Embed Size (px)
Citation preview

Biomimetic Total Synthesis of Natural
Products
Thesis submitted for the degree of Doctor of Philosophy
Hiu Chun Lam
Bsc (Hons.) Chemistry
Department of Chemistry
University of Adelaide
Aug, 2017

II
To my family

III
Declaration
I certify that this work contains no material which has been accepted for the award of any
other degree or diploma in my name, in any university or other tertiary institution and, to the
best of my knowledge and belief, contains no material previously published or written by
another person, except where due reference has been made in the text. In addition, I certify
that no part of this work will, in the future, be used in a submission in my name, for any other
degree or diploma in any university or other tertiary institution without the prior approval of
the University of Adelaide and where applicable, any partner institution responsible for the
joint-award of this degree.
I give consent to this copy of my thesis, when deposited in the University Library, being
made available for loan and photocopying, subject to the provisions of the Copyright Act
1968.
I also give permission for the digital version of my thesis to be made available on the web,
via the University’s digital research repository, the Library Search and also through web
search engines, unless permission has been granted by the University to restrict access for a
period of time.
I acknowledge the support I have received for my research through the provision of an
Australian Government Research Training Program Scholarship
Hiu Chun Lam Date

IV
Acknowledgements
First, I would like to thank my supervisor Dr. Jonathan George for his guidance during my
PhD. I remember when I first joined the George group during my 2nd year of undergraduate
studies, Jonathan personally trained me in the laboratory. His passion and enthusiasm in
organic chemistry has influenced me greatly during my research studies, and his constant
presence in the laboratory has motivated me to work hard. In addition, Jonathan is
extraordinarily generous to send me to conferences in Europe and Australia, where I could
present my work and learn chemistry. For all the reasons above, my PhD experience has been
superb and it is my pleasure to work with Jonathan.
Next, I would like to thank the George group, for being supportive throughout my PhD. The
regular Wednesday group lunch and Friday drinking sessions have always been enjoyable. To
Kevin, thank you for being my mentor. To Justin, thanks for your help with the hyperjapone
project. To Henry, thank you for helping me with the verrubenzospirolactone project. I would
also like to thank Aaron where we worked on the rhodonoid project together. To all the new
additions of the George group (Aaron, Lauren, Stefania and Laura), I wish you all the best in
your PhD. To the prodigy JP, I also wish you good luck in your future postgraduate studies. I
would like to specifically thank Kevin and Justin, for their company on my roller coaster
research journey. Their encouragement has always been helpful. I will cherish the good times
we had inside and outside of the laboratory.
I would like to thank Professor Andrew Abell to allow me to use his group’s HPLC and
polarimeter. To Professor Chris Sumby, thank you for running all the single crystal X-ray
crystallography. To the Sumby/Doonan group (particular Michael, Alex, Natasha and Rob),
thanks for examining the crystals after we have recrystallized them in the laboratory. I would
like to thank eResearch SA to grant my access to the supercomputer Tizard and the database
for theoretical calculations. To Professor Greg Metha and Dr. David Huang and their groups,
thank you for teaching me to perform the theoretical calculations. To Phil, thank you for
running the NMR machines and mass spectrometers. To Dr. Justin Chalker from Flinders
University, thank you for giving us chemicals for my research project. I would like to thank
the University of Adelaide, it is my privilege to study for a PhD here.
At the end, I would like to thank my family for their support, specifically my parents Kent
and Daisy, my sister Elva for their unconditional love.

V
Abstract
This thesis describes several syntheses of natural products. The overall synthetic approach is
to mimic how these secondary metabolites could be derived in Nature, where we aim to gain
insights into the biosynthesis of these natural products from the syntheses.
The first synthesis of hyperjapones A-I was achieved by an oxidative hetero-Diels-Alder
reaction. The transformation of hyperjapone A to hyperajaponols A and C was achieved via
an epoxidation and an acid-catalysed rearrangement cascade reaction, forming 4 stereocenters
and 2 rings in 1 step.
The first synthesis of verrubenzospirolactone was achieved from a Diels-Alder reaction of the
polyene in water. Capillobenzopyranol, the proposed biosynthetic precursor of
verrubenzospirolactone was also synthesized and converted into verrubenzospirolactone by
mirroring our proposed biosynthetic pathway.
The first synthesis of rhodonoids C and D, and murrayakonine D was achieved. The key
biomimetic step was the acid catalysed rearrangement of an epoxide, forming 3 stereocenters
and 2 rings in 1 step.
The biomimetic total synthesis of yezo’otogirin C was achieved via an oxidative radical
cyclization cascade reaction, forming 2 rings, 2 stereocenters, 1 C=C bond, 1 C-C bond and 1
C-O bond in 1 step.

VI
List of abbreviations
ºC degree Celsius
Å Angstrom 1H Hydrogen-1 13C Carbon-13
18-crown-6 1,4,7,10,13,16-Hexaoxacyclooctadecane
Ac acetyl
AIBN azobisisobutyronitrile
aq. aqueous
atm atmospheric
Bn benzyl
br broad
Bu butyl
c concentration for specific optical rotation measurements
CAN ceric ammonium nitrate
cm-1 wavenumber
conc. correlation spectroscopy
CSA 1-(S)-(+)-10-camphorsulfonic acid
DBU 1,8-diazobicycloundec-7-ene
DDQ 2,3-dichloro-5,6-dicyano-para-benzoquinone
DIBAL-H diisobutylaluminium hydride
DMF dimethylformamide
DMSO dimethyl sulfoxide
dr diastereomeric ratio
ESI electrospray ionization
epi epimer
equiv. equivalents
Et ethyl
g grams
h hours
HMBC heteronuclear multiple bond correlation spectroscopy
HPLC high performance liquid chromatography
HRMS high resolution mass spectrometry
HSQC heteronuclear single quantum correlation spectroscopy

VII
Hz Hertz
hν light
i-Pr isopropyl
IR infrared
J coupling constant
KHMDS potassium hexamethyldisilazide
KO-tBu potassium tert-butoxide
LDA lithium diisopropylamine
m-CPBA meta-chloroperoxybenzoic acid
Me methyl
MHz megahertz
min minutes
Mp melting point
Ms mesyl
NBS N-bromosuccinimide
n-BuLi n-butyllithium
NMO N-methylmorpholine
NMR nuclear magnetic resonance
NOESY Nuclear Overhauser Effect Spectroscopy
Nu nucleophile
o-DCB 1,2-dichlorobenzene
o-quinone methide ortho-quinone methide
p-TsOH para-toluenesulfonic acid
PCC pyridinium chlorochromate
Pd2(dba)3 tris(dibenzylideneacetone)dipalladium (0)
PDC pyridinium dichromate
Pd/C palladium on activated carbon
PhI(OAc)2 (Diacetoxyiodo)benzene
PhMe toluene
P(o-tol)3 Tri(o-tolyl)phosphine
ppm part per million
Rf retention factor

VIII
Rh2[(R)-RTAD]4 tetrakis[(R)-(–)-(1-adamantyl)-(N-
phthalimido)acetate]dirhodium (II)
rt room temperature
SN1 unimolecular nucleophilic substitution
SN2 bimolecular nucleophilic substitution
TBAF tetrabutylammonium fluoride
TBAB tetrabutylammonium bromide
TBAI tetrabutylammonium iodide
TBDPS tert-butyldiphenylsilyl
TBS tert-butyldimethylsilyl
TEMPO 2,2,6,6-tetramethyl-1-piperidinyloxy
TFA trifluoroacetic acid
Tf trifluoromethanesulfonate
THF tetrahydrofuran
TLC thin layer chromatography
TMS trimethylsilyl
TPAP tetrapropylammonium perruthenate
w/w mass percentage

IX
Table of Contents
Declaration ............................................................................................................................. IIIAcknowledgements ................................................................................................................ IVAbstract ................................................................................................................................... VList of abbreviations .............................................................................................................. VI Chapter 1 - General Introduction
1.1. Natural products synthesis ........................................................................................... 11.2. Biomimetic total synthesis of natural products .......................................................... 41.3. References ...................................................................................................................... 7 Chapter 2 - Biomimetic Total Synthesis of Hyperjapones A-I, and Hyperjaponols A and C2.1. Introduction ................................................................................................................... 82.1.1. Diels-Alder reaction ..................................................................................................... 82.1.2. Chemistry of humulene (2.11) ..................................................................................... 92.1.3. Chemistry of caryophyllene (2.30) ............................................................................. 112.1.4. Isolation of hyperjapones and hyperjaponols ............................................................. 142.1.5. Proposed biosynthesis of hyperjapone A (2.49) and hyperjaponols A-C (2.54–2.56)152.2. Results and discussion ................................................................................................. 172.2.1. Synthesis of norflavesone (2.58) ................................................................................ 172.2.2. Biomimetic total synthesis of hyperjapones B (2.50) & D (2.52) .............................. 182.2.3. Investigation on the hetero-Diels-Alder reaction ....................................................... 192.2.4. Biomimetic total synthesis of hyperjapone A (2.49) .................................................. 222.2.5. Biomimetic total synthesis of hyperjaponol C (2.56) ................................................ 222.2.6. Biomimetic total synthesis of hyperjaponol A (2.54) ................................................ 252.2.7. Biomimetic total synthesis of hyperjapones C (2.51) and E (2.53) ........................... 282.2.8. Isolation of hyperjapones F to I (2.87–2.90) .............................................................. 302.2.9. Biomimetic total synthesis of hyperjapones F and G (2.87 & 2.88) .......................... 302.2.10. Biomimetic total synthesis of hyperjapone H (2.89) ................................................ 312.2.11. Biomimetic total synthesis of hyperjapone I (2.90) ................................................. 312.2.12. Preliminary theoretical calculations of the transition state of cationic alkene cyclization/1,2-shift ................................................................................................................. 322.3. Summary ...................................................................................................................... 342.4. References .................................................................................................................... 362.5. Experimental ................................................................................................................ 382.5.1. General methods ......................................................................................................... 382.5.2. Experimental procedures ............................................................................................ 392.5.3. NMR spectra .............................................................................................................. 612.5.4. Tables of 1H and 13C NMR data ............................................................................... 1192.5.5. Single crystal X-ray data .......................................................................................... 1292.5.6. Computational Data .................................................................................................. 1312.5.7. References ................................................................................................................ 142

X
Chapter 3 - Biomimetic Total Synthesis of Verrubenzospirolactone3.1. Introduction ............................................................................................................... 1433.1.1. Diels-Alder reaction of furan ................................................................................... 1433.1.2. Furan oxidation ........................................................................................................ 1433.1.3. Syntheses and reactions of 2H-chromene ................................................................ 1453.1.4. Isolation of verrubenzospirolactone ......................................................................... 1483.1.5. Aims of this study .................................................................................................... 1503.2. Results and discussion ............................................................................................... 1513.2.1. Synthesis of aldehyde 3.48 ....................................................................................... 1513.2.2. Synthesis of the Horner-Wadsworth-Emmons reagent 3.47 .................................... 1533.2.3. Biomimetic total synthesis of verrubenzospirolactone (3.38) .................................. 1533.2.4. Synthesis of capillobenzopyranol (3.39) and its oxidation ...................................... 1583.2.5. Biomimetic total synthesis of verrubenzospirolactone (3.38) .................................. 1603.2.6. Bioinspired cascade reaction .................................................................................... 1623.3. Summary .................................................................................................................... 1673.4. References .................................................................................................................. 1693.5. Experimental .............................................................................................................. 1713.5.1. General methods ....................................................................................................... 1713.5.2. Experimental procedures .......................................................................................... 1723.5.3. NMR spectra ............................................................................................................ 2023.5.4. Table of 1H and 13C NMR data ................................................................................ 2583.5.5. Single crystal X-ray data .......................................................................................... 2593.5.6. References ................................................................................................................ 263Chapter 4 - Biomimetic Total Synthesis of Rhodonoids C and D, and Murrayakonine D
4.1. Introduction ............................................................................................................... 2644.1.1. Isolation of rhodonoids and murrayakinone D ......................................................... 2644.1.2. Total synthesis of (±)-rhodonoids A (4.1) and B (4.2) by Hsung ............................ 2654.1.3. Proposed biosynthesis of rhodonoids C and D ......................................................... 2674.1.4. Epoxide cyclisation reaction in the synthesis of siccanin (4.28) by Trost ............... 2684.2. Results and discussion ............................................................................................... 2704.2.1. Biomimetic total synthesis of rhodonoids C and D .................................................. 2704.2.2. Investigation on the reactivity of the epoxide 4.18 .................................................. 2734.2.3. Synthesis of mahanimbine (4.5) ............................................................................... 2764.2.4. Biomimetic total synthesis of murrayakonine D (4.6) ............................................. 2774.2.5. Biomimetic total synthesis of rhodonoids C and D reported by Hsung ................... 2784.3. Summary .................................................................................................................... 2794.4. References .................................................................................................................. 2804.5. Experimental .............................................................................................................. 2814.5.1. General methods ....................................................................................................... 2814.5.2. Experimental procedures .......................................................................................... 2824.5.3. NMR spectra ............................................................................................................ 3064.5.4. Tables of 1H and 13C NMR data ............................................................................... 3464.5.5. Single crystal X-ray data .......................................................................................... 3504.5.6. References ................................................................................................................ 352

XI
Chapter 5 - Biomimetic Total Synthesis of Yezo'otogirin C
5.1. Introduction ............................................................................................................... 3535.1.1. Reductive radical cyclization ................................................................................... 3535.1.2. Oxidative radical cyclization .................................................................................... 3535.1.3. Isolation of yezo’otogirins A-C ................................................................................ 3555.1.4. Proposed biosynthesis of yezo’otogirn A (5.19) ...................................................... 3565.1.5. Previous biomimetic total synthesis of yezo’otogirin A (5.19) ............................... 3575.1.6. Previous bioinspired total synthesis of yezo’otogirin C (5.21) by Lee .................... 3585.1.7. Aims of this project .................................................................................................. 3595.2. Results and discussion ............................................................................................... 3605.2.1. Synthesis of 6-epi-pre-yezo’otogirin C (5.37) ......................................................... 3605.2.2. Synthesis of yezo’otogirin C (5.21) from 6-epi-pre-yezo’otogirin C (5.37) ............ 3615.2.3. Synthesis of yezo’otogirin C (5.21) from pre-yezo’otogirin C (5.38) ..................... 3625.2.4. Improved total synthesis of yezo’otogirin C (5.21) reported by Lee. ...................... 3655.3. Summary .................................................................................................................... 3665.4. References .................................................................................................................. 3675.5. Experimental .............................................................................................................. 3685.5.1. General methods ....................................................................................................... 3685.5.2. Experimental procedures .......................................................................................... 3695.5.3. NMR spectra ............................................................................................................ 3815.5.4. Table of 1H and 13C NMR data ................................................................................ 3925.5.5. References ................................................................................................................ 393

1
1. Introduction
1.1. Natural products synthesis
Mankind has achieved great discoveries due our curious and adventurous nature, from
looking for new species in rain forests and deep oceans, to searching for new subatomic
particles in the Large Hadron Collider. For organic chemists, a primary interest is in naturally
occurring small molecules. Nature has a great library of these secondary metabolites and we
have only discovered a fraction of it. Every week, there are isolation reports of new natural
products, varying in structural complexity and biological activity. These organic molecules
are not derived by chance or randomness, but by selective pressure in Nature. Therefore, each
natural product has its purpose and role that is crucial to the hosts (plants, bacteria, fungi).
These species are often limited in resources (precursors or reagents, narrow range of
temperature etc.), but still manage to generate molecules with intriguing and fascinating
structures.1 To organic chemists, the occurrence of these natural products is an intimidating
challenge presented by Nature, and we wonder if we could synthesize them in laboratory.
100 years ago, Professor Robert Burns Woodward was born, who later dedicated his career to
natural products synthesis and opened a new era in organic chemistry.2 He synthesized
numerous natural products, including cholesterol (1.1)3 and chlorophyll a (1.2) (Figure 1.1).4
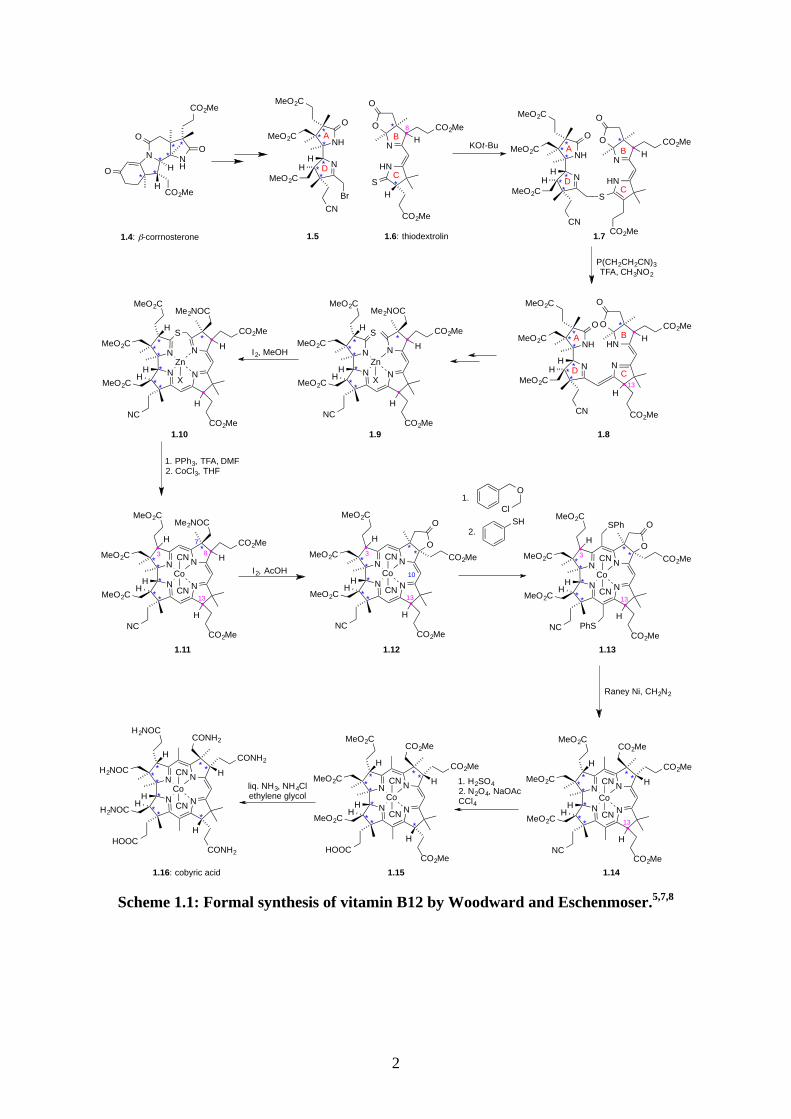
One of the Woodward’s syntheses is the formal synthesis of vitamin B12 (1.3).5 Vitamin B12
(1.3) is the most structurally complex of all vitamins; the corrin ring contains 9 stereocenters
and 6 of them are contiguous. Woodward and Eschenmoser, along with 99 researchers,
together took 11 years to accomplish this great achievement.
Figure 1.1: Selected examples of Woodward’s syntheses.3,4,5,6,7,8
N N
NN
H2NOC
H
H2NOC
HH
H2NOC
H2NOC
H
H
CONH2CONHisopropanolphosphate
ribosedimethylbenzimidazole
Co
CN **
*****
**
1.3: vitamin B12
CONH2
HO
H
HH
H N
N N
N
O
Mg
CO2Me
O O
1.2: chlorophyll a1.1: cholesterol
2
Scheme 1.1: Formal synthesis of vitamin B12 by Woodward and Eschenmoser.5,7,8
N NH
OO
CO2Me
H
CO2MeH
O
1.4: β-corrnosterone
****
**
NH
O
MeO2C
MeO2C
NHH
MeO2C
CNBr
D
A***
***
N
O
O
HCO2Me
HN
SH
B
C
*
*
1.6: thiodextrolin1.5
KOt-BuNH
O
MeO2C
MeO2C
NHH
MeO2C
CN
D
A***
***
N
O
O
HCO2Me
HN
S
B
C
*
CO2Me
CO2Me
P(CH2CH2CN)3TFA, CH3NO2
NH
O
MeO2C
MeO2C
NHH
MeO2C
CN
D
A***
***
HN
O
O
HCO2Me
N
B
C
*
H
CO2Me
N N
NN
MeO2C
H
MeO2C
HH
MeO2C
Me2NOC
H
H
CO2MeNC
Zn
*
****
**
X
S CO2Me
N N
NN
MeO2C
H
MeO2C
HH
MeO2C
Me2NOC
H
H
CO2MeNC
Zn
*
****
**
X
S CO2Me
I2, MeOH
N N
NN
MeO2C
H
MeO2C
HH
MeO2C
Me2NOC
H
H
CO2MeNC
Co
*
****
* CN
CO2Me3 8
13CN
1. PPh3, TFA, DMF2. CoCl3, THF
N N
NN
MeO2C
H
MeO2C
HH
MeO2C
H
CO2MeNC
Co
*
****
* CN3
13CN
O
O
CO2Me*
N N
NN
MeO2C
H
MeO2C
HH
MeO2C
H
CO2MeNC
Co
*
****
* CN3
13CN
O
O
CO2Me*
PhS
SPh
N N
NN
MeO2C
H
MeO2C
HH
MeO2C
H
CO2MeNC
Co
*
****
* CN
13CN
*CO2Me
H
CO2Me
*
Raney Ni, CH2N2
O
ClSH
2.
1.
1. H2SO42. N2O4, NaOAcCCl4N N
NN
MeO2C
H
MeO2C
HH
MeO2C
H
CO2MeHOOC
Co
*
****
* CN
CN
*CO2Me
H
CO2Me
*liq. NH3, NH4Clethylene glycol
*
1.16: cobyric acid
N N
NN
H2NOC
H
H2NOC
HH
H2NOC
H
CONH2HOOC
Co
*
****
* CN
CN
*CONH2
H
CONH2
*
*
I2, AcOH
1.7
1.10 1.9 1.8
1.11 1.12 1.13
1.15 1.14
10
8
13
7

3
The synthesis of B12 began from preparation of β-corrnosterone (1.4) in 32 steps, which
possessed 6 contiguous asymmetric carbon atoms and the A-D ring of B12. β-Corrnosterone
(1.4) was then converted into 1.5, which was ready to be coupled with thiodextrolin (1.6).
Thiodextrolin (1.6) was prepared in 21 steps, and it existed as two diastereoisomers differing
on the stereocenter C-8 in ring B. The two diastereoisomers could be separated but the C-8
stereocenter was readily epimerised. Hence, thiodextrolin (1.6) was used as a mixture in the
coupling step. The thiolactam of thiodextrolin (1.6) was deprotonated using KOt-Bu, which
then attacked the bromide 1.5 to give 1.7. 1.7 was transformed into 1.8 by TFA which united
the A-D ring and the B-C ring with a C-C bond. However, in this process, the
stereochemistry at C-13 on ring C was lost. 1.8 was then converted into 1.9 which contained
a thiolactam and a terminal alkene. Oxidative cyclization of 1.9 using I2 afforded 1.10. 1.10
was then rearranged to the corrin ring by TFA and the Zn metal center was replaced by Co
ion to give 1.11. During the transformation of the thioether 1.10 to the corrin ring, the
stereocenter C-3 on ring A was lost. Thus, a total of 3 stereocenters in 1.11 were lost in the
sequence of these reactions and 1.11 was in fact in a mixture of 8 diastereoisomers. 1.11
underwent an oxidative cyclization to give 1.12 which contained a lactone. The formation of
the lactone corrected the stereochemistry at C-8 because of the amide at C-7 dictated the
stereochemistry at C-8 during the cyclization. The lactone ring also provided steric hindrance
to avoid substitution occurring at C-10 in the next step. The substitution was conducted using
benzyl chloromethyl ether, followed by substitution of chloride to phenylthiol ether which
gave 1.13. The presence of phenylthiol ether allowed the separation of the mono-substituted
and the di-substituted compounds. The phenylthiol ethers 1.13 was then reduced to methyl
group by Raney Ni; in the same step, the lactone on ring C was also cleaved and methylated
to give the methyl ester 1.14. At this stage, HPLC and high pressure liquid-liquid partition
chromatography were used to isolate 1.14 with the desired stereochemistry at C-3. The two
diastereoisomers differing at C-13 could be separated at this point but it was not important
because in the next step, when the cyanide group was hydrolysed into an amide group using
H2SO4, the stereocenter C-13 would be epimerised and hence the two diastereoisomers of
1.14 (at C-13) were used in the hydration step. The amide with the correct stereochemistry
would then be isolated from HPLC, followed by conversion to carboxylic acid 1.15 using
nitrogen tetroxide. All methyl esters on 1.15 were converted into amide by liquid NH3 with
catalytic NH4Cl to give cobryic acid (1.16). Cobryic acid (1.16) was previously converted
into Vitamin B12 (1.3) by Friedrich.9 This concluded the formal synthesis of B12 by
Woodward and Eschenmoser.5,7,8

4
Recently, I was lucky to meet Professor Ian Fleming and Professor Leon Ghosez, and they
were very kind to share their stories, particularly about working with Professor Woodward as
postdoctoral researchers. Their memorable time included discussing chemistry with Professor
Woodward, and witnessing him tremendously excited (no less than the researchers) when
someone produced perfect crystals.
Today, I am grateful to work with my supervisor Dr. Jonathan George, and had experienced
great satisfaction when we synthesized and isolated natural products in the laboratory,
especially when the NMR data from the isolation and the synthetic sample is perfectly
matched. I believe this is the ultimate motivation for organic chemists to pursue total
synthesis.
1.2. Biomimetic total synthesis of natural products
A natural product can be synthesized by numerous possible pathways, and we are only
limited by our imagination. Our group is interested in synthesizing these organic molecules
by mimicking how they could be derived in Nature, specifically if they are generated from a
pre-disposed, non-enzymatic biosynthesis. Assuming the biosynthetic hypothesis is correct,
organic chemists should be able to reproduce the chemistry in a laboratory setting.
Scheme 1.2: Biomimetic total synthesis of tropinone (1.20) reported by Robinson.10
Sir Robert Robinson reported the first example of a biomimetic total synthesis 100 years ago
in the synthesis of tropinone (1.20), which was prepared from a three-component one-pot
synthesis (Scheme 1.2). Not only does the molecular complexity increase dramatically in one
step, but the connection of the new rings and the relative stereochemistry in the molecule are
all correctly installed in this reaction.
Thus, the initial phase of biomimetic synthesis would require speculation on how rings and
stereocenters of natural products are derived from their corresponding biosynthetic precursors.
Subsequently, these compounds would be synthesized while mirroring the proposed
biosynthesis using simple chemical reagents. At the end of the synthesis, organic chemists
would gain insights onto the biosynthetic pathways of these natural products, as a feedback to
the biosynthetic proposal.
COH
COH
1.17: succinaldehyde
MeNH2
1.18: methylamine
O
CO2H
CO2H1.19: 3-oxopentanedioic acid
N
O
heat +CO2+H2O
1.20: tropinone

5
While the biosynthetic precursors may not be synthesized using a biomimetic approach, we
aim to develop concise methods to construct those molecules. For instance, we would avoid
unnecessary redox reactions, and minimise functional group and protecting group
manipulations. Since the report of tropinone synthesis by Robinson, organic chemists have
been utilising this philosophy and strategy to synthesize natural products, as the biomimetic
approach is arguably the quickest and most economical way to access a natural product. One
recent example is the biomimetic total synthesis of homodimericin A (1.28). (±)-
Homodimericin A (1.28) was isolated by Clardy in 2016 from a pair of species, bacteria
Streptomyces sp. 4231 and fungi Trichoderma harzianum.11 Homodimericin A (1.28) was
isolated as a racemate and hence suggesting it is likely to be derived via a pre-disposed, non-
enzymatic biosynthesis. As outlined in our proposed biosynthesis (Scheme 1.3), oxidation of
1.21 could give quinone 1.22. Dimerisation of 1.22 via a Michael reaction could afford 1.23,
followed by a second Michael reaction to give 1.24. Oxidation of 1.24 could give 1.25, which
then undergoes intramolecular Diels-Alder reaction to give 1.26. Tautomerisation of the enol
1.26 (highlighted in blue) could afford 1.27. Intramolecular aldol reaction of 1.27 could then
afford homodimericin A (1.28). The isolation chemists also proposed a similar biosynthesis
with a different order of Michael reaction and oxidation but the overall proposal is the
same.11 In June 2017, the biomimetic total synthesis of homodimericin A (1.28) was reported
by three independent groups (Tang12, Wang13, Yang14). Each reported a unique way to
synthesize the monomer 1.21, but the chemistry to achieve homodimericin A (1.28) was
identical. This showcases biomimetic total synthesis of natural products is highly robust, and
also remarkably attractive to organic chemists as shown in all three reports.
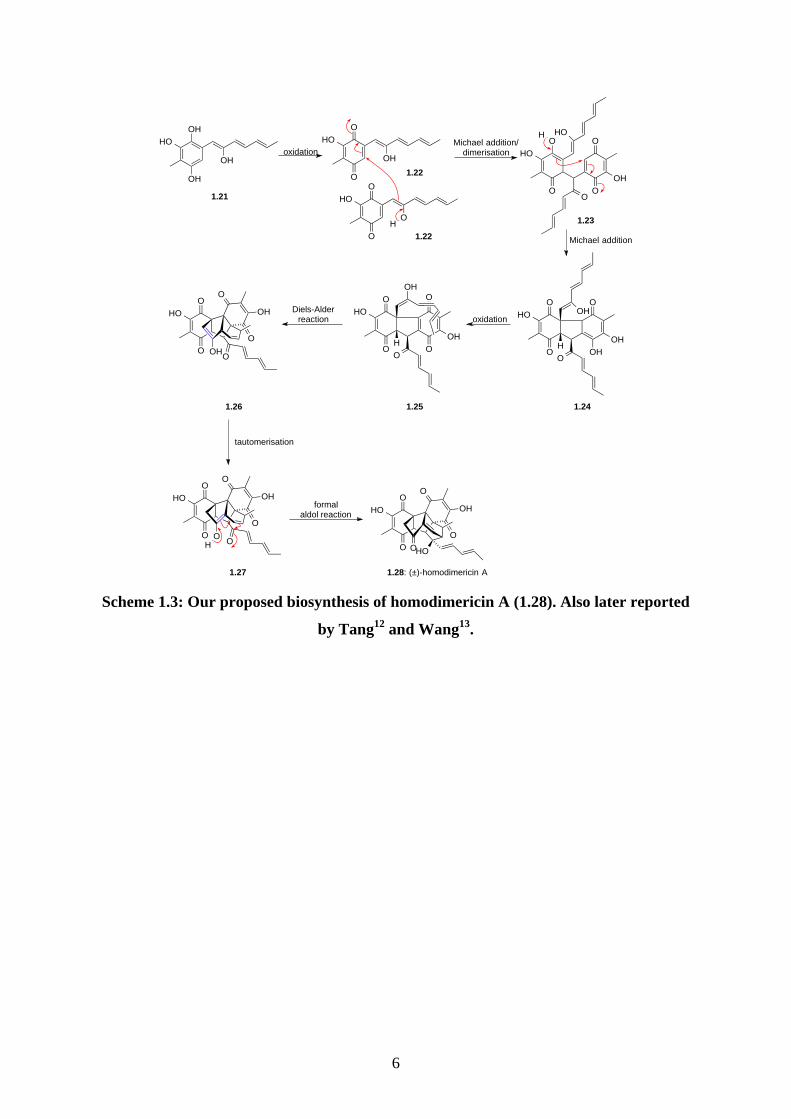
6
Scheme 1.3: Our proposed biosynthesis of homodimericin A (1.28). Also later reported
by Tang12 and Wang13.
OH
OH
HO
OHoxidation
O
O
HO
OH
O
O
HO
OH
O
O
HO
OO
O
OH
HOH
OOH
O
OH
OHO
OH
OH
OO
O
OH
OHO
OH
OH
O
O
HO
O
O
OH
HOO
O
O
HO
O
O
OH
OOH
Michael addition/dimerisation
Michael addition
oxidationDiels-Alder
reaction
formalaldol reaction
1.21
1.22
1.22
1.23
1.241.25
1.27 1.28: (±)-homodimericin A
O
O
HO
O
O
OH
OOH
1.26
tautomerisation

7
1.3. References
1. Nicolaou, K. C.; Vourloumis, D.; Winssinger, N.; Baran, P. S. Angew. Chem. Int. Ed., 2000, 39, 44.
2. Halford, B. Chemical and Engineering News, 2017, 95. 3. Woodward, R. B.; Sondheimer, F.; Taub, D. J. Am. Chem. Soc., 1951, 73, 3548. 4. Woodward, R. B.; Ayer, W. A.; Beaton, J. M.; Bickelhaupt, F.; Bonnett, R.;
Buchschacher, P.; Closs, G. L.; Dutler, H.; Hannah, J.; Hauck, F. P.; Ito, S.; Langemann, A.; Legoff, E.; Leimgruber, W.; Lwowski, W.; Sauer, J.; Valenta, Z.; Volz, H. J. Am. Chem. Soc., 1960, 82, 3800.
5. Woodward, R. B. Angew. Chem. Int. Ed., 1963, 75, 871. 6. Woodward, R. B. Science, 1966, 153, 487. 7. Woodward, R. B. Pure and applied chemistry. Chimie pure et appliquee, 1968, 17,
519. 8. Woodward, R. B. Pure and applied chemistry. Chimie pure et appliquee, 1971, 25,
283. 9. Friedrich, W.; Gross, G.; Bernhauer, K.; Zeller, P. Helv. Chim. Acta., 1960, 43, 704. 10. Robinson, R. J. Chem. Soc., Trans., 1917, 111, 762. 11. Mevers, E.; Sauri, J.; Liu, Y.; Moser, A.; Ramadhar, T. R.; Varlan, M.; Williamson, R.
T.; Martin, G. E.; Clardy, J. J Am Chem Soc, 2016, 138, 12324. 12. Feng, J.; Lei, X.; Guo, Z.; Tang, Y. Angew. Chem. Int. Ed., 2017, 56, 7895. 13. Ma, D.; Liu, Y.; Wang, Z. Angew. Chem. Int. Ed., 2017, 56, 7886. 14. Huang, J.; Gu, Y.; Guo, K.; Zhu, L.; Lan, Y.; Gong, J.; Yang, Z. Angew. Chem. Int.
Ed., 2017, 56, 7890.

8
Chapter 2
Biomimetic Total Synthesis of Hyperjapones A-I, Hyperjaponols A and C
2.1. Introduction
2.1.1. Diels-Alder reaction
Figure 2.1: An illustration of Diels-Alder reaction.
The Diels-Alder reaction is a classic reaction in organic synthesis, where by a diene and a
dienophile undergo a concerted cycloaddition to give a 6-membered ring (Figure 2.1).1 There are
numerous examples in the literature where Diels-Alder reactions are involved in natural product
syntheses.2,3,4 For example, in the total synthesis of (–)-furaquinocin C (2.6) reported by Smith5
(Scheme 2.1), two silyl enol ethers were generated from the deprotonation of the ketone 2.1 to give
diene 2.3. Diene 2.3 and dienophile 2.4 then underwent a Diels-Alder reaction to give intermediate
2.5, followed by elimination, rearomatization and desilylation to give (–)-furaquinocin C (2.6) in 1
step.
Scheme 2.1: Total synthesis of (–)-furaquinocin C (2.6) by Smith.5
A reaction is classified as a hetero-Diels-Alder reaction when it involves one or more heteroatoms
during the cycloaddition. An example is the total synthesis of variecolortide A (2.10) by Trauner6
(Scheme 2.2), where the heterodiene hydroxyviorcristin (2.7) and the dienophile isoechinulin B (2.8)
underwent a hetero-Diels-Alder reaction to afford intermediate 2.9. 2.9 was oxidized by air in the
same pot to give (±)-variecolortide A (2.10).6 More recent examples of hetero-Diels-Alder reactions
in natural product synthesis have been summarised elegantly by Heravi.7
"diene""dienophile"
Diels-Alder reaction
O
OMeO
O
OH
2.6: (−)-furaquinocin C51%
O
OMeO
O
OTMS
Br
TMSOO
OMeO
O
OTMS
TMSO
BrDiels-Alderreaction
elimination/rearomatization/
desilylation
2.5 2.32.4
O
OTMS
TMSO
2.3
O
OTMS
O
2.2
O
O
O
2.1
LDA, TMSClTHF, –78 °C LDA, TMSCl

9
Scheme 2.2: Total synthesis of variecolortide A (2.10) by Trauner.6
2.1.2. Chemistry of humulene (2.11)
Figure 2.2: Structure of humulene (2.11).
Humulene (2.11) is a cyclic sesquiterpene natural product containing three alkenes (Figure 2.2).
Each alkene possesses different reactivity, as shown in an epoxidation study by Fujita (Scheme 2.3).
When humulene (2.11) was treated with one equivalent of m-CPBA, the major product of the
reaction was epoxide 2.12. The results from this study suggested that the Δ1,2-alkene in humulene
(2.11) is the most reactive, followed by the Δ8,9-alkene and lastly the Δ4,5-alkene.8 The hypothesis is
in good agreement with the results from the diepoxidation of humulene (2.11), which afforded
epoxide 2.15 (Scheme 2.3).
Scheme 2.3: Epoxidation of humulene (2.11) by Fujita.8
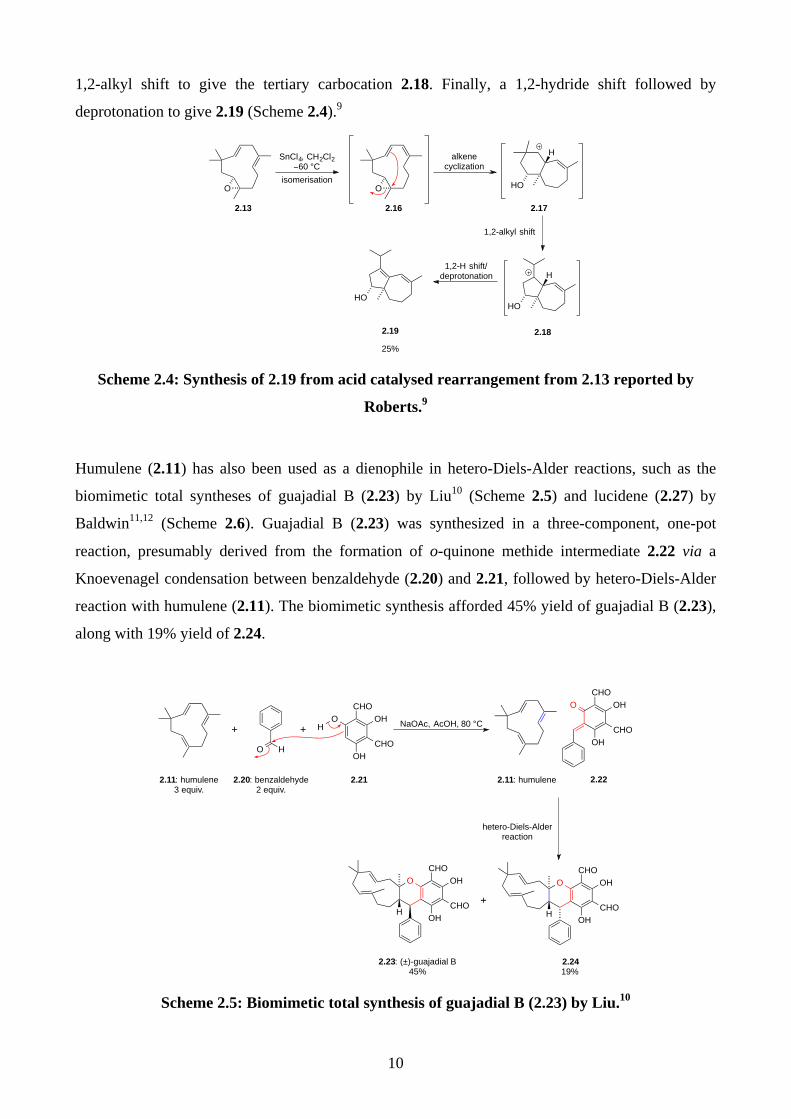
In addition, humulene epoxide 2.13 has been shown to undergo an acid catalysed rearrangement
reaction. Roberts reported a synthesis of 2.19 by treating humulene epoxide 2.13 with SnCl4 in
CH2Cl2 at –60 ºC. The proposed mechanism starts from the isomerisation of alkene 2.13 to give
2.16, followed by an alkene cyclisation to form a 6-membered ring 2.17, which then undergoes a
O
OOH
HO
OH
HNNH
O
O
HN
2.10: (±)-variecolortide A48%
O
OHOH
HO
O
HNNH
O
O
HN
oxidation/tautomerisation
O
OHOH
HO
O
HNNH
O
O
HN
2.7: hydroxyviorcristin
2.8: isoechinulin B
Diels-Alder reaction
2.9
o-DCB, air, 180 °C
1
25
4
89
2.11: humulene
45 21
8 9
OO
O++
m-CBPA (1 eq.)CH2Cl2,−10 °C to rt
m-CBPA (2 eq.)CH2Cl2,−10 °C to rt
2.15 2.12 2.13 2.142.11: humulene(75%) (14%) (5%)(30%)
O
O45 2
1
8 9
10
1,2-alkyl shift to give the tertiary carbocation 2.18. Finally, a 1,2-hydride shift followed by
deprotonation to give 2.19 (Scheme 2.4).9
Scheme 2.4: Synthesis of 2.19 from acid catalysed rearrangement from 2.13 reported by
Roberts.9
Humulene (2.11) has also been used as a dienophile in hetero-Diels-Alder reactions, such as the
biomimetic total syntheses of guajadial B (2.23) by Liu10 (Scheme 2.5) and lucidene (2.27) by
Baldwin11,12 (Scheme 2.6). Guajadial B (2.23) was synthesized in a three-component, one-pot
reaction, presumably derived from the formation of o-quinone methide intermediate 2.22 via a
Knoevenagel condensation between benzaldehyde (2.20) and 2.21, followed by hetero-Diels-Alder
reaction with humulene (2.11). The biomimetic synthesis afforded 45% yield of guajadial B (2.23),
along with 19% yield of 2.24.
Scheme 2.5: Biomimetic total synthesis of guajadial B (2.23) by Liu.10
1,2-H shift/deprotonation
HOHO
H
H
HO
1,2-alkyl shift
2.13 2.17
2.182.19
25%
O
SnCl4, CH2Cl2−60 °C
2.16
Oisomerisation
alkenecyclization
O
H
CHOOH
CHOOH
2.23: (±)-guajadial B45%
O
H
CHOOH
CHOOH
2.2419%
+
2.11: humulene3 equiv.
OCHO
OH
CHOOH
2.11: humulene 2.22
hetero-Diels-Alderreaction
O
CHO
OHCHO
OHO
2.20: benzaldehyde2 equiv.
2.21
NaOAc, AcOH, 80 °C++ H
H

11
Similarly, lucidene (2.27) was synthesized by heating 2.25 and humulene (2.11) in a sealed tube,
where 2.25 first underwent elimination to give o-quinone methide intermediate 2.26, followed by
hetero-Diels-Alder reactions on the Δ1,2-alkene and Δ8,9-alkene of humulene (2.11) to give lucidene
(2.27) (Scheme 2.6). Both monoadduct 2.29 and isolucidene (2.28) were also isolated in the same
reaction.11,12 The synthesis of lucidene (2.27) and guajadial B (2.28) indicates the hetero-Diels-
Alder reaction between humulene (2.11) and o-quinone-methide is highly diastereoselective and
regioselective.
Scheme 2.6: Biomimetic total synthesis of lucidene (2.27) by Baldwin.11,12
2.1.3. Chemistry of caryophyllene (2.30)
Figure 2.3: Structure of caryophyllene (2.30).
Caryophyllene (2.30) is an enantiopure bicyclic sesquiterpene natural product with two alkenes
(Figure 2.3). Caryophyllene (2.30) could also undergo epoxidation, where the Δ4,5-alkene can be
oxidized spontaneously under air to give caryophyllene oxide (2.31).13 The trans-Δ4,5-alkene in the
9-membered ring of caryophyllene (2.30) is highly reactive because the ring strained in
caryophyllene (2.30) would be released after the reaction. Furthermore, caryophyllene oxide (2.31)
could also rearrange under reductive conditions to give 2.33 as the predominate product (Scheme
2.7).14
O
O
H
H
O
O
hetero-Diels-Alderreaction
O
O
H
H
O
H
HO
HO
2.252.05 equiva.2.11: humulene
xylene, 170 °Cseal tube+
2.26
1
2
98
2.27: (±)-lucidene 2.28: (±)-isolucidene2.29
++
17%, 2.27/2.28 = 2.5:128%
H
H
2.30: caryophyllene
4
58
15

12
Scheme 2.7: Reductive acid-catalysed rearrangement reaction of caryophyllene oxide (2.31)
by Barrero.14
Alternatively, caryophyllene oxide (2.31) could be ring opened by tetracyanoethylene
(NC)2C=C(CN)2 to give 2.37 and 2.38 (Scheme 2.8). The authors hypothesized that the low energy
π* orbital in (NC)2C=C(CN)2 could interact with the electron-rich epoxide, followed by
fragmentation of epoxide to generate alkenes 2.37 and 2.38.15
Scheme 2.8: Cleavage of caryophyllene oxide (2.31) using (NC)2C=C(CN)2 reported by
Marcias-Sanchez.15
Unsurprisingly, caryophyllene (2.30) has also been used as a dienophile in hetero-Diels-Alder
reactions. In the synthesis of cytosporolide model 2.41 by the George group, o-quinone methide
2.40 was generated from 2.39 and HC(OEt)3, which then underwent a hetero-Diels-Alder reaction
with caryophyllene (2.30) to give 2.41 (Scheme 2.9).16 This synthesis of 2.41 supported the
structural reassignment of cytosporolides A–C.16 The total synthesis of cytosporolide A (2.45) was
later completed by Takao (Scheme 2.10), in which the key step was the hetero-Diels-Alder reaction
between a highly functionalised diene 2.43 and dienophile 2.42 to give 2.44.17 Deprotection of 2.44
gave cytosporolide A (2.45) in 63% over two steps.
H
H
2.30: caryophyllene
H
H
O
H
H
O
OH
H
H OH
H
H OH
H
H OH
H
H+++
Cp2TiCl2, Mn, 2,4,6-collidineTMSCl, THF, rt
+
55%, 2.31:2.32, d.r. 4:1
90%, 2.33:2.34:2.35:2.36, d.r. 20:9:5:1
m-CPBA, CH2Cl2, rt
2.31: caryophyllene oxide 2.32
2.33 2.34 2.35 2.36
H
H
O
H
H
OH
H
H
OH
71%13%
(NC)2C=C(CN)2acetone, reflux
+
2.31 2.37 2.38

13
Scheme 2.9: Synthesis of cytosporolide model 2.41 by the George group.16
Scheme 2.10: Total synthesis of (+)-cytosporolide A (2.45) by Takao.17
In a similar fashion, caryophyllene (2.30) was also incorporated in the syntheses of guajadial (2.46)
and psidial A (2.47) by Lee and coworkers.18 o-Quinone methide 2.22 was generated from
Knoevenagel condensation of benzaldehyde (2.20) and 2.21, followed by a hetero-Diels-Alder
reaction with caryophyllene (2.30) to give a mixture of guajadial (2.46) and psidial A (2.47),
alongside 2.48 (Scheme 2.1).18
Scheme 2.11: Biomimetic total synthesis of guajadial (2.46) and psidial A (2.47) by Lee and
coworkers.18
The 9-membered ring in caryophyllene (2.30) is flexible and it can adopt different conformations.
Calculations from Toma suggested a 33:44:23 ratio of the βα:αα:ββ conformations in caryophyllene
O
H
H
H
O
OH
MeO O
H
O
O
OH
MeO O
H
H
2.30: caryophyllene 2.40 2.41
hetero-Diels-Alderreaction
MeO O
OH
HO
HO HC(OEt)3, TFA100 °C
2.3953%
O
H
HO
H
O
OH
HO O
OH OMeH
C7H15
OH
O
O
OH
HO O
H
OH
2.43
OMe
OBz
TESO
C7H15
2.42
hetero-Diels-Alderreaction
O
HOH
H
O
OH
HO O
OBz OMeH
C7H15
OTES
1. DIBAL, CH2Cl2, –78 °C2. HF·pyridine, THF, rt
63% over 2 steps
2.45: (+)-cytosporolide A2.44
O
H
H
H
CHOOH
CHOOH
O
H
H
H
CHOOH
CHOOH
O
H
H
H
CHOOH
CHOOH
++
OCHO
OH
CHOOHH
H
2.30: caryophyllene
hetero-Diels-Alderreaction
2.22
CHO
OHCHO
OHHO
OH
H
2.30: caryophyllene 2.20: benzaldehyde 2.21
5% w/w PTS (aq)100 °C
+
*PTS = PEG-600/α-Tocopherol-based diester of Sebacic acid
2.46: guajadial 2.47: psidial A 2.48
25%, 2.46:2.47:2.48, d.r. 3:1:1
H
+
1'1'1'
19
19
19

14
(2.30) (Figure 2.4).19 2.48 is presumably generated from the less abundant ββ caryophyllene
conformer, whereas guajadial (2.46) and psidial A (2.47) are derived from the more abundant βα
and αα conformers. Note that reacting with βα and αα conformers does not change the relative
stereochemistry between C-1 and C-9 with respect to the Δ4,5-alkene in caryophyllene (2.30). The
stereochemistry difference at C-1’ in guajadial (2.46) and psidial A (2.47) can be derived from the
steric/kinetic effect of the E/Z isomer of o-quinone methide 2.22.
Figure 2.4: Conformers of caryophyllene (2.30) and theirs theoretical population calculated
by Toma.19
2.1.4. Isolation of hyperjapones and hyperjaponols
Figure 2.5: Hyperjapones A-E (2.49–2.53)20 and hyperjaponols A-C (2.54–2.56).21
Hyperjapones A-E (2.49–2.53) are structurally related meroterpenoids that were isolated from
Hypericum japonicum by Xu (Figure 2.5).20 Hyperjapone A (2.49) is a racemic molecule containing
an 11/6/6 ring system, whereas all hyperjapones B-E (2.50–2.53) are enantiopure and possess a
4/9/6/6 ring system. Shortly after the isolation of hyperjapones, Zhang reported the isolation of
hyperjaponols A-C (2.54–2.56) from the same plant (Figure 2.5).21 Hyperjaponol C (2.56) contains
a 5/7/6/6 ring system with an unusual trans-isodaucane structure, and is the most stereochemically
complex among the three hyperjaponols. The racemic nature of hyperjaponols (2.54–2.56) implies
the presence of a non-enzymatic, highly pre-disposed biosynthesis.
βα33%
αα44%
ββ23%
αβ0%
H
H
2.30: caryophyllene
19
4
5 19
45 1
4
5
91
9
45
19
45
O
OH
O
OH
(±)-2.49: hyperjapone A
O
OH
O
OH
H
H
O
OH
O
OH
H
H
2.50: hyperjapone B 2.52: hyperjapone D
O
OH
O
OH
H
H
O
OH
O
OH
H
H
2.51: hyperjapone C
2.53: hyperjapone E
H
HO
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
O
OH
O
OHHO
(±)-2.55: hyperjaponol B

15
2.1.5. Proposed biosynthesis of hyperjapone A (2.49) and hyperjaponols A-C (2.54–
2.56)
Scheme 2.12: Proposed biosynthesis of hyperjaponols A-C (2.54–2.56).
We propose the biosynthesis of hyperjaponols A-C (2.54–2.56) is derived from hyperjapone A
(2.49) (Scheme 2.12). Our proposed biosynthetic pathway begins from the trimethylation of
acylphloroglucinol 2.57, which gives norflavesone (2.58), a natural product isolated from a
flowering plant Lepsospermum scoparium.22 Oxidation of norflavesone (2.58) would give a
heterodiene intermediate 2.59, which undergoes a Diels-Alder reaction with humulene (2.11) to
form hyperjapone A (2.49). From the examples shown in the synthesis of lucidene (2.27)11,12 and
guajadial B (2.23),10 we predict that the hetero-Diels-Alder reaction would take place onto the Δ1,2-
alkene of humulene (2.11).
The X-ray crystal structure of hyperjapone A (2.49) shows that the Δ8,9-alkene adopting a
conformation such that one face of the alkene is exposed, while the other face is not accessible due
to the steric hindrance of the macrocycle.20 Therefore, the epoxidation of hyperjapone A (2.49)
should take place on the exposed face of Δ8,9-alkene to give 2.60. Epoxide 2.60 could be ring-
opened by acid to give tertiary carbocation 2.61, followed by cyclization onto the Δ4,5-alkene to
form a 6/7 membered ring system and a secondary carbocation 2.62.9 We predict that the
cyclization of 2.61 should be diastereoselective based on the geometry and the conformation of the
macrocycle of 2.61. A stereoselective 1,2-alkyl shift of 2.62 could generate the tertiary carbocation
2.63. The loss of a proton from the methyl group in carbocation 2.63 could afford hyperjaponol C
H
O O
OH O
HO OH
OH
HO O
OH O
2.58: norflavesone 2.11: humulene 2.59
hetero-Diels-Alderreaction
2.49: (±)-hyperjapone A
trimethylation oxidation
humulene (XX)
epoxidation
1,2-alkyl shift
cation-alkene cyclization
H
2.57
concerted, asynchronous cation-alkene cyclization/1,2-alkyl shift
HO
H
O
HOH
O
O
2.60
HO
HH
OH
O
O
2.61
HO
HO
HH
OH
O
O
HO H
2.62
2.63
HO OH
O
O
HO H
HH H
O OH
O
O
HO H
2.56: (±)-hyperjaponol C
HH
ring-opening of epoxide
− H
− H
HO
HH
OH
O
O
2.54: (±)-hyperjaponol A 2.55: (±)-hyperjaponol B
H
HO
HH
OH
O
O
HOH
HO
HH
OH
O
O
HO
2.49: (±)-hyperjapone A
+
O
8 9
5 4
12

16
(2.56), while deprotonation of carbocation 2.61 would generate hyperjaponol A (2.54) or
hyperjaponol B (2.55).
In regards to the alkene cyclisation of carbocation 2.61 and the 1,2-alkyl shift in our proposed
biosynthesis of hyperjaponol C (2.56), it is possible that these processes might occur in a concerted,
asynchronous rearrangement, as supported by Tantillo’s hypothesis.23,24 Tantillo’s investigation into
the cationic rearrangement reactions of terpene natural products using theoretical calculations often
show that these rearrangements are not stepwise, but rather occur in a concerted manner.24 For
example, the proposed biosynthesis of presilphiperfolanol (2.67) involves a 1,2-alkyl shift/alkene
cyclisation of 2.64 to presilphiperfolanyl cation (2.66) (Scheme 2.13).25 Tantillo’s calculations
suggested the secondary carbocation 2.65 was not involved in the biosynthesis, but rather a 1,2-shift
and alkene cyclization of 2.64 occurred simultaneously to give 2.67 via a single transition state.23
We have also consulted Tantillo on the biosynthesis of hyperjaponol C (2.54), and he predicted that
it is likely to occur in a concerted, asynchronous rearrangement.
Scheme 2.13: Proposed biosynthesis of (–)-presilphiperfolanol (2.67).23,25
H
H H HH1,2-alkylshift
alkenecyclization
asynchronous, concerted rearrangment
2.662.652.64
HH
2.67: (–)-presilphiperfolanol
OHH2O
1,3-hydride shift/

17
2.2. Results and discussion
2.2.1. Synthesis of norflavesone (2.58)
Scheme 2.14: Acylation of phloroglucinol (2.68).
The synthesis began with the Friedel-Crafts acylation of phloroglucinol (2.68) using AlCl3 and i-
PrCOCl in PhNO2 at 65 ºC,26 the reaction worked smoothly and can be scaled up to 10 g (Scheme
2.14). We have also lowered the amount of isobutyryl chloride from 2 equiv. to 1.2 equiv. without
any loss of yield.
Scheme 2.15: Dearomative trimethylation reported by Nguyen.27
Scheme 2.16: Synthesis of norflavesone (2.58).
Nguyen and coworkers27 have reported a dearomative trimethylation on 2.69 using KOt-Bu and
MeI (Scheme 2.15), which was applied to acylphloroglucinol 2.57 and afforded norflavesone (2.58)
in good yield (Scheme 2.16). We also investigated whether using fewer equivalents of base and MeI
could give dimethylated acylphloroglucinol 2.71. Interestingly, we did not observe 2.71, but a lower
yield of trimethylated acylphloroglucinol 2.58. We hypothesize after the first methylation of 2.57,
the reactivity of the molecule increases until the third methylation has completed. This observation
was different from the prenylation of acylphloroglucinol 2.57 reported by George, where he
observed a mixture of diprenylated 2.72/2.73 and triprenylated acylphloroglucinol 2.74 (Scheme
2.17).28
HO OH
OH
HO OH
OH O2.68: phloroglucinol 2.57
i-PrCOCl, AlCl3PhNO2, 65 °C
87%
HO O
OH O
2.70
MeI, KOt-BuMeOH, reflux
HO
OH
2.69
72%
OH
O
HO O
OH O
2.58: norflavesone
MeI, KOt-Bu, MeOH, 65 °C
HO OH
OH O
2.57
79%
HO OH
OH O
HO O
OH O
not observed
2.71

18
Scheme 2.17: Dearomative prenylation reported by George28.
2.2.2. Biomimetic total synthesis of hyperjapones B (2.50) & D (2.52)
In the biomimetic total synthesis of hyperguinone B (2.77) reported by George, 2.75 was oxidized
by PhI(OAc)2 and TEMPO to give a heterodiene intermediate 2.76, which underwent a 6π
electocyclization to give hyperguinone B (2.77) (Scheme 2.18). We hoped that in a similar fashion,
norflavesone (2.58) could be oxidized to give the heterodiene intermediate 2.59, then undergoes a
hetero-Diels-Alder reaction with humulene (2.11) or caryophyllene (2.30).
Scheme 2.18: Biomimetic total synthesis of hyperguinone B (2.77) by George28.
To our delight, reaction with caryophyllene (2.30) and norflavesone (2.58) in the presence of
PhI(OAc)2 and TEMPO gave a 2.5:1 mixture of hyperjapones B (2.50) and D (2.52) in 26% overall
yield (Scheme 2.19). The ratio of hyperjapones B and D was in good agreement with the population
of conformers in caryophyllene (2.30), where the predominant βα and αα conformers reacted with
the heterodiene intermediate 2.59 to give hyperjapone B (2.50), while the less abundant ββ
conformer gave hyperjapone D (2.52).19 We found that the mixture of hyperjapones B and D were
impossible to separate by flash column chromatography or HPLC. Interestingly, we observed the
hyperjapones B (2.50) and D (2.52) mixture was not soluble in most organic solvents, where
hyperjapone B (2.50) could be selectively recrystallized from MeOH. Unfortunately, the filtrate still
contained a 1:1 mixture of hyperjapones B (2.50) and D (2.52). We have attempted to recrystallise
the hyperjapones B (2.50) and D (2.52) mixture with various other solvents, but to no avail. At this
point, we did not pursue further on the purification of hyperjapone D (2.52). The optical rotation of
hyperjapone B (2.50) is [!]!!" +10º (c 0.62, MeOH), which matches with the natural product [!]!!"
+5º (c 0.15, MeOH), and hence confirmed the absolute configuration of hyperjapone B (2.50).
HO O
OH O
39%
prenyl bromide, KOt-BuMeOH, 65 °C
HO OH
OH O
2.57
HO OH
OH O
HO O
OH O
30% 10%
2.742.72 2.73
+ +
HO O
OH O
PhI(OAc)2, TEMPO, THF−78 °C to rt O O
OH O
O O
OH O
2.77: hyperguinone B2.762.75
6π electrocyclization
73%

19
Scheme 2.19: Biomimetic total synthesis of hyperjapones B (2.50) and D (2.52).
2.2.3. Investigation on the hetero-Diels-Alder reaction
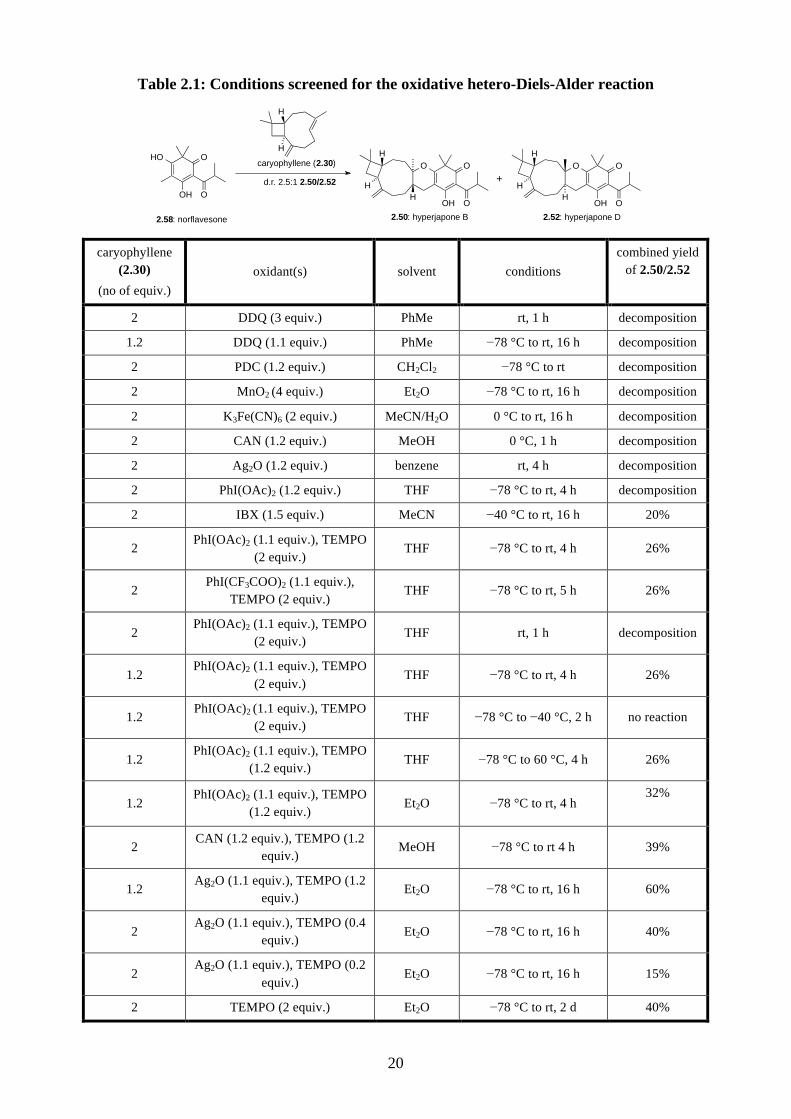
We then further investigated the oxidation of norflavesone (2.58) by screening different conditions
in an attempt to improve the yield for the oxidative cycloaddition (Table 2.1). First, we discovered
the reaction did not proceed at –78 °C to –40 °C, and the starting material would decompose if the
reaction started at room temperature. It suggests that the heterodiene 2.59 would be formed between
–40 °C and room temperature. We also performed a few control experiments to show that a
stoichiometric amount of TEMPO is essential for this oxidation. We have also attempted to improve
the reaction by screening various co-oxidants. For instance, Ag2O has been used as an oxidant to
generate an o-quinone methide 2.79 in the total synthesis of schefflone (2.80) (Scheme 2.20).29
From the collective results, we found that the combination of Ag2O and TEMPO gave the highest
overall yield of 60%.
Scheme 2.20: Biomimetic total synthesis of schefflone (2.80) using Ag2O as an oxidant by
Lei.29
2.58: norflavesone
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.50: hyperjapone B 2.52: hyperjapone D
26%, d.r. 2.5:1 2.50/2.52
H
H
caryophyllene (2.30)PhI(OAc)2, TEMPO
THF, –78 °C to rtHO O
OH O
O
MeO
OOMe
OOMe
OMe
OMe
OMe
2.80: schefflone
O
MeO
O
MeO
OOMe
OMe
OMeOMe
OMe
OHOMe
Ag2O, benzene, rt
72%
hetero-Diels-Alderreaction
2.782.79
20
Table 2.1: Conditions screened for the oxidative hetero-Diels-Alder reaction
caryophyllene (2.30)
(no of equiv.) oxidant(s) solvent conditions
combined yield of 2.50/2.52
2 DDQ (3 equiv.) PhMe rt, 1 h decomposition
1.2 DDQ (1.1 equiv.) PhMe −78 °C to rt, 16 h decomposition
2 PDC (1.2 equiv.) CH2Cl2 −78 °C to rt decomposition
2 MnO2 (4 equiv.) Et2O −78 °C to rt, 16 h decomposition
2 K3Fe(CN)6 (2 equiv.) MeCN/H2O 0 °C to rt, 16 h decomposition
2 CAN (1.2 equiv.) MeOH 0 °C, 1 h decomposition
2 Ag2O (1.2 equiv.) benzene rt, 4 h decomposition
2 PhI(OAc)2 (1.2 equiv.) THF −78 °C to rt, 4 h decomposition
2 IBX (1.5 equiv.) MeCN −40 °C to rt, 16 h 20%
2 PhI(OAc)2 (1.1 equiv.), TEMPO
(2 equiv.) THF −78 °C to rt, 4 h 26%
2 PhI(CF3COO)2 (1.1 equiv.),
TEMPO (2 equiv.) THF −78 °C to rt, 5 h 26%
2 PhI(OAc)2 (1.1 equiv.), TEMPO
(2 equiv.) THF rt, 1 h decomposition
1.2 PhI(OAc)2 (1.1 equiv.), TEMPO
(2 equiv.) THF −78 °C to rt, 4 h 26%
1.2 PhI(OAc)2 (1.1 equiv.), TEMPO
(2 equiv.) THF −78 °C to −40 °C, 2 h no reaction
1.2 PhI(OAc)2 (1.1 equiv.), TEMPO
(1.2 equiv.) THF −78 °C to 60 °C, 4 h 26%
1.2 PhI(OAc)2 (1.1 equiv.), TEMPO
(1.2 equiv.) Et2O −78 °C to rt, 4 h
32%
2 CAN (1.2 equiv.), TEMPO (1.2
equiv.) MeOH −78 °C to rt 4 h 39%
1.2 Ag2O (1.1 equiv.), TEMPO (1.2
equiv.) Et2O −78 °C to rt, 16 h 60%
2 Ag2O (1.1 equiv.), TEMPO (0.4
equiv.) Et2O −78 °C to rt, 16 h 40%
2 Ag2O (1.1 equiv.), TEMPO (0.2
equiv.) Et2O −78 °C to rt, 16 h 15%
2 TEMPO (2 equiv.) Et2O −78 °C to rt, 2 d 40%
2.58: norflavesone
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.50: hyperjapone B 2.52: hyperjapone D
d.r. 2.5:1 2.50/2.52
H
H
caryophyllene (2.30)HO O
OH O

21
Here, we proposed two possible oxidation pathways for the formation of heterodiene 2.59 (Scheme
2.21). The first pathway involves a single electron oxidation of TEMPO to a reactive species 2.81,
which then reacts with norflavesone (2.58) via a hydride transfer to give heterodiene 2.59. The
second pathway suggests norflavesone (2.58) undergoing a single electron oxidation by TEMPO to
give a radical intermediate 2.82, which then further oxidized to heterodiene 2.59. From the control
experiment where 2 equiv. of TEMPO was used for the oxidative hetero-Diels-Alder reaction, and
40% yield of hyperjapones B (2.50) and D (2.52) was obtained, we propose the second oxidation
pathway is more plausible.
Proposed mechanism 1:
Proposed mechanism 2:
Scheme 2.21: Proposed oxidation mechanism of norflavesone (2.58) by TEMPO.
NO
[O]NO
O O
OH O
+O O
OH O
NOH
TEMPO-H
+
TEMPO 2.58: norflavesone 2.59
H
H
2.81
2.58: norflavesone
HO O
OH O
NO
TEMPOO O
OH O
[O]O O
OH O
NOH
TEMPO-H
2.82 2.59
e.g. Ag2O,PhI(OAc)2,
CAN,TEMPO

22
2.2.4. Biomimetic total synthesis of hyperjapone A (2.49)
Scheme 2.22: Biomimetic total synthesis of hyperjapone A (2.49).
With the optimized conditions, we moved to the total synthesis of hyperjapone A (2.49). By
changing the dienophile to humulene (2.11), the oxidative hetero-Diels-Alder reaction afforded
hyperjapone A (2.49) in 32% yield (Scheme 2.22). The yield was almost halved from the reaction
with caryophyllene (2.30). This was expected as the Δ4,5-alkene in caryophyllene (2.30) is more
reactive than the Δ1,2-alkene in humulene (2.11). For example, caryophyllene (2.30) can be oxidized
by air readily while humulene (2.11) cannot.13
Nonetheless, the hetero-Diels-Alder reaction was chemoselective and regioselective as we did not
observe any reaction on the Δ4,5 and Δ8,9-alkenes. The reaction was also diastereoselective, where
the relative stereochemistry was pre-determined by the Z-configuration of the Δ1,2-alkene of
humulene (2.11). We then investigated whether it was possible for a second hetero-Diels-Alder
reaction to take place on the Δ8,9-alkene by reducing the equivalents of humulene (2.11), as shown
in the biomimetic total synthesis of lucidene (2.27) by Baldwin.11,12 However, we observed
hyperjapone A (2.49) was the only product. We have also heated the reaction in a sealed tube, but
no second Diels-Alder reaction was observed.
2.2.5. Biomimetic total synthesis of hyperjaponol C (2.56)
With hyperjapone A (2.49) in hand, we moved onto the total synthesis of hyperjaponols A to C.
Treating hyperjapone A (2.49) with m-CPBA in standard conditions gave epoxide 2.60 (Scheme
2.23). The epoxidation was chemoselective, as there was no epoxidation on the Δ4,5-alkene. A
similar observation was reported in the diepoxidation on humulene (2.11) by Fujita.8
HO O
OH O2.58
humulene (2.11)TEMPO, Ag2O
THF, −78 °C to rtO
OH
O
OH
(±)-2.49: hyperjapone A
32%
1
25
4
8 9
23
Scheme 2.23: Epoxidation of hyperjapone A (2.49).
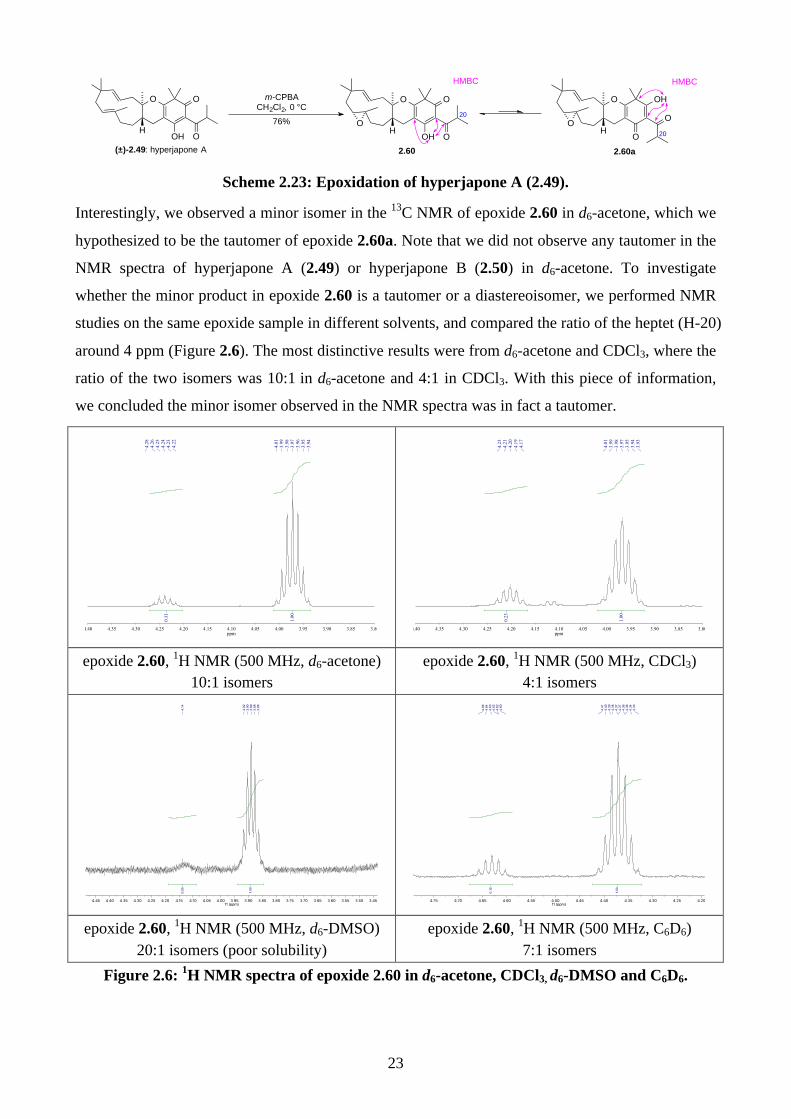
Interestingly, we observed a minor isomer in the 13C NMR of epoxide 2.60 in d6-acetone, which we
hypothesized to be the tautomer of epoxide 2.60a. Note that we did not observe any tautomer in the
NMR spectra of hyperjapone A (2.49) or hyperjapone B (2.50) in d6-acetone. To investigate
whether the minor product in epoxide 2.60 is a tautomer or a diastereoisomer, we performed NMR
studies on the same epoxide sample in different solvents, and compared the ratio of the heptet (H-20)
around 4 ppm (Figure 2.6). The most distinctive results were from d6-acetone and CDCl3, where the
ratio of the two isomers was 10:1 in d6-acetone and 4:1 in CDCl3. With this piece of information,
we concluded the minor isomer observed in the NMR spectra was in fact a tautomer.
epoxide 2.60, 1H NMR (500 MHz, d6-acetone)
10:1 isomers epoxide 2.60, 1H NMR (500 MHz, CDCl3)
4:1 isomers
epoxide 2.60, 1H NMR (500 MHz, d6-DMSO)
20:1 isomers (poor solubility) epoxide 2.60, 1H NMR (500 MHz, C6D6)
7:1 isomers Figure 2.6: 1H NMR spectra of epoxide 2.60 in d6-acetone, CDCl3, d6-DMSO and C6D6.
O
OH
O
OH
(±)-2.49: hyperjapone A
O
OH
O
OH
O
2.60
m-CPBA CH2Cl2, 0 °C
76%
O
O
OH
OH
O
2.60a
HMBC HMBC
20
20

24
We then focused on the acid-catalysed rearrangement of the epoxide 2.60 to hyperjaponols A to C.
We first tried p-TsOH in CH2Cl2 at room temperature, which gave hyperjaponol C (2.56) in 43%
yield, presumably derived from the cationic alkene-cyclization/1,2-alkyl shift cascade reaction
(Scheme 2.24). The reaction could also be done with catalytic p-TsOH (0.1 equiv.) with no loss of
yield.
Scheme 2.24: Biomimetic total synthesis of hyperjaponol C (2.56).
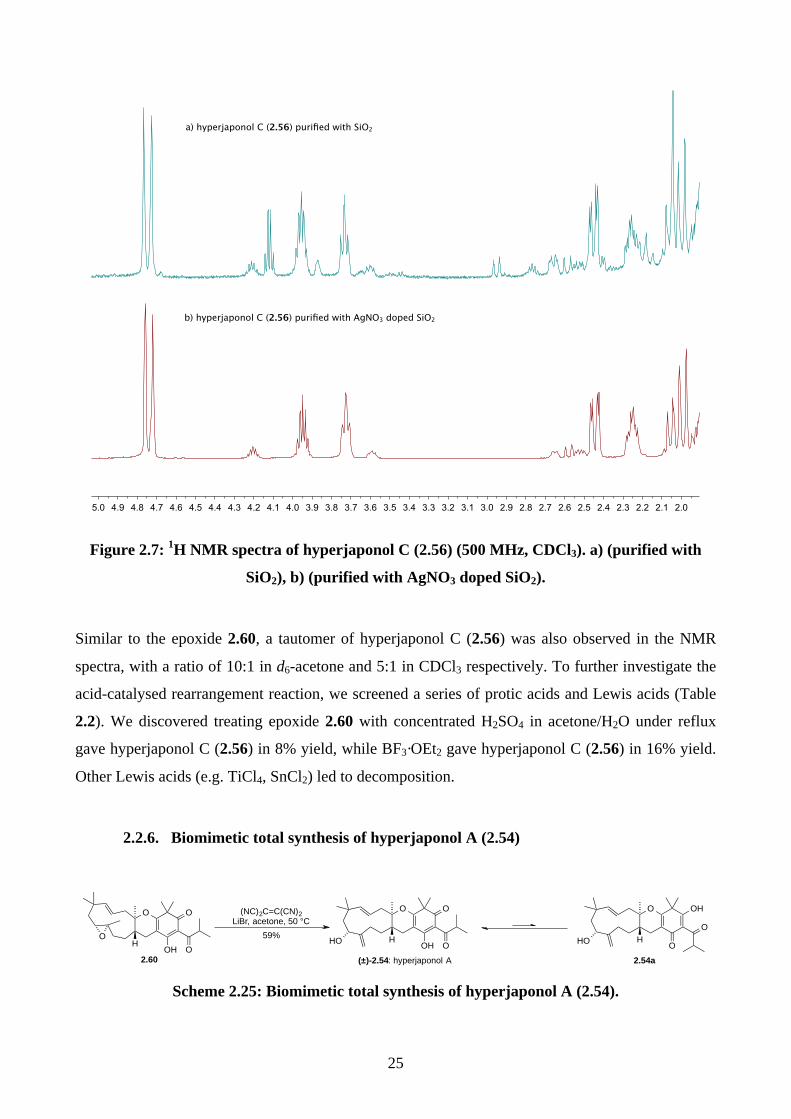
In the acid-catalysed rearrangement reaction, we observed impurities that shared similar Rf to
hyperjaponol C (2.56), which could not be separated by column chromatography. We read a
purification modification of column chromatography involving doping AgNO3 with SiO2,30,31 which
is an underutilized trick that is used more often in natural product isolation than in organic synthesis.
It is believed that the Ag+ can interact with the π electrons in alkenes, which differentiates organic
molecules with varied alkene systems and thus providing a secondary interaction in silica.30,31 This
modified purification worked perfectly and we obtained pure hyperjaponol C (2.56) in 43% yield
(Figure 2.7).
O
OH
O
OH
O
2.60
p-TsOH·H2O CH2Cl2, rt
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
43%
O
O
OH
OH
H
HO
2.56a
HMBC HMBC
25
Figure 2.7: 1H NMR spectra of hyperjaponol C (2.56) (500 MHz, CDCl3). a) (purified with
SiO2), b) (purified with AgNO3 doped SiO2).
Similar to the epoxide 2.60, a tautomer of hyperjaponol C (2.56) was also observed in the NMR
spectra, with a ratio of 10:1 in d6-acetone and 5:1 in CDCl3 respectively. To further investigate the
acid-catalysed rearrangement reaction, we screened a series of protic acids and Lewis acids (Table
2.2). We discovered treating epoxide 2.60 with concentrated H2SO4 in acetone/H2O under reflux
gave hyperjaponol C (2.56) in 8% yield, while BF3·OEt2 gave hyperjaponol C (2.56) in 16% yield.
Other Lewis acids (e.g. TiCl4, SnCl2) led to decomposition.
2.2.6. Biomimetic total synthesis of hyperjaponol A (2.54)
Scheme 2.25: Biomimetic total synthesis of hyperjaponol A (2.54).
O
OH
O
OH
O
2.60
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
(NC)2C=C(CN)2LiBr, acetone, 50 °C
59%
O
O
OH
OHHO
2.54a

26
We then investigated the use of (NC)2C=C(CN)2 for the ring opening reaction of epoxide 2.60, as it
has been used in a similar reaction with caryophyllene oxide (2.31) reported by Marcias-Sanchez.15
We were delighted to observe hyperjaponol A (2.54) in 59% yield in this reaction (Scheme 2.25).
We believed hyperjaponol B (2.55) could be generated from similar conditions. Our hypothesis was
the formation of the endocyclic alkene would be favoured under thermal conditions. However,
when a higher boiling point solvent pent-2-one was used, it gave hyperjaponol A (2.54) in a lower
yield (42%) and other solvents led to decomposition (Table 2.2). Unfortunately, we have yet to
observe hyperjaponol B (2.55) in our synthetic work.
Table 2.2: Conditions screened for the acid-catalysed rearrangement reaction
reagents solvent conditions product, yield
(NC)2C=C(CN)2 (0.2 equiv.), LiBr (5 equiv.)
acetone rt no reaction
(NC)2C=C(CN)2 (0.2 equiv.), LiBr (5 equiv.)
acetone 50 °C, 2 h hyperjaponol A (2.54),
59%
(NC)2C=C(CN)2 (0.2 equiv.), LiBr (5 equiv.)
pentan-2-one reflux, 2.5 h hyperjaponol A (2.54),
42%
(NC)2C=C(CN)2 (0.2 equiv.), LiBr (5 equiv.)
PhMe reflux, 1 d decomposition
(NC)2C=C(CN)2 (0.2 equiv.), LiBr (5 equiv.)
EtOH reflux, 4 h decomposition
(NC)2C=C(CN)2 (0.2 equiv.), LiBr (5 equiv.)
CH2Cl2 rt, 2 d no reaction
(NC)2C=C(CN)2 (0.1 equiv.)
DMSO rt, 16 h no reaction
(NC)2C=C(CN)2 (0.1 equiv.)
DMSO reflux, 16 h decomposition
1 M HCl CHCl3 rt, 2 h no reaction
1 M HCl CHCl3 reflux, 3 h no reaction
conc. HCl CHCl3 rt, 3 h hyperjaponol A (2.54),
30%
conc. HCl CHCl3 reflux, 2 h hyperjaponol A (2.54),
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
O
OH
O
OH
H
HO
or
2.56: hyperjaponol C
O
OH
O
OH
O
2.60
conditions
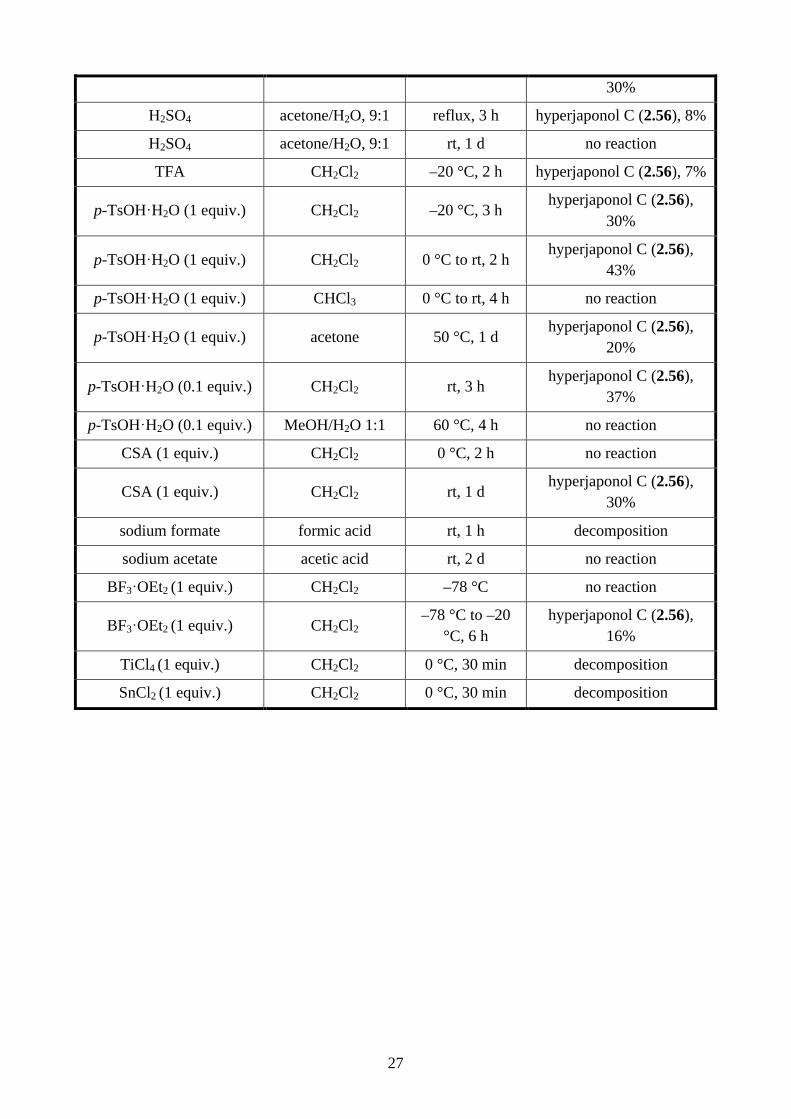
27
30%
H2SO4 acetone/H2O, 9:1 reflux, 3 h hyperjaponol C (2.56), 8%
H2SO4 acetone/H2O, 9:1 rt, 1 d no reaction
TFA CH2Cl2 –20 °C, 2 h hyperjaponol C (2.56), 7%
p-TsOH·H2O (1 equiv.) CH2Cl2 –20 °C, 3 h hyperjaponol C (2.56),
30%
p-TsOH·H2O (1 equiv.) CH2Cl2 0 °C to rt, 2 h hyperjaponol C (2.56),
43%
p-TsOH·H2O (1 equiv.) CHCl3 0 °C to rt, 4 h no reaction
p-TsOH·H2O (1 equiv.) acetone 50 °C, 1 d hyperjaponol C (2.56),
20%
p-TsOH·H2O (0.1 equiv.) CH2Cl2 rt, 3 h hyperjaponol C (2.56),
37%
p-TsOH·H2O (0.1 equiv.) MeOH/H2O 1:1 60 °C, 4 h no reaction
CSA (1 equiv.) CH2Cl2 0 °C, 2 h no reaction
CSA (1 equiv.) CH2Cl2 rt, 1 d hyperjaponol C (2.56),
30%
sodium formate formic acid rt, 1 h decomposition
sodium acetate acetic acid rt, 2 d no reaction
BF3·OEt2 (1 equiv.) CH2Cl2 –78 °C no reaction
BF3·OEt2 (1 equiv.) CH2Cl2 –78 °C to –20
°C, 6 h hyperjaponol C (2.56),
16%
TiCl4 (1 equiv.) CH2Cl2 0 °C, 30 min decomposition
SnCl2 (1 equiv.) CH2Cl2 0 °C, 30 min decomposition

28
In the synthesis of hyperjaponol C (2.56), both epoxidation and acid-catalysed rearrangement
reactions were robust and clean, so it was natural to attempt a one pot reaction.32 Indeed, the
reaction went extraordinarily well. After the addition of m-CPBA to hyperjapone A (2.49), the
reaction was monitored by TLC carefully; when all of the hyperjapone A (2.49) was consumed,
catalytic p-TsOH was added. The reaction went to completion overnight and gave 33% yield of
hyperjaponol C (2.56), which was approximately the combined yield of the two separate steps
(Scheme 2.26). Alternatively, m-CPBA and p-TsOH could be added simultaneously to hyperjapone
A (2.49) in CH2Cl2 and gave hyperjaponol C (2.56) in 26% yield.
Scheme 2.26: One pot epoxidation and acid-catalysed rearrangement reaction of hyperjapone
A (2.49) to give hyperjaponol C (2.56).
2.2.7. Biomimetic total synthesis of hyperjapones C (2.51) and E (2.53)
Scheme 2.27: Total synthesis of norisoleptospermone (2.84).
After the total synthesis of hyperjaponols A (2.54) and C (2.56), we moved onto the synthesis of
hyperjapones C (2.51) and E (2.53). The synthesis started from Friedel-Crafts acylation of
phloroglucinol (2.68) with AlCl3 and (S)-EtCH(Me)COCl.33 The reaction went smoothly and gave
acylphloroglucinol 2.83 in good yield. Trimethylation of acylphloroglucinol 2.83 by KOt-Bu and
MeI gave norisoleptospermone (2.84) in 69% yield (Scheme 2.27), which is also a natural product
isolated from a flowering plant Lepsospermum scoparium.22
m-CPBA, CH2Cl20 °C then
p-TsOH·H2O, rtO
OH
O
OH
(±)-2.49: hyperjapone A
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
33%
orm-CPBA, p-TsOH·H2O
CH2Cl2 rt26%
HO OH
OH
HO OH
OH O
2.68: phloroglucinol 2.83
(S)-EtCH(Me)COCl, AlCl3PhNO2, 65 °C H
71%
HO O
OH O2.84: norisoleptospermone
MeI, KOt-BuMeOH, 65 °C H
69%

29
Scheme 2.28: Biomimetic total synthesis of hyperjapones C (2.51) and E (2.53).
With norisoleptospermone (2.84) in hand, we proceeded to the oxidative hetero-Diels-Alder
reaction with caryophyllene (2.30). Under the optimized conditions, the reaction gave a 2.5:1
mixture of hyperjapones C (2.51) and E (2.53) in 61% yield (Scheme 2.28). We could not separate
the two isomers by flash column chromatography or HPLC.
Scheme 2.29: Biomimetic total synthesis of hyperjapones analogues 2.85 and 2.86.
Since we believe the biosynthesis of hyperjapones A-E is highly predisposed and non-enzymatic,
the hyperjapone analogues 2.85 and 2.86 derived from norisoleptospermone (2.84) and humulene
(2.11) could be two undiscovered natural products. The addition of TEMPO and Ag2O to
noisoleptospermone (2.84) and humulene (2.11) in THF gave a 1:1 mixture of 2.85 and 2.86 with
35% overall yield (Scheme 2.29).
HO O
OH O
2.84: norisoleptospermone
H O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.51: hyperjapone C 2.53: hyperjapone E
H H
61%, d.r. 2.5:1 2.51/2.53
H
H
caryophyllene (2.30)TEMPO, Ag2O
THF, –78 °C to rt
2.85 2.86
+O
OH
O
OH
O
OH
O
OH
HHHO O
OH O
H
35%, d.r. 1:1 2.85/2.86
humulene (2.11)TEMPO, Ag2O
THF, –78 °C to rt
2.84: norisoleptospermone
undiscovered natural products?

