Embed Size (px)
Citation preview

IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIUS009322055B2
lI2j United States PatentJanaway et al.
(10) Patent No. : IJS 9,322,055 B2(45) Date of Patent: Apr. 26, 2016
(54) SYSTEM AND METHOD FOR DETERMININGCOPIES-PER-UNIT-VOLUME USING PCRAND FLOW CONTROL OF DROPLETS
(56) References Cited
U.S. PATENT DOCUMENTS
(75) Inventors: Gordon A. Janaway, Hayward, CA(US); Mark Andersen, Carlsbad, CA(US); Kornelija Zgonc, Carlsbad, CA(US); Michael Pallas, San Bruno, CA(US); Marcin Sikora, Foster City, CA(US); Casey McFarland, San Francisco,CA (US); Ferrier N. Le, San Jose, CA(US); Haopeng Wang, Carlsbad, CA(US); Jian Gong, San Marcos, CA (US);Gothami Padmabandu, San Diego, CA(US)
(73) Assignee: Life Technologies Corporation,Carlsbad, CA (US)
( * ) Notice: Subject to any disclaimer, the term of thispatent is extended or adjusted under 35U.S.C. 154(b) by 47 days.
(21) Appl. No 4 14/009, 304
(22) PCT Filed: Mar. 30, 2012
(86) PCT Noz PCT/US2012/031533
) 371 (c)(I),(2), (4) Date: Oct. 1, 2013
(87) PCT Pub. No 4 WO2012/135667
PCT Pub. Date: Oct. 4, 2012
(65) Prior Publication Data
US 2014/0248623 Al Sep. 4, 2014
Related U.S.Application Data
(51) Int. Cl.C12Q 1/68 (2006.01)C12P 19/34 (2006.01)
(52) U.S. Cl.CPC ...................................... C12Q 1/686 (2013.01)
(58) Field of ClassiTication SearchCPC ............... C12Q I/686; C12Q 2537/16; C12Q
2563/143; C12Q 2563/159; C12Q 2565/626See application file for complete search history.
(60) Provisional application No. 61/470, 713, filed on Apr.I, 2011, provisional application No. 61/481, 085, filedon Apr. 29, 2011.
2005/0042639 Al * 2/2005 Knapp et al. . ... 435/6
FOREIGN PATENT DOCUMENTS
WOWOWOWO
98/470032007/0247982007/1494322010/117461
10/19983/2007
12/200710/2010
OTHER PUBLICATIONS
Kojima et al. , "PCR amplification from single DNA molecules on
magnetic beads in emulsion; application for high-throughput screen-
ing of transcription factor targets, "Nucleic Acids Research, 2005,vol. 33, No. 17, eI50, pp. 1-9.*International Search Report and Written Opinion of the ISA forInternational Appl. No. PCT&S2012/031533 dated Jun. 12, 2012.Curcio, Mario et al. , "Continuous segmented-Flow PolymeraseChain Reaction for High-Throughput Miniaturized DNA Amplifica-tion", Anal. Chem, vol. 75l ll, American Chemical Society, 2003, 1-7.Dorfman, Kevin D, et al. , "Contamination-free Continuous FlowMicrofluidic Polymerase Chain Reaction for Quantitative and Clini-cal Applications", Analytical Chemistry, American Chemical Soci-ety, vol. 77, No. 11, Jun. 2005, 3700-3704.
* cited by examiner
Primary Examiner Young J Kim
(57) ABSTRACT
22 Claims, 34 Drawing Sheets
Methods and systems for quantification of a target nucleicacid in a sample are provided. The method includes forminga plurality ofdiscrete sample portions. Each of the plurality ofdiscrete sample portions comprising a portion of the sample,and a reaction mixture. The method further includes ampli-fying the plurality of discrete sample portions to form a plu-rality of discrete processed sample portions. At least onediscrete processed sample portion containing nucleic acidamplification reaction products. Fluorescence signals aredetected from the at least one of the plurality of discreteprocessed sample portions to determine a presence of the atleast one target nucleic acid. The method also includes deter-mining the respective volumes of the plurality of the pluralityof discrete processed sample portions, and estimating thenumber of copies-per-unit-volume of the at least one targetnucleic acid in the sample. Estimating the number of copies-per-unit-volume is based on the number of discrete processedsample portions determined to contain the at least one targetnucleic acid therein.

U.S. Patent Apr. 26, 2016 Sheet 1 of 34 US 9,322,055 B2
Q3
g
LL Q LU
Q —H(o ~ ~Qu@
~KBQ~ Q Z. CL
QE
CL
LU ~Qr~&&(LU(IE + M
CL
~ (s & 9&- U Q &H Q LU U
L~ (0LU CL
P.LU U CLK Clb
LU
UC, LU
CO
QCL
CU LU
I-. =
Q C3 &Q ~U
& ((E w
Q D
LLI Q LU
CL
~ LU ~Q CU
L'- + Z&g K Q
g ((I LU
CO LU ~U '.
UCL
(3 LLI
(OI ULU QEL
Q U ~~~ Q)
(I
CL
CL
LLILLI
Z.
(0
I- !'
LLI CC' LU
I- CC I
Z.
D LU Z-H X Q LLI
QZ&QQW—Ci Q g
D
~ LU LLI
LLI g LLI
CL Q + ZQ Ki lÃ
LL ~Q@Q E- E- g
Q w~ Q CL
~ VLLI
ZQCCLU CU~~m@ ~ C3
fÃ!—(CE
—cr Q(o + +

U.S. Patent Apr. 26, 2016 Sheet 2 of 34 US 9,322,055 B2
LU
fQ
D 003 gerLU —DC3 &-!-
&COQk- CD U-
03~Qg
QHOD
~ CD LUQ + CDUz:&Z LU KQ Q CL
"L U)
0 CL" Q&KCL" DgUZ:Q
D- r-—Z Q
CD
Q CDU CD
gjQ
Q D
C!
!—Q
~ UU
Q CO
Z Q
CD ~QQ ~~ Ã
—CL
C!
(OLU
QQLU
CL CL
LU ~LU
Z.
Q
LU QCD
Z.
CQ
Z.
RCoLU

U.S. Patent Apr. 26, 2016 Sheet 3 of 34 US 9,322,055 B2
e:

U.S. Patent Apr. 26, 2016 Sheet 4 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 5 of 34 US 9,322,055 B2
0
U.S. Patent Apr. 26, 2016 Sheet 6 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 7 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 8 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 9 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 10 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 11 of 34 US 9,322,055 B2
8

U.S. Patent Apr. 26, 2016 Sheet 12 of 34 US 9,322,055 B2
C
6
eg
8 8 8
U.S. Patent Apr. 26, 2016 Sheet 13 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 14 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 15 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 16 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 17 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 18 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 19 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 20 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 21 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 22 of 34 US 9,322,055 B2

U.S. Patent Apr. 26, 2016 Sheet 23 of 34 US 9,322,055 B2
U!
8
2
0U!
UI
Q
4J
II
LA M ILA LA LA M& LA LAN ~ N
LA C3 ILA U! UA LAC3

U.S. Patent Apr. 26, 2016 Sheet 24 of 34 US 9,322,055 B2
LA
CJCA
LI J
\U
LA\U
LA
\U
LA
Ig
2
N
LA
\U
Li. L;L.
LA C3 LA
r-LA LA CJ LA LA LA C) LA LA
O'J
LA LA
C3

U.S. Patent Apr. 26, 2016 Sheet 25 of 34 US 9,322,055 B2
I~ LI
LA M UA
F) LJ
LA LA Cl LA LA LA C3 UA LA
r- L-.i r-U! LA
C3

U.S. Patent Apr. 26, 2016 Sheet 26 of 34 US 9,322,055 B2
X
LA
LA
r-
:V V
L j
LA
2
C
LA C3 LA LA LA M LA LA LAI
C3 LA LA LA LA
r~C3

U.S. Patent Apr. 26, 2016 Sheet 27 of 34 US 9,322,055 B2
OV:V
LA
LA
LA CI IA
Cd
I
LA LA C3 LA LA LA M LA LA LA LA
r- m IC3

U.S. Patent Apr. 26, 2016 Sheet 28 of 34 US 9,322,055 B2
I
LA lA W lA LA LA W LA LA LA LA

U.S. Patent Apr. 26, 2016 Sheet 29 of 34 US 9,322,055 B2
re
rA
GV r J
N
Q
4J
LI! OIU
II I II
LA LA UJ iH UJ LA
r-. ~ AJ ~ r-LA H LA
II
ILA LA UA
C3

U.S. Patent Apr. 26, 2016 Sheet 30 of 34 US 9,322,055 B2
Lj j
OU V
Lj j
L-A
Q
4J
LA C3 LAI
LA LA M LA LA LA C3 LA LA LA
CJC)

U.S. Patent Apr. 26, 2016 Sheet 31 of 34 US 9,322,055 B2
GV
LA
LA
LA C3 LA LA LA C) LA LA LA C3 LAN ~ N
LA LA LA
O'JC3

U.S. Patent Apr. 26, 2016 Sheet 32 of 34 US 9,322,055 B2
Uj
N
Uj
LA IW LA
CA C'0
I
UA LA W LA LA LA KI LA LA
C'4
UA LA
IUC3

U.S. Patent Apr. 26, 2016 Sheet 33 of 34 US 9,322,055 B2
LI I
LI I
\U
IjI!:~l
UJ
U I
LI I
SIg
F
NCl
Cl
LrI
LrI LrI UrJ W UrJ LrI U I W LrI LrI
( J

