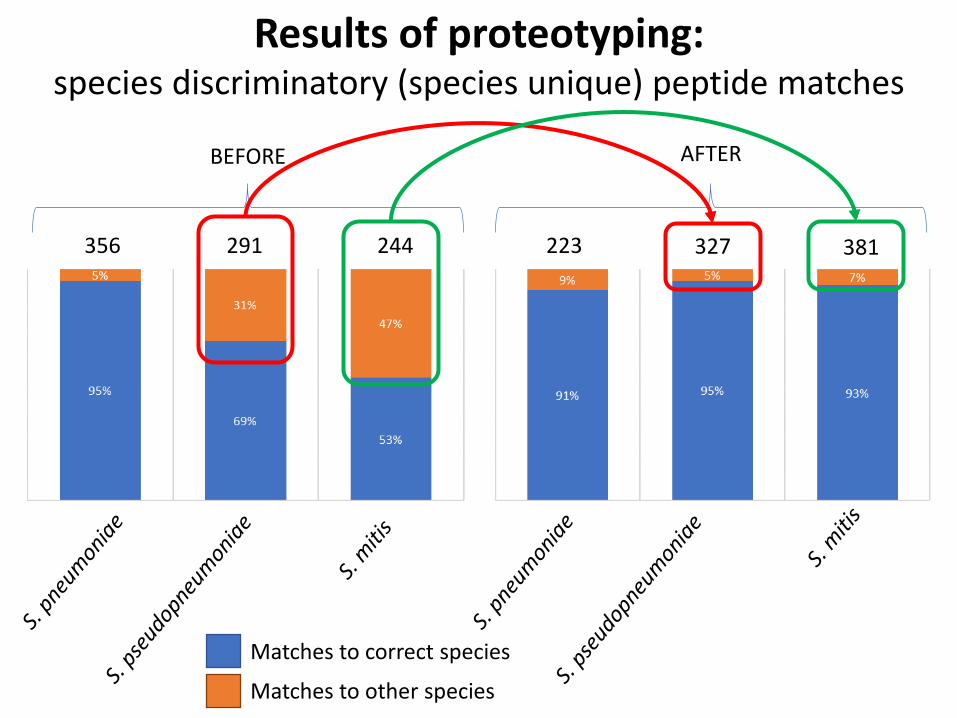
Results of proteotyping:species discriminatory (species unique) peptide matches
Matches to correct species
Matches to other species
356 291 244
BEFORE
223 327 381
AFTER
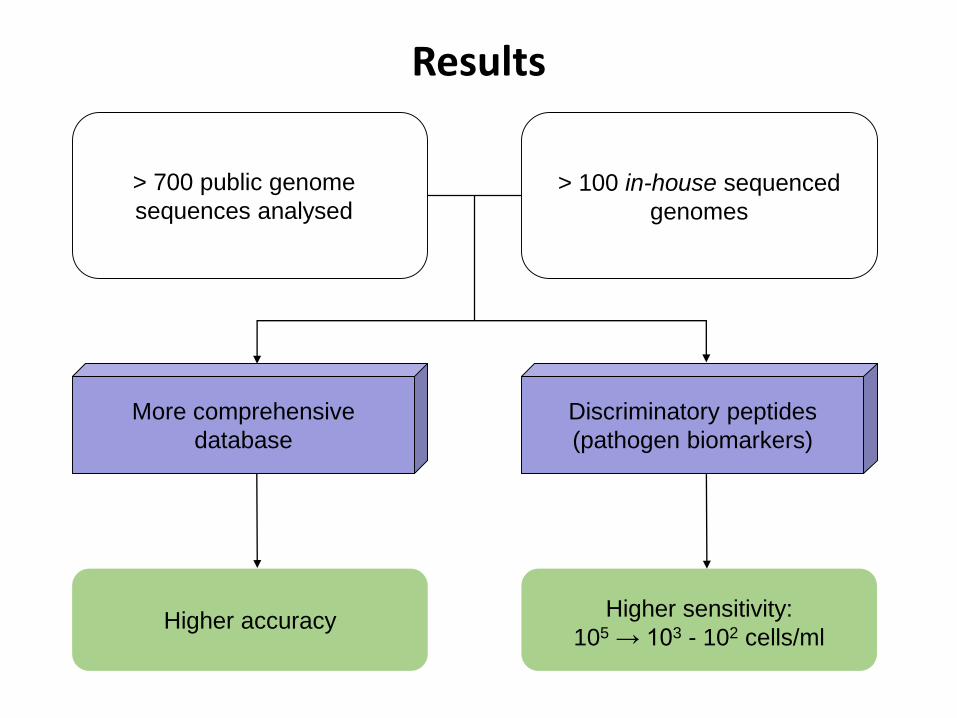
> 700 public genome
sequences analysed
> 100 in-house sequenced
genomes
More comprehensive
database
Discriminatory peptides
(pathogen biomarkers)
Higher accuracyHigher sensitivity:
105 → 103 - 102 cells/ml
Results
