Embed Size (px)
Citation preview

Using Pictorial Structures to Identify Proteins in X-ray Crystallographic Electron Density Maps
Frank DiMaio [email protected] Shavlik [email protected] N. Phillips, Jr. [email protected]
ICML Bioinformatics Workshop21 August 2003
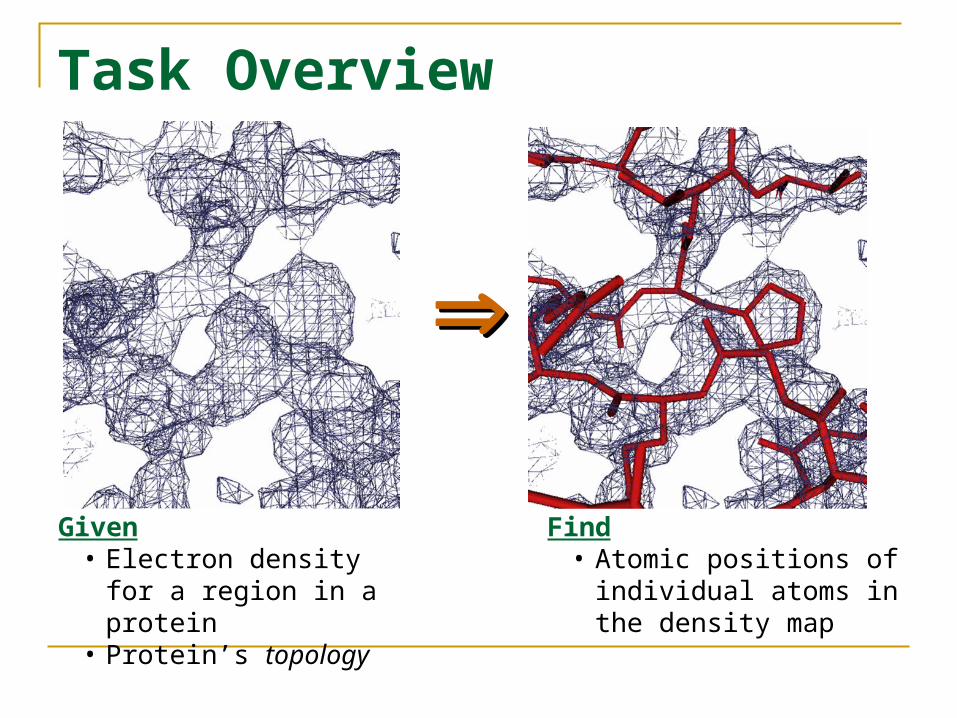
Task Overview
Given • Electron density for a
region in a protein• Protein’s topology
Find• Atomic positions of
individual atoms in the density map
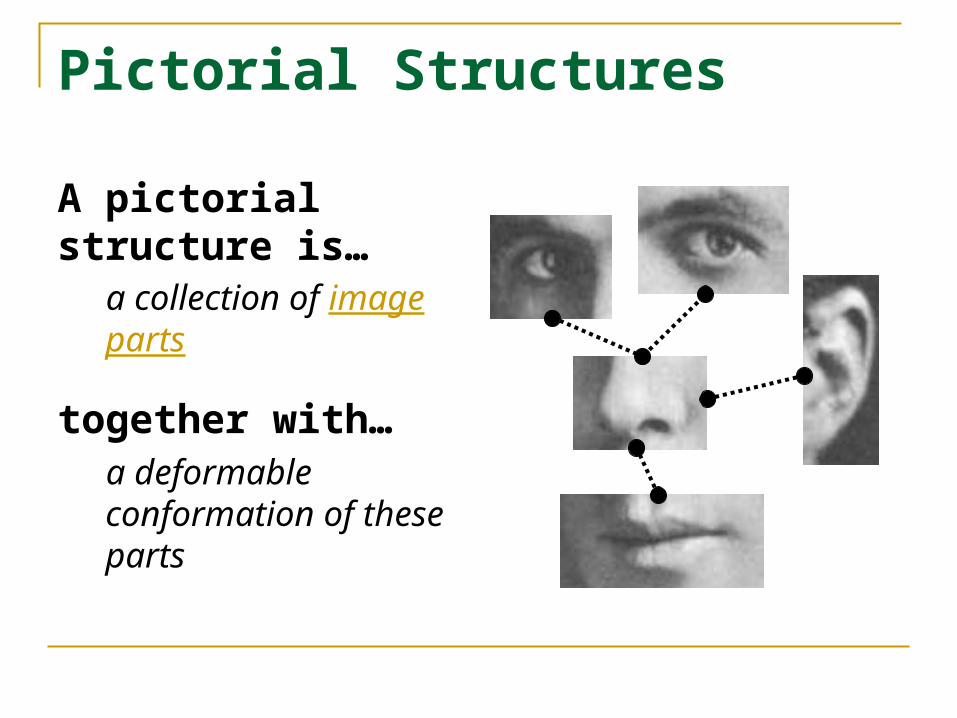
Pictorial Structures
A pictorial structure is…
a collection of image parts
together with…a deformable conformation of these parts
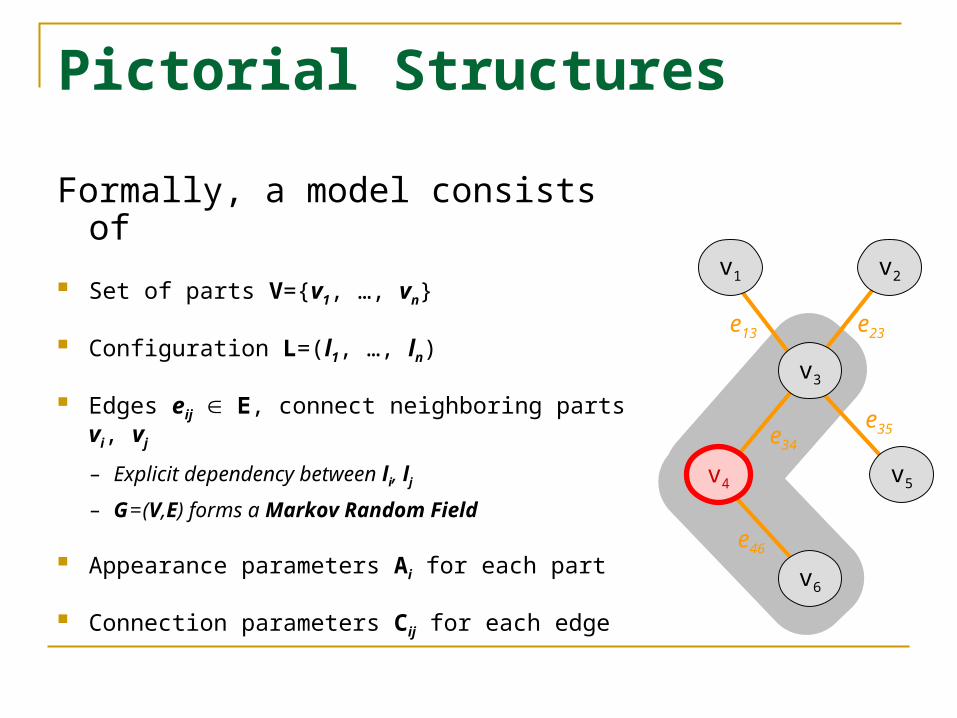
Pictorial Structures
Formally, a model consists of
Set of parts V={v1, …, vn}
Configuration L=(l1, …, ln)
