Embed Size (px)
Citation preview

1071-9
http://d
From t
dren
Address
Pitts
amy
Update on Nuclear Mitochondrial Genesand Neurologic DisordersAmy Goldstein, MD, Poonam Bhatia, MD, and Jodie M. Vento, MGC, CGC
091/12/$-see f
x.doi.org/10.1
he Division of
’s Hospital of
reprint requ
burgh, 4401
.goldstein@ch
The majority of primary mitochondrial disorders are due to nuclear gene mutations, notaberrations within the mitochondrial genome. The nervous system is frequently involveddue to its high-energy demands. Many nonspecific neurologic symptoms may be present inmitochondrial disease; however, there are well-recognized red flags that should alert theclinician to the possibility of mitochondrial disease. There is an ever increasing number ofnuclear gene mutations discovered that play a role in primary mitochondrial disease and itsneurologic symptomatology. Neurologists need to be aware of the wide neurologicpresentation, the red-flag symptoms, and the nuclear gene mutations involved in thepathophysiology of mitochondrial disease to diagnose and manage this patient population.Semin Pediatr Neurol 19:181-193 C 2012 Elsevier Inc. All rights reserved.
IntroductionPrimary mitochondrial diseases are a group of clinically and
genetically heterogeneous disorders that result in decreased
energy production of the respiratory chain (RC) in the formof adenosine triphosphate (ATP). The etiology of primary
mitochondrial disease is complex and can include abnorm-
alities in RC function due to structural or assemblyproteins, translation of the mitochondrial deoxyribonucleic
acid (mtDNA), and maintenance of the mtDNA. Primary
mitochondrial disease may be classified clinically bysymptoms or as a defined syndrome or by biochemical or
molecular, or both defects. The same clinical phenotype
may be caused by a variety of genetic etiologies, and thesame mutation can lead to a wide phenotypic spectrum,
even within the same family. For example, a child may have
epilepsy, developmental delay, and hypotonia and undergoa skin biopsy for evaluation. The RC testing may reveal an
isolated complex I deficiency, which may be due to one outof more than 30 known genetic mutations causing complex
I deficiency. In younger children, the RC testing may be
completely normal in tissue, despite harboring a mutationknown to cause complex I deficiency. Current technology
allows for massively parallel sequencing of many nuclear
mitochondrial genes with a genetic mutation discovery inabout 20%-25% of selected patients.1 With the rapid
ront matter & 2012 Elsevier Inc. All rights reserved.
016/j.spen.2012.09.005
Child Neurology, Department of Pediatrics, Chil-
Pittsburgh of UPMC, Pittsburgh, PA.
ests to Amy Goldstein, MD, Children’s Hospital of
Penn Avenue, Pittsburgh, PA 15224. E-mail:
p.edu
evolution of molecular genetic testing, the ability to detect
a pathologic mutation through next-generation sequencingwould dramatically improve our ability to arrive at a
genetic diagnosis. Therefore, classification is complicated
and a combined clinical, biochemical, and geneticapproach is needed. We have organized this paper based
on the neurologic phenotypes as this would be most
helpful to the practicing clinician. However, the betterknown biochemical and molecular defects with heteroge-
neous phenotypes would be highlighted as well, providing
insight into the genetic classification of these disorders.
Neurologic SymptomsIn primary mitochondrial disease, the organ systems
requiring the most ATP to function properly present withevidence of energy failure. Therefore, the energy-
demanding nervous system is frequently affected, especially
in childhood. Brain and muscle, along with vision andhearing, are typically involved, bringing that child to the
attention of the child neurologist.
Mitochondrial disease should be included in the differ-ential diagnosis for almost any neurologic symptom. The
suspicion should be increased when 3 or more organ
systems are involved without a known etiology. Mitochon-drial disease has a wide spectrum of presentations with any
symptom, any organ, any age, and any inheritance pattern.2
However, common presenting symptoms are stroke-likeepisodes, headache, seizures, psychomotor regression,
ataxia, and encephalopathy.3
181

Table 1 Red-Flag Neurologic Symptoms
Neurologic Symptomor Sign
Red-flag Signs
Stroke Nonvascular distribution; MRI/ADC map shows a mixture of hyperintensity andhypointensity
Basal ganglia lesions Bilateral symmetric (characteristic of Leigh syndrome); also with brainstem lesionsEncephalopathy-hepatopathy Precipitated by valproic acid exposure; associated hepatic failureEpilepsy Epilepsia partialis continua (EPC), myoclonus, and status epilepticusCognitive decline Regression with illnessAtaxia Associated with epilepsy or other systemic symptoms; neuroimaging may show cerebellar
atrophy, white matter lesions, and basal ganglia lesionOcular signs Optic nerve atrophy, ophthalmoplegia, and ptosis; retinopathySensorineural hearing loss At early age, accompanied by other systemic symptoms
ADC ¼ apparent diffusion coefficient.
A. Goldstein, P. Bhatia, and J.M. Vento182
Symptoms most suggestive of mitochondrial disease are
typically progressive or recurrent, but may have partial
recovery after a decline. Disease symptoms may be pre-cipitated by a metabolic stressor, such as infection, fasting,
surgery, or any medication that may be toxic to the
mitochondria. The neurologic symptom may be the pre-senting feature of the disease, but they are often a part of a
multisystem clinical picture, and therefore, the clinician
must be aware of other systemic symptoms.Although any neurologic symptom could be the pre-
senting sign of mitochondrial disease, there have been well-
established neurologic red-flag symptoms that should alert theclinician to a possible diagnosis of mitochondrial disease
(Table 1).4,5 Other common neurologic symptoms or
findings include myopathy, neuropathy, and leukoencepha-lopathy. Exercise intolerance that is out of proportion to
weakness is concerning for mitochondrial disease. The
systemic symptoms to be aware of include cardiac conduc-tion disorders such as Wolff-Parkinson-White syndrome,
cardiomyopathy, hepatopathy, renal involvement including
Fanconi syndrome orglomerulopathy, diabetes mellitus orpancreatic exocrine failure, bone marrow failure, and
gastrointestinal dysmotility or chronic intestinal pseudo-
obstruction.
Stroke-Like Lesions in NonvascularDistributionMetabolic strokes are the result of a significant, sudden
compromise in energy production. Unlike vascular strokes,they do not follow a vascular territory and usually occur in
younger individuals. Although these lesions are most
typical of mtDNA point mutations causing mitochondrialencephalomyopathy with lactic acidosis and stroke-like
episodes, nuclear gene mutations, such as polymerase
gamma (POLG), can cause secondary mtDNA changes,such as mtDNA depletion and multiple deletions, leading
to a metabolic stroke-like presentation.6 The neuroimaging
apparent diffusion coefficient map can show a mixture ofhypointensity and hyperintensity, suggestive of both cyto-
toxic and vasogenic edema.7
Case example: A 16-year-old female presents with acutevision loss and constant left leg twitching. The result of
electroencephalogram (EEG) is consistent with epilepsia
partialis continua (EPC) with epileptiform discharges from
the right centro-parieto-temporal and right occipito-temporal region. Brain magnetic resonance imaging (MRI)
reveals T2 prolongation in right occipital, pulvinar, pre-
frontal gyrus, cerebellum, and splenium of corpus callo-sum. Result for testing of mtDNA point mutations is
negative. Further nuclear diagnostic testing reveals com-
pound heterozygosity of 2 known pathogenic mutationsin POLG.
