Embed Size (px)
Citation preview

Class 02: Whole genome sequencing

The seminal papers
www.cs.arizona.edu/people/gene/#papers
• ``Is Whole Genome Sequencing Feasible?''
• ``Whole-Genome DNA Sequencing''
• ``A Whole-Genome Assembly of Drosophila''

Shotgun sequencing
• Multiply target sequence
• Break sequences into random fragments
• Sort by size, discard big and small pieces
• ‘Insert into bacterial virus (‘vector’)
• Infect bacterial, and let it reproduce, ‘cloning’ the insert
• ‘Read’ the insert

Definitions
• G – length of target sequence
• L – avg length of read
• R – number of sequencing reads
• N – base pairs sequences = RL
• I – avg length of clone inset
• c – N/G = avg sequence coverage
• m – RI/2G, avg clone or map coverage

Problems
• Incomplete coverage
• Sequencing errors (< .01, avg)
• Unknown orientation
• Repeated sequences

Repeat problem
• Repeats vary in length, number, fidelity• Length: few bp to thousands• Number: highly variable, even by individual• Fidelity: sometimes 1-2% variation, or less
(multiple copies, pseudogenes)• Long, infrequent, hi-fi repeats are the biggest
problem

Overlap phase
• Compare every read (in both orientations) to every other
• Accept weighted agreement, bounded by fixed epsilon
• Exact solution is tractable
• Result is overlap graph, with each read a node, each overlap an edge

Layout phase
• Determine pairs which position each fragment
• In graph theoretic terms, find a spanning forest
• Optimal spanning forest is NP-hard
• Variation on greedy is commonly used

Consensus phase
• Problem: find consensus of multiple alignment of reads
• Initially, use overlaps in the spanning forest
• Apply one of several algorithms to refine this
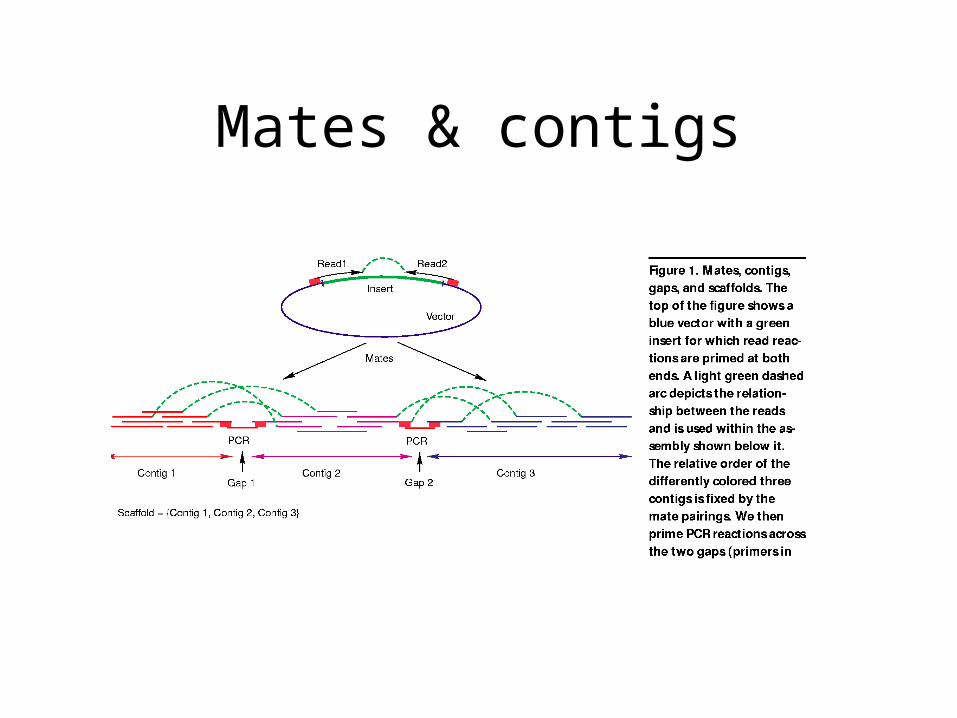
Mates & contigs

‘Double-barreled’ shotgun
• Choose inserts of length at least two ‘reads’
• Sequence both ends (we know their relative orientation and distance)
• Used to order and orient contigs
• Use a supplementary process to fill in the gaps between contigs

Clone by clone (HGP)

Whole genome assembly
• Mates can resolve short repeats
• Problem when you ‘exit’ the repeat: you don’t know which is right
• Resolve using a mate pair which has a read in the unique flanking sequence

Whole genome (illustr)





