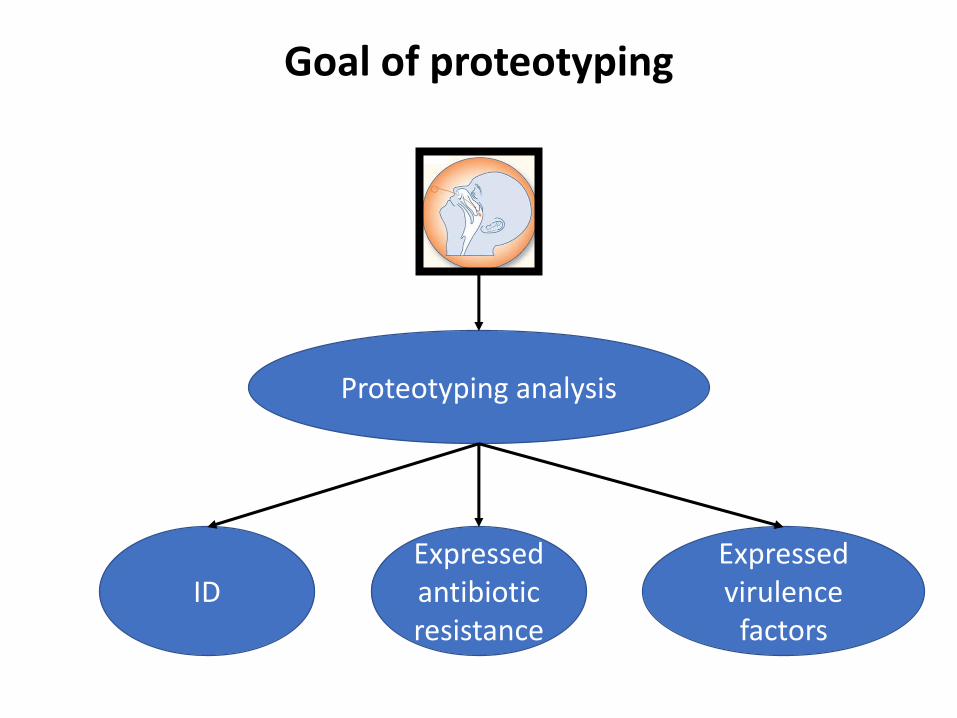
Goal of proteotyping
Proteotyping analysis
IDExpressed antibiotic resistance
Expressed virulence
factors
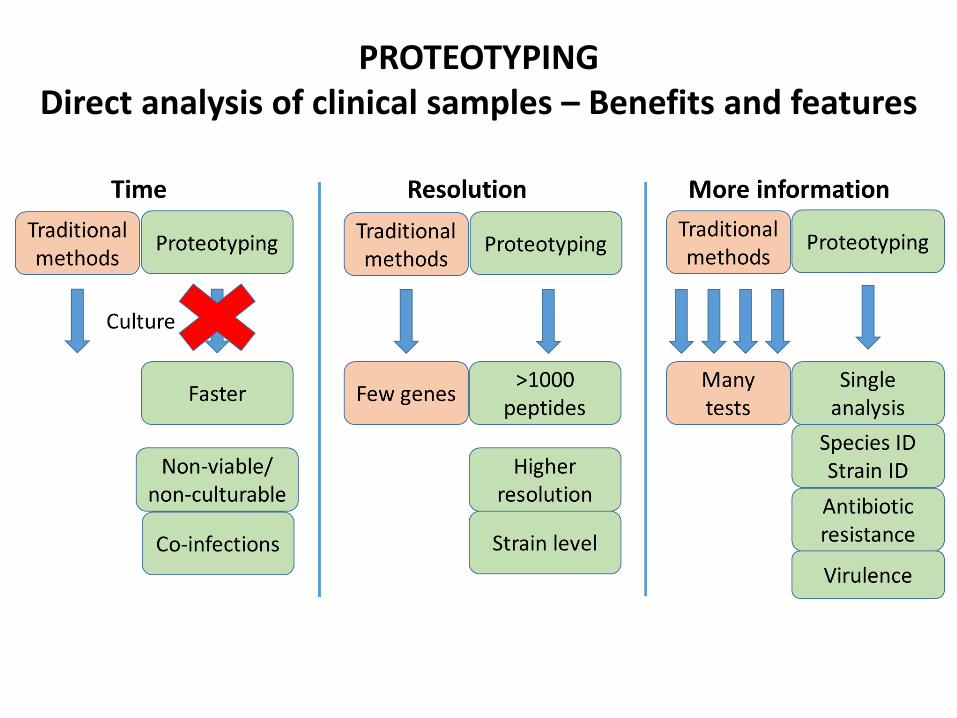
PROTEOTYPINGDirect analysis of clinical samples – Benefits and features

Proteotyping depends on having a high quality
genome database
Conclusions
Several genomes of each species are necessary
More effort should be put on sequencing more
species diversity, including type strains!