30
2.2.8. Isolation of hyperjapones F to I (2.87–2.90)
Figure 2.8: Structures of hyperjapones F to I (2.87–2.90).
After we published our work on the total synthesis of hyperjapones and hyperjaponols,34 the
isolation of hyperjapones F-I (2.87–2.90) was reported by Xu (Figure 2.8).35 These natural products
were also isolated from Hypericum japonicum, and they are presumably biosynthesized from the
hetero-Diels-Alder reaction with 2.59. Note the absolute configurations of hyperjapones H (2.89)
and I (2.90) were not determined from the isolation. Therefore, we aimed to synthesize both
enantiomers of hyperjapones H (2.89) and I (2.90) and determine the absolute configuration of the
natural products.
2.2.9. Biomimetic total synthesis of hyperjapones F and G (2.87 & 2.88)
Scheme 2.30: Biomimetic Total synthesis of hyperjapones F (2.87) and G (2.88).
Similar to our previous synthesis of hyperjapones, norflavesone (2.58) was oxidised and reacted
with (–)-sabinene (2.91) to give a 1:1 mixture of hyperjapones F (2.87) and G (2.88) (Scheme 2.30).
O
OH
O
O2.89: (+)-hyperjapone H
OH
O
O
2.90: (+)-hyperjapone I
O
H
O
OH
O
O2.87: (–)-hyperjapone F
O
OH
O
O2.88: (+)-hyperjapone G
O O
OH O
2.59
H+
2.91: (–)-sabinene (–)-2.92: (–)-(β)-pinene
(+)-2.92: (+)-(β)-pinene
(–)-2.93: (–)-(α)-pinene
(+)-2.93: (+)-(α)-pinene
hetero-Diels-Alder reaction
*absolute configurations of hyperjapones H (2.89) and I (2.90)were not determined from isolation
HO O
OH O2.58: norflavesone
(−)-sabinene (2.91)Ag2O, TEMPO
THF, −78 °C to rtO
OH
O
O2.87: hyperjapone F
13%
H
O
OH
O
O2.88: hyperjapone G
+
1:1

31
The yield of this reaction was only 13%, as the terminal alkene of (–)-sabinene (2.91) is less
reactive compared to the strained trans-alkenes of caryophyllene (2.30) and humulene (2.11).
2.2.10. Biomimetic total synthesis of hyperjapone H (2.89)
Scheme 2.31: Biomimetic total synthesis of hyperjapone H (2.89).
Interestingly, the oxidative hetero-Diels-Alder reaction did not proceed as well with (β)-pinene
(2.92) using Ag2O and TEMPO. The reaction gave hyperjapone H (2.89) as well as some complex
side products, which were difficult to separate by column chromatography. The AgNO3 doped SiO2
column chromatography did not work on this occasion. Looking back at the Table 2.1, we repeated
the reaction using our second-best conditions with CAN and TEMPO in MeOH. To our delight,
treating (+)-(β)-pinene (+)-2.92 and norflavesone (2.58) with CAN and TEMPO gave a 25% yield
of (+)-hyperjapone H (+)-2.89; while using (–)-(β)-pinene (–)-2.92 gave a 27% yield of (−)-
hyperjapone H (−)-2.89 (Scheme 2.31). The optical rotations of (+)-2.89 and (−)-2.89 are [!]!!"
+24º (c 1.0, MeOH) and [!]!!" −20° (c 1.0, MeOH). The natural sample has an optical rotation of
[!]!!" +3° (c 0.12, MeOH). Since the racemic (β)-pinene (2.92) is naturally occurring, we suspect
that natural hyperjapone H (2.92) is a racemate, if not a scalemic mixture.
2.2.11. Biomimetic total synthesis of hyperjapone I (2.90)
Scheme 2.32: Biomimetic total synthesis of hyperjapone I (2.90).
HO O
OH O
2.58: norflavesone
(−)-(β)-pinene (−)-2.92CAN, TEMPO
MeOH, −78 °C to rt O
OH
O
O(−)-2.89: (−)-hyperjapone H
27%
(+)-(β)-pinene (+)-2.92CAN, TEMPO
MeOH, −78 ºC to rtO
OH
O
O(+)-2.89: (+)-hyperjapone H
25%
HO O
OH O
2.58: norflavesone
(−)-(α)-pinene (−)-2.93CAN, TEMPO
MeOH, −78 ºC to rt
OH
O
O
(−)-2.90: (−)-hyperjapone I
5%
O
H
(+)-(α)-pinene (+)-2.93CAN, TEMPO
MeOH, −78 ºC to rt
OH
O
O
(+)-2.90: (+)-hyperjapone I
8%
O
H

32
In the synthesis of hyperajapone I (2.90), we again observed inseparable side products using Ag2O
and TEMPO, and hence we chose to use CAN and TEMPO for the reaction. The yield was 8% with
(+)-(α)-pinene (+)-2.93 and 5% with (–)-(α)-pinene (+)-2.93 (Scheme 2.32). The yield of this
reaction was relatively low, due to the poor reactivity of the tri-substituted alkene in (α)-pinene
(2.93). The optical rotations are [!]!!" = +95° (c 1.0, MeOH) for (+)-2.90 and [!]!!" = −98° (c 1.0,
MeOH) for (−)-2.90. The natural product has an optical rotation of [!]!!" = +51° (c 0.13, MeOH).
Therefore, we suspect that natural hyperjapone I (2.90) is possibly a scalemic mixture, or the natural
sample might be impure. We also managed to recrystallise hyperjapone I (2.90) from MeOH and
obtained an X-ray structure to confirm the relative stereochemistry (Figure 2.9).
Figure 2.9: X-ray structure of hyperjapone I (2.90).
2.2.12. Preliminary theoretical calculations of the transition state of cationic alkene-
cyclization/1,2-shift
To investigate the concerted, asynchronous cationic alkene-cyclization/1,2-alkyl shift hypothesis,
we performed calculations on each proposed carbocation intermediate and their corresponding
transition states.
From the diagram (Figure 2.10), the energy of the first carbocation 2.61 is the highest, and the
energy of the final carbocation 2.63 is the lowest. It suggests the overall reaction profile is
enthalpically favoured.
On the left of the diagram, it represents the asynchronous cation-alkene cyclization/1,2-alkyl shift,
where 2.61 rearranges to 2.63 in a single transition state. On the right side of the diagram, it
illustrates the stepwise process: the alkene-cyclization of 2.61 gives 2.62, followed by 1,2-alkyl
shift to give 2.63.
The calculation shows that the asynchronous cation-alkene cyclization/1,2-alkyl shift is a
reasonable pathway. In the transition state 2.61–2.63, the distance between C-4 and C-9 is 1.71 Å
(from 3.85 Å in 2.61), which shows partial single bond character (1.54 Å for a C-C single bond).

33
And the double bond between C-4 and C-5 is elongated to 1.41 Å (from 1.33 Å in 2.61). And the
ring is slightly twisted, which is in position for the 1,2-alkyl shift.
However, the calculation for the stepwise process is problematic. First, in the optimised structure of
2.62, the distance between C-4 to C-9 is 1.84 Å, which is slightly longer than a single bond,
suggesting it is unlikely for 2.62 to have a 6/7 membered ring structure. In addition, the transition
state from 2.61 to 2.62 could not be calculated. Moreover, in the transition state 2.62–2.63, the
distance between C-4 and C-9 is elongated to 2.02 Å, which suggests the single bond between C-4
and C-9 is broken. Therefore, it is not a simple 1,2-alkyl shift to reach 2.63 from 2.62. To conclude,
our calculations agreed with Tantillo’s hypothesis, suggesting the concerted, asynchronous process
is favoured over the stepwise process.23,24
Figure 2.10: Energy diagram of the carbocation rearrangements. All ground states were
calculated from 6-31G+(d,p), with Density Functional Theory m062x. The transition states
were calculated from 6-31+G(d), with Density Functional Theory m062x.
–32.66 kJ mol-1
–209.48 kJ mol-1
transitin state 2.61-2.63
+116.13 kJ mol-1
no transition from 2.61 to 2.62
–207.64 kJ mol-1
+146.95 kJ mol-1
transition state 2.62-2.63
O
OH
O
OH
O
OH
O
OH
H
HO
HO O
OH
O
OH
H
HO
49
5
49
2.63
2.61
2.62

34
2.3. Summary
We have developed a concise biomimetic total synthesis of hyperjapones A-I via an oxidative
hetero-Diels-Alder reaction. We have also converted hyperjapone A into hyperjaponol C via an
epoxidation and an acid-catalysed rearrangement cascade reaction, which generated 2 rings, and 4
stereocenters in 1 step. The overall synthesis is protecting group free with good pot economy
(Figure 2.11).36,37 It is also noteworthy that the total synthesis mirrored our biosynthetic proposal
precisely.
The synthesis of hyperjaponol C highlights the reactivity of all 3 alkenes in humulene, where each
alkene is reacted sequentially in a chemoselective, stereoselective and regioselective manner:
hetero-Diels-Alder reaction on the Δ1,2-alkene, epoxidation on the Δ8,9-alkene and the acid-
catalysed rearrangement on the Δ4,5-alkene. The synthesis suggests a highly predisposed
biosynthesis of hyperjaponol C.
Figure 2.11: Summary of the biomimetic total synthesis of hyperjaponol C (2.56).
The syntheses of hyperjapones B, H and I confirmed the absolute configuration of these natural
products. We plan to distribute both enantiomers of hyperjapones H and I, epoxide 2.60 and
hyperjapones analogues 2.85/2.86 to the isolation chemists. We hope the synthesis of hyperjapones
and hyperjaponols could help identify and characterise natural products that are yet to be isolated.
After our publication on the synthesis of hyperjapones and hyperjaponols, there were a few reports
on the isolation and biomimetic total synthesis of structurally related natural products.38,39,40 These
natural products were synthesized via a hetero-Diels-Alder reaction with caryophyllene or
humulene (Figure 2.12).
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol CMeI
OH
OHHO
Cl
O
4 steps7% overall yield
6 C-C bonds6 stereocentres
3 rings
35
Figure 2.12: Isolation and synthesis of frutescones by Wang38, rhodomentones A and B by
Qiu39 and rhodomyrtials A and B by Luo and Kong40.
∗∗
O
H
H
HO
OHO
OMe
*S, 2.96: frutescone E *R, 2.97: frutescone F
OMe
2.98: (±)-frutescone G
∗∗
O
H
H
H
O
*S, 2.94: frutescone B *R, 2.95: frutescone C
OMe
O
H
H
OO
2.99: rhodomentone A
O
H
H
OO
2.100: rhodomentone B
OH
OH
∗∗
O
H
H
O
O
HO
O
O
*S, 2.101: rhodomyrtial A*R, 2.102: rhodomyrtial B

36
2.4. References
1. Clayden, J.; Greeves, N.; Warren, S. G., Organic chemistry. Oxford University Press: Oxford; New York, 2012.
2. Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem. Int. Ed., 2002, 41, 1668.
3. Snyder, S. A.; Kontes, F. Isr. J. Chem., 2011, 51, 378. 4. Nicolaou, K. C.; Snyder, S. A. Proceedings of the National Academy of Sciences of the
United States of America, 2004, 101, 11929. 5. Smith, A. B.; Sestelo, J. P.; Dormer, P. G. J. Am. Chem. Soc., 1995, 117, 10755. 6. Kuttruff, C. A.; Zipse, H.; Trauner, D. Angew. Chem. Int. Ed., 2011, 50, 1402. 7. Heravi, M. M.; Ahmadi, T.; Ghavidel, M.; Heidari, B.; Hamidi, H. RSC Adv., 2015, 5,
101999. 8. Zigon, N.; Hoshino, M.; Yoshioka, S.; Inokuma, Y.; Fujita, M. Angew. Chem. Int. Ed., 2015,
54, 9033. 9. Bryson, I.; Roberts, J. S.; Sattar, A. Tetrahedron Lett., 1980, 21, 201. 10. Gao, Y.; Wang, G. Q.; Wei, K.; Hai, P.; Wang, F.; Liu, J. K. Org. Lett., 2012, 14, 5936. 11. Adlington, R. M.; Baldwin, J. E.; Pritchard, G. J.; Williams, A. J.; Watkin, D. J. Org. Lett.,
1999, 1, 1937. 12. Rodriguez, R.; Moses, J. E.; Adlington, R. M.; Baldwin, J. E. Org. Biomol. Chem., 2005, 3,
3488. 13. Steenackers, B.; Campagnol, N.; Fransaer, J.; Hermans, I.; De Vos, D. Chem. Eur. J., 2015,
21, 2146. 14. Barrero, A. F.; Herrador, M. M.; del Moral, J. F. Q.; Arteaga, P.; Sanchez, E. M.; Arteaga, J.
F.; Piedra, M. Eur. J. Org. Chem., 2006, 3434. 15. Collado, I. G.; Hanson, J. R.; MaciasSanchez, A. J. Tetrahedron, 1996, 52, 7961. 16. Spence, J. T. J.; George, J. H. Org. Lett., 2011, 13, 5318. 17. Takao, K.; Noguchi, S.; Sakamoto, S.; Kimura, M.; Yoshida, K.; Tadano, K. J. Am. Chem.
Soc., 2015, 137, 15971. 18. Lawrence, A. L.; Adlington, R. M.; Baldwin, J. E.; Lee, V.; Kershaw, J. A.; Thompson, A.
L. Org. Lett., 2010, 12, 1676. 19. Clericuzio, M.; Alagona, G.; Ghio, C.; Toma, L. J. Org. Chem., 2000, 65, 6910. 20. Yang, X. W.; Li, Y. P.; Su, J.; Ma, W. G.; Xu, G. Org. Lett., 2016, 18, 1876. 21. Hu, L. Z.; Zhang, Y.; Zhu, H. C.; Liu, J. J.; Li, H.; Li, X. N.; Sun, W. G.; Zeng, J. F.; Xue,
Y. B.; Zhang, Y. H. Org. Lett., 2016, 18, 2272. 22. Killeen, D. P.; Larsen, L.; Dayan, F. E.; Gordon, K. C.; Perry, N. B.; van Klink, J. W. J. Nat.
Prod., 2016, 79, 564. 23. Tantillo, D. J. Chem. Soc. Rev., 2010, 39, 2847. 24. Tantillo, D. J. Nat. Prod. Rep., 2011, 28, 1035. 25. Hong, A. Y.; Stoltz, B. M. Angew. Chem. Int. Ed., 2014, 53, 5248. 26. Crombie, L.; Jones, R. C. F.; Palmer, C. J. J. Chem. Soc., Perkins Trans. 1, 1987, 317. 27. Nguyen, N. T.; Pham, V. C.; Litaudon, M.; Gueritte, F.; Bodo, B.; Nguyen, V. T.; Nguyen,
V. H. Tetrahedron, 2009, 65, 7171. 28. George, J. H.; Hesse, M. D.; Baldwin, J. E.; Adlington, R. M. Org. Lett., 2010, 12, 3532. 29. Liao, D. H.; Li, H. H.; Lei, X. G. Org. Lett., 2012, 14, 18. 30. Mander, L. N.; Williams, C. M. Tetrahedron, 2016, 72, 1133. 31. Williams, C. M.; Mander, L. N. Tetrahedron, 2001, 57, 425. 32. Markwell-Heys, A. W.; Kuan, K. K. W.; George, J. H. Org. Lett., 2015, 17, 4228. 33. Fobofou, S. A. T.; Franke, K.; Porzel, A.; Brandt, W.; Wessjohann, L. A. J. Nat. Prod.,
2016, 79, 743.

37
34. Lam, H. C.; Spence, J. T. J.; George, J. H. Angew. Chem. Int. Ed., 2016, 55, 10368. 35. Li, Y. P.; Yang, X. W.; Xia, F.; Yan, H.; Ma, W. G.; Xu, G. Tetrahedron Lett., 2016, 57,
5868. 36. Gaich, T.; Baran, P. S. J. Org. Chem., 2010, 75, 4657. 37. Hayashi, Y. Chem. Sci., 2016, 7, 866. 38. Hou, J. Q.; Guo, C.; Zhao, J. J.; He, Q. W.; Zhang, B. B.; Wang, H. J. Org. Chem., 2017, 82,
1448. 39. Liu, H. X.; Chen, K.; Yuan, Y.; Xu, Z. F.; Tan, H. B.; Qiu, S. X. Org. Biomol. Chem., 2016,
14, 7354. 40. Zhang, Y. L.; Chen, C.; Wang, X. B.; Wu, L.; Yang, M. H.; Luo, J.; Zhang, C.; Sun, H. B.;
Luo, J. G.; Kong, L. Y. Org. Lett., 2016, 18, 4068.

38
2.5. Experimental
2.5.1. General methods
All chemicals used were purchased from commercial suppliers and used as received. All reactions
were performed under an inert atmosphere of N2. All organic extracts were dried over anhydrous
magnesium sulfate. Thin layer chromatography was performed using aluminium sheets coated with
silica gel F254. Visualization was aided by viewing under a UV lamp and staining with ceric
ammonium molybdate or KMnO4 stain followed by heating. All Rf values were measured to the
nearest 0.05. Flash chromatography was performed using 40-63 micron grade silica gel. Melting
points were recorded on a digital melting point apparatus and are uncorrected. Infrared spectra were
recorded using an FT-IR spectrometer as the neat compounds. High field NMR was recorded using
a 500 MHz spectrometer (1H at 500 MHz, 13C at 125 MHz). Solvents used for spectra were d6-
acetone or CDCl3 unless otherwise specified. 1H chemical shifts are reported in ppm on the δ-scale
relative to TMS (δ 0.0) or CDCl3 (δ 7.26) and 13C NMR are reported in ppm relative to CDCl3 (δ
77.00). Multiplicities are reported as (br) broad, (s) singlet, (d) doublet, (t) triplet, (q) quartet, (quin)
quintet, (sext) sextet, (hept) heptet and (m) multiplet. All J-values were rounded to the nearest 0.1
Hz. ESI high resolution mass spectra were recorded on a ESI-TOF mass spectrometer. Optical
rotations were measured on a modular circular polarimeter.

39
2.5.2. Experimental procedures
To a solution of phloroglucinol (2.68) (5.0 g, 39.6 mmol) in PhNO2 (50 mL) at room temperature
was added AlCl3 (21.2 g, 159 mmol) portionwise, followed by slow addition of isopropyl chloride
(4.59 mL, 43.6 mmol). The mixture was heated at 65 ºC for 3.5 h. The solution was cooled to room
temperature, then quenched with 1 M HCl (30 mL) and diluted with MeOH (10 mL). The mixture
was extracted with EtOAc (2 × 50 mL). The combined organic layers were extracted with 1 M
NaOH (2 × 50 mL). The aqueous extracts were acidified by conc. HCl. The aqueous layer was
extracted with EtOAc (3 × 70 mL). The combined organic extracts were washed with brine (200
mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (4:1 → 2:1, petrol/EtOAc gradient elution) to give 2.57 as a
yellow solid (6.75 g, 87%). Data for 2.57 matched that of the published data.1
Rf = 0.45 (1:1, petrol/EtOAc) 1H NMR (500 MHz, CD3OD): δ 5.81 (s, 1H), 3.97 (hept, J = 6.7 Hz, 1H), 1.13 (d, J = 6.8 Hz,
6H). 13C NMR (125 MHZ, CD3OD): δ 211.6, 165.82, 165.75, 104.6, 95.8, 39.9, 19.7.
HO OH
OH
HO OH
OH O2.68: phloroglucinol 2.57
i-PrCOCl, AlCl3PhNO2, 65 °C
87%

40
To a solution of 2.57 (2.84 g, 14.4 mmol) in MeOH (30 mL) at room temperature was added KOt-
Bu (6.00 g, 53.6 mmol) and MeI (2.69 mL, 43.2 mmol). The solution was heated at 65 °C for 3 h.
The reaction was cooled to room temperature, then acidified by 1 M aqueous HCl solution (40 mL).
The aqueous solution was extracted with EtOAc (2 × 30 mL). The combined organic extracts were
washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified
by flash column chromatography on SiO2 (4:1, petrol/EtOAc) to give norflavesone 2.58 as a white
gum (2.61 g, 79%). Data for 2.58 matched that of the isolation data.2
Rf = 0.40 (1:1, petrol/EtOAc)
IR (neat): 3171, 2981, 2929, 1643, 1575, 1514, 1462, 1376, 1334, 1223, 1181, 1165 cm-1. 1H NMR (500 MHz, d6-DMSO): δ 19.13 (br s, 1H), 3.90 (hept, J = 6.8 Hz, 1H), 1.78 (s, 3H), 1.29
(s, 6H), 1.04 (d, J = 6.8 Hz, 6H). 13C NMR (125 MHz, d6-DMSO): δ 206.5, 195.8, 189.1, 176.1, 103.5, 101.8, 48.5, 34.6, 24.3,
18.9, 7.4.
HRMS (ESI): calculated for C13H17O4 237.1132 [M−H]-, found 237.1135.
HO O
OH O2.58: norflavesone
MeI, KOt-BuMeOH, 65 °C
HO OH
OH O2.57
79%

41
To a solution of 2.58 (1.80 g, mmol), and α-humulene (2.11) (1.24 g, 6.07 mmol) in Et2O (30 mL)
at −78 °C was added Ag2O (1.97 g, 6.49 mmol) and TEMPO (1.42 g, 9.10 mmol). The reaction was
stirred at −78 °C for 1 h, then warmed to room temperature and stirred for 16 h. The solution was
filtered and concentrated in vacuo. The residue was purified by flash column chromatography on
SiO2 (1:0 → 30:1, petrol/EtOAc gradient elution) to give (±)-hyperjapone A (2.49) as a white solid
(860 mg, 32%).
Mp: 174 – 175 °C
Rf = 0.65 (5:1, petrol/EtOAc)
IR (neat): 2968, 2934, 2861, 1656, 1619, 1509, 1473, 1381, 1265, 1190, 1158, 1103 cm-1. 1H NMR (500 MHz, d6-acetone): δ 19.26 (br s, 1H), 5.23 (d, J = 15.7 Hz, 1H), 5.11 (dd, J = 12.0,
3.2 Hz, 1H), 5.04 (ddd, J = 15.9, 10.5, 2.7 Hz, 1H), 3.98 (hept, J = 6.8 Hz, 1H), 2.78 (m, 1H), 2.56
(d, J = 14.6 Hz, 1H), 2.46 (dd, J = 14.7, 10.6 Hz, 1H), 2.24 (d, J = 12.5 Hz, 1H), 2.12 (dd, J = 12.7,
7.2 Hz, 1H), 1.92 (t, J = 12.2 Hz, 1H), 1.85 – 1.83 (m, 2H), 1.75 (dd, J = 13.0, 4.5 Hz, 1H), 1.65 (s,
3H), 1.45 – 1.40 (m, 1H), 1.35 (s, 3H), 1.29 (s, 3H), 1.25 – 1.20 (m, 1H), 1.17 (s, 3H), 1.11 (d, J =
6.0 Hz, 3H), 1.09 (d, J = 5.6 Hz, 3H), 1.05 (s, 3H), 1.03 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 207.9, 196.7, 189.4, 173.7, 143.8, 137.4, 123.8, 120.7, 103.1,
85.9, 48.9, 42.6, 42.2, 38.8, 38.3, 35.8, 35.6, 30.6, 30.4, 25.2, 24.4, 24.2, 22.5, 20.3, 19.4, 19.2,
17.2.
HRMS (ESI): calculated for C28H41O4 441.2999 [M+H]+, found 441.3000.
HO O
OH O2.58
humulene (2.11)TEMPO, Ag2O
THF, −78 °C to rtO
OH
O
OH
(±)-2.49: hyperjapone A
32%

42
To a solution of (±)-hyperjapone A (2.49) (474 mg, 1.08 mmol) in CH2Cl2 (20 mL) at 0 °C was
added m-CPBA (77%, 265 mg, 1.18 mmol). The reaction was stirred at 0 °C for 1 h, then quenched
with saturated aqueous NaHCO3 solution. The organic layer was separated and the aqueous layer
was extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were washed with brine (50
mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (4:1, petrol/EtOAc) to give epoxide 2.60 as a white solid (493 mg,
76%).
Mp: 220 – 223 °C
Rf = 0.20 (10:1, petrol/EtOAc)
IR (neat): 2958, 2933, 2871, 1703, 1658, 1623, 1517, 1473, 1380, 1316, 1237, 1195 cm-1.
10:1 mixture of tautomers in d6-acetone
Data for major tautomer 2.60: 1H NMR (500 MHz, d6-acetone): δ 19.23 (s, 1H), 5.53 (dd, J = 15.7, 1.2 Hz, 1H), 5.32 (ddd, J =
15.7, 10.1, 4.0 Hz, 1H), 3.97 (hept, J = 6.7 Hz, 1H), 2.73 (d, J = 10.9 Hz, 1H), 2.69 (d, J = 10.8 Hz,
1H), 2.65 – 2.60 (m, 2H), 2.13 (dd, J = 13.6, 9.0 Hz, 1H), 1.81 – 1.75 (m, 2H), 1.70 (d, J = 13.6 Hz,
1H), 1.62 (dd, J = 14.4, 9.9 Hz, 1H), 1.36 (s, 3H), 1.32 – 1.30 (m, 1H), 1.29 (s, 3H), 1.29 (s, 3H),
1.25 – 1.23 (m, 1H), 1.19 (s, 3H), 1.17 (s, 3H), 1.10 (d, J = 5.2 Hz, 3H), 1.09 (d, J = 5.2 Hz, 3H),
1.04 (s, 3H), 0.99 (dd, J = 13.6, 9.9 Hz, 1H). 13C NMR (125 MHz, d6-acetone): δ 207.9, 196.7, 189.4, 173.7, 142.1, 121.2, 105.1, 103.3, 84.6,
61.9, 60.7, 84.9, 42.8, 41.1, 38.3, 37.6, 36.2, 35.7, 31.3, 26.2, 25.4, 24.4, 24.2, 22.3, 19.8, 19.4,
19.2, 17.6.
O
OH
O
OH
(±)-2.49: hyperjapone A
O
OH
O
OH
O
2.60
m-CPBA CH2Cl2, 0 °C
76%
O
O
OH
OH
O
2.60a

43
Partial data for minor tautomer 2.60a: 1H NMR (500 MHz, d6-acetone): δ 5.51 (d, J = 11.3 Hz, 1H), 4.24 (hept, J = 6.6 Hz, 1H). 13C NMR (125 MHz, d6-acetone): δ 205.8, 199.4 183.1, 166.0, 142.8, 121.5, 108.6, 103.1, 82.7,
61.9, 60.8, 49.0, 42.9, 41.1, 38.3, 37.7, 36.5, 36.2, 30.6, 26.4, 25.3, 24.4, 24.2, 23.2, 19.6, 19.34,
19.29, 17.6.
4:1 mixture of tautomers in CDCl3
Data for major tautomer 2.60: 1H NMR (500 MHz, CDCl3): δ 19.14 (s, 1H), 5.39 (d, J = 15.7 Hz, 1H), 5.23 – 5.18 (m, 1H), 3.97
(hept, J = 6.3 Hz, 1H), 2.73 (dd, J = 16.6, 4.8 Hz, 1H), 2.65 – 2.62 (m, 2H), 2.43 (dd, J = 14.0, 10.9
Hz, 1H), 2.18 (dd, J = 13.4, 9.4 Hz, 1H), 1.82 – 1.78 (m, 2H), 1.75 – 1.67 (m, 1H), 1.60 (td, J =
11.7, 4.7 Hz, 1H), 1.49 – 1.43 (m, 2H), 1.38 (s, 3H), 1.32 (s, 3H), 1.29 (s, 3H), 1.16 (s, 3H), 1.15 (d,
J = 7.9 Hz, 3H), 1.14 (d, J = 5.8 Hz, 3H), 1.11 (s, 3H), 1.05 (s, 3H), 1.03 – 0.99 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 207.8, 196.7, 188.6, 172.6, 142.5, 120.2, 104.7, 102.5, 83.3, 61.8,
60.9, 48.4, 42.5, 40.3, 37.6, 36.9, 35.7, 35.5, 30.9, 25.7, 25.3, 24.2, 23.9, 21.8, 19.7, 19.2, 19.0,
17.4.
Partial data for minor tautomer 2.60a: 13C NMR (125 MHz, CDCl3): δ 210.5, 198.6, 183.4, 165.9, 142.2, 120.5, 107.9, 107.8, 81.8, 61.9,
60.9, 43.3, 42.6, 40.3, 37.7, 36.9, 36.3, 35.7, 30.9, 25.9, 25.2, 24.3, 24.2, 22.5, 19.5, 19.2, 19.1,
17.5.
HRMS (ESI): calculated for C28H41O5 457.2949 [M+H]+, found 457.2946.

44
To a solution of 2.60 (82 mg, 0.18 mmol) in CH2Cl2 (3 mL) at room temperature was added p-
TsOH·H2O (38 mg, 0.20 mmol) and stirred for 1 h. The reaction was quenched with saturated
aqueous NaHCO3 solution. The organic layer was separated and the aqueous layer was extracted
with CH2Cl2 (2 × 5 mL). The combined organic extracts were washed with brine, dried over
MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (doped with 10% w/w AgNO3, 3:1, petrol/EtOAc) to give (±)-
hyperjaponol C (2.56) as a white solid (35 mg, 43%).
Mp: 161 – 163 °C.
Rf = 0.75 (1:1, petrol/EtOAc)
IR (neat): 3448, 2973, 2930, 2869, 1649, 1612, 1570, 1523, 1472, 1451, 1380, 1165 cm-1.
5:1 mixture of tautomers in CDCl3
Data for major tautomer 2.56: 1H NMR (500 MHz, CDCl3): δ 19.08 (s, 1H), 4.77 (s, 1H), 4.73 (s, 1H), 3.96 (hept, J = 6.8 Hz,
1H), 3.73 (t, J = 9.1 Hz, 1H), 2.45 (dd, J = 16.5, 4.9 Hz, 1H), 2.26 (td, J = 11.2, 5.6 Hz, 1H), 2.05
(dd, J = 17.4, 14.3 Hz, 1H), 2.00 (d, J = 16.5 Hz, 1H), 1.95 – 1.85 (m, 3H), 1.83 – 1.80 (m, 1H),
1.74 (dd, J = 10.2, 6.0 Hz, 1H), 1.72 – 1.70 (m, 1H), 1.67 (s, 3H), 1.61 (br s, 1H), 1.52 – 1.41 (m,
2H), 1.30 (s, 3H), 1.28 – 1.27 (m, 1H), 1.26 (s, 3H), 1.15 (d, J = 6.8 Hz, 3H), 1.13 (d, J = 5.9 Hz,
3H), 1.12 (s, 3H), 0.82 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 208.0, 197.1, 188.6, 173.7, 146.0, 111.1, 104.7, 103.6, 84.3, 82.3,
48.5, 47.0, 46.9, 41.4, 41.3, 40.6, 39.3, 36.0, 35.6, 26.2, 25.8, 24.7, 23.4, 20.4, 19.5, 19.14, 19.11,
12.3.
O
OH
O
OH
O
2.60
p-TsOH·H2O CH2Cl2, rt
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
43%
O
O
OH
OH
H
HO
2.56a

45
Data for minor tautomer 2.56a: (partially characterised) 1H NMR (500 MHz, CDCl3): δ 18.56 (br s, 1H), 4.21 (hept, J = 6.8 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 210.5, 198.8, 183.7, 166.9, 146.2, 111.0, 104.7, 103.6, 82.8, 82.3,
48.5, 47.2, 46.9, 43.3, 41.3, 40.8, 39.3, 36.3, 36.0, 26.3, 25.6, 25.5, 23.8, 20.2, 19.5, 19.2, 19.1,
13.4.
HRMS (ESI): calculated for C28H41O5 457.2949 [M+H]+, found 457.2948.
10:1 mixture of tautomers in d6-acetone
Data for major tautomer 2.56: 1H NMR (500 MHz, d6-acetone): δ 19.19 (s, 1H), 4.75 (s, 2H), 3.95 (hept, J = 7.0 Hz, 1H), 3.75
(br s, 1H), 3.73 – 3.71 (m, 1H), 2.42 (dd, J = 16.3, 4.9 Hz, 1H), 2.30 (td, J = 11.5, 5.7 Hz, 1H), 2.07
– 2.05 (overlapped m, 2H), 1.97 – 1.93 (m, 2H), 1.91 – 1.85 (m, 2H), 1.84 – 1.78 (m, 2H), 1.69 (s,
3H), 1.53 – 1.48 (m, 2H), 1.35 – 1.32 (m, 1H), 1.27 (s, 3H), 1.24 (s, 3H), 1.19 (s, 3H), 1.10 (d, J =
7.4 Hz, 3H), 1.08 (d, J = 8.6 Hz, 3H), 0.84 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 208.1, 196.8, 189.4, 174.6, 147.6, 111.0, 105.0, 104.3, 85.4,
82.1, 49.0, 48.0, 47.6, 42.0, 41.2, 40.0, 36.6, 35.8, 26.8, 25.9, 25.3, 23.7, 20.5, 19.5, 19.30, 19.28,
12.6.

46
To a solution of of (±)-hyperjapone A (2.49) (220 mg, 0.50 mmol) in CH2Cl2 (20 mL) at 0 °C was
added m-CPBA (77%, 123 mg, 0.55 mmol). The reaction was stirred at 0 °C for 1 h. p-TsOH·H2O
(105 mg, 0.55 mmol) was added. The reaction was warmed to room temperature and stirred for 1 h,
then quenched with saturated aqueous NaHCO3 solution. The organic layer was separated and the
aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were washed
with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (doped with 10% AgNO3, 3:1, petrol/EtOAc) to give (±)-
hyperjaponol C (2.56) as a white solid (75 mg, 33%).
m-CPBA, CH2Cl20 °C then
p-TsOH·H2O, rtO
OH
O
OH
(±)-2.49: hyperjapone A
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
33%
O
O
OH
OH
H
HO
2.56a

47
To a solution of 2.60 (22 mg, 0.048 mmol) in acetone (3 mL) at room temperature was added
tetracyanoethylene (1 mg, 9.6 µmol) and LiBr (21 mg, 0.24 mmol). The solution was heated at 50
°C for 2 h. The reaction was quenched with H2O (5 mL). The aqueous layer was extracted with
Et2O (4 × 10 mL). The combined organic extracts were washed with brine, dried over MgSO4,
filtered and concentrated in vacuo. The residue was purified by flash column chromatography on
SiO2 (2:1, petrol/EtOAc) to give (±)-hyperjaponol A (2.54) as a colourless oil (13 mg, 59%).
Data for 2.54:
Rf = 0.70 (petrol/EtOAc, 1:1, petrol/EtOAc)
IR (neat): 3448, 2956, 2933, 2869, 1731, 1655, 1619, 1595, 1534, 1473, 1382, 1349, 1309 cm-1.
5:1 mixture of tautomers in CDCl3
Data for major tautomer 2.54: 1H NMR (500 MHz, CDCl3): δ 19.16 (s, 1H), 5.39 (d, J = 15.7 Hz, 1H), 5.21 (s, 1H), 5.16 (ddd, J
= 15.4, 9.9, 4.0 Hz, 1H), 4.96 (s, 1H), 3.97 (hept, J = 6.6 Hz, 1H), 3.89 (d, J = 9.0 Hz, 1H), 2.83 (d,
J = 11.6 Hz, 1H), 2.52 (dd, J = 13.8, 3.9 Hz, 1H), 2.48 (dd, J = 13.2, 8.6 Hz, 1H), 2.39 (dd, J =
14.1, 10.0 Hz, 1H), 2.12 (dd, J =13.7, 9.7 Hz, 1H), 2.05 (dd, J = 13.4, 10.1 Hz, 1H), 1.85 – 1.79 (m,
2H), 1.64 – 1.60 (m, 2H), 1.40 – 1.39 (m, 1H), 1.37 (s, 3H), 1.31 (s, 3H), 1.17 (d, J = 8.1 Hz, 3H),
1.15 (d, J = 7.0 Hz, 3H), 1.11 (s, 3H), 1.09 (s, 3H), 1.04 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 207.9, 196.9, 188.6, 172.8, 155.5, 144.4, 120.0, 113.7, 104.7,
102.5, 83.8, 72.9, 49.0, 48.2, 43.7, 37.4, 35.9, 35.5, 32.7, 32.1, 30.6, 25.3, 24.6, 24.1, 22.1, 19.5,
19.2, 19.1.
Data for minor tautomer 2.54a: (partially characterised) 1H NMR (500 MHz, CDCl3): δ 18.59 (s, 1H), 5.37 (d, J = 15.7 Hz, 1H), 5.19 (s, 1H), 4.95 (s, 1H),
4.23 (hept, J = 6.71 Hz, 1H). 13C NMR (12t MHz, CDCl3): δ 210.5, 198.7, 183.6, 166.0, 155.8, 144.1, 120.4, 113.6, 107.8,
102.6, 82.3, 72.8, 49.0, 43.7, 43.3, 47.6, 36.3, 36.0, 32.7, 30.6, 29.9, 25.2, 24.6, 24.3, 22.8, 19.5,
9.29, 19.25.
HRMS (ESI): calculated for C28H41O5 457.2949 [M+H]+, found 457.2947.
O
OH
O
OH
O
2.60
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
(NC)2C=C(CN)2LiBr, acetone, 50 °C
59%
O
O
OH
OHHO
2.54a

48
To a solution of 2.58 (350 mg, 1.47 mmol) and caryophyllene (2.30) (0.66 mL, 2.94 mmol) in Et2O
(10 mL) at −78 °C was added Ag2O (209 mg, 1.76 mmol) and TEMPO (459 mg, 2.94 mmol). The
mixture was stirred for −78 °C for 1 h, then warmed to room temperature over 30 min and stirred at
room temperature for 16 h. The solution was filtered and concentrated in vacuo. The residue was
purified by flash column chromatography on SiO2 (1:0 → 30:1, petrol/EtOAc gradient elution) to
give hyperjapones B (2.50) and D (2.52) as a yellow gum (388 mg, 60%, d.r. 2.5:1). A small sample
of hyperjapones B (2.50) and D (2.52) was recrystallized from MeOH to give hyperjapone B (2.50)
as white crystals.
Mp = 140 − 142 °C
Rf = 0.40 (1:1, petrol/EtOAc)
Data for hyperjapone B (2.50):
IR (neat): 2930, 2870, 1659, 1627, 1519, 1472, 1380, 1368, 1321, 1275, 1245, 1199 cm-1.
[!]!!" = +10.2° (c 0.62, MeOH) 1H NMR (500 MHz, d6-acetone): δ 19.23 (br s, 1H), 4.92 (s, 1H), 4.90 (s, 1H), 3.97 (hept, J = 6.8
Hz, 1H), 2.54 – 2.46 (m, 2H), 2.38 (dd, J = 16.5, 5.1 Hz, 1H), 2.27 – 2.17 (m, 2H), 2.08 – 2.03
(overlapped m, 1H), 1.98 – 1.92 (m, 3H), 1.83 – 1.76 (m, 2H), 1.73 (t, J = 10.4 Hz, 1H), 1.60 (dd, J
= 10.5, 7.6 Hz, 1H), 1.57 – 1.45 (m, 2H), 1.32 (s, 3H), 1.28 (s, 3H), 1.20 (s, 3H), 1.10 (d, J = 5.4
Hz, 3H), 1.08 (d, J = 5.3 Hz, 3H), 1.01 (s, 3H), 0.97 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 207.9, 196.7, 189.4, 173.6, 152.9, 110.7, 105.0, 102.9, 85.4,
54.0, 49.0, 42.8, 37.9, 37.1, 35.9, 35.8, 34.6, 34.2, 33.8, 30.4, 25.4, 25.3, 24.3, 23.4, 22.3, 21.2,
19.33, 19.26.
HRMS (ESI): calculated for C28H41O4 441.2999 [M+H]+, found 441.2996.
2.58
O
OH
O
OH
H
H
O
OH
O
OH
H
H
+2.50: hyperjapone B
2.52: hyperjapone D
60%, d.r. 2.5:1 2.50/2.52
H
H
caryophyllene (2.30)TEMPO, Ag2OTHF, –78 °C to rt
HO O
OH O

49
Data for hyperjapone D (2.52): 1H NMR (500 MHz, d6-acetone): δ 19.24 (br s, 1H), 4.84 (br s, 1H), 4.76 – 4.75 (m, 1H), 4.01 –
3.94 (overlapped m, 1H), 2.79 – 2.75 (overlapped m, 1H), 2.66 (dd, J = 16.5, 5.2 Hz, 1H), 2.54 –
2.46 (overlapped m, 1H), 2.27 – 2.11 (overlapped m, 3H), 1.91 – 1.87 (overlapped m, 1H), 1.84 –
1.74 (overlapped m, 1H), 1.73 – 1.68 (overlapped m, 2H), 1.62 – 1.55 (overlapped m, 3H), 1.53 –
1.46 (overlapped m, 2H), 1.33 (s, 3H), 1.27 (s, 3H), 1.13 (s, 3H), 1.11 – 1.08 (overlapped m, 6H),
1.00 (s, 3H), 0.98 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 207.9, 196.7, 189.3, 173.9, 155.9, 110.3, 105.0, 102.9, 85.6,
57.2, 49.0, 43.0, 39.3, 37.3, 35.8, 35.2, 34.5, 34.2, 33.9, 29.9, 25.4, 24.2, 24.1, 23.3, 22.6, 20.1,
19.4, 19.2.

50
To a solution of phloroglucinol (2.68) (0.95 g, 7.53 mmol) in PhNO2 (10 mL) at room temperature
was added AlCl3 (4.02 g, 30.1 mmol) portionwise, followed by slow addition of (S)-methylbutyric
acyl chloride (1.18 g, 9.79 mmol). The mixture was heated at 65 ºC for 16 h. The solution was
cooled to room temperature, then quenched with 1 M HCl (10 mL) and diluted with MeOH (5 mL).
The mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were extracted
with 1 M NaOH (2 × 10 mL). The aqueous extracts were acidified by conc. HCl. The aqueous layer
was extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with brine,
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (4:1 → 2:1, petrol/EtOAc gradient elution) to give 2.83 as a yellow solid
(1.11 g, 71%). Data for 2.83 matched that of the published data.3
Data for 2.83:
Rf = 0.65 (1:1, petrol/EtOAc)
[!]!!" = +26.8° (c 0.25, CHCl3) 1H NMR (500 MHz, CD3OD): δ 6.05 (s, 2H), 4.10 (sext, J = 6.8 Hz, 1H), 2.06 (dp, J = 14.1, 7.2
Hz, 1H), 1.61 (dp, J = 14.5, 7.4 Hz, 1H), 1.36 (d, J = 6.7 Hz, 3H), 1.15 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHZ, CD3OD): δ 211.4, 165.84, 165.78, 105.2, 95.9, 46.7, 28.1, 17.1, 12.3.
HO OH
OH
HO OH
OH O2.68 2.83
(S)-EtCH(Me)COCl, AlCl3PhNO2, 65 °C H
71%

51
To a solution of 2.83 (700 mg, 3.33 mmol) in MeOH (25 mL) at room temperature was added KOt-
Bu (1.23 g, 10.99 mmol) and MeI (0.69 mL, 10.99 mmol). The reaction was stirred at 65 °C for 5 h,
then cooled to room temperature. The reaction mixture was acidified with 1 M HCl (30 mL), then
extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with brine, dried
over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (4:1 → 2:1, petrol/EtOAc gradient elution) to give norisoleptosperone
(2.84) as a yellow gum (580 mg, 69%). Data for 2.84 matched that of the isolation data.2
Data for 2.84:
Rf = 0.30 (1:1, petrol/EtOAc)
[!]!!" = +19.0° (c 0.87, CHCl3)
IR (neat): 3229, 2969, 2934, 2876, 1648, 1583, 1518, 1473, 1378, 1228, 1180, 1164 cm-1. 1H NMR (500 MHz, d6-DMSO): δ 19.21 (br s, 1H), 3.78 (sext, J = 6.8 Hz, 1H), 1.78 (s, 3H), 1.62
(sext, J = 7.2 Hz, 1H), 1.29 (overlapped m, 7H), 1.03 (d, J = 6.7 Hz, 3H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, d6-DMSO): δ 205.9, 195.9, 189.2, 175.8, 104.3, 101.9, 94.3, 48.5, 40.9,
26.2, 24.4, 24.0, 16.5, 11.7, 7.3.
HRMS (ESI): calculated for C14H21O4 253.1434 [M+H]+, found 253.1431.
HO O
OH O2.84: norisoleptospermone
MeI, KOt-BuMeOH, 65 °C H
69%
HO OH
OH O
H
2.83

52
To a solution of 2.84 (118 mg, 0.40 mmol), and caryophyllene (2.30) (0.18 mL, 0.79 mmol) in Et2O
(5 mL) at −78 °C was added Ag2O (111 mg, 0.48 mmol) and TEMPO (123 mg, 0.79 mmol). The
reaction was stirred at −78 °C for 1 h, then warmed to room temperature and stirred for 3 h. The
solution was filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (1:0 → 50:1, petrol/EtOAc gradient elution) to give hyperjapones C (2.51)
and E (2.53) as a yellow gum (129 mg, 61%, d.r. 2.5:1).
Data for hyperjapone C (2.51):
Rf = 0.60 (5:1, petrol/EtOAc)
IR (neat): 2952, 2930, 2870, 2659, 1627, 1519, 1472, 1380, 1368, 1199 cm-1. 1H NMR (500 MHz, d6-acetone): δ 19.28 (s, 1H), 4.92 (s, 1H), 4.90 (s, 1H), 3.90 – 3.83
(overlapped m, 1H), 2.52 – 2.46 (overlapped m, 2H), 2.38 (dd, J = 16.5, 5.1 Hz, 1H), 2.27 – 2.16
(overlapped m, 2H), 2.27 – 2.16 (overlapped m, 2H), 2.05 (overlapped m, 1H), 1.99 – 1.92
(overlapped m, 1H), 1.86 – 1.68 (overlapped m, 4H), 1.60 (dd, J = 10.5, 7.6 Hz, 1H), 1.58 – 1.54
(overlapped m, 1H), 1.53 – 1.48 (overlapped m, 1H), 1.39 – 1.35 (overlapped m, 1H), 1.32 (s, 3H),
1.27 (s, 3H), 1.20 (s, 3H), 1.09 (d, J = 6.8 Hz, 3H), 1.01 (s, 3H), 0.97 (s, 3H), 0.87 (t, J = 7.4 Hz,
3H). 13C NMR (125 MHz, d6-acetone): δ 207.2, 196.9, 189.4, 173.5, 152.9, 110.7, 105.9, 103.0, 85.4,
54.0, 49.1, 42.8, 42.2, 37.9, 37.1, 35.9, 34.7, 34.2, 33.8, 30.4, 27.4, 25.5, 25.3, 23.9, 23.4, 22.3,
21.2, 17.1, 12.2.
HO O
OH O2.84
H
O
OH
O
OH
H
H
O
OH
O
OH
H
H
+2.51: hyperjapone C
2.53: hyperjapone E
H
H
61%, d.r. 2.5:1 2.51/2.53
H
H
caryophyllene (2.30)TEMPO, Ag2O
THF, –78 °C to rt

53
Data for hyperjapone E (2.53): 1H NMR (500 MHz, d6-acetone): δ 19.31 (br s, 1H), 4.84 (s, 1H), 4.75 (t, J = 1.7 Hz, 1H), 3.90 –
3.83 (overlapped m, 1H), 2.80 – 2.78 (overlapped m, 1H), 2.67 (dd, J = 16.5, 5.2 Hz, 1H), 2.52 –
2.47 (overlapped m, 1H), 2.27 – 2.16 (overlapped m, 2H), 2.12 – 2.08 (m, 1H), 1.98 – 1.87
(overlapped m, 1H), 1.64 – 1.83 (overlapped m, 5H), 1.62 – 1.58 (overlapped m, 1H), 1.53 – 1.46
(overlapped m, 3H), 1.40 – 1.35 (m, 1H), 1.33 (s, 3H), 1.27 (s, 3H), 1.13 (s, 3H), 1.08 (d, J = 6.7
Hz, 3H), 1.00 (s, 3H), 0.98 (s, 3H), 0.91 – 0.88 (overlapped m, 3H). 13C NMR (125MHz, d6-acetone): δ 207.3, 196.8, 189.4, 173.9, 156.0, 110.5, 105.9, 103.0, 85.6,
57.2, 49.0, 43.0, 42.1, 39.4, 39.3, 37.3, 35.2, 34.5, 33.9, 29.9, 27.5, 25.2, 24.4, 24.1, 23.3, 22.6,
20.1, 17.0, 12.2.
HRMS (ESI): calculated for C29H43O4 455.3156 [M+H]+, found 455.3152.

54
To a solution of 2.84 (93 mg, 0.39 mmol), and humulene (2.11) (160 mg, 0.78 mmol) in Et2O (5
mL) at −78 °C was added Ag2O (108 mg, 0.47 mmol) and TEMPO (122 mg, mmol). The reaction
was stirred at −78 °C for 1 h, then warmed to room temperature and stirred for 16 h. The solution
was filtered and concentrated in vacuo. The residue was purified by flash column chromatography
on SiO2 (1:0 → 50:1, petrol/EtOAc gradient elution) to give a mixture of 2.85 and 2.86 as a white
solid (59 mg, 35%, d.r. 1:1).
Rf = 0.60 (5:1, petrol/EtOAc)
IR (neat): 2964, 2934, 2869, 1660, 1627, 1523, 1472, 1382, 1189. 1158 cm-1. 1H NMR (500 MHz, d6-acetone): δ 19.32 (br s, 1H), 5.23 (d, J = 15.9 Hz, 1H), 5.13 – 5.11 (m,
1H), 5.05 (ddd, J = 15.9, 10.6, 2.6 Hz, 1H), 3.87 (pent, J = 6.7 Hz, 1H), 2.78 (s, 1H), 2.58 – 2.55
(m, 1H), 2.46 (dd, J = 14.6, 10.6 Hz, 1H), 2.24 (t, J = 2.26 Hz, 1H), 2.12 (dd, J = 12.7, 7.3 Hz, 1H),
1.93 – 1.88 (m, 1H), 1.84 – 1.80 (m, 2H), 1.76 – 1.69 (m, 2H), 1.65 (s, 3H), 1.45 – 1.38 (m, 2H),
1.35 (s, 3H), 1.29 (s, 3H), 1.23 – 1.20 (m, 1H), 1.17 (s, 3H), 1.09 (t, J = 6.7 Hz, 3H), 1.05 (s, 3H),
1.03 (s, 3H), 0.90 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, d6-acetone): δ 207.3, 196.8, 189.5, 173.7, 143.8, 137.4, 123.8, 120.7, 105.8,
103.2, 85.9, 49.0, 42.2, 42.1, 38.8, 38.3, 35.6, 30.4, 30.1, 29.9, 27.4, 24.9, 24.7, 24.2, 22.5, 20.3,
17.2, 17.0, 12.2.
HRMS (ESI): calculated for C29H43O4 455.3156 [M+H]+, found 455.3153.
2.85
2.86
+
O
OH
O
OH
O
OH
O
OH
H
H
HO O
OH O
H
35%, d.r. 1:1 2.85/2.86
humulene (2.11)TEMPO, Ag2O
THF, –78 °C to rt
2.84

55
To a solution of noflavesone (2.58) (200 mg, 0.84 mmol) and (−)-sabinene (2.91) (75%, 0.27 mL,
1.68 mmol) in anhydrous THF (10 mL) at −78 °C was added Ag2O (233 mg, 1.00 mmol) and
TEMPO (157 mg, 1.00 mmol). The mixture was stirred at −78 °C for 1 h, then warmed to room
temperature and stirred for an addition of 16 h. The reaction was filtered, then concentrated in
vacuo. The residue was purified by flash column chromatography on SiO2 (100:1, petrol/EtOAc) to
give a 1:1 mixture of hyperjapone F (2.87) and hyperjapone G (2.88) as a colourless oil (40 mg,
13%).
Partial Data for 2.87 and 2.88:
Rf = 0.45 (20:1, petrol/EtOAc)
NMR data for hyperjapone F (2.87): 1H NMR (500 MHz, d6-acetone): δ 3.97 (hept, J = 6.7 Hz, 1H), 2.54 – 2.43 (overlapped m, 1H),
2.34 – 2.27 (m, 1H), 2.00 – 1.93 (overlapped m, 1H), 1.90 – 1.84 (overlapped m, 2H), 1.79 – 1.72
(overlapped m, 1H), 1.66 (dd, J = 12.1, 8.1 Hz, 1H), 1.48 – 1.43 (overlapped m, 1H), 1.41 – 1.35
(overlapped m, 1H), 1.33 (s, 3H), 1.30 (s, 3H), 1.09 (d, J = 6.8 Hz, 6H), 1.03 (d, J = 6.9 Hz, 3H),
0.96 (overlapped d, J = 6.8 Hz, 3H), 0.54 (dd, J = 8.3, 5.5 Hz, 1H), 0.48 – 0.47 (overlapped m, 1H). 13C NMR (125 MHz, d6-acetone): δ 208.0, 196.7, 189.6, 174.0, 105.1, 103.5, 49.2, 35.8, 35.3,
33.19, 33.15, 31.2, 28.3, 25.9, 25.5, 24.8, 20.4, 20.0, 19.32, 19.30, 17.2, 13.2.
NMR data for hyperjapone G (2.88): 1H NMR (500 MHz, d6-acetone): δ 3.97 (hept, J = 6.7 Hz, 1H), 2.54 – 2.43 (overlapped m, 2H),
2.00 – 1.93 (overlapped m, 1H), 1.90 – 1.84 (overlapped m, 1H), 1.79 – 1.72 (overlapped m, 2H),
1.62 – 1.56 (m, 1H), 1.41 – 1.35 (overlapped m, 1H), 1.32 – 1.31 (m, 1H), 1.30 (s, 3H), 1.27 (s,
3H), 1.09 (d, J = 6.8 Hz, 6H), 0.96 (overlapped d, J = 6.9 Hz, 3H), 0.91 (d, J = 6.9 Hz, 3H), 0.90 –
0.88 (m, 1H), 0.48 – 0.47 (m, 1H). 13C NMR (125 MHz, d6-acetone): δ 208.0, 196.7, 189.7, 175.2, 105.1, 103.2, 89.2, 49.2, 35.8,
34.4, 33.3, 30.0, 25.3, 25.2, 24.5, 19.83, 19.82, 19.32, 19.30, 16.3, 12.1.
HRMS (ESI): calculated for C23H33O4 373.2373 [M+H]+, found 373.2375.
HO O
OH O2.58: norflavesone
(−)-sabinene (2.91)Ag2O, TEMPO
THF, −78 ºC to rtO
OH
O
O2.87: hyperjapone F
13%
H
O
OH
O
O2.88: hyperjapone G
+
1:1

56
To a solution of norflavesone (2.58) (200 mg, 0.84 mmol) and (+)-(β)-pinene (+)-2.92 (0.16 mL,
1.01 mmol) in MeOH (10 mL) at −78 °C was added ceric ammonium nitrate (552 mg, 1.01 mmol)
and TEMPO (158 mmol, 1.01 mmol). The mixture was stirred at −78 °C for 10 min, then warmed
to room temperature and stirred for 16 h. The reaction was diluted with H2O (10 mL), then
extracted with Et2O (2 × 20 mL). The combined organic extracts were washed with H2O (40 mL),
brine (40 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was partially
purified by flash column chromatography on SiO2 (100:1 → 20:1 gradient elution, petrol/EtOAc).
The product was purified by flash column chromatography on SiO2 (1:1, petrol/CH2Cl2) to give (+)-
hyperjapone H (+)-2.89 as a light yellow oil (78 mg, 25%).
Data for (+)-2.89:
Rf = 0.35 (20:1, petrol/EtOAc)
IR(neat): 2974, 2927, 2870, 1655, 1623, 1518, 1472, 1457, 1386, 1367, 1353, 1251, 1186 cm-1.
[!]!!" = +23.5° (c 1.0, MeOH)
Data for major tautomer: 1H NMR (500 MHz, d6-acetone): δ 19.21 (br s, 1H), 3.97 (hept, J = 6.8 Hz, 1H), 2.37 (t, J = 6.6
Hz, 2H), 2.31 – 2.26 (m, 1H), 2.16 (t, J = 5.1 Hz, 1H), 2.02 – 1.97 (m, 4H), 1.95 – 1.92 (m, 2H),
1.82 (dt, J = 13.8, 6.8 Hz, 1H), 1.67 (d, J = 10.2 Hz, 1H), 1.31 (s, 3H), 1.30 (s, 3H), 1.26 (s, 3H),
1.09 (d, J = 6.8 Hz, 6H), 1.04 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 207.9, 196.7, 189.4, 174.0, 105.0, 103.2, 86.3, 50.4, 49.3,
41.4, 38.9, 35.8, 32.6, 29.1, 27.8, 27.0, 25.4, 25.0, 24.9, 23.4, 19.32, 19.30, 15.8.
Data for minor tautomer: 1H NMR (500 MHz, d6-acetone): δ 18.60 (br s, 1H), 4.25 (hept, J = 6.8 Hz, 1H), 2.37 (t, J = 6.6
Hz, 2H), 2.31 – 2.26 (m, 1H), 2.11 (t, J = 5.1 Hz, 1H), 2.02 – 1.97 (m, 4H), 1.95 – 1.92 (m, 2H),
1.73 (td, J = 13.7, 7.0 Hz, 1H), 1.68 (d, J = 9.8 Hz, 1H), 1.45 (s, 3H), 1.40 (s, 3H), 1.31 (s, 3H),
1.30 (s, 3H), 1.26 (s, 3H), 1.12 (d, J = 7.0 Hz, 6H), 1.03 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 210.9, 199.3, 183.2, 166.3, 108.7, 103.0, 84.3, 50.4, 41.5,
36.5, 35.9, 33.1, 30.6, 29.0, 27.9, 27.2, 25.5, 25.2, 25.1, 23.5, 16.5.
HO O
OH O2.58: norflavesone
(+)-(β)-pinene (+)-2.92CAN, TEMPO
MeOH, −78 ºC to rtO
OH
O
O(+)-2.89: (+)-hyperjapone H
25%

57
HRMS (ESI): calculated for C23H33O4 373.2373 [M+H]+, found 373.2372.
To a solution of norflavesone (2.58) (200 mg, 0.84 mmol) and (−)-(β)-pinene (−)-2.92 (0.16 mL,
1.01 mmol) in MeOH (10 mL) at −78 °C was added ceric ammonium nitrate (552 mg, 1.01 mmol)
and TEMPO (158 mmol, 1.01 mmol). The mixture was stirred at −78 °C for 10 min, then warmed
to room temperature and stirred for 16 h. The reaction was diluted with H2O (10 mL), then
extracted with Et2O (2 × 20 mL). The combined organic extracts were washed with H2O (40 mL),
brine (40 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was partially
purified by flash column chromatography on SiO2 (100:1→20:1 gradient elution, petrol/EtOAc).
The product was purified by flash column chromatography on SiO2 (1:1, petrol/CH2Cl2) to give (−)-
hyperjapone H (−)-2.89 as a light yellow oil (83 mg, 27%).
Data for (−)-2.89:
Rf = 0.35 (20:1, petrol/EtOAc)
IR(neat): 2972, 2929, 2871, 1656, 1624, 1523, 1474, 1387, 1312, 1252, 1193 cm-1.
[!]!!" = −19.7° (c 1.0, MeOH)
Data for major tautomer: 1H NMR (500 MHz, d6-acetone): δ 19.21 (br s, 1H), 3.97 (hept, J = 6.8 Hz, 1H), 2.37 (t, J = 6.6
Hz, 2H), 2.31 – 2.26 (m, 1H), 2.16 (t, J = 5.1 Hz, 1H), 2.01 – 1.97 (m, 4H), 1.96 – 1.92 (m, 2H),
1.82 (dt, J = 13.8, 6.8 Hz, 1H), 1.67 (d, J = 10.2 Hz, 1H), 1.31 (s, 3H), 1.30 (s, 3H), 1.26 (s, 3H),
1.09 (d, J = 6.8 Hz, 6H), 1.04 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 207.9, 196.7, 189.5, 174.0, 105.0, 103.2, 86.3, 50.4, 49.3,
41.4, 38.9, 35.8, 32.7, 29.1, 27.8, 27.0, 25.4, 25.0, 24.9, 23.4, 19.31, 19.29, 15.8.
Data for minor tautomer: 1H NMR (500 MHz, d6-acetone): δ 18.60 (br s, 1H), 4.25 (hept, J = 6.8 Hz, 1H), 2.37 (t, J = 6.6
Hz, 2H), 2.31 – 2.26 (m, 1H), 2.11 (t, J = 5.1 Hz, 1H), 2.01 – 1.97 (m, 4H), 1.96 – 1.92 (m, 2H),
1.73 (td, J = 13.7, 7.0 Hz, 1H), 1.68 (d, J = 9.8 Hz, 1H), 1.45 (s, 3H), 1.40 (s, 3H), 1.31 (s, 3H),
1.30 (s, 3H), 1.26 (s, 3H), 1.12 (d, J = 7.0 Hz, 6H), 1.03 (s, 3H).
HO O
OH O2.58: norflavesone
(−)-(β)-pinene (–)-2.92CAN, TEMPO
MeOH, −78 ºC to rtO
OH
O
O(−)-2.89: (−)-hyperjapone H
27%

58
13C NMR (125 MHz, d6-acetone): δ 210.9, 199.3, 183.2, 166.3, 108.7, 103.0, 84.3, 50.4, 41.5,
36.5, 35.9, 33.1, 30.6, 29.0, 27.9, 27.2, 25.5, 25.2, 25.1, 23.5, 16.5.
HRMS (ESI): calculated for C23H33O4 373.2373 [M+H]+, found 373.2366.

59
To a solution of noflavesone (2.58) (1.00 g, 4.19 mmol) and (+)-(α)-pinene (+)-2.93 (1.33 mL, 8.38
mmol) in MeOH (50 mL) at −78 °C was added ceric ammonium nitrate (2.76 g, 5.03 mmol) and
TEMPO (838 mg, 5.03 mmol). The mixture was stirred at −78 °C for 1 h, then warmed to room
temperature and stirred for 2 d. The reaction was diluted with H2O (50 mL), then extracted with
Et2O (2 × 100 mL). The combined extracts were washed with H2O (100 mL), brine (100 mL), dried
over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (doped with 1% w/w AgNO3, 100:1, petrol/EtOAc) to give (+)-
hyperjapone I (+)-2.90 as a white solid (130 mg, 8%).
Data for (+)-2.90:
Rf = 0.35 (20:1, petrol/EtOAc)
M.p.: 110 – 111 °C
IR(neat): 2972, 2922, 2873, 1650, 1617, 1516, 1472, 1439, 1388, 1343, 1317 cm-1.
[!]!!" = +95° (c 1.0, MeOH)
Data for major tautomer: 1H NMR (500 MHz, d6-acetone): δ 19.31 (br s, 1H), 3.98 (hept, J = 6.8 Hz, 1H), 2.83 – 2.77 (m,
1H), 2.45 (dd, J = 16.2, 1.8 Hz, 1H), 2.39 (dd, J = 16.3, 6.8 Hz, 1H), 2.26 – 2.19 (m, 3H), 1.93 (dd,
J = 9.6, 4.5 Hz, 1H), 1.43 (s, 3H), 1.37 – 1.32 (overlapped m, 1H), 1.33 (s, 3H), 1.29 (s, 3H), 1.28
(s, 3H), 1.15 (s, 3H), 1.11 (d, J = 6.8 Hz, 3H), 1.09 (d, J = 6.7 Hz, 3H), 0.92 – 0.88 (m, 1H). 13C NMR (125 MHz, d6-acetone): δ 207.5, 196.6, 189.9, 174.8, 105.0, 100.4, 87.1, 55.1, 49.2,
41.8, 40.3, 35.8, 35.7, 31.2, 30.1, 28.9, 28.8, 26.2, 23.9, 23.2, 23.0, 19.6, 19.4, 19.3.
Data for minor tautomer: 1H NMR (500 MHz, d6-acetone): δ 18.64 (br s, 1H), 4.27 (hept, J = 6.7 Hz, 1H), 2.73 (dd, J =
17.0, 7.8 Hz, 1H), 1.44 (s, 3H). 13C NMR (125 MHz, d6-acetone): δ 210.7, 199.3, 183.2, 167.2, 105.7, 100.2, 84.9, 55.3, 49.2,
44.0, 36.5, 36.0, 31.7, 26.0, 24.3, 23.1, 20.4, 19.5.
HRMS (ESI): calculated for C23H33O4 373.2373 [M+H]+, found 373.2379.
HO O
OH O2.58: norflavesone
(+)-(α)-pinene (+)-2.93CAN, TEMPO
MeOH, −78 ºC to rt
OH
O
O(+)-2.90: (+)-hyperjapone I
8%
O
H