Edges eij E, connect neighboring parts vi, vj
– Explicit dependency between li, lj
– G = (V,E) forms a Markov Random Field
Appearance parameters Ai for each part
Connection parameters Cij for each edge
v3
v4 v5
v6
v1 v2
e13 e23
e34
e35
e46
v4

Matching Algorithm Overview Want configuration L of model Θ maximizing
P(L|I,Θ) P(I|L,Θ) · P(L|Θ)
P(I|L,Θ) = Πi P(I|li,Θ) =1
Z1e- Σi matchi(li)
P(L|Θ) = Π (vi,vj)E P(li,lj|Cij) =1
Z2e- Σ(vi,vj)E dij(li,lj)
Equivalent to minimizing
Σi matchi(li) + Σ(vi,vj)E dij(li,lj)

Linear-Time Matching Algorithm A Dynamic Programming implementation runs in
quadratic time
Requires tree configuration of parts
Felzenszwalb & Huttenlocher (2000) developed linear-time matching algorithm
Additional constraint on part-to-part cost function dij
Basic “Trick”: Parallelize minimization computation over entire grid using a Generalized Distance Transform
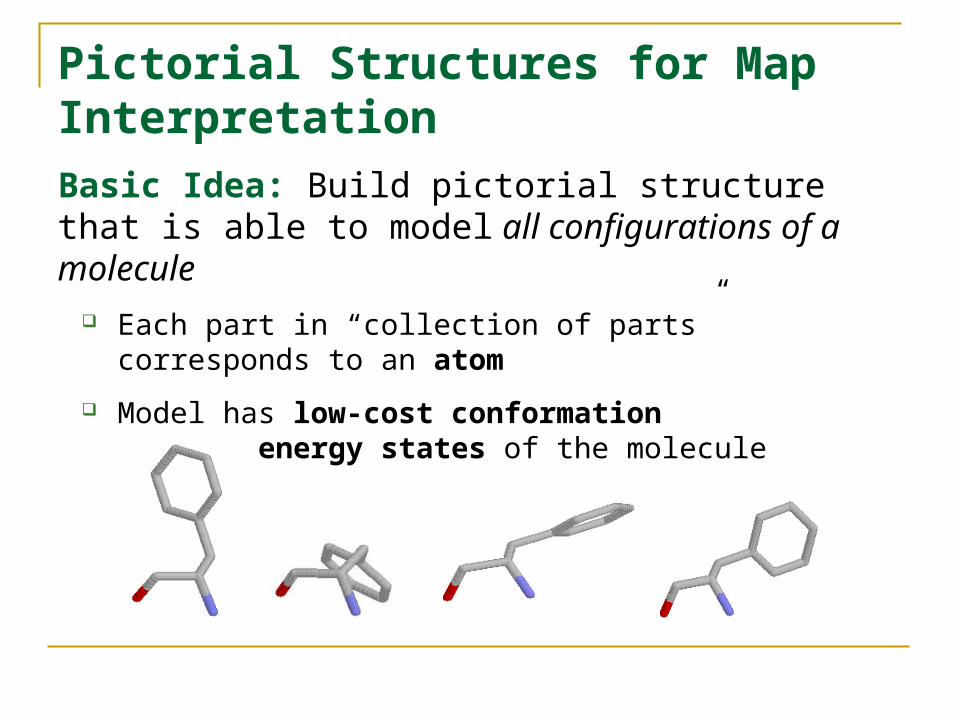
Pictorial Structures for Map InterpretationBasic Idea: Build pictorial structure that is able to model all configurations of a molecule
Each part in “collection of parts” corresponds to an atom
Model has low-cost conformation for low-energy states of the molecule
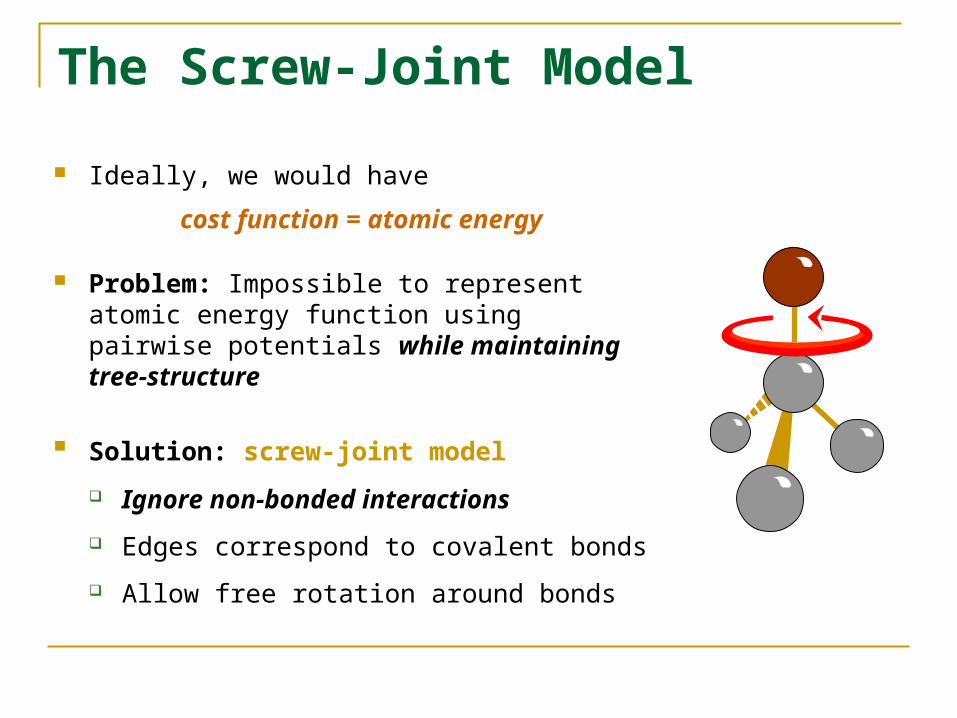
The Screw-Joint Model
Ideally, we would have
cost function = atomic energy
Problem: Impossible to represent atomic energy function using pairwise potentials while maintaining tree-structure
Solution: screw-joint model Ignore non-bonded interactions
Edges correspond to covalent bonds