Databases must be curated!

Hedvig Engström Jakobsson, Roger Karlsson, Lucia Gonzales Siles, Daniel
Jaén Luchoro, Shora Yazdan, Beatriz Piñeiro, Omar AL-Bayati, Sebastian
Feine, Edward Moore
Acknowledgements
Susann Skovbjerg, Per Sikora, Erika Tång Hallbäck, Christina Åhrén,
Nahid Karami, Liselott Svensson & ”the CCUG ladies”
Margarita Gomila, Antonio Busquets, Francisco Aliaga Lozano, Antoni
Bennasar Figueras
Anna Johnning, Erik Kristiansson
Fredrik Boulund, Kaisa Thorell, Lars Engstrand
Anders Karlsson

¡Muchas gracias!Questions?

Supplementaryslides
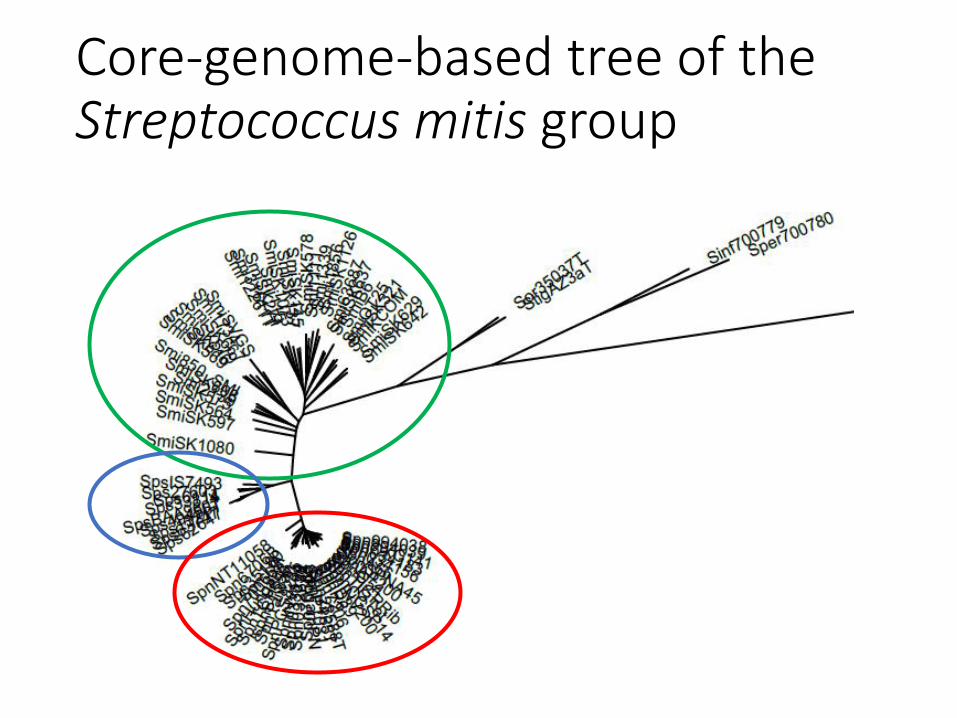
Core-genome-based tree of theStreptococcus mitis group

Taxonomical verification
LC-MS/MS
Match peptides to
sequences
(BLAT)Taxonomic
assignment /
antibiotic
resistance genes
Match
Mass spectra to
peptides
(X!Tandem)
GENOME DBNCBI Taxonomy
DBPEPTIDE DB
ANIb
(JSpecies)
Core genome-based
cluster analysis
Genome sources
All complete genomes from RefSeq