U.S. Patent Apr. 26, 2016 Sheet 34 of 34 US 9,322,055 B2

US 9,322,055 B21
SYSTEM AND METHOD FOR DETERMININGCOPIES-PER-UNIT-VOLUME USING PCR
AND FLOW CONTROL OF DROPLETS
CROSS-REFERENCE TO RELATEDAPPLICATIONS
This application is a National Stage entry from PCT Appli-cation No. PCT/US2012/031533 filed Mar. 30, 2012, whichclaims a benefit under 35 USC )119(e)from U.S.ProvisionalApplication Nos. 61/470, 713,
filed
Ap. I, 2011, and 61/481,085, filed Apr. 29, 2011; all applications are incorporatedherein by reference in their entirety.
BACKGROUND
The present disclosure generally relates to systems andmethods for carrying out digital polymerase chain reaction(dPCR) assays. The disclosure further relates to controllingthe flow of droplets dispersed in a carrier fluid through aconduit.
Digital Polymerase Chain Reaction (dPCR) is a methodthat has been described, for example, in U.S. Pat. No. 6, 143,496 to Brown et al. Results from dPCR can be used to detectand quantify the concentration of rare alleles, to provideabsolute quantitation ofnucleic acid samples, and to measurelow fold-changes in nucleic acid concentration.
dPCR is often performed using apparatus adapted fromconventional qPCR, in which replicates are arrayed in a twodimensional array format including m rows by n columns,i.e., an mxn format. PCR cycling and read-out (end-point orreal-time) generally occurs within the same array. A maxi-mum ofmxn replicates can be processed in a single batch run.Generally, increasing the number of replicates increases theaccuracy and reproducibility of dPCR results.
The (mxn) format in most quantitative polymerase chainreaction (qPCR) platforms is designed for sample-by-assayexperiments, in which PCR results need to be addressable forpost-run analysis. For dPCR, however, the specific position orwell of each PCR result may be immaterial and only thenumber ofpositive and negative replicates per sample may beanalyzed.
The read-out of dPCR, that is, the number of positivereactions and the number of negative reactions, is linearlyproportional to the template concentration, while the read-outof qPCR (signal vs. cycle) is proportional to the log of thetemplate concentration. For this reason, dPCR typically isconstrained to a narrow dynamic range of template input.
SUMMARY
According to various embodiments, the present teachingsprovide methods and systems for quantification of a targetnucleic acid in a sample. The method includes forming aplurality of discrete sample portions. Each of the plurality ofdiscrete sample portions comprising a portion of the sample,and a reaction mixture. The method further includes ampli-
fying the plurality of discrete sample portions to form a plu-rality of discrete processed sample portions. At least onediscrete processed sample portion containing nucleic acidamplification reaction products. Fluorescence signals aredetected from the at least one of the plurality of discreteprocessed sample portions to determine a presence of the atleast one target nucleic acid. The method also includes deter-mining the respective volumes of the plurality of the pluralityof discrete processed sample portions, and estimating thenumber of copies-per-unit-volume of the at least one target
10
20
25
30
35
40
45
50
55
60
65
nucleic acid in the sample. Estimating the number of copies-per-unit-volume is based on the number of discrete processedsample portions determined to contain the at least one targetnucleic acid therein.
In some embodiments, the plurality of discrete sampleportions may comprise sample portions of different sizes orall of the same size. Each of the plurality of discrete sampleportions may comprise a volume of an aqueous reactionmedium at least partially surrounded by a medium that is atleast substantially immiscible with the plurality of discretesample portions. As used herein, sample portions may bereferred to as droplets, sample volumes, or reactions volumes,for example. The medium that is substantially immisciblewith the plurality of discrete sample portions may compriseone or more of a mineral oil, a silicone oil, a paraffin oil, afluorinated fluid, a perfluorinated polyether, and a combina-tion thereof. In some embodiments, the plurality of discretesample portions comprise magnetic beads and the method
may further comprise magnetically focusing the magneticbeads within a flow stream in a flow cytometer. In someembodiments, the plurality of sample portions may comprisenon-magnetic beads, including porous or hollow beads, forexample. The porous or hollow beads may be spherical orcylindrical. One or more of the plurality of sample portionsmay comprise a passive reference dye, a light-scatteringenhancing material, q-dots, a protein, a colloidal metal, col-loidal gold, a reporter dye, a reaction-independent flowmarker, or a combination thereof. In some embodiments, oneor more sample discrete portions of the plurality of sampleportions comprises a primer pair, a nucleotide probe, and aTaq polymerase.
In another embodiment, a system for quantification of atarget nucleic acid in a sample is provided. The systemincludes an emulsion apparatus configured to form a pluralityof discrete sample portions. Each of the plurality of discretesample portions includes a portion of the sample, and a reac-tion mixture. The system further includes an amplificationapparatus configured to amplify the plurality of discretesample portions to form a plurality of discrete processedsample portions, including at least one discrete processedsample portion containing nucleic acid amplification reactionproducts. The system also includes excitation detection appa-ratus configured to detect fluorescence signals from the atleast one of the plurality of discrete processed sample por-tions to determine a presence of the at least one target nucleicacid. Furthermore, the system includes a processor config-ured to estimate the number of copies-per-unit-volume of theat least one target nucleic acid in the sample based on thenumber of discrete processed sample portions determined tocontain the at least one target nucleic acid therein.
BRIEF DESCRIPTION OF THE DRAWINGS
A better understanding of the features and advantages ofthe present disclosure will be obtained by reference to theaccompanying drawings, which are intended to illustrate, notlimit, the present teachings.
FIG. 1 is a flow diagram ofan exemplary method accordingto various embodiments of the present teachings.
FIG. 2 is a flow diagram of another exemplary methodaccording to various embodiments of the present teachings.
FIG. 3 is a schematic diagram of an exemplary system andmethod according to various embodiments of the presentteachings.
FIG. 4 is a block diagram of an exemplary system accord-ing to various embodiments of the present teachings.