60
To a solution of norflavesone (2.58) (1.0 g, 4.19 mmol) and (−)-(α)-pinene (−)-2.93 (1.33 mL, 8.38
mmol) in MeOH (30 mL) at −78 °C was added ceric ammonium nitrate (2.76 g, 5.03 mmol) and
TEMPO (828 mmol, 5.03 mmol). The mixture was stirred at −78 °C for 10 min, then warmed to
room temperature and stirred for 5 d. The reaction was diluted with H2O (20 mL), then extracted
with Et2O (2 × 50 mL). The combined organic extracts were washed with H2O (100 mL), brine (100
mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was partially purified by
flash column chromatography on SiO2 (100:1→50:1 gradient elution, petrol/EtOAc). The product
was purified by flash column chromatography on SiO2 (doped with 1% w/w AgNO3, 100:1,
petrol/EtOAc) to give (−)-hyperjapone I (−)-2.90 as a white solid (81 mg, 5%).
Data for (−)-2.90:
Rf = 0.35 (20:1, petrol/EtOAc)
M.p.: 111 – 113 °C
IR (neat): 2980, 2923, 2869, 1650, 1619, 1514, 1471, 1441, 1388, 1343, 1318, 1251 cm-1.
[!]!!" = −98° (c 1.0, MeOH)
Data for major tautomer: 1H NMR (500 MHz, d6-acetone): δ 19.31 (br s, 1H), 3.97 (hept, J = 6.8 Hz, 1H), 2.83 – 2.78 (m,
1H), 2.45 (dd, J = 16,3m 1.9 Hz, 1H), 2.39 (dd, J = 16.2, 6.8 Hz, 1H), 2.26 – 2.19 (m, 3H), 1.94 –
1.91 (m, 1H), 1.43 (s, 3H), 1.36 – 1.34 (m, 1H), 1.33 (s, 3H), 1.29 (s, 3H), 1.28 (s, 3H), 1.15 (s,
3H), 1.11 (d, J = 6.8 Hz, 3H), 1.09 (d, J = 6.8 Hz, 3H), 0.92 – 0.88 (m, 1H). 13C NMR (500 MHz, d6-acetone): δ 207.5, 196.6, 189.9, 174.8, 105.0, 100.3, 87.1, 55.1, 49.2,
41.8, 40.3, 35.8, 35.7, 31.2, 30.1, 28.9, 28.8, 26.2, 23.9, 23.0, 19.6, 19.4, 19.3.
Data for minor tautomer: 1H NMR (500 MHz, d6-acetone): δ 18.64 (br s, 1H), 4.27 (hept, J = 6.8 Hz, 1H), 2.76 – 2.70 (m,
1H), 1.44 (s, 3H). 13C NMR (500 MHz, d6-acetone): δ 210.7, 199.3, 183.2, 167.1, 105.7, 100.2, 84.9, 55.3, 49.2,
44.0, 36.5, 36.0, 31.7, 26.0, 24.3, 23.1, 20.4, 19.5.
HRMS (ESI): calculated for C23H33O4 373.2373 [M+H]+, found 373.2379.
HO O
OH O2.58: norflavesone
(−)-(α)-pinene (–)-2.93CAN, TEMPO
MeOH, −78 ºC to rt
OH
O
O(−)-2.90: (−)-hyperjapone I
5%
O
H
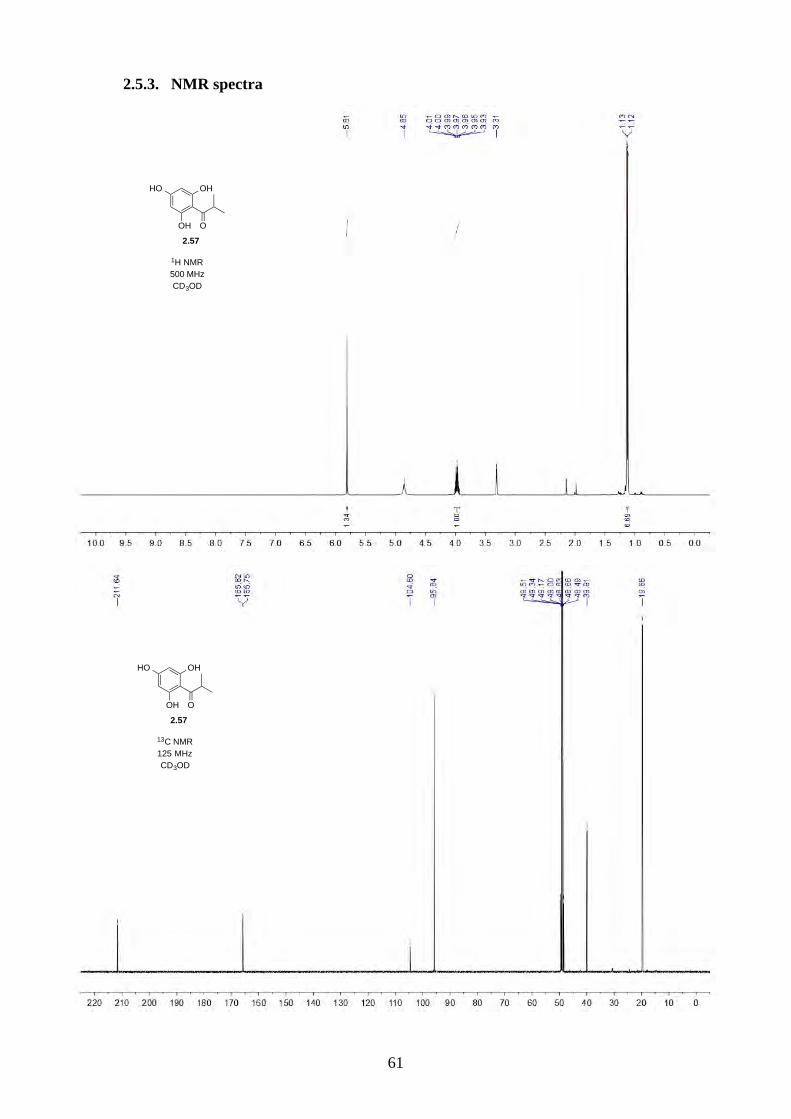
61
2.5.3. NMR spectra
HO OH
OH O
2.57
1H NMR500 MHzCD3OD
HO OH
OH O
2.57
13C NMR125 MHzCD3OD
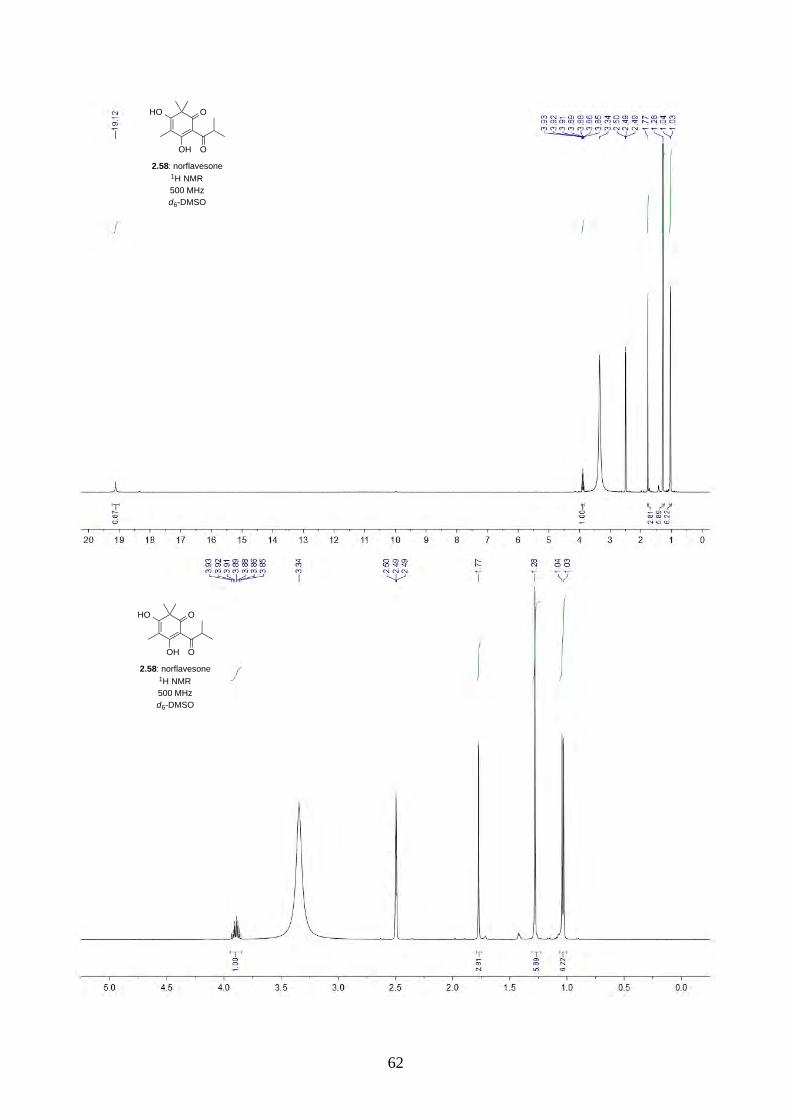
62
HO O
OH O
1H NMR500 MHzd6-DMSO
2.58: norflavesone
HO O
OH O
1H NMR500 MHzd6-DMSO
2.58: norflavesone
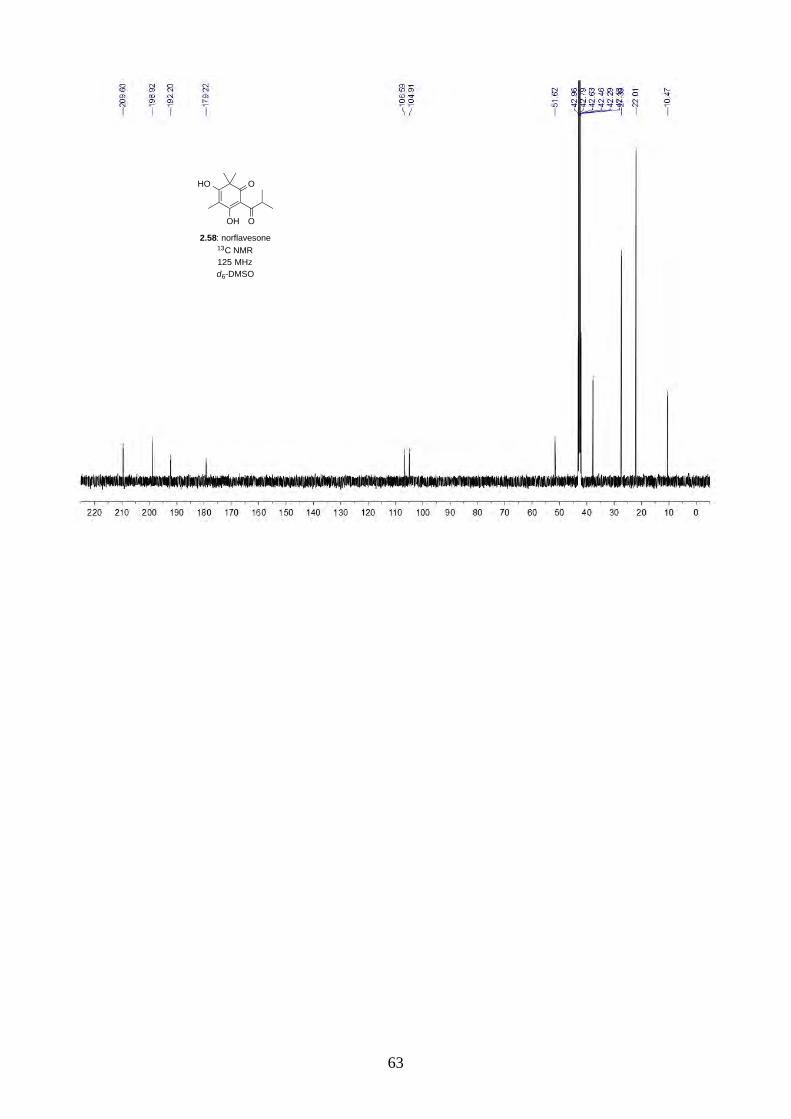
63
HO O
OH O
13C NMR125 MHzd6-DMSO
2.58: norflavesone
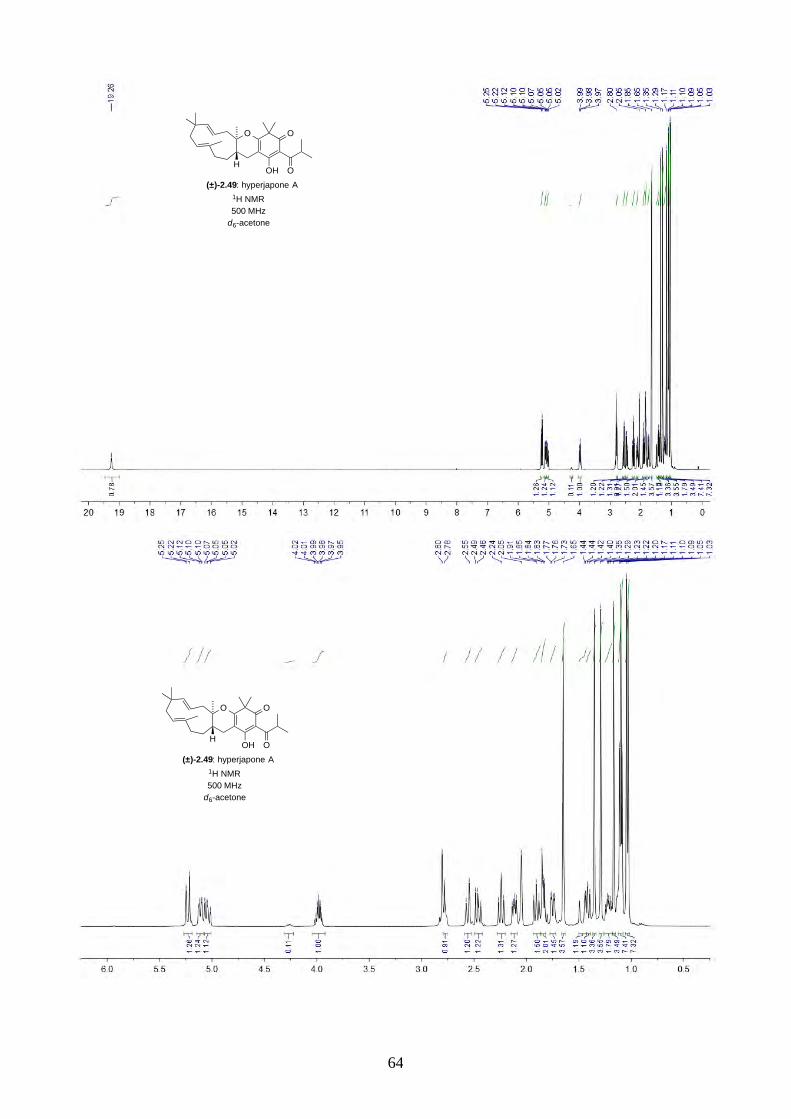
64
O
OH
O
OH
(±)-2.49: hyperjapone A1H NMR500 MHz
d6-acetone
O
OH
O
OH
(±)-2.49: hyperjapone A1H NMR500 MHz
d6-acetone
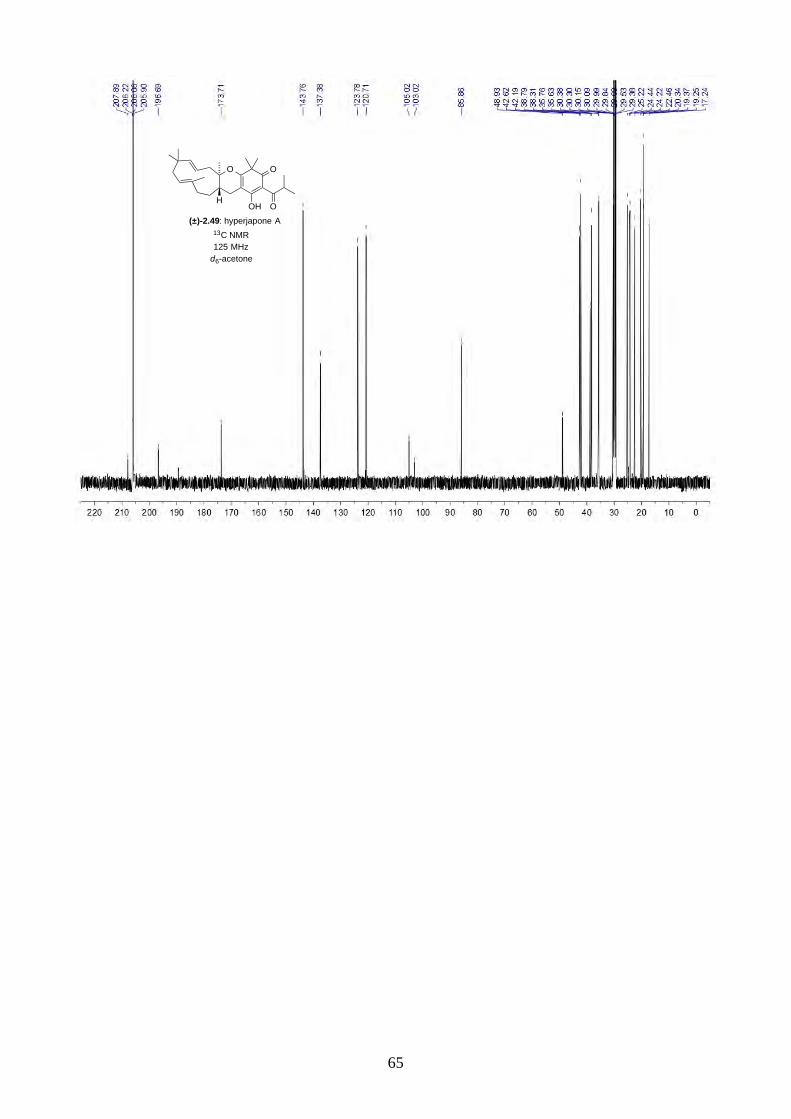
65
O
OH
O
OH
(±)-2.49: hyperjapone A13C NMR125 MHz
d6-acetone
66
O
OH
O
OH
(±)-2.49: hyperjapone ACOSY
500 MHzd6-acetone
O
OH
O
OH
(±)-2.49: hyperjapone AHSQC
500 MHzd6-acetone
67
O
OH
O
OH
(±)-2.49: hyperjapone AHMBC
500 MHzd6-acetone
O
OH
O
OH
(±)-2.49: hyperjapone ANOESY500 MHz
d6-acetone
68
O
OH
O
OH
(±)-2.49: hyperjapone A1H NMR500 MHz
CDCl3
O
OH
O
OH
(±)-2.49: hyperjapone A1H NMR500 MHz
CDCl3
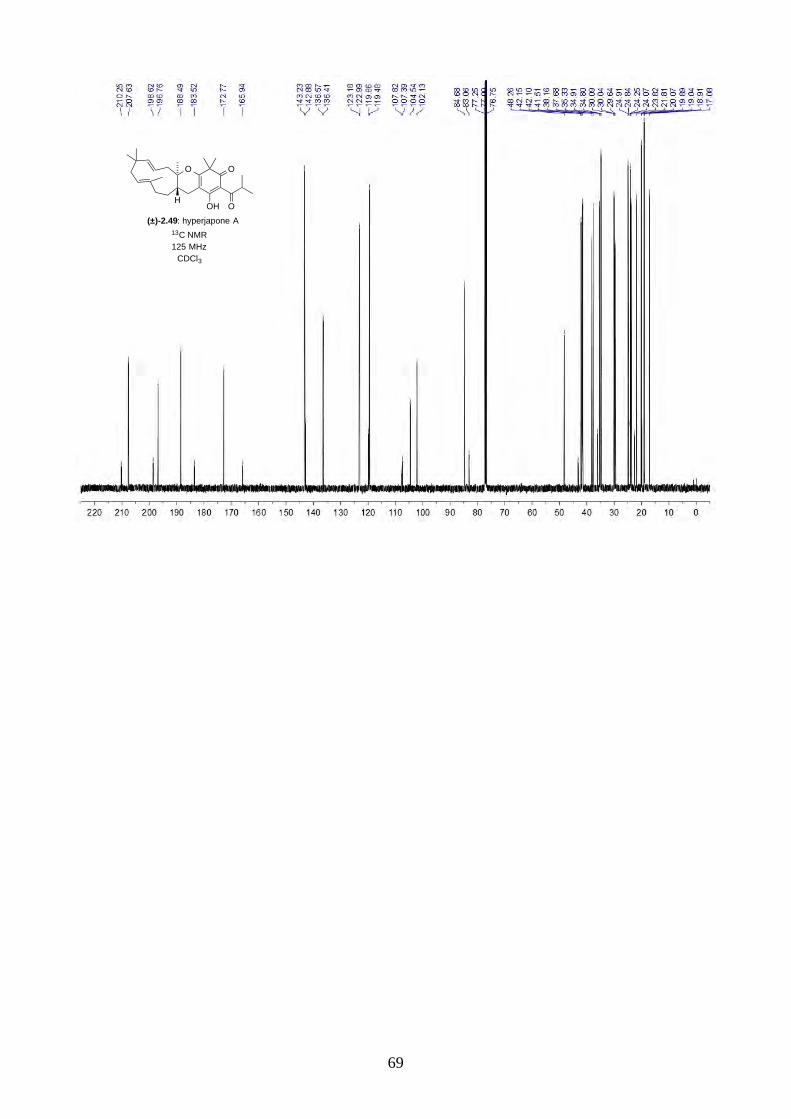
69
O
OH
O
OH
(±)-2.49: hyperjapone A13C NMR125 MHz
CDCl3
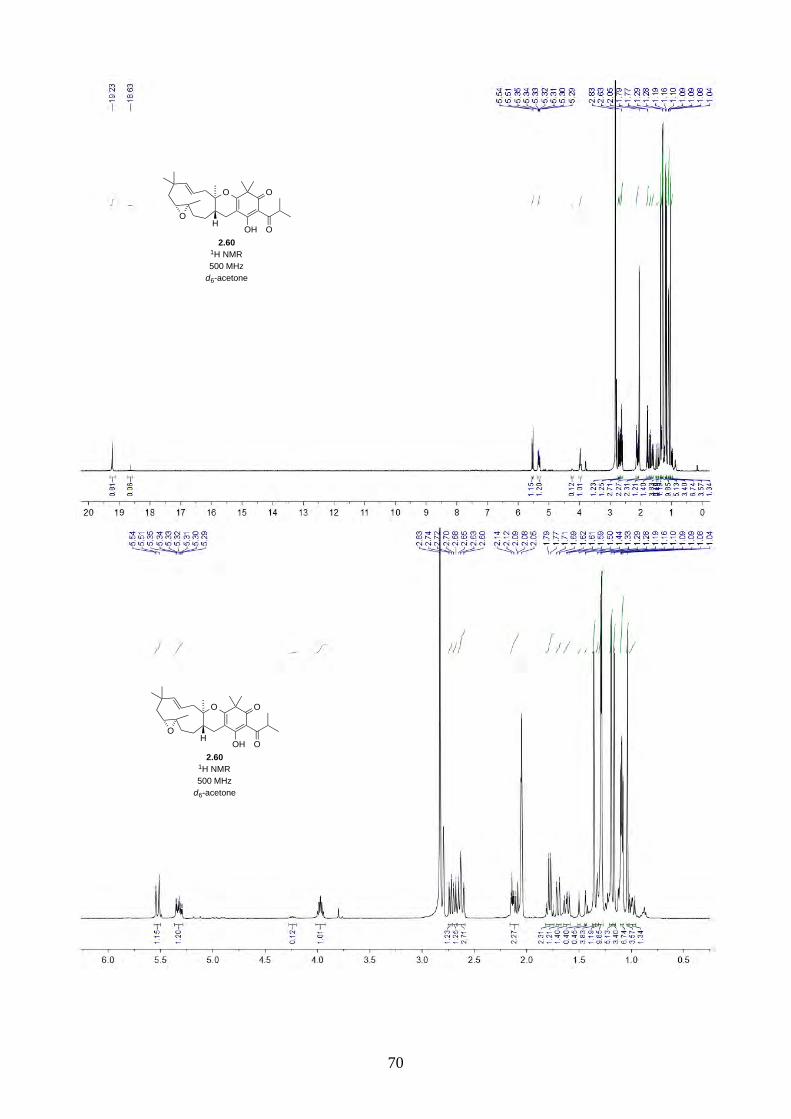
70
O
OH
O
OH
O
1H NMR500 MHz
d6-acetone
2.60
O
OH
O
OH
O
1H NMR500 MHz
d6-acetone
2.60
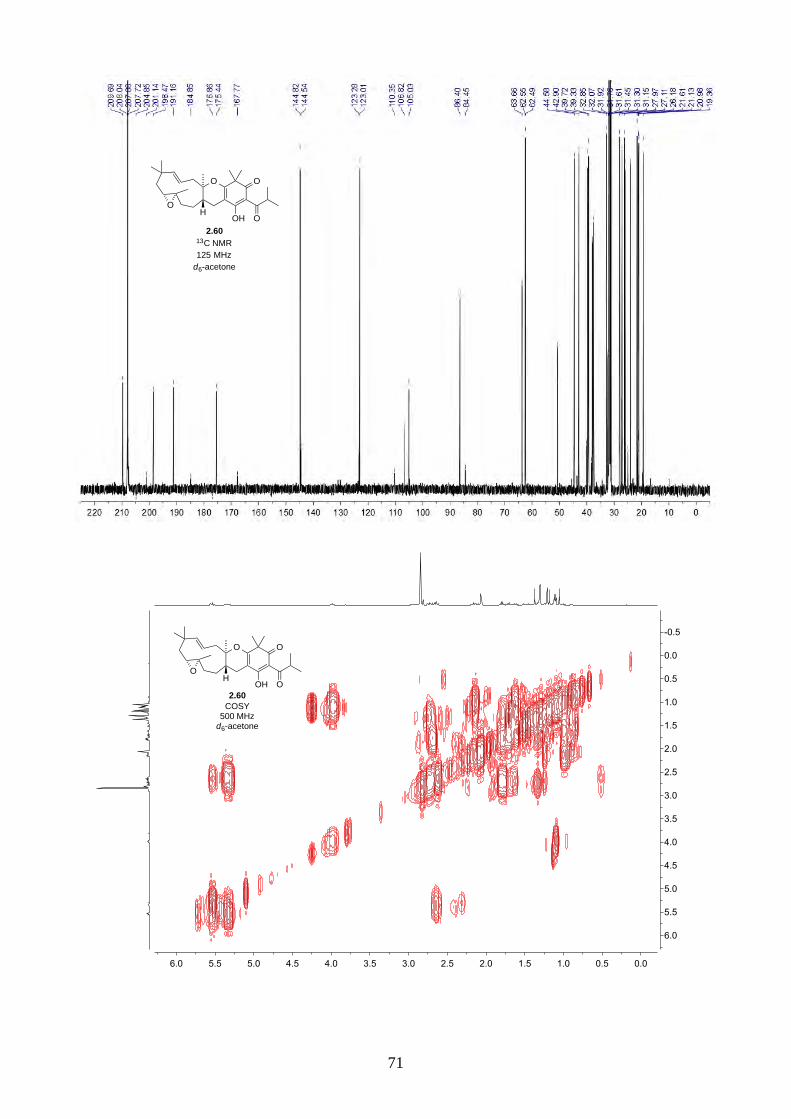
71
O
OH
O
OH
O
13C NMR125 MHz
d6-acetone
2.60
O
OH
O
OH
O
COSY500 MHz
d6-acetone
2.60
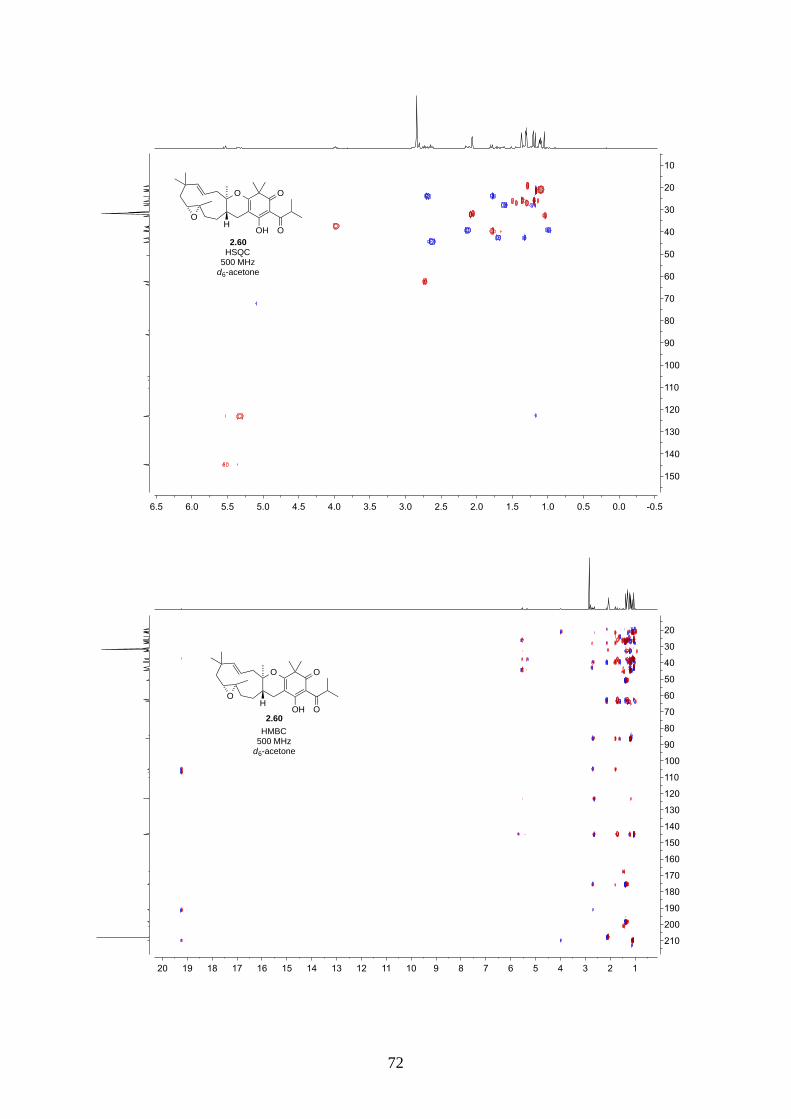
72
O
OH
O
OH
O
HSQC500 MHz
d6-acetone
2.60
O
OH
O
OH
O
HMBC500 MHz
d6-acetone
2.60
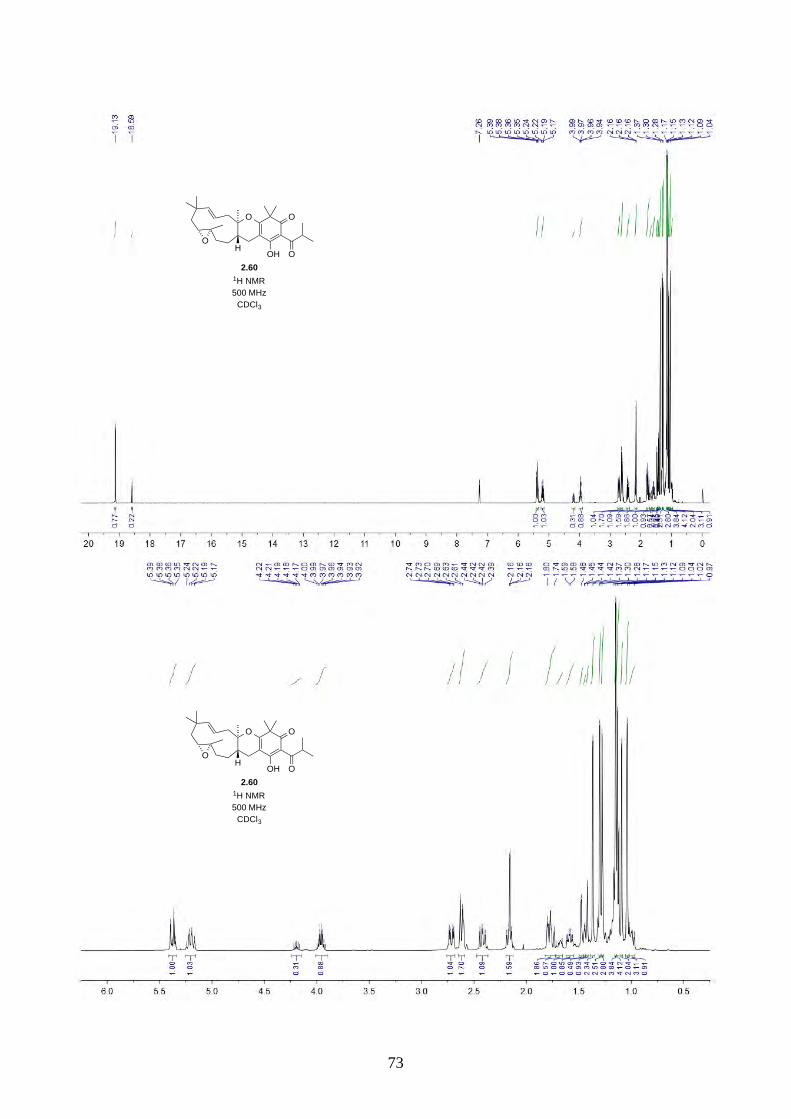
73
O
OH
O
OH
O
1H NMR500 MHz
CDCl3
2.60
O
OH
O
OH
O
1H NMR500 MHz
CDCl3
2.60
74
O
OH
O
OH
O
13C NMR125 MHz
CDCl3
2.60
75
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C1H NMR500 MHz
CDCl3
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C1H NMR500 MHz
CDCl3
76
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C13C NMR125 MHz
CDCl3
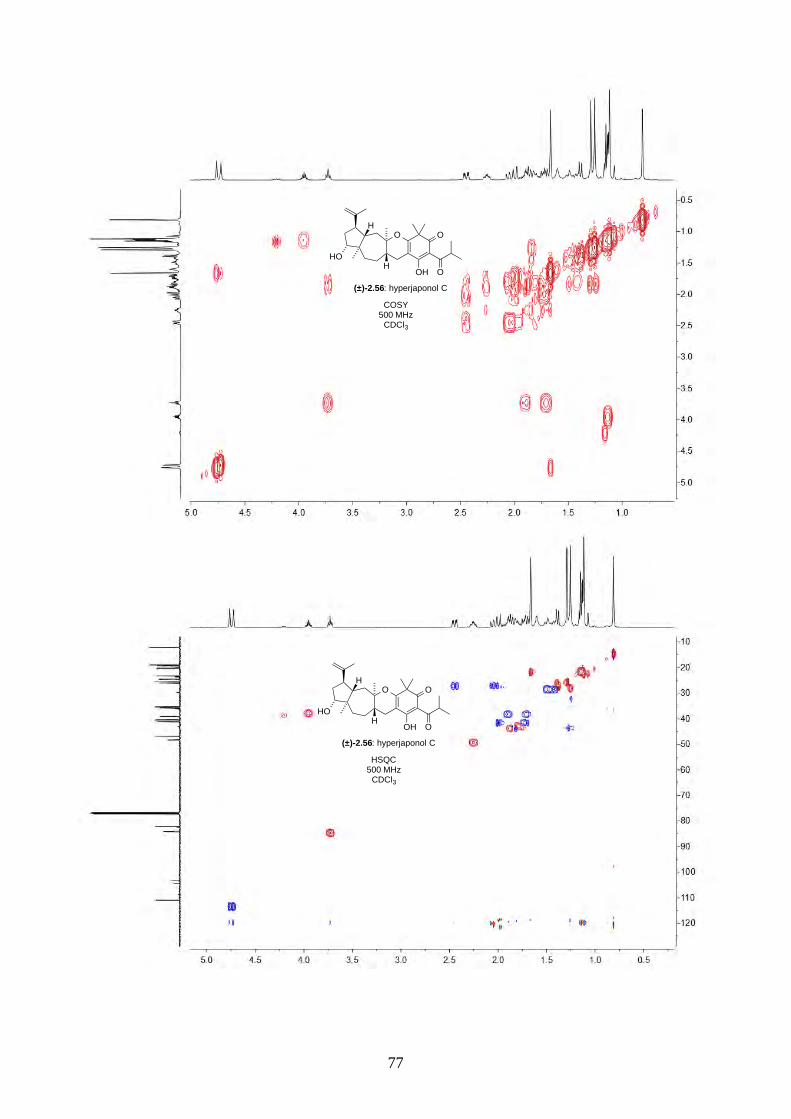
77
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
COSY500 MHz
CDCl3
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
HSQC500 MHz
CDCl3
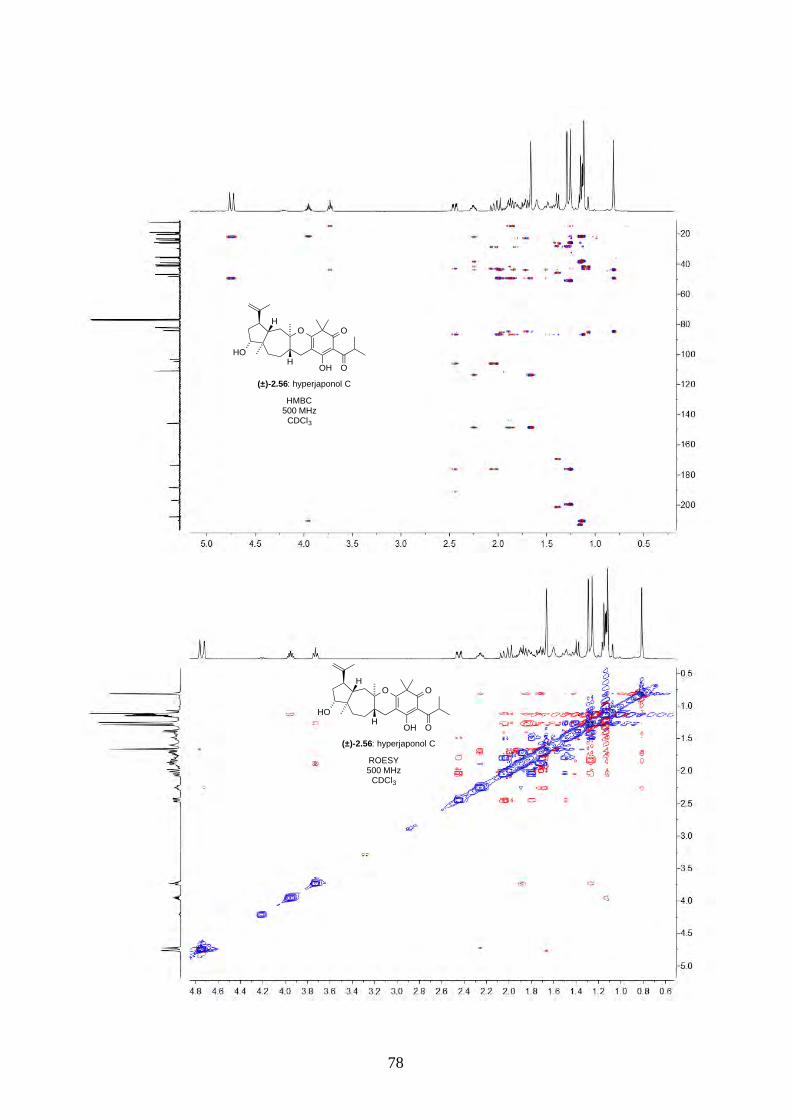
78
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
HMBC500 MHz
CDCl3
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
ROESY500 MHz
CDCl3
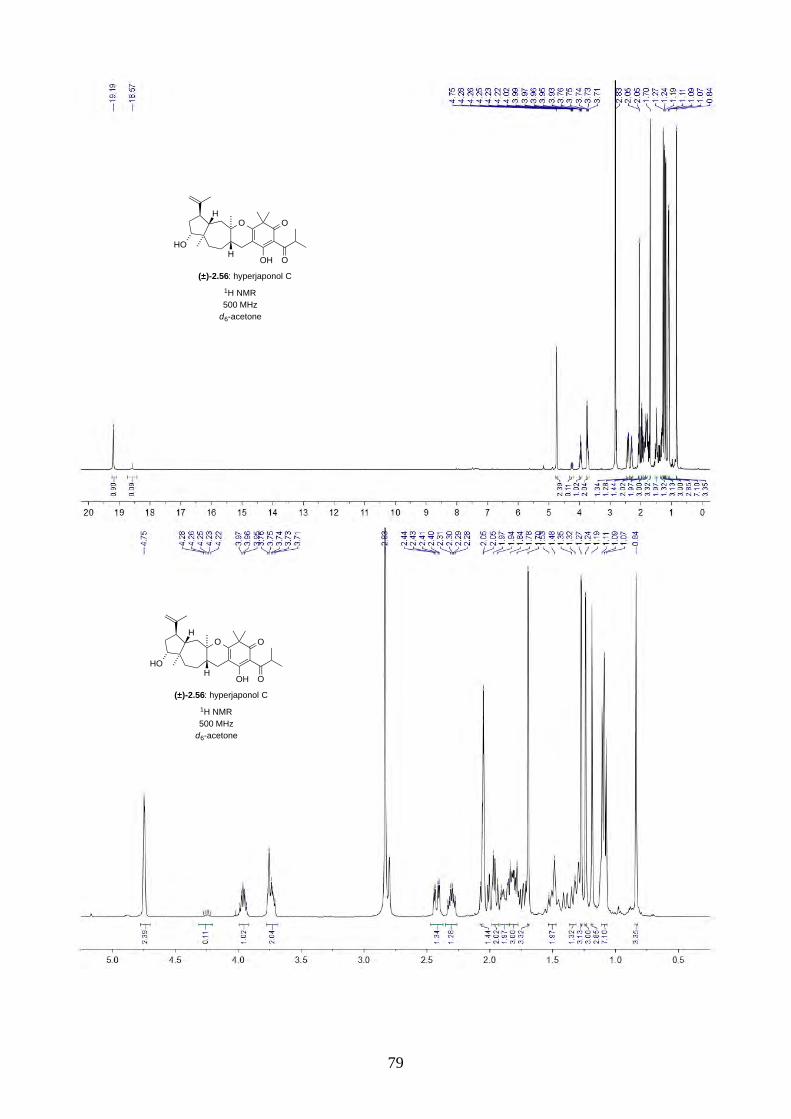
79
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C1H NMR500 MHz
d6-acetone
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C1H NMR500 MHz
d6-acetone
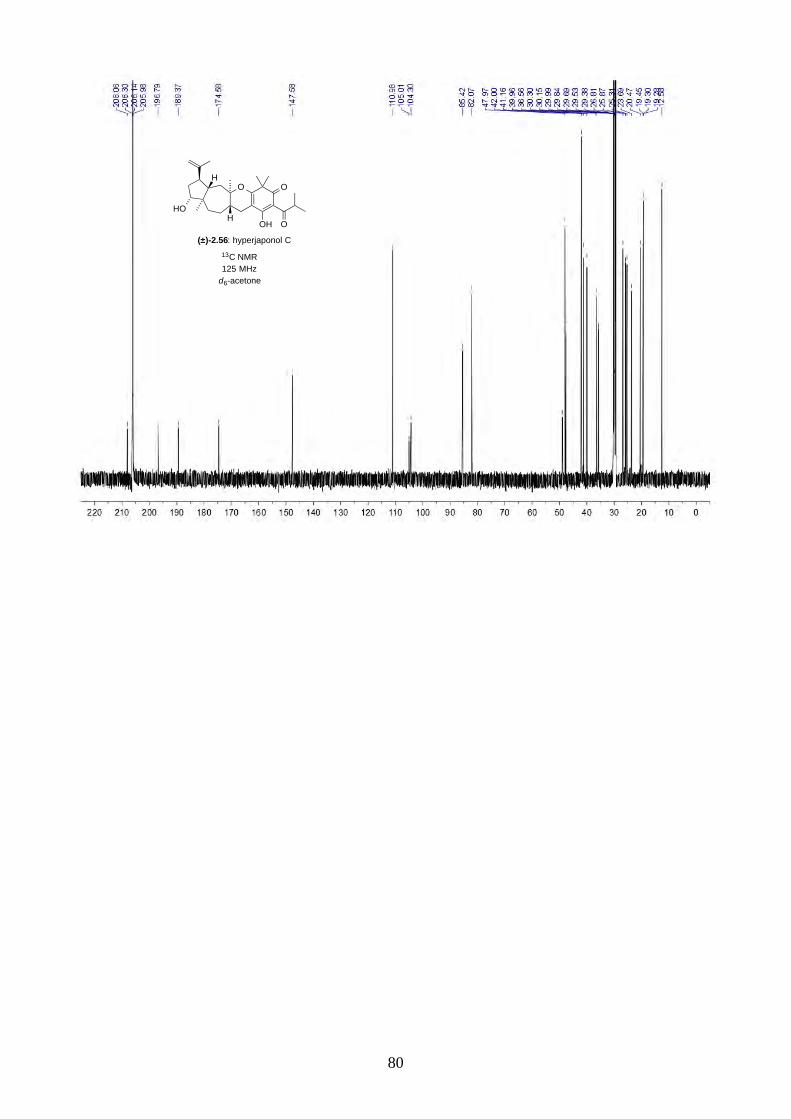
80
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C13C NMR125 MHz
d6-acetone
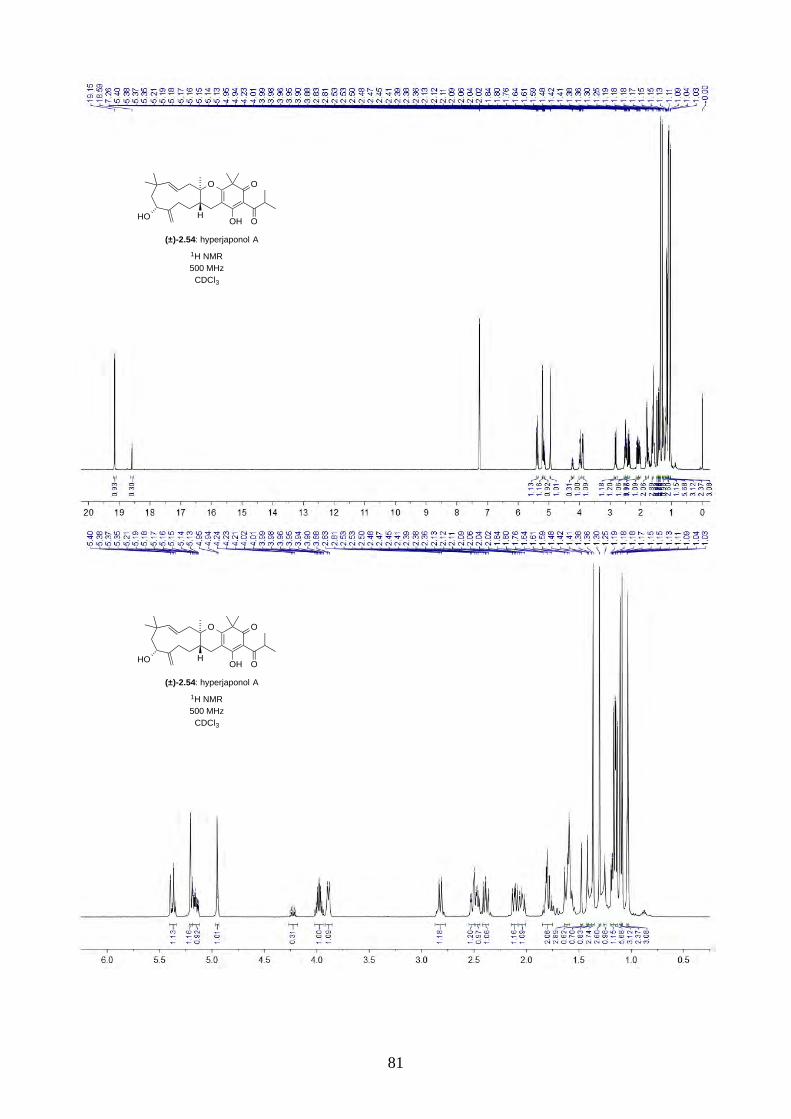
81
O
OH
O
OHHO
(±)-2.54: hyperjaponol A1H NMR500 MHz
CDCl3
O
OH
O
OHHO
(±)-2.54: hyperjaponol A1H NMR500 MHz
CDCl3
82
O
OH
O
OHHO
(±)-2.54: hyperjaponol A13C NMR125 MHz
CDCl3
83
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
COSY500 MHz
CDCl3
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
HSQC500 MHz
CDCl3
84
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
HMBC500 MHz
CDCl3
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
ROESY500 MHz
CDCl3
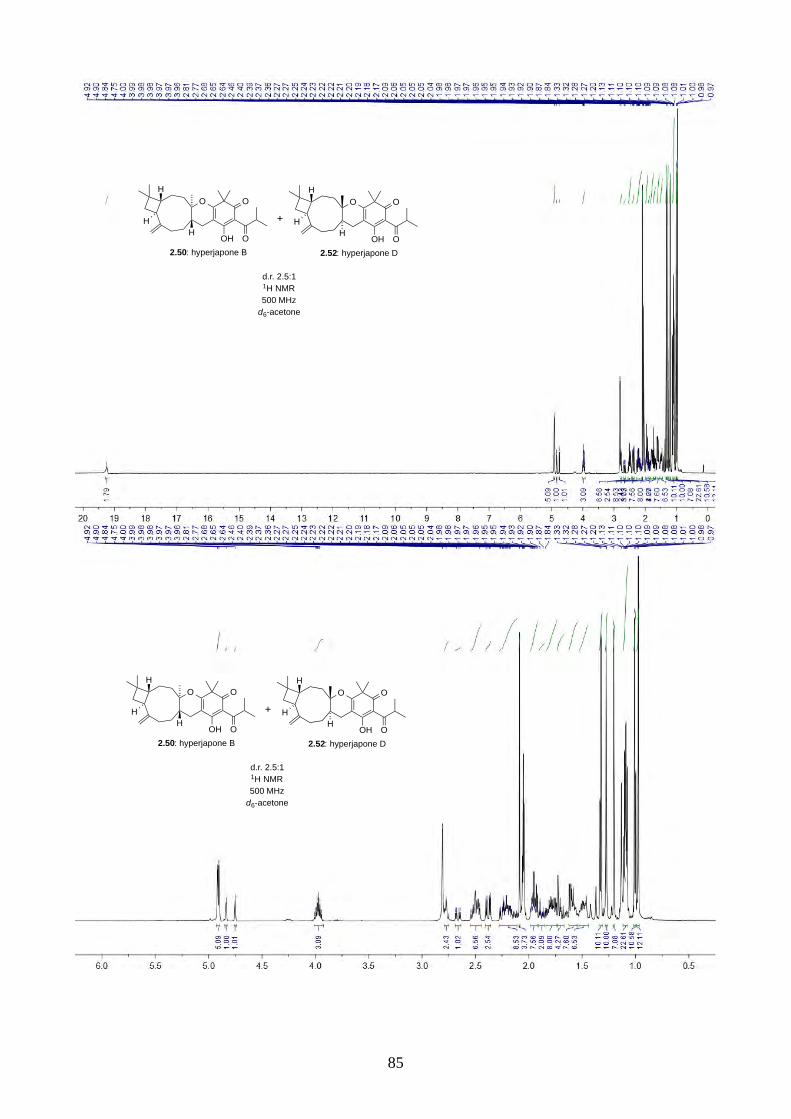
85
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.50: hyperjapone B 2.52: hyperjapone D
d.r. 2.5:11H NMR500 MHz
d6-acetone
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.50: hyperjapone B 2.52: hyperjapone D
d.r. 2.5:11H NMR500 MHz
d6-acetone
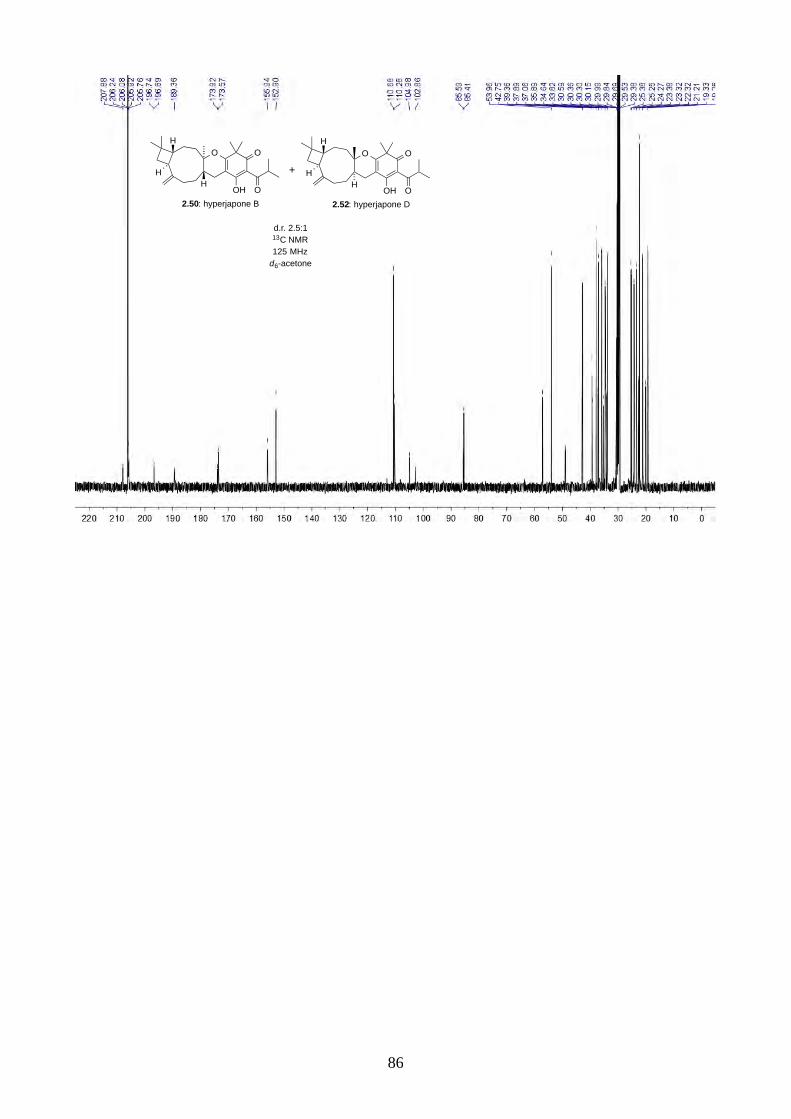
86
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.50: hyperjapone B 2.52: hyperjapone D
d.r. 2.5:113C NMR125 MHz
d6-acetone
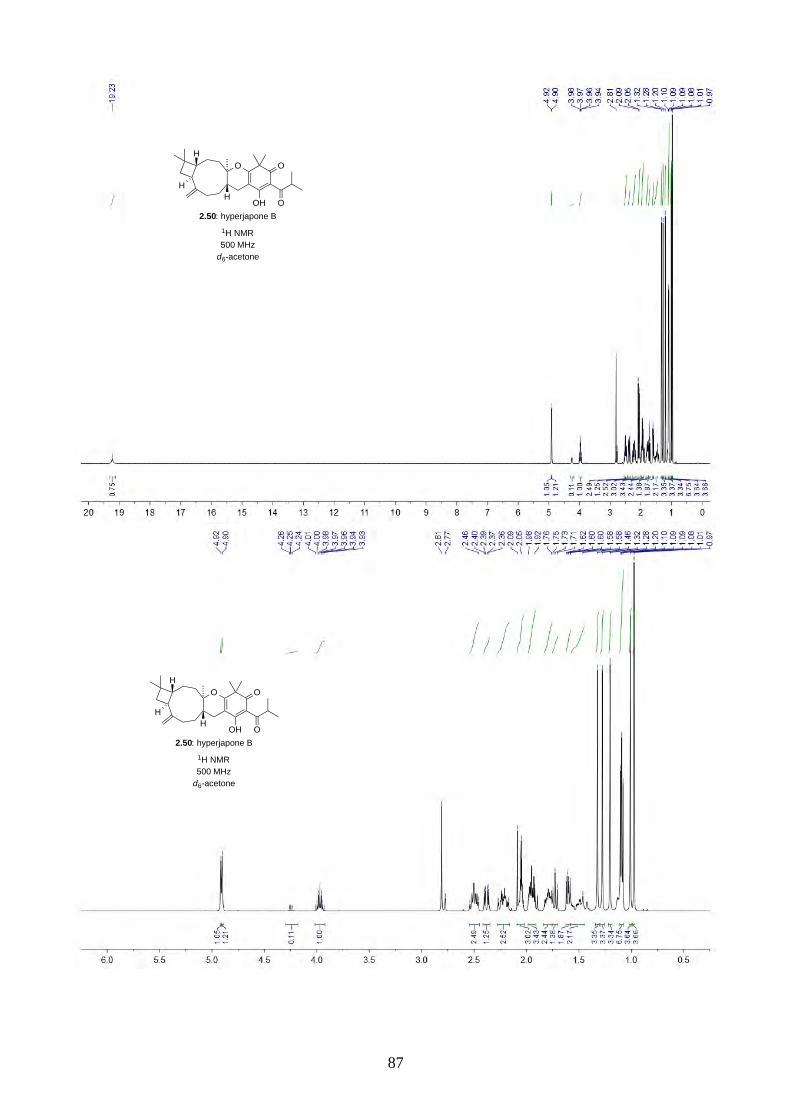
87
O
OH
O
OH
H
H
2.50: hyperjapone B1H NMR500 MHz
d6-acetone
O
OH
O
OH
H
H
2.50: hyperjapone B1H NMR500 MHz
d6-acetone
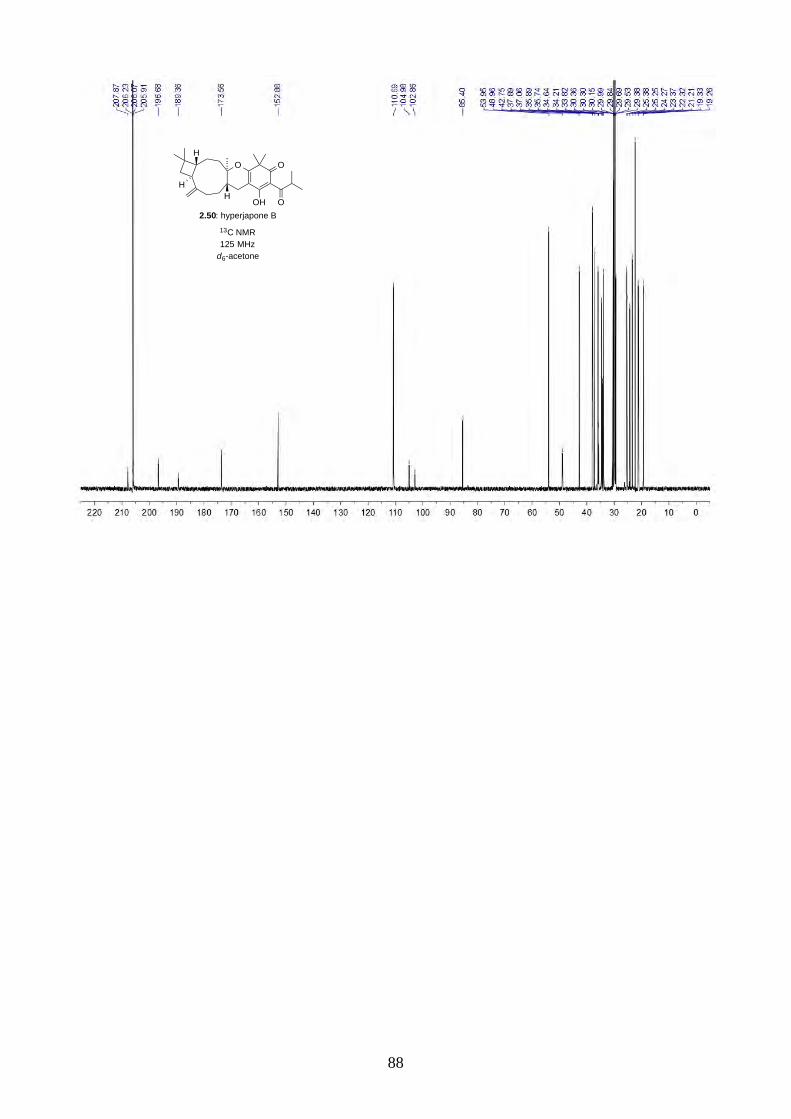
88
O
OH
O
OH
H
H
2.50: hyperjapone B13C NMR125 MHz
d6-acetone
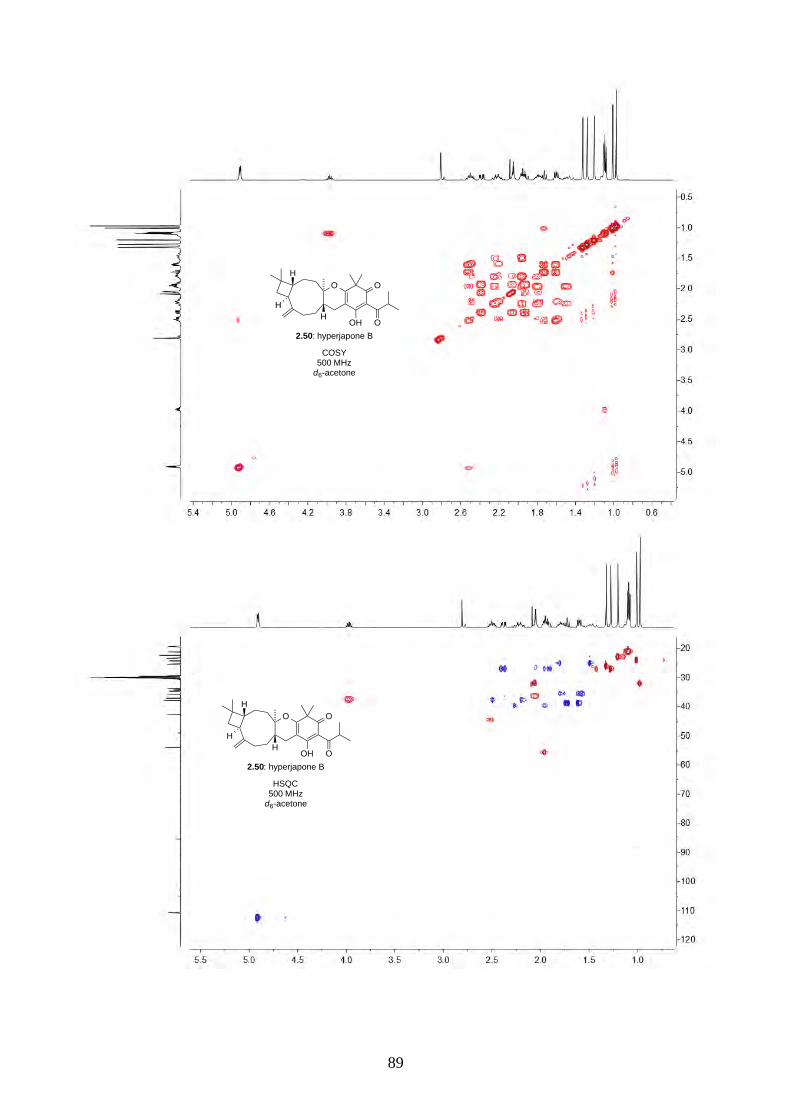
89
O
OH
O
OH
H
H
2.50: hyperjapone B
COSY500 MHz
d6-acetone
O
OH
O
OH
H
H
2.50: hyperjapone B
HSQC500 MHz
d6-acetone
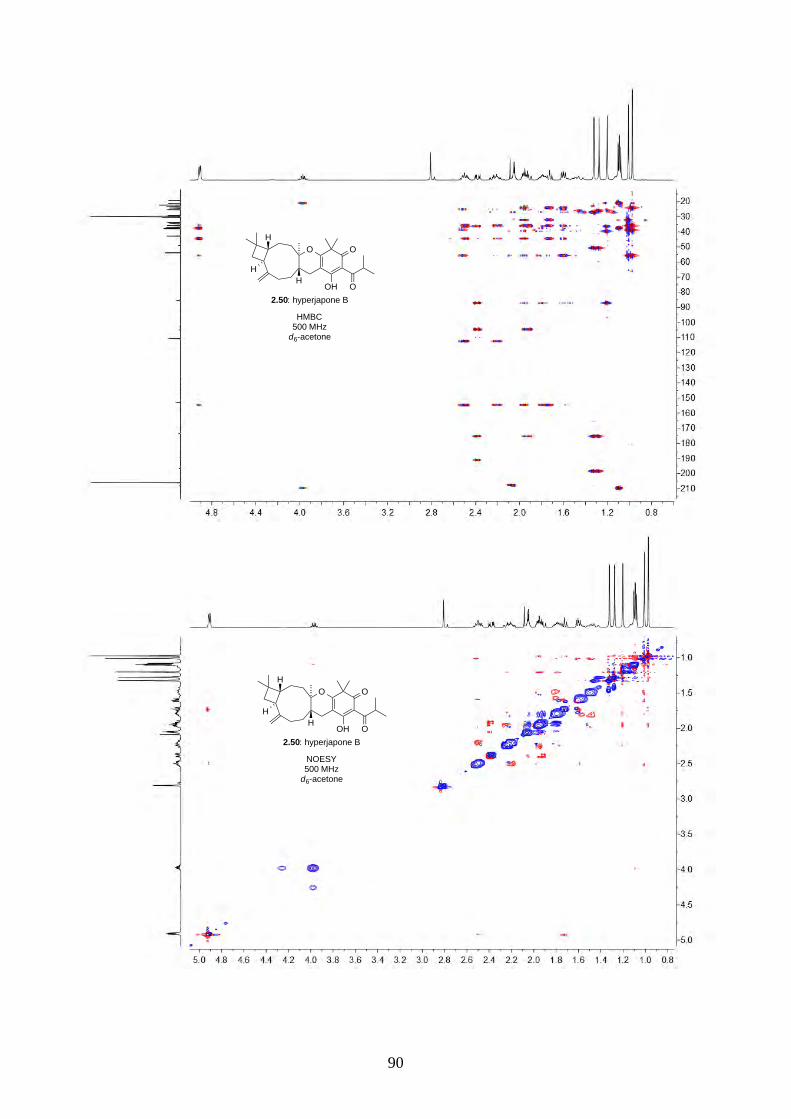
90
O
OH
O
OH
H
H
2.50: hyperjapone B
HMBC500 MHz
d6-acetone
O
OH
O
OH
H
H
2.50: hyperjapone B
NOESY500 MHz
d6-acetone
91
HO OH
OH O
H
1H NMR500 MHzCD3OD
2.83
HO OH
OH O
H
13C NMR125 MHzCD3OD
2.83
92
HO O
OH O
H
(+)-2.84: norisoleptospermone1H NMR500 MHzd6-DMSO
HO O
OH O
H
(+)-2.84: norisoleptospermone1H NMR500 MHzd6-DMSO
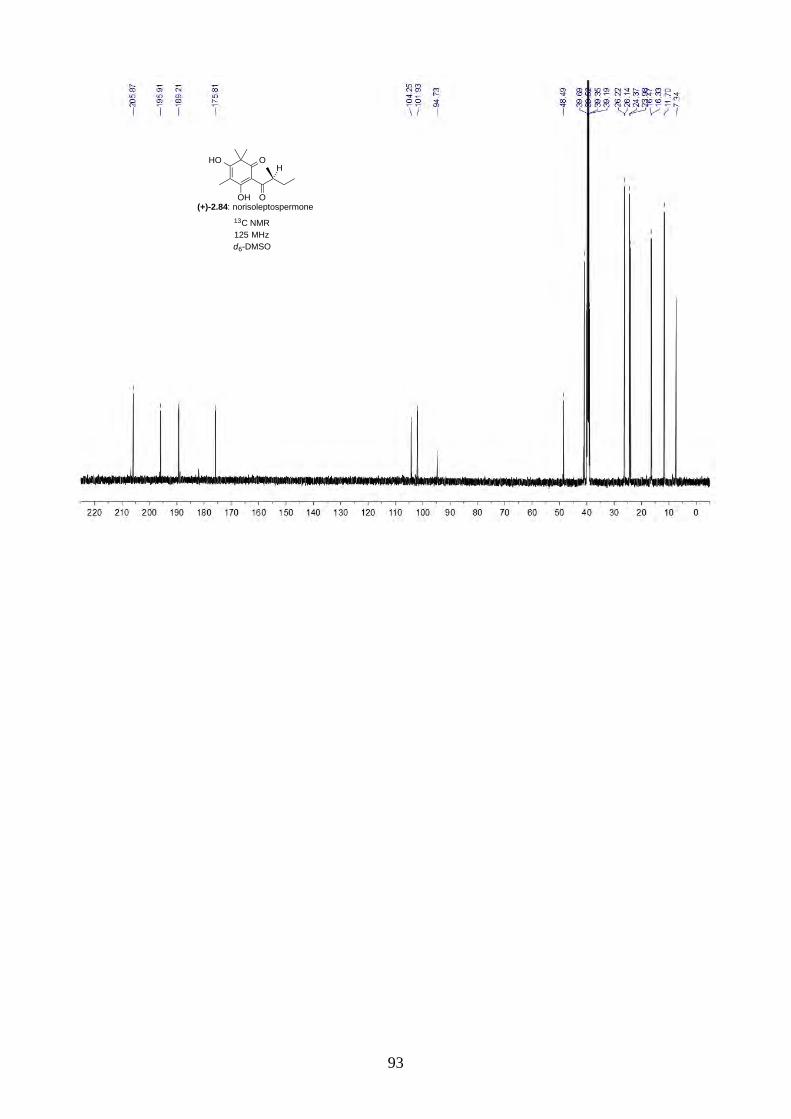
93
HO O
OH O
H
(+)-2.84: norisoleptospermone13C NMR125 MHzd6-DMSO
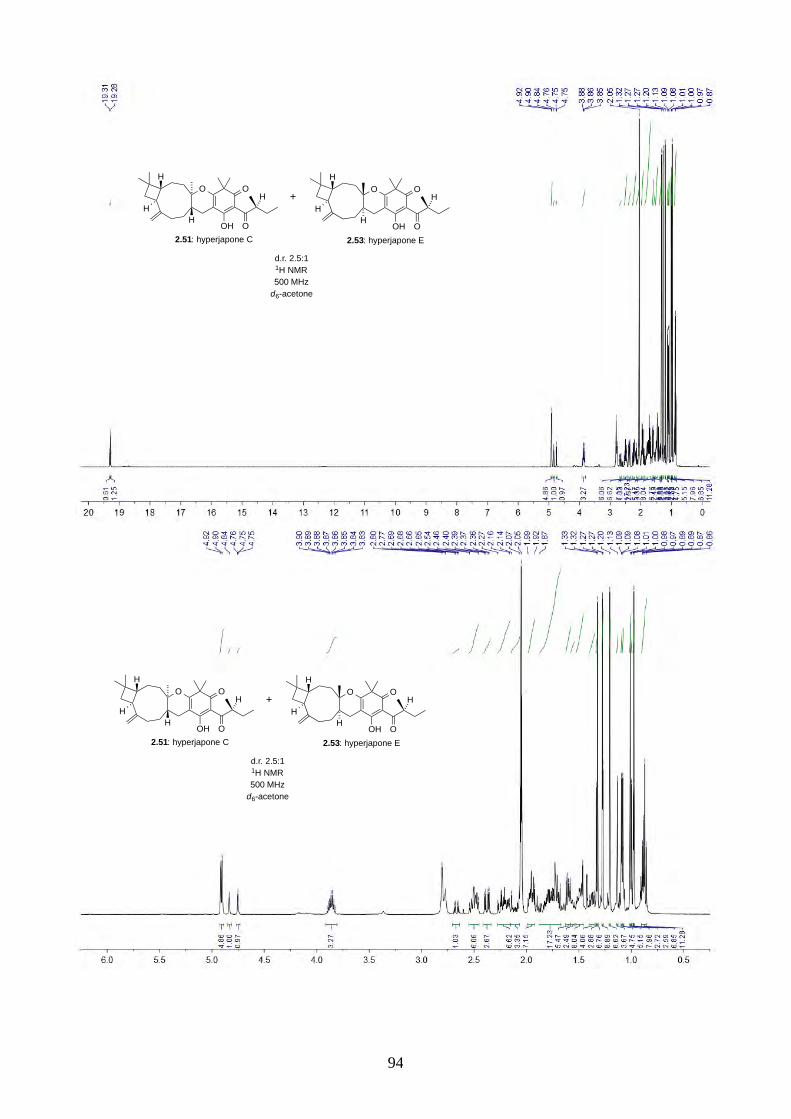
94
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.51: hyperjapone C 2.53: hyperjapone E
H H
d.r. 2.5:11H NMR500 MHz
d6-acetone
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.51: hyperjapone C 2.53: hyperjapone E
H H
d.r. 2.5:11H NMR500 MHz
d6-acetone
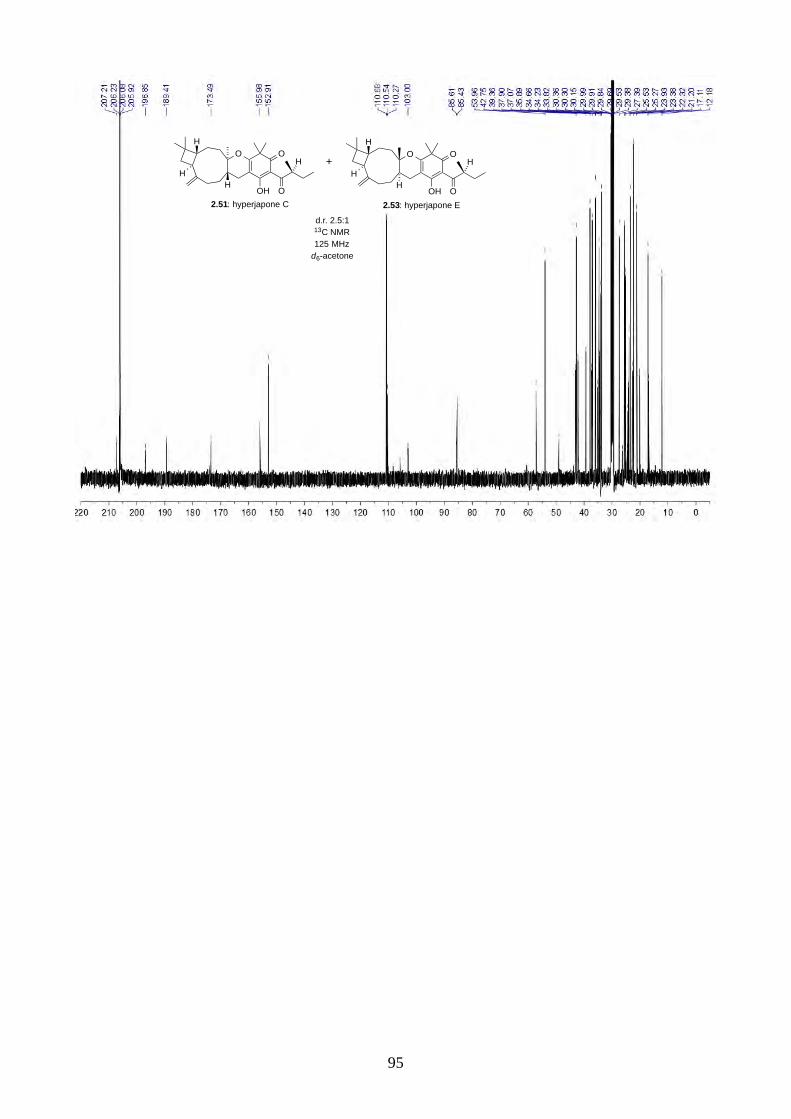
95
d.r. 2.5:113C NMR125 MHz
d6-acetone
O
OH
O
OH
H
H
O
OH
O
OH
H
H+
2.51: hyperjapone C 2.53: hyperjapone E
H H
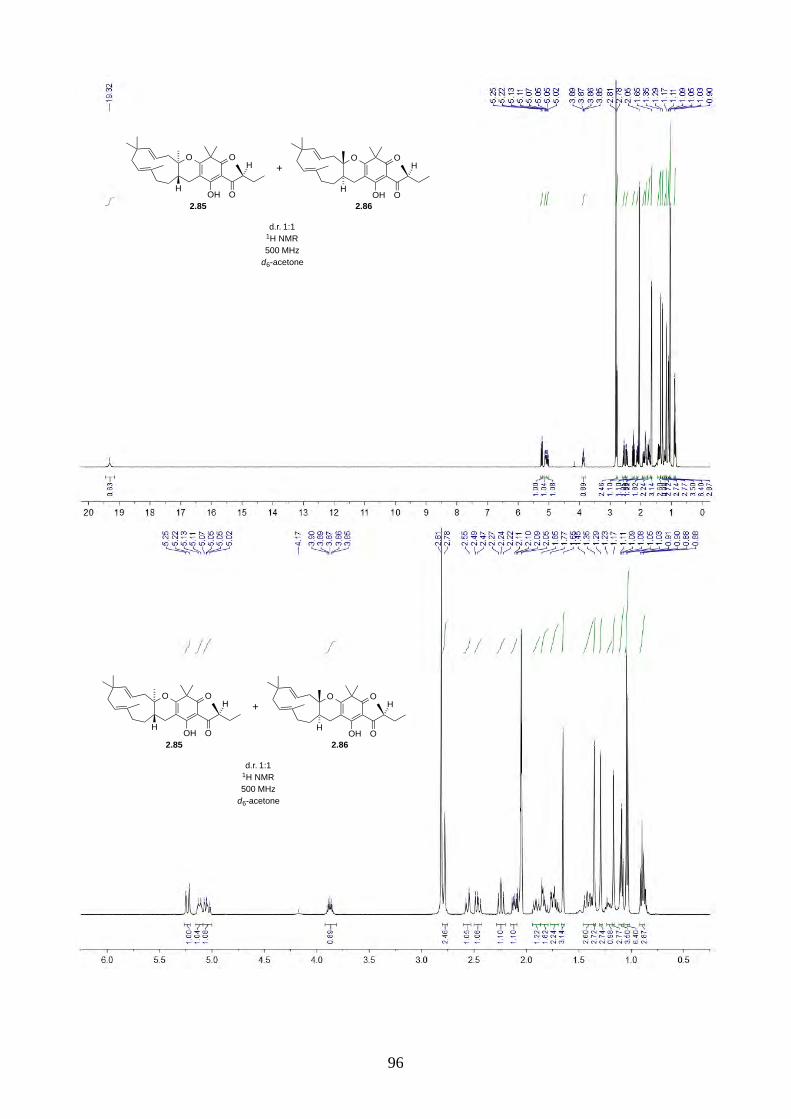
96
2.85 2.86
+O
OH
O
OH
O
OH
O
OH
HH
d.r. 1:11H NMR500 MHz
d6-acetone
2.85 2.86
+O
OH
O
OH
O
OH
O
OH
HH
d.r. 1:11H NMR500 MHz
d6-acetone
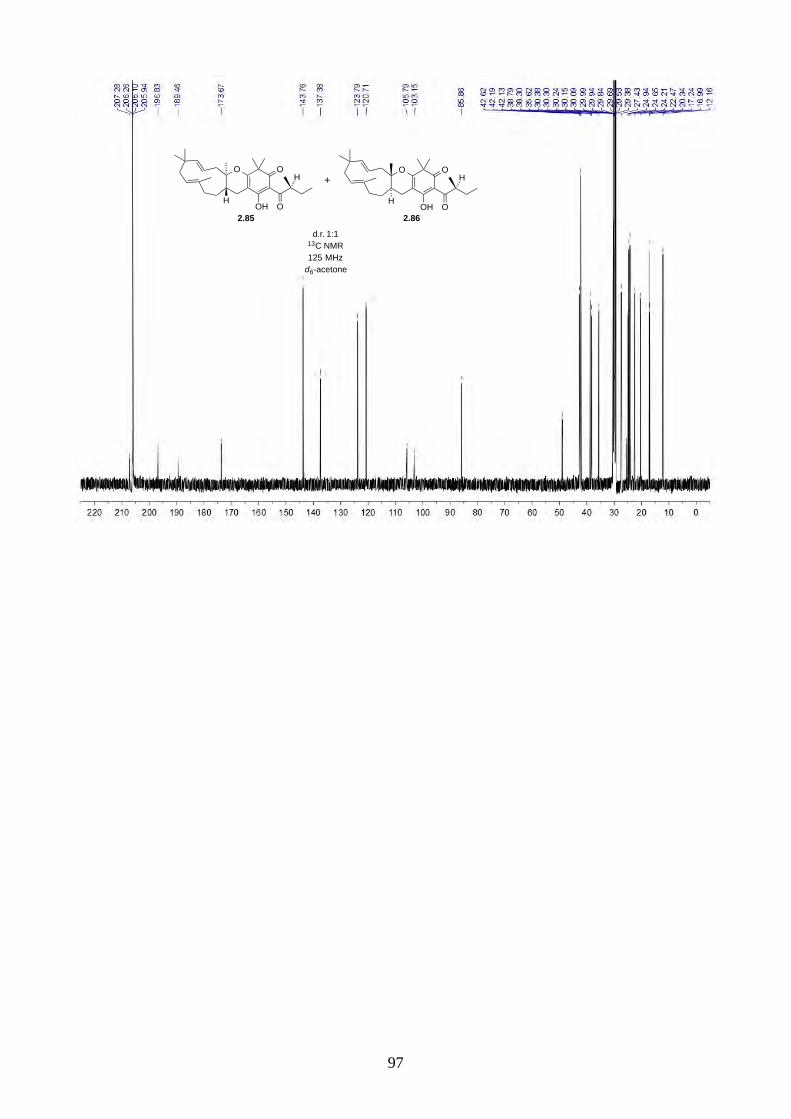
97
2.85 2.86
+O
OH
O
OH
O
OH
O
OH
HH
d.r. 1:113C NMR125 MHz
d6-acetone
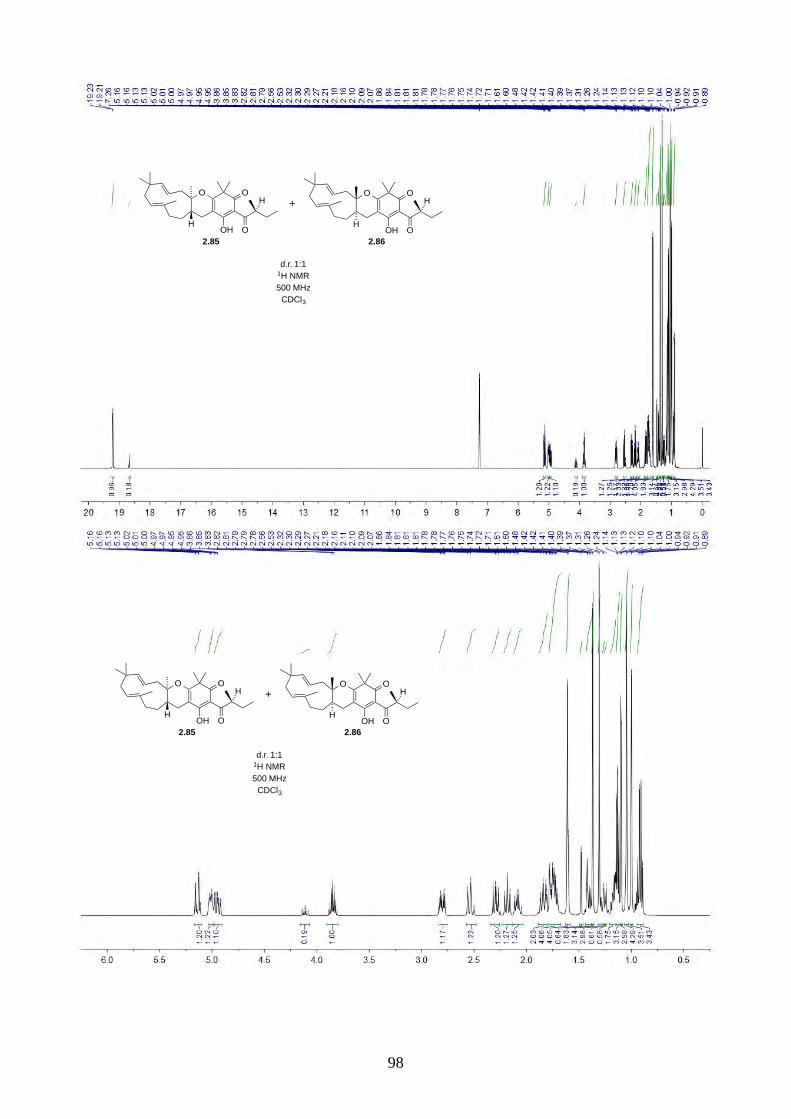
98
2.85 2.86
+O
OH
O
OH
O
OH
O
OH
HH
d.r. 1:11H NMR500 MHz
CDCl3
2.85 2.86
+O
OH
O
OH
O
OH
O
OH
HH
d.r. 1:11H NMR500 MHz
CDCl3
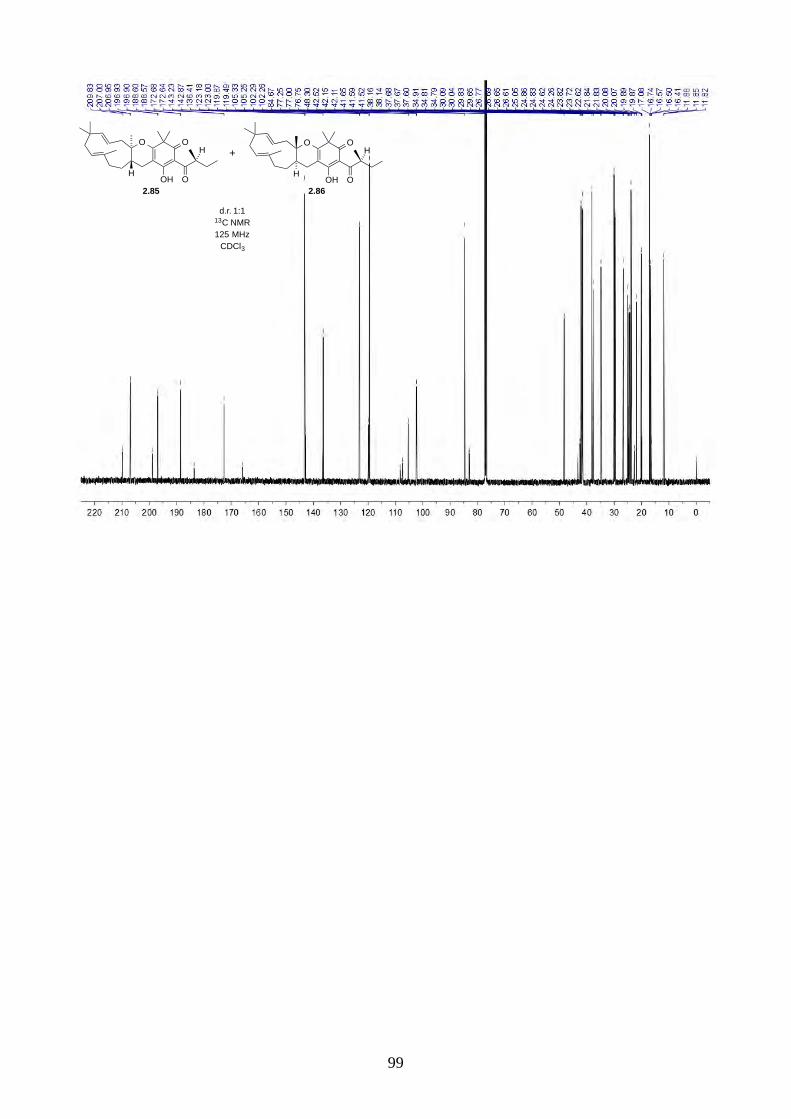
99
2.85 2.86
+O
OH
O
OH
O
OH
O
OH
HH
d.r. 1:113C NMR125 MHz
CDCl3
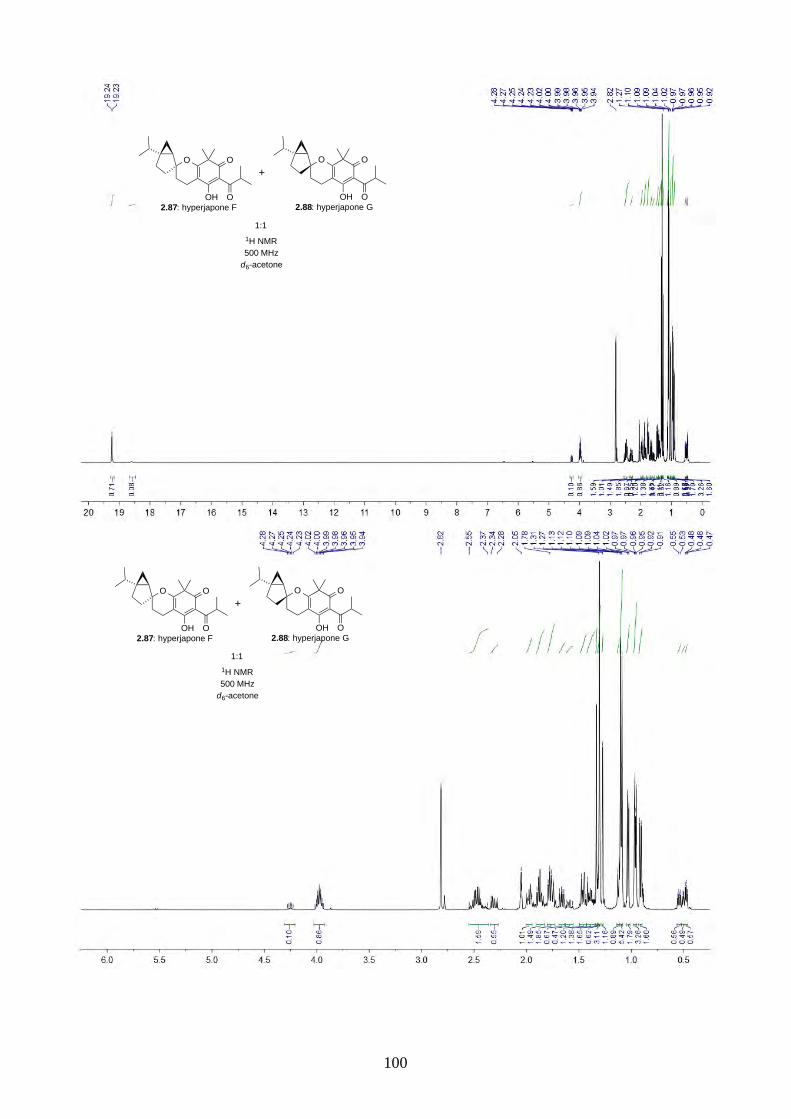
100
O
OH
O
O2.87: hyperjapone F
O
OH
O
O2.88: hyperjapone G
+
1:11H NMR500 MHz
d6-acetone
O
OH
O
O2.87: hyperjapone F
O
OH
O
O2.88: hyperjapone G
+
1:11H NMR500 MHz
d6-acetone
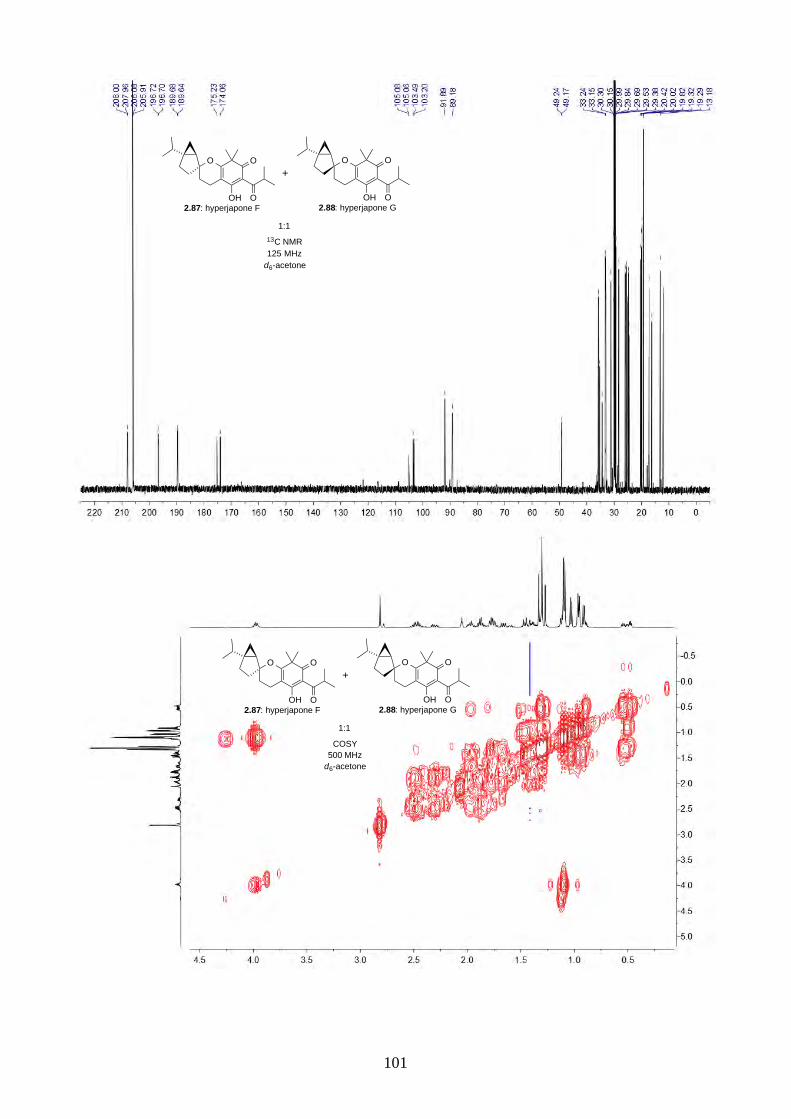
101
O
OH
O
O2.87: hyperjapone F
O
OH
O
O2.88: hyperjapone G
+
1:113C NMR125 MHz
d6-acetone
O
OH
O
O2.87: hyperjapone F
O
OH
O
O2.88: hyperjapone G
+
1:1
COSY500 MHz
d6-acetone
102
O
OH
O
O2.87: hyperjapone F
O
OH
O
O2.88: hyperjapone G
+
1:1
HSQC500 MHz
d6-acetone
O
OH
O
O2.87: hyperjapone F
O
OH
O
O2.88: hyperjapone G
+
1:1
HMBC500 MHz
d6-acetone
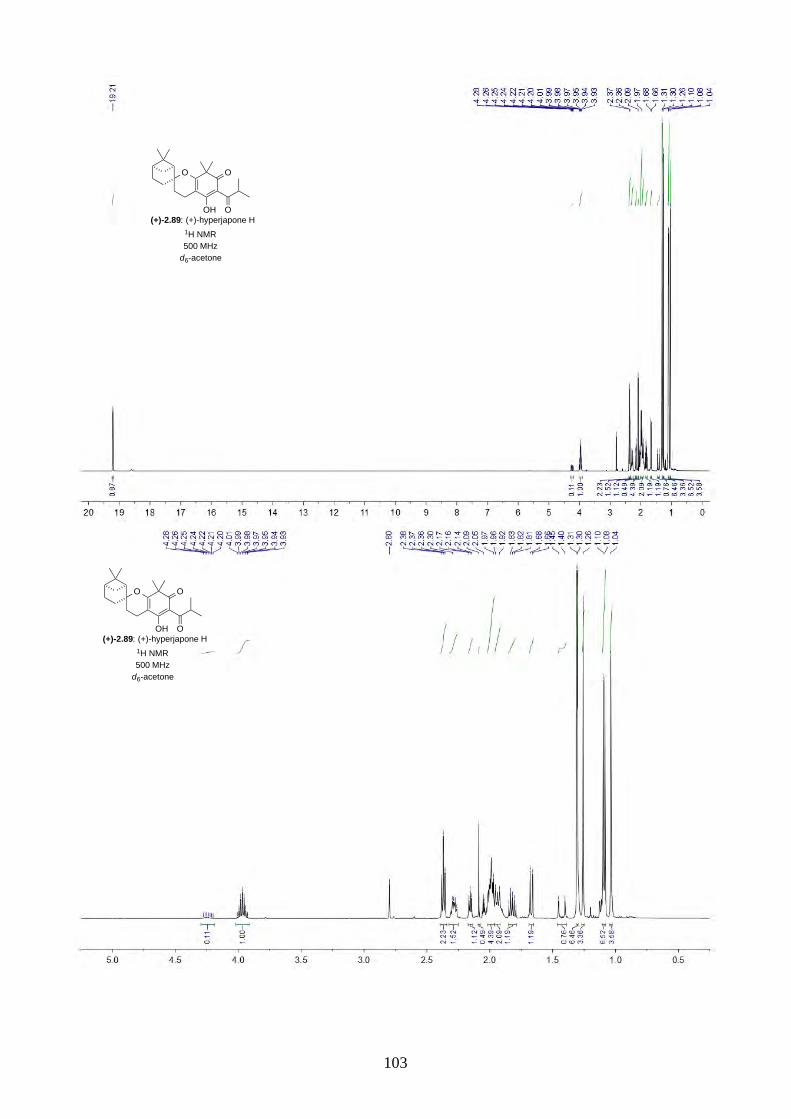
103
O
OH
O
O(+)-2.89: (+)-hyperjapone H
1H NMR500 MHz
d6-acetone
O
OH
O
O(+)-2.89: (+)-hyperjapone H
1H NMR500 MHz
d6-acetone
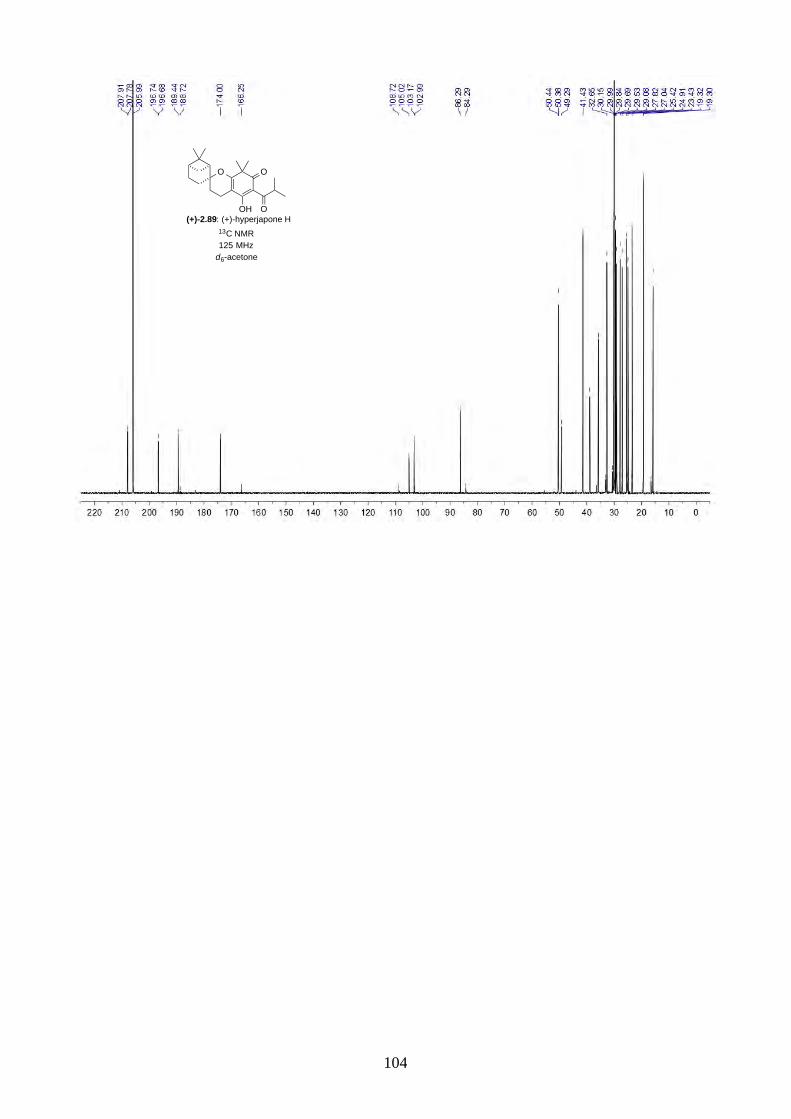
104
O
OH
O
O(+)-2.89: (+)-hyperjapone H
13C NMR125 MHz
d6-acetone
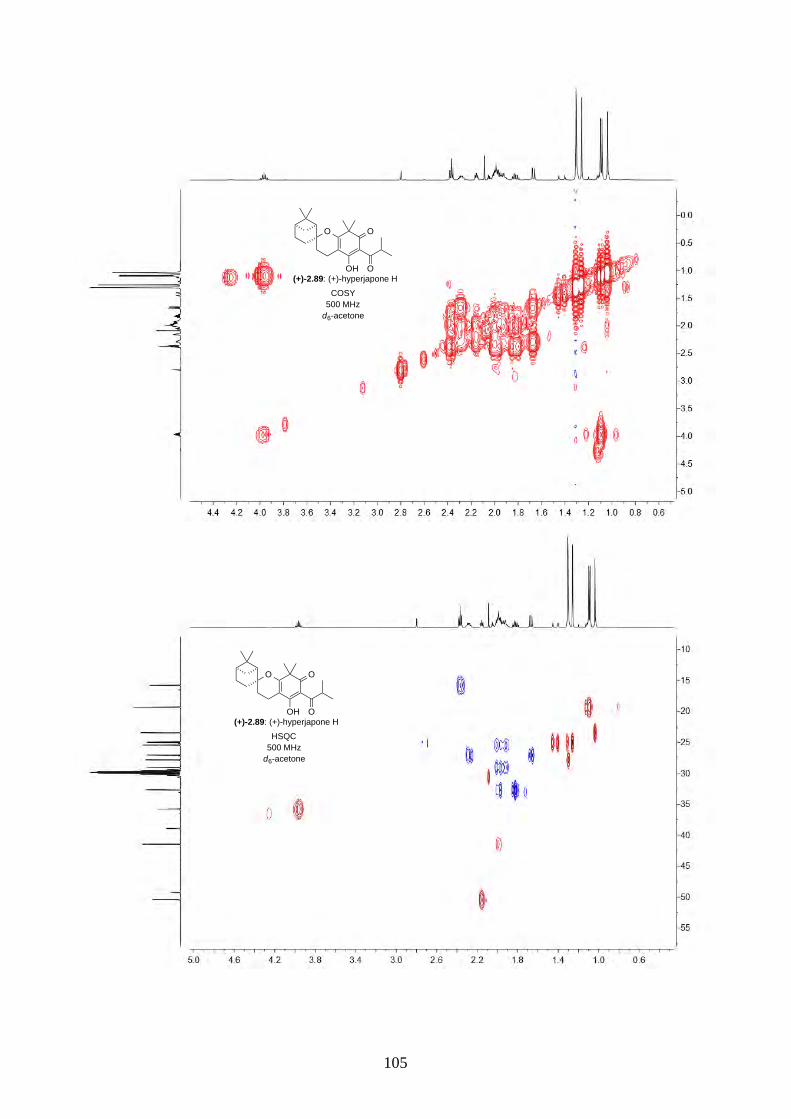
105
O
OH
O
O(+)-2.89: (+)-hyperjapone H
COSY500 MHz
d6-acetone
O
OH
O
O(+)-2.89: (+)-hyperjapone H
HSQC500 MHz
d6-acetone
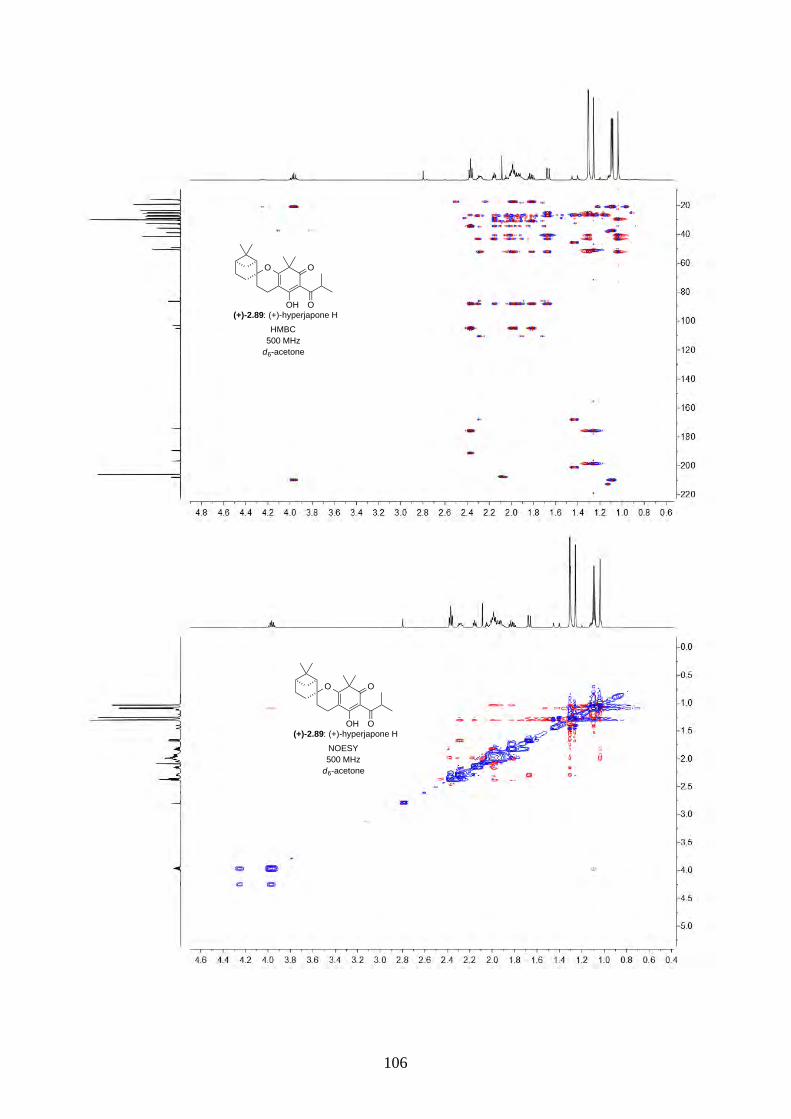
106
O
OH
O
O(+)-2.89: (+)-hyperjapone H
HMBC500 MHz
d6-acetone
O
OH
O
O(+)-2.89: (+)-hyperjapone H
NOESY500 MHz
d6-acetone
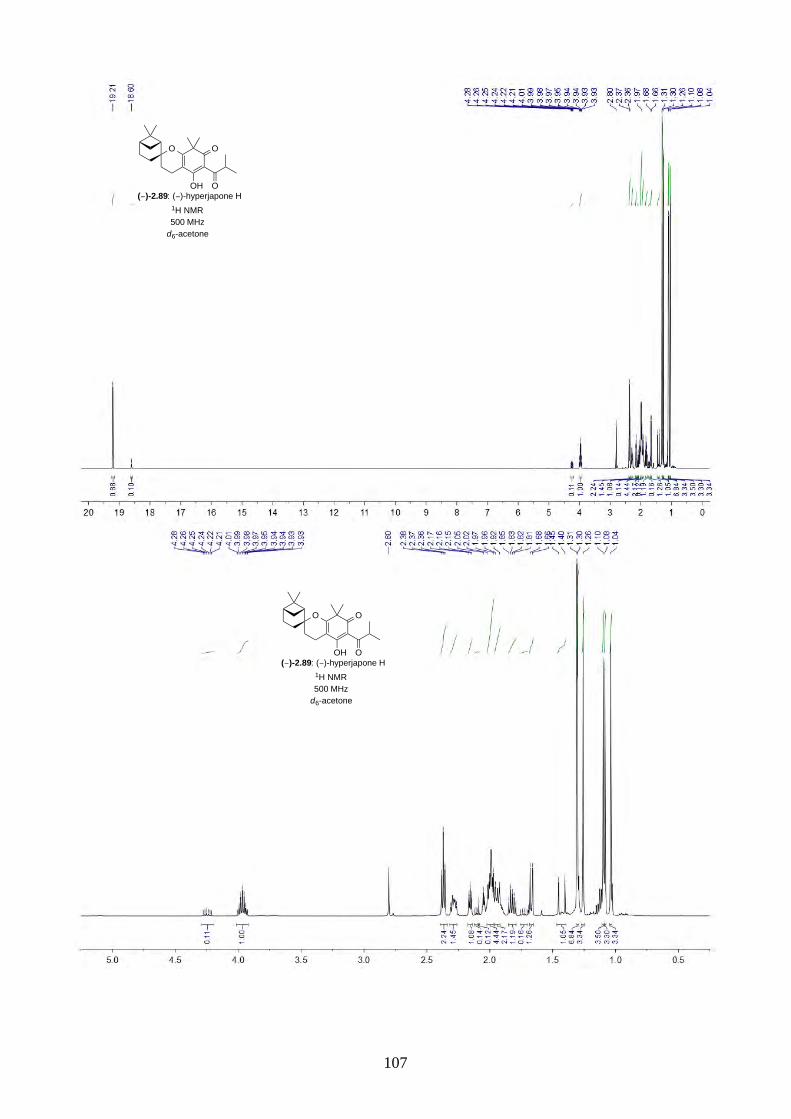
107
O
OH
O
O(−)-2.89: (−)-hyperjapone H
1H NMR500 MHz
d6-acetone
O
OH
O
O(−)-2.89: (−)-hyperjapone H
1H NMR500 MHz
d6-acetone
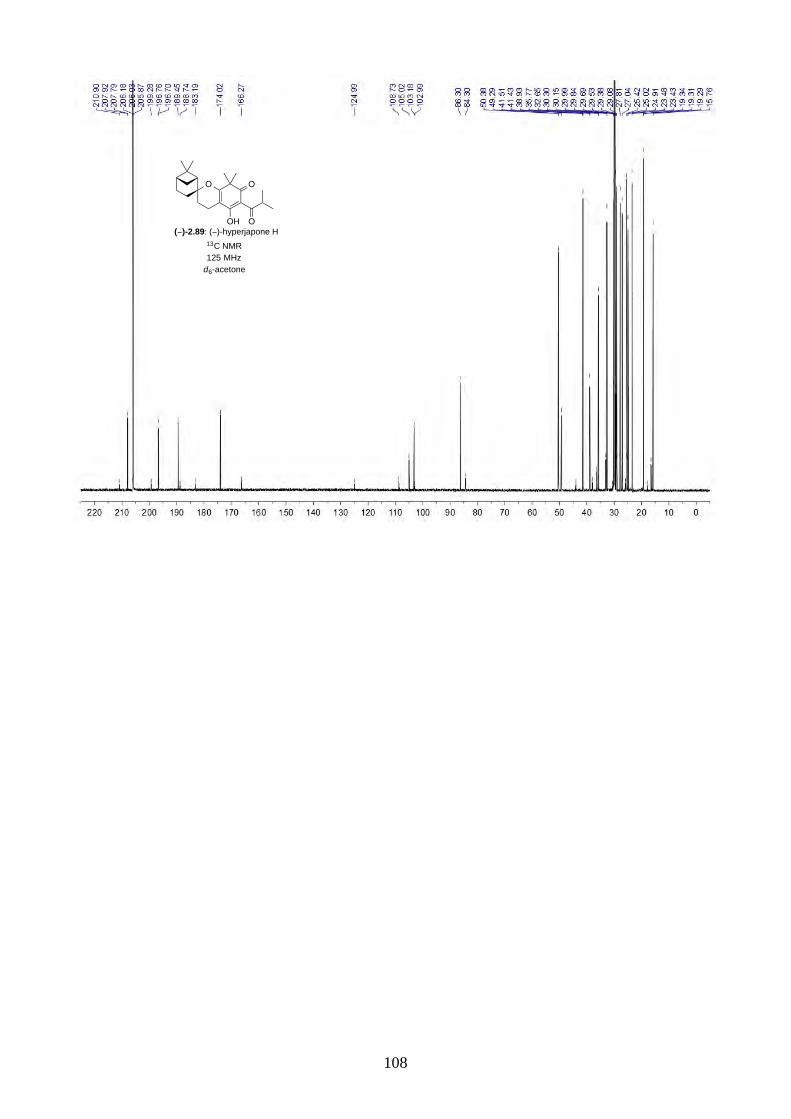
108
O
OH
O
O(−)-2.89: (−)-hyperjapone H
13C NMR125 MHz
d6-acetone
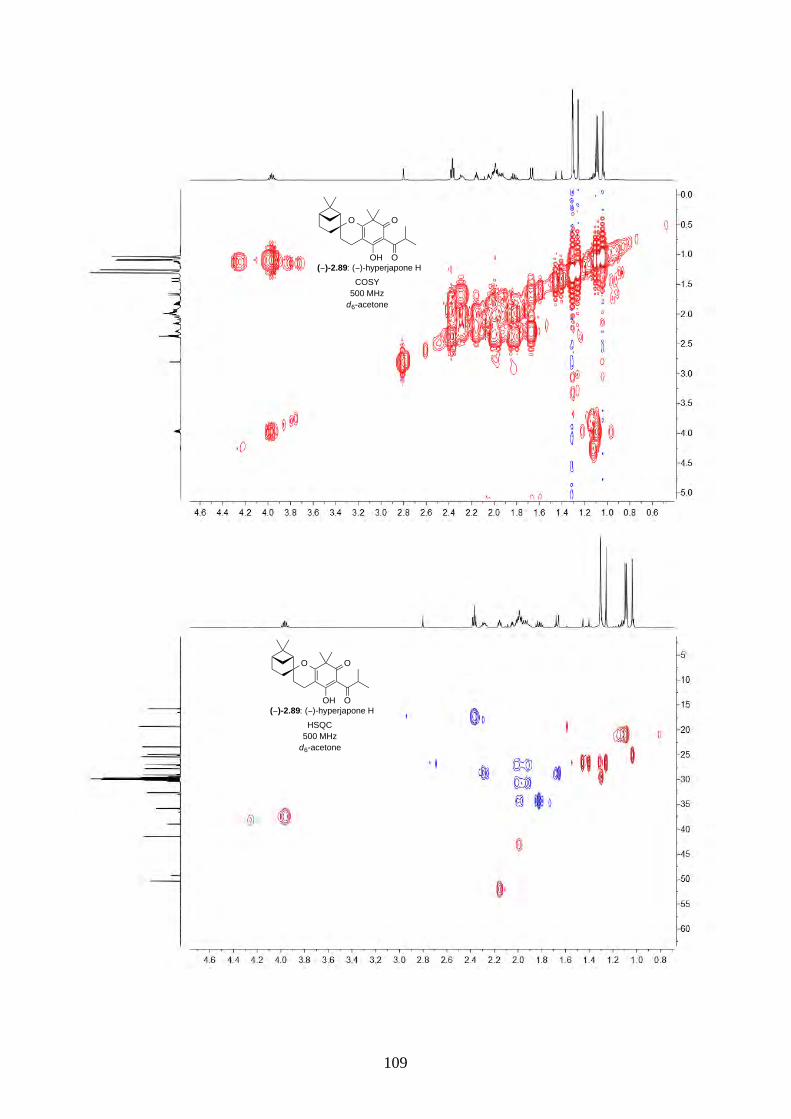
109
O
OH
O
O(−)-2.89: (−)-hyperjapone H
COSY500 MHz
d6-acetone
O
OH
O
O(−)-2.89: (−)-hyperjapone H
HSQC500 MHz
d6-acetone
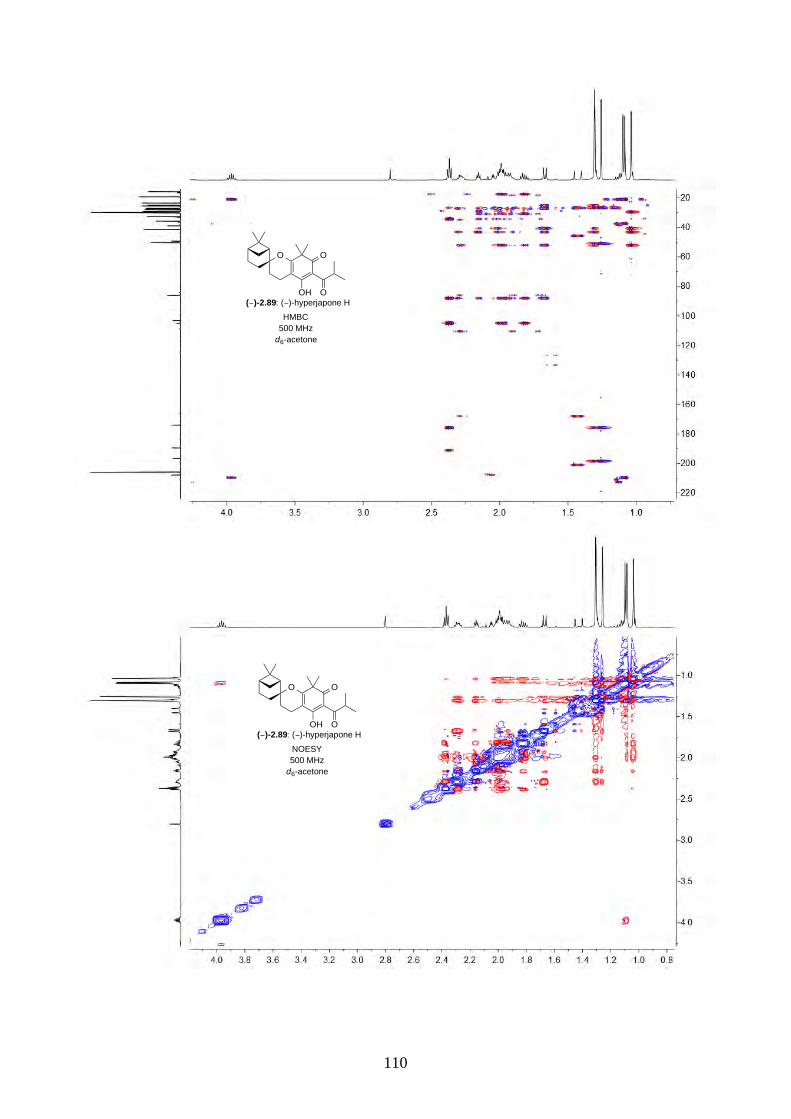
110
O
OH
O
O(−)-2.89: (−)-hyperjapone H
HMBC500 MHz
d6-acetone
O
OH
O
O(−)-2.89: (−)-hyperjapone H
NOESY500 MHz
d6-acetone
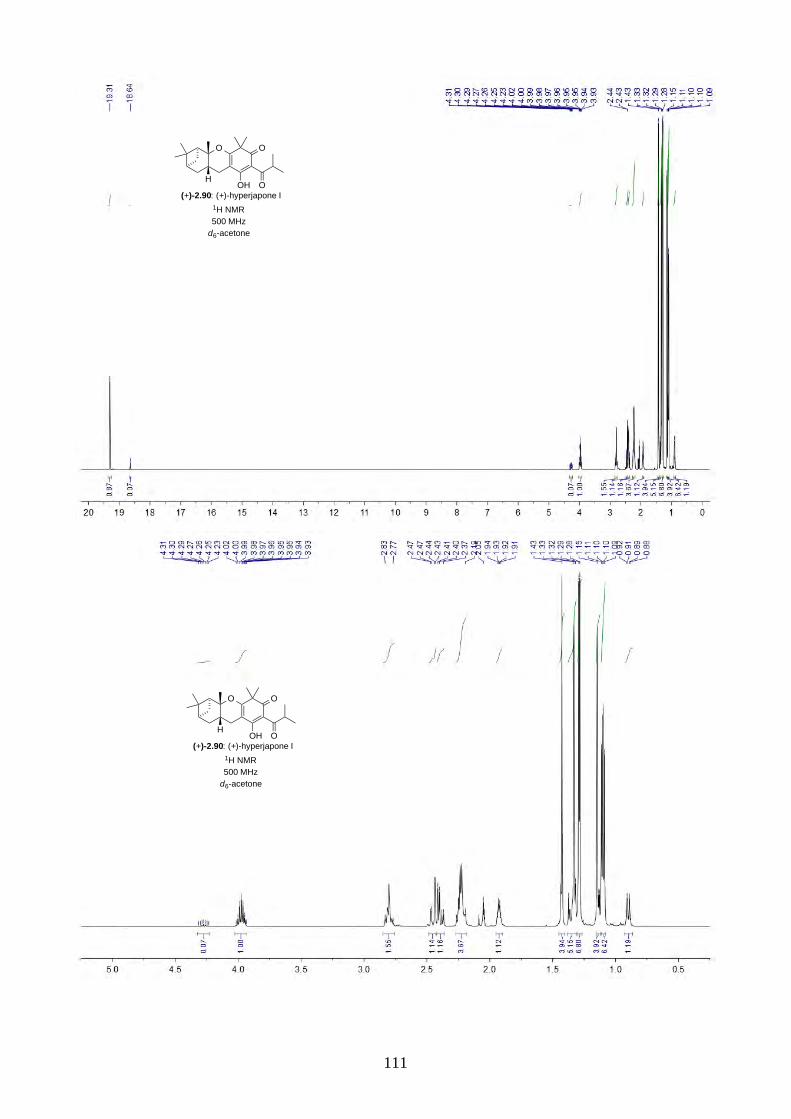
111
OH
O
O(+)-2.90: (+)-hyperjapone I
O
1H NMR500 MHz
d6-acetone
H
OH
O
O(+)-2.90: (+)-hyperjapone I
O
1H NMR500 MHz
d6-acetone
H
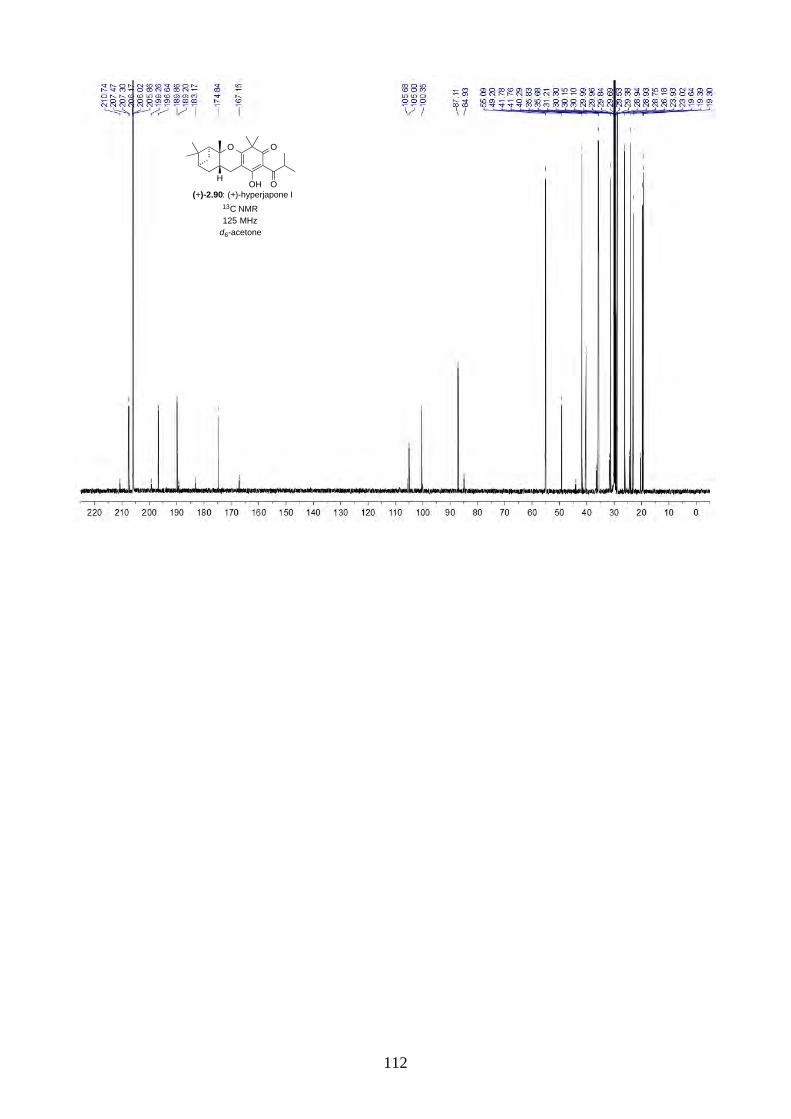
112
OH
O
O(+)-2.90: (+)-hyperjapone I
O
13C NMR125 MHz
d6-acetone
H
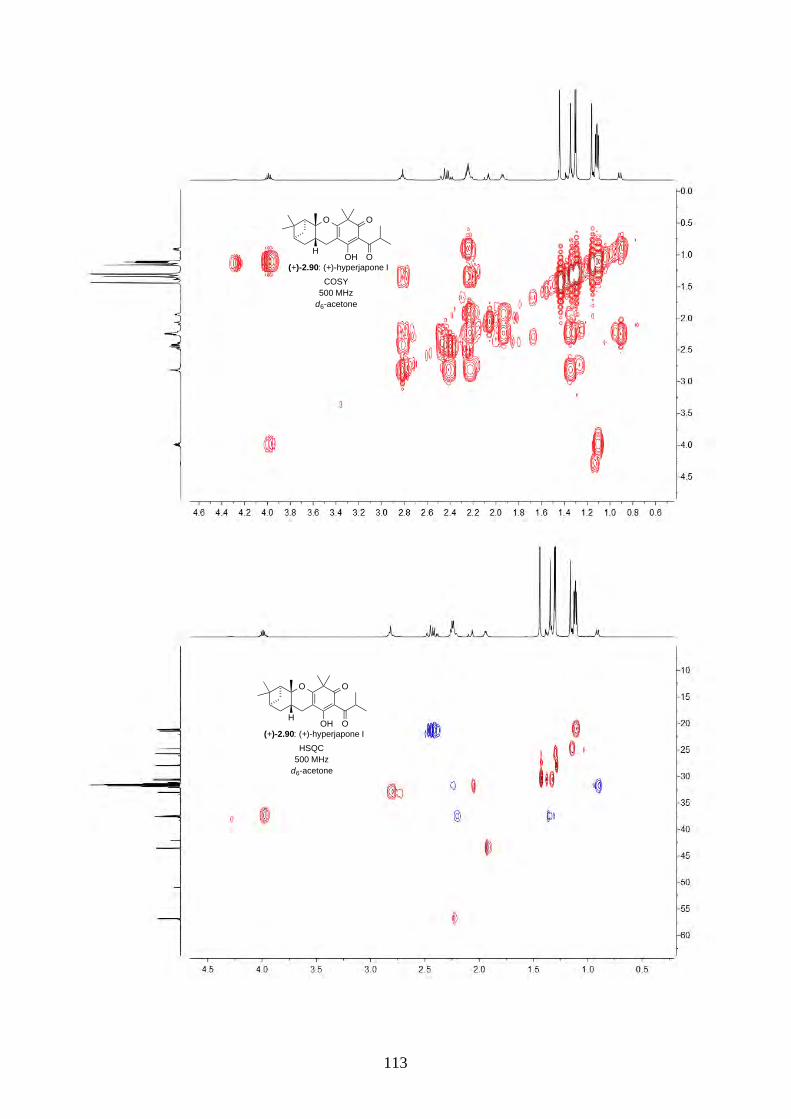
113
OH
O
O(+)-2.90: (+)-hyperjapone I
O
COSY500 MHz
d6-acetone
H
OH
O
O(+)-2.90: (+)-hyperjapone I
O
HSQC500 MHz
d6-acetone
H
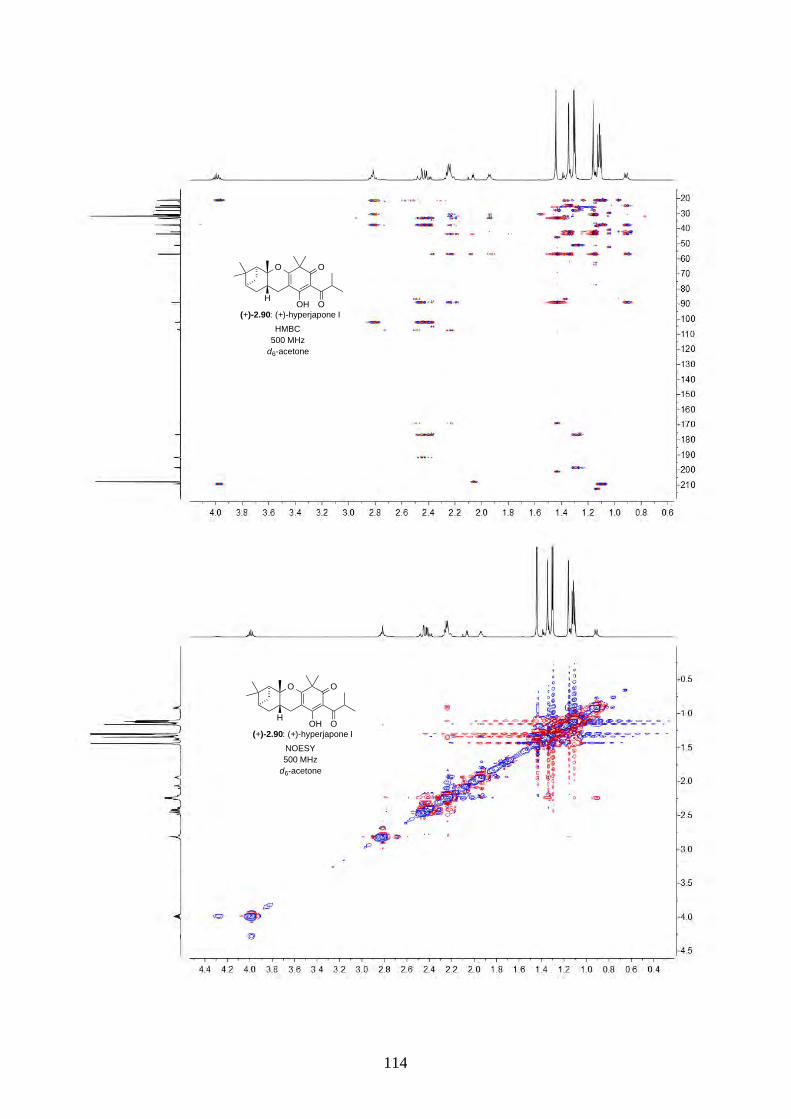
114
OH
O
O(+)-2.90: (+)-hyperjapone I
O
HMBC500 MHz
d6-acetone
H
OH
O
O(+)-2.90: (+)-hyperjapone I
O
NOESY500 MHz
d6-acetone
H
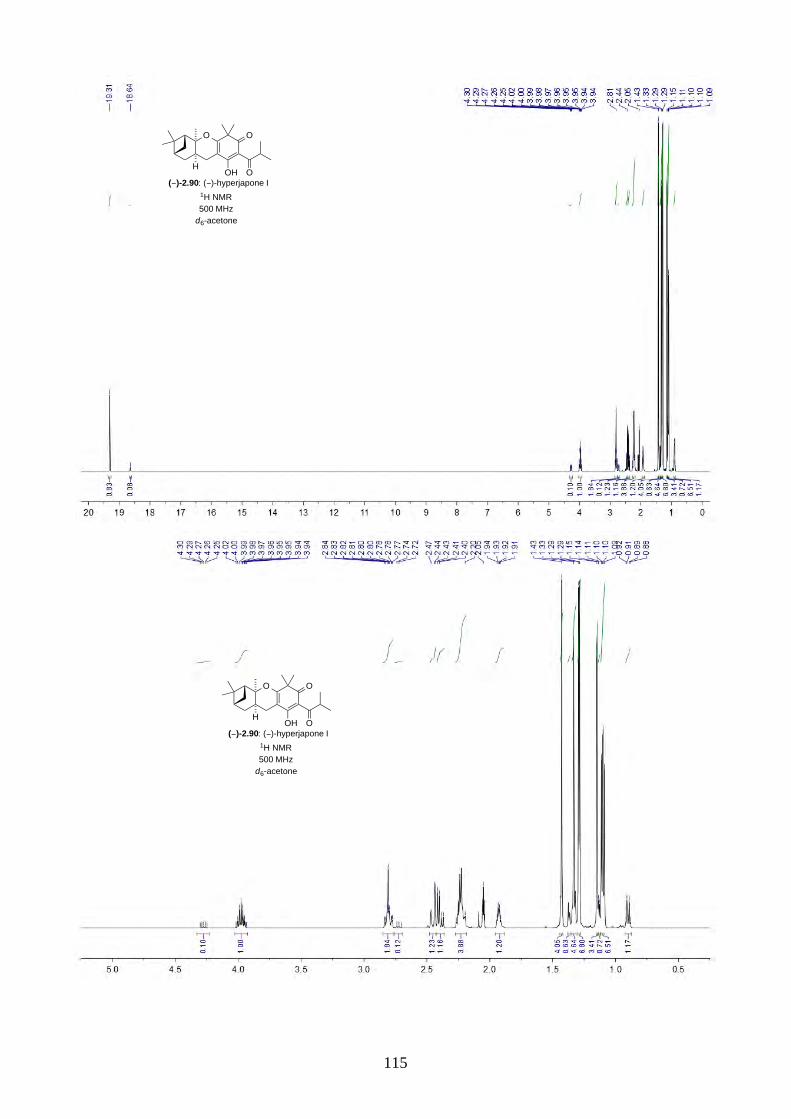
115
OH
O
O(−)-2.90: (−)-hyperjapone I
O
1H NMR500 MHz
d6-acetone
H
OH
O
O(−)-2.90: (−)-hyperjapone I
O
1H NMR500 MHz
d6-acetone
H
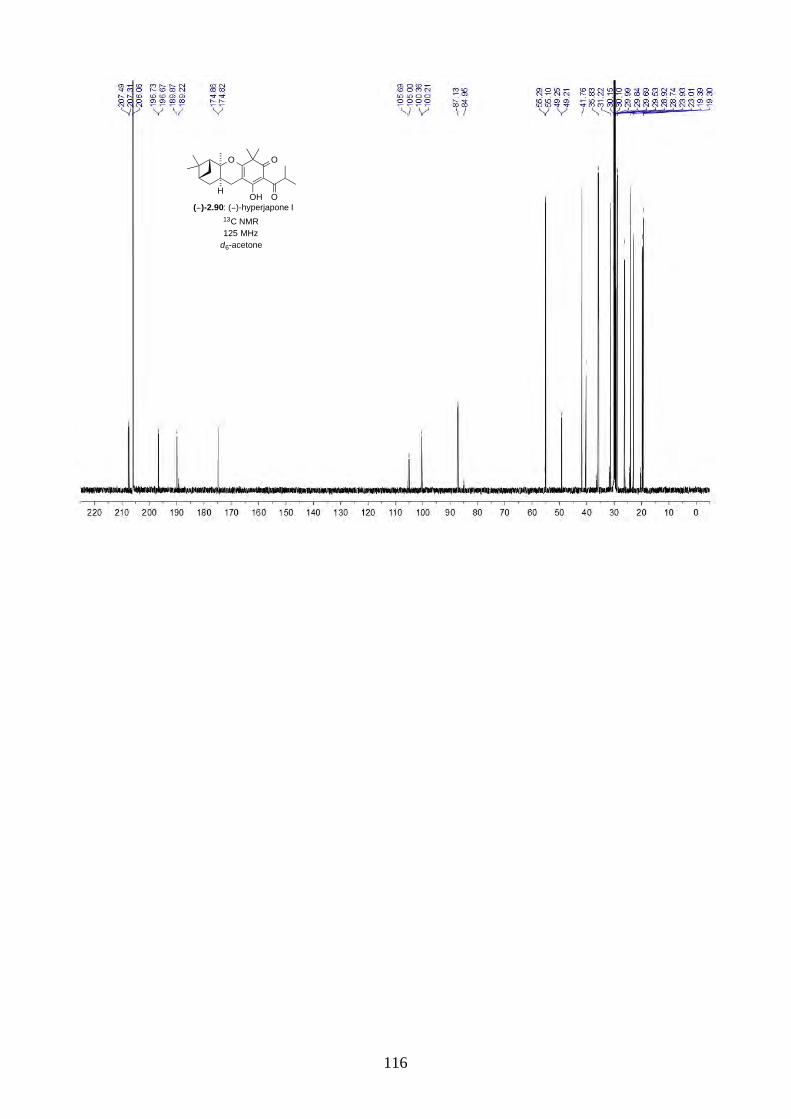
116
OH
O
O(−)-2.90: (−)-hyperjapone I
O
13C NMR125 MHz
d6-acetone
H
117
OH
O
O(−)-2.90: (−)-hyperjapone I
O
COSY500 MHz
d6-acetone
H
OH
O
O(−)-2.90: (−)-hyperjapone I
O
HSQC500 MHz
d6-acetone
H
118
OH
O
O(−)-2.90: (−)-hyperjapone I
O
HMBC500 MHz
d6-acetone
H
OH
O
O(−)-2.90: (−)-hyperjapone I
O
NOESY500 MHz
d6-acetone
H
119
2.5.4. Tables of 1H and 13C NMR data
Comparison of the 1H and 13C NMR spectra of natural and synthetic hyperjapone A (2.49).
Assignment Natural sample 1H NMR,
d6-acetone, 600 MHz
Synthetic sample, 1H NMR,
d6-acetone, 500 MHz
Natural sample 13C NMR,
d6-acetone, 150 MHz
Synthetic sample, 13C NMR,
d6-acetone, 125 MHz
1 104.9 105.0
2 189.3 189.4
3 102.9 103.0
4 2.77, br d (J = 11.8 Hz)
1.82, m
2.77, m
1.85 – 1.83, overlapped m 22.3 22.5
5 1.83, m 1.85 – 1.83, overlapped m 35.5 35.6
6 1.41, t (J = 12.8 Hz)
1.20, m
1.42, dd (J = 15.0, 11.7 Hz)
1.25 – 1.20, m 30.2 30.3
7 2.10, dd (J = 12.8, 7.6 Hz)
1.89, t (J = 12.8 Hz)
2.12, dd (J = 12.8, 7.4 Hz)
1.91, t (J = 12.3 Hz) 38.2 38.3
8 137.3 137.4
9 5.10, d (J = 12.4 Hz) 5.12, dd (J = 11.3, 3.3 Hz) 123.7 123.8
10 2.23, t (J = 12.4 Hz) 2.24, t (J = 12.4 Hz) 42.1 42.2
11 38.7 38.8
12 5.22, d (J = 15.8 Hz) 5.23, d (J = 15.7 Hz) 143.7 143.8
13 5.03, dd (J = 15.8, 10.8 Hz) 5.04, ddd (J = 15.5, 10.6,
2.5 Hz) 120.6 120.7
14 2.55, d (J = 14.3 Hz)
2.45, dd (J = 14.3, 11.2 Hz)
2.56, d (J = 14.8 Hz)
2.46, dd (J = 14.6, 10.7Hz) 42.5 42.6
15 85.5 85.9
16 173.7 173.7
17 48.8 48.9
18 196.6 196.7
19 207.8 207.9
20 3.96, hept (J = 6.8 Hz) 3.98, hept (J = 6.8 Hz) 35.7 35.8
21 1.08, d (J = 6.8 Hz) 1.09, overlapped d (J = 6.3
Hz) 19.2 19.3
22 1.09, d (J = 6.8 Hz) 1.10, overlapped d (J = 6.3 19.3 19.4
O
OH
O
OH
(±)-2.49: hyperjapone A
153
6
10
12
7
11
8
249
13
22
211716
19
14
1520
18
23
2827262425
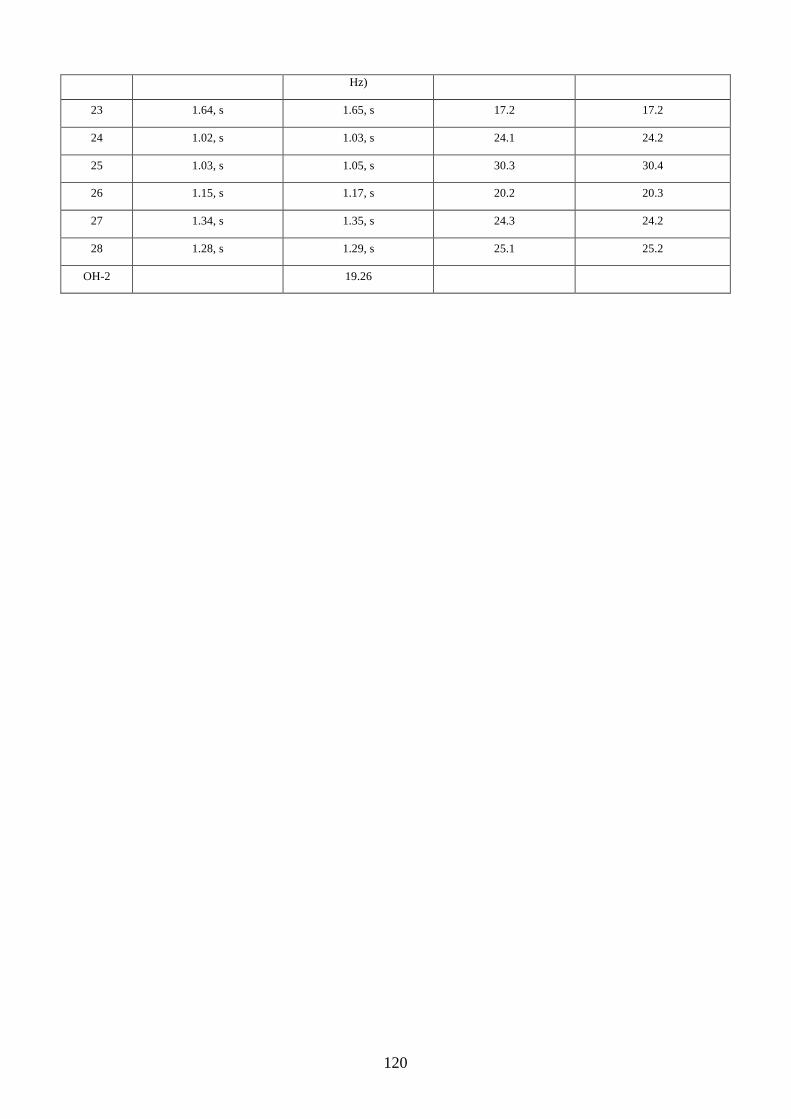
120
Hz)
23 1.64, s 1.65, s 17.2 17.2
24 1.02, s 1.03, s 24.1 24.2
25 1.03, s 1.05, s 30.3 30.4
26 1.15, s 1.17, s 20.2 20.3
27 1.34, s 1.35, s 24.3 24.2
28 1.28, s 1.29, s 25.1 25.2
OH-2 19.26
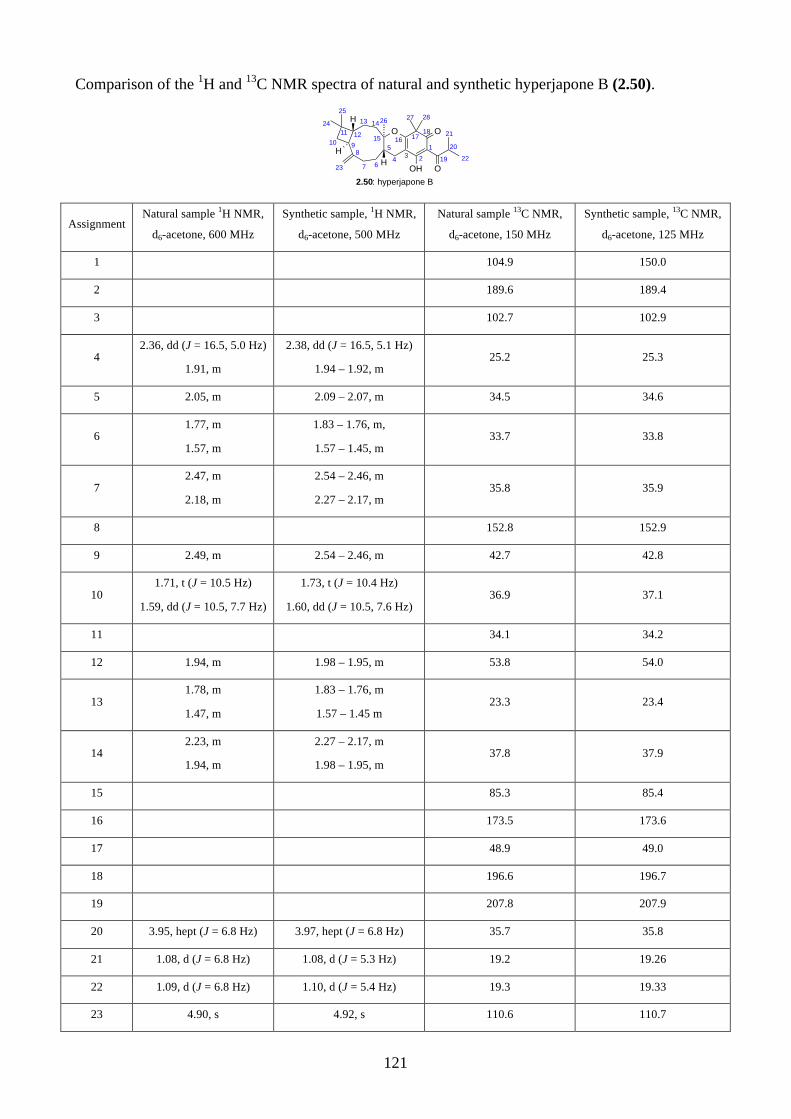
121
Comparison of the 1H and 13C NMR spectra of natural and synthetic hyperjapone B (2.50).
Assignment Natural sample 1H NMR,
d6-acetone, 600 MHz
Synthetic sample, 1H NMR,
d6-acetone, 500 MHz
Natural sample 13C NMR,
d6-acetone, 150 MHz
Synthetic sample, 13C NMR,
d6-acetone, 125 MHz
1 104.9 150.0
2 189.6 189.4
3 102.7 102.9
4 2.36, dd (J = 16.5, 5.0 Hz)
1.91, m
2.38, dd (J = 16.5, 5.1 Hz)
1.94 – 1.92, m 25.2 25.3
5 2.05, m 2.09 – 2.07, m 34.5 34.6
6 1.77, m
1.57, m
1.83 – 1.76, m,
1.57 – 1.45, m 33.7 33.8
7 2.47, m
2.18, m
2.54 – 2.46, m
2.27 – 2.17, m 35.8 35.9
8 152.8 152.9
9 2.49, m 2.54 – 2.46, m 42.7 42.8
10 1.71, t (J = 10.5 Hz)
1.59, dd (J = 10.5, 7.7 Hz)
1.73, t (J = 10.4 Hz)
1.60, dd (J = 10.5, 7.6 Hz) 36.9 37.1
11 34.1 34.2
12 1.94, m 1.98 – 1.95, m 53.8 54.0
13 1.78, m
1.47, m
1.83 – 1.76, m
1.57 – 1.45 m 23.3 23.4
14 2.23, m
1.94, m
2.27 – 2.17, m
1.98 – 1.95, m 37.8 37.9
15 85.3 85.4
16 173.5 173.6
17 48.9 49.0
18 196.6 196.7
19 207.8 207.9
20 3.95, hept (J = 6.8 Hz) 3.97, hept (J = 6.8 Hz) 35.7 35.8
21 1.08, d (J = 6.8 Hz) 1.08, d (J = 5.3 Hz) 19.2 19.26
22 1.09, d (J = 6.8 Hz) 1.10, d (J = 5.4 Hz) 19.3 19.33
23 4.90, s 4.92, s 110.6 110.7
O
OH
O
OH
H
H
2.50: hyperjapone B
153
6
1012
7
11
824
9
13
22
211716
19
14
1520
18
23
28272624
25
122
4.89, s 4.90, s
24 0.99, s 1.01, s 22.3 22.3
25 0.96, s 0.97, s 30.3 30.4
26 1.19, s 1.20, s 21.1 21.2
27 1.26, s 1.28, s 25.3 25.4
28 1.31, s 1.32, s 24.2 24.3
OH-2 19.23
* The 1H NMR spectra data from the isolation was incorrectly referenced at 2.06 ppm for d6-acetone.
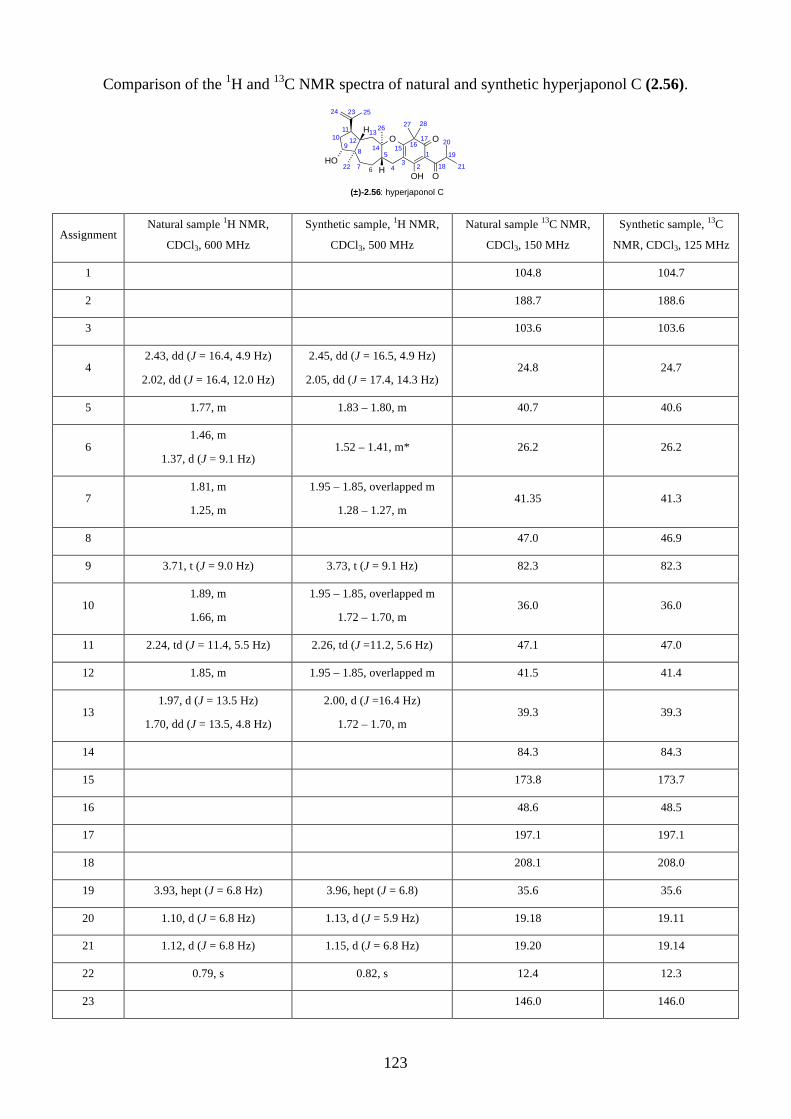
123
Comparison of the 1H and 13C NMR spectra of natural and synthetic hyperjaponol C (2.56).
Assignment Natural sample 1H NMR,
CDCl3, 600 MHz
Synthetic sample, 1H NMR,
CDCl3, 500 MHz
Natural sample 13C NMR,
CDCl3, 150 MHz
Synthetic sample, 13C
NMR, CDCl3, 125 MHz
1 104.8 104.7
2 188.7 188.6
3 103.6 103.6
4 2.43, dd (J = 16.4, 4.9 Hz)
2.02, dd (J = 16.4, 12.0 Hz)
2.45, dd (J = 16.5, 4.9 Hz)
2.05, dd (J = 17.4, 14.3 Hz) 24.8 24.7
5 1.77, m 1.83 – 1.80, m 40.7 40.6
6 1.46, m
1.37, d (J = 9.1 Hz) 1.52 – 1.41, m* 26.2 26.2
7 1.81, m
1.25, m
1.95 – 1.85, overlapped m
1.28 – 1.27, m 41.35 41.3
8 47.0 46.9
9 3.71, t (J = 9.0 Hz) 3.73, t (J = 9.1 Hz) 82.3 82.3
10 1.89, m
1.66, m
1.95 – 1.85, overlapped m
1.72 – 1.70, m 36.0 36.0
11 2.24, td (J = 11.4, 5.5 Hz) 2.26, td (J =11.2, 5.6 Hz) 47.1 47.0
12 1.85, m 1.95 – 1.85, overlapped m 41.5 41.4
13 1.97, d (J = 13.5 Hz)
1.70, dd (J = 13.5, 4.8 Hz)
2.00, d (J =16.4 Hz)
1.72 – 1.70, m 39.3 39.3
14 84.3 84.3
15 173.8 173.7
16 48.6 48.5
17 197.1 197.1
18 208.1 208.0
19 3.93, hept (J = 6.8 Hz) 3.96, hept (J = 6.8) 35.6 35.6
20 1.10, d (J = 6.8 Hz) 1.13, d (J = 5.9 Hz) 19.18 19.11
21 1.12, d (J = 6.8 Hz) 1.15, d (J = 6.8 Hz) 19.20 19.14
22 0.79, s 0.82, s 12.4 12.3
23 146.0 146.0
O
OH
O
OH
H
HO
(±)-2.56: hyperjaponol C
153
6
10 12
7
11
8
24
9
13
22 21
1716
1914 15
20
18
23
282726
24 25
124
24 1.64, s 1.67, s 19.5 19.5
25 4.74, s
4.70, s
4.77, s
4.73, s 111.2 111.1
26 1.10, s 1.12, s 20.4 20.4
27 1.27, s 1.30, s 23.5 23.4
28 1.23, s 1.26, s 25.9 25.8
OH-2 19.08, s
OH-9 1.61, br s
* the doublet at 1.37 ppm reported in literature should be 2 methyl groups at 1.40 and 1.38 ppm
from the minor tautomer, corresponding to carbon at 23.8 and 25.5 ppm.
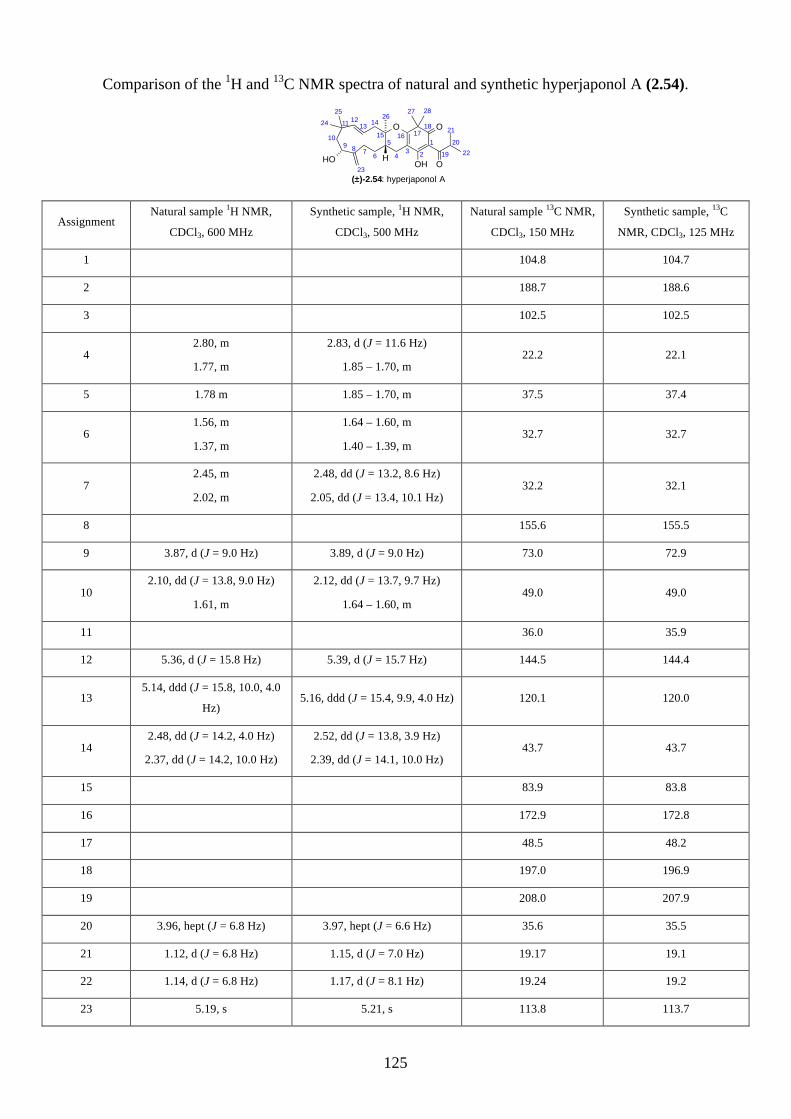
125
Comparison of the 1H and 13C NMR spectra of natural and synthetic hyperjaponol A (2.54).
Assignment Natural sample 1H NMR,
CDCl3, 600 MHz
Synthetic sample, 1H NMR,
CDCl3, 500 MHz
Natural sample 13C NMR,
CDCl3, 150 MHz
Synthetic sample, 13C
NMR, CDCl3, 125 MHz
1 104.8 104.7
2 188.7 188.6
3 102.5 102.5
4 2.80, m
1.77, m
2.83, d (J = 11.6 Hz)
1.85 – 1.70, m 22.2 22.1
5 1.78 m 1.85 – 1.70, m 37.5 37.4
6 1.56, m
1.37, m
1.64 – 1.60, m
1.40 – 1.39, m 32.7 32.7
7 2.45, m
2.02, m
2.48, dd (J = 13.2, 8.6 Hz)
2.05, dd (J = 13.4, 10.1 Hz) 32.2 32.1
8 155.6 155.5
9 3.87, d (J = 9.0 Hz) 3.89, d (J = 9.0 Hz) 73.0 72.9
10 2.10, dd (J = 13.8, 9.0 Hz)
1.61, m
2.12, dd (J = 13.7, 9.7 Hz)
1.64 – 1.60, m 49.0 49.0
11 36.0 35.9
12 5.36, d (J = 15.8 Hz) 5.39, d (J = 15.7 Hz) 144.5 144.4
13 5.14, ddd (J = 15.8, 10.0, 4.0
Hz) 5.16, ddd (J = 15.4, 9.9, 4.0 Hz) 120.1 120.0
14 2.48, dd (J = 14.2, 4.0 Hz)
2.37, dd (J = 14.2, 10.0 Hz)
2.52, dd (J = 13.8, 3.9 Hz)
2.39, dd (J = 14.1, 10.0 Hz) 43.7 43.7
15 83.9 83.8
16 172.9 172.8
17 48.5 48.2
18 197.0 196.9
19 208.0 207.9
20 3.96, hept (J = 6.8 Hz) 3.97, hept (J = 6.6 Hz) 35.6 35.5
21 1.12, d (J = 6.8 Hz) 1.15, d (J = 7.0 Hz) 19.17 19.1
22 1.14, d (J = 6.8 Hz) 1.17, d (J = 8.1 Hz) 19.24 19.2
23 5.19, s 5.21, s 113.8 113.7
O
OH
O
OHHO
(±)-2.54: hyperjaponol A
153
6
10
12
7
11
824
9
13
22
211716
19
14
1520
18
23
282726
24
25
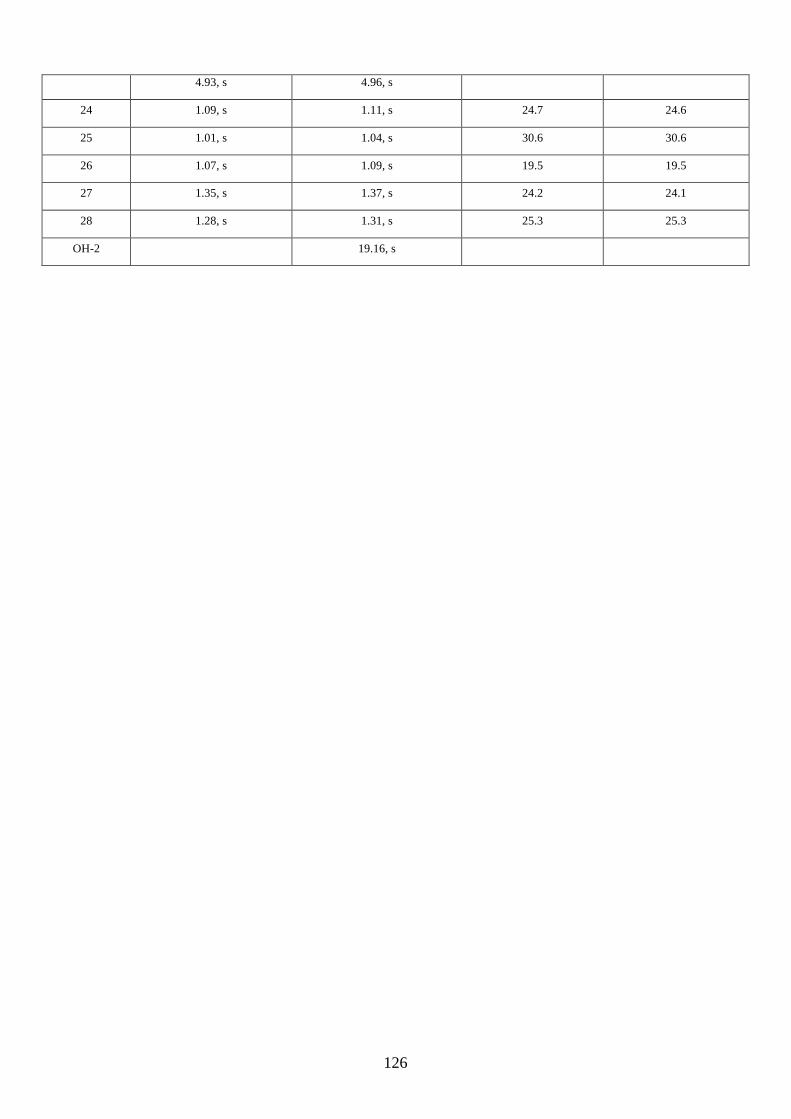
126
4.93, s 4.96, s
24 1.09, s 1.11, s 24.7 24.6
25 1.01, s 1.04, s 30.6 30.6
26 1.07, s 1.09, s 19.5 19.5
27 1.35, s 1.37, s 24.2 24.1
28 1.28, s 1.31, s 25.3 25.3
OH-2 19.16, s
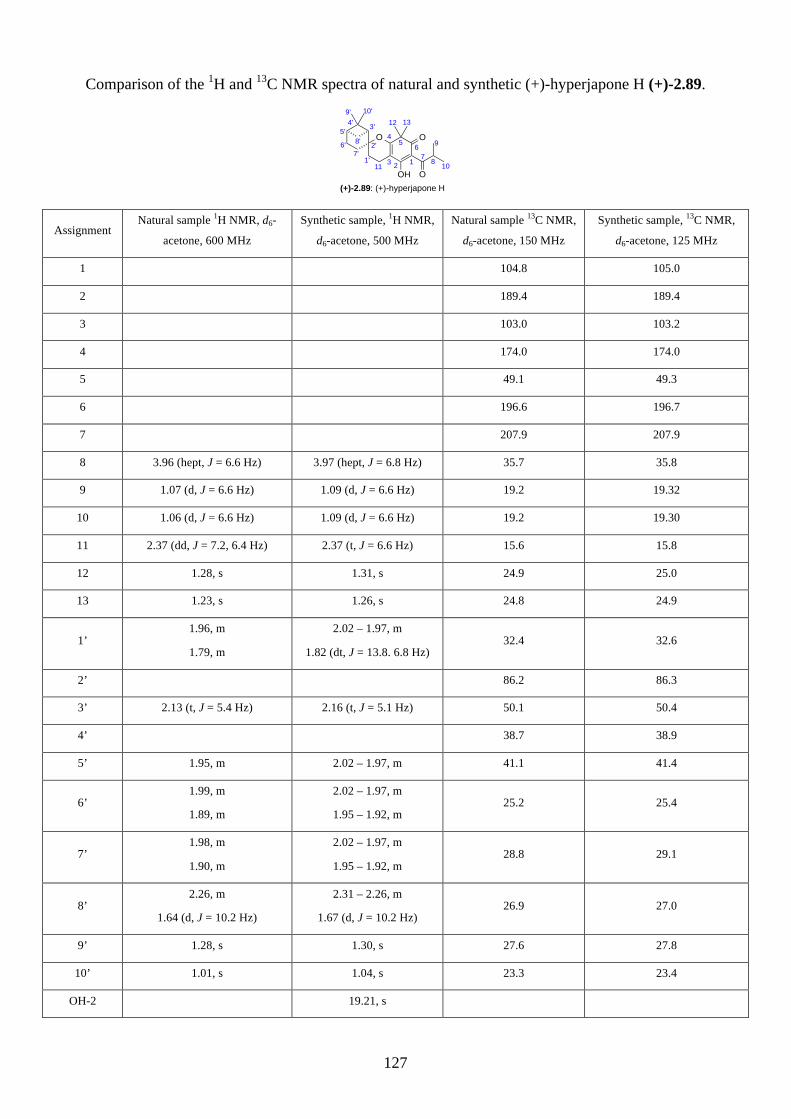
127
Comparison of the 1H and 13C NMR spectra of natural and synthetic (+)-hyperjapone H (+)-2.89.
Assignment Natural sample 1H NMR, d6-
acetone, 600 MHz
Synthetic sample, 1H NMR,
d6-acetone, 500 MHz
Natural sample 13C NMR,
d6-acetone, 150 MHz
Synthetic sample, 13C NMR,
d6-acetone, 125 MHz
1 104.8 105.0
2 189.4 189.4
3 103.0 103.2
4 174.0 174.0
5 49.1 49.3
6 196.6 196.7
7 207.9 207.9
8 3.96 (hept, J = 6.6 Hz) 3.97 (hept, J = 6.8 Hz) 35.7 35.8
9 1.07 (d, J = 6.6 Hz) 1.09 (d, J = 6.6 Hz) 19.2 19.32
10 1.06 (d, J = 6.6 Hz) 1.09 (d, J = 6.6 Hz) 19.2 19.30
11 2.37 (dd, J = 7.2, 6.4 Hz) 2.37 (t, J = 6.6 Hz) 15.6 15.8
12 1.28, s 1.31, s 24.9 25.0
13 1.23, s 1.26, s 24.8 24.9
1’ 1.96, m
1.79, m
2.02 – 1.97, m
1.82 (dt, J = 13.8. 6.8 Hz) 32.4 32.6
2’ 86.2 86.3
3’ 2.13 (t, J = 5.4 Hz) 2.16 (t, J = 5.1 Hz) 50.1 50.4
4’ 38.7 38.9
5’ 1.95, m 2.02 – 1.97, m 41.1 41.4
6’ 1.99, m
1.89, m
2.02 – 1.97, m
1.95 – 1.92, m 25.2 25.4
7’ 1.98, m
1.90, m
2.02 – 1.97, m
1.95 – 1.92, m 28.8 29.1
8’ 2.26, m
1.64 (d, J = 10.2 Hz)
2.31 – 2.26, m
1.67 (d, J = 10.2 Hz) 26.9 27.0
9’ 1.28, s 1.30, s 27.6 27.8
10’ 1.01, s 1.04, s 23.3 23.4
OH-2 19.21, s
123
45
67
8
9
1011
O
OH
O
O(+)-2.89: (+)-hyperjapone H
2'
3'4'5'
6'7'
8'
9' 10'
1'
12 13
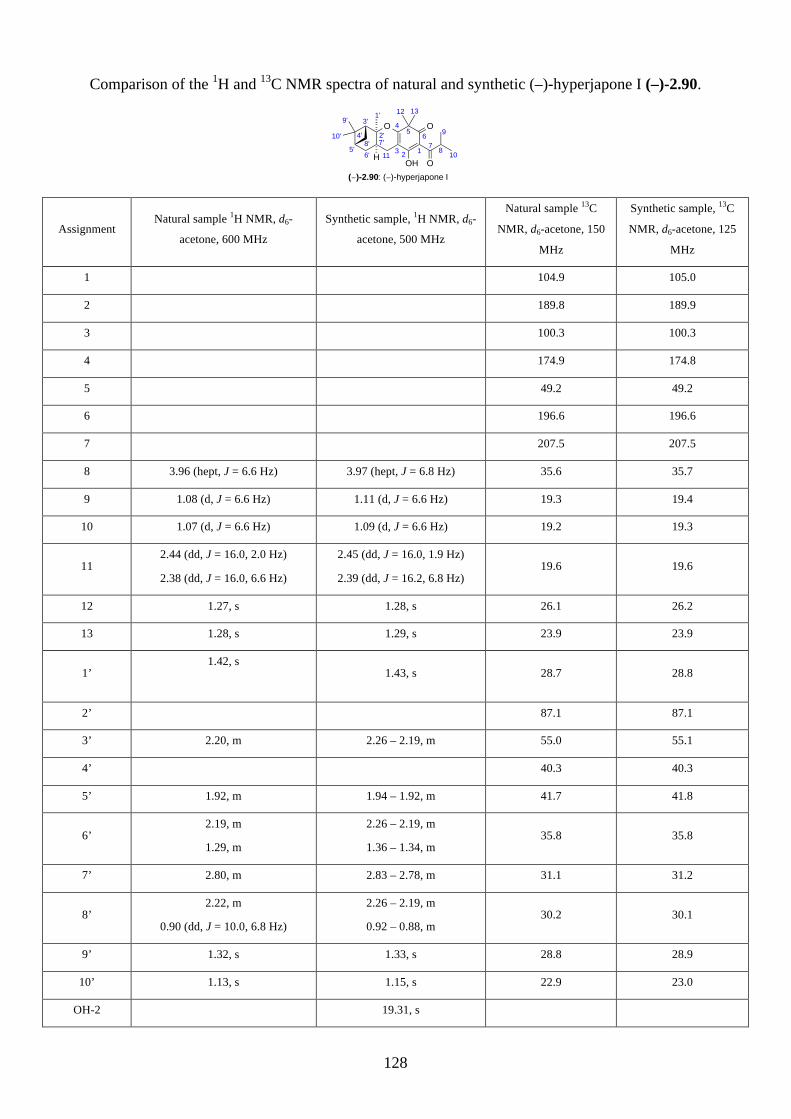
128
Comparison of the 1H and 13C NMR spectra of natural and synthetic (–)-hyperjapone I (–)-2.90.
Assignment Natural sample 1H NMR, d6-
acetone, 600 MHz
Synthetic sample, 1H NMR, d6-
acetone, 500 MHz
Natural sample 13C
NMR, d6-acetone, 150
MHz
Synthetic sample, 13C
NMR, d6-acetone, 125
MHz
1 104.9 105.0
2 189.8 189.9
3 100.3 100.3
4 174.9 174.8
5 49.2 49.2
6 196.6 196.6
7 207.5 207.5
8 3.96 (hept, J = 6.6 Hz) 3.97 (hept, J = 6.8 Hz) 35.6 35.7
9 1.08 (d, J = 6.6 Hz) 1.11 (d, J = 6.6 Hz) 19.3 19.4
10 1.07 (d, J = 6.6 Hz) 1.09 (d, J = 6.6 Hz) 19.2 19.3
11 2.44 (dd, J = 16.0, 2.0 Hz)
2.38 (dd, J = 16.0, 6.6 Hz)
2.45 (dd, J = 16.0, 1.9 Hz)
2.39 (dd, J = 16.2, 6.8 Hz) 19.6 19.6
12 1.27, s 1.28, s 26.1 26.2
13 1.28, s 1.29, s 23.9 23.9
1’ 1.42, s
1.43, s 28.7 28.8
2’ 87.1 87.1
3’ 2.20, m 2.26 – 2.19, m 55.0 55.1
4’ 40.3 40.3
5’ 1.92, m 1.94 – 1.92, m 41.7 41.8
6’ 2.19, m
1.29, m
2.26 – 2.19, m
1.36 – 1.34, m 35.8 35.8
7’ 2.80, m 2.83 – 2.78, m 31.1 31.2
8’ 2.22, m
0.90 (dd, J = 10.0, 6.8 Hz)
2.26 – 2.19, m
0.92 – 0.88, m 30.2 30.1
9’ 1.32, s 1.33, s 28.8 28.9
10’ 1.13, s 1.15, s 22.9 23.0
OH-2 19.31, s
123
45
67
8
9
1011
O
OH
O
O(−)-2.90: (−)-hyperjapone I
2'
3'
4'
5'6'
7'8'
9'
10'
1' 12 13
H
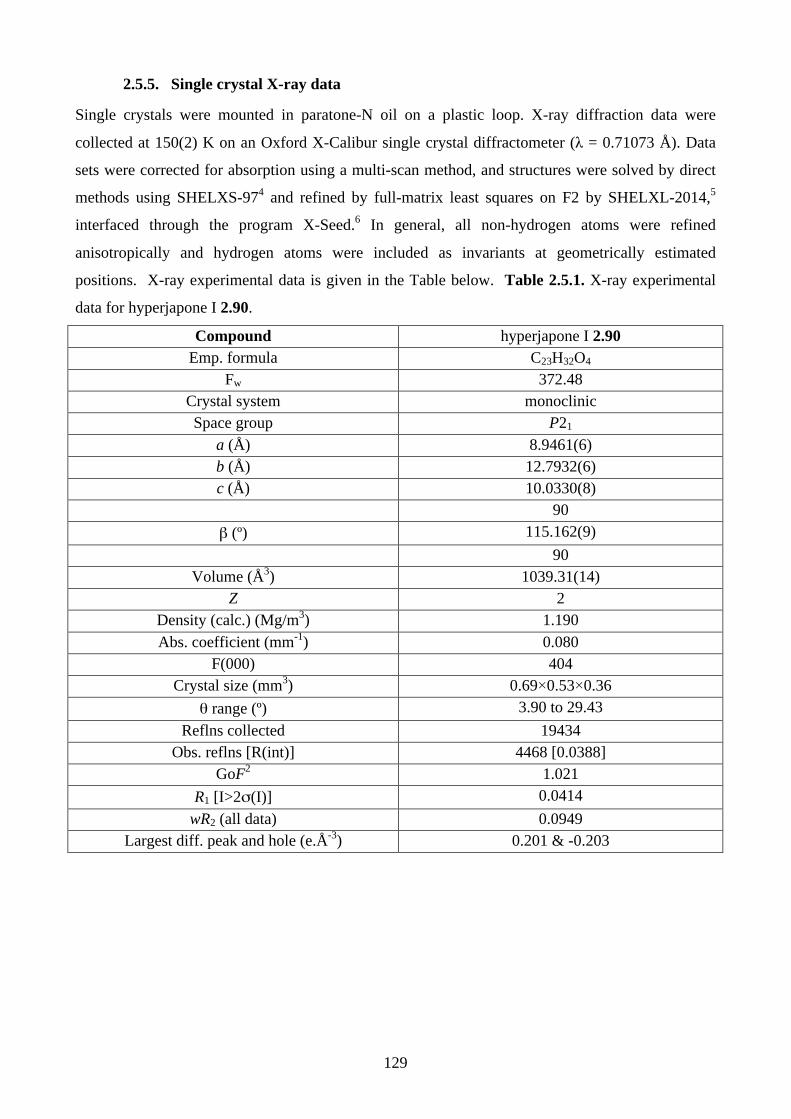
129
2.5.5. Single crystal X-ray data
Single crystals were mounted in paratone-N oil on a plastic loop. X-ray diffraction data were
collected at 150(2) K on an Oxford X-Calibur single crystal diffractometer (λ = 0.71073 Å). Data
sets were corrected for absorption using a multi-scan method, and structures were solved by direct
methods using SHELXS-974 and refined by full-matrix least squares on F2 by SHELXL-2014,5
interfaced through the program X-Seed.6 In general, all non-hydrogen atoms were refined
anisotropically and hydrogen atoms were included as invariants at geometrically estimated
positions. X-ray experimental data is given in the Table below. Table 2.5.1. X-ray experimental
data for hyperjapone I 2.90.
Compound hyperjapone I 2.90 Emp. formula C23H32O4
Fw 372.48 Crystal system monoclinic Space group P21
a (Å) 8.9461(6) b (Å) 12.7932(6) c (Å) 10.0330(8)
90 β (º) 115.162(9)
90 Volume (Å3) 1039.31(14)
Z 2 Density (calc.) (Mg/m3) 1.190 Abs. coefficient (mm-1) 0.080
F(000) 404 Crystal size (mm3) 0.69×0.53×0.36
θ range (º) 3.90 to 29.43 Reflns collected 19434
Obs. reflns [R(int)] 4468 [0.0388] GoF2 1.021
R1 [I>2σ(I)] 0.0414 wR2 (all data) 0.0949
Largest diff. peak and hole (e.Å-3) 0.201 & -0.203
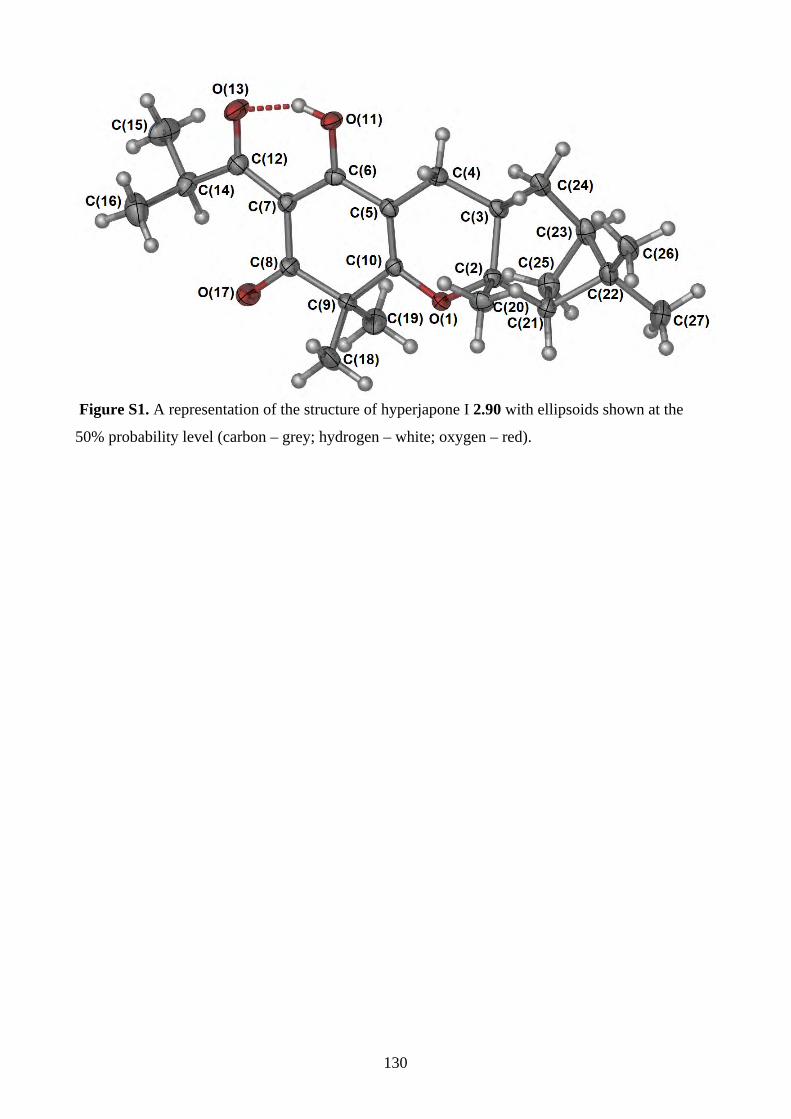
130
Figure S1. A representation of the structure of hyperjapone I 2.90 with ellipsoids shown at the
50% probability level (carbon – grey; hydrogen – white; oxygen – red).