Leigh Syndrome (Bilateral Symmetric BasalGanglia Lesions)Leigh syndrome is named after Denis Leigh (pronouncedLee), a British neuropathologist, who described subacute
necrotizing encephalomyelopathy in a 7-month-old in 1951.8
Historically, it has been a finding seen at autopsy, but isnow commonly identified on neuroimaging of the brain
with calcifications on computed tomography or MRI with
typical focal, bilateral, and symmetric T2 hyper- T1hypointense lesions with a characteristic involvement of
periaqueductal gray, pons, midbrain, brainstem, cerebel-
lum, basal ganglia, and rarely white matter involvement.9,10
Symptoms and MRI findings can fluctuate based on
metabolic stress with typical worsening seen during acute
illness, fasting, or surgery.Symptoms of Leigh syndrome include encephalopathy
with global developmental delay, seizures, basal ganglia or
brainstem dysfunction with prominent movement disorder,dysphagia and feeding difficulties, somnolence, nystagmus,
respiratory involvement including apnea, and ataxia. The
movement disorder can include choreathetosis, dyskinesia,and dystonia and can be difficult to treat. Feeding
difficulties and increased calorie burn from constant move-
ments often lead to failure to thrive. Many of these childrenare at a high risk for aspiration and should be monitored
for swallowing difficulties with a consideration on feeding
tube placement. Seizures are present in 40% of patientswith Leigh syndrome. Other neurologic symptoms include
neuropathy, myopathy, and optic atrophy. Systemic symp-
toms may include diabetes, short stature, cardiomyopathy,anemia, renal failure, vomiting, and diarrhea.11 Some

Genetics for the neurologist 183
patients may be diagnosed with cerebral palsy or hypoxic-
ischemic encephalopathy and have a relatively static course,
although usually Leigh syndrome is a progressive disease.12
Leigh syndrome is typically seen more in the pediatric
population, but reports of adults with Leigh syndrome are
seen throughout the literature.13,14 Adults may presentwith different features than those seen in the pediatric
population, such as more prominent cranial nerve dysfunc-
tion and cerebellar and long-tract signs instead of devel-opmental delay, failure to thrive, elevated lactate level,
imaging of the abnormalities in basal ganglia, and COX
deficiency.15
Leigh syndrome is caused by both nuclear- and mito-
chondrial-encoded mutations, including pyruvate dehydro-
genase complex deficiency, RC complex deficiency (inparticular, SURF1, an assembly factor for complex IV),
and PDSS2, causing primary coenzyme Q10 (CoQ10)
deficiency.16,17
Case example: a 5-year-old with long-standing cerebral
palsy and seizure disorder presents to the emergency room
with difficulty in breathing. He acutely develops a feverwith temperature of 1041F and becomes encephalopathic.
Biochemical testing shows lactic acidosis. MRI scan of the
brain reveals an increased signal on MRI in bilateral basalganglia. Two weeks later, he is no longer able to track
visually and lost verbal expression. Diagnostic testing
reveals 2 mutations in the autosomal recessive gene,COX10, which is a known cause of complex IV deficiency.
Encephalopathy With HepatopathyEncephalopathy is a nonspecific clinical symptom but
combined with a finding of hepatopathy should increasethe suspicion of mitochondrial disease as well as other
inborn errors of metabolism. The most common causes of
encephalopathy with hepatopathy are the mtDNA deple-tion syndromes (MDDS).18 Alpers-Huttenlocher syndrome,
also known as Alpers or AHS, is one of the MDDS
phenotypes and represents a prototypical nuclear genemutation in POLG, causing a breadth of clinical pheno-
types. The clinical spectrum of POLG is discussed sepa-
rately later. Other MDDS gene mutations that includeencephalopathy-hepatopathy symptoms are DGUOK,
MPV17, TWINKLE, and SUCLG1.19 These hepatocerebral
phenotypes are characterized by early-onset neurologicabnormalities and progressive liver failure. Biochemical
testing reveals hypoglycemia and lactic acidosis, as well
as multiple RC complex deficiencies and mtDNA depletion(reduction in mtDNA copy number).20
Case example: a 3-year-old girl presents acutely with
episodes of altered mental status, abdominal pain withbloating during feeds. EEG during her altered mental status
episode is normal. She has a history of failure to thrive and
refractory gastroesophageal reflux. Her result of neuroima-ging is normal. Her serum lactates are on borderline level,
and her urine organic acids show dicarboxylic acids. Her
gastroenterologist orders a liver biopsy for elevated transa-minases. Liver biopsy demonstrates complex I þ III þ IV
deficiency. Two heterozygous mutations in the autosomal
recessive gene, DGUOK, are determined to be the etiology
of her symptoms.
Cognitive Decline (Mitochondrial Dementia)Regression, or loss of previously achieved milestones, is a
red-flag sign of childhood neurodegenerative disease.
Regression can be slowly progressive or acute whentriggered by mitochondrial stressors. Epilepsy and stroke-
like episodes can also trigger a cognitive decline.
Cognitive impairment may be obvious on clinical exam-ination or may be so focal and well compensated that
neuropsychological testing is necessary to determine any
deficits. The range of cognitive deficits seen in mitochon-drial disease is wide and can have more of a psychiatric
presentation with frank psychosis, acute confusional states,
and behavioral changes or mimic other neurodegenerativedementias seen in Alzheimer or Parkinson disease.21
Childhood POLG and Leigh syndrome have altered mental
status as a common symptom.Dementia often starts with specific focal cognitive deficits
and not a global decline. Dementia may be seen in isolation
at presentation but as with other mitochondrial diseases, ismore likely to be seen with other symptoms. Other central
nervous system symptoms that often accompany dementia
include epilepsy, stroke-like episodes, weakness, spasticity,movement disorders, ataxia, weakness, and migraine head-
ache. Result of neuroimaging may be normal or may show
global atrophy, basal ganglia calcifications, or areas ofincreased T2-weighted signal in the basal ganglia or white
matter.21
The nuclear mitochondrial diseases that have cognitivedecline as a common symptom include mitochondrial neu-
rogastrointestinal encephalomyopathy (MNGIE); POLG-
related disorders (various phenotypes); progressive externalophthalmoplegia (PEO; due to POLG, TWINKLE, TK2, or
ANT1); leukoencephalopathy with brainstem and spinal cord
involvement and lactate level elevation; Charcot-Marie-Toothdisease type 2; Leigh syndrome (due to SURF1); diabetes
insipidus, diabetes mellitus, optic atrophy, and deafness also
called Wolfram syndrome; and Mohr-Tranebjaerg (due toDDP1) also called deafness and dystonia syndrome.22
Cognitive impairment with decline is a frequent feature
of childhood and adult mitochondrial disease, most likelydue to Leigh syndrome in the younger age group. The
cognitive impairment progresses with duration of disease
and is seen as a symptom more frequently when the resultof neuroimaging shows abnormal. Treatment is supportive,
but CoQ10 deficiency should be investigated to offer
specific treatment.23
Case example: a 17-year-old male presents with a history
of myopathy. Previous muscle biopsy revealed COX-
negative fibers; biochemical testing confirms complex IVdeficiency. He begins to have difficulty in reading compre-
hension, attention, and memory. MRI and EEG reports are
normal. Neuropsychological testing is performed to deter-mine specific deficits and to recommend school

A. Goldstein, P. Bhatia, and J.M. Vento184
accommodations. Sequencing of the mtDNA genome in
muscle is normal, and further nuclear genetic analysis does
not reveal pathogenic mutations in known nuclear-encodedcomplex IV genes.
EpilepsyThe brain has an extremely high-energy requirement and is
therefore frequently involved in children who have mito-chondrial disease. Epilepsy is the main childhood mani-
festation of mitochondrial encephalopathy.24 Epilepsy may
be the presenting feature of mitochondrial disease, but asseen with other neurologic symptoms, it is often part of a
multisystem clinical picture. Other features, such as cog-
nitive decline, may evolve over time but be interpreted asan epileptic encephalopathy. Common findings in epilepsy
that should raise the suspicion of mitochondrial disease
include EPC, myoclonus, and status epilepticus, especiallyif the seizures are explosive in onset.24 Associated systemic
symptoms commonly seen with mitochondrial epilepsy
include sensorineural hearing loss, retinopathy, cardiomyo-pathy or arrhythmia, diabetes mellitus, hepatopathy, and
renal tubulopathy.24
Seizures are quite common in mitochondrial disease,with a reported incidence of 35%-60% in patients with
biochemical or molecularly confirmed mitochondrial dis-
ease.24 In a retrospective study, patients with idiopathicepilepsy admitted to the pediatric epilepsy monitoring unit
were reviewed for previous testing and 28% had biochem-
ical abnormalities suggestive of mitochondrial dysfunction,and in those with multifocal epileptiform discharges,
biochemical abnormalities were seen in 75%.25
Patients with epilepsy and mitochondrial disease havemixed seizure types. One review of patients with confirmed
RC defects revealed a spectrum of epilepsy phenotypes
ranging from Ohtahara syndrome to Landau-Kleffner syn-drome, with seizure types ranging from generalized to
partial. Seizure onset was in young children, and the
neuroimaging of the majority showed abnormality.26
Another review of patients with seizures and mitochon-
drial disease demonstrated that seizures were preceded by
multisystemic symptoms, such as global delay, failure tothrive, or ataxia in the majority of patients, and most had
multiple seizure types. The epilepsy phenotypes ranged
from neonatal refractory status epilepticus with multiorganfailure to infantile spasms, myoclonic epilepsy, recurrent
status epilepticus, and EPC. For one-third of the patients,
serum and urine biochemical testing was completelynormal and diagnosis was made on liver biopsy RC testing.