US 9,322,055 B2
FIG. 5A depicts a plurality of discrete sample portionsaccording to various embodiments of the present teachings.
FIG. 5B depicts a plurality of discrete processed sampleportions according to various embodiments of the presentteachings.
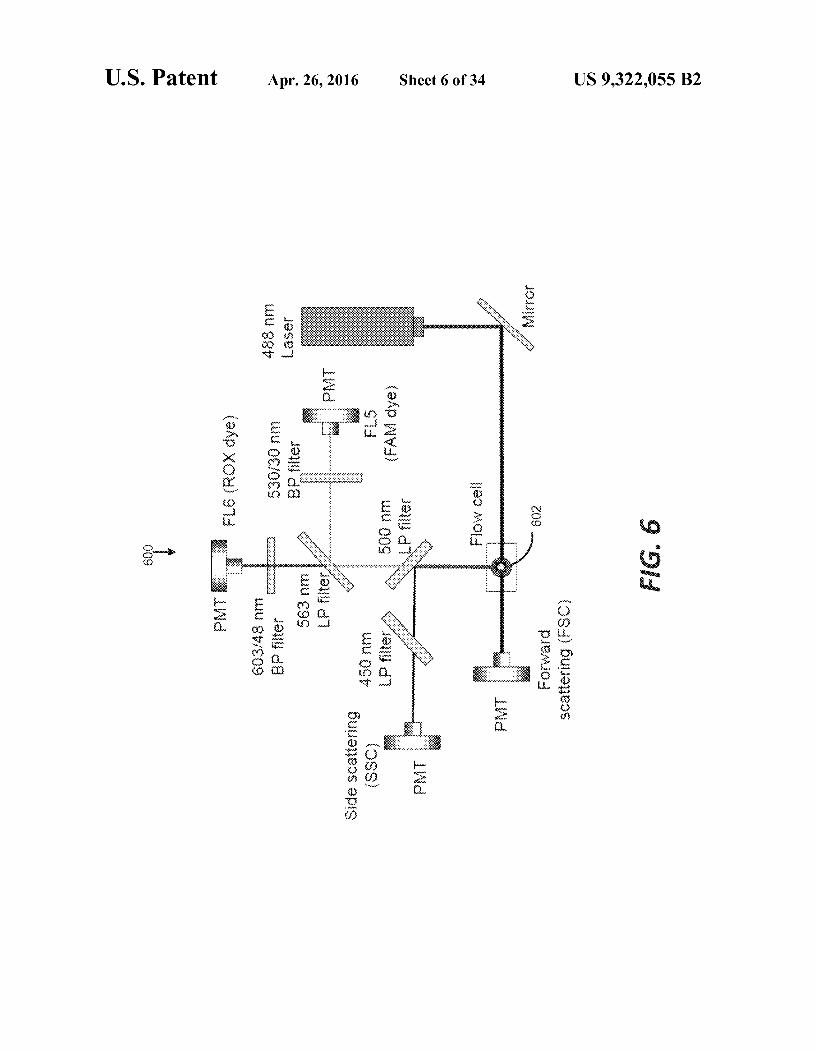
FIG. 6 illustrates an exemplary optical setup using flowcytometry to analyze a plurality of discrete sample portionsaccording to various embodiments of the present teachings.
FIG. 7A illustrates flow data of samples that included notemplate control according to various embodiments of thepresent teachings.
FIG. 7B illustrates flow data of samples including a tem-plate control according to various embodiments of the presentteachings.
FIGS. SA and SB illustrate a three layered emulsionaccording to various embodiments of the present teachings.
FIG. 9 illustrates a method of preparing a sample accordingto various embodiments of the present teachings.
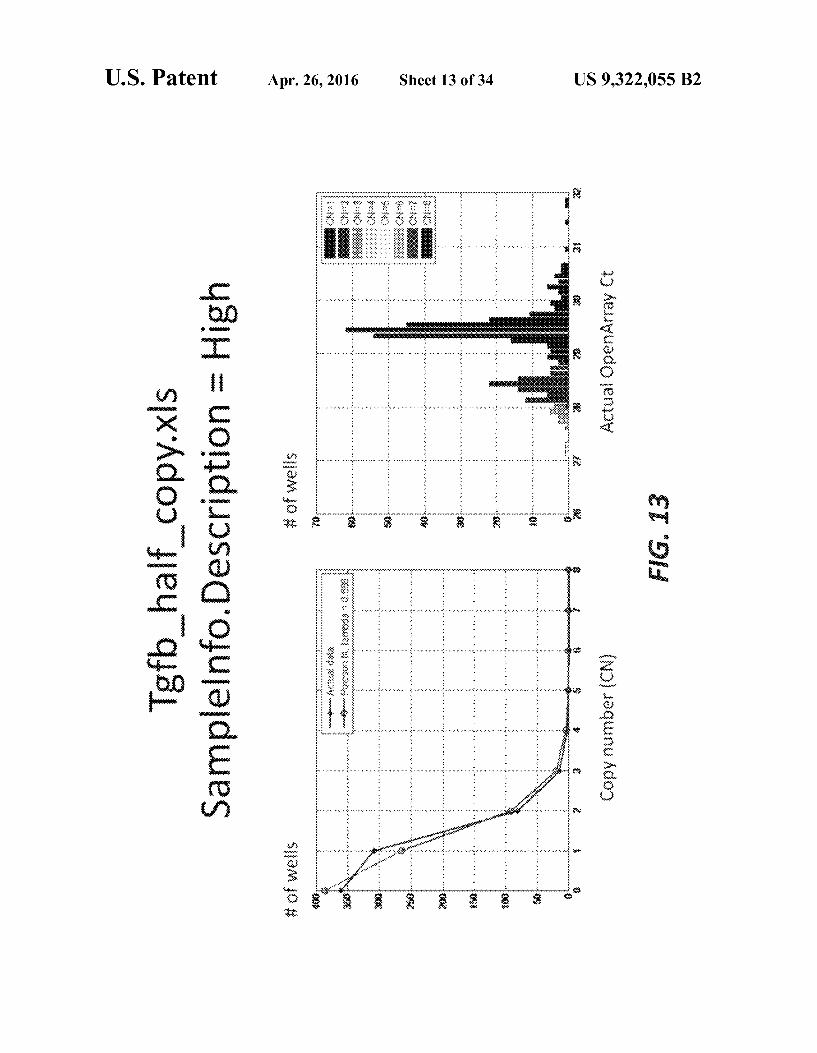
FIGS. 10-15 illustrate exemplary plots showing a relation-ship between C and copy number according to variousembodiments of the present teachings.
FIGS. 16-21 illustrate exemplary modeled C plots versusactual C plots.
FIGS. 22-33 illustrate exemplary C distributions accord-ing to various embodiments of the present teachings.
FIG. 34 illustrates an exemplary histogram for determiningPCR efficiency according to various embodiments of thepresent teachings.
DETAILED DESCRIPTION
The systems and methods described herein provides adPCR system and method that does not rely on the fixed(mxn) format and instead uses droplet flow control to greatlyincreases the number ofdPCR replicates that can be obtainedwhile also obtaining a tally of positive and negative resultsuseful for dPCR. Unlike fixed format approaches, the accu-racy and precision of the dPCR result can be tuned as desired,simply by varying the observation time and flow rate to obtainthe required number of replicates.
In various embodiments, the methods and systemsdescribed herein may be used to detect other biological com-ponents of interest. These biological components of interestmay be any suitable biological target including, but are notlimited to, DNA sequences (including cell-free DNA), RNAsequences, genes, oligonucleotides, molecules, proteins,biomarkers, cells (e.g. , circulating tumor cells), or any othersuitable target biomolecule.
Furthermore, in addition to dPCR, the methods and sys-tems in various embodiments may be used in applications,such as fetal diagnostics, PCR, qPCR, dPCR, allele-specificPCR, asymmetric PCR, ligation-mediated PCR, multiplexdPCR, nested PCR, bridge PCR, genome walking, viraldetection and quantification standards, genotyping, sequenc-ing validation, mutation detection, detection of geneticallymodified organisms, rare allele detection, and copy numbervariation.
As used herein, droplets may be referred to as sampleportions, sample volumes, or reactions volumes, for example.
In some embodiments, a flow cytometer, for example, anacoustic flow cytometer can be used to control the flow ofaqueous droplets dispersed in a substantially immiscible car-rier fluid, through a conduit.
According to various embodiments, analysis of a samplemay include preparing uniform or variously-sized sampleportions. The sample portions are amplified so that the sampleportions contain the target nucleic acid. Amplification may be
6
10
16
20
26
30
36
40
46
60
66
60
66
performed by polymerase chain reactions (PCR) with targetconcentration near terminal dilution. According to variousembodiments, amplification may also be performed by iso-thermal amplification, thermal convention, infrared mediatedthermal cycling, or helicase dependent amplification, forexample.
According to various embodiments, detection of a targetmay be, but is not limited to, fluorescence detection, detectionof positive or negative ions, pH detection, voltage detection,or current detection, alone or in combination, for example.
The volume of the sample portions may be known. If thesample portions are different sizes, the volume of the sampleportions may need to be determined. The positive and nega-tive reactions within the plurality of sample portions arecounted. More particularly, the number of sample portionsthat contain successful amplification ofthe target nucleic acidare counted. The sizes and the positive and negative reactionsmay be determined by imaging, for example. The average
copy number per reaction is estimated. The estimation may bemade using a Poisson distribution. Then, the target copynumber per unit volume in the starting sample is estimated.This exemplary method is generally shown in the flow chartdepicted in FIG. 1.
As shown in FIG. 1, the method may comprise forming aplurality of discrete sample portions in step 102. Each of theplurality of a discrete sample portions may include a portionof the sample, and reaction reagents, such as PCR reagents.The sample may also be diluted. In some embodiments theplurality of discrete sample portions may be reactor droplets.The plurality of discrete sample portions may also be porousbeads or magnetic beads, for example. The discrete sampleportions may be a plurality of sizes, a uniform size, or differ-ent predetermined sizes.
The method may further comprise amplification of theplurality of discrete sample portions to form a plurality ofprocessed sample portions in step 104. At least one of theplurality of discrete processed sample portions containnucleic acid amplification reaction products. The amplifica-tion may be by subjecting the reactor droplets to thermal
cycling in various embodiments.Then, the method may include detecting fluorescence sig-
nals from the processed sample portions to determine a pres-ence of at least one target nucleic acid, as in step 106.In otherwords, the plurality of discrete processed sample portionsmay be determined to be positive or negative for amplifica-tion. According to various embodiments, detection ofa targetmay be, but is not limited to, fluorescence detection, detectionof positive or negative ions, pH detection, voltage detection,or current detection, alone or in combination, for example.
The method further includes determining the respectivevolumes of the plurality of discrete processed sample por-tions in step 10S. The volumes may be determined fromimaging the plurality of discrete processed sample portions.In various embodiments, the imaging may be done by posi-tioning the plurality ofdiscrete sample portions in the field ofview of an imaging apparatus. This may be used where theplurality of discrete sample portions are a plurality of sizes,which may be referred to as a polydisperse emulsion. In otherembodiments, the discrete sample portions may be of a uni-form size and the volume may be known. In yet other embodi-ments, the discrete sample portions may be a known numberof different sizes, which may be referred to as an multi-monodispersed emulsion. For example, the discrete sample por-tions may be two different sizes. In other embodiments, thediscrete sample portions may be three different sizes, for

US 9,322,055 B2
example. In this way, by determining which size each of theplurality of discrete sample portions is, the volume may bedetermined.
According to various embodiments, a method is providedto create monodisperse reverse emulsions using T-junctions,as described for example, in U.S. Patent Application Publi-
cation No. US 2007/0141593 Al to Lee et al. , which is incor-porated herein in its entirety by reference. There would thus
be no need to estimate the size ofthe discrete sample portions.Some or all of these workflow steps may be integrated into asingle closed system.
The polydisperse emulsions achieved by the present sys-tems and methods are relatively easy and cheap to make, anddo not require a consumable. Due to the difference in volumeof the discrete sample portions volumes, they create approxi-mately 3OM greater dynamic range of sample input for dPCRwithout sample dilution. Acoustic focusing allows massivedroplet throughput and low shear stress to stabilize the plu-
rality of discrete sample portions in a flow. Also, acousticfocusing has flow speed flexibility that allows a significant
signal to noise increase by varying flow speeds. This processallows very high numbers of discrete sample portions to beread, increasing the accuracy, precision, and dynamic rangeof dPCR.
The method may further include estimating the number oftarget template copies-per-unit-volume of the at least onetarget nucleic acid in the sample based on the number ofdiscrete processed sample portions determined to contain theat least one target nucleic acid there, in step 110.As such, thequantity of the target nucleic acid may be determined.
In various embodiments, the method may also compriseaveraging the results to improve accuracy and precision and/
or estimating the target copy number per unit volume in thestarting sample. Such methods may increase the dynamicrange of dPCR and provides alternative analysis methods.
One exemplary method of this type is depicted in the flowchart shown in FIG. 2. As shown in FIG. 2, the method mayfirst comprise generating a plurality of discrete sample por-tions. More particularly, generating the plurality of discretesample portions includes combining reaction componentsthat include sample, assay, and PCR reagents, with a substan-tially immiscible continuous phase liquid, to form a mixture.The mixture is emulsified to form the plurality of discretesample portions. In various embodiments, the plurality ofdiscrete sample portions is an emulsion of reactor dropletsdispersed in the continuous phase liquid.
Amplification of the plurality of discrete sample portionsmay be accomplished by thermal cycling to form a pluralityof discrete processed sample portions. The plurality of dis-crete sample portions may be flowed through a conduit be foreamplification. In other embodiments, the plurality of discreteprocessed sample portions, after amplification, may beflowed the conduit. The plurality of discrete processedsample portions are assessed to determine if the discretesample portions are positive or negative for amplification.The volumes of the processed sample portions are estimated.Then, the number of target template copies-per-unit-volumein the sample may be estimated.
FIG. 3 is a schematic diagram of an exemplary system andmethod according to various embodiments of the presentteachings. In a first step of the method, an emulsion apparatus20 is provided where an emulsion 22 is prepared comprisinga plurality of discrete sample portions 24. With reference toFIG. 4, an exemplary dPCR system 400 is illustrated, includ-ing emulsion apparatus 20 for forming a plurality of discretesample portions. In some embodiments, the discrete sample
6
10
16
20
26
30
36
40
46
60
66
60
66
portions 24 may be droplets of an aqueous sample dispersedin a substantially immiscible carrier 26, for example, in afluorinated liquid.
FIG. 5A illustrates target nucleic acids 500 containedwithin discrete sample portions ofvarious sizes. Emulsion 22is then amplified. In some embodiments, the amplification isaccomplished by an amplification apparatus 402, referringback to FIG. 4, thermally cycling such that the discretesample portions containing a target nucleic acid are replicatedwithin the discrete sample portion. The amplification appa-ratus may be a PCR instrument. In another example, theamplification apparatus may be a serpentine thermal cycler.In some embodiments, after amplifying, emulsion 22 is thenmoved into and through a conduit 2S in the direction of flow
shown by flow arrow 30, in a flow cytometry station 32. Inconduit 2S, the discrete sample portions 24 may be separated,for example, to form a single-file line of sample portions.Within, at an exit of, or adjacent an exit of, conduit 2S, an
excitation detection apparatus 34 can be located where eachdiscrete sample portion is illuminated by an excitation source36 and fluorescence that may result from the illuminationmay be detected, indicating positive amplification of the tar-
get nucleic acid.FIG. 5B illustrates discrete sample portions with positive
amplification 502 as well as discrete sample portions withnegative amplification 504. The plurality of discrete sampleportions may contain a reference dye only, contain no refer-ence dye and no target nucleic acid, or contain a reference dyeand a target nucleic acid.
Following excitation and detection by an excitation detec-tion apparatus 404, with reference again back to FIG. 4, a plotofthe results can be generated as shown graphing the detectedtarget nucleic acid signal against the detected reference sig-nal. The excitation and detection apparatus 404 can compriseone or more of the excitation and detection componentsdescribed, for example, in U.S. Patent Application Publica-tion No. US 2007/0141593 Al to Lee et al. , which is incor-porated herein in its entirety by reference.
In an exemplary embodiment, a TAQMAN fluorescentprobes qPCR reaction with target cDNA is overlayed with a5-fold higher volume of mineral oil in a 96-well plate. Aninverse emulsion may be created in an ultrasonic bath. Theplurality of discrete sample portions formed using thismethod comprises sample portions from about I fL to aboutI pL in volume. The plurality ofdiscrete sample portions maybe thermally cycled in the plate until a plateau of replicates isproduced in the positive discrete sample portions. With ref-erence back to FIG. 4, an optical imager 404 may be used toimage the plurality of discrete sample portions in order todetermine size and volume of the discrete sample portions.For example, the discrete sample portions may be analyzedusing an optical camera, or flow cytometry, to estimate thesize (volume) and to determine positivity. Instead of estimat-ing the copy number per droplet, however, the user deter-mines the droplet volume that delivered a certain frequency ofpositivity. For example, one could ask what discrete sampleportion size delivered positive amplification reactions at a66% frequency. If I pL discrete sample portions delivered thisfrequency, then the initial target concentration would be 1.7x10-12M (1.7M). If I fL discrete sample portions deliveredthis frequency, then the initial target concentration would be1.7x10-9 (1.7 nM). In an alternative embodiment, the discretesample portions positivity frequency may be plotted by dis-crete sample portions size. The slope of a portion of theresulting curve may be used to estimate the initial discretesample portion concentration.