131
2.5.6. Computational Data
Molecular geometries, energies, and free energies of reactants, products, and transition states
were computed by density functional theory using the Gaussian 09 (Revision D.01) software
package. Geometries were optimized in the gas phase at the M062X/6-31G(d,p) for stable state
or M062X/6-31G(d) for transition state. The optimized geometries were identified as stable or
transition states by the number of imaginary frequencies (0 or 1, respectively).
132
Carbocation 2.61 1 1 C -5.43592700 0.98919800 0.85568100 C -5.72333100 -0.51974700 0.67173500 C -3.95043400 1.17753400 0.65603800 C -5.63509300 -1.08927000 -0.81401300 C -3.33951300 1.74420600 -0.38390100 C -1.86775500 1.79863600 -0.70089700 C -0.83485400 0.93374700 0.04754600 C -4.42100600 -1.91966800 -0.83245600 C -3.11297300 -1.35167600 -1.17783800 C -2.06796600 -1.40377100 -0.01998700 C -0.81141600 -0.57174300 -0.34097400 C -4.56179600 -3.34464000 -0.54491600 C -5.78540800 1.32223200 2.31808400 C -6.30654800 1.84421000 -0.06700000 C -0.82237000 1.16407500 1.56392000 H -3.35535200 0.72544100 1.44610700 H -5.52823700 -0.25172800 -1.51410800 O 0.38831600 1.51421700 -0.47566900 C 1.56059300 0.87883600 -0.26260000 C 1.62635100 -0.40303700 0.14903900 C 0.39449000 -1.23872500 0.33125200 C 2.74975700 1.76204900 -0.49777200 C 4.10541300 1.03175800 -0.37047400 C 4.14165400 -0.32721200 0.16148600 C 2.92785600 -0.99646200 0.39422800 C 2.75081300 2.86539400 0.58449700 C 2.66466500 2.39655100 -1.89675800 C 5.36658200 -1.06086300 0.45922100 C 6.74738300 -0.50360100 0.21403600 C 7.05980300 -0.66128800 -1.28630500 C 7.78857300 -1.21988000 1.07135000 H -0.64590400 -0.59596100 -1.42836900 O 5.09914100 1.66455400 -0.69572500 O 2.89892400 -2.21683300 0.84473100 O 5.27399600 -2.22724200 0.91349300 H 6.72940700 0.56505200 0.44042300 H 8.76880900 -0.76588500 0.90374400 H 7.55235900 -1.14633100 2.13630200 H 7.84701600 -2.28011800 0.81378800 H 8.05395700 -0.25523300 -1.49096600 H 7.05886100 -1.72101300 -1.56183300 H 6.34023700 -0.12268000 -1.90622200 H 3.93304900 -2.47070000 0.96736700 H 3.60980200 3.51848600 0.41469500 H 1.83062000 3.45247400 0.52398000 H 2.83377500 2.43396500 1.58774500 H 2.62922300 1.62710000 -2.67450600 H 1.77135600 3.01999900 -1.97565300 H 3.55405500 3.00812800 -2.05609400 H 0.19232400 -1.41242100 1.39846200 H 0.56182500 -2.22821200 -0.10740200 H -1.12498100 2.18810700 1.79696200 H -1.47890900 0.47599700 2.10220700 H 0.18749100 1.00886200 1.95234400 H -1.53048900 2.83691000 -0.58158300 H -1.74156200 1.59143100 -1.77298400 H -3.95353000 2.23577500 -1.14036800 H -1.76461100 -2.44560300 0.12259200 H -2.52276000 -1.08120200 0.91942200 H -2.72462700 -2.00961500 -1.97383100 H -3.21562800 -0.34209200 -1.56900900