Allow free rotation around bonds
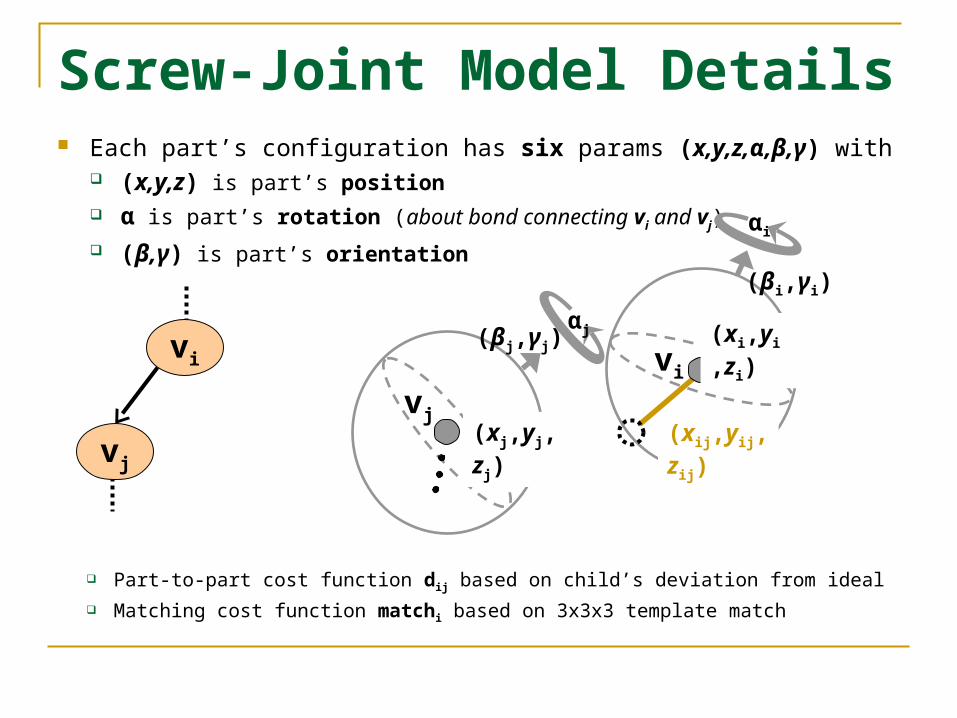
Screw-Joint Model Details Each part’s configuration has six params (x,y,z,α,β,γ) with
(x,y,z) is part’s position α is part’s rotation (about bond connecting vi and vj)
(β,γ) is part’s orientation
vi
vj
vi
vj(xij,yij,zij)
(βi,γi)
(βj,γj) (xi,yi,zi)
(xj,yj,zj)
αj
αi
Part-to-part cost function dij based on child’s deviation from ideal
Matching cost function matchi based on 3x3x3 template match

Pictorial Structures for Map Interpretation
Ideally, we would … Build pictorial structure for the entire protein Run the matching algorithm to get best layout
However, computationally infeasible
Instead, we use two-phase algorithm that …a) computes best backbone trace
b) computes best sidechain conformation(current focus)