US 9,322,055 B2
Furthermore, dPCR system 400 can include one or moreprocessors, such as a processor 40S.As such, generating anyplots or determining the estimating the number ofcopies-per-unit-volume of the at least one target nucleic acid in thesample based on the number of discrete processed sampleportions determined to contain the at least one target nucleicacid therein, according to embodiments of the present teach-ings, may be calculated by processor 40S. Processor 40S canbe implemented using a general or special purpose processingengine such as, for example, a microprocessor, controller orother control logic. Processor 40S may be included in a sepa-rate computing system, or in any one of optical imager 404,excitation detection apparatus 404, amplification apparatus402, or emulsion apparatus 20.
It will be appreciated that, for clarity purposes, the abovedescription has described embodiments of the invention withreference to different functional units and processors. How-
ever, it will be apparent that any suitable distribution of func-tionality between different functional units, processors ordomains may be used without detracting from the invention.For example, functionality illustrated to be performed byseparate processors or controllers may be performed by thesame processor or controller. Hence, references to specificfunctional units are only to be seen as references to suitablemeans for providing the described functionality, rather thanindicative of a strict logical or physical structure or organiza-tion.Forming Discrete Sample Portions
According to various embodiments, the present teachingsprovide a system including an emulsion apparatus, and amethod that uses the same. The emulsion apparatus generatesa plurality of discrete sample portions. The emulsion appara-tus may generate the plurality of discrete sample portions byvarious methods, such as shaking, stirring, sonicating,extruding, or shear electowetting, for example. In someembodiments, the emulsion apparatus may be a sonicator, avortexer, or a plate shaker. Emulsification parameters, such asemulsification method, strength/power, time, oil/surfactant
chemistry, viscosity, concentration, aqueous phase composi-tion, and water-to-oil ratio, for example, may be optimized toproduce desired sizes for the discrete sample portions. Insome embodiments, the discrete sample portions have adiameter of between 10to 150 pm and a volume ofbetween IpL to I nL.
Exemplary systems for methods of preparing and process-ing emulsions that may be used according to the presentteachings include those described in U.S. patent applicationSer. No. 12/756, 547, filed Apr. 8, 2010, to Lau et al. for"System and method for preparing and using bulk emulsion, "which is incorporated herein in its entirety by reference.Exemplary systems for methods of processing and thermallycycling emulsions that may be used according to the presentteachings include those described in U.S. patent applicationSer. No. 12/756, 783, filed Apr. 8, 2010, to Liu et al. for"System comprising dual-sided thermal cycler and emulsionPCR in a pouch, " which is also incorporated herein in itsentirety by reference.
The method may further comprise diluting the sample toform a diluted sample and forming the plurality of discretesample portions from the diluted sample. Dilution may com-prise terminally diluting the sample to achieve an average ofless than one of the at least one target nucleic acid moleculesper sample portion. In some embodiments, the method furthercomprises: serially diluting different portions of the sample
by different respective dilution ratios; dividing each serially
5
10
15
20
25
30
35
40
45
50
55
60
65
diluted portion into a plurality of aliquots; and processingeach of the plurality of aliquots of each of the serially dilutedportions.
According to various embodiments, the components of theplurality of discrete sample portions may be provided in amulti-well plate. Forming the plurality of discrete sampleportions may include emulsifying an aqueous sample with amedium that is at least substantially immiscible with thesample. In some embodiments, the emulsifying may com-prise mixing the aqueous sample with the medium that is atleast substantially immiscible with the sample in the multi-well plate, sonicating the aqueous sample with the mediumthat is at least substantially immiscible with the sample in themulti-well plate, shaking the aqueous sample in the mediumthat is at least substantially immiscible with the sample in themulti-well plate, or stirring the aqueous sample in themedium that is at least substantially immiscible with thesample in the multi-well plate. Emulsification to form a plu-rality of discrete sample portions may also take place in thepresence of a surfactant, so that the kinetic stability of theemulsion increases. Some surfactants which may be used, butare not limited to, are Span80, STF9, ABIL EM90, and DCBY11-030,for example.
In various embodiments, polydisperse emulsions may begenerated. Generally, polydisperse emulsions are less difft-cult to make, minimally handled, can be formed in batches,and greatly increase the dynamic range of dPCR. Further-more, a small reaction chambers allow analysis withoutsample dilution that can introduce error. For example, heat,shaking, sonic energy, ultrasonic baths, combinationsthereof, and the like can be used to produce emulsions, forexample, to process batches of emulsions in 96-well, 384-well plates, or cell culture plates without the need for anyspecial consumables to physically touch the samples. In otherembodiments, a plate may be used based on the amplificationapparatus. This greatly reduces the chance of cross-contami-nation. In addition, polydisperse emulsions typically vary involume from about I fL to 10 pL, eliminating the need todilute samples to achieve terminal dilutions. Since forming anemulsion may result in very small discrete sample portions,the method may further include removing very small dropletsthat may be problematic to detection or amplification.
In another embodiment, multi-mono dispersed emulsionsmay be formed. A multi-mono dispersed emulsion mayinclude two or more sizes of discrete sample portions, wherethe sizes are known or predetermined. For example, a multi-mono dispersed emulsion may contain three different sizes ofdiscrete sample portions that are substantially the same sizeas three different predetermined sizes. Substantially meanswithin+/ —10%of the predetermined size. By determining theplurality of discrete sample portions which size of the differ-ent predetermined sizes each discrete sample portion is, thevolume of each discrete sample portion can be determined.Multi-mono dispersed emulsions may vary in volume fromabout I fL to 10 pL. In other words, each droplet can bebinned into a predetermined size. In this way, the dynamicrange can be increased and an optical imager may be simpli-fied.
Any of a variety of substantially or totally immisciblefluids can be used as the carrier fluid. By immiscible what ismeant is immiscible with respect to the aqueous sample drop-lets. The substantially or totally immiscible fluid can com-prise, for example, paraffin oil, mineral oil, silicone oil, aperfluorinated polyether (PFPE), other fluorinated fluids, flu-
orinated solvents, combinations thereof, and the like. Somespecific fluids that can be used as a carrier fluids includeGALDEN HT170 available from Solvay Solexis of West