133
H -5.31187100 -3.53110300 0.23046700 H -5.03358600 -3.75583200 -1.45814500 H -5.08784100 -1.09331600 1.36063600 H -6.75475600 -0.75691900 0.96098200 H -6.12540800 2.90491600 0.12764800 H -6.11950400 1.67734300 -1.13215100 H -7.36682100 1.64904900 0.12572000 H -5.56762100 2.37383600 2.52265500 H -6.84699300 1.14790200 2.51861000 H -5.19981400 0.71429400 3.01567400 H -3.61761200 -3.85453900 -0.36049900 O -6.72798700 -1.91271300 -1.11471900 H -7.54013500 -1.39149200 -1.15501400 EM062X/6-31G+(d,p)
-3851472.657 kJ mol-1
134
Carbocation 2.62 1 1 C -5.71490800 1.11846900 0.55012100 C -6.26843600 0.42463700 -0.71067000 C -4.23782700 0.83097400 0.58980200 C -5.54727600 -0.91802900 -0.96453500 C -3.49693000 0.71605300 -0.57500700 C -2.02308500 1.00025200 -0.78287200 C -0.88656400 0.26544600 -0.05821600 C -4.14805600 -0.97456300 -0.27682800 C -3.13088300 -1.74731700 -1.16075600 C -1.77163900 -2.13167900 -0.56569300 C -0.62850200 -1.11995500 -0.67507500 C -4.20492300 -1.67079300 1.10147400 C -6.46876100 0.68669200 1.81780200 C -5.80926300 2.66079600 0.42122700 C -1.00388400 0.27519800 1.46380900 H -3.72642200 0.81620600 1.55312000 H -5.38350600 -1.04029300 -2.04474600 O 0.22517200 1.12259600 -0.39574200 C 1.48245300 0.63620800 -0.22937500 C 1.73704800 -0.67412100 -0.05123500 C 0.65296200 -1.71308900 -0.08733200 C 2.51887400 1.71954500 -0.24278000 C 3.97047600 1.19917700 -0.13063600 C 4.20892700 -0.21571300 0.13096700 C 3.11335600 -1.09417300 0.15998500 C 2.28559300 2.63345500 0.98004600 C 2.40240400 2.53742700 -1.54192700 C 5.52844800 -0.79708400 0.36352800 C 6.80713600 0.00253200 0.30623200 C 7.17224600 0.20174700 -1.17747700 C 7.93182300 -0.70576900 1.05835900 H -0.44725800 -0.92659100 -1.74302100 O 4.85582700 2.03408100 -0.24431100 O 3.26674600 -2.36674700 0.37792500 O 5.61110600 -2.02793700 0.58805400 H 6.61365000 0.98745200 0.73775100 H 8.83086100 -0.08441000 1.03263000 H 7.66658200 -0.88554800 2.10359800 H 8.16221800 -1.67090500 0.60096100 H 8.09330200 0.78655900 -1.24550500 H 7.34593500 -0.76792500 -1.65541900 H 6.38974900 0.73884300 -1.71717600 H 4.32020200 -2.48156800 0.51440200 H 3.04371400 3.41977300 0.97761700 H 1.29246000 3.08731100 0.92556800 H 2.37024000 2.07028400 1.91573600 H 2.53715700 1.89901100 -2.42079000 H 1.42346700 3.01862700 -1.60159900 H 3.18390300 3.29905500 -1.54389800 H 0.46884700 -2.11706200 0.91847000 H 0.98379000 -2.56362100 -0.69206300 H -1.24521700 1.28262800 1.81701200 H -1.75600300 -0.42620100 1.82885500 H -0.04872200 -0.01263100 1.91077600 H -1.91219900 2.06669700 -0.54445000 H -1.81985800 0.91458200 -1.85444000 H -4.05311500 0.91502700 -1.49043800 H -1.43905500 -3.02206300 -1.11081200 H -1.88120000 -2.46516100 0.47110700 H -3.69098800 -2.67318900 -1.35618500 H -3.00514000 -1.25475100 -2.13168600

135
H -5.04348200 -1.36059700 1.71682400 H -4.33406600 -2.73615000 0.89625400 H -6.16006700 1.07636000 -1.58376200 H -7.34113700 0.24621900 -0.59165900 H -5.44053200 3.15982900 1.32217300 H -5.25204800 3.03262800 -0.44363600 H -6.86133200 2.93145200 0.29546100 H -5.90300000 0.92192900 2.72522900 H -7.41368100 1.23405500 1.86723400 H -6.71630000 -0.37627000 1.81358700 H -3.28597700 -1.54344300 1.66626700 O -6.25823400 -2.02825800 -0.46011200 H -6.99556500 -2.24397300 -1.04348700 EM062X/6-31G+(d,p)
-3851505.321 kJ mol-1
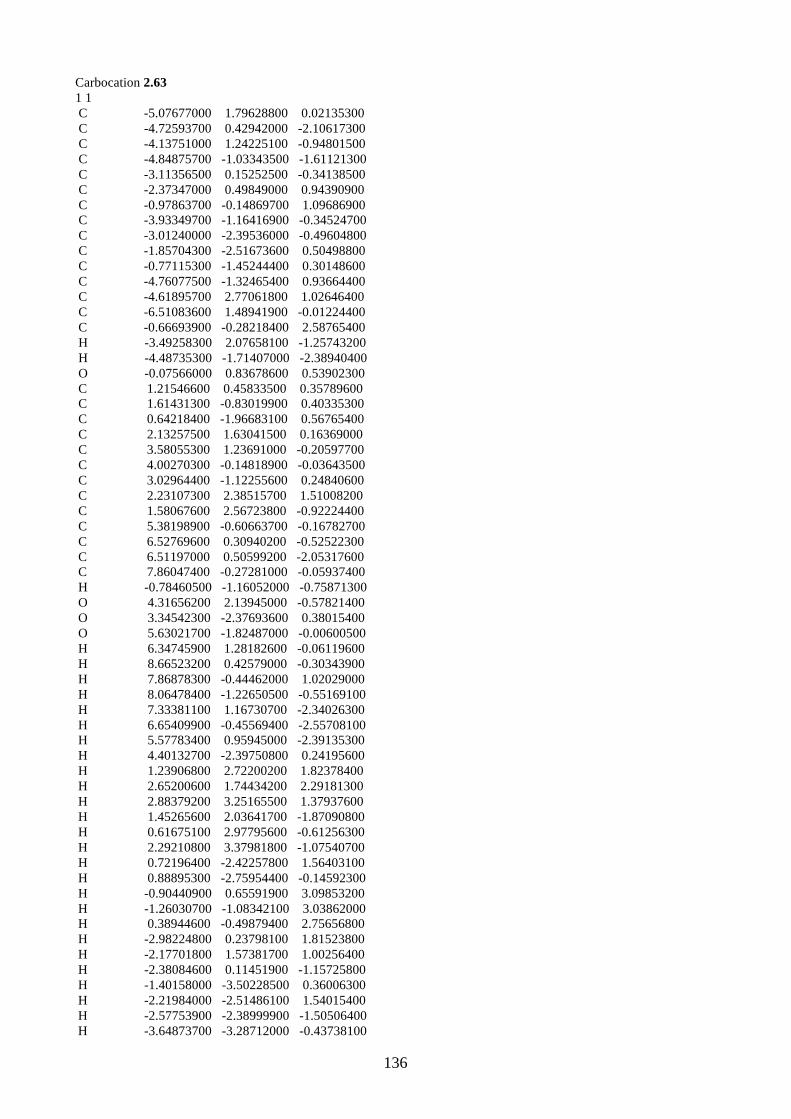
136
Carbocation 2.63 1 1 C -5.07677000 1.79628800 0.02135300 C -4.72593700 0.42942000 -2.10617300 C -4.13751000 1.24225100 -0.94801500 C -4.84875700 -1.03343500 -1.61121300 C -3.11356500 0.15252500 -0.34138500 C -2.37347000 0.49849000 0.94390900 C -0.97863700 -0.14869700 1.09686900 C -3.93349700 -1.16416900 -0.34524700 C -3.01240000 -2.39536000 -0.49604800 C -1.85704300 -2.51673600 0.50498800 C -0.77115300 -1.45244400 0.30148600 C -4.76077500 -1.32465400 0.93664400 C -4.61895700 2.77061800 1.02646400 C -6.51083600 1.48941900 -0.01224400 C -0.66693900 -0.28218400 2.58765400 H -3.49258300 2.07658100 -1.25743200 H -4.48735300 -1.71407000 -2.38940400 O -0.07566000 0.83678600 0.53902300 C 1.21546600 0.45833500 0.35789600 C 1.61431300 -0.83019900 0.40335300 C 0.64218400 -1.96683100 0.56765400 C 2.13257500 1.63041500 0.16369000 C 3.58055300 1.23691000 -0.20597700 C 4.00270300 -0.14818900 -0.03643500 C 3.02964400 -1.12255600 0.24840600 C 2.23107300 2.38515700 1.51008200 C 1.58067600 2.56723800 -0.92224400 C 5.38198900 -0.60663700 -0.16782700 C 6.52769600 0.30940200 -0.52522300 C 6.51197000 0.50599200 -2.05317600 C 7.86047400 -0.27281000 -0.05937400 H -0.78460500 -1.16052000 -0.75871300 O 4.31656200 2.13945000 -0.57821400 O 3.34542300 -2.37693600 0.38015400 O 5.63021700 -1.82487000 -0.00600500 H 6.34745900 1.28182600 -0.06119600 H 8.66523200 0.42579000 -0.30343900 H 7.86878300 -0.44462000 1.02029000 H 8.06478400 -1.22650500 -0.55169100 H 7.33381100 1.16730700 -2.34026300 H 6.65409900 -0.45569400 -2.55708100 H 5.57783400 0.95945000 -2.39135300 H 4.40132700 -2.39750800 0.24195600 H 1.23906800 2.72200200 1.82378400 H 2.65200600 1.74434200 2.29181300 H 2.88379200 3.25165500 1.37937600 H 1.45265600 2.03641700 -1.87090800 H 0.61675100 2.97795600 -0.61256300 H 2.29210800 3.37981800 -1.07540700 H 0.72196400 -2.42257800 1.56403100 H 0.88895300 -2.75954400 -0.14592300 H -0.90440900 0.65591900 3.09853200 H -1.26030700 -1.08342100 3.03862000 H 0.38944600 -0.49879400 2.75656800 H -2.98224800 0.23798100 1.81523800 H -2.17701800 1.57381700 1.00256400 H -2.38084600 0.11451900 -1.15725800 H -1.40158000 -3.50228500 0.36006300 H -2.21984000 -2.51486100 1.54015400 H -2.57753900 -2.38999900 -1.50506400 H -3.64873700 -3.28712000 -0.43738100

137
H -5.30830100 -0.41781800 1.22053200 H -5.50325800 -2.11269100 0.80852300 H -4.01355500 0.48655100 -2.93445100 H -5.68264100 0.80369100 -2.47963300 H -6.90321600 2.19671100 -0.76999500 H -6.73674200 0.48525600 -0.38404200 H -7.01514200 1.72173900 0.92769000 H -5.38293100 3.53559400 1.19934800 H -4.54453000 2.21257800 1.97656400 H -3.65135000 3.21865400 0.80894900 H -4.12212000 -1.58948500 1.78155400 O -6.19113500 -1.36980000 -1.29217400 H -6.62290500 -1.77105600 -2.05445100 EM062X/6-31G+(d,p)
-3851566.010 kJ mol-1
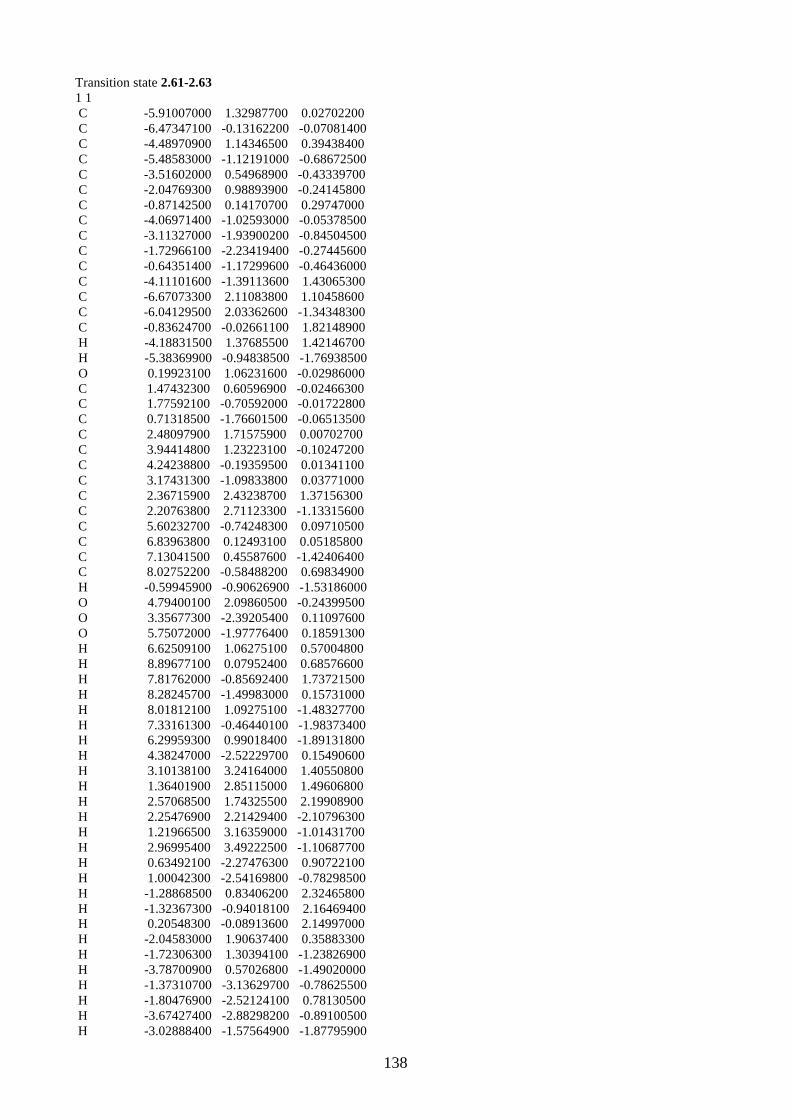
138
Transition state 2.61-2.63 1 1 C -5.91007000 1.32987700 0.02702200 C -6.47347100 -0.13162200 -0.07081400 C -4.48970900 1.14346500 0.39438400 C -5.48583000 -1.12191000 -0.68672500 C -3.51602000 0.54968900 -0.43339700 C -2.04769300 0.98893900 -0.24145800 C -0.87142500 0.14170700 0.29747000 C -4.06971400 -1.02593000 -0.05378500 C -3.11327000 -1.93900200 -0.84504500 C -1.72966100 -2.23419400 -0.27445600 C -0.64351400 -1.17299600 -0.46436000 C -4.11101600 -1.39113600 1.43065300 C -6.67073300 2.11083800 1.10458600 C -6.04129500 2.03362600 -1.34348300 C -0.83624700 -0.02661100 1.82148900 H -4.18831500 1.37685500 1.42146700 H -5.38369900 -0.94838500 -1.76938500 O 0.19923100 1.06231600 -0.02986000 C 1.47432300 0.60596900 -0.02466300 C 1.77592100 -0.70592000 -0.01722800 C 0.71318500 -1.76601500 -0.06513500 C 2.48097900 1.71575900 0.00702700 C 3.94414800 1.23223100 -0.10247200 C 4.24238800 -0.19359500 0.01341100 C 3.17431300 -1.09833800 0.03771000 C 2.36715900 2.43238700 1.37156300 C 2.20763800 2.71123300 -1.13315600 C 5.60232700 -0.74248300 0.09710500 C 6.83963800 0.12493100 0.05185800 C 7.13041500 0.45587600 -1.42406400 C 8.02752200 -0.58488200 0.69834900 H -0.59945900 -0.90626900 -1.53186000 O 4.79400100 2.09860500 -0.24399500 O 3.35677300 -2.39205400 0.11097600 O 5.75072000 -1.97776400 0.18591300 H 6.62509100 1.06275100 0.57004800 H 8.89677100 0.07952400 0.68576600 H 7.81762000 -0.85692400 1.73721500 H 8.28245700 -1.49983000 0.15731000 H 8.01812100 1.09275100 -1.48327700 H 7.33161300 -0.46440100 -1.98373400 H 6.29959300 0.99018400 -1.89131800 H 4.38247000 -2.52229700 0.15490600 H 3.10138100 3.24164000 1.40550800 H 1.36401900 2.85115000 1.49606800 H 2.57068500 1.74325500 2.19908900 H 2.25476900 2.21429400 -2.10796300 H 1.21966500 3.16359000 -1.01431700 H 2.96995400 3.49222500 -1.10687700 H 0.63492100 -2.27476300 0.90722100 H 1.00042300 -2.54169800 -0.78298500 H -1.28868500 0.83406200 2.32465800 H -1.32367300 -0.94018100 2.16469400 H 0.20548300 -0.08913600 2.14997000 H -2.04583000 1.90637400 0.35883300 H -1.72306300 1.30394100 -1.23826900 H -3.78700900 0.57026800 -1.49020000 H -1.37310700 -3.13629700 -0.78625500 H -1.80476900 -2.52124100 0.78130500 H -3.67427400 -2.88298200 -0.89100500 H -3.02888400 -1.57564900 -1.87795900
139
H -4.96211100 -0.95278500 1.95921000 H -4.20453000 -2.47779700 1.51247200 H -6.74748000 -0.48989500 0.92611500 H -7.39668500 -0.07929300 -0.66004500 H -5.51623800 2.99350300 -1.34971600 H -5.66802500 1.42525500 -2.17112900 H -7.10190600 2.23061600 -1.52371900 H -6.34136300 3.15392200 1.14459700 H -7.74030600 2.10775900 0.87625000 H -6.53757500 1.66224700 2.09473100 H -3.20511100 -1.08364700 1.94962000 O -5.91106000 -2.44567800 -0.44212200 H -6.67200100 -2.66746200 -0.99866300 EM062X/6-31G+(d)
-3851356.534 kJ mol-1
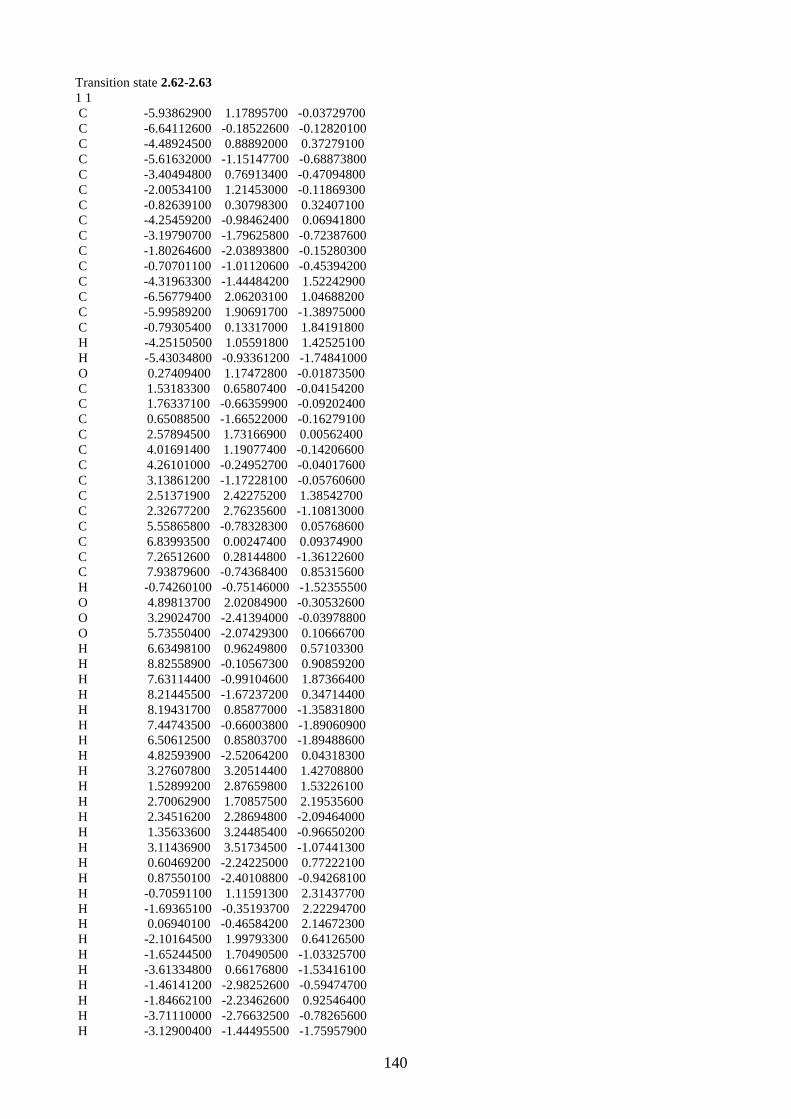
140
Transition state 2.62-2.63 1 1 C -5.93862900 1.17895700 -0.03729700 C -6.64112600 -0.18522600 -0.12820100 C -4.48924500 0.88892000 0.37279100 C -5.61632000 -1.15147700 -0.68873800 C -3.40494800 0.76913400 -0.47094800 C -2.00534100 1.21453000 -0.11869300 C -0.82639100 0.30798300 0.32407100 C -4.25459200 -0.98462400 0.06941800 C -3.19790700 -1.79625800 -0.72387600 C -1.80264600 -2.03893800 -0.15280300 C -0.70701100 -1.01120600 -0.45394200 C -4.31963300 -1.44484200 1.52242900 C -6.56779400 2.06203100 1.04688200 C -5.99589200 1.90691700 -1.38975000 C -0.79305400 0.13317000 1.84191800 H -4.25150500 1.05591800 1.42525100 H -5.43034800 -0.93361200 -1.74841000 O 0.27409400 1.17472800 -0.01873500 C 1.53183300 0.65807400 -0.04154200 C 1.76337100 -0.66359900 -0.09202400 C 0.65088500 -1.66522000 -0.16279100 C 2.57894500 1.73166900 0.00562400 C 4.01691400 1.19077400 -0.14206600 C 4.26101000 -0.24952700 -0.04017600 C 3.13861200 -1.17228100 -0.05760600 C 2.51371900 2.42275200 1.38542700 C 2.32677200 2.76235600 -1.10813000 C 5.55865800 -0.78328300 0.05768600 C 6.83993500 0.00247400 0.09374900 C 7.26512600 0.28144800 -1.36122600 C 7.93879600 -0.74368400 0.85315600 H -0.74260100 -0.75146000 -1.52355500 O 4.89813700 2.02084900 -0.30532600 O 3.29024700 -2.41394000 -0.03978800 O 5.73550400 -2.07429300 0.10666700 H 6.63498100 0.96249800 0.57103300 H 8.82558900 -0.10567300 0.90859200 H 7.63114400 -0.99104600 1.87366400 H 8.21445500 -1.67237200 0.34714400 H 8.19431700 0.85877000 -1.35831800 H 7.44743500 -0.66003800 -1.89060900 H 6.50612500 0.85803700 -1.89488600 H 4.82593900 -2.52064200 0.04318300 H 3.27607800 3.20514400 1.42708800 H 1.52899200 2.87659800 1.53226100 H 2.70062900 1.70857500 2.19535600 H 2.34516200 2.28694800 -2.09464000 H 1.35633600 3.24485400 -0.96650200 H 3.11436900 3.51734500 -1.07441300 H 0.60469200 -2.24225000 0.77222100 H 0.87550100 -2.40108800 -0.94268100 H -0.70591100 1.11591300 2.31437700 H -1.69365100 -0.35193700 2.22294700 H 0.06940100 -0.46584200 2.14672300 H -2.10164500 1.99793300 0.64126500 H -1.65244500 1.70490500 -1.03325700 H -3.61334800 0.66176800 -1.53416100 H -1.46141200 -2.98252600 -0.59474700 H -1.84662100 -2.23462600 0.92546400 H -3.71110000 -2.76632500 -0.78265600 H -3.12900400 -1.44495500 -1.75957900

141
H -5.21260900 -1.08375900 2.03845000 H -4.33614300 -2.54062200 1.52160100 H -7.53137900 -0.13484900 -0.76266800 H -6.96717500 -0.51570400 0.86756000 H -5.38717800 2.81706900 -1.37447800 H -5.66388800 1.28427300 -2.22657800 H -7.02978500 2.19909200 -1.59576000 H -6.07045700 3.03585300 1.10689100 H -7.62182000 2.23917100 0.81313900 H -6.51556800 1.58482300 2.03193300 H -3.44952400 -1.13209600 2.09846400 O -5.99396500 -2.50141300 -0.64085600 H -6.60243200 -2.65765000 0.09754200 EM062X/6-31G+(d)
-3851358.369 kJ mol-1

142
2.5.7. References
1. Crombie, L.; Jones, R. C. F.; Palmer, C. J. J. Chem. Soc., Perkins Trans. 1, 1987, 317. 2. Killeen, D. P.; Larsen, L.; Dayan, F. E.; Gordon, K. C.; Perry, N. B.; van Klink, J. W. J.
Nat. Prod., 2016, 79, 564. 3. Fobofou, S. A. T.; Franke, K.; Porzel, A.; Brandt, W.; Wessjohann, L. A. J. Nat. Prod.,
2016, 79, 743. 4. Sheldrick, G. M. Acta Crystallogr A, 1990, 46, 467. 5. Sheldrick, G. M. Acta Crystallographica a-Foundation and Advances, 2015, 71, 3. 6. Barbour, L. J. J. Supramol. Chem., 2003, 1, 189.

143
Chapter 3
Biomimetic Total Synthesis of Verrubenzospirolactone
3.1. Introduction
3.1.1. Diels-Alder reaction of furan
Scheme 3.1: Diels-Alder reaction between furan (3.1) and maleic anhydride (3.3).1,2
Furan (3.1) is a 5-membered aromatic compound that can behave as a diene in Diels-Alder
reactions (Scheme 3.1), as first reported by Diels and Alder in the reaction with maleic
anhydride (3.2).1 The product 3.3 was later elucidated by Woodward (Scheme 3.1).2 Since
then, there have been numerous reports on the Diels-Alder reaction of furans in organic
synthesis.3
3.1.2. Furan oxidation
Diels-Alder reactions are not limited to carbogenic systems. For instance, furan (3.1) could
also undergo a Diels-Alder reaction with singlet oxygen 3.4 to form an endoperoxide 3.5,
which could be ring opened by a nucleophile to give peroxide 3.6 (Scheme 3.2). An example
of this chemistry is the synthesis of leucosceptroid O (3.11) by Magauer (Scheme 3.3).4
Singlet oxygen was generated using a photosensitizer (rose bengal)5 and reacted with
leucosceptroid A (3.7) to give endoperoxide 3.8. Intramolecular ring opening of 3.8 would
give peroxide 3.9, followed by Kornblum-DeLaMare rearrangement6 of 3.10 to give
leucosceptroid O (3.11).
Scheme 3.2: Diels-Alder reaction between furan (3.1) with singlet oxygen 3.4.
O
3.1: furan
O OO
3.2: maleic anhydride
OOO
OH
Hexo-3.3
+
O
3.1: furan
1O O
3.4: singlet oxygen
Diels-Alder OO O
3.5
NuO OOHNu
3.6
+

144
Scheme 3.3: Biomimetic synthesis of leicosceptroid O (3.11) by Magauer.4
Alternatively, endoperoxide 3.5 could undergo Kornblum-DeLaMare rearrangement
promoted by base to give γ-hydroxybutenolide 3.12. This reaction was reported by Faulkner
in the oxidation of 3.13 to 3.14 (Scheme 3.4).7 Faulkner observed i-Pr2NEt gave the best
yield for this reaction relative to other amine bases (e.g. 2,2,6,6-tetramethylpiperidine).8,9
Scheme 3.4: Kornblum-DeLaMare rearrangement promoted by i-Pr2NEt.7
H
OHO
OO
H
O
HOO
H
OHO
OO
H
O
OAcO
H
N
Ac2O, pyridine
H
OHO
OO
H
O
O
H
OHO
H
O
HO
OO
O
H
OHO
H
O
HO
O
Rose bengal, O2, hνMeOH, –78 °C
Kornblum-DeLaMarerearrangement
3.7: leucosceptroid A
3.11: leucosceptroid O26%
3.8 3.9
3.10
OO O
3.5
O OHO
3.12
i-Pr2NEt Hα β
γ
γ-hydroxybutenolide
O
OH
O
OH
O
HO
rose bengal, i-Pr2NEt–78 °C, CH2Cl2
80%
3.13 3.14

145
Furans can also be oxidized to γ-hydroxybutenolides directly by alternative reagents. For
example, 3.15 has been oxidized to γ-hydroxybutenolide 3.16 by m-CPBA (Scheme 3.5).10
3.18 has been oxidized by PCC to give γ-hydroxybutenolide intermediate followed by
elimination to give 3.19 (Scheme 3.6).11
Scheme 3.5: Furan oxidation by m-CPBA reported by Wiesner.10
Scheme 3.6: Furan oxidation by PCC reported by Sha.11
3.1.3. Syntheses and reactions of 2H-chromene
Figure 3.1: Structure of 2H-chromene 3.20 and 4H-chromene 3.21.12,13
Chromene is an aromatic ring fused with a pyran that can be either a 2H-chromene 3.20 or
4H-chromene 3.21 (Figure 3.1).12,13 An early example of a 2H-chromene synthesis was
reported by Nummy in 1951. The lactone in 3-methyl-coumarin 3.22 was attacked by
MeMgBr, followed by elimination of 3.23 to give the o-quinone methide reactive
intermediate 3.24. 3.24 underwent 6π-electrocyclization to give chromene 3.25 (Scheme
3.7).14
OHO
OH H
O
H H
H
HH
O
OHO
OH H
O
H H
H
HH
O
OHO
OH H
O
H H
H
HH
O
m-CBPA, NaOAc, AcOHCH2Cl2, rt
NaBH4MeOH/H2O, rt
71% over 2 steps
O
HO
O
3.15 3.16 3.17
OO OTBSH OO OTBSPCC, celite, CH2Cl2, rt
65%
3.18 3.19
O
O1
2
34
O1
2
34
3.202H-chromene
3.214H-chromene

146
Scheme 3.7: Synthesis of 2H-chromene 3.25 reported by Nummy.14
A classic example of biomimetic synthesis using a chromene is the synthesis of deoxybruceol
analogue 3.31 reported by Crombie.15 Starting from resorcinol 3.26 with citral (3.27),
Knoevenagel condensation gave o-quinone methide 3.28, followed by 6π-electrocyclization
to give chromene 3.29. Base-promoted tautomerisation of chromene 3.29 gave o-quinone
methide 3.30, which then underwent an intramolecular Diels-Alder reaction to give 3.31
(Scheme 3.8). Chromene 3.29 was also found to undergo a [2+2] cycloaddition under light to
give 3.33.
O OMeMgBr
OH
OO
elimination
6π-electrocyclization
3.22 3.23
3.25 3.24
1 2
OH
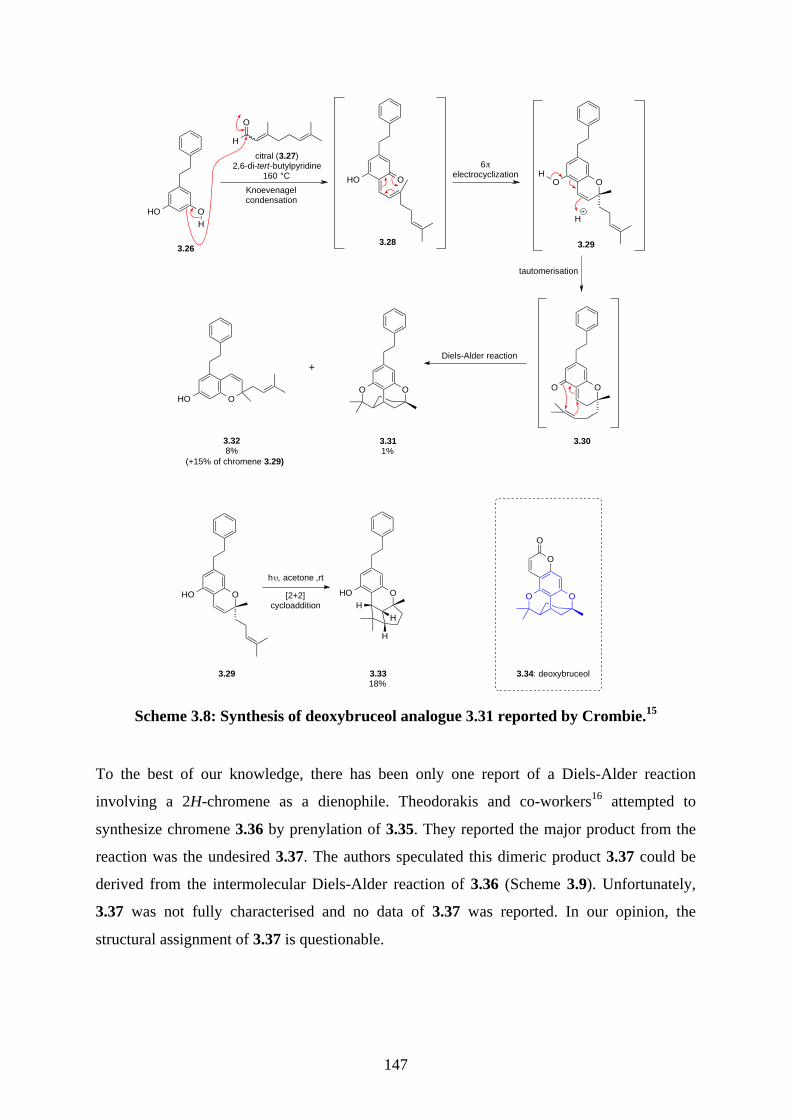
147
Scheme 3.8: Synthesis of deoxybruceol analogue 3.31 reported by Crombie.15
To the best of our knowledge, there has been only one report of a Diels-Alder reaction
involving a 2H-chromene as a dienophile. Theodorakis and co-workers16 attempted to
synthesize chromene 3.36 by prenylation of 3.35. They reported the major product from the
reaction was the undesired 3.37. The authors speculated this dimeric product 3.37 could be
derived from the intermolecular Diels-Alder reaction of 3.36 (Scheme 3.9). Unfortunately,
3.37 was not fully characterised and no data of 3.37 was reported. In our opinion, the
structural assignment of 3.37 is questionable.
HO O
O
O OH
H
O O
H
citral (3.27)2,6-di-tert-butylpyridine
160 °C
3.311%
HO O
H
HH
[2+2]cycloaddition
3.29
3.30
(+15% of chromene 3.29)
hυ, acetone ,rt
3.3318%
Diels-Alder reaction
3.26
HO O
3.28
Knoevenagelcondensation
6πelectrocyclization
tautomerisation
HO O
3.29
HO O
+
3.328%
3.34: deoxybruceol
H
OO
O
O
OO

148
Scheme 3.9: Diels-Alder reaction with chromene reported by Theodorakis.16
3.1.4. Isolation of verrubenzospirolactone
Figure 3.2: Verrubenzospirolactone and its related natural products.17,18,19
(+)-Verrubenzospirolactone (3.38) and (+)-capillobenzopyranol (3.39) were isolated from the
soft coral Sinularia verruca by Gustafson and Yan in 2016 (Figure 3.2).19 (+)-
Capillobenzopyranol (3.39) was previously isolated from Sinularia capillosa along with
furanquinone (3.40) by Duh in 2010.18 Furanoquinone (3.40) and furanoquinol (3.41) were
first isolated from Sinularia capillosa by Coll.17 From the structural similarities among these
four natural products and their co-isolation, we propose there is a biosynthetic link that
connects all of these meroterpenoids.
OO
OMe
MeO
O OMe
MeOOH
OMe
OMeO OH
OMe
OMeO OH
NaH, prenyl bromidePhMe, –30 °C
3.35 3.36
3.3755%
(not characterised)
Diels-Alder reaction/dimerisation
+20% of chromene 3.36
OHO
OMeO OMe
MeOOH
3.36
OHO
OMe
MeO
O OMe
MeOOH
3.37
tautormerisation
MeO
O
OH
H H
H
O
O
O
OH
O
3.39: capillobenzopyranol3.38: verrubenzospirolactone
OH
OH
OO
O
O
3.41: furanoquinol3.40: furanoquinone

149
Scheme 3.10: Proposed biosynthesis of verrubenzospirolactone (3.38).
Our proposed biosynthesis begins from oxidation of furanoquinol (3.41) to give
furanoquinone (3.40).20 Tautomerisation of 3.40 could give o-quinone methide 3.42 which
undergoes 6π-electrocyclization to give capillobenzopyranol (3.39).14 Capillobenzopyranol
(3.39) could undergo a Diels-Alder reaction with singlet oxygen to give endoperoxide 3.43.5
Ring opening of endoperoxide 3.43 by H2O could give peroxide 3.44, which can undergo the
Kornblum-DeLaMare rearrangement to give γ-hydroxybutenolide 3.45,6 followed by a
stereoselective dehydration to give Z-3.46. Intramolecular Diels-Alder reaction of Z-3.46
would give verrubenzospirolactone (3.38). It is crucial that the alkene in 3.46 is in Z-
configuration, which will dictate the relative stereochemistry of the spirocycle of
verrubenzospirolactone (3.38).
OH
OH
O
3.41: furanoquinol
O
O
O
3.40: furanoquinone
O
OH
O
H
O
OH
O
3.39: capillobenzopyranol
O
OH
O
O
OH
O
O
OH
O
OH
H H
H
O
O
- H2O
intramolecularDiels-Alder reaction
O
HOOO
1O2
Z Z
Eexo TS
OO
H2O
O
OH
O
HOO
OH
3.44
3.45
oxidation tautomerisation
6π-electrocyclization
ring opening of endoperoide
Kornblum-DeLaMarerearrangement
elimination
3.42
3.43
3.46 3.38: verrubenzospirolactone

150
3.1.5. Aims of this study
Scheme 3.11: Retrosynthetic analysis of 3.46.
The aims of this project were to synthesize 3.46 from relatively simple starting materials. In
addition, we planned to investigate the intramolecular Diels-Alder reaction of 3.46 to give
verrubenzospirolactone (3.38), which would be the first example to use 2H-chromene as a
dienophile in a Diels-Alder reaction. In addition, we would like to investigate the synthesis of
capillobenzopyranol (3.39) and then convert it into verrubenzospirolactone (3.38) via our
proposed biosynthetic pathway. 3.46 could be accessed through a Horner-Wadsworth-
Emmons reaction between 3.47 and 3.48. Aldehyde 3.48 could be derived from methyl
hydroquinone (3.49) and citral (3.27).
O
OH
O O OH
OH
O
HO OP
O
OEtEtO
Z-3.46 3.49: methylhydroquinone 3.27: citral3.47
O
OH
OH
3.48

151
3.2. Results and discussion
3.2.1. Synthesis of aldehyde 3.48
Scheme 3.12: Synthesis of chromene 3.54 reported by Gembus.21
Scheme 3.13: Synthesis of chromene 3.55.
The synthesis began with the union of methyl hydroquinone (3.49) and citral (3.27) via
Knoevenagel condensation followed by 6π-electrocyclization to form chromene 3.55
(Scheme 3.13). This reaction was quite challenging for various reasons. First, hydroqinone
(3.50) is not a good substrate for chromene synthesis as shown in the example reported by
Gembus,21 where only 40% yield of chromene 3.54 was isolated in this step (Scheme 3.12).
In addition, there could be regioselectivity issues with methyl hydroquinone (3.49) as a
substrate. Indeed, a mixture of side products (3.56, 3.57 and 3.58) was observed from the
reaction (Scheme 3.13). The purification was difficult and we attempted to separate the
mixtures with AgNO3 doped SiO222,23 but it was unsuccessful. We eventually discovered
using 1:1 petrol/CH2Cl2 as eluent gave the best separation, where our desired chromene 3.55
would elute first, followed by co-elution of 3.56, 3.57 and 3.58. Subsequent purifications of
the remaining mixture of side products allowed us to isolate pure samples of 3.56, 3.57 and
3.58 for characterisation. To date, our optimal conditions allow this reaction to run on a 8 g
O
OH
OMe
MeO+
3.50: hydroquinone 3.51
pyridine, reflux, 6 d
O
OH3.5440%
H
O
OH3.53
O
OH
OMe
H
Knoevenagel condensation
6π-electrocyclization
3.52
O
OH
OH
OH
O
H
PhB(OH)2, AcOHPhMe, 110 °C
3.49: methyl hydroquinone
3.27: citral
3.5514%
O
OHO
OH
H
H+ + +
O
OH
H
H
3.56 3.57 3.58

152
scale and consistently give 14% yield of our desired chromene 3.55. We could have designed
a longer synthetic route to selectively synthesize chromene 3.55, but considering both citral
(3.27) and methyl hydroquinone (3.49) are quite cheap ($0.13 for 1 g of citral, $0.25 for 1 g
of methyl hydroquinone), we could access chromene 3.55 from this faster and more
economical 1-step approach.
Scheme 3.14: Riley oxidation of chromene 3.55.
The next step was a Riley oxidation24 of chromene 3.55, which gave alcohol 3.59 in 33%
yield, along with 7% yield of aldehyde 3.48 and 35% yield of recovered starting material 3.55
(Scheme 3.14). We attempted to drive the reaction to completion by leaving it overnight,
which led to the consumption of chromene 3.55. However, we also observed a complex
mixture of products where alcohol 3.59 or aldehyde 3.48 could not be isolated. Therefore, we
concluded our best strategy was to leave the reaction for 2 to 3 h, then isolate 3.59 and 3.48.
Scheme 3.15: Oxidation of alcohol 3.59 to aldehyde 3.48.
The oxidation of alcohol 3.59 to aldehyde 3.48 was also problematic. We attempted the
oxidation using various reagents including Dess-Martin periodinane, TPAP/NMO, PCC,
MnO2, PhI(OAc)2/TEMPO,25 we observed no aldehyde 3.48 in these conditions. Eventually,
we discovered alcohol 3.59 could be oxidized to 3.48 by Swern oxidation, and it was crucial
to limit the amount of oxalyl chloride to 1 equiv. to avoid complications of the reaction.
During the Swern reaction, we observed solid residue when the Swern reagent was first
added to alcohol 3.59. It was essential to warm the reaction to room temperature and allowed
this solid to dissolve in solution, before cooling back to –78 ºC and adding Et3N. We
O
OH3.55
O
OH
CH2OH
3.59
33% (+ 7% of 3.48)
SeO2, t-BuOOHCH2Cl2, rt
O
OH
CHO
+
3.48
O
OH
CH2OH
3.59
O
OH
CHO
3.48
(COCl)2, DMSO, Et3NCH2Cl2, −78 °C to rt
66%

153
discovered if the solid was not fully dissolved before addition of Et3N, no aldehyde 3.48
would be formed. Otherwise, the reaction gave aldehyde 3.48 in 66% yield consistently.
3.2.2. Synthesis of the Horner-Wadsworth-Emmons reagent 3.47
Scheme 3.16: Synthesis of Horner-Wadsworth-Emmons reagent 3.47.26
With the aldehyde 3.48 in hand, our focus moved onto the synthesis of Horner-Wadsworth-
Emmons reagent 3.47, following a procedure from Li26 with slight modifications. For the
bromination reaction, Li used NBS and benzoyl peroxide (BPO) in CCl4,26 we substituted
BPO with AIBN as a radical initiator;27 we have also used benzene over CCl4 because of
limited access to CCl4 in Australia. The bromination of 3.60 gave 3.61 in 97% yield. The next
step was a Michaelis-Arbuzov reaction28 of 3.61 in neat P(OEt)3, which gave 3.47 in good
yield (Scheme 3.16).26 These reactions could be done on 10 g scale.
3.2.3. Biomimetic total synthesis of verrubenzospirolactone (3.38)
Scheme 3.17: Horner-Wadsworth-Emmons reaction between 3.48 and 3.47.
For the Horner-Wadsworth-Emmons reaction, we initially used KOt-Bu and LDA as base but
the yield was relatively poor (45% and 35% respectively). After switching to n-BuLi, the
reaction proceeded smoothly and gave E-3.46 in 79% yield as a mixture of E/Z isomers. The
E/Z ratio ranged from 3.3:1 to 4.5:1. The stereochemistry of the E/Z isomers could be
confirmed by NOESY analysis (Figure 3.3).
OO BrOONBS, AIBN
benzene, reflux
97%
3.613.60
OO PO
OEtOEt
P(OEt)3, 110 °C
88%
3.47
O
OH
HH
O
H
O
EO
OH
CHO
OO PO
OEtOEt3.47 =
3.47, n-BuLiTHF, −78 °C to rt
79%
E/Z = 4.5:13.48 3.46
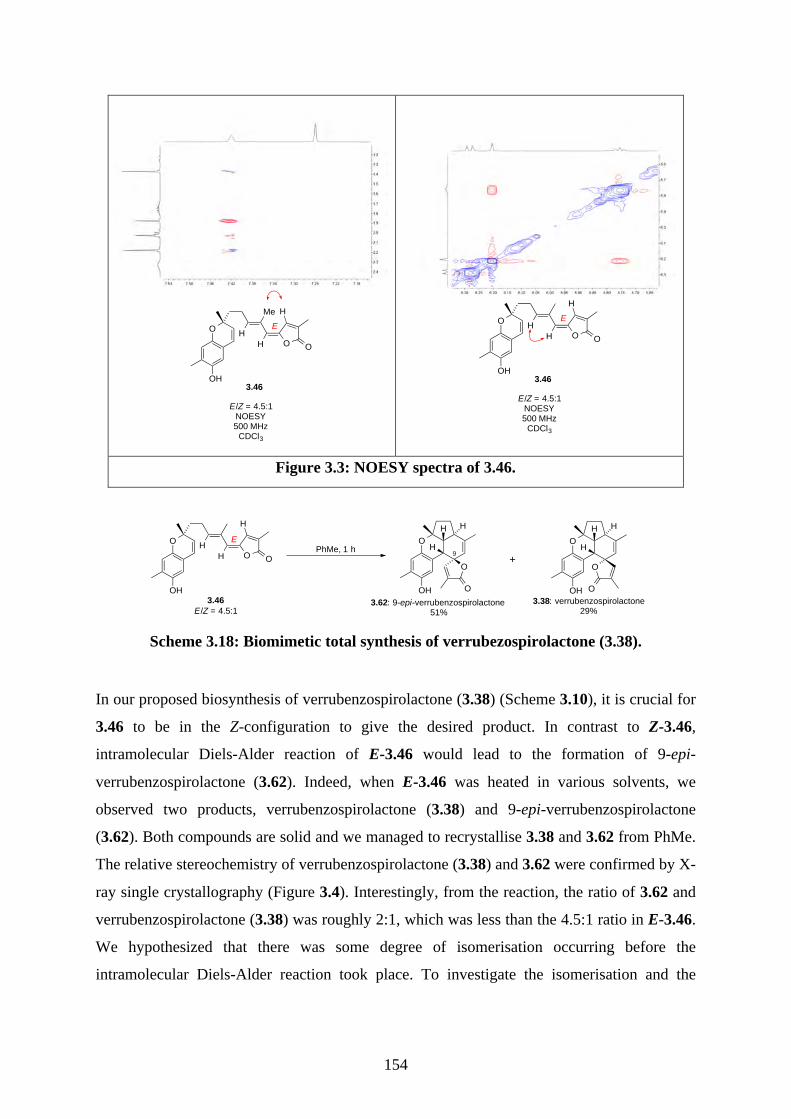
154
Figure 3.3: NOESY spectra of 3.46.
Scheme 3.18: Biomimetic total synthesis of verrubezospirolactone (3.38).
In our proposed biosynthesis of verrubenzospirolactone (3.38) (Scheme 3.10), it is crucial for
3.46 to be in the Z-configuration to give the desired product. In contrast to Z-3.46,
intramolecular Diels-Alder reaction of E-3.46 would lead to the formation of 9-epi-
verrubenzospirolactone (3.62). Indeed, when E-3.46 was heated in various solvents, we
observed two products, verrubenzospirolactone (3.38) and 9-epi-verrubenzospirolactone
(3.62). Both compounds are solid and we managed to recrystallise 3.38 and 3.62 from PhMe.
The relative stereochemistry of verrubenzospirolactone (3.38) and 3.62 were confirmed by X-
ray single crystallography (Figure 3.4). Interestingly, from the reaction, the ratio of 3.62 and
verrubenzospirolactone (3.38) was roughly 2:1, which was less than the 4.5:1 ratio in E-3.46.
We hypothesized that there was some degree of isomerisation occurring before the
intramolecular Diels-Alder reaction took place. To investigate the isomerisation and the
O
OH
Me
HH
O
H
O
E
E/Z = 4.5:1NOESY500 MHz
CDCl3
3.46
O
OH
HH
O
H
O
E
E/Z = 4.5:1NOESY500 MHz
CDCl3
3.46
PhMe, 1 hO
OH
HH
O
H
O
E O
OH
H H
H
O
O
O
OH
H H
H
O
O3.38: verrubenzospirolactone
29%3.62: 9-epi-verrubenzospirolactone
51%
+
E/Z = 4.5:13.46
9

155
Diels-Alder reaction, we screened a series of conditions, using different solvents, temperature
and time (Table 3.1).
verrubenzospirolactone (3.38) 9-epi-verrubenzospirolactone (3.62)
Figure 3.4: X-ray structures of 3.38 and 3.62.
Table 3.1: Conditions for isomerisation and Diels-Alder reaction of 3.46.
Starting Material Conditions Results
E-3.46 Z-3.46 solvent temp. time E-3.46 Z-3.46 3.62 3.38 % conversion
4.5 1 PhMe 110 °C 1 h 0 0 1.8 1 100%
4.5 1 PhMe rt (16 h); then 110 °C (1 h) 0 0 1 1 100%
4.5 1 CHCl3 rt 1 week 1 5 4 1.2 46%
4.5 1 CHCl3 rt 2 weeks 1 6.3 8.8 5 65%
3.3 1 CHCl3 30 °C 16 h 1.7 5.5 2 1 29%
3.3 1 CHCl3 30 °C 90 h 1 3.9 3.8 2.1 55%
3.3 1 CHCl3 50 °C 16 h 1 6.5 7.8 6.6 66%
3.3 1 H2O 30 °C 16 h 2.6 2 1.7 1 37%
3.3 1 H2O 30 °C 90 h 1 4 6.6 4.2 68%
4.5 1 H2O 40 °C 16 h 1.3 2.2 3.9 1 58%
3.3 1 H2O 50 °C 16 h 1 2.5 4 3 67%
4.5 1 H2O 50 °C 40 h 0 0 1 1.1 100%
*From the Horner–Wadsworth-Emmons reaction, the E/Z ratio of 3.46 varied from 3.3:1 to
4.5:1. Ratios of the starting materials and reaction products were determined by 1H NMR
analysis of the mixtures.
O
OH
HH
O
H
O
E O
OH
H H
H
O
O
O
OH
H H
H
O
O
+conditions
E/Z = 4.5:13.46 3.62 3.38
9

156
As shown in Table 3.1, we observed isomerisation and Diels-Alder reaction occurred in all
three solvents investigated. We were also delighted to observe the reaction occurred in H2O, a
biologically relevant solvent. At 30 ºC in H2O, 68% of E-3.46 was converted to 3.38 and 3.62
in 90 h; when the temperature was elevated to 50 ºC, we observed 100% conversion in 40 h.
Considering Sinularia verruca is a soft coral from tropical oceans, it is possible for sea water
to be heated up to slightly above room temperature and promote such reaction. Therefore, we
believe the biosynthesis of verrubenzospirolactone (3.38) could occur spontaneously instead
of catalysed by Diels-Alderase enzymes.29,30 However, from collective results across all
conditions, there was no strong evidence to suggest H2O catalysing the Diels-Alder reaction
faster than organic solvents.31,32
Scheme 3.19: Attempted synthesis of Still-Gennari reagent 3.63.
Since the Horner-Wadsworth-Emmons reaction between 3.48 and 3.47 predominately gave
E-3.46, we were curious of the possible outcome of a Still-Gennari modification of the
Horner-Wadsworth-Emmons reaction, which would expect to give the desired Z-3.46.33 We
attempted to synthesize the Still-Gennari reagent 3.63 using a variety of bases (DBU,34
NaH,35 Cs2CO3,36 KOtBu, n-BuLi) but all conditions gave no reaction or decomposition
(Scheme 3.19).
Scheme 3.20: Isomerisation of E-3.46 by Pd(II) catalyst.37
With our failed attempt to synthesize the Still-Gernnari reagent 3.63, the next objective was
to optimize the isomerisation of E-3.46 into Z-3.46 without giving any Diels-Alder product.
We first investigated the isomerisation using conventional methods (e.g. light38, I239) but all
led to decomposition of E-3.46. Spencer have reported an isomerisation of Z-alkenes to the
OO Br
3.61
OO PO
OCH2CF3
OCH2CF3
base
3.63
OPH(OCH2CF3)2
Pd(MeCN)2Cl2, DMF, rt
E/Z = 4.5:1
O
OH
HH
O
H
O
E O
OH
HH
O O
H
Z
Z/E = 7.5:1
79%
3.46 3.46

157
E-alkenes using catalytic Pd(MeCN)2Cl2.37 To our delight, this condition isomerised E-3.46
into Z-3.46 in a ratio of 7.5:1 favoured the Z-3.46 (Scheme 3.20).
Scheme 3.21: Biomimetic total synthesis of verrubenzospirolactone (3.38) from Z-3.46.
We then studied the intramolecular Diels-Alder reaction of Z-3.46, and observed that
verrubenzospirolactone (3.38) was the predominant product in both PhMe and H2O (Scheme
3.21). The ratio of 3.38 and 3.62 was similar to the ratio in Z-3.46, which suggests there was
no further isomerisation of 3.46.
Scheme 3.22: Attempted synthesis of verrubenzospiralactone (3.38) by Knoevenagel
condensation and Diels-Alder reaction.
We have also investigated the synthesis of verrubenzospirolactone (3.38) from Knoevenagel
condensation between 3.48 and 3.60 to generate Z-3.46 in situ, followed by intramolecular
Diels-Alder reaction to give verrubenzospirolactone (3.38). However, the reaction was not
selective as we observed both 3.62 and 3.38 in low yield (Scheme 3.22).
PhMe, 110 ̊ C
O
OH
HH
O O
H
ZO
OH
H H
H
O
O
O
OH
H H
H
O
O
3.38: verrubenzospirolactone
+
Z/E = 7.5:1
H2O, 50 ̊ C
3.46
12% 69%
3.62: 9-epi-verrubenzospirolactone
10% 61%
piperidine, t-BuOH, refluxO
OH
CHO
3.48
OO
3.60 O
OH
H H
H
O
O
O
OH
H H
H
O
O
3.38: verrubenzospirolactone (25%)3.62 (8%)
+

158
3.2.4. Synthesis of capillobenzopyranol (3.39) and its oxidation
Scheme 3.23: Total synthesis of capillobenzopyranol (3.39).
After the synthesis of verrubenzospirolactone (3.38) from Z-3.46, we were interested to
synthesize capillobenzopyranol (3.39), the proposed biosynthetic precursor of
verrubenzospirolactone (3.38), and attempt to convert capillobenzopyranol (3.39) into
verrubenzospirolactone (3.38) via our proposed biosynthetic pathway (Scheme 3.10). A
convenient approach to access capillobenzopyranol (3.39) was from a reduction of polyene
3.46. Consulting the literature, there are a few reaction conditions known to reduce furanones
to furans.4,40,41 To our delight, reduction of 3.46 with LiAlH4 gave capillobenzopyranol (3.39)
in 52% yield (Scheme 3.23). NMR of 3.39 matched from the isolation data.18
Scheme 3.24: Oxidation of 3.39 to γ-methoxybutenolide 3.65.
Our next objective was to oxidize capillobenzopyranol (3.39) with singlet oxygen. When
capillobenzopyranol (3.39) was exposed to light in the presence of rose bengal and O2, the
reaction gave peroxide 3.64, which we presumed the endoperoxide 3.43 was ring opened by
MeOH. Kornblum-DeLaMare rearrangement of 3.64 with Ac2O and pyridine gave γ-
methoxybutenolide 3.65 (Scheme 3.24).42 The yield of the two steps was quite low due to the
instability of 3.64 and the unselective esterification on the free phenol of 3.64 by Ac2O and
pyridine. Nonetheless, elimination of MeOH would allow access of polyene 3.46. However,
with our best effort, heat, acids or bases gave no reaction.
Our attention returned to the oxidation of capillobenzopyranol (3.39), to explore other
possibilities, for instance if we could change the methoxy group to a hydroxy group, we
could further convert it into a better leaving group (e.g. -OMs or -OTf). Therefore, we
O
OH
O
3.39: capillobenzopyranol
O
OH
O O LiAlH4, Et2O, 0 °C
52%
E
3.46
O
OH
O
O
OMeAc2O, pyridine, rt
21% over 2 steps
O
OH
O
3.39: capillobenzopyranol
O2, rose bengalMeOH, hν, 0 ºC
O
OH
O
OOH
OMe
3.64 3.651:1 d.r.

159
changed the solvent from MeOH to water for the oxidation. Surprisingly, we did not observe
any product but slow decomposition of capillobenzopyranol (3.39). We thought perhaps
water was not a strong enough nucleophile to attack the endoperoxide, and we chose a
stronger nucleophile (hydroxide) in the reaction. However, only decomposition was observed.
We also suspected capillobenzopyranol (3.39) was not soluble in H2O, so we introduced co-
solvents (e.g. DMSO, DMF, MeCN, THF etc.) but all gave no reaction.
Literature also showed i-Pr2NEt could be used as a base to promote Kornblum-DeLaMare
rearrangement of an endoperoxide to give a γ-hydroxybutenolide in one pot.4,8,9,43 However
we observed slow decomposition using this reagent. We also tested different combinations of
solvent and base but with no success. In addition, we have investigated a different
photosensitiser, TPP, but to no avail.4,44,45,46
Scheme 3.25: Undesired oxidation of capillobenzopyranol (3.39).
We then investigated a direct oxidation (using DDQ47, PCC11, m-CPBA48 and NBS49) of
furan 3.39 to γ-hydroxybutanolide 3.45, but all conditions led to decomposition. In one
reaction with PDC, we isolated 3.66 as a 2:1 mixture of diastereoisomers (Scheme 3.25). 3.66
could be derived from an oxidation of the phenol.
We went back to the literature and we found Clive50,51 and Salomon52 reported the use of
NaClO2 to oxidize furan to γ-hydroxybutenolide.50,51 It was an accidental discovery for Clive
and co-workers as they were using NaClO2 for a standard Pinnick oxidation, and there was no
oxidation on the aldehyde but on the furan.51 To our delight, using NaClO2 in t-BuOH/H2O,
capillobenzopyranol (3.39) was converted into γ-hydroxybutenolide 3.45 as a 1:1 mixture of
diastereoisomers in 46% overall yield (Scheme 3.26).
PDC, DMF, rt
16%
O
OH
O
3.39 3.66
O
O
OHO
2:1 d.r.

160
Scheme 3.26: Oxidation of capillobenzopyranol (3.39).
3.2.5. Biomimetic total synthesis of verrubenzospirolactone (3.38)
Scheme 3.27: Total synthesis of verrubenzospirolactone (3.38) from 3.45.
To our surprise, γ-hydroxybutenolide 3.45 was quite stable for standard elimination
conditions. We tried bases (e.g. i-Pr2NEt, i-Pr2NH, Et3N) or acids (e.g. p-TsOH, HCl, H2SO4)
or heat, but it gave no reaction. We also attempted to convert the hydroxy group into -OMs or
-OTf but it gave no reaction or decomposition when the temperature was elevated. Finally,
with Ac2O and pyridine, we managed to eliminate the hydroxy group to give Z-3.67.
However, the elimination reaction was not selective and esterification of phenol was
observed. Standard hydrolysis of acetate group with K2CO3 gave Z-3.46 as a single
stereoisomer. Only Z-3.46 was observed without trace of E-3.46 (see Figure 3.5), thus
suggesting a highly stereoselective elimination of 3.45. Heating polyene Z-3.46 in PhMe
gave verrubenzospirolactone (3.38) in quantitative yield. From this result, we proposed 3.45
and Z-3.46 are undiscovered natural products.
O
OH
O
3.39: capillobenzopyranol
NaClO2, NaH2PO4t-BuOH/H2O, rt
O
OH
O
O
OH
3.45
46%
1:1 d.r.
O
OH
O Ac2O, pyridine, rtO
OAc
O
O
3.67
46%
single stereoiosmer
OOH
3.45
Z
PhMe, reflux
100%
O
OH
H H
H
O
O
3.38: verrubenzospirolactone
K2CO3, MeOH, rt
O
OH
O
O
3.46
70%
Z
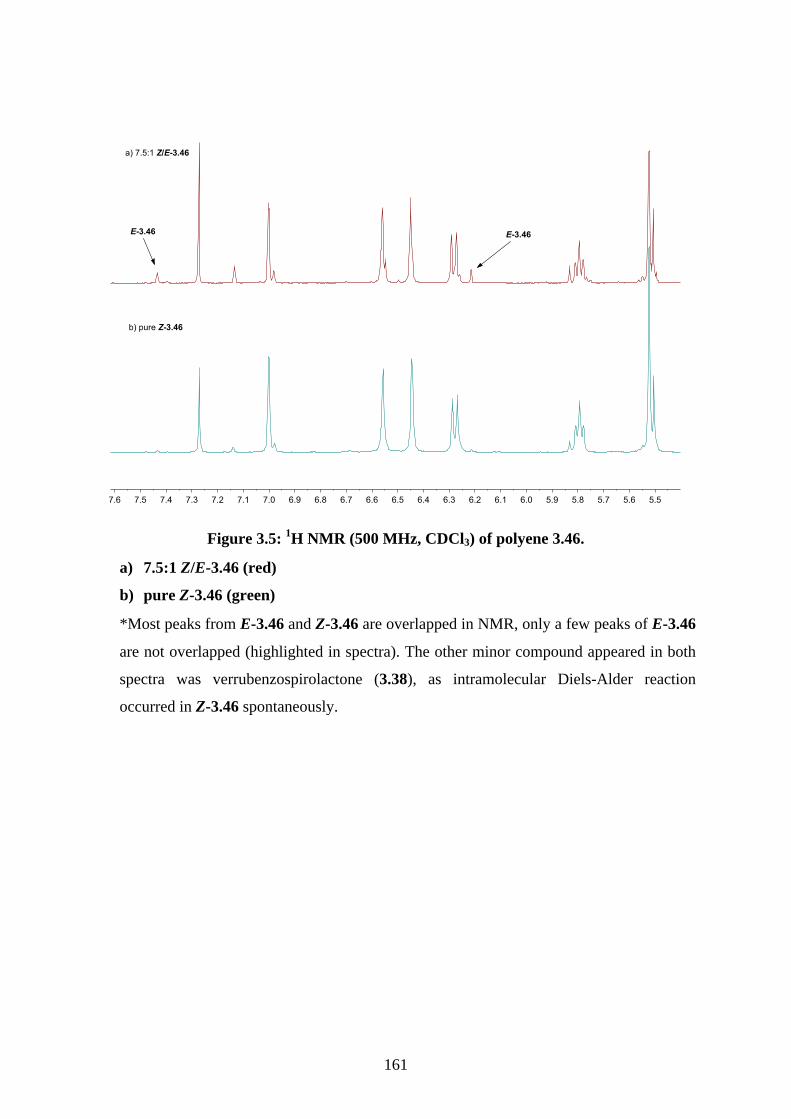
161
Figure 3.5: 1H NMR (500 MHz, CDCl3) of polyene 3.46.
a) 7.5:1 Z/E-3.46 (red)
b) pure Z-3.46 (green)
*Most peaks from E-3.46 and Z-3.46 are overlapped in NMR, only a few peaks of E-3.46
are not overlapped (highlighted in spectra). The other minor compound appeared in both
spectra was verrubenzospirolactone (3.38), as intramolecular Diels-Alder reaction
occurred in Z-3.46 spontaneously.

162
3.2.6. Bioinspired cascade reaction
After the biomimetic total synthesis of verrubenzospirolactone (3.38), we wanted to
synthesize verrubenzospirolactone (3.38) by a cascade reaction (Scheme 3.28): Knoevenagel
condensation between methyl hydroquinone (3.49) and polyene 3.68 could give ortho-
quinone methide 3.69, followed by oxa-6π-electrocyclization to form chromene 3.46, and
finally an intramolecular Diels-Alder reaction would generate verrubenzospirolactone (3.38).
Scheme 3.28: Potential cascade reaction to give verrubenzospirolactone (3.38).
However, our previous synthesis of chromene 3.55 showed that there were issues with the
selectivity and reactivity of methylhydroquinone (3.49). Hence, we decided to synthesize an
alternative molecule containing the verrubenzospirolactone skeleton. First, we decided to
substitute methylhydroquinone (3.49) with phloroglucinol (3.70), which is highly reactive
and symmetrical. In addition, when we examined the X-ray structure of 9-epi-
verrubenzospirolactone (3.62) (Figure 3.6), the aromatic proton at H-3’ is in close proximity
to the C-10 of furanone. When the aromatic ring is replaced with phloroglucinol (3.70), the
free hydroxyl group at C-3’ could potentially undergo an intramolecular oxa-Michael
reaction onto the C-10 of the furanone after the intramolecular Diels-Alder reaction which
would add one more step on top of the original cascade reaction proposal (Scheme 3.29).
Therefore, we chose to use polyene E-3.68 over Z-3.68 for the bioinspired cascade reaction,
as this would lead to the desired stereochemistry which allow the final Michael reaction.
O
O
OO
O
H
OO
intramolecularDiels-Alder reaction
exo TS
Z
Z
3.49: methylhydroquinone
Knoevenagel condensation
3.46
Z-3.68
OH
HO
O
OH
H H
H
O
O
3.38: verrubenzospirolactone
H
O
OH
O
O
Z
3.69
oxa-6π-electrocyclization
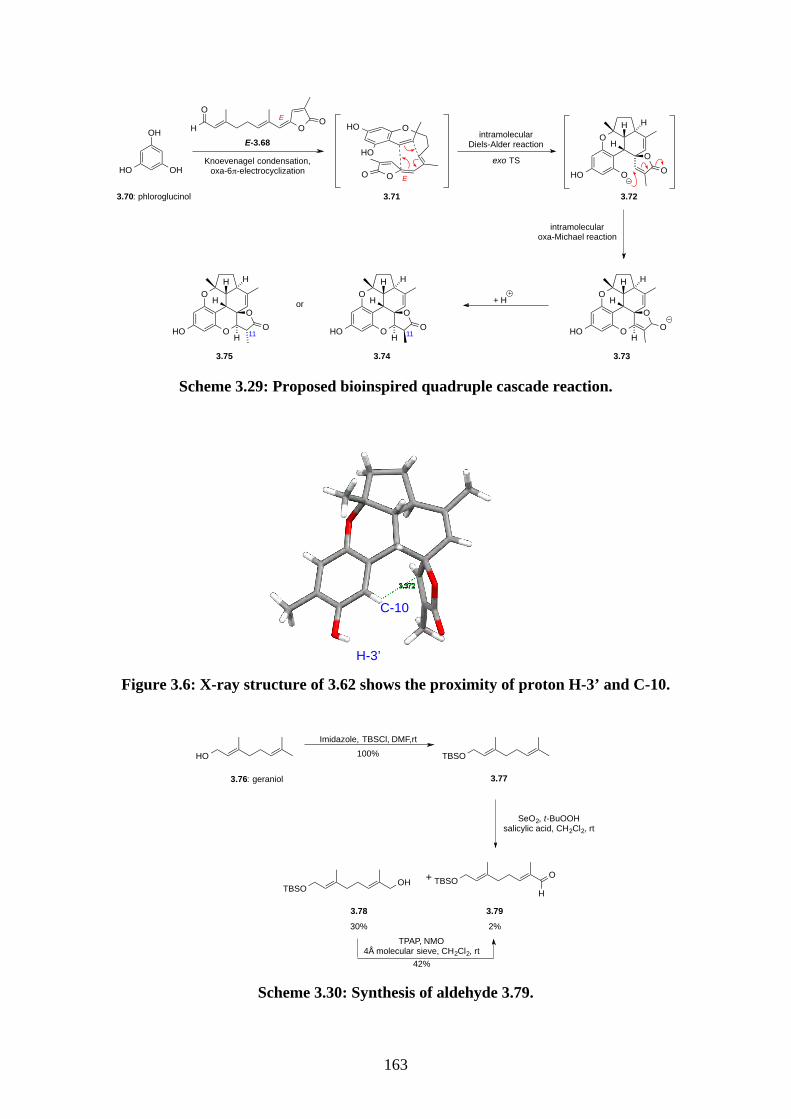
163
Scheme 3.29: Proposed bioinspired quadruple cascade reaction.
Figure 3.6: X-ray structure of 3.62 shows the proximity of proton H-3’ and C-10.
Scheme 3.30: Synthesis of aldehyde 3.79.
OH
OHHO
O
H
H
HO
H
O
OO
OHO
OO
O
H
H
HO
H
O
O
HO
O
H OO
HO
intramolecularDiels-Alder reaction
exo TS
intramolecularoxa-Michael reaction
E
E
3.70: phloroglucinol
Knoevenagel condensation, oxa-6π-electrocyclization
11
3.71 3.72
3.74
O
H
H
HO
H
O
O
HO
11
3.75
or
E-3.68
O
H
H
HO
H
O
O
HO
3.73
+ H
HO
3.76: geraniol
Imidazole, TBSCl, DMF,rt100% TBSO
3.77
SeO2, t-BuOOHsalicylic acid, CH2Cl2, rt
TBSO
3.78
OH TBSO
3.79
O+H
2%30%
TPAP, NMO4Å molecular sieve, CH2Cl2, rt
42%
H-3’
C-10

164
To synthesize aldehyde 3.68, we started by protecting geraniol (3.76) with a TBS group to
afford 3.77, followed by Riley oxidation to give 3.78.53 At first, we conducted the allylic
oxidation using standard procedure53 but we found the reaction gave a complex mixture of
products. Consulting the literature, we found salicylic acid was often used as an additive for
this reaction.54,55,56 We then added salicylic acid to the reaction mixture and it gave a
reasonable yield of alcohol 3.78 and aldehyde 3.79, alongside 25% of recovered starting
material 3.77. Alcohol 3.78 was then converted to aldehyde 3.79 by Ley oxidation (Scheme
3.30).
Scheme 3.31: Bioinspired cascade synthesis of 3.74 & 3.75.
Horner-Wadsworth-Emmons reaction between 3.79 and 3.47 gave 3.80 as a 4:1 mixture of
E/Z isomers, which could not be separated by column chromatography (Scheme 3.31). We
proceeded with the synthesis by removing the TBS group with TBAF to give 3.81, followed
by Ley oxidation to give 3.68 in 70% overall yield with a 4:1 ratio of E/Z isomers. At this
point, there was some separation between E-3.68 and Z-3.68 by column chromatography
where we could purify E-3.68 as a single stereoisomer (Scheme 3.31).
With the aldehyde E-3.68 in hand, we investigated the bioinspired cascade reaction. We first
used our previous chromene synthesis conditions with PhB(OH)2 and AcOH,57,58 but the
reaction was unsuccessful and degradation of E-3.68 was observed (Table 3.2). Consulting
the literature, Ca(OH)2 has been used as an alternative milder reagent for chromene synthesis
with phloroglucinol (3.70).59,60 To our delight, when E-3.68 and phloroglucinol (3.70) were
heated with Ca(OH)2 in EtOH, a 3:1 mixture of 3.74/3.75 with 46% overall yield was isolated.
The difference between 3.74 and 3.75 was in the stereochemistry at C-11. The relative
TBSOO
3.47, n-BuLiTHF, –78 °C to rt
85%
TBAF, THF, rt
80%
3.79 3.80E/Z = 4:1
3.81E/Z = 4:1
TPAP, NMO, 4Å MS, CH2Cl2, rt
48%
H
E
E-3.68single stereoisomer
OO PO
OEtOEt
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
+
3.74:3.75
Ca(OH)2, EtOHreflux
46%
1111
3:1 d.r.
OO
TBSO EO
OHO
EO
OH
O
OH
OHHO
3.70: phloroglucinol
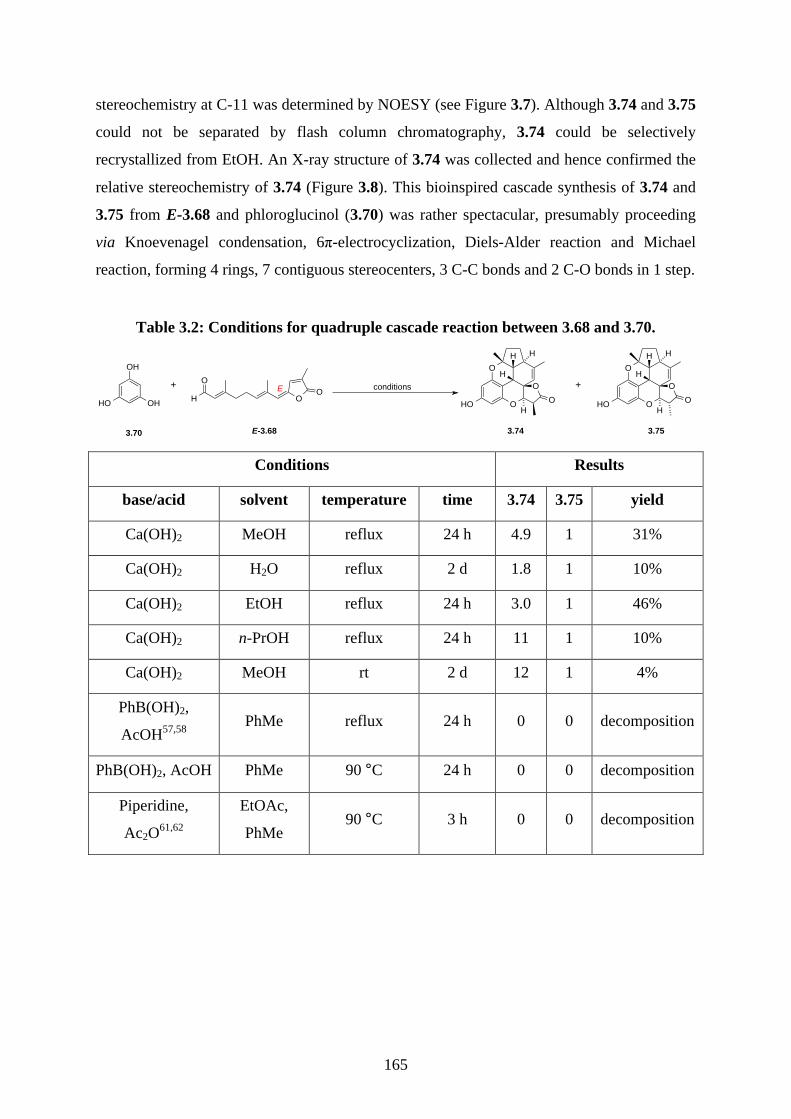
165
stereochemistry at C-11 was determined by NOESY (see Figure 3.7). Although 3.74 and 3.75
could not be separated by flash column chromatography, 3.74 could be selectively
recrystallized from EtOH. An X-ray structure of 3.74 was collected and hence confirmed the
relative stereochemistry of 3.74 (Figure 3.8). This bioinspired cascade synthesis of 3.74 and
3.75 from E-3.68 and phloroglucinol (3.70) was rather spectacular, presumably proceeding
via Knoevenagel condensation, 6π-electrocyclization, Diels-Alder reaction and Michael
reaction, forming 4 rings, 7 contiguous stereocenters, 3 C-C bonds and 2 C-O bonds in 1 step.
Table 3.2: Conditions for quadruple cascade reaction between 3.68 and 3.70.
Conditions Results
base/acid solvent temperature time 3.74 3.75 yield
Ca(OH)2 MeOH reflux 24 h 4.9 1 31%
Ca(OH)2 H2O reflux 2 d 1.8 1 10%
Ca(OH)2 EtOH reflux 24 h 3.0 1 46%
Ca(OH)2 n-PrOH reflux 24 h 11 1 10%
Ca(OH)2 MeOH rt 2 d 12 1 4%
PhB(OH)2,
AcOH57,58 PhMe reflux 24 h 0 0 decomposition
PhB(OH)2, AcOH PhMe 90 °C 24 h 0 0 decomposition
Piperidine,
Ac2O61,62
EtOAc,
PhMe 90 °C 3 h 0 0 decomposition
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
E-3.68
conditions
OH
OHHO
+E+
3.70 3.74 3.75
OO
H
O
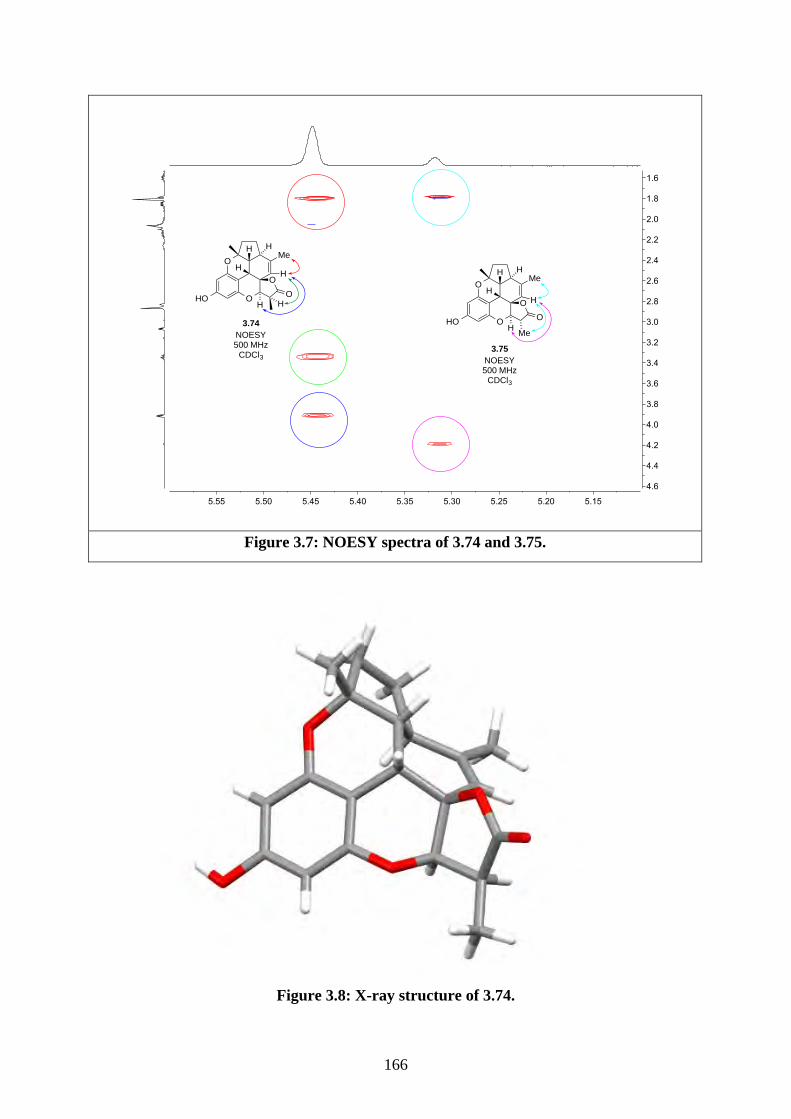
166
Figure 3.7: NOESY spectra of 3.74 and 3.75.
Figure 3.8: X-ray structure of 3.74.
OMe
H
H
HO
H
O
O
HO
Me
H
NOESY500 MHz
CDCl3
3.75
OMe
H
H
HO
H
O
O
HO
H
H
NOESY500 MHz
CDCl3
3.74

167
3.3. Summary
We have developed a concise total synthesis of verrubenzospirolactone from commercially
available materials. The overall synthetic strategy was protecting group free with good step
economy (Figure 3.9).63 Although the undesired E-3.46 was synthesized, thermal
isomerisation gave Z-3.46, followed by intramolecular Diels-Alder reaction to afford
verrubenzospirolactone. The structures of verrubenzospirolactone and 9-epi-
verrubenzospirolactone were confirmed by X-ray crystallography. In addition, E-3.46 was
isomerised by Pd(CN)2Cl2 to give predominant Z-3.46 which then converted into
verrubenzospirolactone. The transformation from E-3.46 and Z-3.46 to
verrubenzospirolactone occurred in H2O at mild temperature suggesting that a Diels-Alderase
may not be involved in the biosynthesis.
Figure 3.9: Summary of biomimetic total synthesis of verrubenzospirolactone.
We have completed a total synthesis of capillobenzopyranol by reducing E-3.46, and
successfully oxidized capillobenzopyranol to γ-hydroxybutenolide 3.45. Dehydration of γ-
hydroxybutenolide gave Z-3.67, and hydrolysis of acetate group gave the proposed
biosynthetic precursor Z-3.46 as a single stereoisomer. Z-3.46 underwent intramolecular
Diels-Alder reaction to give verrubenzospirolactone (3.38) in quantitative yield (Figure 3.10).
This synthesis suggested a highly predisposed biosynthesis of verrubenzospirolactone. We
then proposed 3.45 and Z-3.46 are undiscovered natural products. We are in the process of
sending γ-hydroxybutenolide 3.45 and Z-3.46 to the isolation chemists to investigate whether
they could be found from the coral extracts.
OH
OH
O
H O
OH
H H
H
O
O
5 steps
O
O
4 C-C bonds5 stereocentres
3 rings
verrubenzospirolactone
PO
OEtOEt

168
Figure 3.10: Summary of the synthesis of verrubenzospirolactone from
capillobenzopyranol.
The biomimetic synthesis of verrubenzospirolactone also inspired a quadruple cascade reaction, in
which the molecular complexity increases drastically, i.e. 7 stereocenters, 4 rings, 3 C-C bonds, 2 C-O
bonds were generated from 2 achiral molecules in 1 step (Figure 3.11). It is an unusual cascade
reaction, where each of the reaction is unique (unlike a radical cascade or Diels-Alder cascade
reaction). However, the reaction did produce a minor diastereoisomer (differing at C-11), which was
derived after the oxa-Michael reaction. The structure of the major isomer was confirmed by X-ray
crystallography.
Figure 3.11: Summary of the bioinspired quadruple cascade reaction.
O
OH
O
3.39: capillobenzopyranol
O
OH
O Oreduction
E
E-3.46
O
OH
O
O
OH
3.45
oxidationO
OH
O
O
Z-3.46
elimination/hydrolysis
O
OH
H H
H
O
O
3.38: verrubenzospirolactone
Z
intramolecularDiels-Alder
reaction
isomerisation/intramolecular
Diels-Alderreaction
E
O
H
H
HO
H
O
O
HO
3.74E-3.683.70: phloroglucinol
OH
OHHO
Knoevenagel condensation/6π-electrocyclization/Diels-Alder reaction/
Michael addition
quadruple cascade reaction4 rings
7 contiguous stereocenters3 C-C bonds2 C-O bondsin one step
11
3:1 d.r. (Me-11)
OO
H
O

169
3.4. References
1. Diels, O.; Alder, K. Ber. Dtsch. Chem. Ges., 1929, 62, 554. 2. Woodward, R. B.; Baer, H. J. Am. Chem. Soc., 1948, 70, 1161. 3. Schindler, C. S.; Carreira, E. M. Chem. Soc. Rev., 2009, 38, 3222. 4. Hugelshofer, C. L.; Magauer, T. J. Am. Chem. Soc., 2015, 137, 3807. 5. Gollnick, K.; Griesbeck, A. Tetrahedron, 1985, 41, 2057. 6. Kornblum, N.; Delamare, H. E. J. Am. Chem. Soc., 1951, 73, 880. 7. Kernan, M. R.; Faulkner, D. J. J. Org. Chem., 1988, 53, 2773. 8. Patil, S. N.; Stephens, B. E.; Liu, F. Tetrahedron, 2008, 64, 10831. 9. Teijeira, M.; Suarez, P. L.; Gomez, G.; Teran, C.; Fall, Y. Tetrahedron Lett., 2005, 46,
5889. 10. Wiesner, K.; Tsai, T. Y. R.; Jin, H. Helv. Chim. Acta., 1985, 68, 300. 11. Peng, S. Z.; Sha, C. K. Org. Lett., 2015, 17, 3486. 12. Pratap, R.; Ram, V. J. Chem. Rev., 2014, 114, 10476. 13. Ferreira, S. B.; da Silva, F. D.; Pinto, A. C.; Gonzaga, D. T. G.; Ferreira, V. F. J.
Heterocyclic Chem., 2009, 46, 1080. 14. Nummy, W. R.; Tarbell, D. S. J. Am. Chem. Soc., 1951, 73, 1500. 15. Crombie, L. W.; Crombie, W. M. L.; Firth, D. F. J. Chem. Soc., Perkins Trans. 1,
1988, 1263. 16. Tisdale, E. J.; Vong, B. G.; Li, H. M.; Kim, S. H.; Chowdhury, C.; Theodorakis, E. A.
Tetrahedron, 2003, 59, 6873. 17. Bowden, B. F.; Coll, J. C.; Desilva, E. D.; Decosta, M. S. L.; Djura, P. J.; Mahendran,
M.; Tapiolas, D. M. Aust. J. Chem., 1983, 36, 371. 18. Cheng, S. Y.; Huang, K. J.; Wang, S. K.; Wen, Z. H.; Chen, P. W.; Duh, C. Y. J. Nat.
Prod., 2010, 73, 771. 19. Yuan, W. P.; Cheng, S. M.; Fu, W. T.; Zhao, M.; Li, X. B.; Cai, Y. P.; Dong, J. Y.;
Huang, K. X.; Gustafson, K. R.; Yan, P. C. J. Nat. Prod., 2016, 79, 1124. 20. Rosa, C. P.; Kienzler, M. A.; Olson, B. S.; Liang, G.; Trauner, D. Tetrahedron, 2007,
63, 6529. 21. Gembus, V.; Sala-Jung, N.; Uguen, D. Bull. Chem. Soc. Jpn., 2009, 82, 843. 22. Williams, C. M.; Mander, L. N. Tetrahedron, 2001, 57, 425. 23. Mander, L. N.; Williams, C. M. Tetrahedron, 2016, 72, 1133. 24. Riley, H. L.; Friend, N. A. C. J. Chem. Soc., 1932, 2342. 25. Williams, D. R.; Atwater, B. A.; Bawel, S. A.; Ke, P.; Gutierrez, O.; Tantillo, D. J.
Org. Lett., 2014, 16, 468. 26. Yang, P.; Yao, M.; Li, J.; Li, Y.; Li, A. Angew. Chem. Int. Ed., 2016, 55, 6964. 27. Hugelshofer, C. L.; Magauer, T. Angew. Chem. Int. Ed., 2014, 53, 11351. 28. Bhattacharya, A. K.; Thyagarajan, G. Chem. Rev., 1981, 81, 415. 29. Anagnostaki, E. E.; Zografos, A. L. Org. Lett., 2013, 15, 152. 30. Stocking, E. M.; Williams, R. M. Angew. Chem. Int. Ed., 2003, 42, 3078. 31. Narayan, S.; Muldoon, J.; Finn, M. G.; Fokin, V. V.; Kolb, H. C.; Sharpless, K. B.
Angew. Chem. Int. Ed., 2005, 44, 3275. 32. Butler, R. N.; Coyne, A. G. Chem. Rev., 2010, 110, 6302. 33. Still, W. C.; Gennari, C. Tetrahedron Lett., 1983, 24, 4405. 34. Gavara, L.; Petit, C.; Montchamp, J. L. Tetrahedron Lett., 2012, 53, 5000. 35. Brittelli, D. R. J. Org. Chem., 1981, 46, 2514.