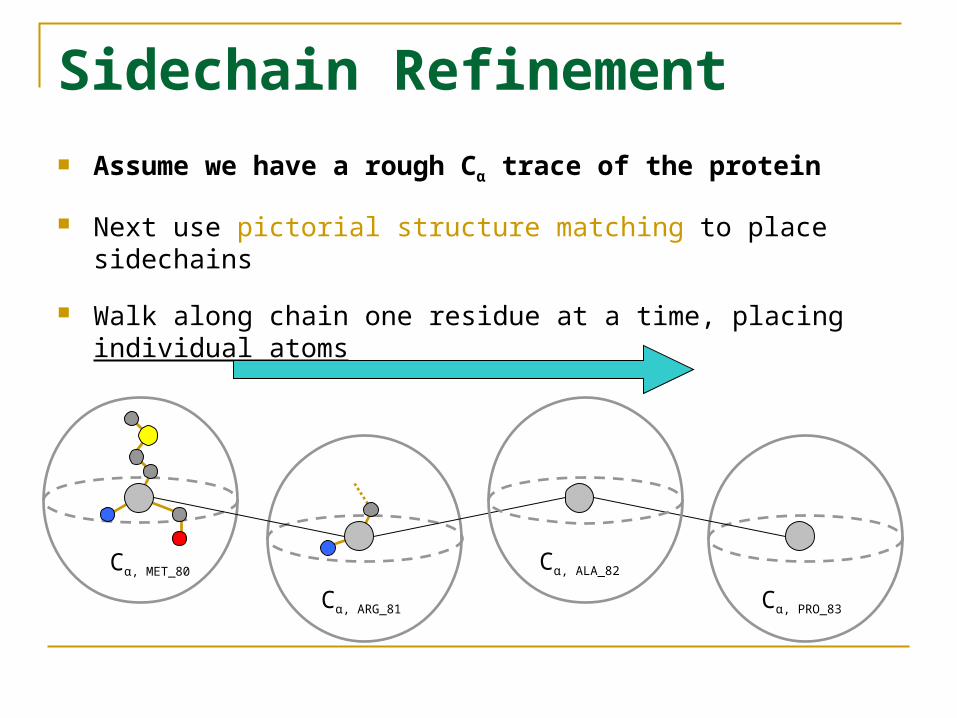
Sidechain Refinement Assume we have a rough Cα trace of the protein
Next use pictorial structure matching to place sidechains
Walk along chain one residue at a time, placing individual atoms
Cα, MET_80
Cα, ARG_81
Cα, ALA_82
Cα, PRO_83
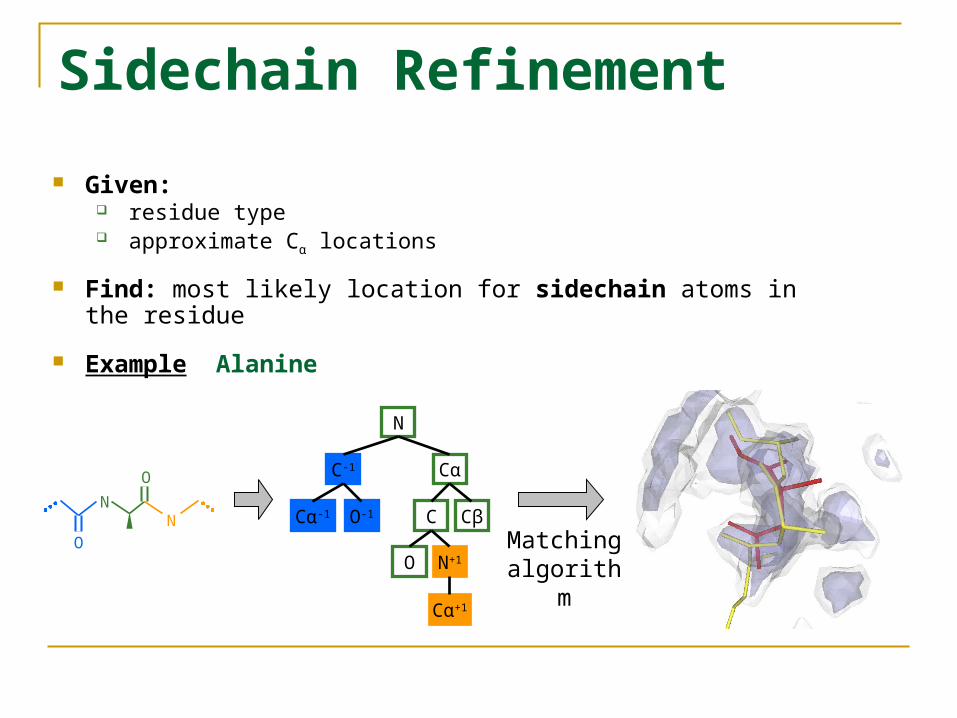
Sidechain Refinement
Given: residue type approximate Cα locations
Find: most likely location for sidechain atoms in the residue
Example Alanine
N
C-1 Cα
Cα-1 O-1 C Cβ
O
Cα+1
N+1
O
N NO Matching
algorithm
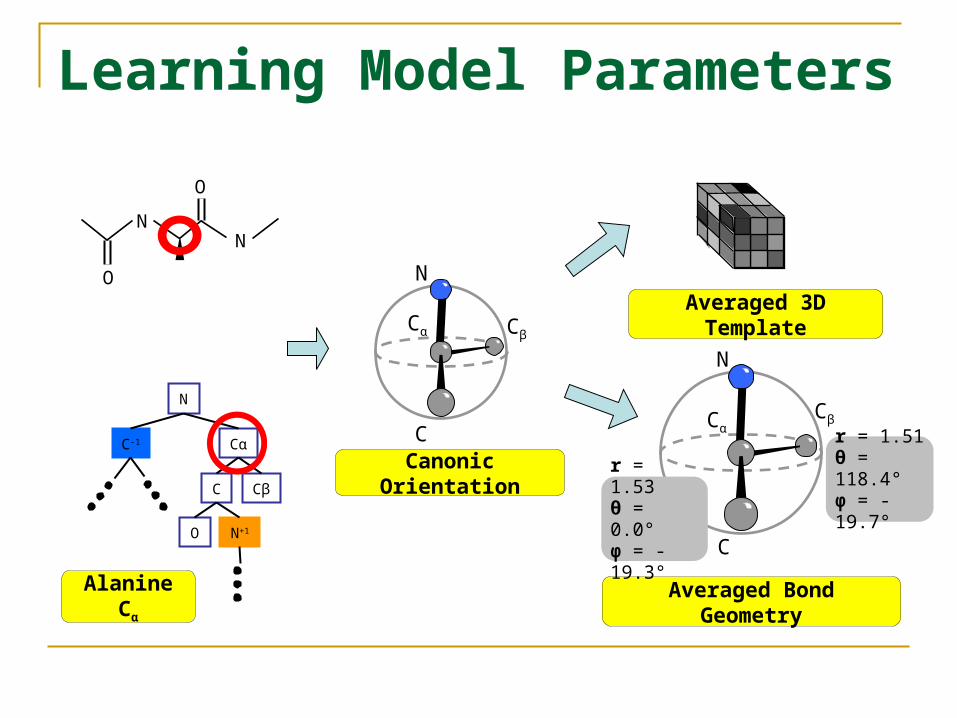
Learning Model Parameters
O
N N
OC
Cα
N
CβAveraged 3D Template
Averaged Bond Geometry
Canonic Orientation
N
C-1 Cα
C Cβ
O N+1
Alanine Cα
C
Cα
N
Cβ
r = 1.53θ = 0.0°φ = -19.3°
r = 1.51θ = 118.4°φ = -19.7°