These authors suggested that epilepsy was a poor prog-
nostic sign in this population, with a 45% mortality rate,and half of their patients dying within 9 months of the
onset of their epilepsy.27
Common mitochondrial etiologies of epilepsy includePOLG, other disorders of mtDNA maintenance (PEO1,
RRM2B, and SUCLA2), complex I deficiency, complex
deficiencies caused by disordered assembly (FOXRED1,BCS1L, SCO2, and TMEM70) or abnormal protein
structure (NDUFV1, NDUFS4, NDUFA1, SDHA, and
MTATP6), disorders of CoQ10 biosynthesis (PDSS2,
COQ2, COQ6, COQ9, and ADCK3), disorders of mito-chondrial translation (RARS2 and TFSM), or disordered
mitochondrial import of small molecules (SLC25A22).24
Myoclonic epilepsy in mitochondrial disease, in particu-lar caused by POLG mutations, can mimic a progressive
myoclonic epilepsy syndrome (such as neuronal ceroid-
lipofuscinosis) due to other associated symptoms, whichinclude cognitive decline, ataxia, motor incoordination,
and pigmentary retinopathy. POLG-related myoclonic epi-
lepsy has been shown to be more resistant to therapy withantiepileptic drugs compared with others, such as complex
I assembly factor gene mutations.24 Therefore, determining
the underlying etiology can have important implications fortreatment.
The treatment of mitochondrial epilepsy can be difficult
and require multiple antiepileptic drugs. The ketogenic dietmay be considered and appears to be helpful in terms of
reducing seizure frequency, but further studies are needed
to determine whether a subgroup of patients wouldrespond better than another. While the ketogenic has been
utilized in different epilepsy types and different RC
diseases, significant side effects, such as metabolic acidosisand persistent hypoglycemia, have been reported.28-31. In
particular, the ketogenic diet is specifically contraindicated
in pyruvate carboxylase deficiency.32 The use of valproicacid (VPA) has been very controversial. Avoidance of VPA-
induced hepatotoxicity and concern for Alpers syndrome
existed long before the POLG gene was discovered.Prospective testing for POLG of patients before the use of
VPA has been suggested by several groups.33,34
Case example: a 17-month-old with liver failure isreferred for neurologic examination. He had normal devel-
opment and neuroimaging. VPA was started after his
second seizure, when he could not tolerate his first antic-onvulsant. Within 3 months, he developed fulminant
hepatic failure. Molecular analysis of POLG revealed 2
heterozygous mutations in trans.
AtaxiaMitochondrial ataxias can present at any age and are
clinically characterized by a cerebellar syndrome of ataxia,
dysarthria, and nystagmus.35 Sensory ataxia from peripheralneuropathy or spinal cord involvement may also be present.
Cerebellar atrophy can be found as the neuroimaging
correlates with this symptom. Additional neuroimagingcharacteristics on MRI include involvement of the basal
ganglia and white matter or increased T2-weighted signal in
the cerebellar cortex.35 Imaging may be normal despite thepresence of cerebellar signs on examination. Other systemic
symptoms that accompany mitochondrial ataxia may include
encephalomyopathy, epilepsy, muscle weakness, regression,hearing loss, ophthalmoplegia, short stature, ataxia, optic
atrophy, increased muscle tone, and stroke-like episodes.35
Nuclear mitochondrial syndromes in which ataxia maybe present include Leigh syndrome; sensory ataxic

Genetics for the neurologist 185
neuropathy, dysarthria, ophthalmoplegia; spinocerebellar
ataxia and epilepsy (SCAE); Alpers-Huttenlocher syn-
drome; X-linked sideroblastic anemia with ataxia;infantile-onset spinocerebellar ataxia; mitochondrial reces-
sive ataxia syndrome (MIRAS); myoclonus epilepsy myo-
pathy sensory ataxia (MEMSA); and leukoencephalopathywith brainstem and spinal cord involvement with lactic
acidosis.36 Ataxia is less frequently seen in chronic PEO
(CPEO) (both recessive and dominant forms); MNGIE;diabetes insipidus, diabetes mellitus, optic atrophy, and
deafness; CoQ10 deficiency; autosomal dominant optic
atrophy and deafness, dilated cardiomyopathy with ataxia;and pyruvate dehydrogenase complex (PDC) deficiency.36
Friedreich ataxia is a secondary mitochondrial disease due
to mutations in frataxin leading to RC deficiencies. Clinicalfindings include cerebellar ataxia, spasticity, cardiomyopa-
thy, dysarthria, and diabetes.37
Case example: a 16-year-old boy presents with intermit-tent difficulty in walking. His examination reveals ataxia
and nystagmus, with mild dysarthria. His MRI report shows
cerebellar atrophy. The serum lactate level was elevated 5times greater than the upper limit of normal, with elevated
pyruvate. Skin fibroblasts reveal PDC deficiency.
Ocular SymptomsPatients with mitochondrial disease should be monitored ona regular basis by an ophthalmologist familiar with findings
associated with mitochondrial disease. The evaluation
should include an ophthalmologic examination, includingvisual acuity testing and a fundoscopic examination, to
screen for optic neuropathy and pigmentary retinopathy.38,39
Visual field testing can reveal abnormalities due tocerebral strokes or lesions by detecting field cuts and signs
of optic neuropathy. The external examination should
detect and monitor ptosis and ophthalmoplegia, whichcan be seen in CPEO due to POLG, POLG2, ANT1, or
TWINKLE mutations.40,41 Optic atrophy and retinal degen-
eration can be seen due to the metabolically active retinalpigment epithelium and retinal ganglion cell layer. Visual
evoked potentials may be abnormal in mitochondrial
disease where the axons of the retinal ganglion cells areinvolved, but the photoreceptors are spared (result of
electroretinography is normal). Electroretinography can
detect cone dysfunction, which has been reported in somenuclear mitochondrial diseases.42
Mitochondrial ptosis can have asymmetric onset and be
slowly progressive with little diurnal variation. When it isaccompanied by pigmentary retinopathy changes or PEO,
mitochondrial disease should be suspected over myasthenia
gravis or other conditions. Pigmentary retinopathy inmitochondrial disease has a perimacular distribution, with-
out drusen, and should not affect vision.42
Case example: a 15-year-old boy with a history ofgeneralized epilepsy and short stature presents with
migraine headaches and difficulty concentrating in school.
On neurologic examination, his eyelids appear to bedroopy. His parents concur that he has appeared more
sleepy than usual. He is seen by an ophthalmologist, and
ptosis is confirmed. Tissue biopsy reveals complex IV
deficiency. Mutations are identified in the autosomalrecessive gene, TWINKLE (C10orf2).
Sensorineural Hearing LossImpaired hearing is a common clinical finding in mito-chondrial disease, due to the high-energy demands of the
auditory apparatus. Seldom seen in isolation, the hearing
loss is usually seen in the context of a multisystem disorder,and in fact, the hearing loss may be missed due to other
clinical symptoms needing attention. Patients with mito-
chondrial disease should undergo testing for hearing losson a routine basis. The hearing loss is amenable to treat
with amplified devices, such as hearing aids and cochlear
implantation. Some of the nuclear mitochondrial genemutations that cause hearing loss include the MDDS
involving SUCLA2, SUCLG1, and TWINKLE.41
Case example: a 4-year-old boy presents with cerebralpalsy and developmental delay. Brain MRI is consistent
with Leigh syndrome, and skin biopsy showed complex IV
deficiency. A complex IV nuclear panel in blood revealedmutations in SURF1. With a diagnosis of mitochondrial
disease, he had brainstem auditory evoked responses and
was found to have bilateral hearing loss. He received acochlear implant, and his encephalopathy has improved.
Genetic Classification ofMitochondrial DiseasesThe clinical suspicion of mitochondrial disease based onthe above-mentioned red-flag neurologic symptoms opens
up a diagnostic odyssey for both the clinician and the
patient. Biochemical screening tests may include serumlactate, pyruvate, amino acids, acylcarnitine profile, carni-
tine battery, CoQ10 levels, and urine organic acids. Tissue
sampling may include skin biopsy for further biochemicalanalysis of RC defects or defects in pyruvate metabolism
(PDC and pyruvate carboxylase). Muscle or liver biopsy can
be analyzed for RC defects, mtDNA copy number to lookfor depletion syndromes, mtDNA sequencing to look for
mutations or deletions, and CoQ10 quantitation.