US 9,322,055 B210
Deptford, N.J., other GALDEN HT liquids available fromSolvay Solexis, FC-40 available from 3M Company of St.Paul, Minn. , and other FLUORINERT liquids availablefrom 3M Company of St. Paul, Minn.
In some embodiments, forming the plurality of discretesample portions may include diluting a sample to near thesingle molecule limit, and combining or mixed the samplewith all reagents needed for a PCR. A good input quantity oftemplate may comprise the dilution limit where each aqueousreactor volume contains I or 0 target nucleic acid molecules,for example, such that the Poisson parameter, X, is close to orequal to 1. If desired, in order to determine an optimal dilu-
tion, an initial quantification using, for example, a QUBITsystem (available from the Qubit Systems Inc. , Kingston,Ontario, Canada) may be used. Unlike lower-replicate dPCRembodiments, the system and method ofthe present teachingscan accommodate a much larger deviation from an ideal X=Iinput concentration.Emulsification and Sampling
In various embodiments, a method to prevent evaporationof the aqueous droplets in water-in-fluorocarbon emulsions isprovided. In this way, errors in sampling an emulsion of aplurality of discrete sample portions are minimized. Discretesample portions, in this case aqueous droplets, are formed byadding hydrocarbon oil into a water-fluorocarbon mixtureand agitating the three phases together. As such, after emul-sification the hydrocarbon oil forms a layer covering theaqueous droplets to prevent evaporation.
Density of aqueous phase is around I g/cc. Density offluorocarbon (FC) fluid is about 1.6-1.8 g/cc. Thus, in water-in-fluorocarbon emulsions, aqueous droplets form a layer ontop of FC fluid due to the density difference. Without a layerto cover the aqueous droplets, the aqueous droplets mayevaporate during storage. Furthermore, evaporation becomesmore severe if the emulsion is heated, for example, in thethermal cycles of the PCR process. Droplets may shrink orevaporate completely, inhibiting biological reactions insidethe droplets and making post-amplification droplet sizingdifficult.
According to various embodiments, hydrocarbon oil maybe layered on top ofthe layer ofaqueous droplets. The densityof hydrocarbon oil is about 0.8 g/cc. Thus, the hydrocarbonoil is immiscible with both water and fluorocarbon. Thehydrocarbon oil layer reduces evaporation of the aqueousdroplets. However, if the hydrocarbon oil is added after emul-sification, this extra step increases complexity of the processand time. Furthermore, ifmany parallel emulsion vessels (e g.sealed PCR wells) need to be opened to add hydrocarbon oil,it may increase the chance of cross contamination amongvessels.
According to various embodiments, hydrocarbon oil isadded into the vessels together with a water-fluorocarbonfluid mixture before the emulsification or agitation. Thusthere are three phases existing in the vessel. The FC phasecontains the fluorocarbon fluid and a surfactant (fluorosur-factant) to make water-in-fluorocarbon emulsion. In oneexample, the fluorocarbon fluid is HFE7500 (3M), the fluo-rosurfactant is a block copolymer containing hydrophilicpoly(ethylene glycol) (PEG) block and fluorophilic poly (per-fluoropropylene ether) (PFPE) block, the hydrocarbon oil is aheavy mineral oil (Sigma-Aldrich 330760), and the aqueousphase includes sodium chloride (NaCI) solution 100 mM indistilled water. The aqueous phase contains water and bio-logical reagents. The hydrocarbon (HC) phase contains onlyneat hydrocarbon oil, but no surfactant. The hydrocarbon oilis agitated together with water-fluorocarbon fluid mixture.After the agitation the aqueous phase is broken into small
5
10
15
20
25
30
35
40
45
50
55
60
65
aqueous droplets stabilized by the fluorosurfactant and theseaqueous droplets form a layer on top ofthe fluorocarbon fluid.The hydrocarbon oil forms a layer on top of the aqueousdroplet layer preventing the evaporation of the aqueous drop-lets.
In one example, the vessel may be a MICROAMP Opti-cal 96-Well Reaction Plate (Applied Biosystems, 4316813)with MICROAMP Clear Adhesive Film (Applied Biosys-tems, 4306311).Tube strips can also be used, e.g. MolecularBioProducts PCR 8-Tube Strips.
EXAMPLE I
50 uL HFE7500 with 2 wt % fluorosurfactant, 10 uL NaCIand 40uLheavymineral oil were addedinthis orderinto eachwell on the plate. The plate was sealed by the adhesive film.When a tube strip is used, the tubes are sealed with cap strip.
The plate or the tube strip was then shaken by an oscillatingmixer (RETSCH MM301) with appropriate adapters. Theoperating parameter used in this example was 15 Hz for Imin.
After shaking, the plate was unloaded from the mixer and
kept in the upright position. A layer formed above theHFE7500. This layer contains the aqueous droplets stabilized
by the fluorosurfactant. A clear mineral oil layer was formedon top the droplet layer, preventing the evaporation of aque-ous droplets. This three-layered emulsion assembly in a tube-strip is depicted in FIG. SA.
The plate or the tube strip with the emulsion sample wasthermocycled between 95' C. and 60' C. for at least 40 cyclesand no significant evaporation was observed. This three-lay-ered assembly in a tube-strip after thermocycles is depicted inFIG. SB.Sampling
According to other embodiments, a method to sample theemulsion for subsequent analysis of the individual droplets isprovided.
As described above, the aqueous droplets form a layer ontop of the fluorocarbon fluid and are covered by the mineraloil. For subsequent analysis of the individual aqueous drop-lets, it is desirable to sample only the aqueous droplets and thefluorocarbon fluid, and to remove the mineral oil from thesample. An exemplary method is described below, with ref-erence to FIG. 9.
In step I, a capillary is inserted into a tube. The bottom endof the capillary is in the fluorocarbon phase and close to thebottom of the tube. In step 2, the fluorocarbon fluid 902 iswithdrawn by applying negative pressure inside the capillary.The fluorocarbon fluid 902 goes into the capillary, followed
by the aqueous droplets 904, and then the mineral oil 906.Next, the negative pressure is stopped when only a small
amount of mineral oil 906 enters the capillary.In step 3, the capillary is in the upright position. Because
the aqueous droplets 904 have much lower density than thefluorocarbon fluid 902, they will float up until reaching the
top of the fluorocarbon fluid 902. The mineral oil 906 insidethe capillary tube forms a "plug" under the fluorocarbon fluid,but it does not
float
u. Thus the aqueous droplets 904 and themineral oil 906 are separated automatically. Additionally,aqueous droplets 904 may be sorted by size because largeaqueous droplets have much higher rising velocity thansmaller aqueous droplets. In some embodiments, a variableamount of backflow can be applied to prevent small dropletsfrom migrating up the capillary.

US 9,322,055 B212
In step 4, sampling, the system can be backflushed toremove residual droplets and mineral oil.
EXAMPLE 25
In this example, the emulsion sample described above inExample I is used to test the sampling procedure. The emul-
sion held in an individual well on a 96-well PCR plate. Thevolume of the fluorocarbon fluid (HFE 7500) plus that of theaqueous droplet layer is 60 uL. The volume of the hydrocar-bon oil (heavy mineral oil) is 40 uL. Thus, the total samplevolume in the well is 100 uL.
A GASTIGHT syringe (Hamilton ¹81320) is usedtogether with a syringe pump (Nexus 3000) to provide nega-
15tive and positive pressure. The inner wall of a capillary (I.D.I mm, length 10mm) is coated with hydrophobic fluoropoly-mer coating. This is to prevent the wetting ofaqueous dropletson the inner wall. An open end of the capillary is connected tothe syringe needle via flexible silicone tubing.
In step I, the capillary was kept upright and the other openend is inserted into the well containing the 3-layered emul-
sion assembly. The position of this end was inside the flu-orocarbo fluid and close to the bottom ofthe tube, as shown inthe attached sketch. 25
In step 2, the "withdraw" mode of the syringe pump wasused to create negative pressure. The total volume to be with-
drawn was set to be 70 uL and volumetric rate is set to 10uL/min. At the end of this step all the fluorocarbon fluid andall the droplets were collected into the capillary, and only asmall fraction ofmineral oil (about 10uL) was also collected.The pump was then stopped and no more oil was collected.
In step 3, the capillary was kept upright for I min. It wasseen that majority of the aqueous droplets float to the top of
35the fluorocarbon fluid, and the bottom section of the fluoro-carbon fluid became free of droplets. The mineral oil "plug"stayed under the fluorocarbon fluid.
In step 4, the "infuse" mode of the syringe pump was usedto apply positive pressure inside the capillary. The volume to 4obe infused was set to be 15 uL and the volumetric rate was 10uL/min.
The mineral oil was pushed out of the capillary, and somefluorocarbon fluid was also pushed out. At the end of step 4,the sample inside the capillary contained only aqueous drop- 45
lets and fluorocarbon fluid. The mineral oil was separatedfrom the sample.
In the embodiments described above, a polydispersedwater-in-fluorocarbon emulsion is created directly inside acommon 96-well PCR plate covered by a lighter oil without 5o
an additional step. Aqueous droplets can are sampled, sortedand analyzed by buoyancy without flow.
As described, embodiments of these methods reduce theevaporation ofaqueous droplets. Furthermore, the cover oil isadded together with aqueous reagents and fluorocarbon fluid 55
before emulsification. Eliminating extra step to open theemulsion vessel to add covering oil after emulsification isavoided. Moreover, the covering oil can be removed from theemulsion sample and will not interfere with sample analysis.
In other embodiments, the plurality of discrete sample 6o
portions may include using non-magnetic beads, magneticbeads, or a combination thereof may be used in the emulsionmix. In some embodiments, the plurality of sample portionsmay comprise non-magnetic beads, including porous or hol-low beads, for example. The porous or hollow beads may be 65
spherical or cylindrical. In some embodiments, no beads areused.
Detection of Positive or Negative AmplificationAccording to various embodiments, flow cytometry (FC) is
used to count the number of droplets exhibiting specific fluo-
rescence signals due to, for example, fluorescently-labelednucleotide probes or antibody tags. The flow cytometry sys-tem can provide a fluid flow of suspended reactor dropletsthrough an analysis region in which an excitation and detec-tion system may be used to serially count the number ofdroplets with one or more specific fluorescent emission char-
acteristics. Multi-color FC systems may be used for labelingor tagging droplets and/or target molecules with differentmarkers.
TAQMAN fluorescent probes, SYBR, LUX, or otherreal-time fluorescence detection methods may be used. Ifbeads and SYBR are used, and the method further com-prises a flow cytometry step, SYBR may subsequently beincluded in the running buffer of a flow cytometer to bind andlabel beads containing dsDNA. Ifbeads and TAQMAN areused, probes may be attached to the beads following cleavage.In some embodiments, labeled primers, such as LUX prim-
ers, may be used to label beads, and the method may allowmultiplexing without a need for using a dye-loaded buffer ina flow cytometry step.
According to various embodiments, a passive reference orspectator reference can be used in the method, for example,included in the aqueous amplification reagent mix. For bead-based approaches, the passive reference may be made toattach to the bead during or after the amplification, forexample, prior to breaking the emulsion. In an exemplaryembodiment, a biotinylated fluorescent ROX™dye is usedwith a streptavidin-terminated bead. For non-bead-basedemulsions, a water-soluble dye, such as ROX™dye, may beused.
The present teachings expand the capabilities of dPCR bygreatly increasing the number of replicates that may be pro-cessed in a given time period when compared to current dPCRmethods. The improvements may be use available instrumen-tation and overcome the limitations of current mxn dPCRconfigurations. In some embodiments, the present teachingsprovide serial processing using conventional flow cytometrysystems applied to dPCR. In some embodiments, the systemand method processes micelles containing PCR reactants anddispersed in a substantially immiscible carrier fluid. In someembodiments, magnetic focusing is used in conjunction withflow cytometry and post-read, downstream collecting andprocessing of single droplets is enabled.
According to various embodiments, emulsion PCR(emPCR) is used and may involve a massively parallel PCRamplification of nucleic acid samples in aqueous reactorsimmersed in a substantially immiscible continuous phase, forexample, in a non-soluble oil medium. The method may beused, for example, to prepare poly-disperse collections ofreactor droplets or libraries for analysis.
According to various embodiments, fluorescent-activatedcell sorting is used such that, following analysis, various fluidmanipulation methods divert droplets with specific fluores-cence signatures to different locations or chambers for enrich-ment or other further processing.
According to the present teachings, a system and methodare provided for using emulsion PCR with a flow cytometryread-out to provide a digital PCR answer. Unlike conven-tional flow cytometry systems and methods that process cells,the "cells" in the present flow cytometer system are insteadindividual PCR reactant-containing droplet reactors, some ofwhich may comprise replicates of PCR reactions, alsoreferred to as positive results.