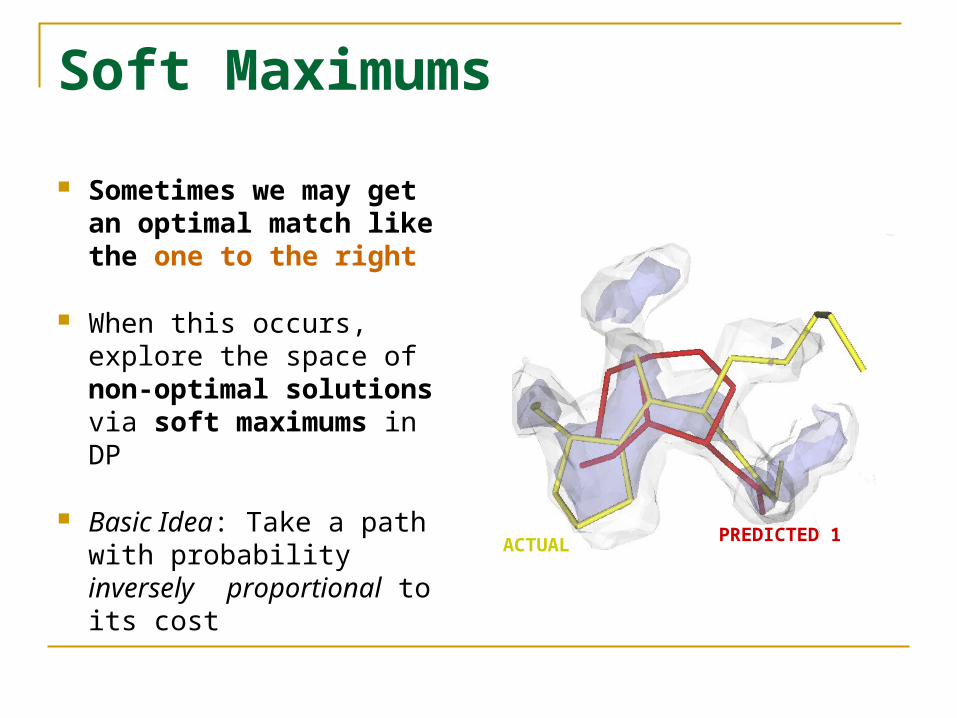
Soft Maximums
Sometimes we may get an optimal match like the one to the right
When this occurs, explore the space of non-optimal solutions via soft maximums in DP
Basic Idea: Take a path with probability inversely proportional to its cost
ACTUAL PREDICTED 1
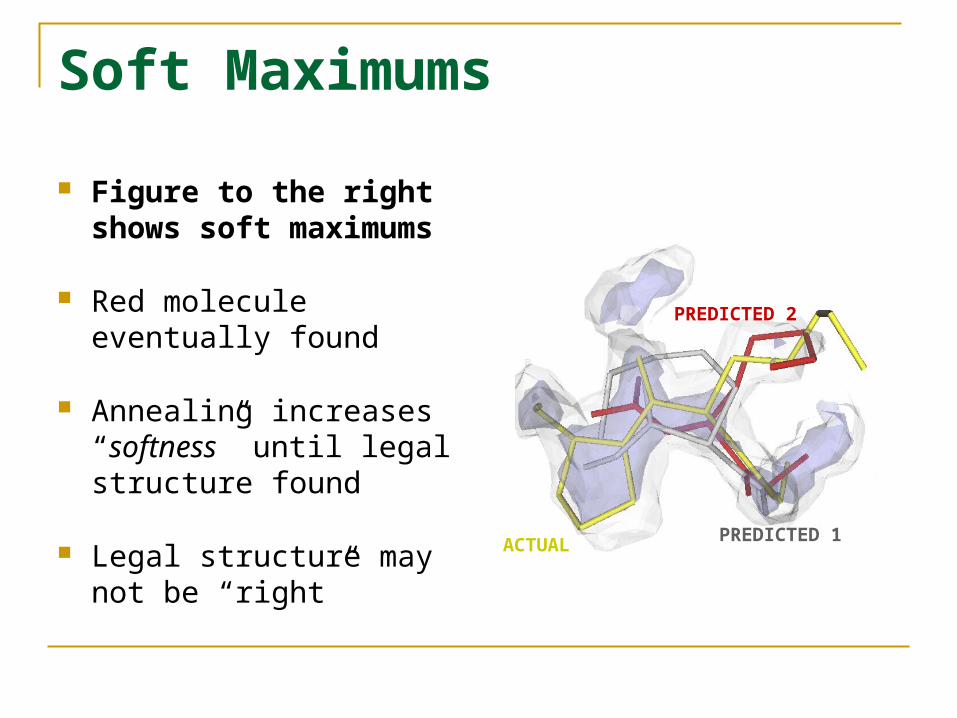
Soft Maximums
Figure to the right shows soft maximums
Red molecule eventually found
Annealing increases “softness” until legal structure found
Legal structure may not be “right”
ACTUAL PREDICTED 1
PREDICTED 2

Results
Only sidechain refinement implemented & tested Experimental Methodology
Assume Cα’s known to within 2Å
Trained on 1.7 Å resolution protein, tested on 1.9 Å resolution protein