Additional genomes from GenBank
In-house sequenced CCUG strains
Trypsin digestion of proteins
‘Proteotyping’ – Workflow (Open approach)
AMR genes
Proteotyping pipeline (TCUP, Boulund et al. 2017)
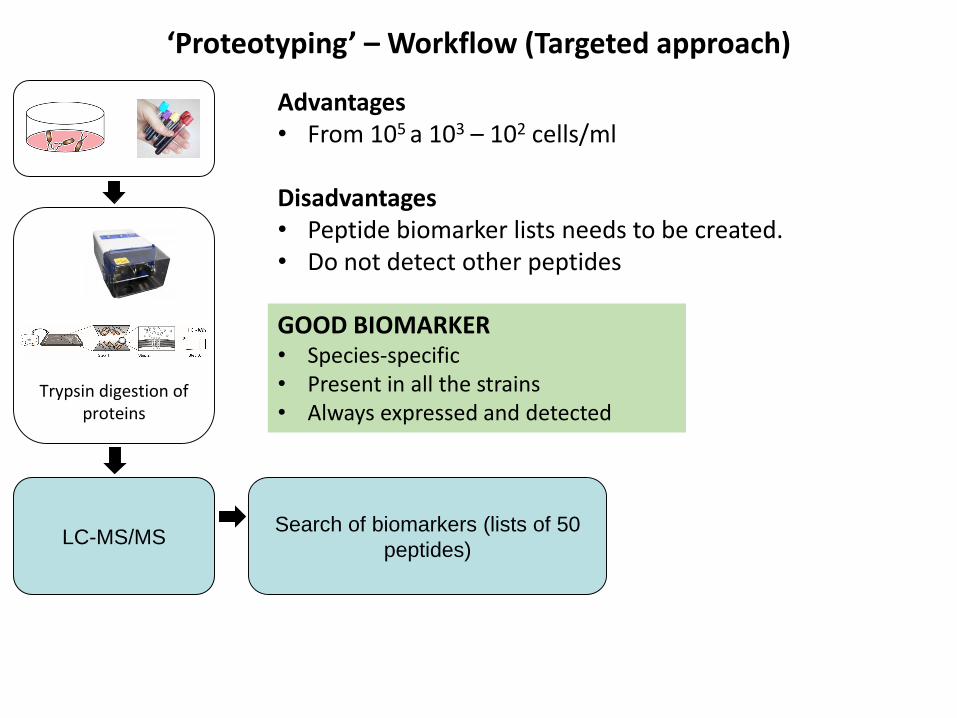
LC-MS/MSSearch of biomarkers (lists of 50
peptides)
Trypsin digestion of proteins
‘Proteotyping’ – Workflow (Targeted approach)
Advantages• From 105 a 103 – 102 cells/ml
Disadvantages• Peptide biomarker lists needs to be created.• Do not detect other peptides
GOOD BIOMARKER• Species-specific• Present in all the strains• Always expressed and detected
http://nanoxisconsulting.com/services-2/lpi-technology-2.html
LPI FlowCell

Minimal ancestry principle
Genus
Species
Family
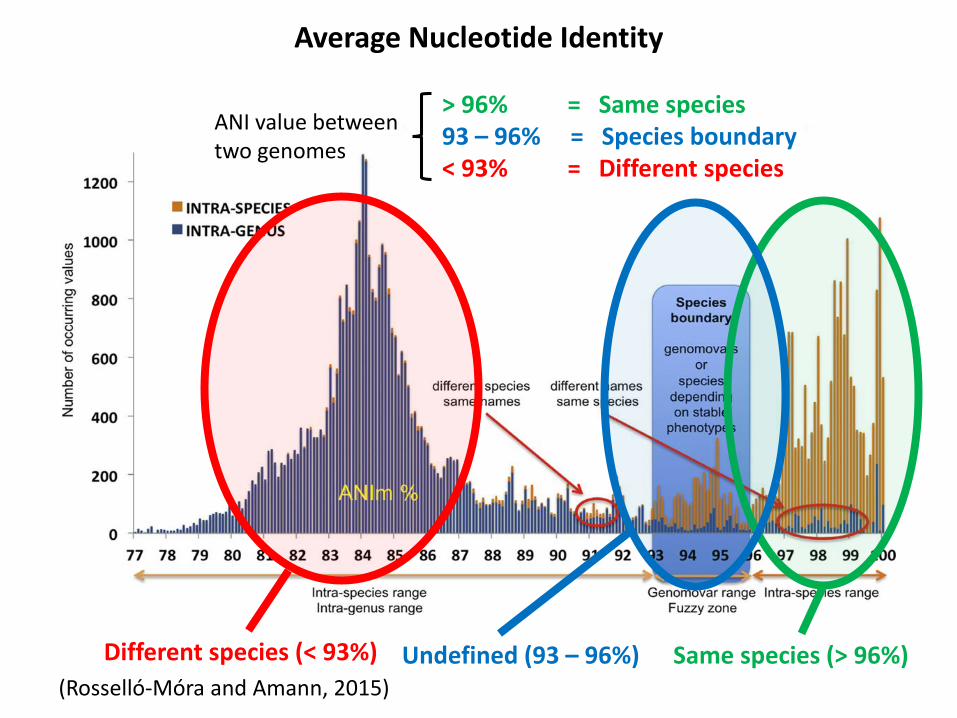
Different species (< 93%) Same species (> 96%)(Rosselló-Móra and Amann, 2015)
Undefined (93 – 96%)
> 96% = Same species93 – 96% = Species boundary< 93% = Different species
ANI value between two genomes
Average Nucleotide Identity