170
36. Cohen, R. J.; Fox, D. L.; Eubank, J. F.; Salvatore, R. N. Tetrahedron Lett., 2003, 44, 8617.
37. Yu, J.; Gaunt, M. J.; Spencer, J. B. J. Org. Chem., 2002, 67, 4627. 38. Singh, K.; Staig, S. J.; Weaver, J. D. J. Am. Chem. Soc., 2014, 136, 5275. 39. Dickinson, R. G.; Lotzkar, H. J. Am. Chem. Soc., 1937, 59, 472. 40. Boegman, N.; During, F.; Garbers, C. F. Chem. Commun. (London), 1966, 600. 41. Morand, P.; Salvator, J.; Kayser, M. M. J. Chem. Soc. Chem. Comm., 1982, 458. 42. Seah, K. Y.; Macnaughton, S. J.; Dallimore, J. W. P.; Robertson, J. Org. Lett., 2014,
16, 884. 43. Patil, S. N.; Liu, F. J. Org. Chem., 2008, 73, 4476. 44. Chen, I. T.; Baitinger, I.; Schreyer, L.; Trauner, D. Org. Lett., 2014, 16, 166. 45. Ferreiro-Mederos, L.; Lanners, S.; Henchiri, H.; Fekih, A.; Hanquet, G. Nat. Prod.
Res., 2009, 23, 256. 46. Furuichi, N.; Hara, H.; Osaki, T.; Mori, H.; Katsumura, S. Angew. Chem. Int. Ed.,
2002, 41, 1023. 47. Yuan, C. C.; Du, B. A.; Yang, L.; Liu, B. J. Am. Chem. Soc., 2013, 135, 9291. 48. Arenas, C.; Rodriguezhahn, L. Phytochemistry, 1990, 29, 2953. 49. Shimizu, S.; Nakamura, S.; Nakada, M.; Shibasaki, M. Tetrahedron, 1996, 52, 13363. 50. Clive, D. L. J.; Ou, L. Tetrahedron Lett., 2002, 43, 4559. 51. Clive, D. L. J.; Minaruzzaman; Ou, L. G. J. Org. Chem., 2005, 70, 3318. 52. Annangudi, S. P.; Sun, M. J.; Salomon, R. G. Synlett, 2005, 1468. 53. Hu, T.; Corey, E. J. Org. Lett., 2002, 4, 2441. 54. Umbreit, M. A.; Sharpless, K. B. J. Am. Chem. Soc., 1977, 99, 5526. 55. Otera, J.; Misawa, H.; Onishi, T.; Suzuki, S.; Fujita, Y. J. Org. Chem., 1986, 51, 3834. 56. Lee, J.; Parker, K. A. Org. Lett., 2012, 14, 2682. 57. Chauder, B. A.; Lopes, C. C.; Lopes, R. S. C.; da Silva, A. J. M.; Snieckus, V.
Synthesis, 1998, 279. 58. Pettigrew, J. D.; Cadieux, J. A.; So, S. S. S.; Wilson, P. D. Org. Lett., 2005, 7, 467. 59. Zheng, S. Y.; Li, X. P.; Tan, H. S.; Yu, C. H.; Zhang, J. H.; Shen, Z. W. Eur. J. Org.
Chem., 2013, 1356. 60. Saimoto, H.; Yoshida, K.; Murakami, T.; Morimoto, M.; Sashiwa, H.; Shigemasa, Y.
J. Org. Chem., 1996, 61, 6768. 61. Luo, G. Y.; Wu, H.; Tang, Y.; Li, H.; Yeom, H. S.; Yang, K.; Hsung, R. P. Synthesis,
2015, 47, 2713. 62. Wu, H.; Hsung, R. P.; Tang, Y. J. Org. Chem., 2017, 82, 1545. 63. Gaich, T.; Baran, P. S. J. Org. Chem., 2010, 75, 4657.

171
3.5. Experimental
3.5.1. General methods
All chemicals used were purchased from commercial suppliers and used as received. All reactions
were performed under an inert atmosphere of N2. All organic extracts were dried over anhydrous
magnesium sulfate. Thin layer chromatography was performed using aluminium sheets coated with
silica gel F254. Visualization was aided by viewing under a UV lamp and staining with ceric
ammonium molybdate or KMnO4 stain followed by heating. All Rf values were measured to the
nearest 0.05. Flash chromatography was performed using 40-63 micron grade silica gel. Melting
points were recorded on a digital melting point apparatus and are uncorrected. Infrared spectra were
recorded using an FT-IR spectrometer as the neat compounds. High field NMR spectra were
recorded using a 500 MHz spectrometer (1H at 500 MHz, 13C at 125 MHz). Solvent used for spectra
were CDCl3 unless otherwise specified. 1H chemical shifts are reported in ppm on the δ-scale
relative to TMS (δ 0.0) and 13C NMR are reported in ppm relative to CDCl3 (δ 77.00). Multiplicities
are reported as (br) broad, (s) singlet, (d) doublet, (t) triplet, (q) quartet, (quin) quintet, (sext) sextet,
(hept) heptet and (m) multiplet. All J-values were rounded to the nearest 0.1 Hz. ESI high
resolution mass spectra were recorded on a ESI-TOF mass spectrometer.

172
3.5.2. Experimental procedures
To a suspension of methylhydroquinone (3.49) (8.0 g, 64.4 mmol), citral (3.27) (16.5 mL, 96.7
mmol), PhB(OH)2 (9.4 g, 77.3 mmol) and AcOH (4.4 mL, 77.3 mmol) in PhMe (300 mL) was
heated at reflux for 24 h using a Dean-Stark apparatus. The reaction was cooled to room
temperature, filtered through SiO2 and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (10:1, petrol/EtOAc) which gave a 2.6:1:1:1 mixture of
3.55:3.56:3.57:3.58, followed by a second flash column chromatography purification on SiO2 (1:1,
petrol/CH2Cl2) to give chromene 3.55 as a yellow oil (2.4 g, 14%). 3.56, 3.57 and 3.58 were later
eluted, with only a few of fractions that were pure for characterisation.
Data for 3.55:
Rf = 0.65 (3:1, petrol/EtOAc)
IR (neat): 3388, 2969, 2915, 1656, 1496, 1452, 1377, 1257, 1178 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.56 (s, 1H), 6.40 (s, 1H), 6.23 (d, J = 9.8 Hz, 1H), 5.52 (d, J = 9.8
Hz, 1H), 5.10 – 5.07 (m, 1H), 4.58 (s, 1H), 2.17 (s, 3H), 2.14 – 2.05 (m, 2H), 1.73 – 1.67 (m, 1H),
1.66 (s, 3H), 1..59 – 1.57 (m, 1H), 1.57 (s, 3H), 1.35 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 147.5, 146.8, 131.7, 129.9, 124.7, 124.3, 122.6, 119.8, 118.3,
112.7, 78.2, 41.0, 26.1, 15.8, 22.9, 17.7, 16.0.
HRMS (ESI): calculated for C17H23O2 259.1693 [M+H]+, found 259.1691.
Data for 3.56:
Rf = 0.60 (3:1, petrol/EtOAc)
IR (neat): 3360, 2968, 2923, 2855, 1592, 1465, 1376, 1312, 1194 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.47 (d, J = 2.8 Hz, 1H), 6.32 (d, J = 3.0 Hz, 1H), 6.25 (d, J = 9.8
Hz, 1H), 5.58 (d, J = 9.8 Hz, 1H), 5.11 – 5.08 (m, 1H), 4.41 (s, 1H), 2.13 (s, 3H), 2.11 – 2.07 (m,
2H), 1.68 – 1.67 (m, 1H), 1.66 (s, 3H), 1.65 – 1.63 (m, 1H), 1.57 (s, 3H), 1.36 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 148.6, 145.1, 131.7, 130.9, 126.5, 124.4, 123.0, 121.5, 117.2,
110.4, 78.0, 41.0, 26.0, 25.8, 22.8, 17.7, 15.6.
HRMS (ESI): calculated for C17H23O2 259.1693 [M+H]+, found 259.1696.
O
OH
OH
OH
O
H
PhB(OH)2, AcOHPhMe, reflux
3.49: methyl hydroquinone
3.27: citral
3.5514%
O
OHO
OH
H
H+ + +
O
OH
H
H
3.56 3.57 3.58

173
Data for 3.57 and 3.58:
Rf = 0.65 (3:1, petrol/EtOAc)
IR (neat): 3388, 2975, 2832, 1499, 1456, 1413, 1367, 1274, 1186 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.74 (s, 1H), 6.68 (s, 1H), 6.58 (s, 1H), 6.54 (s, 1H), 5.84 – 5.82
(m, 2H), 4.45 (s, 1H), 4.43 (s, 1H), 3.46 (s, 1H), 3.11 (d, J = 10.7 Hz, 1H), 2.17 (s, 3H), 2.15 (s,
3H), 2.10 – 2.09 (m, 2H), 2.02 – 1.98 (m, 1H), 1.92 (dd, J = 17.6, 5.5 Hz, 1H), 1.88 – 1.78 (m, 3H),
1.72 (s, 3H), 1.69 (s, 3H), 1.68 – 1.63 (m, 1H), 1.60 – 1.52 (m, 3H), 1.40 (m, 6H), 1.38 – 1.26 (m,
3H), 1.25 (s, 3H), 1.14 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 147.5, 147.13, 147.05, 145.7, 135.3, 135.1, 123.1, 122.09, 122.05,
119.10, 119.08, 114.4, 111.9, 111.3, 107.5, 75.6, 60.6, 44.9, 43.9, 39.5, 34.2, 32.3, 31.0, 30.5, 30.1,
28.1, 27.7, 26.7, 25.8, 25.5, 25.1, 24.8, 23.7, 23.6, 23.0, 21.2, 20.7, 20.0, 19.4, 17.8, 15.7, 14.3.

174
To a solution of 3.55 (2.28 g, 8.83 mmol) in CH2Cl2 (150 mL) at room temperature was added SeO2
(196 mg, 1.76 mmol) and t-BuOOH (5.5 M, 5.8 mL, 31.7 mmol). The reaction was stirred at room
temperature for 2 h, then quenched with saturated Na2SO3 aqueous solution (50 mL). The aqueous
layer was separated and extracted with CH2Cl2 (50 mL). The combined organic extracts were
washed with brine (100 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue
was purified by flash column chromatography on SiO2 (6:1 → 2:1, petrol/EtOAc gradient elution)
to give alcohol 3.59 as a yellow oil (803 mg, 33%), further elution gave aldehyde 3.48 as a yellow
oil (167 mg, 7%).
Data for alcohol 3.59:
Rf = 0.40 (1:1, petrol/EtOAc)
IR (neat): 3357, 2976, 2924, 2857, 1669, 1625, 1497, 1458, 1427, 1376, 1265, 1177 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.55 (s, 1H), 6.42 (s, 1H), 6.27 (d, J = 9.8 Hz, 1H), 5.52 (d, J = 9.8
Hz, 1H), 5.40 (t, J = 7.11 Hz, 1H), 4.32 (s, 1H), 3.97 (s, 2H), 2.22 – 2.13 (overlapped m, 2H), 2.18
(s, 3H), 1.76 – 1.66 (m, 2H), 1.63 (s, 3H), 1.36 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 147.6, 146.7, 134.9, 129.7, 126.2, 124.8, 122.8, 119.6, 118.2,
112.7, 78.1, 69.1, 40.7, 26.3, 22.6, 16.1, 13.8.
HRMS (ESI): calculated for C17H22O3Na 297.1461 [M+Na]+, found 297.1456.
Data for aldehyde 3.48:
Rf = 0.30 (5:1, petrol/EtOAc)
IR (neat): 3390, 2972, 2925, 1671, 1638, 1496, 1458, 1427, 1377, 1265, 1178 cm-1. 1H NMR (500 MHz, CDCl3): δ 9.35 (s, 1H), 6.54 (s, 1H), 6.51 (dt, J = 7.36, 1.38 Hz, 1H), 6.44 (s,
1H), 6.29 (d, J = 9.8 Hz, 1H), 5.50 (d, J = 9.8 Hz, 1H), 5.23 (s, 1H), 2.55 – 2.45 (m, 2H), 2.18 (s,
3H), 1.89 – 1.77 (m, 2H), 1.70 (s, 3H), 1.39 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 195.8, 155.3, 148.0, 146.3, 139.3, 128.8, 125.3, 123.9, 119.2,
118.2, 112.8, 77.8, 39.5, 26.4, 24.3, 16.1, 9.2.
HRMS (ESI): calculated for C17H21O3 273.1485 [M+H]+, found 273.1482.
O
OH3.55
O
OH
CH2OH
3.59
33% (+ 7% of 3.48)
SeO2, t-BuOOHCH2Cl2, rt
O
OH
CHO
+
3.48

175
To a solution of DMSO (0.34 mL, 4.73 mmol) in CH2Cl2 (30 mL) in −78 °C was added oxalyl
chloride (0.2 mL, 2.37 mmol). The mixture was stirred at −78 °C for 10 min, then added to a
solution of 3.59 (590 mg, 2.15 mmol) in CH2Cl2 (10 mL) at −78 °C. The solution was warmed to
room temperature for 10 min, then cooled to −78 °C. Et3N (1.79 mL, 12.9 mmol) was added at −78
°C. The mixture was warmed to room temperature over 20 min, then quenched with H2O (30 mL).
The aqueous layer was separated and extracted with CH2Cl2 (30 mL). The combined organic
extracts were washed with brine (100 mL), dried over MgSO4, filtered and concentrated in vacuo.
The residue was purified by flash column chromatography on SiO2 (4:1, petrol/EtOAc) to give 3.48
as a yellow oil (386 mg, 66%). Data for 3.48 matched that previously obtained.
O
OH
CH2OH
3.59
O
OH
CHO
3.48
(COCl)2, DMSO, Et3NCH2Cl2, −78 °C to rt
66%

176
To a solution of 3.60 (5.04 g, 51.4 mmol) in benzene (100 mL) at room temperature was added
NBS (10.1 g, 56.5 mmol) and AIBN (83 mg, 0.51 mmol). The reaction was heated at reflux for 2 h,
then cooled to room temperature and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (3:1, petrol/EtOAc) to give 3.61 as a yellowish oil (8.85 g, 97%).
Data of 3.61 matched from literature.1
Data for 3.61:
Rf = 0.45 (3:1, petrol/EtOAc)
IR (neat): 3320, 1745, 1667, 1440, 1349, 1208, 1096, 1009 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.20 (s, 1H), 6.83 (s, 1H), 2.01 (s, 3H).
OO BrOO
NBS, AIBNbenzene, reflux
97%
3.613.60

177
To a solution of 3.61 (8.85 g, 50.0 mmol) was added in P(OEt)3 (8.57 mL, 50.0 mmol). The mixture
was stirred at 110 °C for 3 h. The crude was purified by flash column chromatography on SiO2 (1:3,
petrol/EtOAc) to give 3.47 as a colourless oil (10.3 g, 88%). Data of 3.47 matched from literature.2
Data for 3.47:
Rf = 0.15 (1:3, petrol/EtOAc)
IR (neat): 2987, 2933, 2913, 1767, 1654, 1479, 1446, 1394, 1371, 1243, 1225, 1164 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.19 (s, 1H), 5.16 (d, J = 15.5 Hz, 1H), 4.25 – 4.08 (m, 4H), 1.98
(s, 3H), 1.37 (t, J = 7.5 Hz, 3H), 1.32 (dd, J = 13.9, 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.4 (d, J = 1.5 Hz), 142.8 (d, J = 6.2 Hz), 131.3 (d, J = 8.2 Hz),
76.4 (d, J = 166.0 Hz), 64.4 (d, J = 7.0 Hz), 64.0 (d, J = 7.0 Hz), 16.4 (d, J = 5.6 Hz), 10.8 (d, J =
2.1 Hz).
OO PO
OEtOEt
OO Br P(OEt)3, 110 °C
88%
3.473.61

178
To a solution of 3.47 (1.0 g, 4.27 mmol) in THF (20 mL) at −78 °C was added n-BuLi (2.0 M
solution in cyclohexane, 1.96 mL, 3.93 mmol). The reaction was stirred and gradually warmed to
room temperature over 15 min. A solution of 3.48 (427 mg, 1.57 mmol) in THF (10 mL) was added
at room temperature. The mixture was stirred at room temperature for 2 h. The reaction was
quenched with 1 M HCl (20 mL). The aqueous layer was separated and extracted with Et2O (20
mL). The combined organic extracts were washed with brine (30 mL), dried over MgSO4, filtered
and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (4:1,
petrol/EtOAc) to give an inseparable mixture of E-3.46 and Z-3.46 (4.5:1) as a yellow oil (435 mg,
79%).
Rf = 0.35 (3:1, petrol/EtOAc)
IR (neat): 3397, 2972, 2923, 1725, 1620, 1598, 1497, 1458, 1426, 1371, 12487, 1174, 1060 cm-1.
HRMS (ESI): calculated for C22H25O4 353.1747 [M+H]+, found 353.1744.
Data for E-3.46: 1H NMR (500 MHz, CDCl3): δ 7.42 (s, 1H), 6.55 (s, 1H), 6.43 (s, 1H), 6.28 (d, J = 9.8 Hz, 1H),
6.21 (s, 1H), 5.76 (t, J = 7.5 Hz, 1H), 5.52 – 5.50 (m, 1H), 4.42 (s, 1H), 2.39 – 2.30 (m, 2H), 2.18
(s, 3H), 2.03 (s, 3H), 1.88 (s, 3H), 1.81 – 1.70 (m, 2H), 1.37 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 170.7, 147.8, 146.9, 146.6, 139.6, 135.1, 130.8, 130.1, 129.3,
124.9, 123.1, 119.53, 119.45, 118.3, 112.7, 77.9, 40.4, 26.4, 23.8, 16.1, 15.5, 11.0.
Data for Z-3.46: 1H NMR (500 MHz, CDCl3): δ 6.99 (s, 1H), 6.55 (s, 1H), 6.43 (s, 1H), 6.27 (d, J = 9.8 Hz, 1H),
5.79 (t, J = 7.5 Hz, 1H), 5.52 – 5.50 (m, 2H), 4.80 (s, 1H), 2.38 – 2.30 (m, 2H), 2.18 (s, 3H), 2.01
(s, 3H), 1.99 (s, 3H), 1.80 – 1.70 (m, 2H), 1.37 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.9, 147.8, 146.6, 145.2, 139.7, 139.4, 129.3, 127.2, 125.0,
123.0, 119.6, 119.5, 118.3, 117.5, 112.7, 77.9, 40.3, 26.3, 23.7, 16.1, 15.1, 15.1.
O
OH
HH
O
H
O
EO
OH
CHO
OO PO
OEtOEt3.47 =
3.47, n-BuLiTHF, −78 °C to rt
79%
E/Z = 4.5:13.48 3.46

179
1H NMR (500 MHz, d6-DMSO): δ 8.70 (s, 1H), 7.43 (s, 1H), 6.47 (s, 1H), 6.45 (s, 1H), 6.32 (d, J
= 9.8 Hz, 1H), 5.85 (t, J = 7.6, 1H), 5.78 (s, 1H), 5.61 (d, J = 9.8 Hz, 1H), 2.25 – 2.21 (m, 2H), 2.03
(s, 3H), 1.91 (s, 3H), 1.90 (s, 3H), 1.69 – 1.66 (m, 2H), 1.28 (s, 3H). 13C NMR (125 MHz, d6-DMSO): δ 170.7, 148.9, 144.8, 144.7, 140.5, 139.1, 131.3, 129.1, 126.1,
124.6, 122.7, 118.7, 117.6, 116.6, 112.0, 77.2, 39.3, 25.6, 23.0, 16.0, 14.6, 10.1.

180
To a solution of 3.47 (354 mg, 1.51 mmol) in THF (10 mL) at 0 ºC was added KOt-Bu (141 mg,
1.26 mmol). The mixture was stirred at 0 ºC for 20 min, followed by addition of 3.48 (137 mg, 0.50
mmol) in THF (5 mL). The reaction was stirred at 0 ºC for 30 min, then warmed to room
temperature and stirred for 1 h, then quenched with 1 M HCl (10 mL). The organic layer was
separated and the aqueous layer was extracted with EtOAc (2 × 10 mL). The combined organic
extracts were washed with brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo.
The residue was purified by flash column chromatography on SiO2 (4:1, petrol/EtOAc) to give an
inseparable mixture of E-3.46 and Z-3.46 (4.5:1) as a yellow oil (75 mg, 45%). Data of E-3.46 and
Z-3.46 matched previously obtained.
To a solution of 3.47 (250 mg, 1.07 mmol) in THF (5 mL) at –78 ºC was added LDA (2.0 M, 0.54
mL, 1.07 mmol). The mixture was stirred at –78 ºC for 5 min, warmed to room temperature and
stirred for 15 min, then cooled back to –78 ºC. 3.48 (97 mg, 0.36 mmol) in THF (5 mL) was added
to the mixture. The reaction was stirred at –78 ºC for 30 min, then warmed to room temperature and
stirred for 3 h, then quenched with saturated NH4Cl solution (10 mL). The organic layer was
separated and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic
extracts were washed with brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo.
The residue was purified by flash column chromatography on SiO2 (4:1, petrol/EtOAc) to give an
inseparable mixture E-3.46 and Z-3.46 (4:1) as a yellow oil (44 mg, 35%). Data of E-3.46 and Z-
3.46 matched previously obtained.
O
OH
HH
O
H
O
EO
OH
CHO
OO PO
OEtOEt3.47
3.47, KOt-BuTHF, −78 °C to rt
45%
E/Z = 4.5:13.48 3.46
O
OH
HH
O
H
O
EO
OH
CHO
OO PO
OEtOEt3.47 =
3.47, LDATHF, −78 °C to rt
35%
E/Z = 4.5:13.48 3.46

181
A solution of a 4.5:1 mixture of E-3.46 and Z-3.46 (133 mg, 0.377 mmol) in PhMe (8 mL) was
heated at reflux for 1 h. The solution was cooled to room temperature and the resultant precipitate
was collected by vacuum filtration to give 3.62 (68 mg, 51%) as a white solid. The filtrate was
concentrated in vacuo and the residue was purified by flash column chromatography on SiO2 (4:1,
petrol/EtOAc) to give verrubenzospirolactone (3.38) as a white solid (39 mg, 29%).
Data for 3.62:
Rf = 0.55 (petrol/EtOAc, 3:1)
IR (neat): 3348, 2965, 1737, 1656, 1627, 1516, 1462, 1442, 1418, 1372, 1191 cm-1.
MP: 220 – 224 °C 1H NMR (500 MHz, d6-DMSO): δ 8.73 (s, 1H), 6.79 (d, J = 1.5 Hz, 1H), 6.45 (s, 1H), 6.33 (s,
1H), 6.04 – 6.03 (m, 1H), 3.46 (d, J = 9.22 Hz, 1H), 3.32 (s, 3H), 2.54 – 2.48 (m, 1H), 2.06 – 2.01
(m, 1H), 1.99 (s, 3H), 1.97 – 1.91 (m, 2H), 1.82 (dd, J = 12.1, 9.3 Hz, 1H), 1.75 (s, 3H), 1.68 (d, J =
1.5 Hz, 3H), 1.58 – 1.51 (m, 1H). 13C NMR (125 MHz, d6-DMSO): δ 173.2, 151.6, 149.0, 145.4, 143.5, 126.6, 123.7, 122.0, 119.8,
117.8, 114.8, 88.6, 81.8, 48.5, 41.3, 38.7, 37.3, 23.7, 22.4, 19.2, 15.7, 10.1. 1H NMR (500 MHz, CDCl3): δ 6.57 (s, 1H), 6.53 (s, 1H), 6.39 (s, 1H), 5.54 (s, 1H), 5.12 (s, 1H),
3.60 (d, J = 9.7 Hz, 1H), 2.62 – 2.56 (m, 1H), 2.18 (dd, J = 7.1, 1.7 Hz, 1H), 2.15 (s, 3H), 2.06 –
1.99 (m, 2H), 1.84 (dd, J = 12.2, 9.8 Hz, 1H), 1.80 (s, 3H), 1.78 (s, 3H), 1.60 – 1.51 (m, 1H), 1.22
(s, 3H). 13C NMR (125 MHz, CDCl3): δ 175.1, 151.2, 148.5, 147.1, 143.8, 128.0, 124.6, 122.2, 120.8,
118.8, 115.0, 89.7, 82.7, 49.7, 42.4, 39.5, 37.9, 24.6, 23.0, 19.6, 15.9, 10.4.
HRMS (ESI): calculated for C22H25O4 353.1747 [M+H]+, found 353.1751.
Data for verrubenzospirolactone (3.38):
Rf = 0.40 (petrol/EtOAc, 3:1)
IR (neat): 3385, 2929, 1730, 1663, 1500, 1437, 1412, 1189 cm-1
MP: 189 – 193 °C
O
OH
HH
O
H
O
E O
OH
H H
H
O
O
O
OH
H H
H
O
O
3.38: verrubenzospirolactone (29%)3.62 (51%)
+PhMe, reflux
E/Z = 4.5:13.46

182
1H NMR (500 MHz, CDCl3): δ 7.31 (s, 1H), 7.11 (d, J = 1.4 Hz, 1H), 6.54 (s, 1H), 5.08 (s, 1H),
4.88 (s, 1H), 3.24 (d, J = 4.6 Hz, 1H), 2.59 – 2.53 (m, 1H), 2.15 (s, 3H), 2.12 – 2.09 (m, 1H), 2.01 –
1.98 (m, 1H), 1.96 (d, J = 1.3 Hz, 3H), 1.94 – 1.91 (m, 1H), 1.81 (dd, J = 11.9, 4.8 Hz, 1H), 1.67 (s,
3H), 1.40 – 1.36 (m, 1H), 1.33 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 174.2, 153.5, 147.6, 146.5, 143.2, 128.2, 124.6, 120.1, 119.8,
117.0, 115.6, 89.2, 82.1, 50.3, 40.9, 39.2, 36.7, 25.2, 24.0, 20.4, 15.8, 10.6.
HRMS (ESI): calculated for C22H25O4 353.1747 [M+H]+, found 353.1744.

183
A solution of a 4.5:1 mixture of E-3.46 and Z-3.46 (135 mg, 0.383 mmol) in PhMe (8 mL) was
stirred at room temperature for 16 h. The solution was heated reflux for a further 1 h. The solution
was cooled to room temperature and the resultant precipitate was collected by vacuum filtration to
give 3.62 (57 mg, 42%) as a white solid. The filtrate was concentrated in vacuo and the residue was
purified by flash column chromatography on SiO2 (4:1, petrol/EtOAc) to give
verrubenzospirolactone (3.38) as a white solid (54 mg, 40%). Data for 3.62 and 3.38 matched that
previously obtained.
A suspension of a 4.5:1 mixture of E-3.46 and Z-3.46 (142 mg, 0.403 mmol) in H2O (20 mL) was
stirred at 50 °C for 40 h. The mixture was cooled to room temperature and extracted with EtOAc (3
× 10 mL). The combined organic extracts were washed with brine (20 mL) dried over MgSO4,
filtered and concentrated in vacuo. The residue was suspended in toluene and the solid was
collected by vacuum filtration to give 3.62 (38 mg, 38%) as a white solid. The filtrate was
concentrated in vacuo and the residue was purified by flash column chromatography on SiO2 (4:1,
petrol/EtOAc) to give verrubenzospirolactone (3.38) as a white solid (58 mg, 41%). Data for 3.62
and 3.38 matched that previously obtained.
PhMe, rt, 16 h, then reflux, 1 h
O
OH
HH
O
H
O
E O
OH
H H
H
O
O
O
OH
H H
H
O
O3.38: verrubenzospirolactone (40%)3.62 (42%)
+
E/Z = 4.5:13.46
H2O, 50 ̊ CO
OH
HH
O
H
O
E O
OH
H H
H
O
O
O
OH
H H
H
O
O3.38: verrubenzospirolactone (41%)3.62 (38%)
+
E/Z = 4.5:13.46

184
To a solution of a 4.5:1 mixture of E-3.46 and Z-3.46 (254 mg, 0.721 mmol) in DMF (20 mL) was
added Pd(MeCN)2Cl2 (27 mg, 0.10 mmol) at room temperature. The mixture was stirred at room
temperature for 40 h. The mixture was quenched with H2O and extracted with EtOAc (3 × 10 mL).
The combined organics were washed with brine (20 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (4:1,
petrol/EtOAc) to give an inseparable 7.5:1 mixture of Z-3.46 and E-3.46 as a yellow oil (201 mg,
79%). Data for Z-3.46 and E-3.46 matched that previously obtained.
A solution of a 7.5:1 mixture of Z-3.46 and E-3.46 (108 mg, 0.306 mmol) in PhMe (7 mL) was
heated at reflux for 1 h. The solution was cooled to room temperature and concentrated in vacuo.
The residue was purified by flash column chromatography on SiO2 (6:1 → 2:1, petrol/EtOAc
gradient elution) to give 3.62 as a white solid (13 mg, 12%). Further elution gave
verrubenzospirolactone (3.38) as a white solid (75 mg, 69%). Data for 3.62 and 3.38 matched that
previously obtained.
A suspension of a 7.5:1 mixture of Z-3.46 and E-3.46 (110 mg, 0.312 mmol) in H2O (15 mL) was
stirred at 50 °C for 60 h. The mixture was cooled to room temperature and extracted with EtOAc (3
× 10 mL). The combined organic extracts were washed with brine (20 mL) dried over MgSO4,
filtered and concentrated in vacuo. The residue was purified by flash column chromatography on
SiO2 (6:1 → 2:1, petrol/EtOAc gradient elution) to give 3.62 as a white solid (11 mg, 10%). Further
Pd(MeCN)2Cl2, DMF, rt
E/Z = 4.5:1
O
OH
HH
O
H
O
E O
OH
HH
O O
H
Z
Z/E = 7.5:1
79%
3.46 3.46
PhMe, refluxO
OH
HH
O O
H
ZO
OH
H H
H
O
O
O
OH
H H
H
O
O
3.38: verrubenzospirolactone (69%)3.62 (12%)
+
Z/E = 7.5:13.46
H2O, 50 ̊ CO
OH
HH
O O
H
ZO
OH
H H
H
O
O
O
OH
H H
H
O
O
XX: verrubenzospirolactone (61%)XX (10%)
+
Z/E = 7.5:1XX

185
elution gave verrubenzospirolactone (3.38) as a white solid (67 mg, 61%). Data for 3.62 and 3.38
matched that previously obtained.
To a solution of 3.48 (10 mg, 0.04 mmol) in t-BuOH (3 mL) at room temperature was added 3.60
(12 mg, 0.12 mmol) and piperidine (0.02 mL, 0.19 mmol). The reaction was stirred at reflux for 2 d,
then cooled to room temperature. The reaction was quenched with 1 M HCl (5 mL), then extracted
with Et2O (2 × 10 mL). The combined organic extracts were dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (5:1,
petrol/EtOAc) to give 3.62 as a white solid (1 mg, 8%). Further elution gave
verrubenzospirolactone (3.38) as a white solid (3 mg, 25%). Data of 3.62 and 3.38 matched
previously obtained.
To a solution of 3.48 (17 mg, 0.06 mmol) in MeOH (10 mL) at room temperature was added 3.60
(19 mg, 0.19 mmol) and piperdine (0.02 mL, 0.19 mmol). The reaction was stirred at reflux for 12
h, then cooled to room temperature and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (3:1, petrol/EtOAc) to give verrubenzospirolactone (3.38) as a
white solid (3 mg, 14%). Data of 3.38 matched previously obtained.
piperdine, t-BuOH, refluxO
OH
CHO
3.48
OO
3.60 O
OH
H H
H
O
O
O
OH
H H
H
O
O
3.38: verrubenzospirolactone (25%)3.62 (8%)
+
piperdine, MeOH, refluxO
OH
CHO
3.48
OO
3.60 O
OH
H H
H
O
O
3.38: verrubenzospirolactone
14%

186
A suspension of a 4.5:1 mixture of E-3.46 and Z-3.46 (1.67 g, 4.74 mmol) in Et2O (60 mL) at 0 °C
was added LiAlH4 (540 mg, 14.2 mmol). The reaction was stirred at room temperature at 0 °C for 1
h, then quenched with 1 M HCl (75 mL). The organic extract was separated and the aqueous layer
was extracted with Et2O (2 × 50 mL). The combined organic extracts were washed with brine (50
mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (8:1, petrol/EtOAc) to give capillobenzopyranol (3.39) as a
colourless oil (840 mg, 52%).
Data for 3.39:
Rf = 0.35 (3:1, petrol/EtOAc)
IR (neat): 3416, 2924, 1497, 1457, 1426, 1371, 1175, 1114, 918, 871 cm-1. 1H NMR (500 MHz, CDCl3) δ 7.05 (s, 1H), 6.55 (s, 1H), 6.40 (s, 1H), 6.24 (d, J = 9.8 Hz, 1H),
5.85 (s, 1H), 5.52 (d, J = 9.8 Hz, 1H), 5.20 (td, J = 7.1, 1.1 Hz, 1H), 4.37 (s, 1H), 3.20 (s, 2H), 2.21
– 2.09 (m, 2H), 2.17 (s, 3H), 1.97 (d, J = 1.0 Hz, 3H), 1.76 – 1.62 (m, 2H), 1.57 (s, 3H), 1.35 (s,
3H). 13C NMR (125 MHz, CDCl3) δ 154.3, 147.4, 146.7, 137.7, 132.0, 129.7, 126.4, 124.5, 122.4,
120.5, 119.6, 118.1, 112.5, 108.8, 77.9, 40.7, 38.4, 26.0, 22.8, 15.9, 15.8, 9.8.
HRMS (ESI): calculated for C22H28O3Na 363.1931 [M+Na]+, found 363.1935.
O
OH
O
3.39: capillobenzopyranol
O
OH
O O LiAlH4, Et2O, 0 °C
52%
E
3.46

187
To a solution of capillobenzopyranol (3.39) (98 mg, 0.29 mmol) in MeOH (8 mL) at 0 °C was
added rose bengal (1 mg). The solution was stirred under O2 with 500 Wlamp for 2 h. The solution
was concentrated in vacuo to give 3.64 (116 mg) which was unstable to chromatography and used
in next step without purification.
Partial data for 3.64:
Rf = 0.10 (3:1, petrol/EtOAc) 1H NMR (500 MHz, CDCl3): δ 8.81 (s, 1H), 6.56 (s, 1H), 6.43 (s, 1H), 6.26 (d, J = 9.8 Hz, 1H),
5.60 (s, 1H), 5.52 (d, J = 9.8 Hz, 1H), 5.16 (q, J = 7.3 Hz, 1H), 4.40 (d, J = 9.1 Hz, 3H), 3.49 (s,
1H), 3.21 (s, 3H), 2.50 � 2.41 (m, 2H), 2.18 (s, 3H), 2.16 � 2.06 (m, 2H), 1.79 (s, 3H), 1.72 � 1.56
(m, 2H), 1.62 (s, 3H), 1.35 (s, 3H). 13C NMR (125MHz, CDCl3): δ 147.4, 146.7, 137.08, 137.07, 130.3, 129.7, 129.6, 129.52, 129.51,
129.38, 129.37, 124.6, 124.5, 122.5, 119.6, 118.2, 118.1, 114.2, 114.1, 112.51, 112.47, 110.63,
110.62, 77.9, 77.8, 50.6, 48.68, 48.65, 40.7, 40.6, 26.0, 25.9, 22.9, 22.8, 17.62, 17.58, 15.9, 12.12,
12.10.
O
OH
O
3.39: capillobenzopyranol
O2, rose bengalMeOH, hν, 0 °C
O
OH
O
OOH
OMe
3.64

188
To a solution of 3.64 (116 mg, 0.29 mmol) in pyridine (5 mL) was added Ac2O (25 mg, 0.25 mmol)
at room temperature and stirred for 20 min. The mixture was diluted with EtOAc (15 mL) and
washed sequentially with H2O (10 mL), saturated CuSO4 solution (2 x 10 mL), brine (10 mL), dried
over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography
on SiO2 (4:1, petrol/EtOAc) to give 3.65 (1:1 mixture of diastereoisomers) as a colourless oil (23
mg, 24% over 2 steps).
Data for 3.65:
Rf = 0.15 (3:1, petrol/EtOAc)
IR (neat): 3430, 2925, 1747, 1457, 1427, 1178, 1141, 965, 872, 759 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.67 (dd, J = 3.2, 1.5 Hz, 1H), 6.55 (s, 1H), 6.42 (s, 1H), 6.26 (d, J
= 9.8 Hz, 1H), 5.52 – 5.49 (m, 1H), 5.20 (t, J = 6.8 Hz, 1H), 4.44 (s, 1H), 3.19 (s, 3H), 2.59 – 2.48
(m, 2H), 2.18 (s, 3H), 2.16 – 2.04 (m, 2H), 1.91 (d, J = 1.6 Hz, 3H), 1.71 – 1.56 (m, 2H), 1.61 (s,
3H), 1.34 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.3, 147.5, 146.6, 146.0, 133.5, 130.99, 130.96, 129.6, 129.5,
128.7, 128.6, 124.6, 122.60, 122.56, 119.5, 118.11, 118.09, 112.48, 112.46, 109.1, 77.9, 77.8, 51.0,
46.94, 46.93, 40.54, 40.47, 29.7, 26.1, 26.0, 22.91, 22.86, 17.58, 17.56, 15.9, 10.46, 10.45.
HRMS (ESI): calculated for C23H28O5Na 407.1829 [M+H]+, found 407.1832.
O
OH
O
O
OMeAc2O, pyridine, rt
21% over 2 steps
O
OH
O
OOH
OMe
3.64 3.65d.r. 1:1

189
To a solution of capillobenzopyranol (3.39) (48 mg, 0.14 mmol) in DMF (4 mL) was added PDC
(80 mg, 0.21 mmol) at room temperature and stirred for 2 h. The mixture was quenched with H2O
(10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic extracts were washed with
brine (3 x 20 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified
by flash chromatography on SiO2 (6:1 � 4:1 gradient elution, petrol/EtOAc) to give 3.66 (2:1
mixture of diastereoisomers) as a colourless oil (8 mg, 16%).
Data for 3.66:
Rf = 0.30 (3:1, petrol/EtOAc)
IR (neat): 3382, 2926, 1674, 1620, 1368, 1258, 1058, 1006, 950, 892 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.05 (s, 1H), 6.55 (s, 1H), 6.43 (d, J = 10.1 Hz, 1H), 6.13 (d, J =
10.2 Hz, 1H), 5.91 (s, 1H), 5.84 (s, 1H), 5.13 (t, J = 6.8 Hz, 1H), 3.17 (s, 2H), 2.79 (s, 1H), 1.97 (s,
3H), 1.98 � 1.87 (m, 2H), 1.90 (s, 3H), 1.70 � 1.59 (m, 2H), 1.54 (s, 3H), 1.53 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 186.8, 154.2, 153.9, 147.2, 147.1, 142.5, 141.6, 140.1, 140.0,
137.8, 137.7, 134.4, 132.2, 126.9, 126.0, 121.6, 120.5, 120.4, 120.3, 120.2, 109.1, 108.8, 88.5, 88.3,
76.6, 42.6, 40.5, 38.41, 38.36, 28.4, 27.0, 22.7, 22.2, 16.0, 15.9, 15.4, 9.8.
HRMS (ESI): calculated for C22H27O4 355.1904 [M+H]+, found 355.1901.
PDC, DMF, rt
16%
O
OH
O
3.39 3.66
O
O
OHO
d.r. 2:1

190
To a solution of capillobenzopyranol (3.39) (50 mg, 0.15 mmol) in t-BuOH (2.5 mL) and H2O (0.5
mL) was added NaH2PO4.2H2O (34 mg, 0.22 mmol) and NaClO2 (39 mg, 0.44 mmol) was added at
room temperature and stirred for 1 h. The mixture was diluted with H2O (10 mL) and extracted with
EtOAc (3 x 10 mL). The combined organic extracts were washed with brine (20 mL), dried over
MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography on
SiO2 (2:1, petrol/EtOAc) to give 3.45 (1:1 mixture of diastereoisomers) as a colourless oil (25 mg,
46%).
Data for 3.45:
Rf = 0.10 (2:1, petrol/EtOAc)
IR (neat): 3379, 2924, 1744, 1426, 1177, 982, 872 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.77 (s, 1H), 6.55 (s, 1H), 6.43 (s, 1H), 6.26 (d, J = 9.8 Hz, 1H),
5.50 (d, J = 9.8 Hz, 1H), 5.35 (dd, J = 16.2, 7.6 Hz, 1H), 4.95 (br s, 1H), 4.13 (br s, 1H), 2.53 – 2.44
(m, 2H), 2.22 – 2.16 (m, 2H), 2.17 (s, 3H), 1.89 (d, J = 0.9 Hz, 3H), 1.75 – 1.61 (m, 2H), 1.71 (s,
3H), 1.35 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.91, 171.87, 147.59, 147.57, 147.3, 147.2, 147.1, 146.4, 146.3,
132.8, 132.7, 132.24, 132.22, 129.4, 129.3, 128.8, 128.7, 124.80, 124.78, 122.81, 122.76, 119.4,
118.2, 112.6, 112.5, 104.41, 104.37, 78.0, 77.9, 47.8, 40.41, 40.36, 26.04, 25.98, 23.12, 23.07,
17.57, 17.56, 15.93, 15.91, 10.4.
HRMS (ESI): calculated for C22H27O5 371.1853 [M+H]+, found 371.1851.
O
OH
O
3.39: capillobenzopyranol
NaClO2, NaH2PO4t-BuOH/H2O, rt
O
OH
O
O
OH
3.45
46%
1:1 d.r.

191
To a solution of 3.45 (23 mg, 0.062 mmol) in pyridine (2 mL) was added Ac2O (19 mg, 0.19 mmol)
at room temperature and stirred for 16 h. The reaction was concentrated in vacuo. The residue was
purified by flash chromatography on SiO2 (6:1, petrol/EtOAc) to give 3.67 as a colourless oil (11
mg, 46%).
Data for 3.67:
Rf = 0.40 (3:1, petrol/EtOAc)
IR (neat): 2924, 1746, 1497, 1365, 1193, 1165, 988, 755 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.99 (d, J = 1.4 Hz, 1H), 6.63 (s, 1H), 6.62 (s, 1H), 6.29 (d, J = 9.9
Hz, 1H), 5.79 (t, J = 7.5 Hz, 1H), 5.52 (d, J = 9.6 Hz, 1H), 5.51 (s, 1H), 2.36 – 2.28 (m, 2H), 2.29
(s, 3H), 2.09 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H), 1.83 – 1.77 (m, 1H), 1.74 – 1.68 (m, 1H), 1.39 (s,
3H). 13C NMR (125 MHz, CDCl3): δ 171.5, 169.6, 150.4, 145.1, 142.6, 139.5, 138.9, 132.3, 130.7,
129.0, 127.0, 122.5, 119.2, 119.1, 118.0, 117.1, 78.4, 40.4, 26.6, 23.5, 20.7, 16.2, 15.0, 10.4.
HRMS (ESI): calculated for C24H24O5Na 417.1672 [M+Na]+, found 417.1668.
O
OH
O Ac2O, pyridine, rtO
OAc
O
O
3.67
46%
single stereoiosmer
OOH
3.45
Z

192
To a solution of 3.67 (49 mg, 0.12 mmol) in MeOH (4 mL) was added K2CO3 (68 mg, 0.49 mmoL)
at rt. The mixture was stirred at room temperature for 30 min. The mixture was quenched with 1 M
HCl (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic extracts were washed
with brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was
purified by flash chromatography on SiO2 (4:1, petrol/EtOAc) to give Z-3.46 as a colourless oil (30
mg, 70%).
Data for Z-3.46:
Rf = 0.35 (3:1, petrol/EtOAc)
IR (neat): 3397, 2972, 2923, 1725, 1620, 1598, 1497, 1458, 1426, 1371, 1248, 1174, 1060 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.99 (s, 1H), 6.55 (s, 1H), 6.44 (s, 1H), 6.27 (d, J = 9.8 Hz, 1H),
5.79 (t, J = 7.5 Hz, 1H), 5.52 (s, 1H), 5.51 (d, J = 9.8 Hz, 1H), 4.80 (s, 1H), 2.34 – 2.28 (m, 2H),
2.18 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H), 1.80 – 1.68 (m, 2H), 1.37 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.9, 147.8, 146.6, 145.2, 139.7, 139.4, 129.3, 127.2, 124.9,
123.0, 119.6, 119.5, 118.3, 117.5, 112.7, 77.9, 40.3, 26.3, 23.7, 16.1, 15.13, 15.10.
HRMS (ESI): calculated for C22H25O4 353.1747 [M+H]+, found 353.1744.
K2CO3, MeOH, rtO
OH
O
O
3.46
70%
ZO
OAc
O
O
3.67
Z

193
A solution of Z-3.46 (30 mg, 0.085 mmol) in PhMe (3 mL) was heated at reflux for 1 h. The
solution was cooled and concentrated in vacuo to yield verrubenzospirolactone 3.38 as a white solid
(30 mg, 100%). Data for 3.38 matched that previously obtained.
O
OH
O
O
3.46
PhMe, reflux100%
O
OH
H H
H
O
O
3.38: verrubenzospirolactone
Z

194
To a solution of geraniol (3.76) (10.0 g, 64.8 mmol) in DMF (100 mL) at room temperature was
added Imidazole (8.82 g, 13.0 mmol) and TBSCl (12.7 g, 8.42 mmol). The reaction was stirred at
room temperature for 2 d, then diluted with H2O (50 mL) and extracted with Et2O (2 × 100 mL).
The combined organic extracts were washed with H2O (200 mL), brine (200 mL), dried over
MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (20:1, petrol/EtOAc) to give 3.77 as a yellow oil (17.4 g, 100%). Data of
3.77 matched from literature.3
Data for 3.77:
Rf = 0.80 (10:1, petrol/EtOAc)
IR (neat): 2968, 2928, 2861, 1719, 1651, 1455, 1378, 1060 cm-1. 1H NMR (500 MHz, CDCl3): δ 5.30 (t, J = 6.3 Hz, 1H), 5.10 (t, J = 6.2 Hz, 1H), 4.19 (d, J = 6.2
Hz, 2H), 2.09 (dd, J = 14.6, 7.1 Hz, 2H), 2.00 – 1.99 (m, 2H), 1.68 (s, 3H), 1.62 (s, 3H), 1.60 (s,
3H), 0.91 (s, 9H), 0.07 (s, 3H), 0.07 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 136.9, 131.5, 124.4, 124.1, 60.4, 39.5, 26.4, 26.0, 25.7, 18.4, 17.7,
16.3, -5.0.
HO
3.76: geraniol
Imidazole, TBSCl, DMF,rt
100% TBSO3.77

195
To a solution of 3.77 (10.0 g, 37.2 mmol) in CH2Cl2 (100 mL) at room temperature was added
salicylic acid (514 mg, 3.72 mmol), t-BuOOH (5 ~ 6 M, 11 mL, 55.0 mmol) and SeO2 (413 mg,
3.72 mmol). The reaction was stirred at room temperature for 17 h, then quenched with saturated
NaHCO3 solution (100 mL). The organic layer was separated and the aqueous layer was extracted
with CH2Cl2 (2 × 100 mL). The combined organic extracts were dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (20:1 →
10:1, petrol/EtOAc gradient elution) to give 3.77 as a yellowish oil (2.45 g, 25%). Further elution
gave 3.79 as a yellowish oil (200 mg, 2%). Further elution gave 3.78 as a yellowish oil (3.05 g,
33%). Data of 3.78 and 3.79 match from literature.4,5
Data for 3.78:
Rf = 0.25 (10:1, petrol/EtOAc)
IR (neat): 3404, 2972, 2857, 1718, 1645, 1457, 1380, 1063 cm-1. 1H NMR (500 MHz, CDCl3): δ 5.39 (t, J = 6.9 Hz, 1H), 5.31 (t, J = 6.3 Hz, 1H), 4.19 (d, J = 6.3
Hz, 2H), 3.99 (br s, 2H), 2.16 (dd, J = 14.9, 7.4 Hz, 2H), 2.06 – 2.03 (m, 2H), 1.67 (s, 3H), 1.63 (s,
3H), 0.91 (s, 9H), 0.07 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 136.4, 135.0, 125.9, 124.7, 69.0, 60.3, 39.1, 26.0, 25.8, 25.7, 16.3,
13.7, –5.1.
Data for 3.79:
Rf = 0.50 (10:1, petrol/EtOAc)
IR (neat): 2972, 2941, 1738, 1370, 1229, 1219 cm-1. 1H NMR (500 MHz, CDCl3): δ 9.39 (s, 1H), 6.47 (t, J = 7.1 Hz, 1H), 5.35 (t, J = 6.3 Hz, 1H), 4.20
(d, J = 6.2 Hz, 2H), 2.48 (q, J = 7.2 Hz, 2H), 2.20 (t, J = 7.6 Hz, 2H), 1.75 (s, 3H), 1.66 (s, 3H),
0.90 (s, 9H), 0.07 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 195.2, 153.9, 139.5, 135.2, 125.7, 60.2, 37.8, 27.1, 26.0, 18.4,
16.3, 9.2, –5.1.
TBSO
3.77
SeO2, t-BuOOHsalicylic acid, CH2Cl2, rt
TBSO3.78
OHTBSO
3.79
O+
H
2%30%
TBSO3.7725%
+

196
To a solution of 3.78 (3.05 g, 10.7 mmol) in CH2Cl2 (300 mL) at room temperature was added
NMO (1.88 g, 16.1 mmol), 4Å molecular sieve (3.0 g) and TPAP (198 mg, 0.54 mmol). The
reaction was stirred at room temperature for 16 h, then filtered through a pad of SiO2 and washed
with CH2Cl2 (200 mL). The filtrate was concentrated in vacuo to give 3.79 as a yellowish oil (1.27
g, 42%). Data of 3.79 matched previously obtained.
TBSO
3.78
TPAP, NMO4Å molecular sieve, CH2Cl2, rt
TBSO3.79
OOH42%
H

197
To a solution of 3.47 (1.55 g, 6.60 mmol) in anhydrous THF (10 mL) at −78 °C was added n-BuLi
(2.0 M solution in cyclohexane, 3.30 mL, 6.60 mmol). The mixture was stirred at −78 °C for 30
min. A solution of 3.79 (932 mg, 3.30 mmol) in anhydrous THF (20 mL) was added. The reaction
was stirred at −78 °C for 15 min, then warmed to room temperature and stirred for 1 h. The reaction
was quenched with saturated NH4Cl aqueous solution. The aqueous layer was separated and
extracted with Et2O (2 × 30 mL). The combined organic extracts were washed with brine (50 mL),
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (10:1 → 5:1 gradient elution, petrol/EtOAc) to give 3.80 as an inseparable
4:1 mixture of regioisomers as colourless oil (1.02 g, 85%).
Data for 3.80:
Rf = 0.65 (petrol/EtOAc, 5:1)
IR (neat): 2952, 2929, 2857, 1777, 1671, 1475, 1463, 1384, 1257, 1058 cm-1.
Data for E-3.80 (major) 1H NMR (500 MHz, CDCl3): δ 7.44 (s, 1H), 6.22 (s, 1H), 5.73 (t, J = 7.2, 1H), 5.32 (t, J = 5.7 Hz,
1H), 4.19 (d, J = 6.1 Hz, 2H), 2.34 – 2.30 (m, 2H), 2.11 (t, J = 7.5 Hz, 2H), 2.03 (s, 3H), 1.92 (s,
3H), 1.63 (s, 3H), 0.89 (s, 9H), 0.06 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 170.5, 146.8, 139.0, 135.8, 134.9, 130.7, 130.0, 125.2, 119.2,
60.2, 38.6, 26.9, 26.0, 16.3, 15.5, 10.9, –5.1.
Data for Z-3.80 (minor) 1H NMR (500 MHz, CDCl3): δ 6.99 (s, 1H), 5.77 – 5.75 (m, 1H), 5.53 (s, 1H), 5.32 (t, J = 5.7 Hz,
1H), 4.19 (d, J = 6.1 Hz, 2H), 2.34 – 2.30 (m, 2H), 2.11 (t, J = 7.5 Hz, 2H), 2.05 (s, 3H), 2.00 (s,
3H), 1.63 (s, 3H), 0.89 (s, 9H), 0.06 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 171.5, 145.1, 139.5, 138.9, 135.9, 135.1, 132.3, 125.1, 117.2,
60.2, 39.0, 26.8, 25.9, 16.3, 15.0, 10.5, -5.1.
HRMS (ESI): calculated for C21H34O3K 401.1909 [M+K]+, found 401.1911.
TBSOO
3.47, n-BuLiTHF, –78 °C to rt
85%
3.80E/Z = 4:1
E
3.79H
OO PO
OEtOEt
OO
TBSO

198
To a solution of 4:1 mixture of 3.80 (1.02 g, 2.81 mmol) in anhydrous THF (10 mL) at room
temperature was added TBAF (1.0 M in THF, 3.10 mL, 3.10 mmol). The reaction was stirred at
room temperature for 1.5 h, then diluted with H2O (10 mL). The aqueous layer was separated and
extracted with Et2O (2 × 10 mL). The combined organic extracts were washed with brine (20 mL),
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (1:1, petrol/EtOAc) to give 3.81 as an inseparable 4:1 mixture of
regioisomers as colourless oil (557 mg, 80%).
Data for 3.81:
Rf = 0.30 (1:1, petrol/EtOAc)
IR (neat): 3432, 2926, 1757, 1668, 1625, 1446, 1382, 1266 cm-1.
Data for E-3.81 (major): 1H NMR (500 MHz, CDCl3): δ 7.45 (s, 1H), 6.23 (s, 1H), 5.73 (t, J = 7.3 Hz, 1H), 5.44 (t, J = 6.8
Hz, 1H), 4.17 (d, J = 6.9 Hz, 2H), 2.36 – 2.30 (m, 2H), 2.16 – 2.13 (m, 2H), 2.04 (s, 3H), 1.93 (s,
3H), 1.70 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 170.5, 146.9, 138.60, 138.57, 134.9, 130.8, 130.1, 124.1, 119.1,
59.3, 38.7, 26.9, 16.3, 15.5, 10.9.
Data for Z-3.81 (minor): 1H NMR (500 MHz, CDCl3): δ 7.00 (s, 1H), 5.79 (t, J = 7.0 Hz, 1H), 5.54 (s, 1H), 5.44 (t, J = 6.8
Hz, 1H), 4.17 (d, J = 6.9 Hz, 2H), 2.36 – 2.30 (m, 2H), 2.16 – 2.13 (m, 2H), 2.05 (s, 3H), 2.00 (s,
3H), 1.70 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.6, 145.2, 139.5, 138.5, 135.1, 132.3, 127.2, 124.1, 117.0,
59.4, 38.6, 26.8, 16.2, 15.2, 10.5.
HRMS (ESI): calculated for C15H20O3Na 271.1305 [M+Na]+, found 271.1299.
3.80E/Z = 4:1
TBAF, THF, rt
80%
3.81E/Z = 4:1
E EO
OHO
OO
TBSO

199
To a solution of 3.81 (557 mg, 2.24 mmol) in CH2Cl2 (10 mL) at room temperature was added
NMO (394 mg, 3.36 mmol), 4Å molecular sieve (557 mg) and tetrapropylammonium perruthenate
(39 mg, 0.11 mmol). The reaction was stirred at room temperature for 4 h, then concentrated in
vacuo. The residue was purified by flash column chromatography on SiO2 (2:1, petrol/EtOAc) to
give 4:1 mixture of E-3.68 and Z-3.68 (386 mg, 70%) as a yellow oil. The mixture was purified by
flash column chromatography on SiO2 (2:1, petrol/EtOAc) to give E-3.68 as yellow solid (264 mg,
48%).
Data for E-3.68:
Rf = 0.40 (1:1, petrol/EtOAc)
IR (neat): 2984, 2928, 2857, 1752, 1669, 1633, 1444, 1374, 1242, 1195, 1124 cm-1.
Data for E-3.68 (major):
MP: 70 – 73 °C 1H NMR (500 MHz, CDCl3): δ 10.00 (d, J = 7.9 Hz, 1H), 7.42 (s, 1H), 6.20 (s, 1H), 5.88 (d, J =
7.9 HZ, 1H), 5.67 (t, J = 7.2 Hz, 1H), 2.44 – 2.40 (m, 2H), 2.35 – 2.32 (m, 2H), 2.19 (s, 3H), 2.03
(s, 3H), 1.93 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 191.0, 170.3, 162.2, 147.3, 136.5, 134.7, 131.7, 130.5, 127.5,
118.5, 39.6, 26.2, 17.6, 15.5, 10.9.
Data for Z-3.68 (minor): 1H NMR (500 MHz, CDCl3): δ 9.99 (d, J = 7.8 Hz, 1H), 7.00 (s, 1H), 5.89 (d, J = 7.9 Hz, 1H),
5.72 (t, J = 6.9 Hz, 1H), 5.51 (s, 1H), 2.45 – 2.39 (m, 2H), 2.35 – 2.32 (m, 2H), 2.18 (s, 3H), 2.06
(s, 3H), 2.00 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 191.1, 171.4, 162.5, 145.6, 139.4, 136.3, 133.2, 127.8, 127.7,
116.4, 39.7, 26.1, 17.5, 15.2, 10.5.
HRMS (ESI): calculated for C15H19O3 247.1329 [M+H]+, found 247.1324.
3.81E/Z = 4:1
TPAP, NMO, 4Å MS, CH2Cl2, rt
70%
O
OO
3.68E/Z = 4:1
E EHO
OHO

200
To a solution of E-3.68 (26 mg, 0.11 mmol) in MeOH (3 mL) at room temperature was added
phloroglucinol (3.70) (27 mg, 0.21 mmol) and Ca(OH)2 (16 mg, 0.21 mmol). The mixture was
stirred at reflux for 24 h, then cooled to room temperature and quenched with 1 M HCl solution (3
mL). The aqueous layer was then extracted with Et2O (2 × 10 mL). The combined organic extracts
were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (2:1, petrol/EtOAc) to give an inseparable 4.9:1 mixture of 3.74
and 3.75 as a white solid (12 mg, 31%).
Data for 3.74 & 3.75:
Rf = 0.45 (1:1, petrol/EtOAc)
IR (neat): 3415, 2937, 1757, 1712, 1626, 1601, 1501, 1361 cm-1.
Data for 3.74 (major) 1H NMR (500 MHz, d6-acetone): δ 8.35 (s, 1H), 6.08 (d, J = 2.1 Hz, 1H), 6.00 (d, J = 2.2 Hz, 1H),
5.43 (s, 1H), 3.90 (d, J = 5.7 Hz, 1H), 3.36 – 3.30 (m, 1H), 3.05 (d, J = 5.8 Hz, 1H), 2.28 – 2.22 (m,
1H), 2.17 – 2.07 (m, 2H), 2.01 – 1.97 (m, 1H), 1.84 (dd, J = 12.2, 5.9 Hz, 1H), 1.79 (s, 3H), 1.58
(dd, J = 11.7, 7.3 Hz, 1H), 1.40 (s, 3H), 1.33 (d, J = 7.2 Hz, 3H). 13C NMR (125 MHz, d6-acetone): δ 177.6, 159.0, 158.3, 154.3, 144.7, 120.7, 99.8, 99.5, 98.2,
88.1, 85.6, 84.3, 46.2, 40.6, 39.8, 39.3, 32.1, 25.0, 23.8, 20.5, 8.9.
Data for 3.75 (minor) 1H NMR (500 MHz, d6-acetone): δ 8.31 (s, 1H), 6.00 (d, J = 2.2 Hz, 1H), 5.94 (d, J = 2.2 Hz, 1H),
5.30 (s, 1H), 4.18 (d, J = 6.0 Hz, 1H), 3.14 (d, J = 4.8 Hz, 1H), 3.00 – 2.94 (m, 1H), 2.28 – 2.22 (m,
1H), 2.17 – 2.07 (m, 2H), 2.01 – 1.97 (m, 1H), 1.89 (dd, J = 12.3, 4.9 Hz, 1H), 1.77 (s, 3H), 1.62 –
1.50 (m, 1H), 1.44 (s, 3H), 1.41 (d, J = 7.6 Hz, 3H). 13C NMR (125 MHz, d6-acetone): δ 176.3, 159.1, 158.8, 154.8, 145.3, 121.9, 99.5, 98.5, 98.1,
86.0, 84.4, 83.1, 46.3, 42.7, 40.7, 40.6, 31.1, 25.3, 24.8, 20.6, 14.4.
HRMS (ESI): calculated for C21H21O5 353.1394 [M–H]–, found 353.1396.
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
O
OO
E-3.68
Ca(OH)2, MeOHreflux31%
OH
OHHO
+E
4.9:1 d.r. (3.74:3.75)
+
3.70
H