The confirmatory molecular diagnosis of mitochondrialdisease is challenging due to the presence of 2 separate but
interacting genomes: the mtDNA and the nuclear DNA
(nDNA). Both of these genomes must undergo DNAreplication and protein synthesis to generate the complex
(I-V) subunits and assembly factors involved in the RC. The
small, circular mtDNA contributes to only a fraction ofmitochondrial disease. Only 13 subunits of the RC are
mtDNA-encoded, whereas nDNA encodes the remaining
74 subunits of complexes I-V.43 In addition, nDNA alsoencodes CoQ10 and cytochrome c and important subunits
in the RC.44 Overall, nDNA encodes about 1500 proteins
targeted to the mitochondria.45 Therefore, most inheritedmitochondrial disease is nuclear encoded46, and as such,
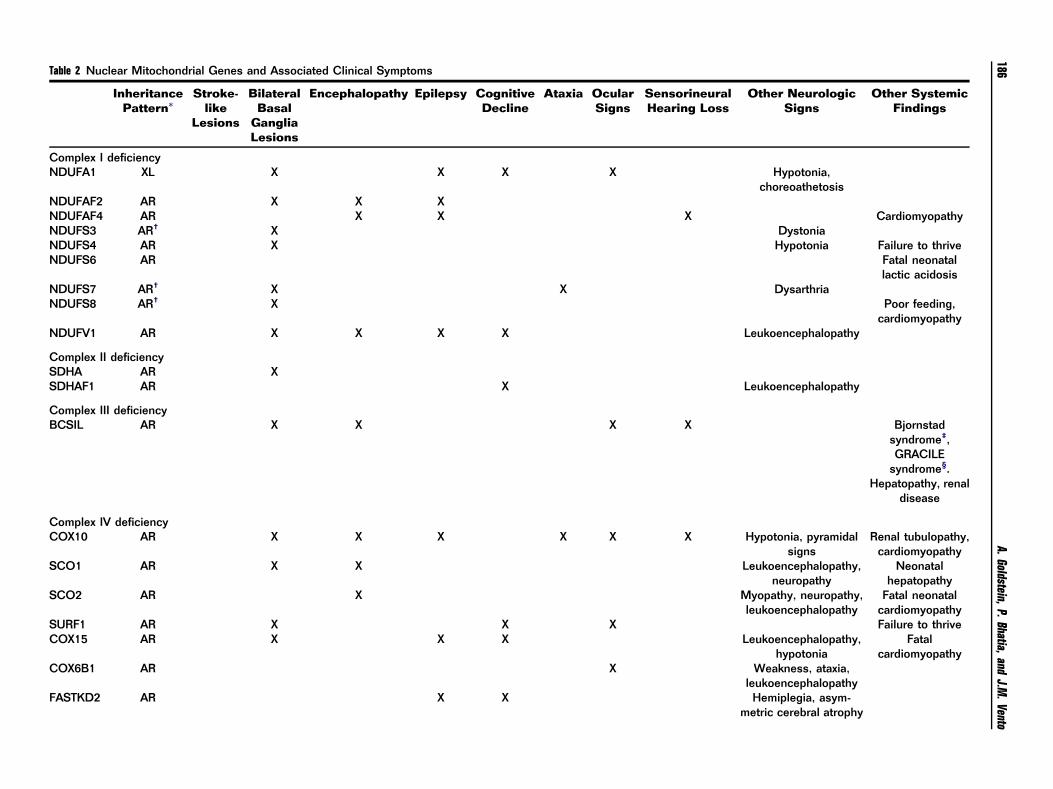
Table 2 Nuclear Mitochondrial Genes and Associated Clinical Symptoms
InheritancePattern*
Stroke-like
Lesions
BilateralBasal
GangliaLesions
Encephalopathy Epilepsy CognitiveDecline
Ataxia OcularSigns
SensorineuralHearing Loss
Other NeurologicSigns
Other SystemicFindings
Complex I deficiencyNDUFA1 XL X X X X Hypotonia,
choreoathetosisNDUFAF2 AR X X XNDUFAF4 AR X X X CardiomyopathyNDUFS3 AR† X DystoniaNDUFS4 AR X Hypotonia Failure to thriveNDUFS6 AR Fatal neonatal
lactic acidosisNDUFS7 AR† X X DysarthriaNDUFS8 AR† X Poor feeding,
cardiomyopathyNDUFV1 AR X X X X Leukoencephalopathy
Complex II deficiencySDHA AR XSDHAF1 AR X Leukoencephalopathy
Complex III deficiencyBCSIL AR X X X X Bjornstad
syndrome‡,GRACILE
syndrome§.Hepatopathy, renal
disease
Complex IV deficiencyCOX10 AR X X X X X X Hypotonia, pyramidal
signsRenal tubulopathy,
cardiomyopathySCO1 AR X X Leukoencephalopathy,
neuropathyNeonatal
hepatopathySCO2 AR X Myopathy, neuropathy,
leukoencephalopathyFatal neonatal
cardiomyopathySURF1 AR X X X Failure to thriveCOX15 AR X X X Leukoencephalopathy,
hypotoniaFatal
cardiomyopathyCOX6B1 AR X Weakness, ataxia,
leukoencephalopathyFASTKD2 AR X X Hemiplegia, asym-
metric cerebral atrophy
A.Goldstein,P.Bhatia,andJ.M
.Vento186

Complex V deficiencyTMEM70 AR X X X X Hypotonia
Cardio-myopa-
thy,IUGR
Coenzyme Q 10 deficiencyPDSS1 AR X X Neuropathy Cardiac valvu-
lopathy, livedoreticularis
PDSS2 AR X X X X Myopathy NephropathyCoQ2 AR X X Myopathy NephropathyCoQ6 X X X NephropathyCoQ9 AR X Myopathy Nephropathy,
cardiomyopathyADCK3 AR X X X X
Mitochondrial DNA depletion syndromesPOLG AD/AR X X X X X X Myopathy, neuropathy,
leukoence phalopathyHepatopathy
RRM2B AD/AR X X X X Myopathy, neuropathy NephropathySUCLA2 AR X X X X Myopathy, dystonia Lactic acidosisSUCLG1 AR X X X Myopathy, dystonia Infantile lactic
acidosisSUCLG2 X X MyopathyC10orf2/
TwinkleAD/AR x X X X X Neuropathy, athetosis Hepatic
involvementDGUOK AR x X Neuropathy Hepatic involve-
ment (neonatalonset, severe)
MPV17 AR x Neuropathy Infantile hepatopa-thy, hypoglycemia
TK2 AR X X Congenital musculardystrophy
TYMP AR X X X Myopathy, neuropathy,leukoence phalopathy
Gastrointestinaldysmotility, anor-
exia, MNGIE
OthersTAZ XL X Weakness Cardiomyopathy
(Barth syndrome)k,neutropenia
OPA1 AD X X X Myopathy, neuropathyOPA3 AD/AR X X Chorea, spastic
paraparesis
Geneticsfor
theneurologist
187
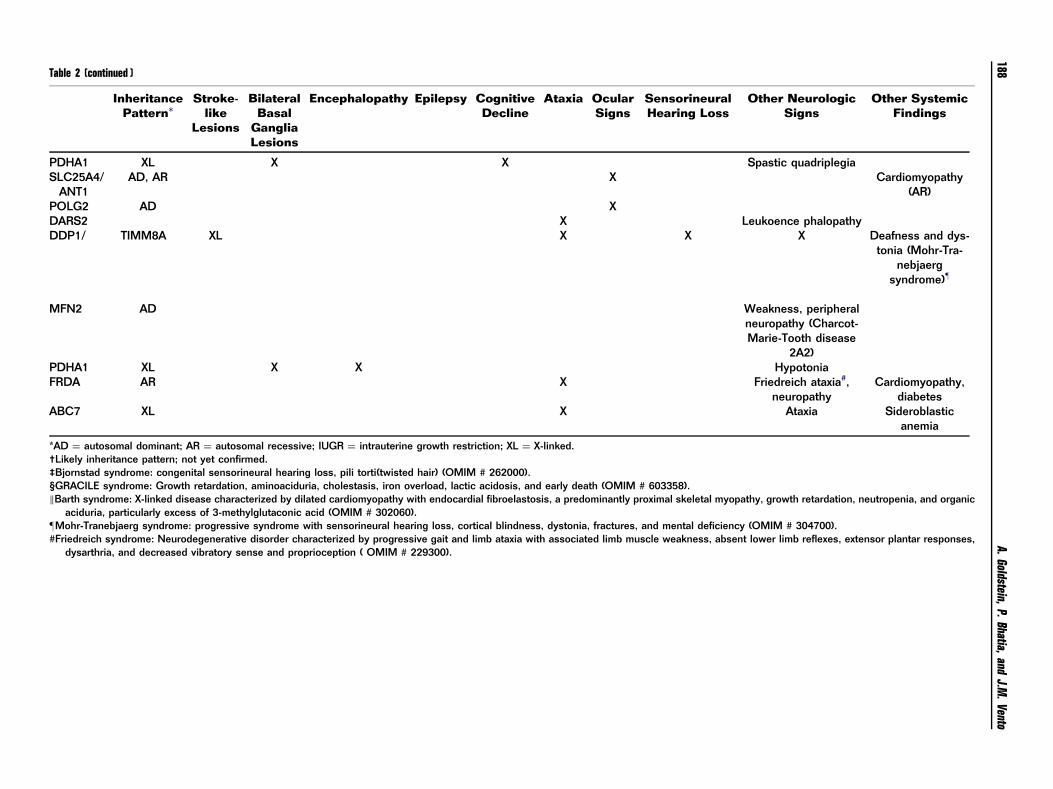
Table 2 (continued )
InheritancePattern*
Stroke-like
Lesions
BilateralBasal
GangliaLesions
Encephalopathy Epilepsy CognitiveDecline
Ataxia OcularSigns
SensorineuralHearing Loss
Other NeurologicSigns
Other SystemicFindings
PDHA1 XL X X Spastic quadriplegiaSLC25A4/
ANT1AD, AR X Cardiomyopathy
(AR)POLG2 AD XDARS2 X Leukoence phalopathyDDP1/ TIMM8A XL X X X Deafness and dys-
tonia (Mohr-Tra-nebjaerg
syndrome)z
MFN2 AD Weakness, peripheralneuropathy (Charcot-Marie-Tooth disease
2A2)PDHA1 XL X X HypotoniaFRDA AR X Friedreich ataxia#,
neuropathyCardiomyopathy,
diabetesABC7 XL X Ataxia Sideroblastic
anemia
*AD ¼ autosomal dominant; AR ¼ autosomal recessive; IUGR ¼ intrauterine growth restriction; XL ¼ X-linked.†Likely inheritance pattern; not yet confirmed.‡Bjornstad syndrome: congenital sensorineural hearing loss, pili torti(twisted hair) (OMIM # 262000).§GRACILE syndrome: Growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, and early death (OMIM # 603358).kBarth syndrome: X-linked disease characterized by dilated cardiomyopathy with endocardial fibroelastosis, a predominantly proximal skeletal myopathy, growth retardation, neutropenia, and organic
aciduria, particularly excess of 3-methylglutaconic acid (OMIM # 302060).zMohr-Tranebjaerg syndrome: progressive syndrome with sensorineural hearing loss, cortical blindness, dystonia, fractures, and mental deficiency (OMIM # 304700).#Friedreich syndrome: Neurodegenerative disorder characterized by progressive gait and limb ataxia with associated limb muscle weakness, absent lower limb reflexes, extensor plantar responses,
dysarthria, and decreased vibratory sense and proprioception ( OMIM # 229300).
A.Goldstein,P.Bhatia,andJ.M
.Vento188

Genetics for the neurologist 189
the inheritance pattern for nuclear mitochondrial genes
follows mendelian laws: autosomal dominant, autosomal
recessive, or X-linked.47
Studies of nDNA can be conducted less invasively than
tissue biopsies through blood and have evolved from