Templates built for ALA, VAL, TYR, LYS
Model Parameters Grid spacing of 0.5 Å within diameter 10 Å sphere Rotational discretization:
12 rotational steps 84 orientations
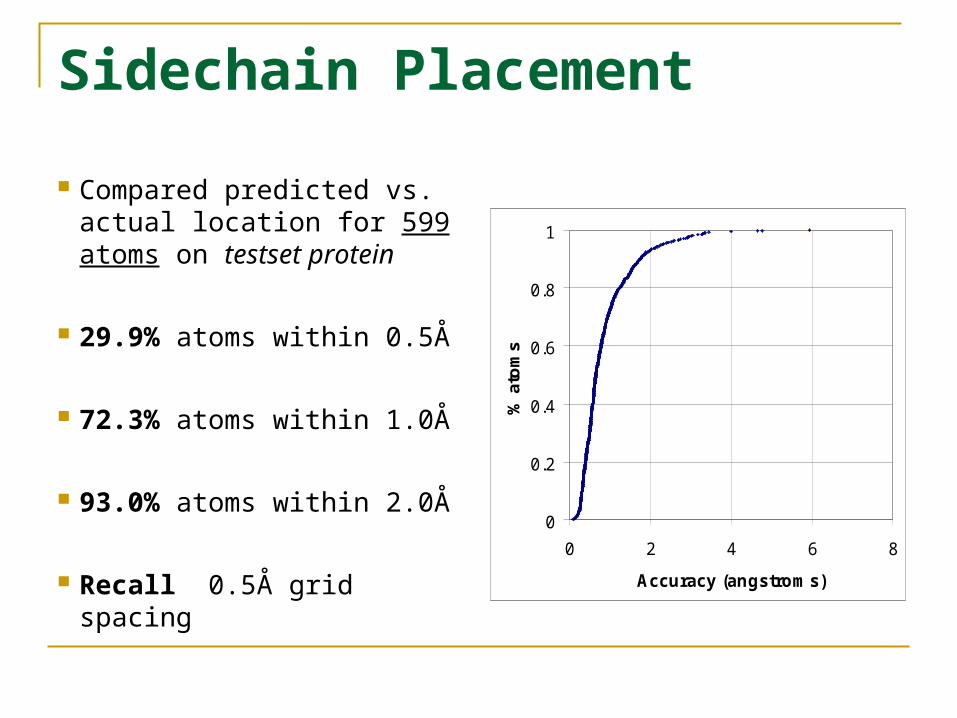
Sidechain Placement
Compared predicted vs. actual location for 599 atoms on testset protein
29.9% atoms within 0.5Å
72.3% atoms within 1.0Å
93.0% atoms within 2.0Å
Recall 0.5Å grid spacing
0
0.2
0.4
0.6
0.8
1
0 2 4 6 8
Accuracy (angstroms)
% a
tom
s
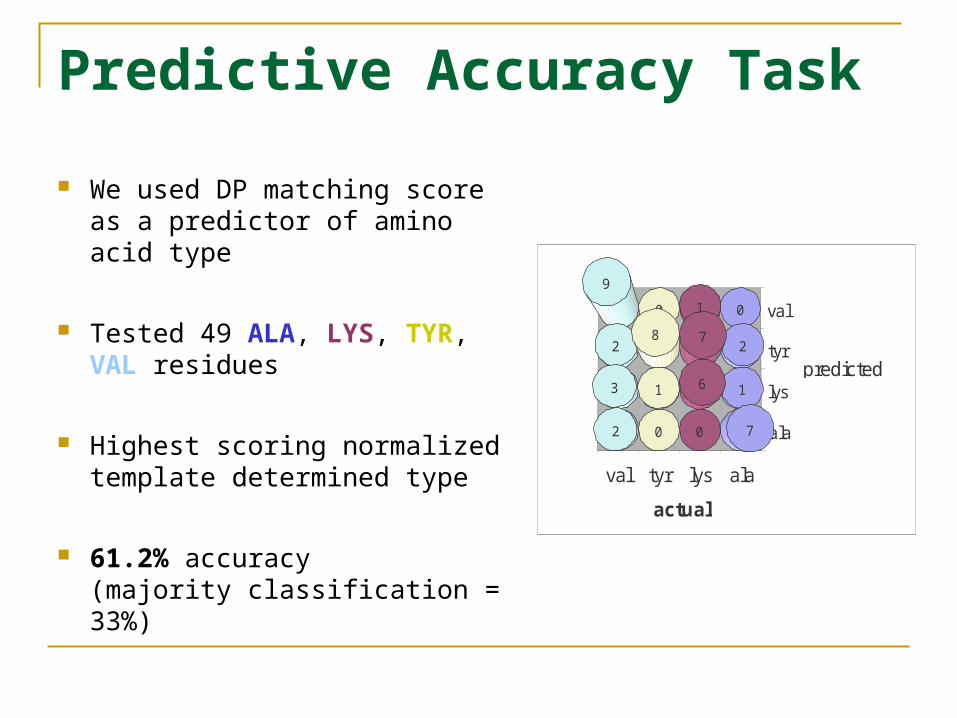
Predictive Accuracy Task
We used DP matching score as a predictor of amino acid type
Tested 49 ALA, LYS, TYR, VAL residues
Highest scoring normalized template determined type
61.2% accuracy (majority classification = 33%)
ala
lys
tyr
val
alalystyrval
0
2
1
7
1
7
6
0
9
2
3
2
0
8
1
0