13US 9,322,055 B2
14In various embodiments, following the formation of the
discrete sample portions, the resulting amplification productscan be introduced into a flow cytometer. This can be accom-plished in any suitable way. For example, when beads areused, the emulsion can be broken with detergent, rapid aque-ous dilution, or another standard emulsion breaking method.An aqueous suspension of the beads can then be introducedinto a flow cytometer in a manner similar to how cells are
typically loaded in a conventional cell manipulating flow
cytometer. In some embodiments, for bead and bead-lessemulsions, lipids may be included in the emulsion followed
by aqueous dilution to result in micelle formation, whereinthe PCR product may be contained, for example, within alipid bilayer. Such a droplet can be referred to as a "PCR cell"that may then be introduced and analyzed in a manner used tointroduce and analyze a conventional biological cell in a flow
cyto meter.Plates of emulsified samples can be bulk thermal cycled
using a standard thermal cycler, for example, using any of themany thermal cyclers available from Applied Biosystems,LLC ofFoster City, Calif. , for example. Furthermore, as usedherein, thermal cycling may include using a thermal cycler,isothermal amplification, thermal convention, infrared medi-ated thermal cycling, or helicase dependent amplification, forexample. In some embodiments, the chip may be integratedwith a built-in heating element.
Amplification may be run to completion to create strongsignals for the reporters present in droplets containing a targetmolecule. In some embodiments, a microfluidic meanderingPCR method may be used or isothermal amplification tech-niques may be used to keep emulsions intact.
According to various embodiments, the method may com-prise subjecting the plurality of discrete sample portions tonucleic acid amplification conditions for carrying out a poly-merase chain reaction, for example, thermal cycling condi-tions.
In some embodiments, the flow cytometer can be set todetect at least two colors, for example, the passive referenceand the indicator dye. In an example, the flow cytometer maybe set up to detect fluorescence from a SYBR dye and froma FAM dye. Using TAQMAN fluorescent probes avail-able from Applied Biosystems, LLC of Foster City, Calif. ,
multiplexing can also be applied generating more than oneprobe signal, for example, to generate signals form FAMVIC, and other dyes. For simplex reactions, 3 types of"cells" should be distinguishable: unlabeled cells from beads/micelles which were not in contact with the sample-PCR mix,cells labeled with the spectator dye only from beads/micelleswhich included PCR mix but no template, and cells labeledwith both dyes, where the bead/micelle was in contact withboth the PCR mix and a target template.
A digital read-out may be calculated as follows. Two-dye"cells" are I (indicating mix+template present), passive dye-only "cells" are 0 (indicating mix but no template), and un-
dyed "cells" are not used in the calculation. The number of 0and I "cells" is used to fit the Poisson equation, and estimatethe number of template molecules present in the originaldiluted sample. From that, the number of template moleculespresent in the original undiluted sample can also be estimated.
According to various embodiments, magnetic beads maybe used in the assay. Magnetic forces may be used to break theemulsion. Cylindrical solenoid fields may be used duringflow cytometry to focus beads into the center of the flowstream, increasing the sensitivity and throughput of the flowsystem. In some embodiments, focusing may be enabled byan acoustic focusing system as described below. Following
5
10
15
20
25
30
35
40
45
50
55
60
65
analysis, variable magnetic fields can be used to direct par-ticular "cells" to an appropriate reservoir.
In some embodiments, if non-magnetic beads are used inthe emulsion, a magnetic passive reference may be includedin the PCR mix. For example, iron nanoparticles may be usedwhich are functionalized for aqueous solubility and covalentattachment to polystyrene beads, silica beads, or other beads.In this way, a magnet can be used to retain beads which areexposed to the reaction mix (true PCR positives and nega-tives) while beads which were not exposed to the mix duringformation may be easily washed away. Such an approach mayalso be used to facilitate clean-up for SOLiD™library prepa-ration.
By using an acoustic focused flow system, as in someembodiments, a method is provided that has very high speedand flexibility. Also very low sheer can help keep the emul-sion intact during the reading process, as opposed to highersheer that might disrupt the emulsion and cause inaccurateresults. The present systems and methods can measure up to1,000,000 discrete sample portions per sample, for example,from about 1,000 to about 1,000,000 discrete sample portionsper sample, from about 10,000 to about 1,000,000 discretesample portions per sample, or from about 100,000 to about1,000,000 discrete sample portions per sample.
Exemplary acoustic flow systems and methods and com-ponents thereof that can be used in the present systems andmethods include those described, for example, in U.S. Pub-lished Patent Applications Nos. US 2009/0050573 Al toWard et al. and US 2009/0178716 A I to Kaduchak et al. , bothof which are incorporated herein in their entireties by refer-ence. Other flow cytometry systems and methods havingcomponents that can be used in the present teachings includethose described, for example, in U.S.Published Patent Appli-cation No. US 2009/013066 Al to Oldham and in U.S. Pat.No. 7,280,207 B2 to Oldham et al. , both of which are alsoincorporated herein in their entireties by reference.Fluorescent-Activated Cell Sorting (FACS)
Determining a presence or an absence of the at least onetarget nucleic acid in each ofthe plurality ofprocessed sampleportions may comprise measuring a fluorescence signal.After determining the presence or absence of the at least onetarget nucleic acid in each ofthe plurality ofprocessed sampleportions, fluorescent-activated cell sorting (FACS) may beused to sort the plurality ofprocessed sample portions accord-ing to the fluorescent signal measured. In some embodiments,the volume of one or more of the processed sample portionsmay be estimated by measuring a fluorescence signal. Thedetermining a presence or an absence may comprise intro-ducing each of the plurality of processed sample portionsindividually into a flow cytometer.
Ifa FACS platform is used, after flow through the analysisregion, a bead/micelle can be segregated according to thefluorescent signals detected. For example, all positive beadscan be shunted, directed, or diverted to a separate reservoir forcollection and subsequent downstream analysis with othermethods, for example, for a sequencing reaction or asequence detection reaction.
The system and method provide the ability to estimate thedroplet volume, as well as the presence of a reporter for PCRamplification. For this purpose, a flow cytometer such as amodified acoustic focusing flow cytometer may be used withan auto sampler. The flow system may autosample from 96- or384-well plates. Acoustic focusing may be sued to enableexcellent focus control and can achieve read speeds of20,000events per second. The size ofa droplet can be measured usingany of a variety of properties of the droplet, for example, bymeasuring laser forward or side scatter properties. In some

15US 9,322,055 B2
16embodiments, a reference dye may optionally be added to theaqueous phase, and the signal from the reference dye may beused to estimate droplet volume. Positive and negative countsare available for each droplet that passes the laser interroga-tion zone in the center of the acoustically focused flow. Highthroughput using parallel acoustic focusing streams may beachieved. Sorting positive droplets may be employed as apreparatory method for DNA sequencing.Non-Flow Cytometry Read-Out Alternatives
In some embodiments, read-out methods after emPCR areused that are alternatives to flow cytometry. For example,emulsion products with beads can be dispersed on a glassslide, for example, containing a thin gel matrix for immobi-lization. The dispersed, fluorescent beads can be read using aSOLiD™platform (available from Applied Biosystems, Fos-ter City, Calif ), a conventional fluorescence microscope, or aCOUNTESS™cell counting system available from Invitro-
gen, Carlsbad, Calif. The presence of passive reference-onlybeads indicates the stochastic limit has been reached, andthese beads do not need to be quantified. Counting the numberof positive beads then provides quantitation of the templatemolecules present in the initial mix. Micelles and/or beadsmay also be read serially using Capillary Electrophoresissystems with polymer (or no polymer), for example, using apolymer specially optimized for such reactors.
According to various embodiments, a system that uses amanual PCR setup is providedthat may use SYBR or TAQ-MAN assays and standard qPCR SuperMixes. The reac-tions may be set up in a standard 96-well plate using typicalPCR volumes, for example, from about 5 pL to about 15 pL,covered with approximately 50 pL of oil or fluorinated fluidfor emulsion PCR. The plate may be sealed using an adhesivecover. More complicated microfluidic reaction assembliesmay also be used.Dilution and Fluorescent-Activated Cell Sorting (FACS)
The present teachings can achieved a throughput of thou-sands of sample droplets per minute. As a result, the dynamicrange of an assay may be expanded and the method can beextended to applications typically reserved for qPCR, such asgene expression, genotyping, and miRNA analysis. Thepresent systems and methods are well suited for small sampleinputs, for example, single cell samples. In some embodi-ments, a fluorescent-activated cell sorting (FACS) system isused and the method may comprise collecting and purifyingselected post-PCR samples from other samples, obviating theneed for user accessibility to and manipulation of samples inplate or array formats. For example, all mutant ampliconsidentified by dPCR may be sorted from wild-type amplicons,for subsequent sequencing.
In some embodiments, the method involves using FACSbefore and after emPCR. Prior to emPCR, the FACS can beused to prepare and isolate single cells (or types of cells) foranalysis. The single cells can be lysed and introduced into theemPCR step for subsequent FC analysis. As an example,circulating tumor cells, as detected by a fluorescent antibody,may be counted and enriched by FACS. The resulting cancercell product can then be analyzed using the dPCR approachdescribed above, that is, emPCR followed by FC/FACS,which provides an extremely low limit of detection for quan-
tifying expression levels in these purified cells.The fraction of beads/micelles containing neither passive
reference nor template may be large if the emulsion PCRprocess is not optimized. With a throughput of 10,000 cellsper minute, if 10% of beads/micelles are in contact with thePCR mix, 60,000 usable digital replicate results may beobtained in one hour of flow cytometer time. Moreover, theemPCR+FC of the present teachings can produce answers
6
10
16
20
26
30
36
40
46
60
66
60
66
beyond typical dPCR results. For example, the method maybe used to simply genotype a sample by measuring the ratio ofthe FAM/ROX cells and the VIC/ROX™ cells. In someembodiments, with such a high throughput, a 3-5 log dynamicrange may be attainable for gene expression or miRNA analy-sis. Using this approach, with 60,000 observations per hour,roughly 1,000-fold differences in expression of two targetsmay be measured. For example, 21,045 cells oftarget A vs. 24cells of target B may be quantified, a 1000-fold difference inexpression levels measured which is absolute in quantity andindependent ofPCR efficiency and other differences betweenthe two targets.Volume Estimation of Discrete Sample Portions
In yet other embodiments of the present teachings, analternative data analysis method for absolute target moleculequantification from heterogeneously sized dPCR reactions, isprovided. The method may comprise determining absolutetarget concentrations from reactions of various volumes. Thereactions may be created with random sizes, randomly ordeliberately. The method is useful in analyzing dPCR datafrom polydisperse emulsions, such as emulsions created withmechanical or sonic energy.
The method may further comprise comparing the pluralityof processed portions to a plurality of standards of knownrespective volumes, for example, a plurality of standards ofknown respective volume that uniformly sized or that are ofdifferent known volumes. The method may further comprisesubjecting a plurality of portions of a standard to the samenucleic acid amplification conditions to form a plurality ofprocessed standards, wherein each of the processed standardsare of a known respective volume, and then comparing theplurality of processed standards to the plurality of processedsample portions. In some embodiments, the plurality ofsample portions have an average of from about 0.1 to about0.8 copy of the target nucleic acid per discrete loaded mixture.The plurality of sample portions may have an average diam-eter of from about 0.3 micrometer (pm) to about 600 pm, or anaverage diameter of from about 1.0 pm to about 100pm, or anaverage volume of from about 0.5 femtoliter (fL) to about Imicroliter (pL), or an average volume of from about 10.0 fL toabout 100 nanoliters (nL).
According to various embodiments, the volume of the dis-crete sample portions is estimated. The present systems andmethods may use an optical imager for using optical scatterproperties, a passive reference dye, optical refraction proper-ties, optical imaging for measurement, optical reflectionproperties, optical absorbance properties, optical transmis-sion properties, a ladder of standard droplet sizes using dif-ferent reference dyes, or a combination thereof to estimatediscrete sample portion volumes. An exemplary opticalimager 600 is depicted in FIG. 6 to detect samples in flow cell602.An optical imager may also be camera configured to takeimages of the discrete sample portions, for example.
In an example, discrete sample portions of first, second,and third volumes of different known respective standardsizes may contain first, second, and third respective detect-ably unique dyes and may be identified and used to scale thesize of the discrete sample portions having unknown volumesizes. The systems and methods of the present teachings mayproduce discrete sample portion sizes of from about 0.3 pm indiameter up to about 1000 pm in diameter, for example, fromabout 0.4 pm in diameter up to about 300 pm in diameter,from about 0.5 pm in diameter up to about 200 pm in diam-
eter, or from about 1.0 pm in diameter up to about 100 pm indiameter. Discrete sample portion volumes ofup to about 1.0pL in size may be produced and processed according to vari-ous embodiments. Discrete sample portion volumes based on