single-gene tests to panels for a particular clinical syndromeor RC defect (eg, complex I deficiency panel) to next-
generation sequencing, which includes hundreds of rele-
vant nuclear mitochondrial genes. With the discovery ofnuclear genes causing mitochondrial disease, clinical phe-
notypes and prognosis are becoming more defined. Table 2
offers an overview of the relevant nuclear mitochondrialgenes and their clinical manifestations.4,24,48,49
The list of nuclear genes associated with mitochondrial
dysfunction is constantly expanding,50 especially with theadvent of commercially available next-generation sequen-
cing. The first identified nuclear gene mutation responsible
for mitochondrial disease was SDHA, which is found insiblings with Leigh syndrome,51 and to date, mutations
have been reported in more than 100 nuclear-encoded
genes.48 In the near future, this number is expected toexpand. Advances in sequencing technology have already
allowed clinicians to better diagnose and treat their
patients, especially in the setting of nuclear mutations ininfantile mitochondrial disease.1
Genes of nDNA are required by the mitochondria for
many functions, including RC complex structural proteinsand assembly factors; mtDNA maintenance (replication,
maintenance, and translation); CoQ10 biosynthesis; main-
tenance of the lipid milieu of the inner mitochondrialmembrane; solute carrier across the inner mitochondrial
membrane; fission and fusion; and mitochondrial biogen-
esis.52,53 Mutations disrupting mtDNA maintenance canproduce single or multiple mtDNA deletions or and cause
the clinical syndromes of MDDS and MNGIE.
Historically, mitochondrial disease has largely beenclassified by clinical symptoms, biochemical testing, and
tissue biopsies. The advent of next-generation sequencing
has enabled molecular diagnosis to be an importantdiagnostic element in the characterization of mitochondrial
syndromes and allowed for more accurate family counsel-
ing. With likely hundreds of nuclear genetic etiologies formitochondrial disease, a classification system is helpful for
neurologists to understand the pathophysiology of clinical
symptoms.
RC DysfunctionNuclear genes are responsible for most of the structural
proteins and assembly factors in the RC. Table 2 sum-
marizes the more common gene mutations grouped byeach RC complex along with the clinical manifestations. A
single nuclear gene mutation can lead to single biochemical
abnormality (ie, NDUFSV1 mutation causing isolatedcomplex I deficiency) or may cause multiple RC complex
deficiencies (ie, POLG mutations causing combined com-
plex I þ IV deficiencies). In pediatric patients withmitochondrial disease, up to 25% of cases have multiple
RC deficiencies, usually due to mutations involving mtDNA
maintenance (POLG), mtDNA deletion (Kearns-Sayre syn-
drome), mitochondrial translation defects, or a CoQ10biosynthesis defect.54
CoQ10 DeficiencyCoQ10, also called ubiquinone, is a potent antioxidant as
well as an electron carrier in the RC between complexes I þII to complex III. CoQ10 is synthesized in the mitochon-
dria and involves at least 12 genes. Defects in the nuclear
genes are inherited in an autosomal recessive manner. Adeficiency of CoQ10 may appear on tissue biopsy analysis
as a combined RC defect involving complexes I þ III or II
þ III.55
Clinically, 6 major phenotypes have been identified13,56:
�
encephalomyopathic form with seizures and ataxia;�
multisystem infantile form with encephalopathy, cardi-omyopathy, and renal failure;
� predominantly cerebellar form with ataxia and cerebellaratrophy;
�
Leigh syndrome with growth retardation; � isolated myopathic form; and�
steroid-resistant nephrotic syndrome.There are 6 primary biosynthetic genes: PDSS1, PDSS2,
COQ2, COQ6, COQ9, and ADCK3 (CABC1 and COQ8);and involvement of 2 other genes: ETFDH and APTX; that
can lead to secondary deficiencies. Testing for the majority
of these genes is clinically available and an importantcondition as primary biosynthetic disorders are amenable
to treat with supplemental CoQ10.56
Defects of mtDNA TranslationThe nDNA encodes proteins that are essential for mtDNAprotein synthesis. A mutation in one of these genes, RARS2,
has been described as a progressive clinical syndrome of
lactic acidosis and pontocerebellar hypoplasia, and severeintractable epilepsy.57 Other translational genes that have
been characterized are included in Table 2.
Disorders of mtDNA MaintenancemtDNA Depletion Syndromes (MDS)MDS are a genetically heterogenous group of disorders with
the same molecular end result of reduced amount of
mtDNA (known as copy number) in specific tissues dueto mutations in genes that affect the mitochondrial nucleo-
tide pools or the mtDNA replication fork and lead to
decreased mtDNA copy number (depletion of mtDNA).58
Symptoms of mtDNA depletion syndrome vary by age but
include specific phenotypes of encephalomyopathy, hepa-
topathy, and isolated myopathy. Infants tend to have moreprominent lactic acidosis, failure to thrive, and hypotonia.
Children tend to have prominent epilepsy and liver
involvement. Muscle weakness tends to have a bimodalpresentation in childhood and then in adulthood.