actual
predicted
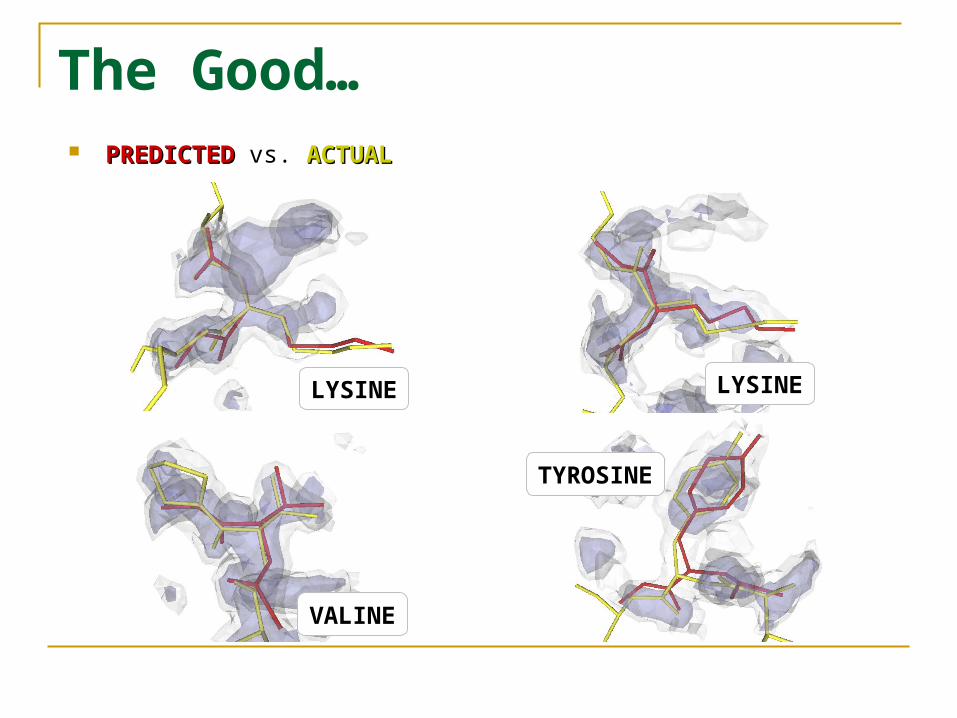
The Good… PREDICTEDPREDICTED vs. ACTUALACTUAL
LYSINELYSINE
VALINE
TYROSINE
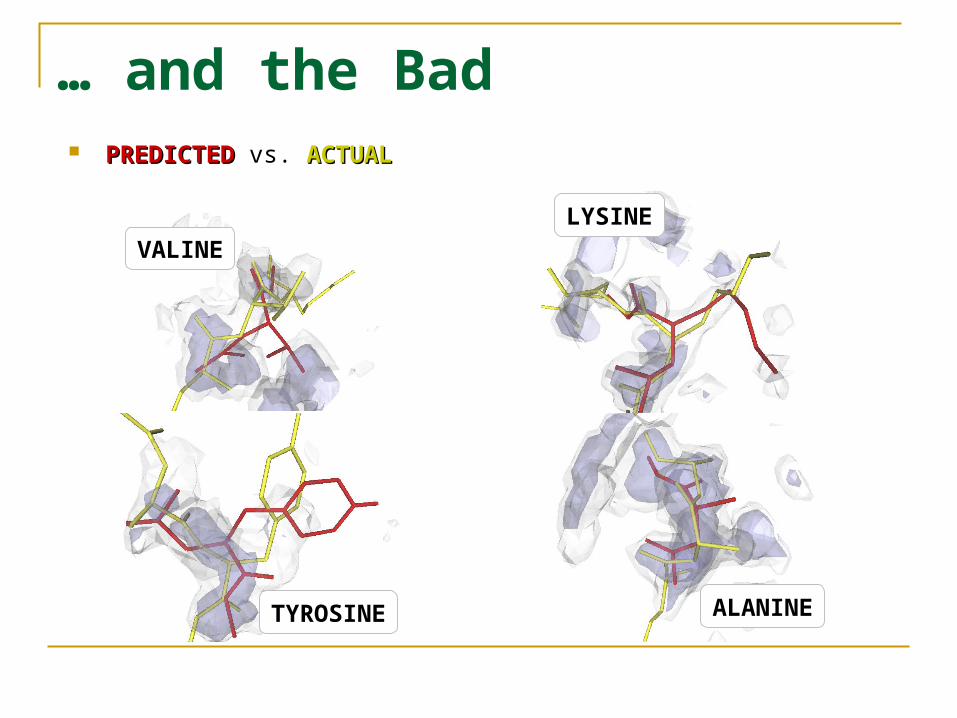
… and the Bad PREDICTEDPREDICTED vs. ACTUALACTUAL
LYSINE
ALANINETYROSINE
VALINE

Future Work
Implement & integrate backbone tracing algorithm, to create complete two-tiered solution
Better strategies to handle illegal molecule configurations perturbation of branches involved in collisions
more accurate representation of atomic energy function, e.g. torsion angle
Better match function … make use of previous work?
More tests (larger training set, higher resolution)

Acknowledgements
NLM grant 1T15 LM007359-01
NLM grant 1R01 LM07050-01
NIH grant P50 GM64598.





