17US 9,322,055 B2
18spherical diameters can be estimated, for example, using aconversion chart such as this one:
Radius diameter volume
0.6 11M
1.4 11M
3 uM6 uM
14 11M
30 11M
60 11M
140 11M
300 11M
600 11M
1.2 11M
2.8 uM6uM
12 11M
28 uM60 11M
120 11M
280 11M
600 11M
120011M
1 fL10 fL
100 fL1 pL
10 pL100 pL
1 nL10 nL
100 nL1 11L
e. co/i
human cell
Measuring the size of each of the plurality of processedsample portions may comprise analyzing each of the pluralityof processed sample portions, and the analyzing may com-prise one or more of measuring or analyzing an index ofrefraction, a light scattering property, a forward light scatter-ing property, a side light scattering property, an opticalabsorption property, an optical transmission property, a peakheight of an optical signal, a peak width of an optical signal,a fluorescent property, a time-of-flight fluorescent property,or a combination thereof. The method may further compriseestimating what size of processed sample portion provides aspecific percentage of processed sample portions of that sizethat test positive for the presence of one or more of the at leastone target nucleic acid, or estimating what size processedsample portion of the differently-sized processed sample por-tions provides a 50% positivity rate with regard to determin-
ing the presence of one or more of the at least one targetnucleic acid.Estimating Number of Copies-Per-Unit-Volume of Sample
According to yet other embodiments of the present teach-ings, a method for estimating the number of copies-per-unit-volume of at least one target nucleic acid in a sample isprovided. The method may comprise: forming a plurality ofdiscrete sample portions each comprising a portion of asample, and a reaction mixture; subjecting each of the plural-ity of discrete sample portions to nucleic acid amplificationconditions to form a plurality of discrete processed sampleportions including at least one discrete processed sampleportion containing nucleic acid amplification reaction prod-ucts; generating and measuring fluorescence signals from atleast some of the plurality of discrete processed sample por-tions; analyzing each of the plurality of discrete processedsample portions individually to determine a presence or anabsence of one or more of the at least one target nucleic acidin each of the plurality of discrete processed sample portionsbased on the fluorescence signals, and to determine therespective volumes of the plurality of discrete reacted mix-tures; and estimating the number of copies-per-unit-volumeof the at least one target nucleic acid in the sample based on
(I) the number of discrete processed sample portions deter-mined to contain one or more of the at least one target nucleicacid present therein, and (2) the determined respective vol-umes. Each of the plurality of discrete sample portions maybe provided in a multi-well plate and the method may furthercomprise emulsifying to form the plurality ofdiscrete sampleportions.
In some embodiments, the method further comprises esti-mating what size of processed sample portion provides aspecific average number of copies-per-unit-volume, or esti-mating what size processed sample portion of the di fferently-sized processed sample portions provides an average of from0.25 to 0.75 copy-per-processed sample portion.
10
15
20
25
30
35
40
45
50
55
60
65
Bayesian Inference to Concentration EstimationAccording to various embodiments, an algorithm may be
used for estimating target concentration within a dPCR sys-tem with multiple discrete sample portions sizes and/or mul-tiple sample dilutions and/or low precision in resolving posi-tive and negative amplification reactions. Typically, dataanalysis methods for digital PCR systems are based on a strictset of assumptions that may not be possible to meet in prac-tice, or may even be undesirable. For example, typicalassumptions may be: all dPCR discrete sample portions haveidentical volume, all dPCR discrete sample portions haveidentical sample dilution, and the distinction between posi-tive and negative discrete sample portions can always bemade with very high precision
If any of the above assumptions are violated in a practicalsystem, by design and/or due to system imperfections, forexample, following standard data analysis methods may leadto suboptimal results.
According to various embodiments, methods ofdata analy-sis may be utilized that explicitly takes into account thatdiscrete sample portions have varying/uncertain volumes andconcentrations, and account for measurements of varyingquality. This may be achieved by estimating the target con-centration through Bayesian inference, although other statis-tical estimation techniques are also appropriate.
Some of the following assumptions may need to be incor-porated into the model statistically linking target concentra-tion to measurements: treating discrete sample portion vol-umes as constants, specified separately for each discretesample portion, or treating discrete sample portions volumesas random variables with known distributions (obtained bycharacterizing or calibrating the system), or treating discretesample portion volumes as random variables with knownconditional distributions, dependent on volume dependentmeasurements obtained at the run time, such as discretesample portion diameter estimate, discrete sample portionarea estimate, passive reference intensity, etc.
One assumption may be treating discrete sample portionconcentrations as constants, specified separately for each dis-crete sample portion, or treating discrete sample portion con-centrations as random variables with known distributions,obtained by characterizing the system and dilution prepara-tion protocol.
Another assumption that may be made is treating end-pointintensity reads as random variables with known "positive"and "negative" distributions, obtained by characterizing themeasurement system.
A goal of Bayesian inference is to produce posterior prob-ability distribution of target concentration, conditional on allavailable information and measurements. Such posterior dis-tribution may then be used to derive maximum likelihoodestimate, unbiased estimate, and/or confidence interval fortarget concentration.
Central to these methods, according to various embodi-ments, is a score function that depends on both the observedresults and a specific value of the target concentration. Thescore function conveys information on how good the agree-ment between a hypothetical value of the concentration andthe actually observed measurements. In other words, a valueof the score function obtained for certain concentration andcertain measurements is a measure of the likelihood that thesemeasurements could arise if the sample under considerationindeed had this concentration.Maximum Likelihood Estimation
In one embodiment, the process of estimating the targetconcentration involves finding the value of concentration forwhich, given a set ofmeasurements, the score function attains

19US 9,322,055 B2
20the maximum value (or appropriately high value; or value
appropriately close to maximum). This search for maximumvalue may be performed by evaluating the score function on apredefined set of candidate solutions and choosing the maxi-mum, a successive approximation method, evaluating theanalytical solution to the maximization problem, or any num-
ber of other maximization method or combination of meth-ods.Unbiased Estimation
In another embodiment, the process ofestimating the targetconcentration involves finding the weighted average of can-didate concentration values (the candidate values comingfrom a predefined or dynamically established set of discreteof continuous concentration values), where the values of thescore function (obtained for the candidate concentrations)serve as the weights in the averaging process. This processcan be achieved by evaluating the score function for all orsubset of candidate values and directly calculating theweighted average; or by evaluating an analytical solution; orby any other appropriate method.Confidence Interval
In addition to producing the estimate of the target concen-tration, a confidence interval may be generated. The confi-dence interval is a range oftarget concentration values, delim-ited by the upper value and lower value, calculated from themeasurements, such that the likelihood that the true targetconcentration value is outside of this range is small. Forexample, a 95%-confidence interval is where the model-based probability of true target concentration being outside ofit is 5%. The confidence interval can be established as therange of target concentration values for which the score func-tion is above certain threshold. The threshold may be dynami-cally calculated. For example, the threshold may be selectedsuch that the sum (or integral) of score function valuesexceeding the threshold is greater by a predefined factor fromthe sum (or integral) of score function values below thethreshold. (for example, if the score function is in fact amodel-based likelihood function and 95%-confidence inter-val is sought, the threshold is selected so that the integral oflikelihoods above the threshold is 95/5 times greater that theintegral of likelihoods below the threshold).Score Function
In some embodiments, the score function can be: a measureof probability; a measure of conditional probability; a mea-sure of likelihood; a logarithm ofprobability. The score func-tion may use the following probabilistic model that expressesthe conditional probability of the positive/negative measure-ments for individual PCR reactions conditioned on the targetconcentration and volumes of the PCR reactions. Alterna-tively, the score function may be a model-based conditionalprobability of fluorescence measurements for individual PCRreactions conditioned on the target concentration and on vol-ume measurements for the PCR reactions.
Under the constraint on the total number ofdroplets, digitalPCR systems with multiple droplet sizes and/or multiplesample dilutions can carry significant dynamic range advan-
tage over system with equally sized and diluted droplets.However, such a system may need much different data analy-sis to determine target concentration in the original sample.For example, the system may need to take into account dis-crete sample portion volumes and dilutions, dilution informa-tion may need to be provided by the user or is system-specific,or discrete sample portion volume information may be eitherprovided or estimated at run time based on a size measure-ment, passive reference intensity, or features of real-timeamplification curve, for example.
6
10
16
20
26
30
36
40
46
60
66
60
66
true target concentration in undiluted sampleN number of droplets
V, true volume of droplet i
C, true dilution coefficient of droplet i (what fraction ofV,is taken up by the undiluted sample)
W, set ofmeasurements or prior information about volumeof droplet i
D, prior information about dilution coefficient of droplet i
X, true number of target molecules in droplet i
Y, plus/minus measurement for droplet i
P(V, i Wd
P(C, i Dd
p(x, i v„c„xlP(Y, ( Xd
P(Y, =0iX, =0l=1 —fp
p(x, i v„c„xlP(Y, i D„W„Xl=
g P(Y, i X;1P(X, i V„C„XlP(C,i DDP(V, i W, )dC, d V,J .(N, =O
P(Y ~D, W, Xl =QP(Y, ~D„W„Xl~=1
N
XML = argmaxg P(V,~D„W„Xl
~=1
Multi-Level Digital PCRIn another embodiment, the number of copies of the target
nucleic acid may be estimated based on real-time measure-ments and C values to discriminate between the number ofstarting template target nucleic acid copies. C values are alsoreferred to as CT values in some examples. In this way, accu-racy and dynamic range of dPCR can be enhanced.
As described in various embodiments above, dPCR isbased at least in part on partitioning the sample into a pluralityof discrete sample portion, which may be viewed as a largenumber of separate PCR reactors. A positive/negative PCRtest is performed on each PCR reactor, whereby each indi-vidual plus/minus test discriminates between zero startingcopies and nonzero starting copies.
According to this embodiment, a method using C valuesfrom each reactor is provided to achieve higher dynamicrange and accuracy than would be possible from simple plus/minus calls.
As described above, a positive/negative data analysis isgenerally described as determining the number of positiveand negative reactions. Then, making the assumption that thenumber of starting template copies per well follows Poissondistribution, it can be inferred the most likely total number ofcopies from the number of positive and negative wells.
According to this embodiment, a C -based dPCR dataanalysis method includes determining a number of negativereactions. For reactions that showed amplification, the mostlikely number of starting template copies from C is deter-mined. The relationship between C and copy number can bedetermined by finding peaks in the C histogram, with right-most peak corresponding to I copy, as illustrated in FIGS.10-15. The method further includes determining the mostlikely total number of template copies by summing up tem-plate copy numbers from individual reactors.