A. Goldstein, P. Bhatia, and J.M. Vento190
Prominent symptoms with increasing age include migraine-
like headaches, ataxia, polyneuropathy, cognitive impair-
ment, psychiatric symptoms, and gastrointestinalsymptoms.41
mtDNA depletion syndromes have become an important
cause of inherited metabolic disorders, especially in chil-dren, but also in adults. The manifestations vary from a
tissue-specific mtDNA depletion to widespread multisyste-
mic disorders. Nine genes are known to underlie this groupof disorders, and many disease genes are still unidentified.
However, the disease mechanisms seem to be intimately
associated with mtDNA replication and nucleotide poolregulation. We review here the current knowledge on the
clinical and molecular genetic features of mtDNA depletion
syndromes. These syndromes have been divided into 3clinical categories: myopathic (TK2), encephalomyopathic
(SUCLA2, SUCLG1, RRM2B, and TYMP), and hepatocer-
ebral (DGUOK, MPV17, POLG, and C10orf2). However,these categories are not discrete, and they can have clinical
overlap, especially as the disease progresses. POLG-related
disorders would be reviewed in most detail, followed by abrief description of the other mitochondrial depletion
syndromes.
POLG-Related DisordersPOLG gene mutations are the most common cause ofinherited mitochondrial diseases in children and adults.
POLG encodes for human POLG, the only polymerase
involved in mtDNA replication and repair; thus, abnorm-alities can lead to mtDNA mutations or multiple deletions
or both. Currently, more than 150 mutations within POLG
are known, with a map of mutations updated on a regularbasis available at /http://tools.niehs.nih.gov/polg/S.59
Strict genotype-phenotype correlation is not possible, as
the clinical presentation is not solely dependent on thePOLG genotype.60,61 Like other mitochondrial disorders,
they involve multiple organ systems, mainly central and
peripheral nervous system, liver, muscle, and other organs,such as the gastrointestinal system.60 Disease progression of
POLG-related disorders is highly unpredictable. Neurologic
manifestations in younger patients mainly consist ofseizures, lactic acidosis, and hepatic failure. However, older
individuals mainly present with myopathy, sensory ataxia,
and CPEO. Neuroimaging findings include an increasedsignal on T2-weighted imaging of the posterior thalami,
dentate nuclei, inferior olives, and occipital cortex.
POLG-related disorders are classified as 6 recognizablephenotypes with symptom overlap, which actually creates a
disease spectrum.
1.
AHS: AHS is the most severe phenotype of the POLG-related disorders.62,63 Many children with AHS havetypical development before the onset of symptoms. AHS
typically presents before 4 years of age, but may presentthrough early adulthood with seizures, developmental
delay with episodic regression, and hepatopathy due to
anticonvulsants. Anticonvulsants, especially VPA, maycause the liver enzymes to elevate, raising the index of
suspicion of POLG. Many mutations in POLG have been
reported and may present in either an autosomal
recessive or dominant fashion. The clinical trial ofrefractory seizures, episodic psychomotor regression,
and a characteristic hepatopathy has been described as
the classic presentation.64 However, the clinical pheno-type and progression are highly variable. The disease
typically starts in infancy or in early childhood with
seizures. Although no seizure semiology is typical forAHS, as the disease progresses, seizures evolve into
more complex and refractory syndromes, such as focal
status epilepticus, EPC, or multifocal myoclonic epi-lepsy. Characteristic EEG findings in the beginning may
be unilateral occipital rhythmic high-amplitude slow
activity and superimposed polyspikes.65 The seizure fociusually shift on EEG with time. The response to antic-
onvulsants is also variable. VPA must be used with
extreme caution when the etiology of seizures isunknown, as it can precipitate liver dysfunction in
AHS.33,66 Cognitive decline is characteristic and may
be the result of neuronal gliosis, refractory seizures, andhigh dosages of various antiepileptic medications. The
characteristic early occipital lobe involvement leads to
migraine with visual auras (in older children) andcortical blindness, and it may also serve as the initial
seizure focus. Other characteristic neurologic symptoms
include hypotonia, sensory ataxia or cerebellar ataxia orboth, extrapyramidal movements, peripheral neuropa-
thy, and progressive spastic paraparesis (due to the
destruction of cerebral cortex). Hepatic involvement isalso highly variable. It can progress to fulminant end-
stage liver disease in few months. The disease progres-
sion can be variable, marked by periods of stability. Lifeexpectancy from onset of symptoms ranges from
3 months to 12 years.60 Brain computed tomography
or MRI may be initially normal or demonstrate gliosis inoccipital lobe regions and generalized atrophy. Cerebel-
lum, basal ganglia, thalamus, and brainstem are sequen-
tially involved.67
2.
MEMSA: MEMSA includes a spectrum of disorders withepilepsy, myopathy (distal and proximal), and ataxia
without ophthalmoplegia.68 It may include some of thepatients previously classified as having MIRAS and
SCAE. Subclinical sensory polyneuropathy leading to
ataxia, starting in teenage years, is usually the first signof the disease. Subsequently, epilepsy develops, begin-
ning as focal but rapidly progressing to refractory
generalized type, accompanied by progressive encepha-lopathy. Myoclonus and myopathy may also be present.
Conspicuous absence of ragged red fibers from muscle
biopsy clearly distinguishes this from myoclonic epi-lepsy with ragged red fibers syndrome.68
3.
Ataxia neuropathy spectrum (ANS): ANS is an auto-somal recessive POLG disorder, in which multipledeletions in mtDNA occur.69 This spectrum, previously
referred to as sensory ataxic neuropathy, dysarthria,
ophthalmoplegia, encompasses several clinical syn-dromes that include ataxia, neuropathy (sensory, motor,

Genetics for the neurologist 191
or mixed), bulbar dysfunction, and PEO without myo-
pathy (in contrast to MEMSA).70 The disease onset is
usually in early teenage years to late third decade. LikeMEMSA, it includes many patients previously classified
as MIRAS or SCAE. Epileptic encephalopathy may be a
component, but disease course is usually less progres-sive. Psychiatric disturbance is common but may be
underrecognized especially in adult-onset mitochondrial
disease. Migrainous headaches may precede symptomsby many years. Other features include myoclonus,
blindness, hearing loss, and varying degree of liver
failure. Muscle pathology is often normal.
4. Childhood myocerebrohepatopathy spectrum (MCHS):MCHS is the rarest form of POLG spectrum. Disease
onset is between first few months of life and 3 years. Thedisease course is rapidly progressive with a fatal out-
come. Neurologic manifestations include developmental
delay, encephalopathy, dementia, myopathy, and hypo-tonia. In contrast to AHS, seizures are not a common
manifestation in MCHS. Other features include failure to
thrive, lactic acidosis, liver failure, renal tubular acido-sis, pancreatitis, cyclic vomiting, and hearing loss.71
Brain MRI can show generalized atrophy. Early disease
onset, hepatopathy, encephalopathy, and a fatal outcomemake this disorder resembles AHS. However, severe
myopathy, specific liver pathology, and nonspecific MRI
brain findings help differentiate MCHS from AHS.63
5.
Autosomal recessive PEO: adult-onset PEO withoutsystemic involvement is the hallmark of autosomal
recessive PEO.72 Molecular genetic confirmation ofPOLG is required for diagnosis, as isolated PEO can
also be seen in thyroid disorders or myasthenia gravis.
PEO may be the first manifestation of other moresystemic disorders in the POLG spectrum like
ANS.73,74 Other associated features may include neuro-
pathy, myopathy, and sensory ataxia.75
6.
Autosomal dominant PEO (adPEO): adPEO is anotheradult-onset disorder, which manifests as progressive
weakness of extraocular eye muscles,72,73 along withproximal myopathy and wasting leading to exercise
intolerance. CPEO þ (CPEO-plus) manifests as sensor-
ineural hearing loss, axonal neuropathy, cerebellarataxia, depression, parkinsonism, hypogonadism,76
and cataracts. Mutations in POLG1, ANT1, or C10orf2
(TWINKLE) may result in multiple mtDNA deletionsleading to this disorder.77
MNGIEMNGIE is an autosomal recessive disorder caused by
mutations in the nuclear TYMP gene coding for the enzymethymidine phosphorylase. Accumulation of thymidine is
toxic and leads to mtDNA instability and RC dysfunction.