201
To a solution of E-3.46 (20 mg, 0.081 mmol) in EtOH (3 mL) at room temperature was added
phloroglucinol (3.70) (31 mg, 0.244 mmol) and Ca(OH)2 (18 mg, 0.244 mmol). The mixture was
stirred at reflux for 24 h, then cooled to room temperature and concentrated in vacuo. The residue
was diluted with 1 M HCl solution (3 mL), then extracted with Et2O (2 × 10 mL). The combined
organic extracts were dried over MgSO4, filtered and concentrated in vacuo. The residue was
purified by flash column chromatography on SiO2 (2:1, petrol/EtOAc) to give an inseparable 3.0:1
mixture of 3.74 and 3.75 as a white solid (13 mg, 46%). Data for 3.74 and 3.75 matched that
previously obtained.
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
O
OO
E-3.68
Ca(OH)2, EtOHreflux46%
OH
OHHO
+E
3:1 d.r. (3.74:3.75)
+
3.70
H
202
3.5.3. NMR spectra
O
OH
3.551H NMR500 MHz
CDCl3
O
OH
3.5513C NMR125 MHz
CDCl3
203
O
OH
3.561H NMR500 MHz
CDCl3
O
OH
3.5613C NMR125 MHz
CDCl3
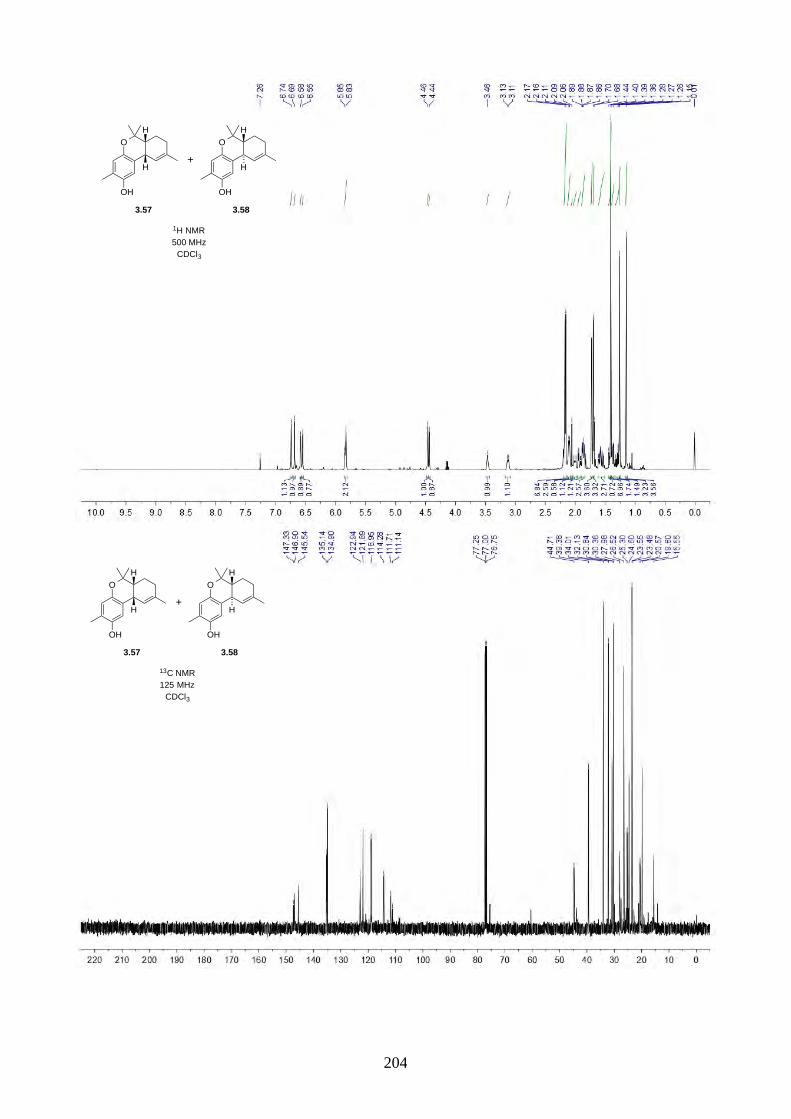
204
O
OH
H
H+
O
OH
H
H
3.57 3.58
1H NMR500 MHz
CDCl3
O
OH
H
H+
O
OH
H
H
3.57 3.58
13C NMR125 MHz
CDCl3
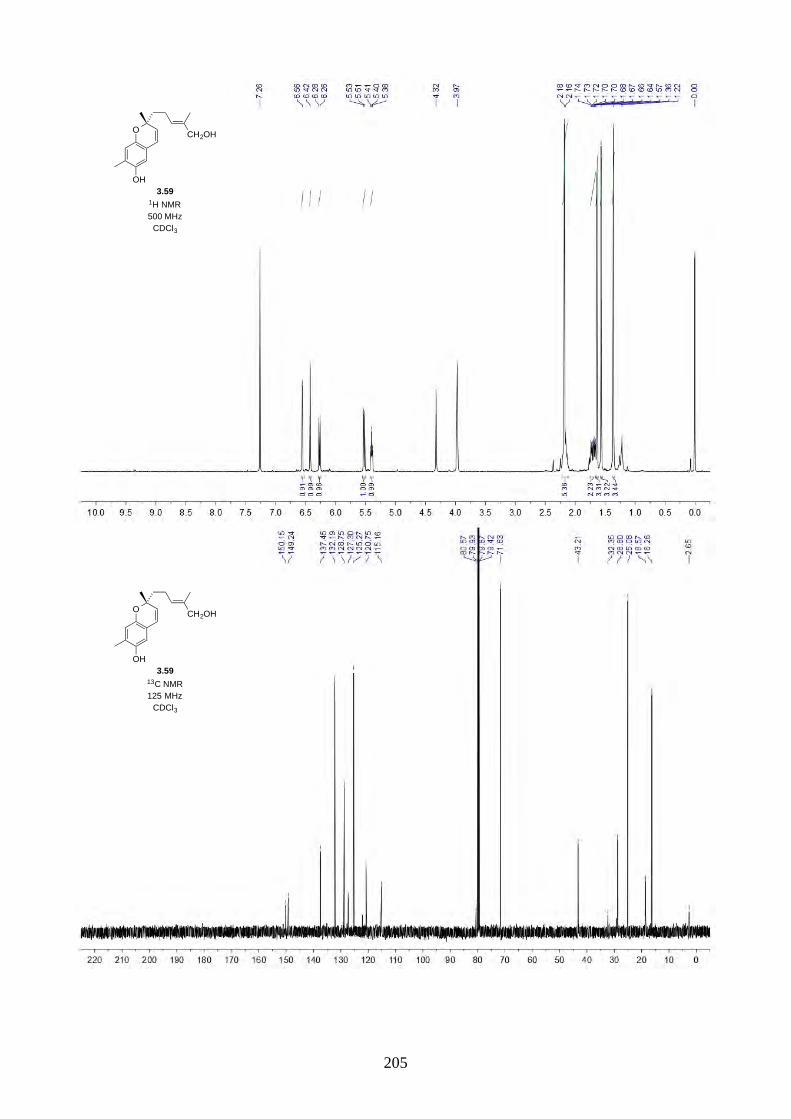
205
O
OH
CH2OH
3.5913C NMR125 MHz
CDCl3
O
OH
CH2OH
3.591H NMR500 MHz
CDCl3
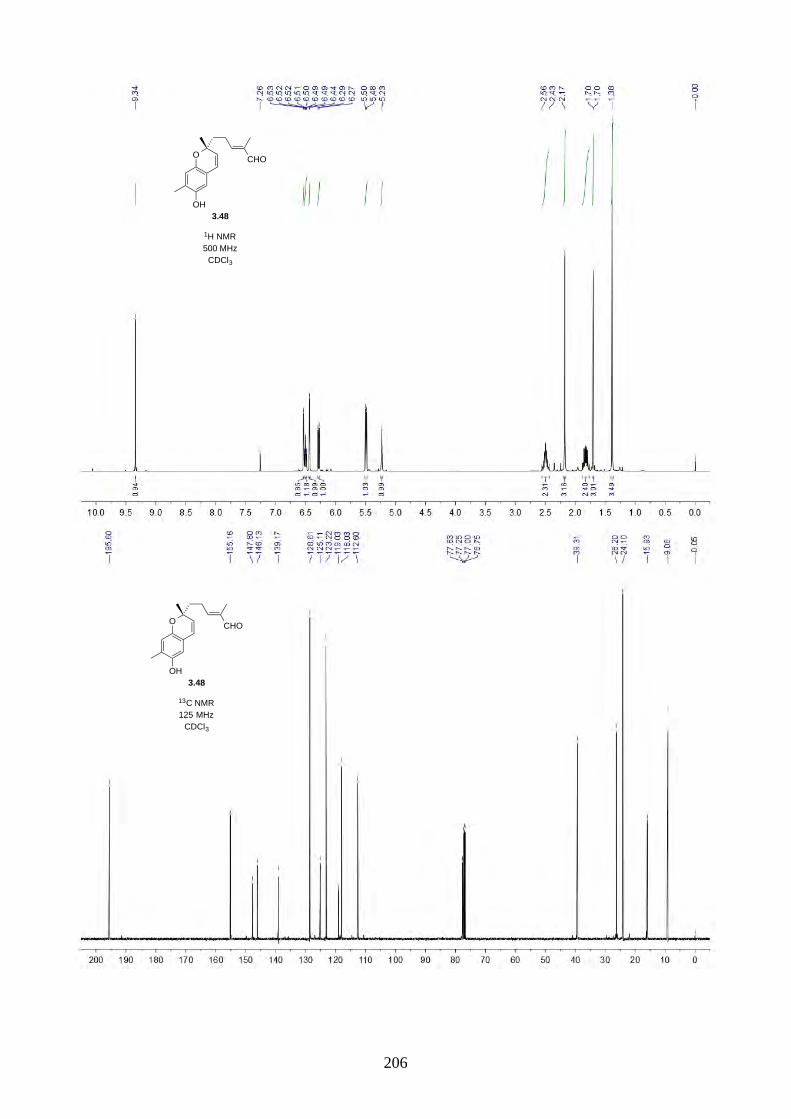
206
O
OH
CHO
3.48
13C NMR125 MHz
CDCl3
O
OH
CHO
3.48
1H NMR500 MHz
CDCl3
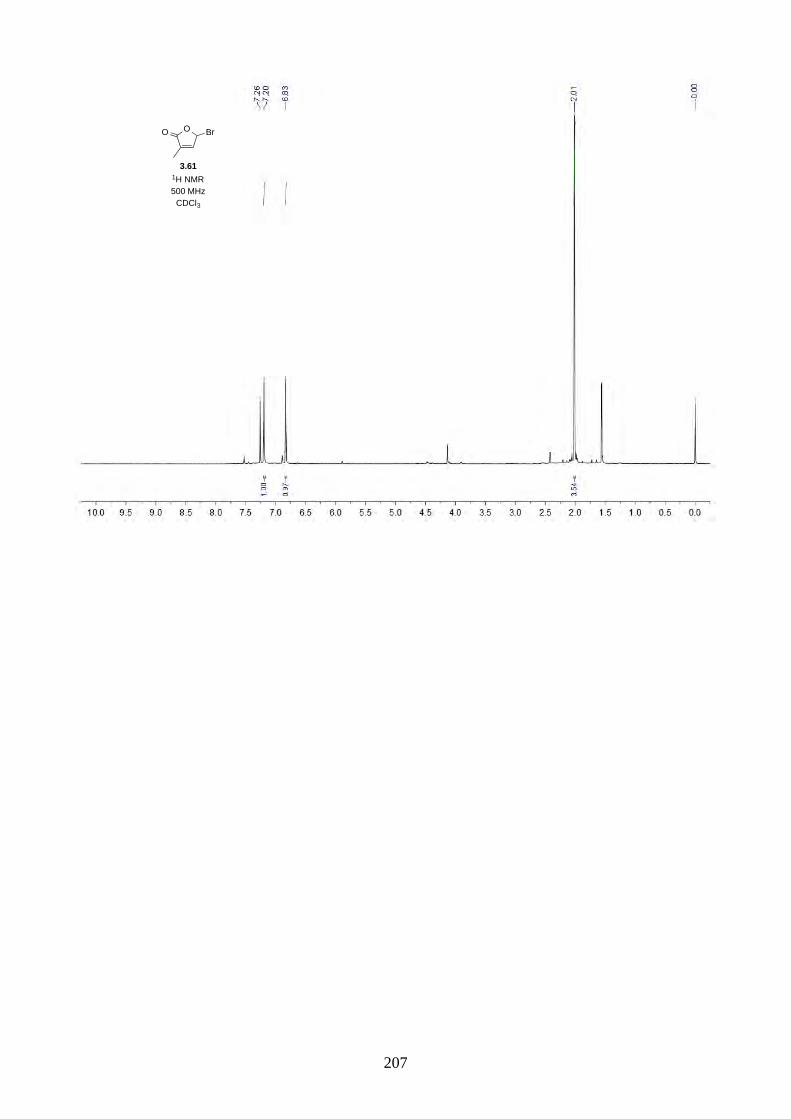
207
OO Br
3.611H NMR500 MHz
CDCl3
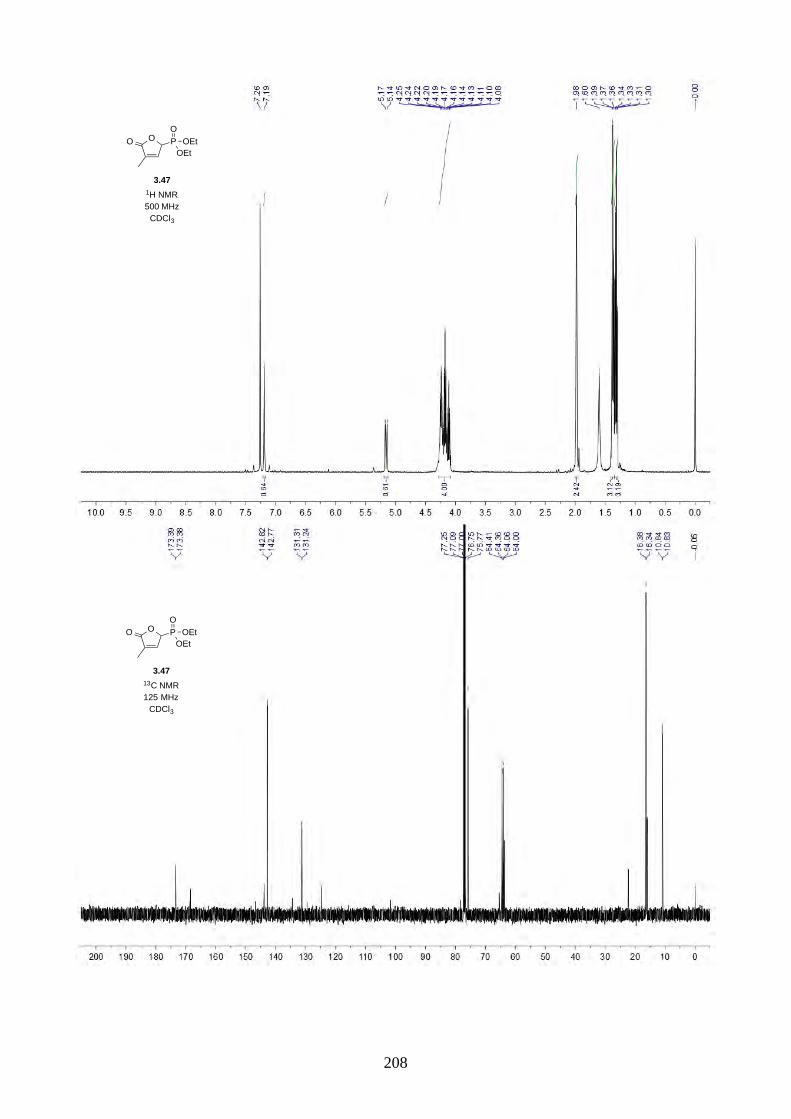
208
OO PO
OEtOEt
3.471H NMR500 MHz
CDCl3
OO PO
OEtOEt
3.4713C NMR125 MHz
CDCl3
209
O
OH
HH
O
H
O
E
E/Z = 4.5:11H NMR500 MHz
CDCl3
3.46
O
OH
HH
O
H
O
E
E/Z = 4.5:113C NMR125 MHz
CDCl3
3.46
210
O
OH
HH
O
H
O
E
E/Z = 4.5:1COSY
500 MHzCDCl3
3.46
O
OH
HH
O
H
O
E
E/Z = 4.5:1HSQC
500 MHzCDCl3
3.46
211
O
OH
HH
O
H
O
E
E/Z = 4.5:1HMBC
500 MHzCDCl3
3.46
O
OH
HH
O
H
O
E
E/Z = 4.5:1NOESY500 MHz
CDCl3
3.46
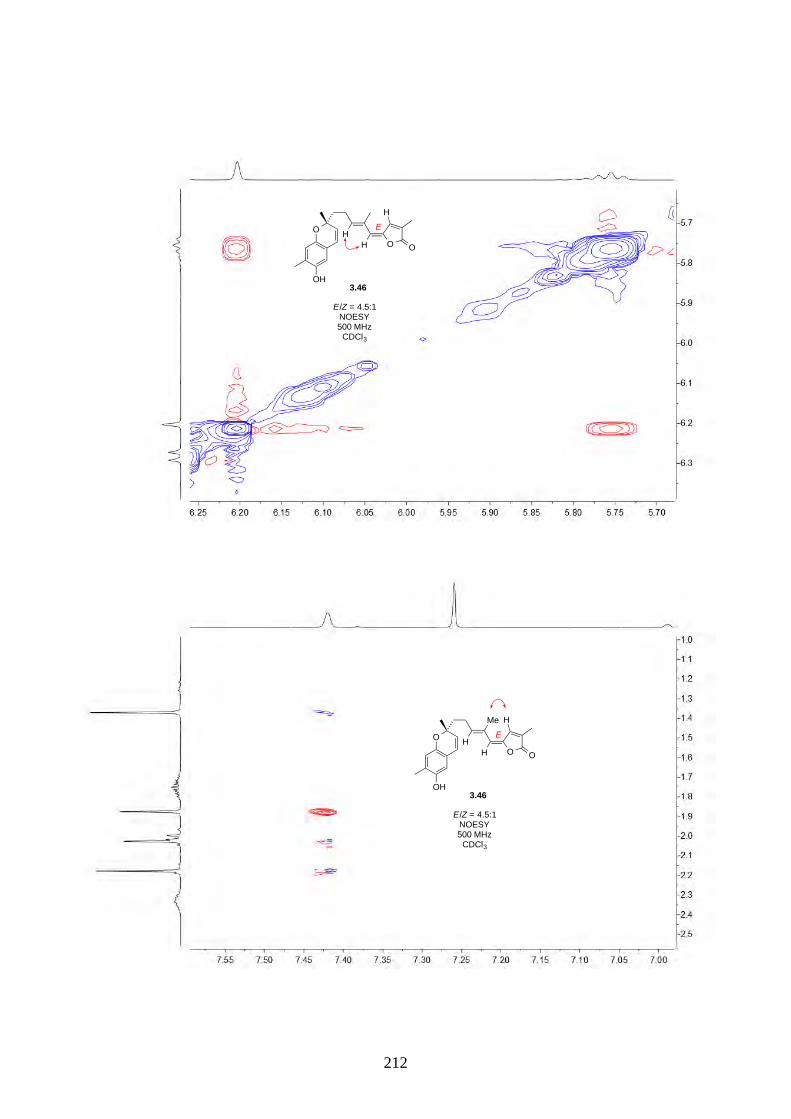
212
O
OH
HH
O
H
O
E
E/Z = 4.5:1NOESY500 MHz
CDCl3
3.46
O
OH
Me
HH
O
H
O
E
E/Z = 4.5:1NOESY500 MHz
CDCl3
3.46
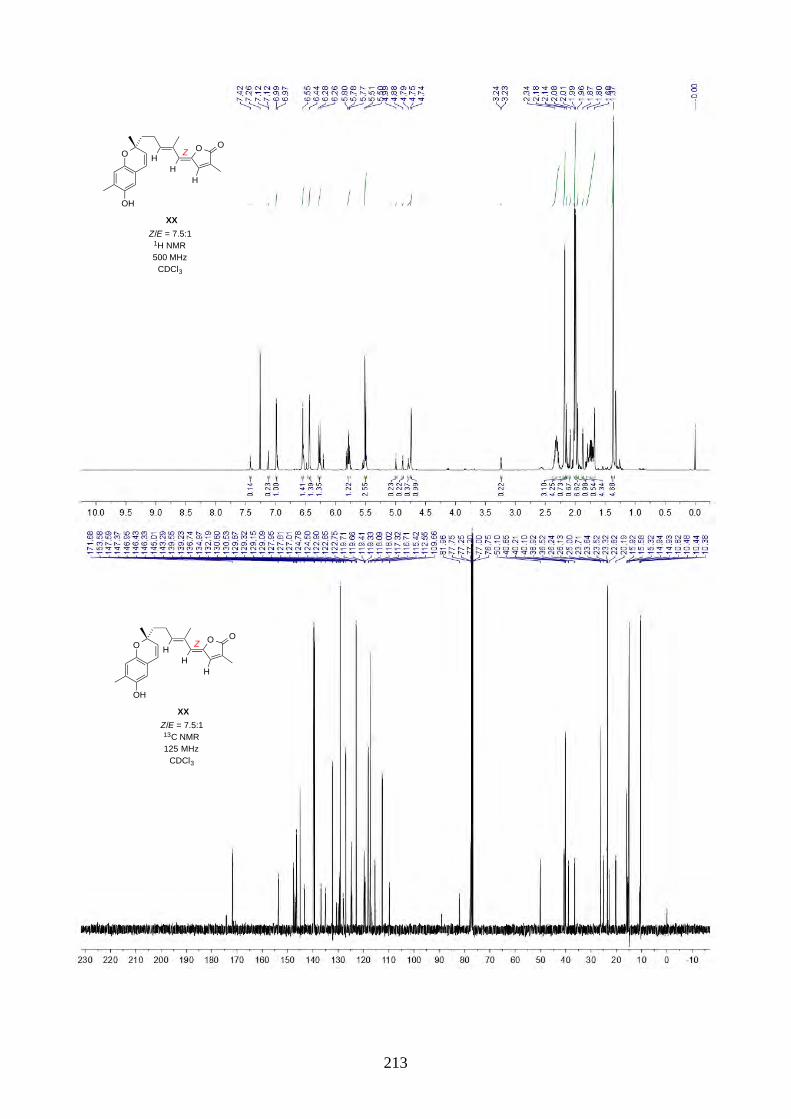
213
O
OH
HH
O O
H
Z
Z/E = 7.5:11H NMR500 MHz
CDCl3
XX
O
OH
HH
O O
H
Z
Z/E = 7.5:113C NMR125 MHz
CDCl3
XX
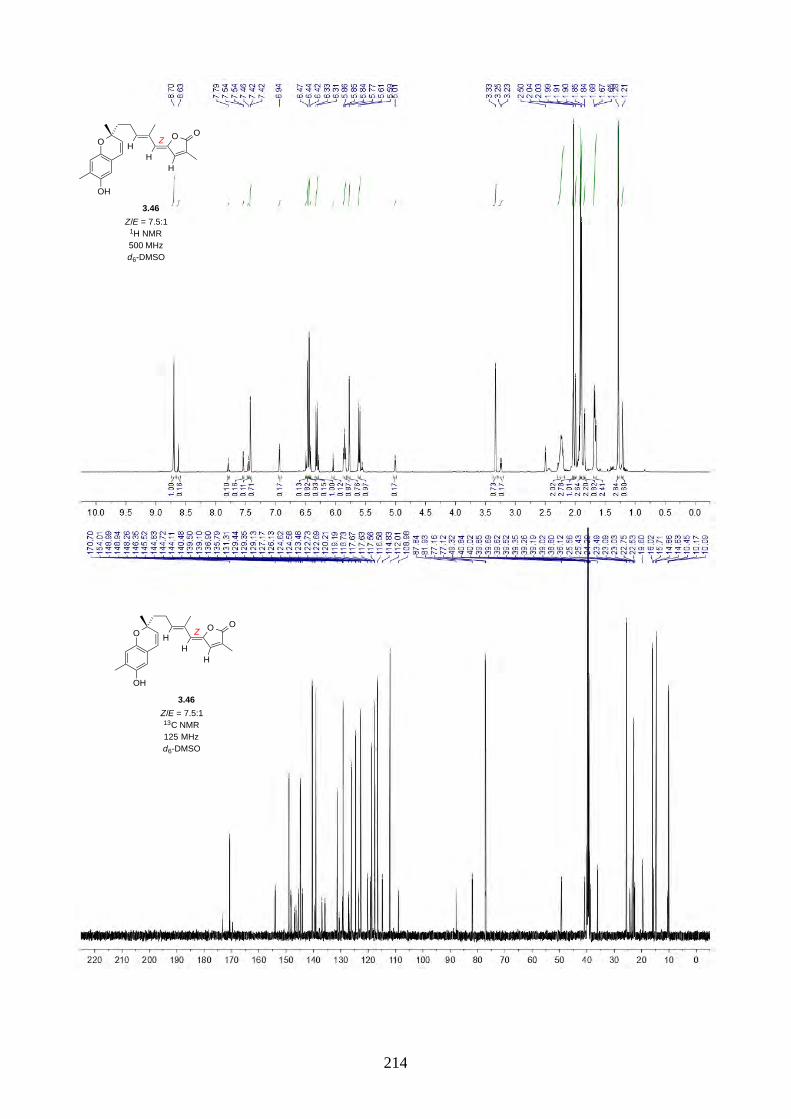
214
O
OH
HH
O O
H
Z
Z/E = 7.5:11H NMR500 MHzd6-DMSO
3.46
O
OH
HH
O O
H
Z
Z/E = 7.5:113C NMR125 MHzd6-DMSO
3.46
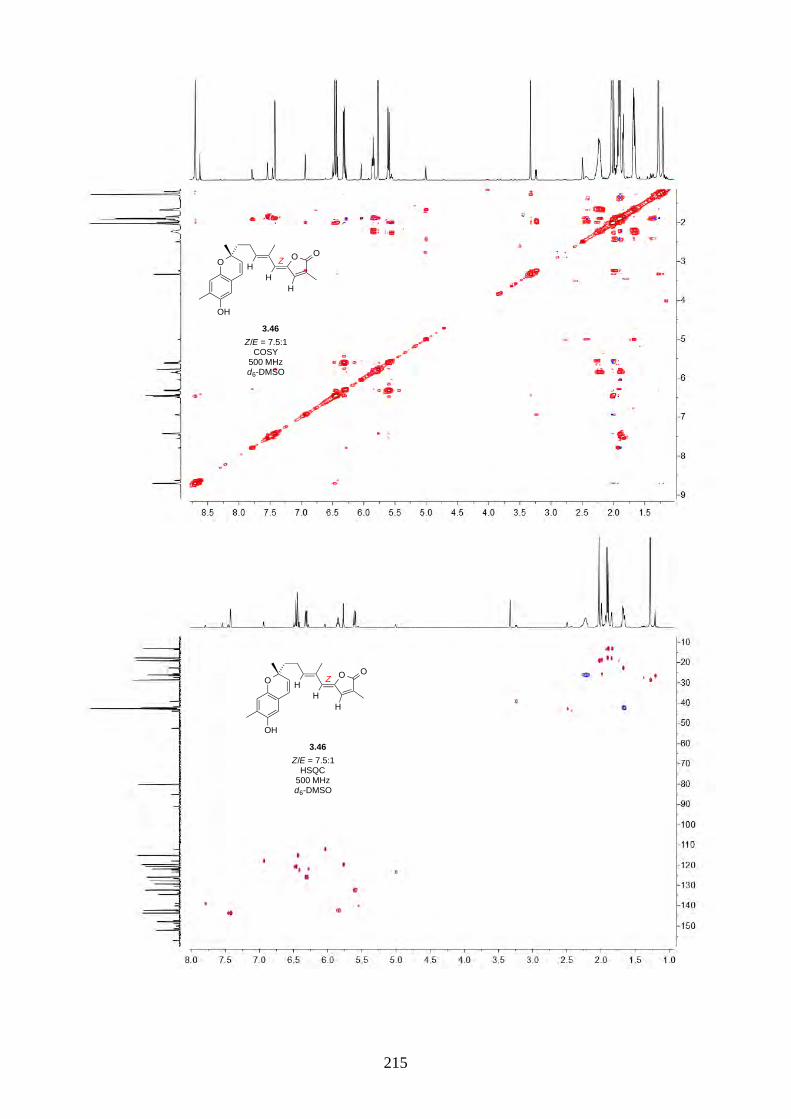
215
O
OH
HH
O O
H
Z
Z/E = 7.5:1COSY
500 MHzd6-DMSO
3.46
O
OH
HH
O O
H
Z
Z/E = 7.5:1HSQC
500 MHzd6-DMSO
3.46
216
O
OH
HH
O O
H
Z
Z/E = 7.5:1HMBC
500 MHzd6-DMSO
3.46
O
OH
HH
O O
H
Z
Z/E = 7.5:1NOESY500 MHzd6-DMSO
3.46
217
O
OH
HH
O O
H
Z
Z/E = 7.5:1NOESY500 MHzd6-DMSO
3.46
218
O
OH
H H
H
O
O
1H NMR500 MHzd6-DMSO
3.62
O
OH
H H
H
O
O
13C NMR125 MHzd6-DMSO
3.62
219
O
OH
H H
H
O
O
COSY500 MHzd6-DMSO
3.62
O
OH
H H
H
O
O
COSY500 MHzd6-DMSO
3.62
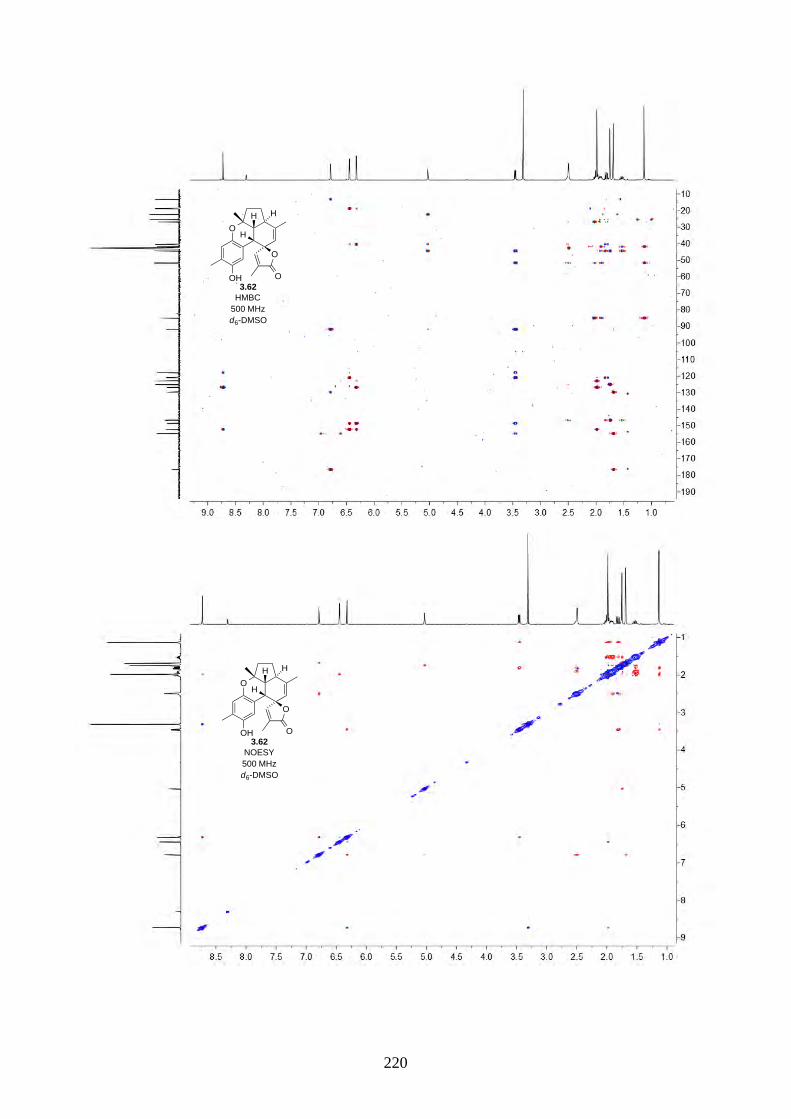
220
O
OH
H H
H
O
O
HMBC500 MHzd6-DMSO
3.62
O
OH
H H
H
O
O
NOESY500 MHzd6-DMSO
3.62
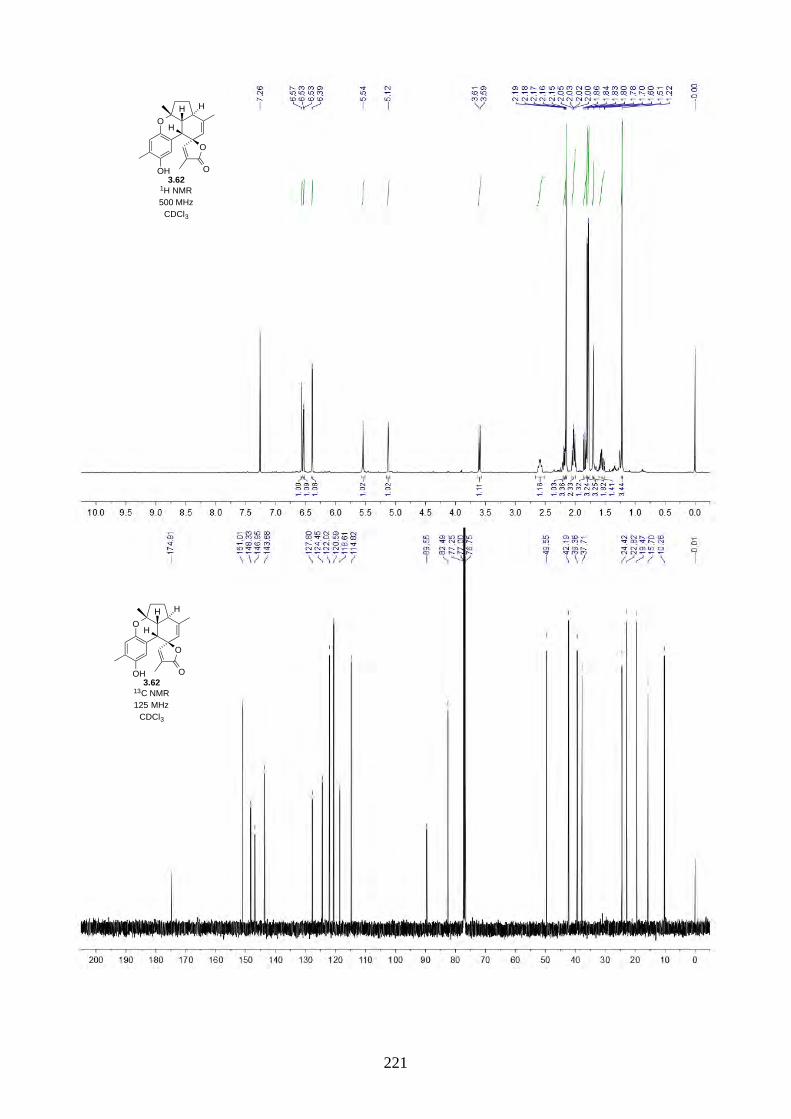
221
O
OH
H H
H
O
O
1H NMR500 MHz
CDCl3
3.62
O
OH
H H
H
O
O
13C NMR125 MHz
CDCl3
3.62
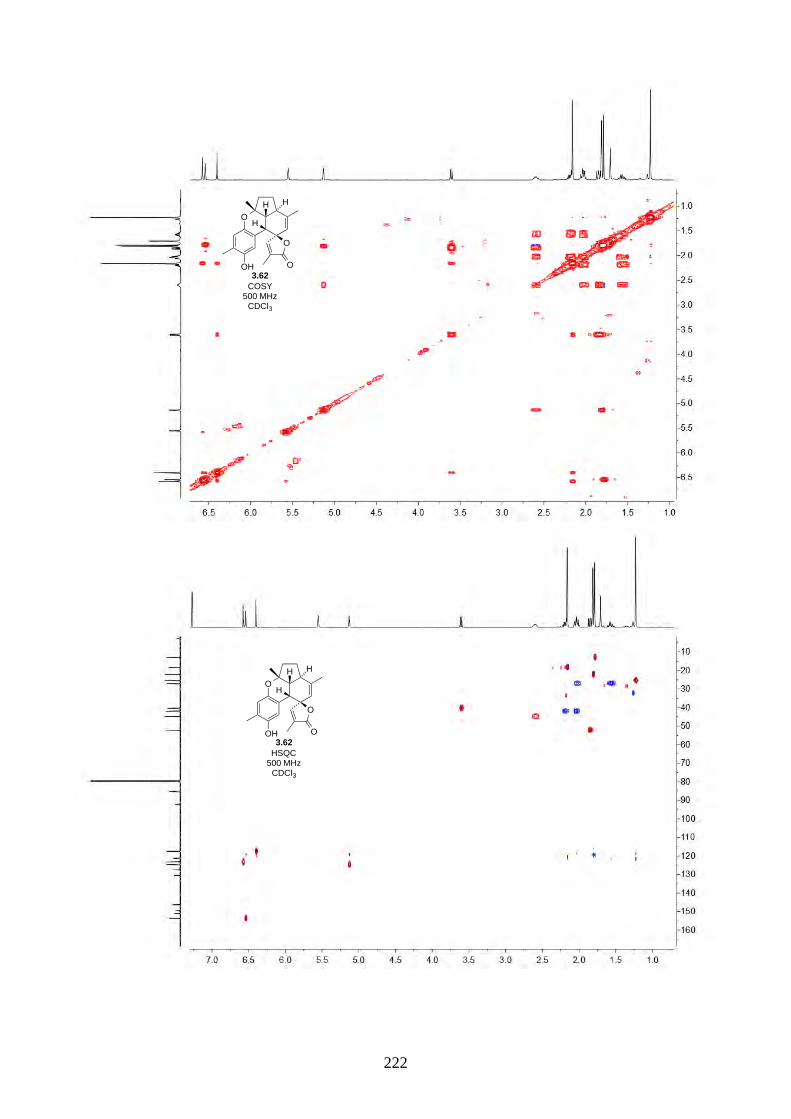
222
O
OH
H H
H
O
O
COSY500 MHz
CDCl3
3.62
O
OH
H H
H
O
O
HSQC500 MHz
CDCl3
3.62
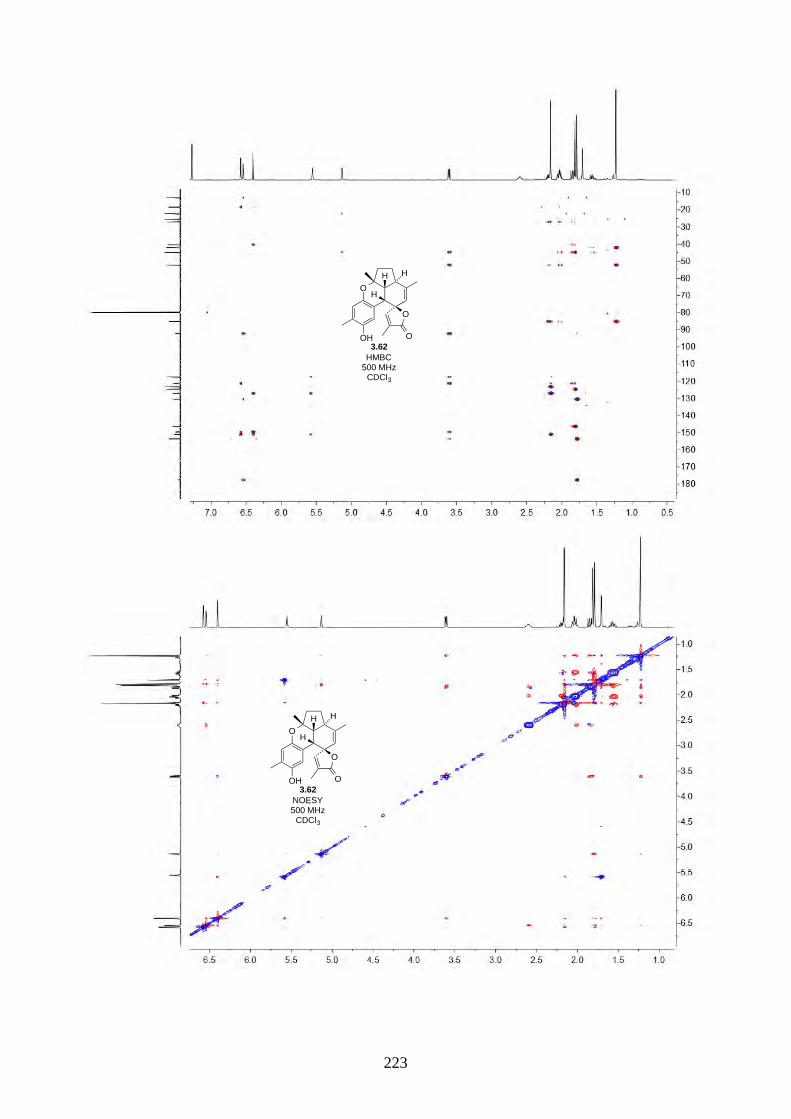
223
O
OH
H H
H
O
O
HMBC500 MHz
CDCl3
3.62
O
OH
H H
H
O
O
NOESY500 MHz
CDCl3
3.62
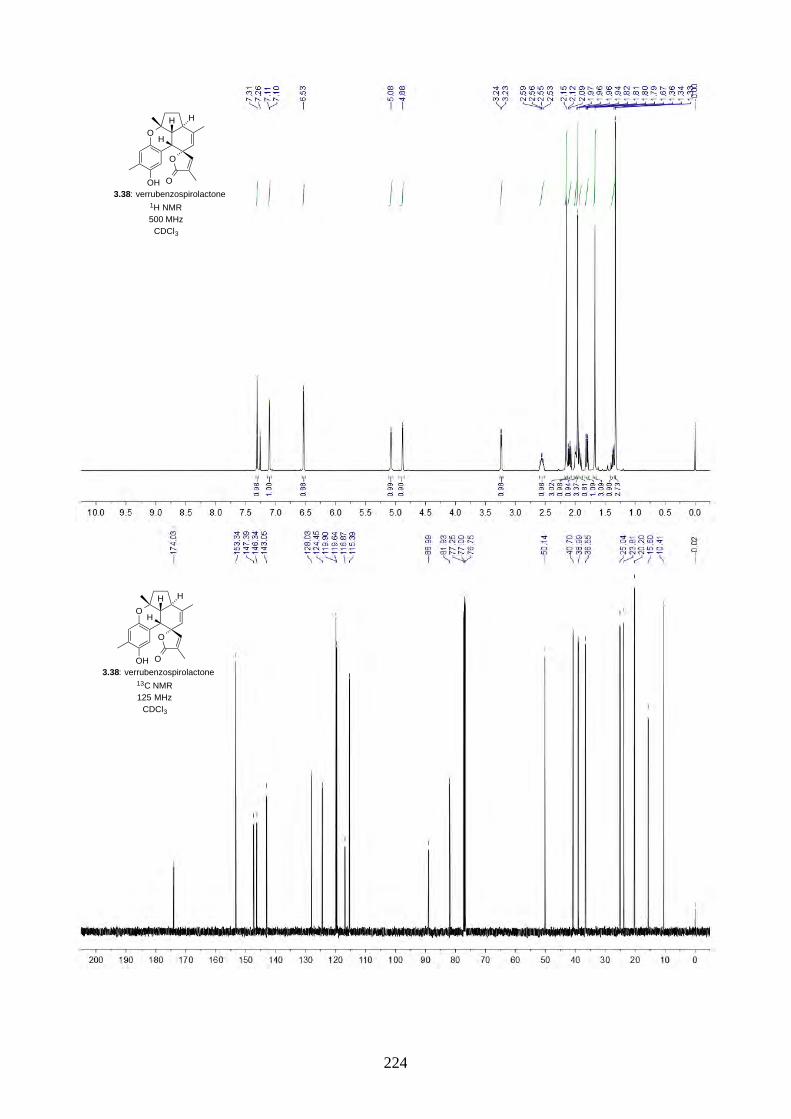
224
O
OH
H H
H
O
O3.38: verrubenzospirolactone
1H NMR500 MHz
CDCl3
O
OH
H H
H
O
O3.38: verrubenzospirolactone
13C NMR125 MHz
CDCl3
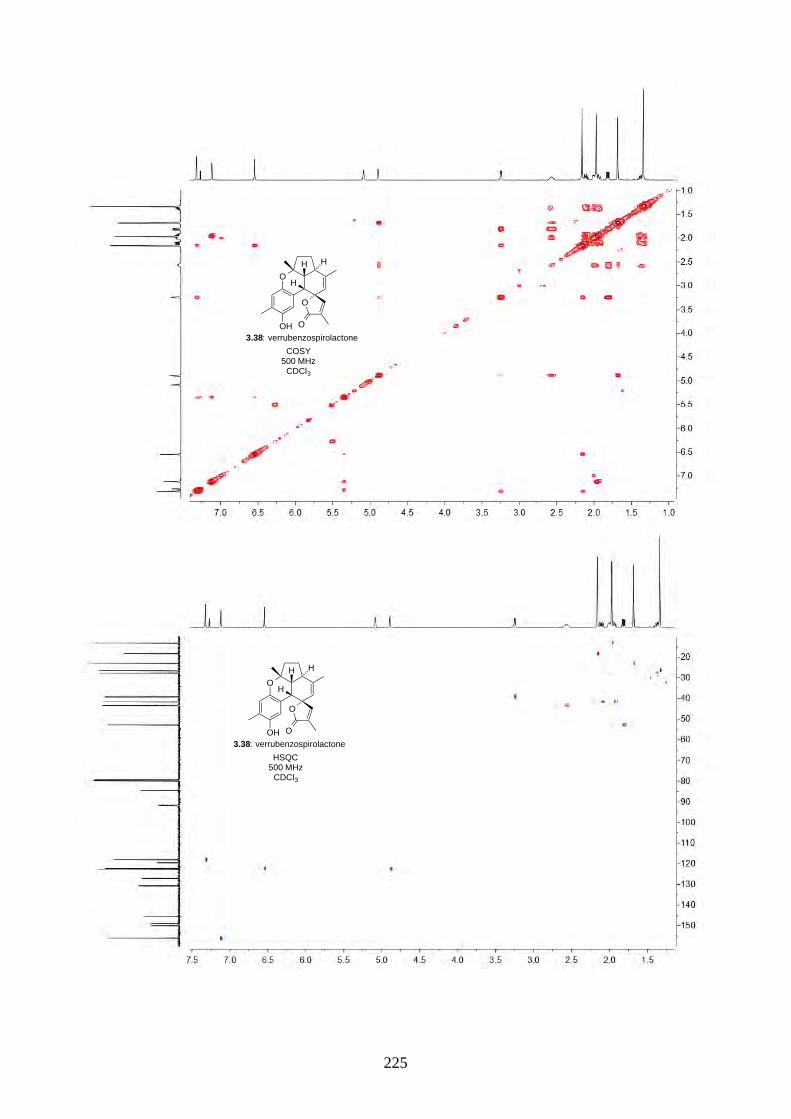
225
O
OH
H H
H
O
O3.38: verrubenzospirolactone
COSY500 MHz
CDCl3
O
OH
H H
H
O
O3.38: verrubenzospirolactone
HSQC500 MHz
CDCl3
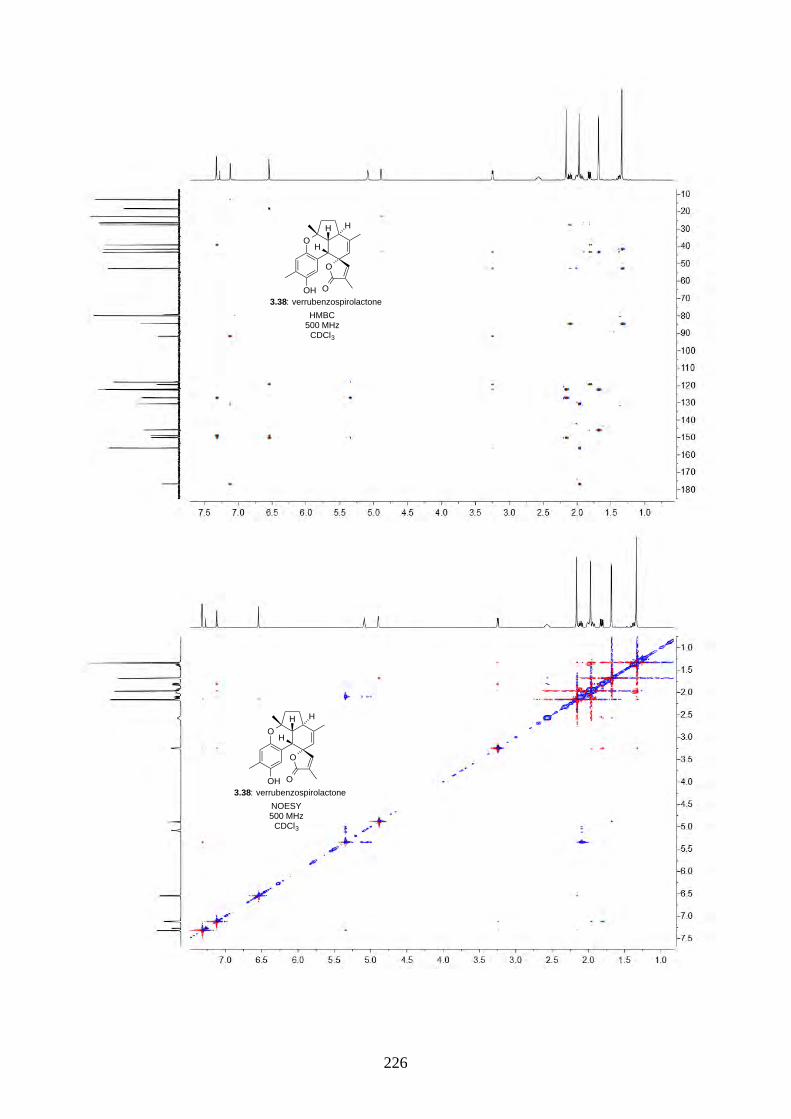
226
O
OH
H H
H
O
O3.38: verrubenzospirolactone
HMBC500 MHz
CDCl3
O
OH
H H
H
O
O3.38: verrubenzospirolactone
NOESY500 MHz
CDCl3
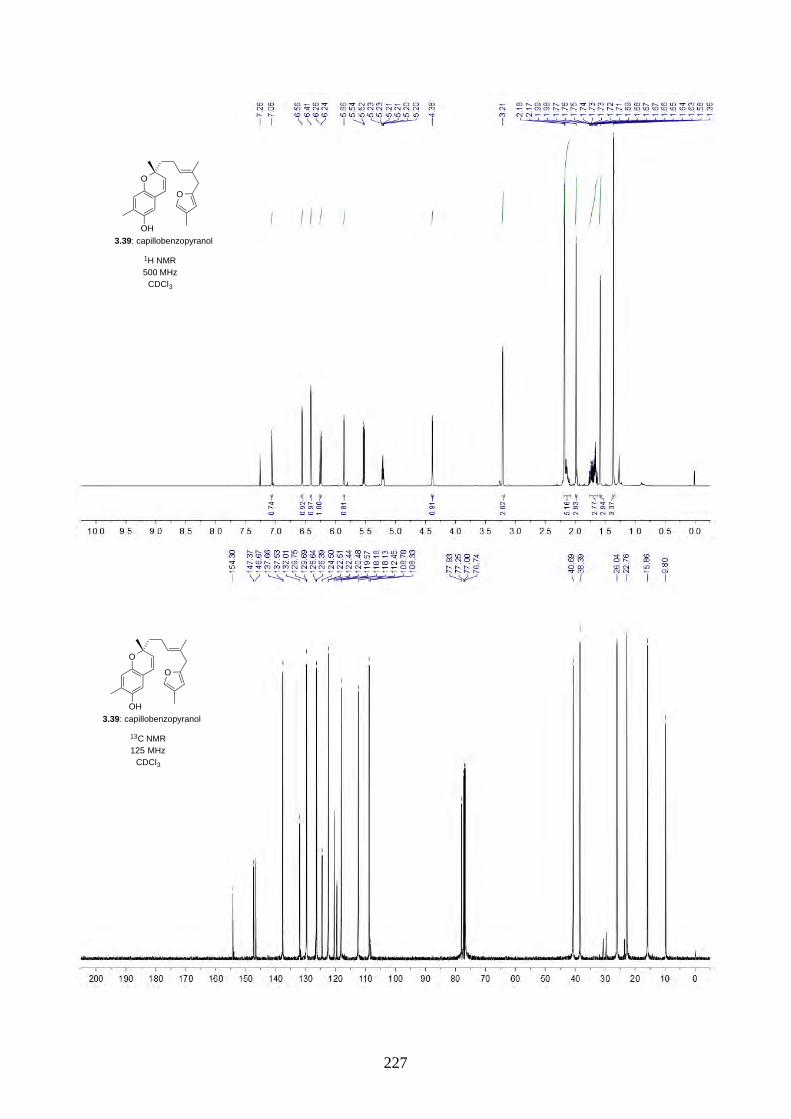
227
O
OH
O
3.39: capillobenzopyranol
1H NMR500 MHz
CDCl3
O
OH
O
3.39: capillobenzopyranol
13C NMR125 MHz
CDCl3
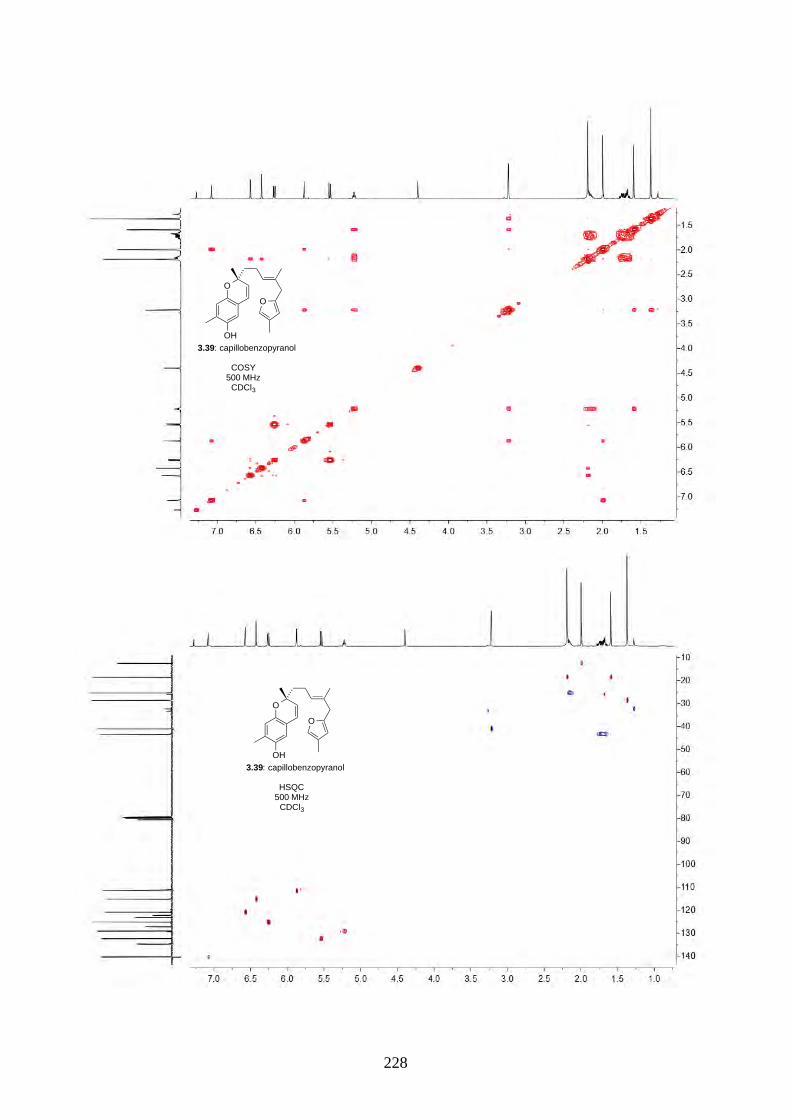
228
O
OH
O
3.39: capillobenzopyranol
COSY500 MHz
CDCl3
O
OH
O
3.39: capillobenzopyranol
HSQC500 MHz
CDCl3
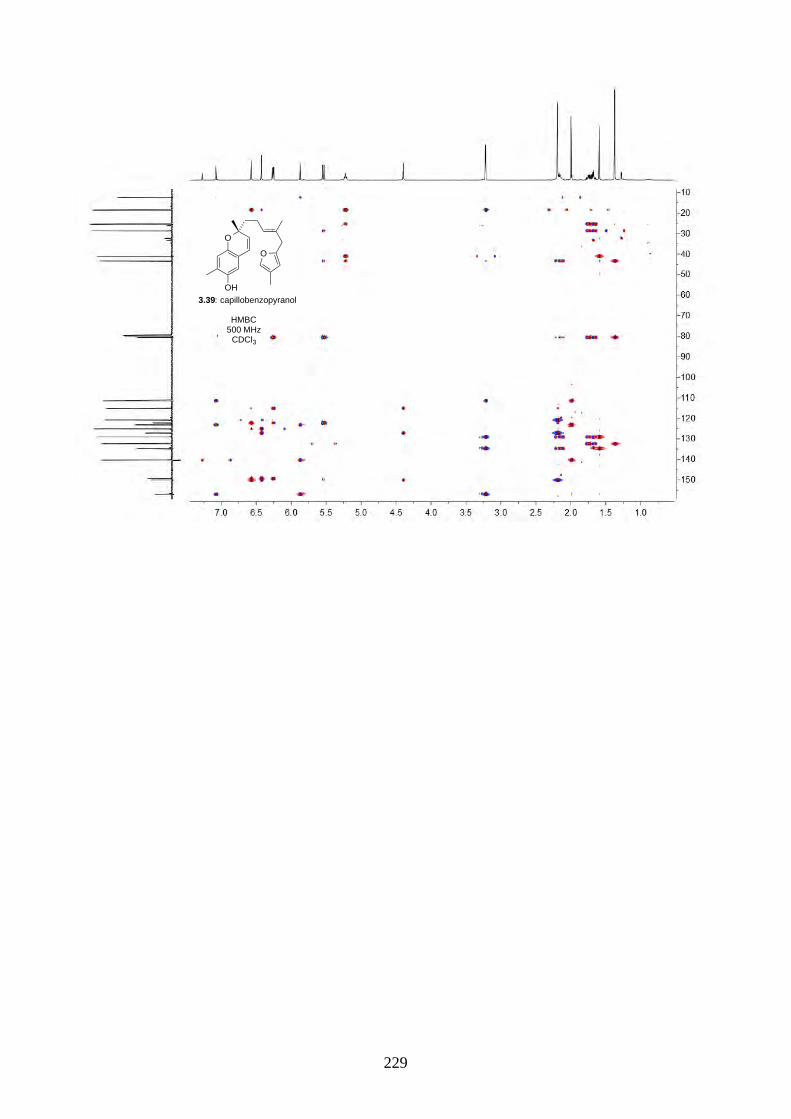
229
O
OH
O
3.39: capillobenzopyranol
HMBC500 MHz
CDCl3
230
O
OH
O
OOH
OMe
3.64
d.r. 1:11H NMR500 MHz
CDCl3
O
OH
O
OOH
OMe
3.64
d.r. 1:113C NMR125 MHz
CDCl3
231
O
OH
O
O
OMe
3.65
d.r. 1:11H NMR500 MHz
CDCl3
O
OH
O
O
OMe
3.65
d.r. 1:113C NMR125 MHz
CDCl3
232
O
OH
O
O
OMe
3.65
d.r. 1:1COSY
500 MHzCDCl3
O
OH
O
O
OMe
3.65
d.r. 1:1HSQC
500 MHzCDCl3
233
O
OH
O
O
OMe
3.65
d.r. 1:1HMBC
500 MHzCDCl3
234
3.66
O
O
OHO
d.r. 2:11H NMR500 MHz
CDCl3
3.66
O
O
OHO
d.r. 2:113C NMR125 MHz
CDCl3
235
3.66
O
O
OHO
d.r. 2:1COSY
500 MHz CDCl3
3.66
O
O
OHO
d.r. 2:1HSQC
500 MHz CDCl3
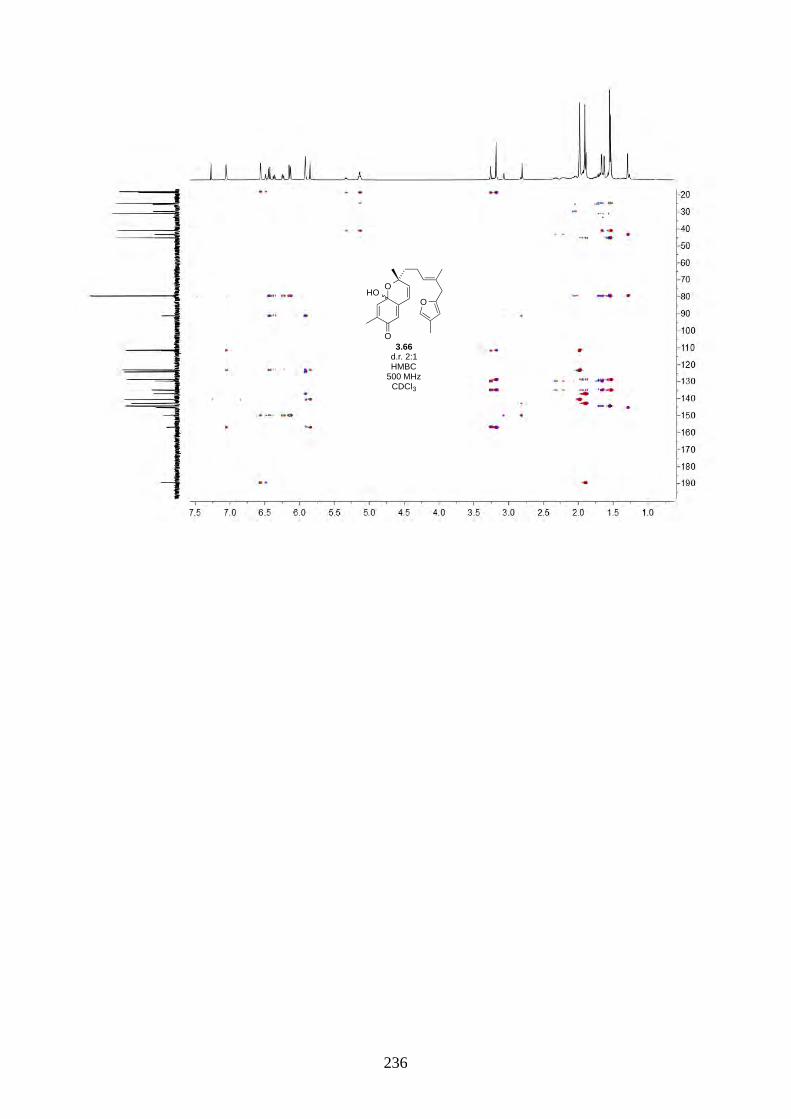
236
3.66
O
O
OHO
d.r. 2:1HMBC
500 MHz CDCl3
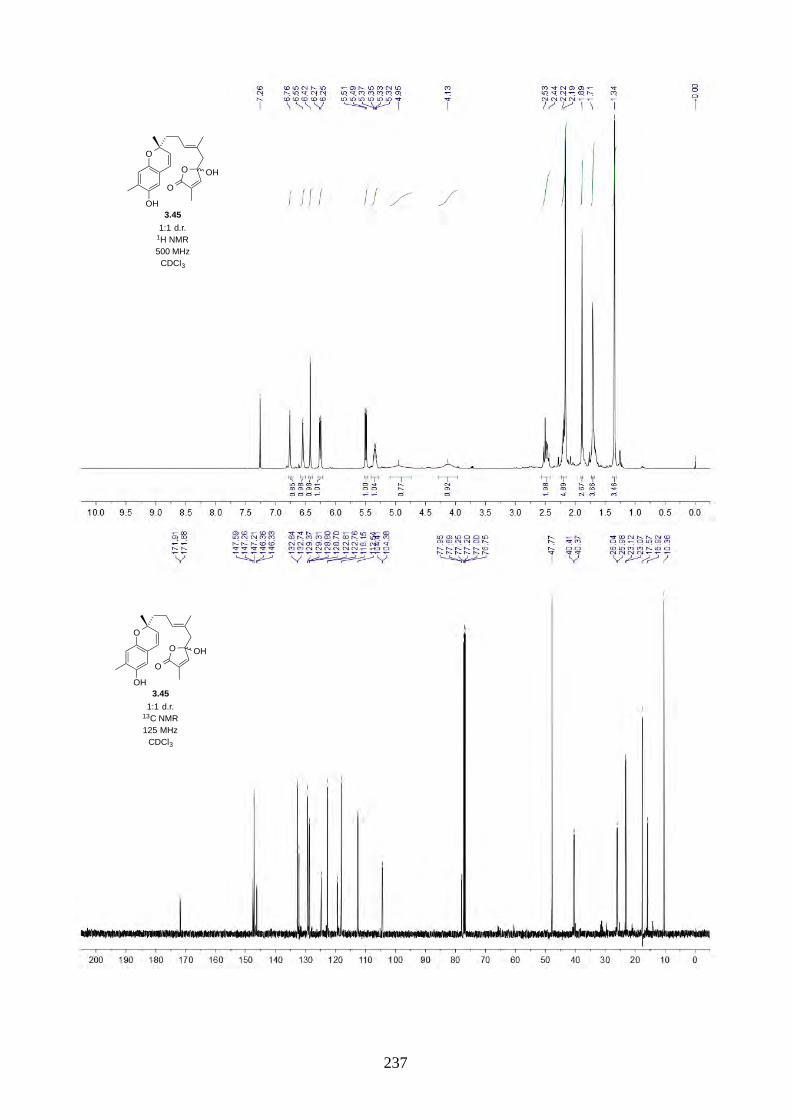
237
O
OH
O
O
OH
3.451:1 d.r.
1H NMR500 MHz
CDCl3
O
OH
O
O
OH
3.451:1 d.r.
13C NMR125 MHz
CDCl3
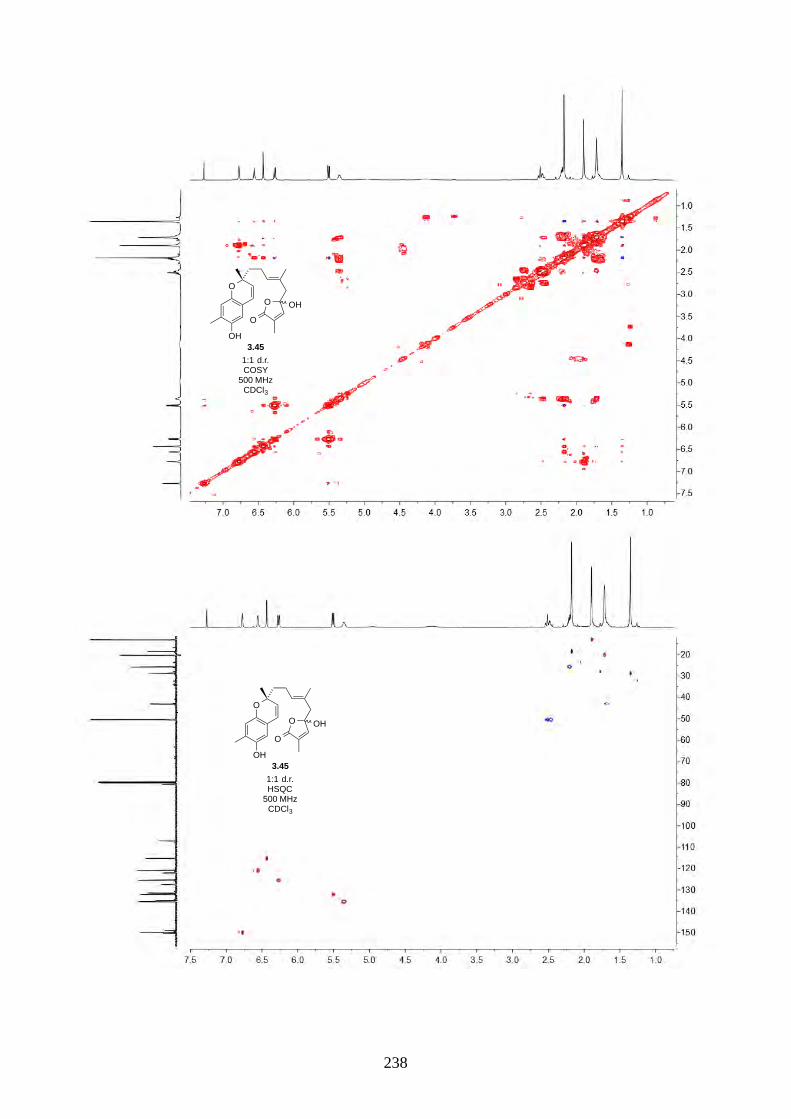
238
O
OH
O
O
OH
3.451:1 d.r.COSY
500 MHzCDCl3
O
OH
O
O
OH
3.451:1 d.r.HSQC
500 MHzCDCl3
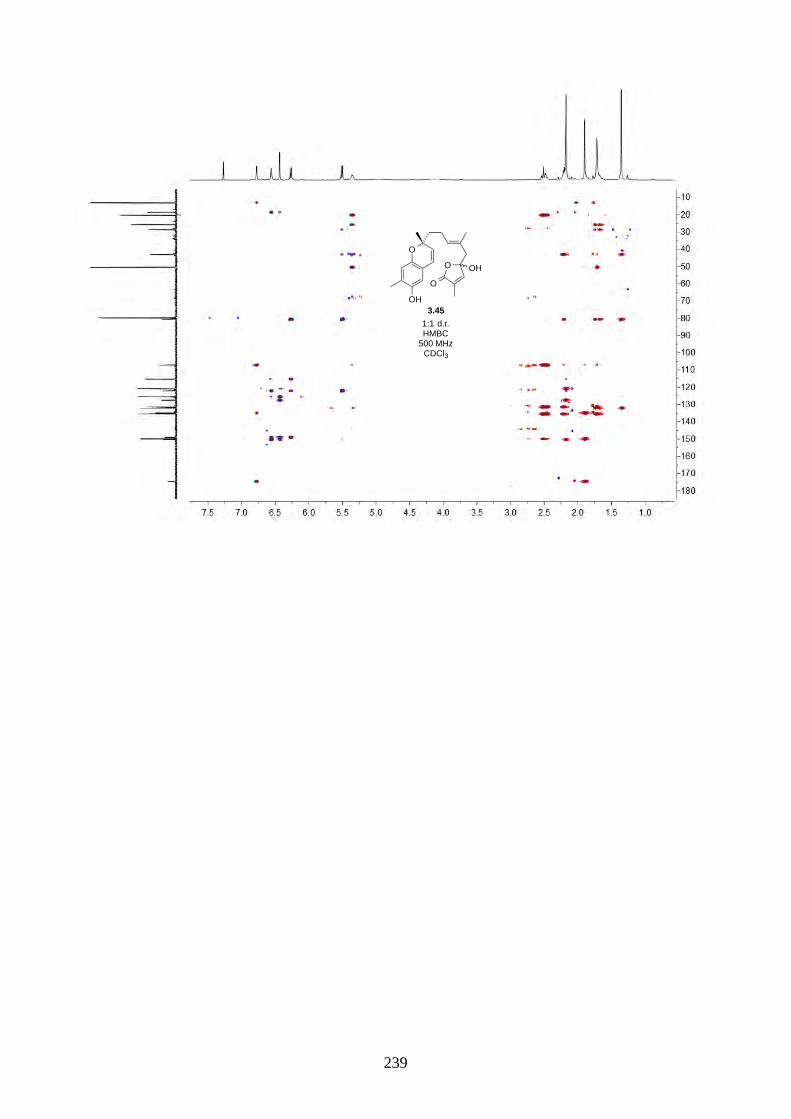
239
O
OH
O
O
OH
3.451:1 d.r.HMBC
500 MHzCDCl3
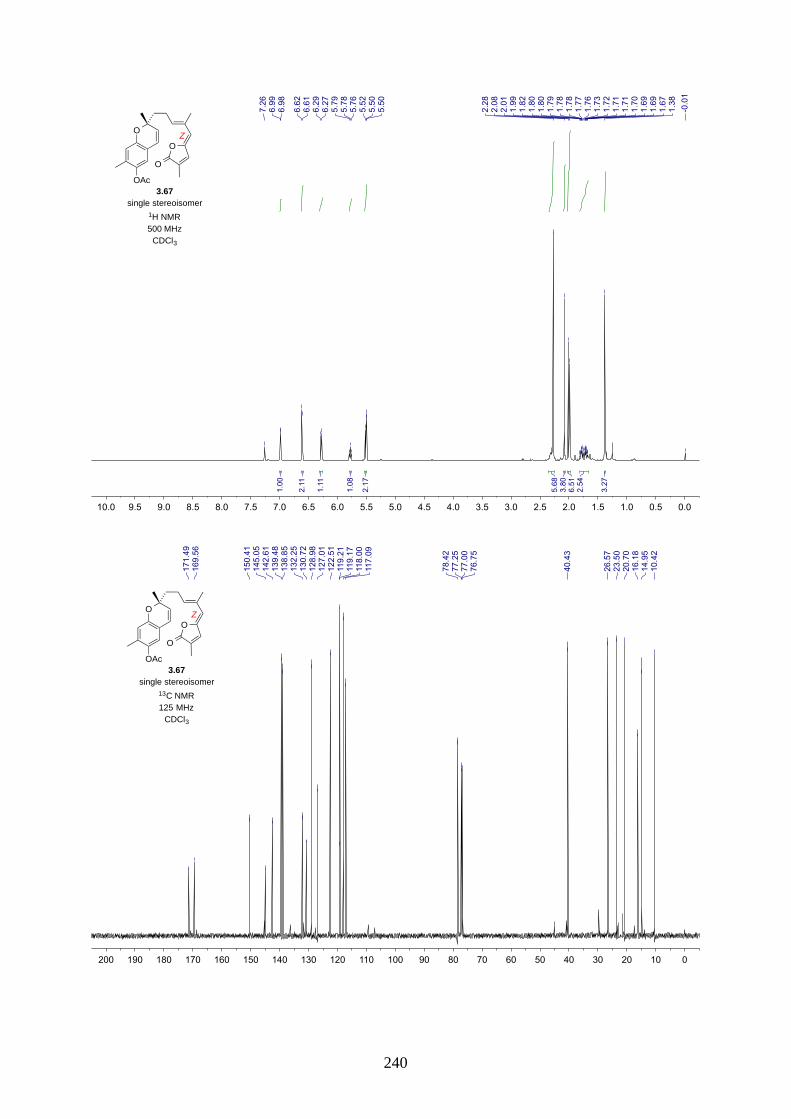
240
O
OAc
O
O
3.67single stereoisomer
Z
1H NMR500 MHz
CDCl3
O
OAc
O
O
3.67single stereoisomer
Z
13C NMR125 MHz
CDCl3
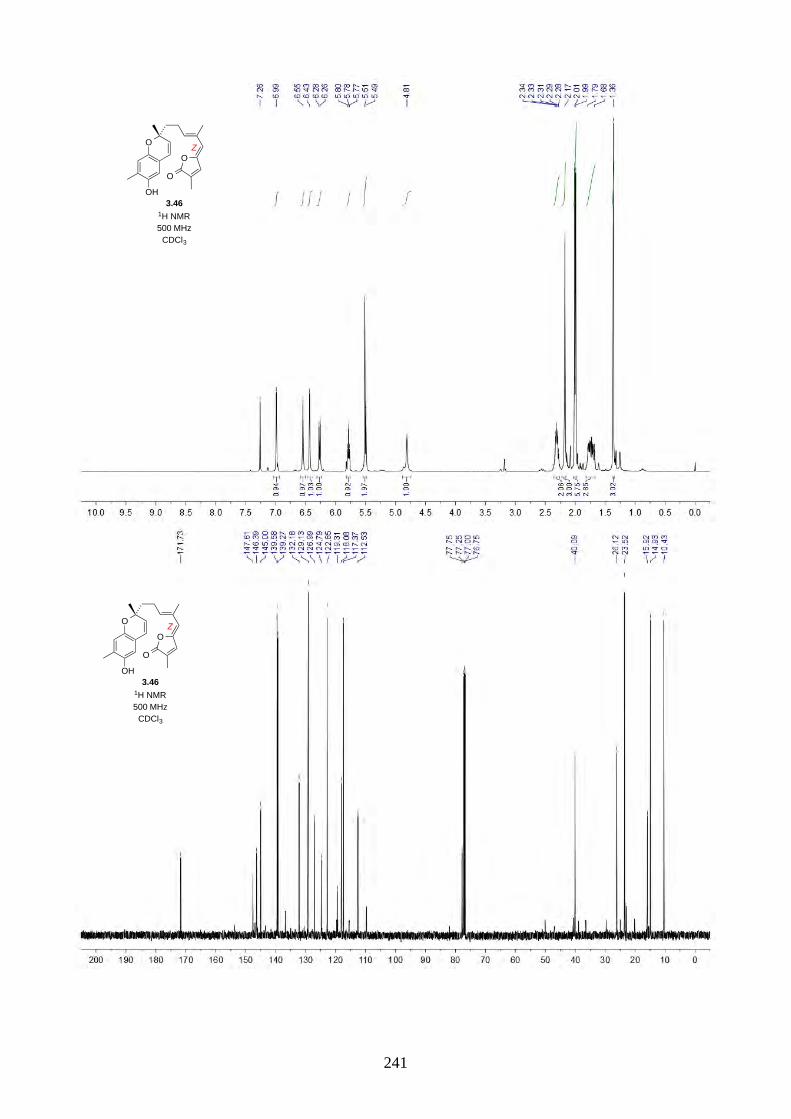
241
O
OH
O
O
3.46
Z
1H NMR500 MHz
CDCl3
O
OH
O
O
3.46
Z
1H NMR500 MHz
CDCl3
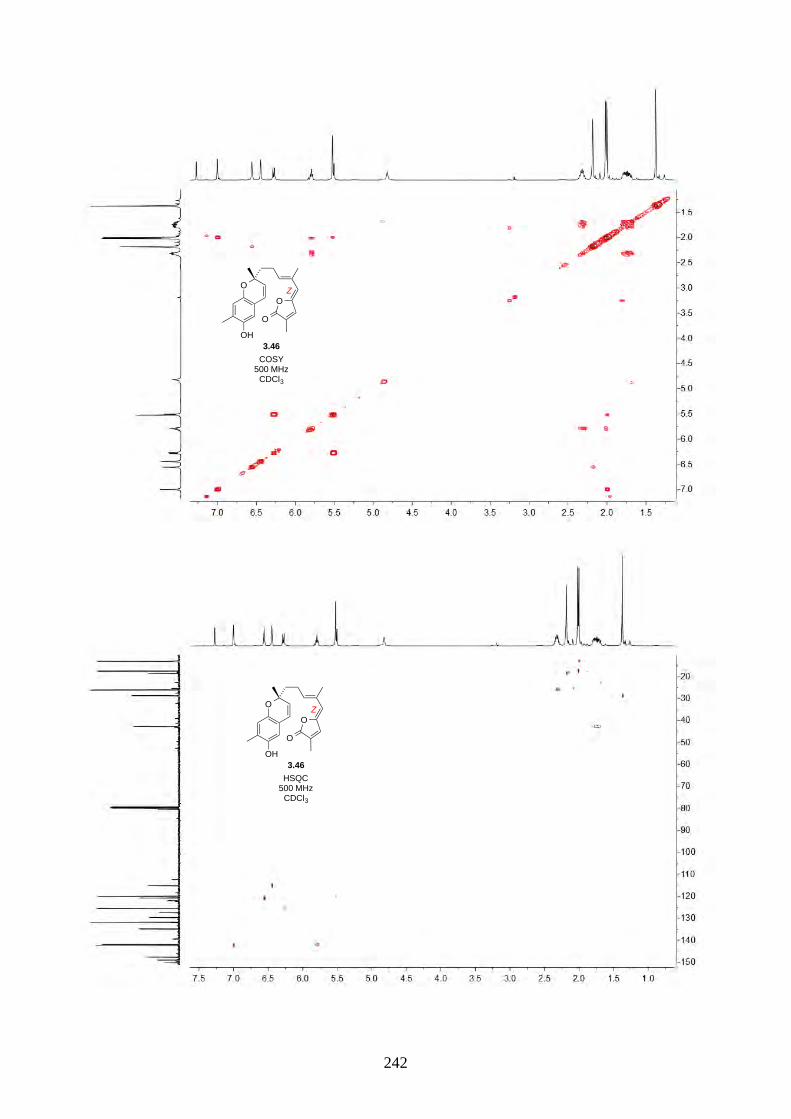
242
O
OH
O
O
3.46
Z
COSY500 MHz
CDCl3
O
OH
O
O
3.46
Z
HSQC500 MHz
CDCl3
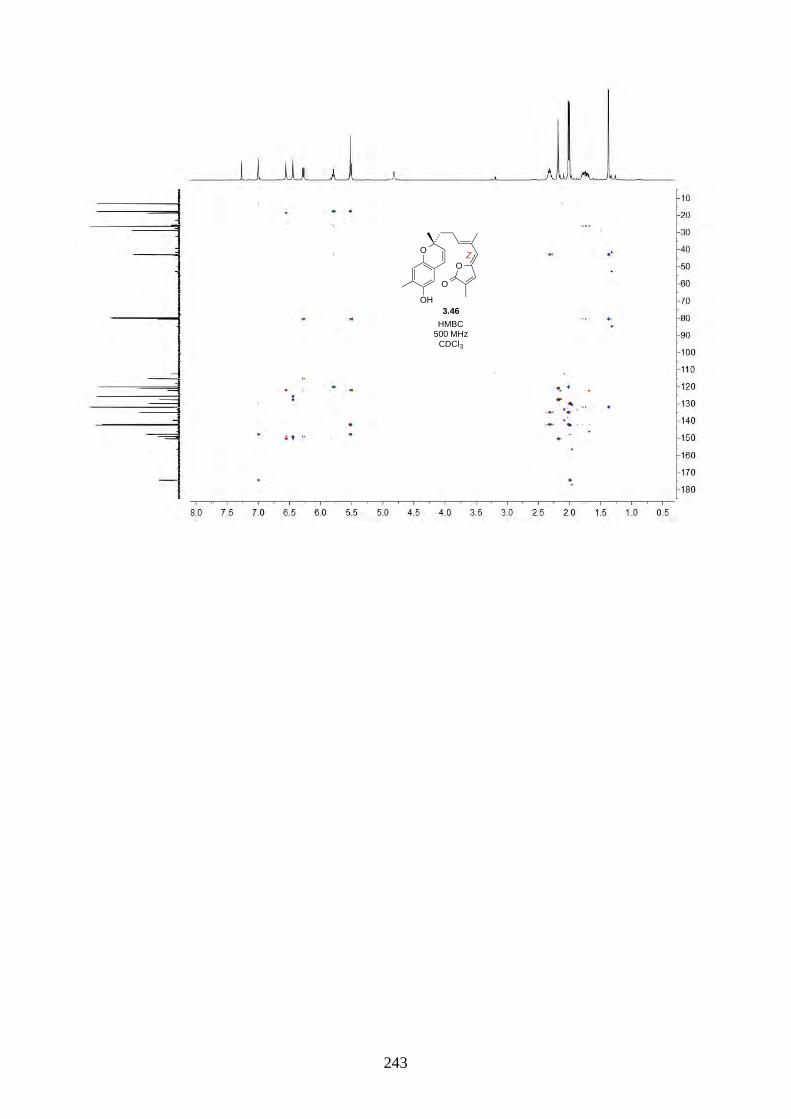
243
O
OH
O
O
3.46
Z
HMBC500 MHz
CDCl3
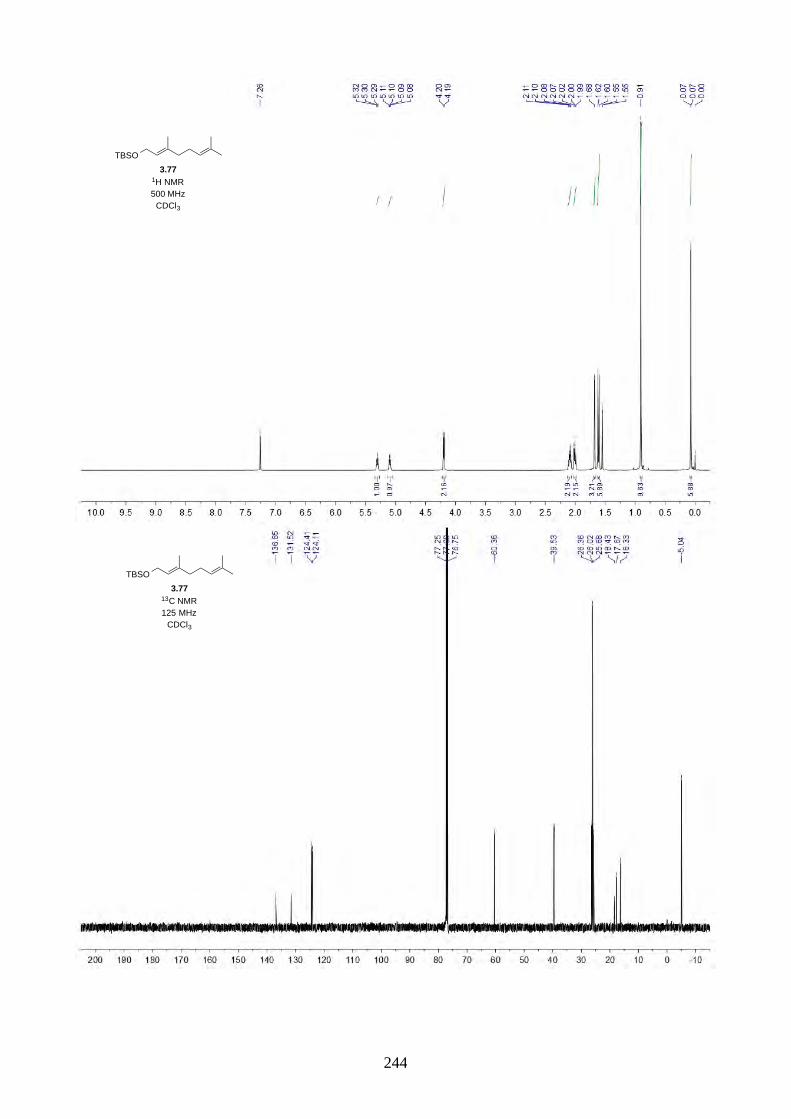
244
TBSO3.77
1H NMR500 MHz
CDCl3
TBSO3.77
13C NMR125 MHz
CDCl3
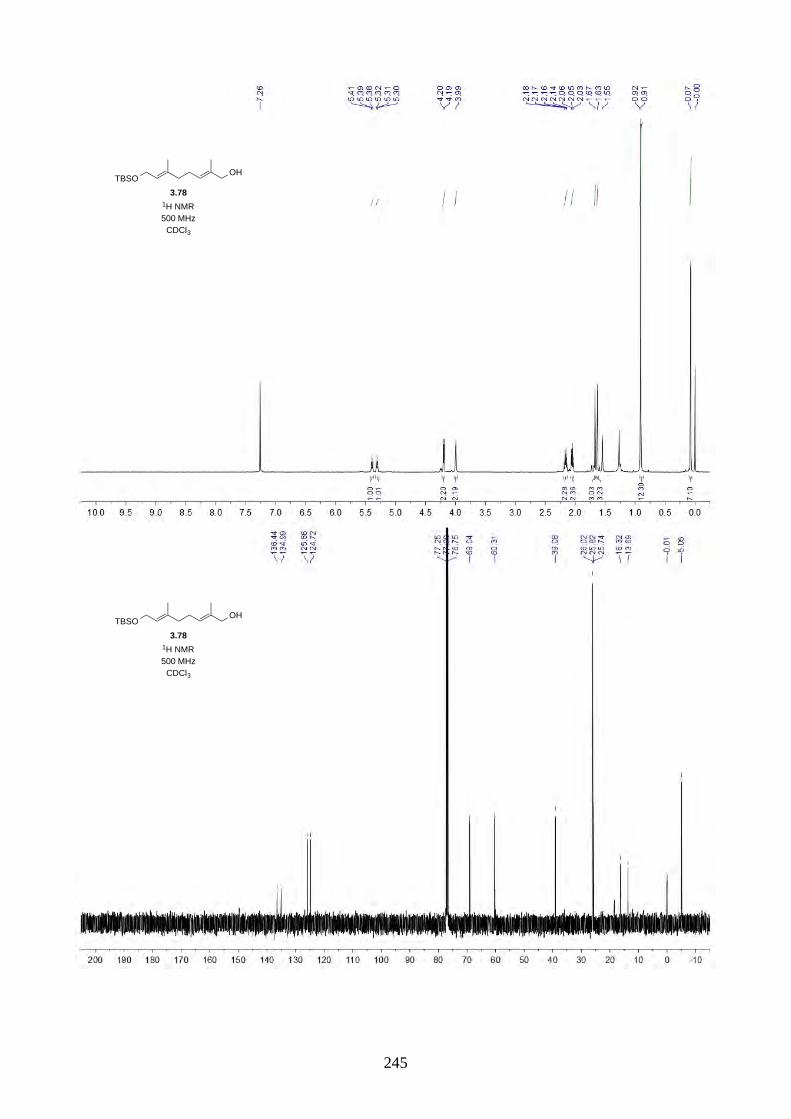
245
TBSO3.78
OH
1H NMR500 MHz
CDCl3
TBSO3.78
OH
1H NMR500 MHz
CDCl3
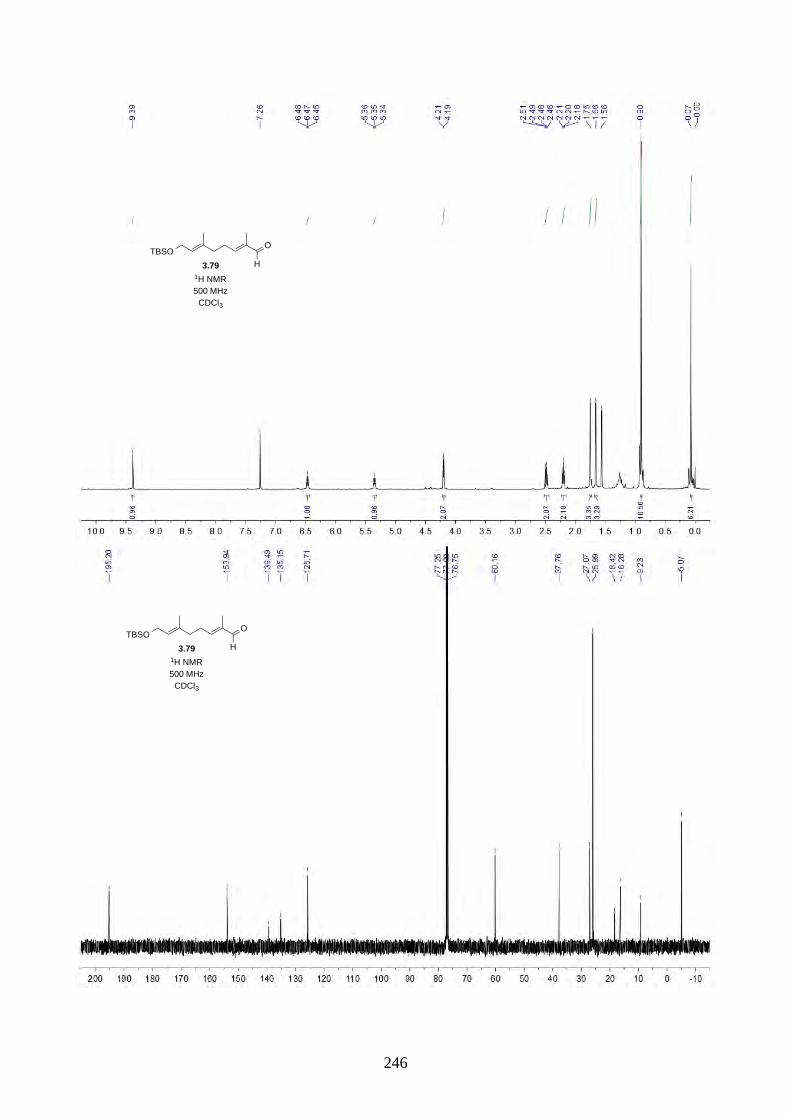
246
TBSO3.79
O
H1H NMR500 MHz
CDCl3
TBSO3.79
O
H1H NMR500 MHz
CDCl3
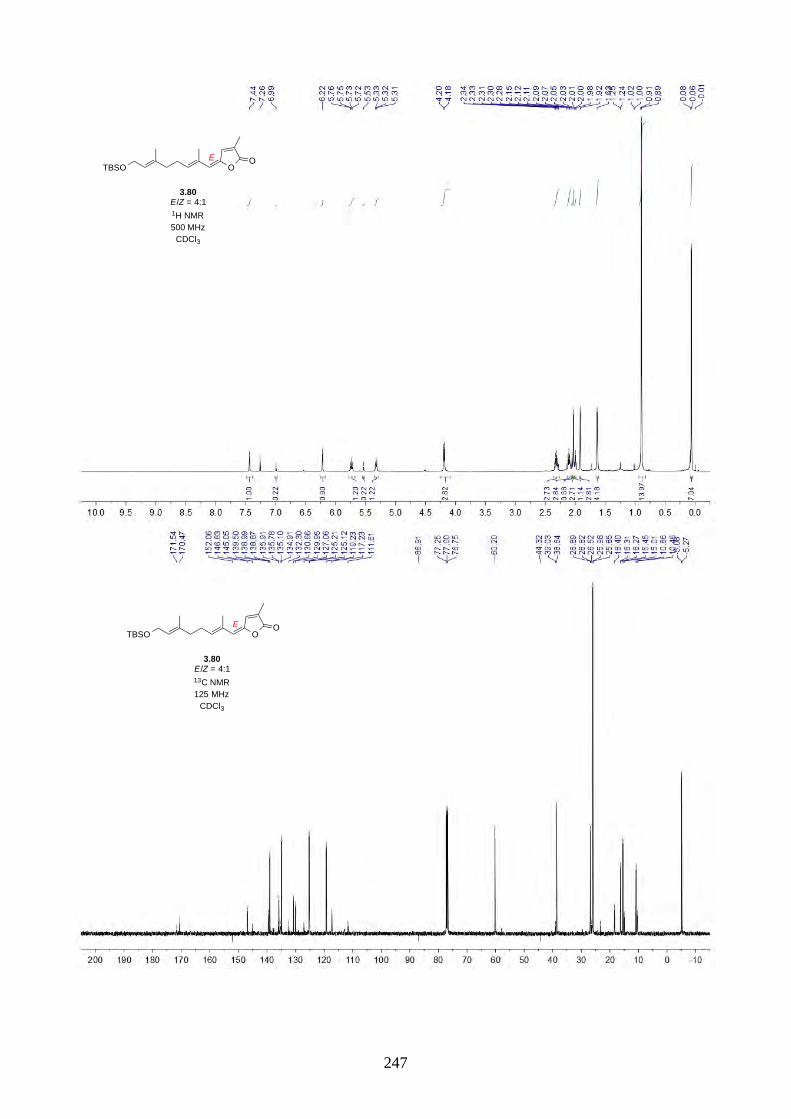
247
1H NMR500 MHz
CDCl3
3.80E/Z = 4:1
EO
OTBSO
13C NMR125 MHz
CDCl3
3.80E/Z = 4:1
EO
OTBSO
248
1H NMR500 MHz
CDCl3
3.81E/Z = 4:1
EO
OHO
13C NMR125 MHz
CDCl3
3.81E/Z = 4:1
EO
OHO
249
1H NMR500 MHz
CDCl3
3.68E/Z = 4:1
EO
OO
H
13C NMR125 MHz
CDCl3
3.68E/Z = 4:1
EO
OO
H
250
1H NMR500 MHz
CDCl3
3.68
EO
OO
H
13C NMR125 MHz
CDCl3
3.68
EO
OO
H
251
COSY500 MHz
CDCl3
3.68
EO
OO
H
HSQC500 MHz
CDCl3
3.68
EO
OO
H
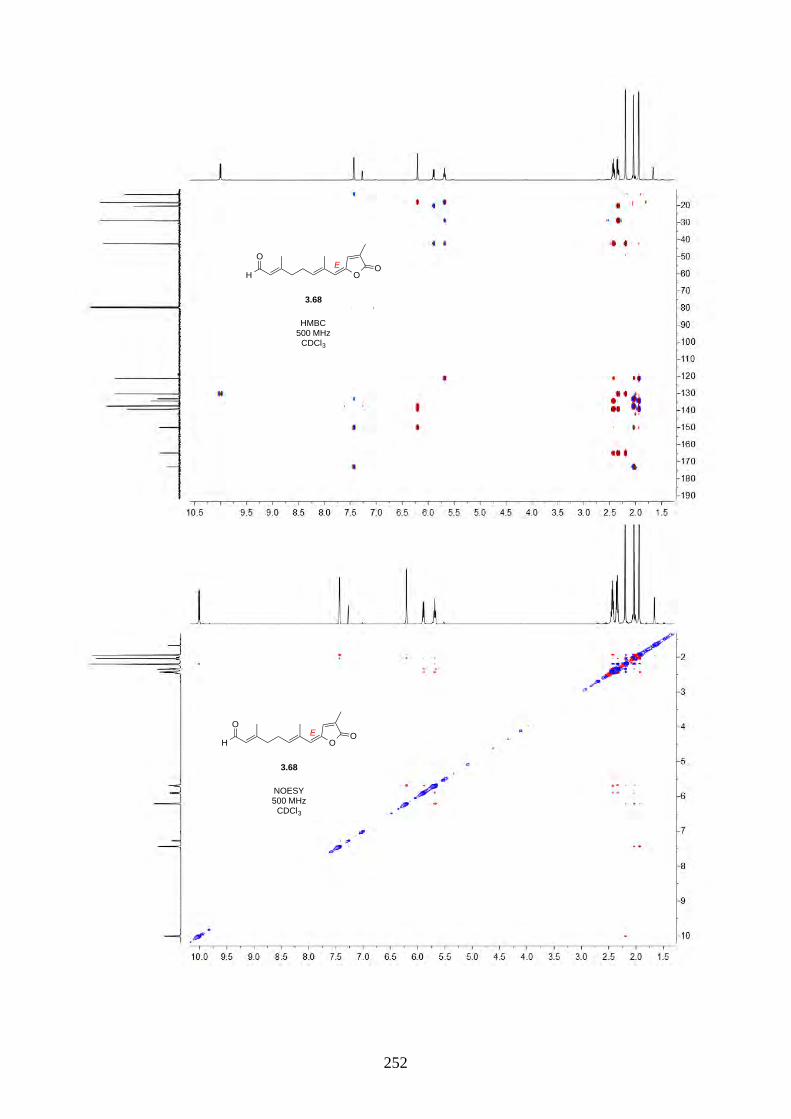
252
HMBC500 MHz
CDCl3
3.68
EO
OO
H
NOESY500 MHz
CDCl3
3.68
EO
OO
H
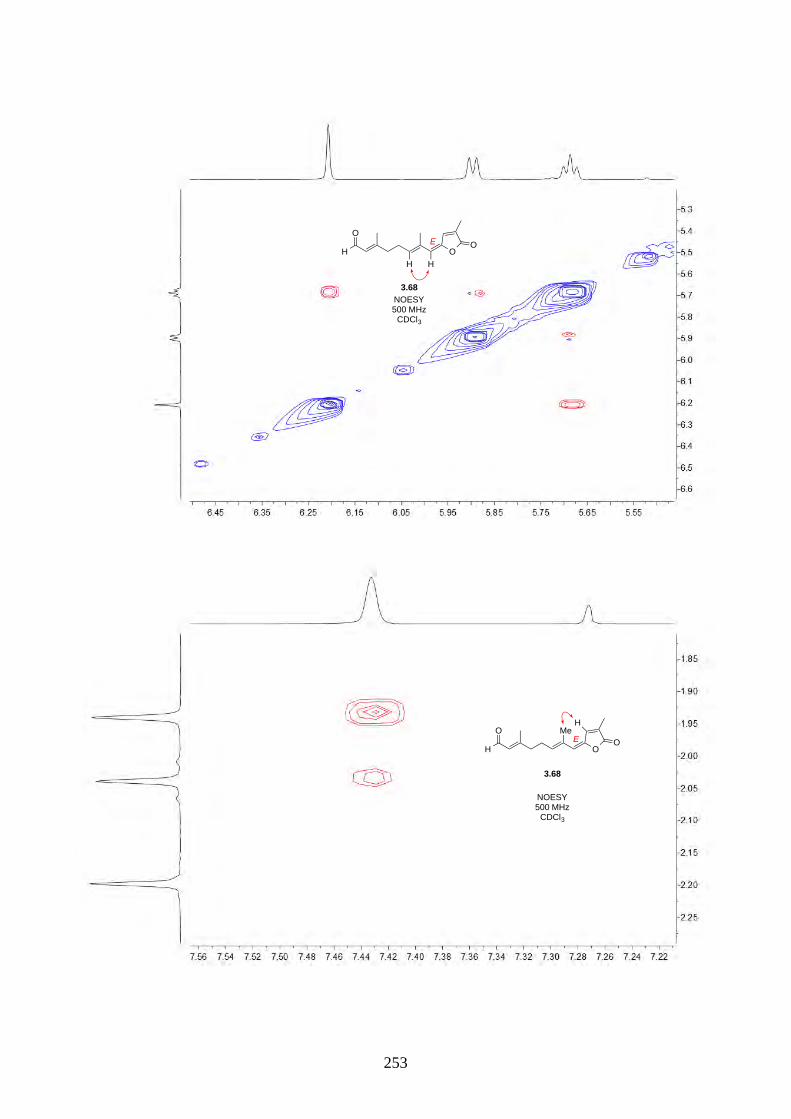
253
NOESY500 MHz
CDCl3
3.68
EO
OO
HH H
NOESY500 MHz
CDCl3
3.68
EO
OO Me
H
H
254
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
+
4.9:1 d.r. (3.74:3.75)1H NMR500 MHz
CDCl3
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
+
3:1 d.r. (3.74:3.75)1H NMR500 MHz
CDCl3
255
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
+
4.9:1 d.r. (3.74:3.75)13C NMR125 MHz
CDCl3
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
4.9:1 d.r. (3.74:3.75)COSY
500 MHzCDCl3
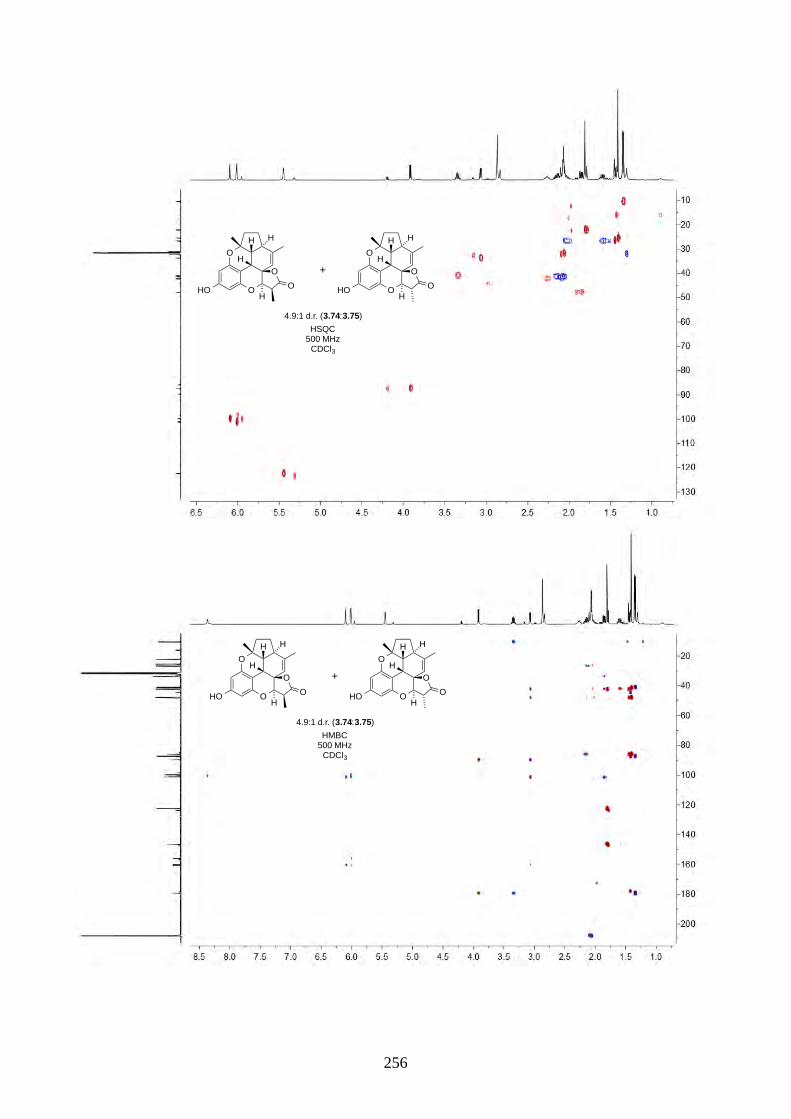
256
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
+
4.9:1 d.r. (3.74:3.75)HSQC
500 MHzCDCl3
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
+
4.9:1 d.r. (3.74:3.75)HMBC
500 MHzCDCl3
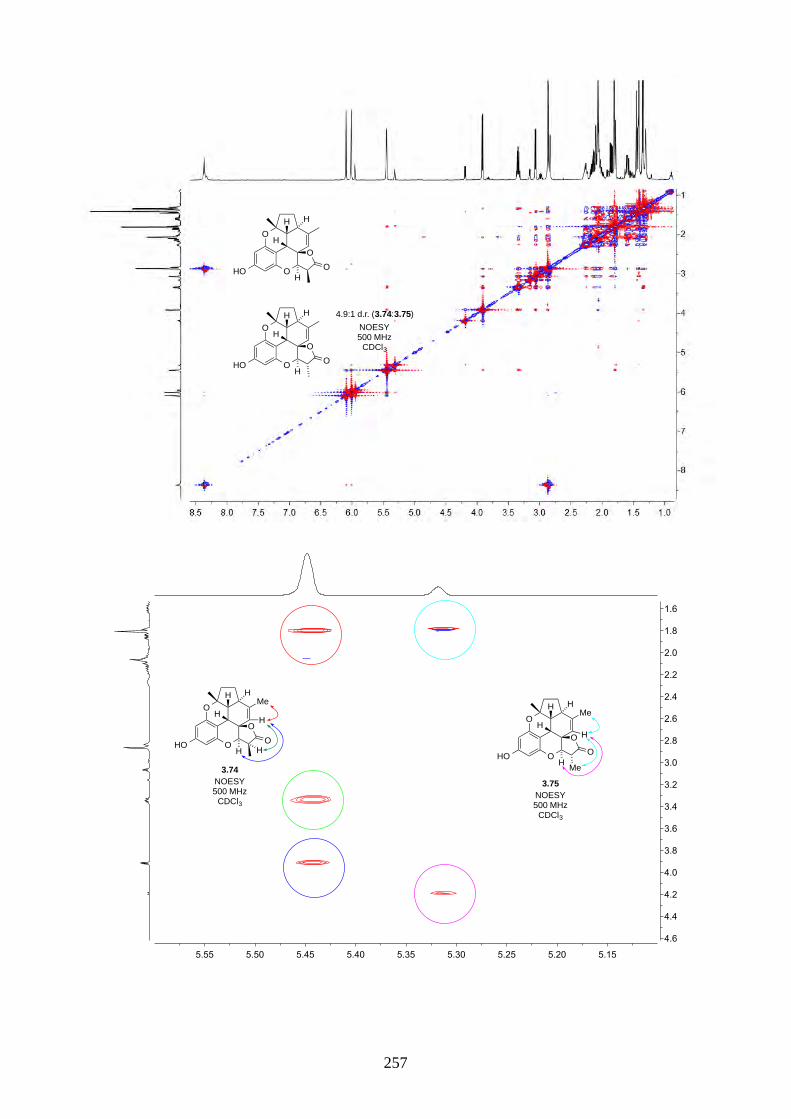
257
O
H
H
HO
H
O
O
HO
O
H
H
HO
H
O
O
HO
4.9:1 d.r. (3.74:3.75)NOESY500 MHz
CDCl3
OMe
H
H
HO
H
O
O
HO
H
H
NOESY500 MHz
CDCl3
3.74
OMe
H
H
HO
H
O
O
HO
Me
H
NOESY500 MHz
CDCl3
3.75
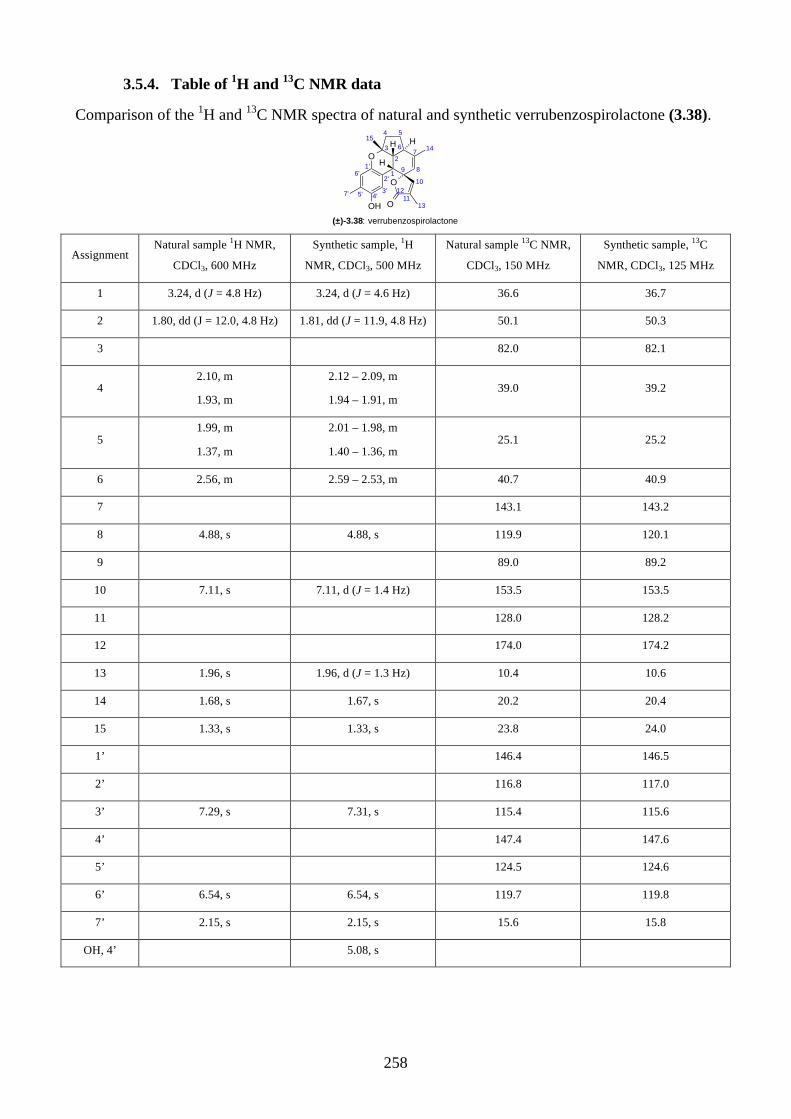
258
3.5.4. Table of 1H and 13C NMR data
Comparison of the 1H and 13C NMR spectra of natural and synthetic verrubenzospirolactone (3.38).
Assignment Natural sample 1H NMR,
CDCl3, 600 MHz
Synthetic sample, 1H
NMR, CDCl3, 500 MHz
Natural sample 13C NMR,
CDCl3, 150 MHz
Synthetic sample, 13C
NMR, CDCl3, 125 MHz
1 3.24, d (J = 4.8 Hz) 3.24, d (J = 4.6 Hz) 36.6 36.7
2 1.80, dd (J = 12.0, 4.8 Hz) 1.81, dd (J = 11.9, 4.8 Hz) 50.1 50.3
3 82.0 82.1
4 2.10, m
1.93, m
2.12 – 2.09, m
1.94 – 1.91, m 39.0 39.2
5 1.99, m
1.37, m
2.01 – 1.98, m
1.40 – 1.36, m 25.1 25.2
6 2.56, m 2.59 – 2.53, m 40.7 40.9
7 143.1 143.2
8 4.88, s 4.88, s 119.9 120.1
9 89.0 89.2
10 7.11, s 7.11, d (J = 1.4 Hz) 153.5 153.5
11 128.0 128.2
12 174.0 174.2
13 1.96, s 1.96, d (J = 1.3 Hz) 10.4 10.6
14 1.68, s 1.67, s 20.2 20.4
15 1.33, s 1.33, s 23.8 24.0
1’ 146.4 146.5
2’ 116.8 117.0
3’ 7.29, s 7.31, s 115.4 115.6
4’ 147.4 147.6
5’ 124.5 124.6
6’ 6.54, s 6.54, s 119.7 119.8
7’ 2.15, s 2.15, s 15.6 15.8
OH, 4’ 5.08, s
O
OH
H H
H
O
O
(±)-3.38: verrubenzospirolactone
1
23
4
14
1311
10
9 8
76
515
7' 5'
6'
3'
2'
12
1'
4'
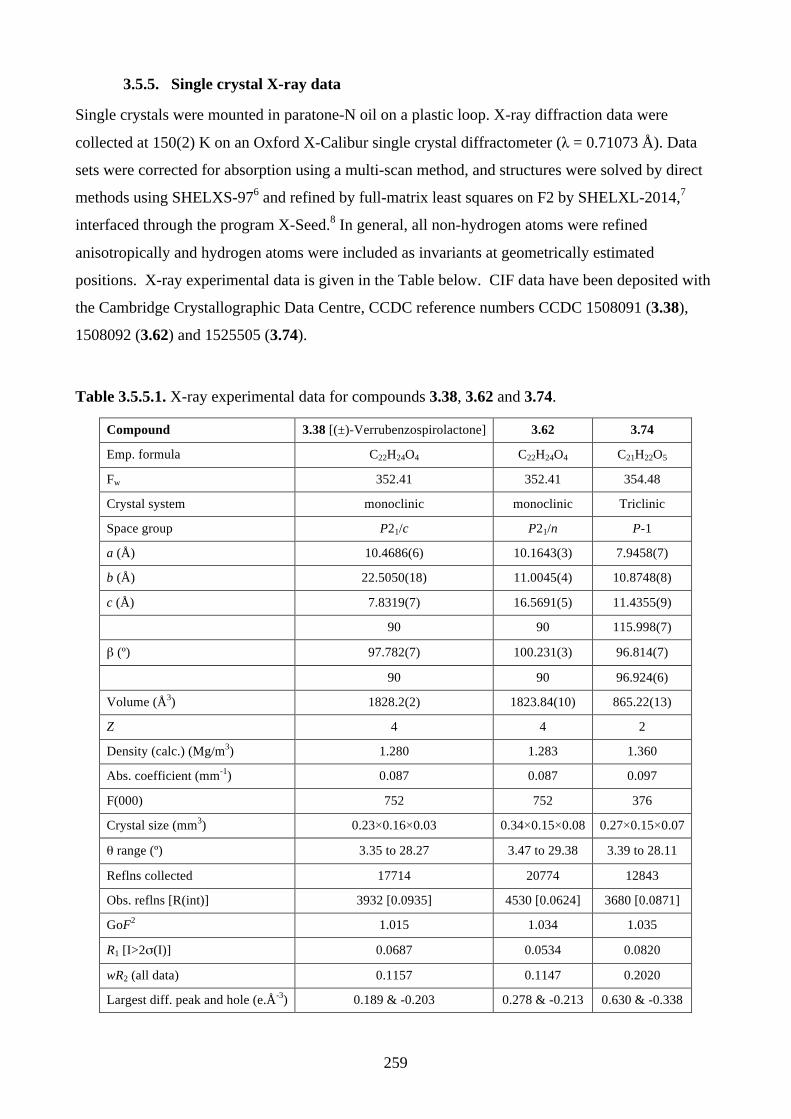
259
3.5.5. Single crystal X-ray data
Single crystals were mounted in paratone-N oil on a plastic loop. X-ray diffraction data were
collected at 150(2) K on an Oxford X-Calibur single crystal diffractometer (λ = 0.71073 Å). Data
sets were corrected for absorption using a multi-scan method, and structures were solved by direct
methods using SHELXS-976 and refined by full-matrix least squares on F2 by SHELXL-2014,7
interfaced through the program X-Seed.8 In general, all non-hydrogen atoms were refined
anisotropically and hydrogen atoms were included as invariants at geometrically estimated
positions. X-ray experimental data is given in the Table below. CIF data have been deposited with
the Cambridge Crystallographic Data Centre, CCDC reference numbers CCDC 1508091 (3.38),
1508092 (3.62) and 1525505 (3.74).
Table 3.5.5.1. X-ray experimental data for compounds 3.38, 3.62 and 3.74.
Compound 3.38 [(±)-Verrubenzospirolactone] 3.62 3.74
Emp. formula C22H24O4 C22H24O4 C21H22O5
Fw 352.41 352.41 354.48
Crystal system monoclinic monoclinic Triclinic
Space group P21/c P21/n P-1
a (Å) 10.4686(6) 10.1643(3) 7.9458(7)
b (Å) 22.5050(18) 11.0045(4) 10.8748(8)
c (Å) 7.8319(7) 16.5691(5) 11.4355(9)
90 90 115.998(7)
β (º) 97.782(7) 100.231(3) 96.814(7)
90 90 96.924(6)
Volume (Å3) 1828.2(2) 1823.84(10) 865.22(13)
Z 4 4 2
Density (calc.) (Mg/m3) 1.280 1.283 1.360
Abs. coefficient (mm-1) 0.087 0.087 0.097
F(000) 752 752 376
Crystal size (mm3) 0.23×0.16×0.03 0.34×0.15×0.08 0.27×0.15×0.07
θ range (º) 3.35 to 28.27 3.47 to 29.38 3.39 to 28.11
Reflns collected 17714 20774 12843
Obs. reflns [R(int)] 3932 [0.0935] 4530 [0.0624] 3680 [0.0871]
GoF2 1.015 1.034 1.035
R1 [I>2σ(I)] 0.0687 0.0534 0.0820
wR2 (all data) 0.1157 0.1147 0.2020
Largest diff. peak and hole (e.Å-3) 0.189 & -0.203 0.278 & -0.213 0.630 & -0.338
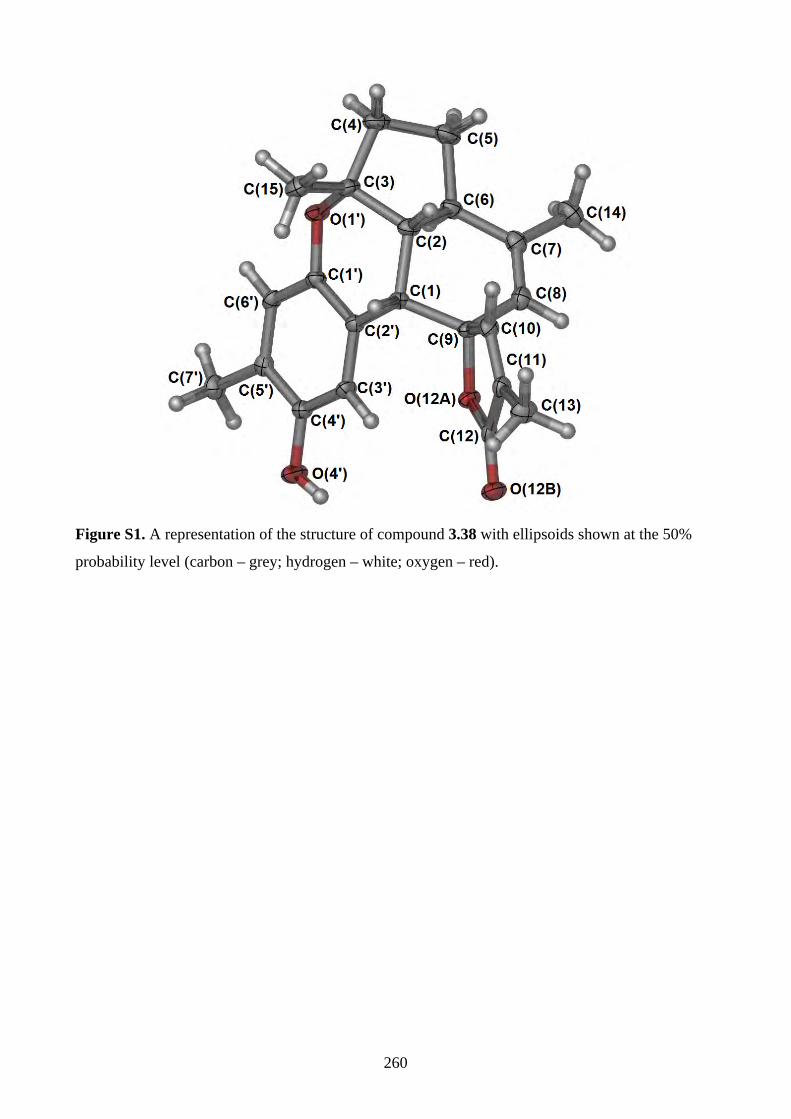
260
Figure S1. A representation of the structure of compound 3.38 with ellipsoids shown at the 50%
probability level (carbon – grey; hydrogen – white; oxygen – red).
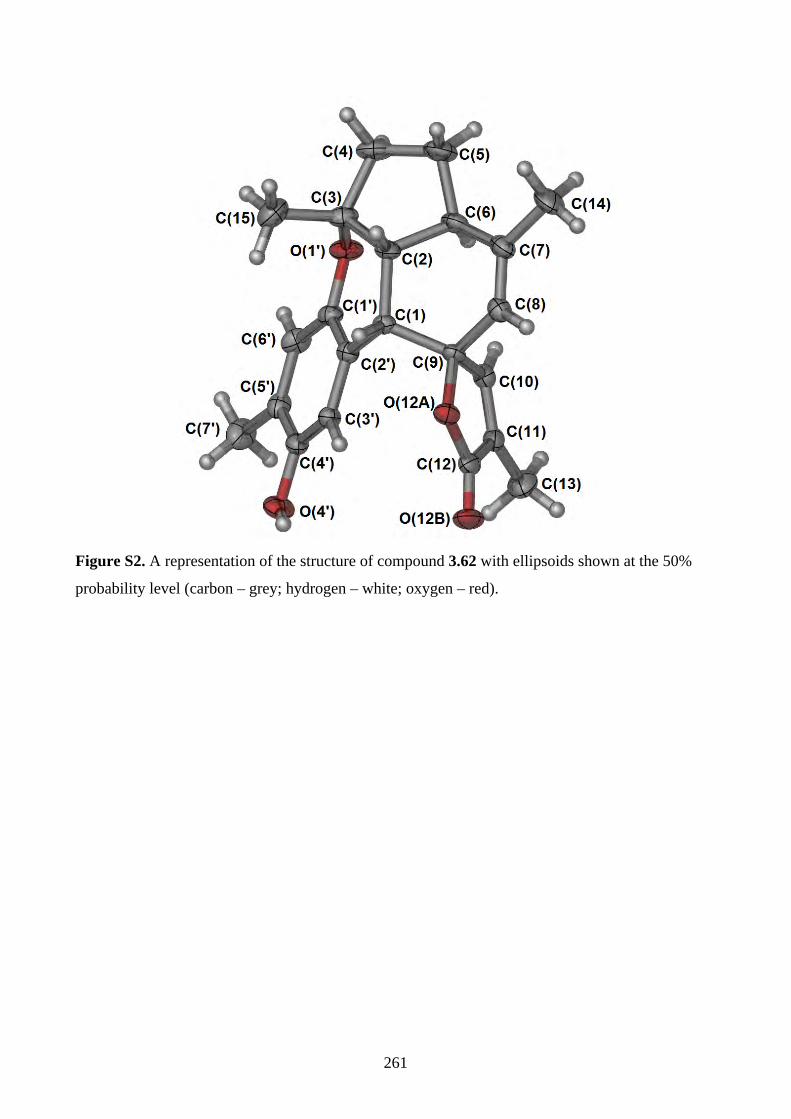
261
Figure S2. A representation of the structure of compound 3.62 with ellipsoids shown at the 50%
probability level (carbon – grey; hydrogen – white; oxygen – red).
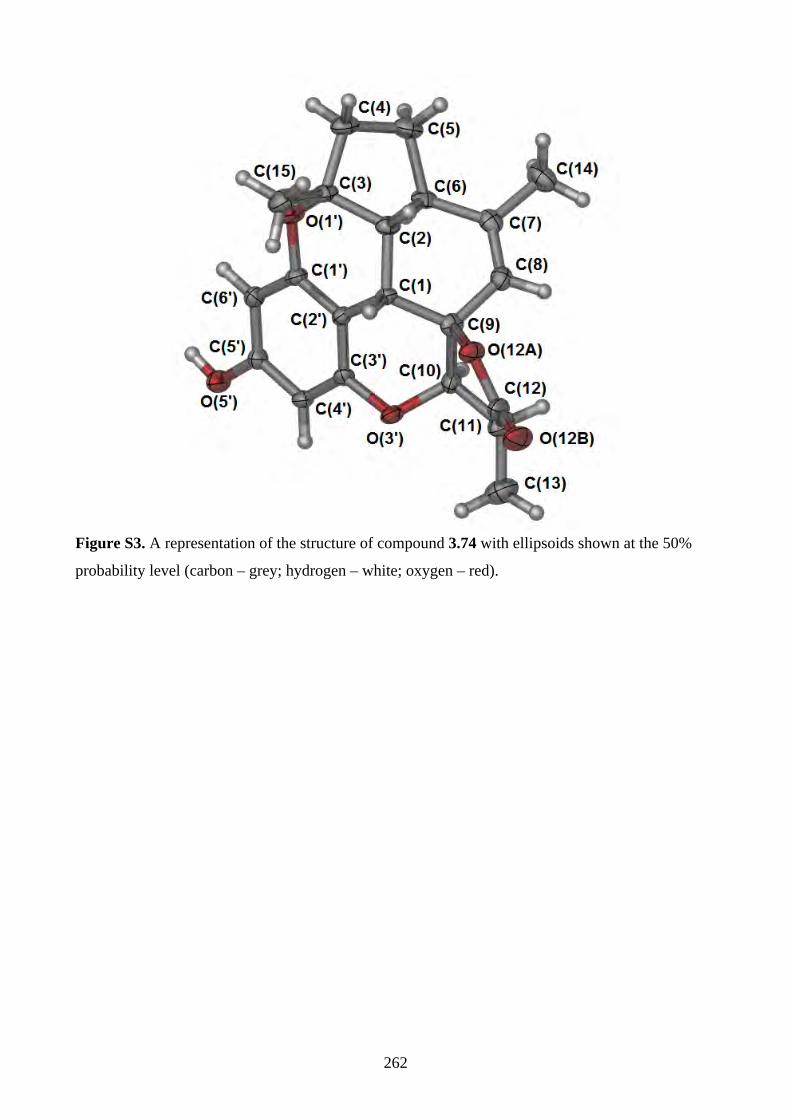
262
Figure S3. A representation of the structure of compound 3.74 with ellipsoids shown at the 50%
probability level (carbon – grey; hydrogen – white; oxygen – red).