21US 9,322,055 B2
22Furthermore, for each well the likelihoods of starting copy
number 0, I, 2, etc, may be determined instead of making a
copy number call. This can be used to determine a totalnumber of copies more precisely with tighter confidenceinterval. Moreover, the process of associating C with copynumber may also assume Poisson copy number distributionand logarithmic spacing between Cqs.
As the result of using Cq-based dPCR with the aboveanalysis according to embodiments described herein, betteraccuracy and dynamic range with the same number of reac-tors, and the same accuracy and dynamic range with lowernumber ofreactors compared to plus/minus-based dPCR maybe achieved.Initial PCR Efficiency Determination from dPCT C Spec-trum
According to another embodiment, a method ofmeasuringPCR efficiency in the first PCR cycle from the Cq spectrum ofa dPCR experiment is provided. The method includes mea-suring the fraction of replicate PCR reactions with one copyof starting template that did not amplify in the first PCR cycle.
PCR efficiency can be described as the percentage of DNAtemplate that produces a copy during a single PCR cycle. PCRefficiency is an important unknown parameter in downstreamanalysis of real-time amplification curves. PCR efficiencycan differ between cycles of PCR, generally decreasing witheach cycle. During real-time PCR, once the amount of PCRtemplate exceeds the detection level, the fluorescence inten-
sity can be used to monitor the rate of change of the templateamplicon and thus to infer the efficiency.
However, this approach cannot be used to directly measurethe efficiency at the initial cycles when the fluorescence isbelow detection level. It is common to make various assump-tions about this initial efficiency, such as assuming 100%initial efficiency, assuming initial efficiency is the same aslater, directly observed efficiency, or to use various modelsthat extrapolate the initial efficiency backward from theobserved efficiency.
According to this embodiment, the method includes usinga target nucleic acid template dilution that targets the averagenumber of starting template copies per reaction close to l.This yields high fraction of reactions to have precisely onestarting copy (assuming a Poisson-distribution across reac-tions). An amplification curve is collected for all reactionsand C values are obtained.
The method may establish the number of reactions A thathad one starting target nucleic acid template copy and that
copy was amplified in the first PCR cycle, and the number ofreactions B that had one starting target nucleic acid templatecopy and that copy was not amplified in the first PCR cycle,but did amplify in subsequent PCR cycles.
The cycle-1 efficiency can be then calculated as A/(A+B),where A and B are determined from the C histogram. This isbased on identification of the histogram peak correspondingto the groups A and B, which is illustrated in FIG. 33.
Furthermore, knowledge ofthe efficiency at the initial PCRcycle can be used as a reagent research and QC tool. Addi-tionally, this method may also enhance our qPCR models
possibly leading to improved accuracy ofqPCR data analysis.Also, as mentioned above, in various embodiments, the
methods and systems described herein may be used to detectother biological components of interest. These biologicalcomponents of interest may include, but are not limited to,cells and circulating tumor cells, for example. Furthermore,in addition to dPCR, the methods and systems in variousembodiments may be used in applications, such as fetal diag-nostics, multiplex DPCR, viral detection, genotyping, andrare allele detection copy number variation.
6
10
16
20
26
30
36
40
46
60
66
60
66
All publications, patents, and patent applications men-tioned in this specification are herein incorporated by refer-ence to the same extent as if each individual publication,patent, or patent application was specifically and individuallyindicated to be incorporated by reference.
While embodiments of the present disclosure have beenshown and described herein, it will be obvious to those skilledin the art that such embodiments are provided by way ofexample only. Numerous variations, changes, and substitu-
tions will now occur to those skilled in the art without depart-ing from the disclosure. It should be understood that variousalternatives to the embodiments of the disclosure describedherein may be employed in practicing the disclosure. It isintended that the following claims define the scope of thedisclosure and that methods and structures within the scope ofthese claims and their equivalents be covered thereby.
The invention claimed is:1.A method for quantification of a target nucleic acid in a
sample, the method comprising:forming a plurality of discrete sample portions, each of the
plurality of discrete sample portions comprising a por-tion of the sample, and a reaction mixture;
amplifying the plurality ofdiscrete sample portions to forma plurality of discrete processed sample portions includ-ing at least one discrete processed sample portion con-taining nucleic acid amplification reaction products;
detecting fluorescence signals from the at least one of theplurality of discrete processed sample portions to deter-mine a presence of the at least one target nucleic acid;
determining the respective volumes of the plurality of dis-crete processed sample portions, wherein the determin-
ing the respective volumes comprises imaging the plu-rality of discrete processed sample portions; and
estimating the number of copies-per-unit-volume of the atleast one target nucleic acid in the sample based on thenumber of discrete processed sample portions deter-mined to contain the at least one target nucleic acidtherein.
2. The method of claim 1, wherein the plurality of discretesample portions comprises discrete sample portions of a plu-rality of sizes.
3. The method of claim 1, wherein the plurality of discretesample portions comprises discrete sample portions of sub-stantially two different sizes.
4. The method of claim 1, wherein the plurality of discretesample portions comprise discrete sample portions of sub-stantially a plurality of predetermined sizes.
5. The method of claim 1, wherein each of the plurality ofsample portions is at least partially surrounded by a mediumthat is at least substantially immiscible with the plurality ofdiscrete sample portions.
6. The method of claim 5, wherein the medium that issubstantially immiscible with the plurality of discrete sampleportions comprises at least one selected from the group con-sisting of: a mineral oil, a silicone oil, a paraffin oil, a fluori-nated fluid, a perfluorinated polyether.
7. The method of claim 1, wherein the plurality of discretesample portions comprises porous beads.
S. The method of claim 1, wherein the plurality of discretesample portions comprises magnetic beads.
9.The method of claim S, further comprising magneticallyfocusing the magnetic beads within a flow stream of immis-cible fluid in a flow cytometer.
10. The method of claim 1, wherein the amplifyingincludes a polymerase chain reaction.

23US 9,322,055 B2
2411.A system for quantification of a target nucleic acid in a
sample, the system comprising:an emulsion apparatus configured to form a plurality of
discrete sample portions, each of the plurality ofdiscretesample portions comprising a portion of the sample, anda reaction mixture;
an amplification apparatus configured to amplify the plu-rality of discrete sample portions to form a plurality ofdiscrete processed sample portions including at least onediscrete processed sample portion containing nucleicacid amplification reaction products;
an excitation detection apparatus configured to detect fluo-rescence signals from the at least one of the plurality ofdiscrete processed sample portions to determine a pres-ence of the at least one target nucleic acid; and
a processor configured to:determine respective volumes of the plurality ofdiscrete
processed sample portions by imaging the plurality ofdiscrete processed sample portions, and
estimate the number of copies-per-unit-volume of the atleast one target nucleic acid in the sample based on thenumber of discrete processed sample portions deter-mined to contain the at least one target nucleic acidtherein.
12. The system of claim 11,wherein the excitation detec-tion apparatus is further configured to determine the respec-tive volumes of the plurality of discrete processed sampleportions.
13.The system of claim 11, further comprising an opticalimager configured to determine the respective volumes of theplurality of discrete processed sample portions.
5
10
15
20
25
14.The system of claim 11,wherein the emulsion appara-tus is configured to form the plurality of discrete sampleportions of a plurality of sizes.
15.The system of claim 11,wherein the emulsion appara-tus is configured to form the plurality of discrete sampleportions of substantially two different sizes.
16.The system of claim 11,wherein the emulsion appara-tus is configured to form the plurality of discrete sampleportions of substantially a plurality of predetermined sizes.
17.The system ofclaim 11,wherein each ofthe plurality ofdiscrete sample portions is at least partially surrounded by amedium that is at least substantially immiscible with theplurality of discrete sample portions.
1S. The system of claim 17, wherein the medium that issubstantially immiscible with the plurality of sample portionscomprises at least one selected from the group consisting of:a mineral oil, a silicone oil, a paraffin oil, a fluorinated fluid,a perfluorinated polyether.
19. The system of claim 11, wherein the plurality of dis-crete sample portions comprises porous beads.
20. The system of claim 11, wherein the plurality of dis-crete sample portions comprises magnetic beads.
21. The system of claim 20, wherein the excitation detec-tion apparatus includes a flow cytometer configured to mag-netically focus the magnetic beads within a flow stream ofimmiscible fluid.
22. The system of claim 11, wherein the amplificationapparatus includes a polymerase chain reaction (PCR) instru-ment.





