Symptoms include severe gastrointestinal dysmotility,cachexia, ptosis and external ophthalmoplegia, peripheral
sensorimotor neuropathy, and a diffuse leukoencephalo-
pathy.78 Diagnosis is made by the detection of elevatedthymidine or decreased thymidine phosphorylase levels in
blood. Confirmatory molecular testing can be performed
with TYMP sequencing. MNGIE is rare, but treatment via
allogenic hematopoetic stem cell transplantation is availableas part of a research protocol, making MNGIE perhaps the
only mitochondrial disorder that is potentially curable.78
Solute Transport Across the InnerMitochondrial MembraneSolute transporters must shuttle specific molecules across
the relatively impermeable inner mitochondrial membrane.SLC25A22 is the mitochondrial glutamate carrier. Muta-
tions in this gene have been associated with neonatal or
early infantile epileptic encephalopathy with burst suppres-sion EEG (Otohara syndrome).79
Symptoms include myoclonic and focal seizures from
first few days of life, microcephaly, hypotonia, and globaldevelopmental delay. Neuroimaging may reveal cerebellum
and corpus callosum hypoplasia, abnormal gyral pattern in
temporoparietal area, and hypomyelination of temporalpoles.24
Another important solute transporter is ANT1
(SLC25A4), which transports ATP out of the mitochondrialmatrix in exchange for adenosine diphosphate. Mutations
in ANT1 can cause adPEO, cardiomyopathy, and skeletal
myopathy.80 Solute transporters are a common, growinggroup of disorders that may be amenable to specific
treatments.
ConclusionsClinical suspicion for nuclear mitochondrial disorders iswarranted when there are neurologic features and multi-
system involvement. Clinical features, biochemical studies,
and neuroradiologic features can help direct diagnostictesting. Determination of a molecular etiology is not always
possible; however, rapidly evolving genomic technology
will greatly improve diagnostic capabilities. Expandedmultigene panels and genomic testing will allow clinicians
to provide more accurate recurrence risk counseling to
families. Additionally, it is expected to improve our under-standing of the spectrum and natural history of the vast
range of mitochondrial disease in time. The advent of
extensive genetic testing may require a clinician to use apatient’s genotype to help determine symptomatology.
Thorough examination, family history, and functional and
biochemical testing will remain critical elements to inter-pret test results and counsel families. Therefore, the
clinician must be aware not only of the clinical diagnostic
signs and the clinical phenotypes of mitochondrial disease,but also of the different diagnostic testing available. We
hope this paper serves as a resource for the clinician who
must gather the clinical information, send or process thebiochemical and molecular data, and arrive at a diagnosis
that not only best fits the clinical symptoms, but allows for
further treatment options and genetic counseling of thefamily in the context of a nuclear mitochondrial disorder.

A. Goldstein, P. Bhatia, and J.M. Vento192
References1. Calvo SE, Compton AG, Hershman SG, et al: Molecular diagnosis of
infantile mitochondrial disease with targeted next-generation sequen-
cing. Science Translational Medicine 4:118ra10, 2012
2. Munnich A, Rotig A, Chretien D, et al: Clinical presentation of
mitochondrial disorders in childhood. Journal of Inherited Metabolic
Disease 19:521-527, 1996
3. Johns DR: Seminars in medicine of the Beth Israel Hospital, Boston.
Mitochondrial DNA and disease. New England journal of medicine
333:638-644, 1995
4. Haas R, Parikh S, Falk M, et al: Mitochondrial disease: A practical
approach for primary care physicians. Pediatrics 120:1326-1333,
2007
5. Parikh S: The neurologic manifestations of mitochondrial disease.
Developmental Disabilities Research Reviews 16:120-128, 2010
6. Deschauer M, Tennant S, Rokicka A, et al: MELAS associated with
mutations in the POLG1 gene. Neurology 68:1741-1742, 2007
7. Ito H, Mori K, Kagami S: Neuroimaging of stroke-like episodes in
MELAS. Brain and Development 33:283-288, 2011
8. Leigh D: Subacute necrotizing encephalomyelopathy in an infant.
Journal of Neurology, Neurosurgery, and Psychiatry 14:216-221, 1951
9. Finsterer J, Kopsa W: Basal Ganglia calcification in mitochondrial
disorders. Metabolic Brain Disease 20:219-226, 2005
10. Farina L, Chiapparini L, Uziel G, et al: MR findings in Leigh syndrome
with COX deficiency and SURF-1 mutations. American Journal of
Neuroradiology 23:1095-1100, 2002
11. Finsterer J: Leigh and Leigh-like syndrome in children and adults.
Pediatric Neurology 39:223-235, 2008
12. Willis TA, Davidson J, Gray RG, et al: Cytochrome oxidase deficiency
presenting as birth asphyxia. Developmental Medicine and Child
Neurology 42:414-417, 2000
13. Van Maldergem L, Trijbels F, DiMauro S, et al: CoenzymeQ-responsive
Leigh’s encephalopathy in two sisters. Annals of Neurology 52:
750-754, 2002
14. Wick R, Scott G, Byard RW: Mechanisms of unexplained death and
autopsy findings in Leigh syndrome (subacute necrotizing encepha-
lomyelopathy). Journal of Forensic and Legal Medicine 14:42-45,
2007
15. Sakushima K, Tsuji-Akimoto S, Niino M, et al: Adult Leigh disease
without failure to thrive. Neurologist 17:222-227, 2011
16. Dahl H: Getting to the nucleus of mitochondrial disorders: identifica-
tion of respiratory chain-enzyme genes causing Leigh syndrome
(Editorial). American Journal of Human Genetics 63:1594-1597,
1998
17. Lopez LC, Schuelke M, Quinzii CM, et al: Leigh syndrome with
nephropathy and CoQ10 deficiency due to decaprenyldiphosphate
synthase subunit 2 (PDSS2) mutations. American Journal of Human
Genetics 79:1125-1129, 2006
18. Dimmock DP, Zhang Q, Dionisi-Vici C, et al: Clinical and molecular
features of mitochondrial DNA depletion due to mutations in
deoxyguanosine kinase. Human Mutation 29:330-331, 2008
19. Spinazzola A, Invernizzi F, Carrara F, et al: Clinical and molecular
features of mitochondrial DNA depletion syndromes. Journal of
Inherited Metabolic Disease 32:143-158, 2009
20. Mandel H, Szargel R, Labay V, et al: The deoxyguanosine kinase gene
is mutated in individuals with depleted hepatocerebral mitochondrial
DNA. Nature Genetics 29:337-341, 2001
21. Finsterer J: Mitochondrial disorders, cognitive impairment and
dementia. Journal of the Neurological Sciences 283:143-148, 2009
22. Finsterer J: Cognitive dysfunction in mitochondrial disorders. Acta
Neurologica Scandinavica 126:1-11, 2012
23. Finsterer J: Cognitive decline as a manifestation of mitochondrial
disorders (mitochondrial dementia). Journal of the Neurological
Sciences 272:20-33, 2008
24. Rahman S: Mitochondrial disease and epilepsy. Developmental Med-
icine and Child Neurology 54:397-406, 2012
25. Parikh S, Cohen BH, Gupta A, et al: Metabolic testing in the pediatric
epilepsy unit. Pediatric Neurology 38:191-195, 2008
26. Lee YM, Kang HC, Lee JS, et al: Mitochondrial respiratory chain
defects: Underlying etiology in various epileptic conditions. Epilepsia
49:685-690, 2008
27. El Sabbagh S, Lebre AS, Bahi-Buisson N, et al: Epileptic phenotypes in
children with respiratory chain disorders. Epilepsia 51:1225-1235,
2010
28. Joshi CN, Greenberg CR, Mhanni AA, et al: Ketogenic diet in Alpers-
Huttenlocher syndrome. Pediatric Neurology 40:314-316, 2009
29. Kang HC, Lee YM, Kim HD, et al: Safe and effective use of the
ketogenic diet in children with epilepsy and mitochondrial respiratory
chain complex defects. Epilepsia 48:82-88, 2007
30. Kossoff EH, Zupec-Kania BA, Amark PE, et al: Optimal clinical
management of children receiving the ketogenic diet: Recommenda-
tions of the International Ketogenic Diet Study Group. Epilepsia
50:304-317, 2009
31. Nangia S, Caraballo RH, Kang HC, et al: Is the ketogenic diet effective
in specific epilepsy syndromes? Epilepsy Research 100:252-257, 2012
32. DeVivo DC, Haymond MW, Leckie MP, et al: The clinical and
biochemical implications of pyruvate carboxylase deficiency. Journal
of Clinical Endocrinology Metabolism 45:1281-1296, 1997
33. Saneto RP, Lee IC, Koenig MK, et al: POLG DNA testing as an
emerging standard of care before instituting valproic acid therapy for
pediatric seizure disorders. Seizure 19:140-146, 2010
34. Stewart JD, Horvath R, Baruffini E, et al: Polymerase g gene POLG
determines the risk of sodium valproate-induced liver toxicity.
Hepatology 52:1791-1796, 2010
35. Poretti A, Wolf NI, Boltshauser E: Differential diagnosis of cerebellar
atrophy in childhood. European Journal of Paediatric Neurology
12:155-167, 2008
36. Finsterer J: Mitochondrial ataxias. Canadian Journal of Neurological
Sciences 36:543-553, 2009
37. Delatycki MB, Williamson R, Forrest SM: Friedreich ataxia: An
overview. Journal of Medical Genetics 37:1-8, 2000
38. Newman NJ: Mitochondrial disease and the eye. Ophthalmology
Clinics of North America 5:405-424, 1992
39. Mullie MA, Harding AE, Petty RKH, et al: The retinal manifestations of
mitochondrial myopathy. Archives of Ophthalmology 103:1825-1830,
1985
40. Agostino A, Valletta L, Chinnery PF, et al: Mutations of ANT 1,
Twinkle, and POLG1 in sporadic progressive external ophthalmople-
gia (PEO). Neurology 60:1354-1356, 2003
41. Suomalainen A, Isohanni P: Mitochondrial DNA depletion
syndromes-many genes, common mechanisms. Neuromuscular Dis-
orders 20:429-437, 2010
42. Ksiazek SM, Peterson PL, Frank RN, et al: Retinopathy in non-Kearns-
Sayre mitochondrial disease. Neurology 40:312, 1990 (suppl 1)
43. Larsson NG, Clayton DA: Molecular genetic aspects of human
mitochondrial disorders. Annual Review of Genetics 29:151-178,
1995
44. DiMauro S, Bonilla E: in Rosenberg RN, Prusiner SB, DiMauro S
(eds.), The Molecular and Genetic Basis of Neurological Disease, ed 2
Boston; Butterworth-Heinemann, 1997, pp 201-235
45. Wong L: Molecular genetics of mitochondrial disorders. Develop-
mental Disabilities Research Reviews 16:154-162, 2010
46. Leonard JV, Schapira AH: Mitochondrial respiratory chain disorders II:
Neurodegenerative disorders and nuclear gene defects. Lancet
355:389-394, 2000
47. Dimauro S, Davidzon G: Mitochondrial DNA and disease. Annals of
Medicine 37:222-232, 2005
48. Koopman WJ, Willems PH, Smeitink JA: Monogenic mitochondrial
disorders. New England Journal of Medicine 366:1132-1141, 2012
49. Calvo SE, Mootha VK: The mitochondrial proteome and human
disease. Annual Review of Genomics and Human Genetics 11:25-44,
2010
50. Tucker EJ, Compton AG, Thorburn DR: Recent advances in the
genetics of mitochondrial encephalopathies. Current Neurology and
Neuroscience Reports 10:277-285, 2010

Genetics for the neurologist 193
51. Bourgeron T, Rustin P, Chretien D, et al: Mutation of a nuclear
succinate dehydrogenase gene results in mitochondrial respiratory
chain deficiency. Nature Genetics 2:144-149, 1995
52. Zhu X, Peng X, Guan MX, et al: Pathogenic mutations of nuclear genes
associated with mitochondrial disorders. Acta Biochimica et Biophy-
sica Sinica (Shanghai) 41:179-187, 2009
53. DiMauro S, Schon EA: Mitochondrial disorders in the nervous system.
Annual Review of Neuroscience 31:91-123, 2008
54. Thorburn DR, Sugiana C, Salemi R, et al: Biochemical and molecular
diagnosis of mitochondrial respiratory chain disorders. Biochimica et
Biophysica Acta 1659:121-128, 2004
55. Ogasahara S, Engel AG, Frens D, et al: Muscle coenzyme Q deficiency
in familial mitochondrial encephalomyopathy. Proceedings of the
National Academy of Sciences of the United States of America
86:2379-2382, 1989
56. Qunzii C, Hirano M: Primary and secondary CoQ10 deficiencies in
humans. BioFactors 37:361-365, 2011
57. Edvardson S, Shaag A, Kolesnikova O, et al: Deleterious mutation in
the mitochondrial arginyl-transfer RNA synthetase gene is associated
with pontocerebellar hypoplasia. American Journal of Human Genet-
ics 81:857-862, 2007
58. Copeland WC: Inherited mitochondrial diseases of DNA replication.
Annual Review of Medicine 59:131-146, 2008
59. Human DNA polymerase gamma mutation database. Available at:
http://tools.niehs.nih.gov/polg/. Accessed July 15, 2012.
60. Cohen BH, Naviaux RK: The clinical diagnosis of POLG disease and other
mitochondrial DNA depletion disorders. Methods 51:364-373, 2010
61. Tang S, Wang J, Lee NC, et al: Mitochondrial DNA polymerase gamma
mutations: an ever expanding molecular and clinical spectrum.
Journal of Medical Genetics 48:669-681, 2011
62. Naviaux RK, Nguyen KV: POLG mutations associated with Alpers
syndrome and mitochondrial DNA depletion. Annals of Neurology
58:491, 2005
63. Nguyen KV, Sharief FS, Chan SS, et al: Molecular diagnosis of Alpers
syndrome. Journal of Hepatology 45:108-116, 2006
64. Harding BN: Progressive neuronal degeneration of childhood with
liver disease (Alpers-Huttenlocher syndrome): A personal review.
Journal of Child Neurology 5:273-287, 1990
65. Engelsen BA, Tzoulis C, Karlsen B, et al: POLG1 mutations cause a
syndromic epilepsy with occipital lobe predilection. Brain 131:
818-828, 2008
66. Bicknese AR, May W, Hickey WF, et al: Early childhood hepatocer-
ebral degeneration misdiagnosed as valproate hepatotoxicity. Annals of
Neurology 32:767-775, 1992
67. Smith JK, Mah JK, Castillo M, et al: Imaging findings in two patients
with Alpers’ syndrome. Clinical Imaging 20:235-237, 1996
68. Van Goethem G, Mercelis R, Lofgren A, et al: Patient homozygous for
a recessive POLG mutation presents with features of MERRF.
Neurology 61:1811-1813, 2003
69. Van Goethem G, Luoma P, Rantamaki M, et al: POLG mutations in
neurodegenerative disorders with ataxia but no muscle involvement.
Neurology 63:1251-1257, 2004
70. Fadic R, Russell JA, Vedanarayanan VV, et al: Sensory ataxic neuro-
pathy as the presenting feature of a novel mitochondrial disease.
Neurology 49:239-245, 1997
71. Wong LJ, Naviaux RK, Brunetti-Pierri N, et al: Molecular and clinical
genetics of mitochondrial diseases due to POLG mutations. Human
Mutation 10:E150-E172, 2008
72. Van Goethem G, Dermaut B, Lofgren A, et al: Mutation of POLG is
associated with progressive external ophthalmoplegia characterized by
mtDNA deletions. Nature Genetics 28:211-212, 2001
73. Lamantea E, Tiranti V, Bordoni A, et al: Mutations of mitochondrial
DNA polymerase gamma are a frequent cause of autosomal dominant
or recessive progressive external ophthalmoplegia. Annals of Neurology
52:211-219, 2002
74. Van Goethem G, Martin JJ, Dermaut B, et al: Recessive POLG
mutations presenting with sensory and ataxic neuropathy in com-
pound heterozygote patients with progressive external ophthalmople-
gia. Neuromuscular Disorders 13:133-142, 2003
75. Milone M, Brunetti-Pierri N, Tang LY, et al: Sensory ataxic neuropathy
with ophthalmoparesis caused by POLG mutations. Neuromuscular
Disorders 18:626-632, 2008
76. Pagnamenta AT, Taanman JW, Wilson CJ, et al: Dominant inheritance
of premature ovarian failure associated with mutant mitochondrial
DNA polymerase gamma. Human Reproduction 21:2467-2473, 2006
77. Fratter C, Gorman GS, Stewart JD, et al: The clinical, histochemical,
and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology
74:1619-1626, 2010
78. Garone C, Tadesse S, Hirano M: Clinical and genetic spectrum of
mitochondrial neurogastrointestinal encephalomyopathy. Brain
134:3326-3332, 2011
79. Molinari F, Raas-Rothschild A, Rio M, et al: Impaired mitochondrial
glutamate transport in autosomal recessive neonatal myoclonic
epilepsy. American Journal of Human Genetics 76:334-339, 2005
80. Echaniz-Laguna A, Chassagne M, Ceresuela J, et al: Complete loss of
expression of the ANT1 gene causing cardiomyopathy and myopathy.
Journal of Medical Genetics 49:146-150, 2012