263
3.5.6. References
1. Ishii, H.; Ishige, M.; Matsushima, Y.; Tohojoh, T.; Ishikawa, T.; Kawanabe, E. J. Chem. Soc., Perkin Trans. 1, 1985, 2353.
2. Yang, P.; Yao, M.; Li, J.; Li, Y.; Li, A. Angew. Chem. Int. Ed., 2016, 55, 6964. 3. Volkert, M.; Uwai, K.; Tebbe, A.; Popkirova, B.; Wagner, M.; Kuhlmann, J.; Waldmann, H.
J. Am. Chem. Soc., 2003, 125, 12749. 4. Cole, K. P.; Hsung, R. P. Org. Lett., 2003, 5, 4843. 5. Hu, T.; Corey, E. J. Org Lett, 2002, 4, 2441. 6. Sheldrick, G. M. Acta Crystallogr A, 1990, 46, 467. 7. Sheldrick, G. M. Acta Crystallographica a-Foundation and Advances, 2015, 71, 3. 8. Barbour, L. J. J. Supramol. Chem., 2003, 1, 189.

264
Chapter 4
Biomimetic Total Synthesis of Rhodonoids C and D, and Murrayakonine D
This project was conducted in collaboration with Mr Aaron Day
4.1. Introduction
4.1.1. Isolation of rhodonoids and murrayakinone D
Figure 4.1: Rhodonoids A-D.1,2
Rhodonoid C (4.3) contains a unique 6/6/5/6 ring system with 4 stereocenters, 3 of which are
contiguous, while rhodonoid D (4.4) has a 6/6/5/5 ring system with 4 contiguous
stereocenters (Figure 4.1). Rhodonoids C and D1 along with biosynthetically related
rhodonoids A (4.1) and B (4.2)2 were isolated from the aerial part of a flowering plant
Rhododendron capitatum by Liao. All rhodonoids were isolated as scalemic mixtures (Table
4.1), suggesting these natural products could be derived from a pre-disposed, non-enzymatic
biosynthesis.
Table 4.1: Ratio of enantiomers of natural rhodonoids A-D.
Ratio of enantiomer + –
rhodonoid A 1 5 rhodonoid B 1 2 rhodonoid C 10 1 rhodonoid D 1 4
Figure 4.2: Murrayakonine D (4.6) and mahanimbine (4.5).3
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
O
OOH
O
HOH
H
O
HOH
H
H
O
4.2: rhodonoid B4.1: rhodonoid A
O
O
NH
4.5: mahanimbine 4.6: murrayakonine D
O
ONH
4.3: rhodonoid C
O
OOH
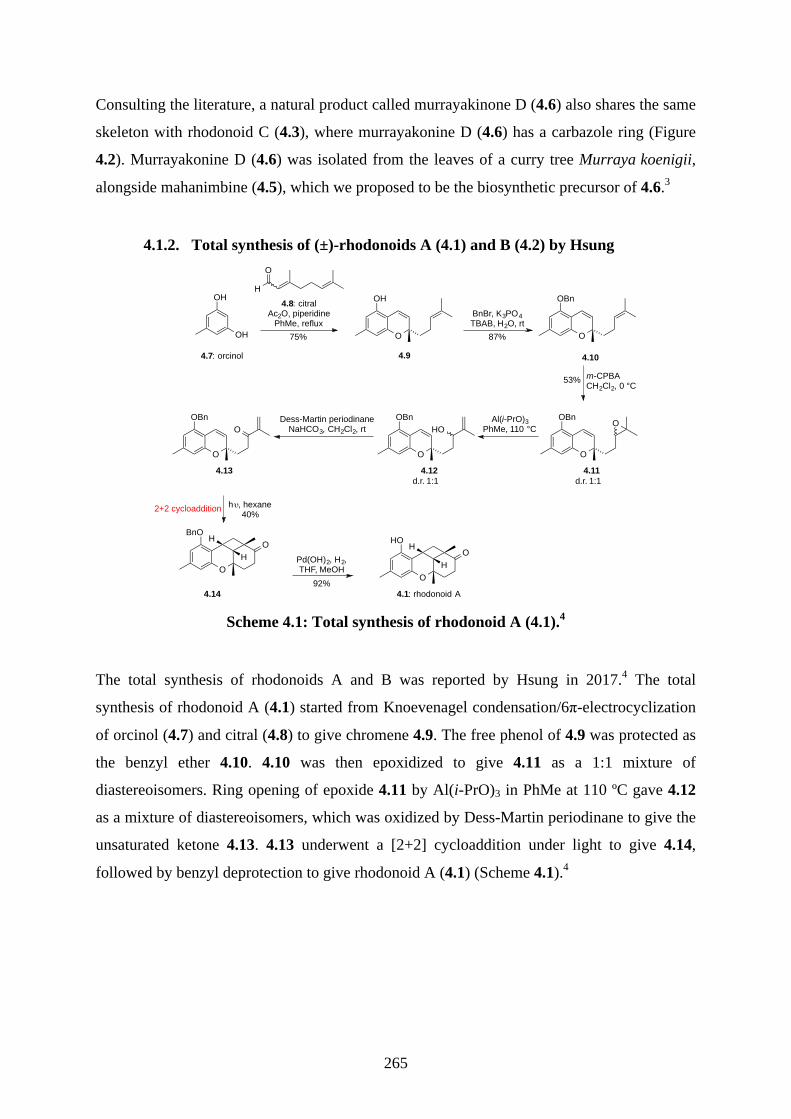
265
Consulting the literature, a natural product called murrayakinone D (4.6) also shares the same
skeleton with rhodonoid C (4.3), where murrayakonine D (4.6) has a carbazole ring (Figure
4.2). Murrayakonine D (4.6) was isolated from the leaves of a curry tree Murraya koenigii,
alongside mahanimbine (4.5), which we proposed to be the biosynthetic precursor of 4.6.3
4.1.2. Total synthesis of (±)-rhodonoids A (4.1) and B (4.2) by Hsung
Scheme 4.1: Total synthesis of rhodonoid A (4.1).4
The total synthesis of rhodonoids A and B was reported by Hsung in 2017.4 The total
synthesis of rhodonoid A (4.1) started from Knoevenagel condensation/6π-electrocyclization
of orcinol (4.7) and citral (4.8) to give chromene 4.9. The free phenol of 4.9 was protected as
the benzyl ether 4.10. 4.10 was then epoxidized to give 4.11 as a 1:1 mixture of
diastereoisomers. Ring opening of epoxide 4.11 by Al(i-PrO)3 in PhMe at 110 ºC gave 4.12
as a mixture of diastereoisomers, which was oxidized by Dess-Martin periodinane to give the
unsaturated ketone 4.13. 4.13 underwent a [2+2] cycloaddition under light to give 4.14,
followed by benzyl deprotection to give rhodonoid A (4.1) (Scheme 4.1).4
OH
OH
4.7: orcinol
H
O
4.8: citralAc2O, piperidine
PhMe, reflux
OH
O
4.9
OBn
O
4.13
O
BnBr, K3PO4TBAB, H2O, rt
O
BnOH
H
O
HOH
H
hυ, hexane40%
OO
Pd(OH)2, H2,THF, MeOH
2+2 cycloaddition
4.1: rhodonoid A4.14
75%
92%
OBn
O
4.10
OBn
O
4.11
O
d.r. 1:1
m-CPBACH2Cl2, 0 °C
87%
53%
OBn
O
4.12
HO
d.r. 1:1
Al(i-PrO)3PhMe, 110 °C
Dess-Martin periodinaneNaHCO3, CH2Cl2, rt

266
Scheme 4.2: Total synthesis of rhodonoid B (4.2).4
The total synthesis of rhodonoid B (4.2) started from chromene Z-4.16, which was
synthesized from orcinol (4.7) and aldehyde 4.15. Z-4.16 then underwent a [2+2]
cycloaddition with Fe(OTf)3 to give 4.17. This reaction was conducted at low temperature, as
the Z-alkene would be readily isomerised and underwent [2+2] cycloaddition to give 12-epi-
4.17. The synthesis continued by functionalising the alkene of 4.17 to an unsaturated ketone
which gave rhodonoid B (4.2) in 8 steps (Scheme 4.2).4
The total synthesis of rhodonoids A (4.1) and B (4.2) was the only example of the synthesis
of rhodonoids. To date, there is no biosynthetic speculation or total synthesis of rhodonoids
C (4.3) and D (4.4) reported in the literature. Our aim is to propose how rhodonids C and D
could be derived in Nature, and to synthesize them via our proposed biosynthetic pathways.
OH
OH
4.7: orcinol
O
HZ
4.15Ac2O, piperidine
PhMe, reflux
OH
O
O
HOH
H
H
O
O
HOH
H
H8 steps
2+2 cycloaddition Fe(OTf)3. CH2Cl2–40 °C
Z
Z-4.16
4.174.2: rhodonoid B
OH
O
E
E-4.16
Fe(OTf)3. CH2Cl2rt
63%d.r. 11:1
O
HOH
H
H
2+2 cycloaddition
12-epi-4.1754%
d.r. 2:1
12
69%
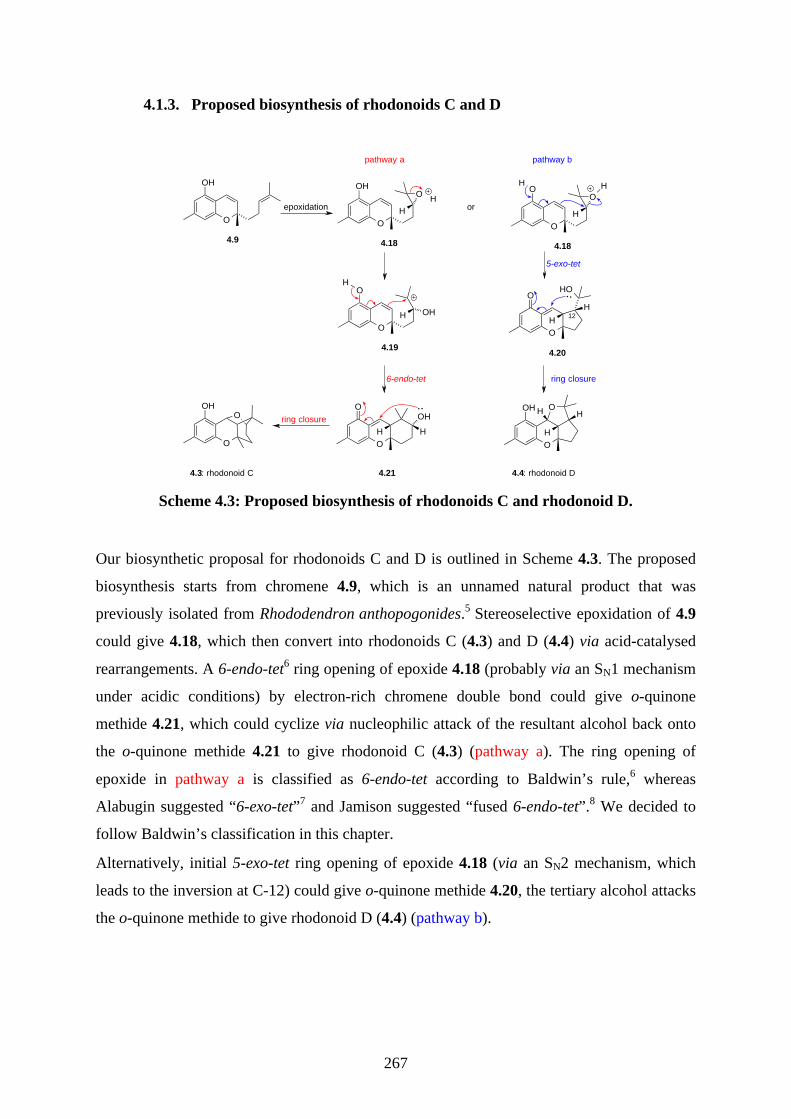
267
4.1.3. Proposed biosynthesis of rhodonoids C and D
Scheme 4.3: Proposed biosynthesis of rhodonoids C and rhodonoid D.
Our biosynthetic proposal for rhodonoids C and D is outlined in Scheme 4.3. The proposed
biosynthesis starts from chromene 4.9, which is an unnamed natural product that was
previously isolated from Rhododendron anthopogonides.5 Stereoselective epoxidation of 4.9
could give 4.18, which then convert into rhodonoids C (4.3) and D (4.4) via acid-catalysed
rearrangements. A 6-endo-tet6 ring opening of epoxide 4.18 (probably via an SN1 mechanism
under acidic conditions) by electron-rich chromene double bond could give o-quinone
methide 4.21, which could cyclize via nucleophilic attack of the resultant alcohol back onto
the o-quinone methide 4.21 to give rhodonoid C (4.3) (pathway a). The ring opening of
epoxide in pathway a is classified as 6-endo-tet according to Baldwin’s rule,6 whereas
Alabugin suggested “6-exo-tet”7 and Jamison suggested “fused 6-endo-tet”.8 We decided to
follow Baldwin’s classification in this chapter.
Alternatively, initial 5-exo-tet ring opening of epoxide 4.18 (via an SN2 mechanism, which
leads to the inversion at C-12) could give o-quinone methide 4.20, the tertiary alcohol attacks
the o-quinone methide to give rhodonoid D (4.4) (pathway b).
OH
O
4.9
OH
O
4.18
H
O H
6-endo-tet
O
O
H H
OH
O
OOH
O
O
4.18
H
OH
O
OH
H
HO
12
O
OH OH
H
H
4.4: rhodonoid D
orepoxidation
pathway a pathway b
5-exo-tet
4.21
4.20
4.3: rhodonoid C
H
O
O
4.19
H OH
H
ring closure
ring closure

268
Scheme 4.4: Proposed biosynthesis of murrayakonine D (4.6).
We also propose the biosynthesis of murrayakonine D (4.6) to be identical with that in
rhodonoid C (4.3) (Scheme 4.4). Starting from mahanimbine (4.5), stereoselective
epoxidation gives 4.22, and ring opening of 4.22 under acidic conditions would give
carbocation 4.23. A 6-endo-tet cyclization would give aza-ortho-quinone methide 4.24, the
resultant alcohol would cyclize back onto the aza-ortho-quinone methide 4.24 to give
murrayakonine D (4.6).
4.1.4. Epoxide cyclization reaction in the synthesis of siccanin (4.28) by Trost
In the total synthesis of siccanin (4.28) by Trost, he initially attempted to rearrange epoxide
4.25 into siccanin (4.28) using Lewis acids via an acid-catalysed cyclization cascade reaction
(Scheme 4.5), which is similar to our pathway a cyclization pathway to give rhodonoid C (4.3)
(Scheme 4.5). However, Trost observed no reaction or decomposition of 4.25. He later
achieved the total synthesis of siccanin (4.28) using a radical cyclization approach (Scheme
4.6).9,10
Scheme 4.5: Attempted acid-catalysed rearrangement of 4.25 to siccanin by Trost.9,10
4.22
H
6-endo-tet
epoxidation
pathway a
O
NH
4.5: mahanimbine
O
NHO
H
O
NH
OHH
O
NH
H
OH
4.6: murrayakonine D
O
ONH
4.23
4.24
O
OMe
H
O
4.25
O
HO
H
O
H
H
4.28: siccanin
BF3·OEt2SnCl2
TiCl2(Oi-Pr)2Yb(OTf)3
FeCl3
no reactionor
decomposition
O
OH
H
O
4.26
LA
O
O
H
O
H
4.27
LA
alkene cyclization

269
When epoxide 4.25 was treated with CpTiIIICl2, 4.33 and 5-epi-methoxysiccanin (4.32) were
observed.9,10 The proposed mechanism began from a single electron reduction of epoxide
4.25 by Ti3+ to give radical intermediate 4.29, followed by a 6-exo-tet cyclization to generate
2 diastereoisomers, 4.30 and 4.31. 4.30 underwent a 6-exo-tet cyclization to give 5-epi-
methoxysiccanin (4.32) in 61% yield, while the desired 4.31 intermediate did not undergo 6-
exo-tet cyclization, but a single electron reduction to give alcohol 4.33 in 20% yield. 4.33 was
then oxidized by I2 and PhI(OAc)2 under light to give 4.34, followed by deprotection using
EtSNa to give siccanin (4.28) (Scheme 4.6). From the total synthesis of siccanin (4.28), we
were interested to apply the radical cyclization methodology into the epoxide cyclization of
4.9 to rhodonoid C (4.3) or D (4.4).
Scheme 4.6: Total synthesis of siccanin (4.28) by Trost.9,10
O
OMe
H
OCp2TiIIICl
reductive ring opening O
OMe
H
OTiIV
O
OMe
H
OTiIV
O
MeO
H
O
H
H
H
4.32: 5-epi-methoxysiccanin
5
O
OMe
H
OTiIV
H
5
O
OMe
HH
HO
6-exo-tet
6-exo-tet
single electronreduction
6-exo-tet
4.29 4.304.25
O
HO
H
O
H
H
4.28: siccanin
5
6-exo-tet
4.314.33
O
MeO
H
O
H
H
4.34
PhI(OAc)2, I2hν, benzene, rt65%
EtSNa, DMF, rt86%
61%
20%

270
4.2. Results and discussion
4.2.1. Biomimetic total synthesis of rhodonoids C and D
Scheme 4.7: Synthesis of epoxide 4.9.
The synthesis began with chromene 4.9, which was prepared from orcinol (4.7) and citral (4.8)
facilitated by EDDA.11 However, it was difficult to purify chromene 4.9 from the unreacted
citral (4.8) by column chromatography. Treatment of the mixture with NaBH4, which reduced
excess citral (4.8) to geraniol, allowed an easier purification of chromene 4.9. Epoxidation of
chromene 4.9 gave a 51% yield of epoxide 4.18 as a 1:1 mixture of diastereoisomers, along
with 27% yield of recovered chromene 4.9 (Scheme 4.7). To our surprise, the epoxidation
never went to completion. We modified the reaction conditions (e.g. excess m-CPBA, longer
reaction time, added NaHCO3, or carefully heating up the reaction) and none gave any
improvement.
Scheme 4.8: Biomimetic total synthesis of rhodonoids C and D.
For the acid-catalysed rearrangement reaction, we first used p-TsOH in CHCl3 and the result
was promising. We isolated rhodonoid C (4.3) in 21% yield, followed by rhodonoid D (4.4)
in 2% yield. There are some drastic changes on the 1H NMR spectra of 4.3 and 4.4. First, we
observed the loss of chromene signals from the epoxide 4.18 at δ 6.64 ppm (H-7) and δ 5.44
ppm (H-8). In addition, the structure of rhodonoid C (4.3) was confirmed by the key doublet
at δ 5.05 ppm (H-7) which coupled to δ 1.78 ppm (H-8). Similarly, the signal at δ 2.82 ppm
OH
OH
H
O
4.8: citralEDDA, PhMe, 110 °C
74%
OH
O
4.7: orcinol 4.9
O
OHO
1:1 d.r.
H
4.18
m-CPBA, CH2Cl2, rt
51%
OH
O
4.9
+
27% recovery
p-TsOH, CHCl3, rt
SnCl4, CHCl3, −60 °C
21%
32%
5%
21%
2%
5%
O
OHO
1:1 d.r.
H
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.18 4.35
78
78
78
12

271
(H-8) which coupled to δ 4.92 ppm (H-7) and 2.56 ppm (H-12) confirmed the structure of
rhodonoid D (4.4).
Ketone 4.35 was also observed from the reaction, presumably derived from ring opening of
epoxide 4.18 under acidic conditions to form carbocation 4.19, followed by a formal 1,2-
hydride shift to give ketone 4.35 (Scheme 4.9). We then conducted an extensive screening
with various Lewis acids and protic acids for this reaction (Table 4.2). We found SnCl4 in
CHCl3 gave the best overall yield with 32% of rhodonoid C (4.3), 5% of rhodonoid D (4.4)
and 21% of 4.35. Only the epoxide with the correct relative stereochemistry could rearrange
to give rhodonoids C and D, so the maximum theoretical yield of this reaction is 50%.
Therefore, to achieve 37% overall yield of rhodonoids is more than acceptable. We then
speculated that ketone 4.35 was likely derived from the non-natural stereoisomer of epoxide
4.18a.
We also observed side products when 1.0 M HCl solution or Lewis acids (TiCl4 or BF3·OEt2)
were used. We proposed under acidic conditions or heat, the epoxide 4.18 might be ring
opened to give carbocation 4.19 and subsequently attacked by Cl– to give 4.36, or attacked by
F– to give 4.37, or attacked by OH– to give 4.38 (Scheme 4.10). Unfortunately, we discovered
bases could not promote the pathway b rearrangement to form rhodonoid D (4.4).
Scheme 4.9: Proposed mechanism for the formation of ketone 4.35.
O
OHO
H
4.18
O
OH
H
4.19
OHacid or heat
O
OHHO
4.35
O
OHO
4.35
deprotonation
d.r. 1:1
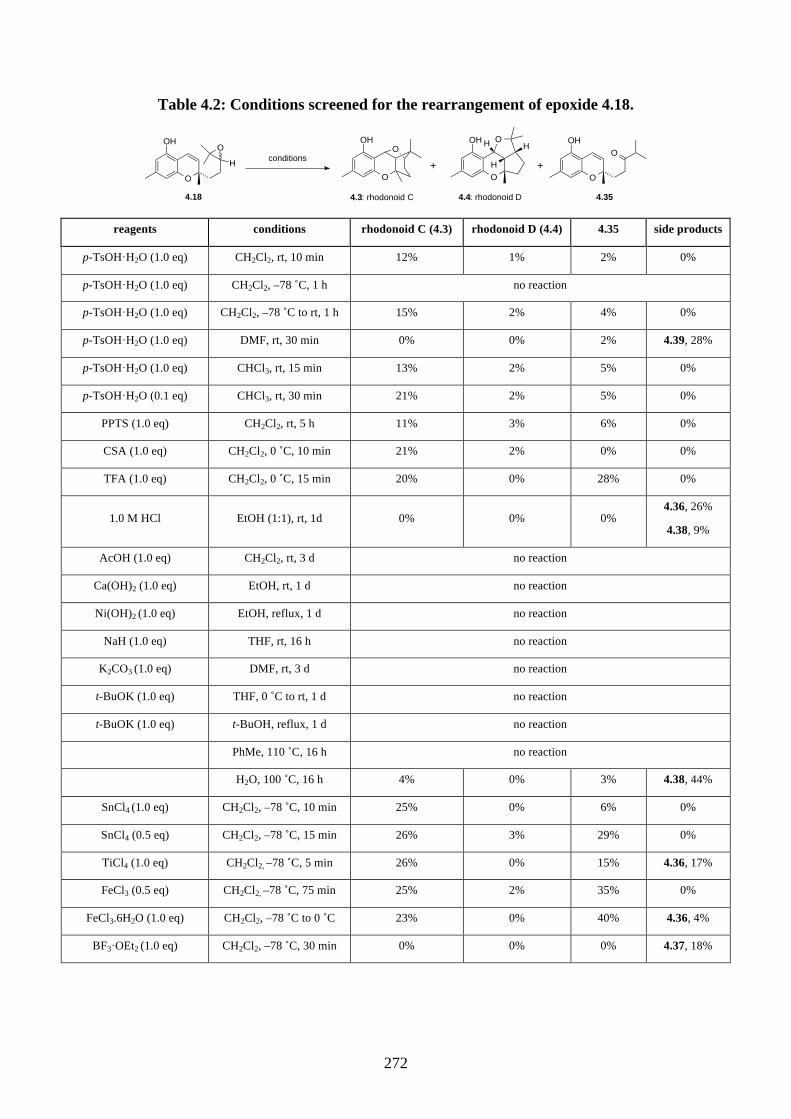
272
Table 4.2: Conditions screened for the rearrangement of epoxide 4.18.
reagents conditions rhodonoid C (4.3) rhodonoid D (4.4) 4.35 side products
p-TsOH·H2O (1.0 eq) CH2Cl2, rt, 10 min 12% 1% 2% 0%
p-TsOH·H2O (1.0 eq) CH2Cl2, –78 ˚C, 1 h no reaction
p-TsOH·H2O (1.0 eq) CH2Cl2, –78 ˚C to rt, 1 h 15% 2% 4% 0%
p-TsOH·H2O (1.0 eq) DMF, rt, 30 min 0% 0% 2% 4.39, 28%
p-TsOH·H2O (1.0 eq) CHCl3, rt, 15 min 13% 2% 5% 0%
p-TsOH·H2O (0.1 eq) CHCl3, rt, 30 min 21% 2% 5% 0%
PPTS (1.0 eq) CH2Cl2, rt, 5 h 11% 3% 6% 0%
CSA (1.0 eq) CH2Cl2, 0 ˚C, 10 min 21% 2% 0% 0%
TFA (1.0 eq) CH2Cl2, 0 ˚C, 15 min 20% 0% 28% 0%
1.0 M HCl EtOH (1:1), rt, 1d 0% 0% 0% 4.36, 26%
4.38, 9%
AcOH (1.0 eq) CH2Cl2, rt, 3 d no reaction
Ca(OH)2 (1.0 eq) EtOH, rt, 1 d no reaction
Ni(OH)2 (1.0 eq) EtOH, reflux, 1 d no reaction
NaH (1.0 eq) THF, rt, 16 h no reaction
K2CO3 (1.0 eq) DMF, rt, 3 d no reaction
t-BuOK (1.0 eq) THF, 0 ˚C to rt, 1 d no reaction
t-BuOK (1.0 eq) t-BuOH, reflux, 1 d no reaction
PhMe, 110 ˚C, 16 h no reaction
H2O, 100 ˚C, 16 h 4% 0% 3% 4.38, 44%
SnCl4 (1.0 eq) CH2Cl2, –78 ˚C, 10 min 25% 0% 6% 0%
SnCl4 (0.5 eq) CH2Cl2, –78 ˚C, 15 min 26% 3% 29% 0%
TiCl4 (1.0 eq) CH2Cl2, –78 ˚C, 5 min 26% 0% 15% 4.36, 17%
FeCl3 (0.5 eq) CH2Cl2, –78 ˚C, 75 min 25% 2% 35% 0%
FeCl3.6H2O (1.0 eq) CH2Cl2, –78 ˚C to 0 ˚C 23% 0% 40% 4.36, 4%
BF3·OEt2 (1.0 eq) CH2Cl2, –78 ˚C, 30 min 0% 0% 0% 4.37, 18%
conditions
O
OHO
H
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.18 4.35

273
Scheme 4.10: Formation of side products from the acid-catalysed rearrangement
reactions.
4.2.2. Investigation on the reactivity of the epoxide 4.18
Scheme 4.11: Study on the reactivity of two diastereoisomers of epoxide 4.18.
To investigate whether the non-natural epoxide could give the ketone 4.35 in the acid-
catalysed rearrangement conditions, we first attempted to separate the two epoxides 4.18a
and 4.18b by HPLC but it was unsuccessful. Alternatively, we intended to separate the two
epoxides chemically, perhaps one isomer would react faster than the other one in the acid
catalysed rearrangement reaction. Therefore, our goal was to stop the reaction halfway before
it went to completion. We chose the SnCl4 conditions in this study because that reaction was
performed at low temperature and the rate of the reaction was quite slow.
We first lowered the equiv. of SnCl4 to 0.5 equiv. (from 1 equiv.), which reduced the rate of
reaction significantly. We monitored the reaction carefully by TLC, and when approximately
O
OHHO
Cl
4.36
O
OHO
H
4.18
O
OH
H
4.19
OHacid or heat
1 M HClEtOH, rt
orFeCl3•6H2OCH2Cl2, rt
orTiCl4, CH2Cl2
–78 °C
d.r. 1:1
H2O, 100 ºC
O
OHHO
OH
4.38d.r. 1:1
p-TsOH, DMF, rt
O
OHO
OH
4.39d.r. 1:1
H O
O
OHHO
F
4.37d.r. 1:1
BF3•OEt3CH2Cl2, –78 °C
d.r. 1:1
4-26%
28%44%
18%
SnCl4, CHCl3, −60 °C 22% 8%3%
d.r. 1:1
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
O
OHO
H
O
OHO
H +
4.18ad.r. 3.3:1
39%
4.18b
4.35
O
OHO
H
O
OHO
H
+
4.18a
4.18b

274
halved of epoxide 4.18 was consumed, the reaction was quenched at –60 ºC. To our delight,
we isolated 22% yield of rhodonoid C (4.3), 3% yield of rhodonoid D (4.4), 8% yield of
ketone 4.35 and 39% yield of recovered starting material. The recovered epoxide 4.18 was in
3.3:1 ratio of the two diastereoisomers. Since we observed a significant conversion of
rhodonoids C (4.3) and D (4.4) from epoxide 4.18, we would assume the major isomer in the
recovered epoxide 4.18 was the non-natural epoxide 4.18a.
Scheme 4.12: Reaction of epoxide 4.18 (diastereomeric ratio of 3.3:1).
Indeed, when 4.18a was treated under the standard conditions, we observed 13% yield of
rhodonoid C (4.3) and 32% yield of ketone 4.35. The ratio of 4.35 to rhodonoid C (4.3) was
3:1 which showed good agreement with the ratio of 4.18a and 4.18b (Scheme 4.12).
Scheme 4.13: One pot reaction of chromene 4.9 to rhodonoids C and D.
We also attempted a one-pot epoxidation and acid-catalysed rearrangement reaction.
However, the result was underwhelming. The TLC showed complex mixtures and little of the
natural products were isolated. The yield from this reaction was much worse than the
combined yield from the two steps synthesis (Scheme 4.13).
Scheme 4.14: Epoxide rearrangement with radical cyclization.9,10
O
OHO
H
O
OHO
H +
4.18a3.3:1 d.r.
SnCl4, CHCl3, −60 °C
13% 32%
O
OHO
4.3: rhodonoid C
+O
OOH
4.18b 4.35
m-CPBA, CH2Cl2, rt;then p-TsOH
5% 4%2%
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.35
OH
O
4.9
Mn, Cp2TiCl2THF, rt
O
OHO
H
4.40d.r. 1:1
O
OH
4.18
OHH
51%8
16
14

275
We also attempted a radical cyclization on epoxide 4.18.9,10 We did not observe the desired
rhodonoid structure but a 1:1 mixture of diastereoisomers 4.40, presumably derived from the
reductive radical cyclization (Scheme 4.14). The relative stereochemistry between C-8 and C-
16 can be confirmed by the NOESY analysis (Figure 4.3).
We also attempted to oxidize alcohol 4.40 into the corresponding ketone for easier
characterisation, however Dess-Martin oxidation, Swern oxidation or Ley oxidation led to
decomposition of 4.40.
Figure 4.3: NOESY of 4.40 (1:1 mixture of diastereoisomers).
4.40a
O
OH MeOH
Me
H
4.40b
O
OH MeOH
Me
H
4.40b
O
OHOH
H H

276
4.2.3. Synthesis of mahanimbine (4.5)
Scheme 4.15: Synthesis of amine 4.43.12
We then shifted our focus onto the total synthesis of murrayakonine D (4.6). We first
prepared mahanimbine (4.5) following a procedure reported by Knolker.12 The synthesis
started with benzyl protection of 2-methyl-5-nitrophenol (4.41). The reaction went smoothly
and gave 4.42 in good yield. However, when the reaction was scaled up to 10 g, we observed
the unreacted BnBr was too difficult to separate from 4.42, and the mixture would be carried
through in the following step where BnBr could then be separated by column
chromatography. Reduction of nitro group 4.42 with Fe gave amine 4.43 in good yield
(Scheme 4.15).
Scheme 4.16: Synthesis of carbazole 4.45.12
For the coupling reaction of amine 4.43, the procedure from Knolker used PhBr, XPhos and
Pd(OAc)2 to give 4.44.12 However, we decided to adapt a procedure from Dethe et al., where
20% of Cu(OAc)2 and PhB(OH)2 were used for this reaction.13 We encountered problems
with this reaction where the yield was around 10% to 20%. In addition, elemental copper was
observed at the end of the reaction, suggesting all the Cu(OAc)2 was reduced. Consulting the
literature, we found examples using stoichiometric or excess of Cu(OAc)2 for similar
reactions with boronic acid.14,15,16 When the amount of Cu(OAc)2 was increased to 2 equiv.,
the yield of this reaction improved to 69%. The next reaction was an intramolecular oxidative
coupling reaction using Pd(OAc)2 in pivalic acid to form carbazole 4.45 in 63% yield
(Scheme 4.16).
NO2
OH
4.41: 2-methyl-5-nitrophenol
BnBr, K2CO3acetone , 56 ºC
NO2
OBn
4.42
Fe, AcOH, 45 ºC
NH2
OBn
4.43
66% over 2 steps
NH2
OBn K2CO3, Cu(OAc)2benzoic acid, PhB(OH)2
PhMe, reflux
HN
OBn
4.444.43
69%
4.45
NH
OBn
K2CO3, Pd(OAc)2pivalic acid, 85 °C
63%

277
Scheme 4.17: Synthesis of mahaimbine (4.5).12
Deprotection of the benzyl group of 4.45 by hydrogenation was surprisingly slow, taking
over 1 day to go to completion. Nonetheless, the reaction gave carbazole 4.46 in good yield.
Mahanimbine (4.5) was synthesized from 4.46 and citral (4.8) with Ti(Oi-Pr)4,12 and we had
to reduce the unreacted citral (4.8) to geraniol with NaBH4 for an easier purification of
mahanimbine (4.5) (Scheme 4.17).
4.2.4. Biomimetic total synthesis of murrayakonine D (4.6)
Scheme 4.18: Biomimetic total synthesis of murrayakonine D (4.6).
Epoxidation of mahanimbine (4.5) gave 4.22 as a 1:1 inseparable mixture of diastereoisomers.
When epoxide 4.22 was treated with p-TsOH, we observed a 7.4:1 ratio of murrayakonine D
(4.6) and the undesired ketone 4.47. Murrayakonine D (4.6) and 4.47 could not be separated
by flash column chromatography (including AgNO3 doped SiO217). To our delight,
murrayakonine D (4.6) is a solid and we triturated the mixture with MeOH to give pure
murrayakonine D (4.6) in 33% yield (Scheme 4.18). We purified the filtrate to get a pure
sample of 4.47 for characterisation. We also repeated the reaction using SnCl4 in CH2Cl2 but
the yield (21% of 4.6) was not as good as with p-TsOH. Unfortunately, we have yet to
observe any product with the rhodonoid D skeleton from this reaction. Perhaps the decreased
nucleophilicity of the carbazole inhibits the pathway b rearrangement (Scheme 4.3). Last but
not least, we managed to recrystallise murrayakonine D (4.6) and collected an X-ray structure
to confirm the relative stereochemistry (Figure 4.4).
4.46
NH
OH
4.45
NH
OBn
H2, Pd/C, CH2Cl2MeOH, rt
88%O
NH
4.5: mahanimbine
4.8: citralTi(Oi-Pr)4, PhMe, −78 ºC to rt;
then NaBH4, MeOH, rt
74%
O
H
m-CPBACH2Cl2, rt
61%O
NH
4.5: mahanimbine
1:1 d.r.
O
NHO
H
p-TsOH, CH2Cl2, rt
SnCl4, CH2Cl2, −78 ºC
4.6: murrayakonine D
O
ONH
33%
21%
O
NHO
+
4.22 4.47

278
Figure 4.4: X-ray structure of murrayakonine D (4.6).
4.2.5. Biomimetic total synthesis of rhodonoids C and D reported by Hsung
Scheme 4.19: Biomimetic total synthesis of rhodonoids C and D by Hsung.18
After we published our work,19 Hsung also reported a synthesis of rhodonoids C and D using
a similar approach (Scheme 4.19).18 The epoxide 4.18 was synthesized with a higher overall
yield by protecting the free phenol with an acetate group. They also discovered heating
epoxide 4.18 in SiO2 gave 14% of rhodonoid C (4.3) and 8% of rhodonoid D (4.4), where the
yield of rhodonoid D (4.4) was improved compared to our synthesis.18
O
OHO
H
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.18
4.35
OH
O
4.9
OAc
O
4.48
Ac2O, DMAPEt3N, CH2Cl2, rt
1. m-CPBACH2Cl2, 0 °C2. K2CO3MeOH, rt
98% over 2 steps85%
SiO2, PhMereflux
14% 8% 26%
d.r. 1:1

279
4.3. Summary
We have developed a concise synthesis of rhodonoids C and D. The overall synthesis of
rhodonoids was protecting group free with good step economy, where rhodonoids C and D
were synthesized from simple starting materials in 3 steps. The key step was the acid
catalysed rearrangement of the epoxide 4.18 forming 2 rings and 3 new stereocenters which
gave both rhodonoids C and D in 1 step (Figure 4.5).
It showcases the power of biomimetic synthesis as the molecular complexity increases in
relative short steps with high stereoselectivity. We have also applied the same methodology
for the synthesis of murrayakonine D. These syntheses of rhodonoids and murrayakonine D
strengthen our belief that the biosynthesis of these natural products is highly pre-disposed and
likely non-enzymatic.
Figure 4.5: Summary of the biomimetic total synthesis of rhodonoids C and D.
OH
OH
4.7: orcinol
H
O
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+O
OOH
3 steps
3 rings4 stereocenters
4.8: citral

280
4.4. References
1. Liao, H. B.; Huang, G. H.; Yu, M. H.; Lei, C.; Hou, A. J. J. Org. Chem., 2017, 82, 1632.
2. Liao, H. B.; Lei, C.; Gao, L. X.; Li, J. Y.; Li, J.; Hou, A. J. Org. Lett., 2015, 17, 5040. 3. Nalli, Y.; Khajuria, V.; Gupta, S.; Arora, P.; Riyaz-Ul-Hassan, S.; Ahmed, Z.; Ali, A.
Org. Biomol. Chem., 2016, 14, 3322. 4. Wu, H.; Hsung, R. P.; Tang, Y. J. Org. Chem., 2017, 82, 1545. 5. Iwata, N.; Kitanaka, S. Chem. Pharm. Bull., 2011, 59, 1409. 6. Baldwin, J. E. J. Chem. Soc. Chem. Comm., 1976, 734. 7. Alabugin, I. V.; Gilmore, K. Chem Commun, 2013, 49, 11246. 8. Vilotijevic, I.; Jamison, T. F. Mar. Drugs, 2010, 8, 763. 9. Trost, B. M.; Shen, H. C.; Surivet, J. P. Angew. Chem. Int. Ed., 2003, 42, 3943. 10. Trost, B. M.; Shen, H. C.; Surivet, J. P. J. Am. Chem. Soc., 2004, 126, 12565. 11. Luo, G. Y.; Wu, H.; Tang, Y.; Li, H.; Yeom, H. S.; Yang, K.; Hsung, R. P. Synthesis,
2015, 47, 2713. 12. Hesse, R.; Gruner, K. K.; Kataeva, O.; Schmidt, A. W.; Knolker, H. J. Chem. Eur. J.,
2013, 19, 14098. 13. Dethe, D. H.; Das, S.; Dherange, B. D.; Mahapatra, S. Chem. Eur. J., 2015, 21, 8347. 14. Chen, S. Y.; Huang, H.; Liu, X. J.; Shen, J. K.; Jiang, H. L.; Liu, H. J. Comb. Chem.,
2008, 10, 358. 15. Elhalem, E.; Bailey, B. N.; Docampo, R.; Ujvary, I.; Szajnman, S. H.; Rodriguez, J. B.
J. Med. Chem., 2002, 45, 3984. 16. Vantourout, J. C.; Miras, H. N.; Isidro-Llobet, A.; Sproules, S.; Watson, A. J. B. J.
Am. Chem. Soc., 2017, 139, 4769. 17. Williams, C. M.; Mander, L. N. Tetrahedron, 2001, 57, 425. 18. Wu, H.; Hsung, R. P.; Tang, Y. Org. Lett., 2017. 19. Day, A. J.; Lam, H. C.; Sumby, C. J.; George, J. H. Org Lett, 2017, 19, 2463.

281
4.5. Experimental
4.5.1. General methods
All chemicals used were purchased from commercial suppliers and used as received. All reactions
were performed under an inert atmosphere of N2. All organic extracts were dried over anhydrous
magnesium sulfate. Thin layer chromatography was performed using aluminium sheets coated with
silica gel F254. Visualization was aided by viewing under a UV lamp and staining with ceric
ammonium molybdate or KMnO4 stain followed by heating. All Rf values were measured to the
nearest 0.05. Flash column chromatography was performed using 40-63 micron grade silica gel.
Melting points were recorded on a digital melting point apparatus and are uncorrected. Infrared
spectra were recorded using an FT-IR spectrometer as the neat compounds. High field NMR spectra
were recorded using a 500 MHz spectrometer (1H at 500 MHz, 13C at 125 MHz). Solvent used for
spectra were CDCl3 unless otherwise specified. 1H chemical shifts are reported in ppm on the δ-
scale relative to TMS (δ 0.0) and 13C NMR are reported in ppm relative to CDCl3 (δ 77.00).
Multiplicities are reported as (br) broad, (s) singlet, (d) doublet, (t) triplet, (q) quartet, (quin)
quintet, (sext) sextet, (hept) heptet and (m) multiplet. All J-values were rounded to the nearest 0.1
Hz. ESI high resolution mass spectra were recorded on a ESI-TOF mass spectrometer.

282
4.5.2. Experimental procedures
To a solution of orcinol (4.7) (15.0 g, 121 mmol) in PhMe (400 mL) at room temperature was added
citral (4.8) (18.4 g, 121 mmol) and ethylenediamine diacetate (440 mg, 2.42 mmol). The reaction
was stirred at reflux for 5 h. The mixture was cooled to room temperature, then concentrated in
vacuo. The residue was redissolved in MeOH (100 mL), followed by addition of NaBH4 (4.58 g,
121 mmol) to reduce unreacted citral (4.8) that is difficult to separate from 4.9. The reaction was
stirred at room temperature for 30 min, then quenched with 1 M HCl solution (200 mL). The
solution was extracted with Et2O (2 × 300 mL). The combined organic extracts were washed with
brine (300 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by
flash column chromatography on SiO2 (8:1, petrol/EtOAc) to give 4.9 as an orange oil (23.1 g,
74%). Data for 4.9 matched that previously reported in the literature.1
Data for 4.9:
Rf = 0.40 (5:1, petrol/EtOAc)
IR (neat): 3387, 2970, 2924, 2857, 1625, 1578, 1509, 1450, 1377, 1330, 1250 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.61 (d, J = 10.0 Hz, 1H), 6.24 (s, 1H), 6.11 (s, 1H), 5.49 (d, J =
10.0 Hz, 1H), 5.10 (t, J = 7.1 Hz, 1H), 4.71 (br s, 1H), 2.20 (s, 3H), 2.13 – 2.07 (m, 2H), 1.72 (dd, J
= 10.7, 5.9 Hz, 1H), 1.66 (s, 3H), 1.66 – 1.63 (m, 1H), 1.58 (s, 3H), 1.37 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 154.1, 151.0, 139.5, 131.6, 127.2, 124.2, 117.0, 109.9, 108.3,
106.7, 78.2, 41.1, 26.2, 25.7, 22.7, 21.5, 17.6.
HRMS (ESI): calculated for C17H21O2 257.1547 [M-H]-, found 257.1539.
OH
OH
H
O
4.8: citralEDDA, PhMe, 110 °C
74%
OH
O
4.7: resocinol 4.9

283
To a solution of 4.9 (5.60 g, 21.7 mmol) in CH2Cl2 (250 mL) at room temperature was added m-
CPBA (77%, 3.92 g, 22.8 mmol). The reaction was stirred at room temperature for 30 min. The
solution was washed sequentially with saturated Na2S2O3 solution (2 × 200 mL) and saturated
NaHCO3 solution (2 × 200 mL). The organic layer was washed with brine (200 mL), dried over
MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (5:1 → 3:1, petrol/EtOAc gradient elution) to give recovered 4.9 as an
orange oil (1.51 g, 27%). Further elution gave epoxide 4.18 (d.r. = 1:1) as a brown oil (3.00 g,
51%).
Data for 4.18:
Rf = 0.35 (5:1, petrol/EtOAc)
IR (neat): 3348, 2969, 2925, 2857, 1624, 1579, 1452, 1452, 1425, 1380, 1329, 1272 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.64 (d, J = 10.0 Hz, 2H), 6.20 (s, 2H), 6.12 (s, 2H), 5.46 (d, J =
10.0 Hz, 1H), 5.43 (d, J = 10.0 Hz, 1H), 5.33 (br s, 2H), 2.77 (t, J = 6.2 Hz, 1H), 2.75 (t, J = 5.8 Hz,
1H), 2.19 (s, 6H), 1.83 (t, J = 8.1 Hz, 2H), 1.75 – 1.66 (m, 6H), 1.38 (s, 3H), 1.37 (s, 3H), 1.30 (s,
3H), 1.29 (s, 3H), 1.25 (s, 3H), 1.24 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 153.9, 153.8, 151.34, 151.32, 139.6, 126.6, 126.3, 117.4, 117.3,
109.6, 109.5, 108.50, 108.46, 106.7, 106.5, 78.0, 77.6, 64.8, 64.5, 59.1, 58.9, 37.9, 37.4, 26.6, 26.1,
24.8, 23.9, 23.6, 21.5, 18.63, 18.57.
HRMS (ESI): calculated for C17H21O3 273.1496 [M-H]-, found 273.1495.
OH
O
4.9
O
OHO
1:1 d.r.
H
4.18
m-CPBA, CH2Cl2, rt
51%
OH
O
4.9
+
27% recovery

284
To a solution of 4.18 (d.r. = 1:1) (685 mg, 2.50 mmol) in CHCl3 (40 mL) at −60 ºC was added
SnCl4 (0.29 mL, 2.50 mmol) dropwise. The reaction was stirred at −60 ºC for 30 min, then
quenched with saturated NaHCO3 solution (50 mL) and warmed to room temperature. The layers
were separated and the organic phase was washed with saturated NaHCO3 solution (2 × 50 mL),
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (5:1 → 3:1, petrol/EtOAc gradient elution) to give rhodonoid D (4.4) as a
white solid (33 mg, 5%). Further elution gave ketone 4.35 as a white solid (144 mg, 21%). Further
elution gave rhodonoid C (4.3) as a white solid (218 mg, 32%).
Data for rhodonoid C (4.3):
Rf = 0.20 (2:1, petrol/EtOAc)
IR (neat): 3259, 2927, 2869, 1628, 1593, 1456, 1421, 1380, 1327, 1266, 1144, 1126 cm-1.
MP: 196 – 198 ºC. 1H NMR (500 MHz, CDCl3): δ 6.30 (s, 1H), 6.26 (s, 1H), 5.64 (br s, 1H), 5.05 (d, J = 4.2 Hz, 1H),
3.85 (s, 1H), 2.23 (s, 3H), 1.87 – 1.81 (m, 2H), 1.73 – 1.69 (m, 2H), 1.64 (s, 3H), 1.49 (dd, J = 14.6,
5.7 Hz, 1H), 1.29 (s, 3H), 1.26 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 155.6, 152.6, 140.2, 109.8, 108.5, 107.8, 82.0, 77.5, 68.9, 51.1,
42.6, 30.1, 27.9, 27.6, 27.0, 23.0, 21.5.
HRMS (ESI): calculated for C17H21O3 273.1496 [M-H]-, found 273.1490.
Data for rhodonoid D (4.4):
Rf = 0.55 (2:1, petrol/EtOAc)
IR (neat): 3435, 2970, 2921, 2853, 1636, 1586, 1460, 1370, 1326, 1299, 1278, 1190 cm-1.
MP: 162 – 165 ºC. 1H NMR (500 MHz, CDCl3): δ 6.92 (s, 1H), 6.35 (s, 1H), 6.29 (s, 1H), 4.92 (d, J = 9.1 Hz, 1H),
2.82 (t, J = 8.5 Hz, 1H), 2.56 (ddd, J = 9.9, 8.0, 3.8 Hz, 1H), 2.23 (s, 3H), 1.85 – 1.78 (m, 1H), 1.75
– 1.72 (m, 1H), 1.67 – 1.61 (m, 1H), 1.48 (s, 3H), 1.44 – 1.40 (m, 1H), 1.33 (s, 3H), 1.29 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 156.1, 151.7, 139.9, 110.0, 109.2, 107.3, 83.2, 82.9, 68.8, 51.6,
51.5, 34.9, 28.0, 27.5, 24.13, 24.07, 21.5.
SnCl4, CHCl3, −60 °C
32% 21%5%
O
OHO
1:1 d.r.
H
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.18 4.35

285
HRMS (ESI): calculated for C17H21O3 273.1496 [M-H]-, found 273.1489.
Data for 4.35:
Rf = 0.50 (2:1, petrol/EtOAc)
IR (neat): 3387, 2970, 2924, 2854, 1698, 1623, 1579, 1510, 1450, 1368, 1329, 1280 cm-1.
MP: 116 – 121 ºC. 1H NMR (500 MHz, CDCl3): δ 6.63 (d, J = 10.0 Hz, 1H), 6.22 (s, 1H), 6.12 (s, 1H), 5.41 (d, J =
10.0 Hz, 1H), 4.71 (br s, 1H), 2.68 – 2.54 (m, 3H), 2.20 (s, 3H), 1.99 – 1.94 (m, 2H), 1.36 (s, 3H),
1.07 (d, J = 7.1 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ 214.8, 153.9, 151.2, 139.7, 126.4, 117.5, 109.7, 108.5, 106.4,
77.9, 41.0, 35.3, 34.9, 26.6, 21.5, 18.33, 18.27.
HRMS (ESI): calculated for C17H23O3 275.1642 [M+H]+, found 275.1642.

286
To a solution of 4.18 (d.r. = 1:1) (66 mg, 0.24 mmol) in CHCl3 (10 mL) at room temperature was
added p-TsOH⋅H2O (4.6 mg, 0.024 mmol). The reaction was stirred at room temperature for 30
min, then quenched with saturated NaHCO3 solution (10 mL). The organic layer was separated and
the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts were
washed with brine (10 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was
purified by flash column chromatography on SiO2 (5:1 → 3:1, petrol/EtOAc gradient elution) to
give rhodonoid D (4.4) as a white solid (1.5 mg, 2%), further elution gave ketone 4.35 as a
colourless oil (3 mg, 5%), further elution gave rhodonoid C (4.3) as a white solid (14 mg, 21%).
Data for 4.3, 4.4 and 4.35 matched that previously obtained.
p-TsOH, CHCl3, rt
21% 5%2%
O
OHO
1:1 d.r.
H
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.18 4.35

287
To a solution of 4.18 (d.r. = 1:1) (2.02 g, 7.24 mmol) in CHCl3 (50 mL) at −60 ºC was added SnCl4
(0.42 mL, 3.62 mmol) dropwise. The reaction was stirred at −60 ºC for 30 min, then quenched with
saturated NaHCO3 solution (50 mL), warmed to room temperature. The mixture was filtered
through a pad of Celite, washed thoroughly with CH2Cl2. The layers were separated and the organic
phase was washed with saturated NaHCO3 solution (2 × 50 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (5:1 →
3:1, petrol/EtOAc gradient elution) to give rhodonoid D (4.4) as a white solid (69 mg, 3%). Further
elution gave ketone 4.35 as a white solid (152 mg, 8%). Further elution gave epoxide 4.18 (d.r. =
3.3:1) as a brown oil (792 mg, 39%). Further elution gave rhodonoid C (4.3) as a white solid (443
mg, 22%). Data for 4.3, 4.4 and 4.35 matched that previously obtained.
NMR data for 4.18a (major diastereomer): 1H NMR (500 MHz, CDCl3): δ 6.63 (d, J = 10.0 Hz, 1H), 6.21 (s, 1H), 6.12 (s, 1H), 5.44 (d, J =
10.0 Hz, 1H), 4.97 (br s, 1H), 2.75 (t, J = 6.2 Hz, 1H), 2.20 (s, 3H), 1.82 (d, J = 8.1 Hz, 1H), 1.74 –
1.64 (m, 3H), 1.38 (s, 3H), 1.28 (s, 3H), 1.23 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 153.9, 151.2, 139.7, 126.4, 117.3, 109.6, 108.4, 106.5, 78.0, 64.6,
58.8, 37.9, 26.6, 24.9, 23.9, 21.5, 18.7.
NMR data for 4.18b (minor diastereomer): 1H NMR (500 MHz, CDCl3): δ 6.63 (d, J = 10.0 Hz, 1H), 6.21 (s, 1H), 6.12 (s, 1H), 5.47 (d, J =
10.0 Hz, 1H), 4.97 (br s, 1H), 2.73 (t, J = 5.9 Hz, 1H), 2.20 (s, 3H), 1.84 (d, J = 8.0 Hz, 1H), 1.74 –
1.64 (m, 3H), 1.38 (s, 3H), 1.29 (s, 3H), 1.24 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 153.8, 151.2, 139.6, 126.7, 117.2, 109.7, 108.4, 106.5, 77.7, 64.4,
58.7, 37.5, 26.1, 24.9, 23.6, 21.5, 18.6.
SnCl4, CHCl3, −60 °C 22% 8%3%
O
OHO
1:1 d.r.
H
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
O
OHO
H
O
OHO
H +
4.18a3.3:1 d.r.
39%
4.18
4.18
4.35

288
To a solution of 4.18a (d.r. = 3.3:1) (99 mg, 0.36 mmol) in CHCl3 (5 mL) at −60 ºC was added
SnCl4 (0.04 mL, 0.36 mmol) dropwise. The reaction was stirred at −60 ºC for 15 min, then
quenched with saturated NaHCO3 solution (5 mL), warmed to room temperature. The aqueous layer
was separated and the organic extract was washed with saturated NaHCO3 solution (2 × 10 mL),
brine (10 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by
flash column chromatography on SiO2 (4:1 → 2:1, petrol/EtOAc gradient elution) to give ketone
4.35 as a white solid (32 mg, 32%). Further elution gave rhodonoid C (4.3) as a white solid (13 mg,
13%). Data for 4.3 and 4.35 matched that previously obtained.
O
OHO
H
O
OHO
H +
4.18a3.3:1 d.r.
SnCl4, CHCl3, −60 °C
13% 32%
O
OHO
4.3: rhodonoid C
+O
OOH
4.18b 4.35

289
To a solution of 4.18 (97 mg, 0.35 mmol) in H2O (10 mL) was heated at reflux for 16 h. The
reaction was cooled to room temperature, then extracted with Et2O (2 × 10 mL). The combined
organic extracts were dried over MgSO4, filtered and concentrated in vacuo. The residue was
purified by flash column chromatography on SiO2 (5:1 → 2:1, petrol/EtOAc gradient elution) to
give 4.35 as a white solid (3 mg, 3%). Further elution gave rhodonoid C (4.3) as a white solid (4
mg, 4%). Further elution gave 4.38 as a white solid (45 mg, 44%). Data of 4.3 and 4.35 matched
previously obtained.
Data for 4.38:
Rf = 0.05 (2:1, petrol/EtOAc)
IR (neat): 3342, 2971, 2929, 2865, 1623, 1578, 1510, 1450, 1421, 1366, 1329 cm-1.
MP: 55 ºC. 1H NMR (500 MHz, CDCl3): δ 6.63 (d, J = 10.0 Hz, 2H), 6.31 (br s, 2H), 6.19 (s, 2H), 6.13 (s,
2H), 5.44 (d, J = 9.9 Hz, 1H), 5.42 (d, J = 9.9 Hz, 1H), 3.37 (t, J = 9.6 Hz, 2H), 2.80 (br s, 2H), 2.45
(br s, 2H), 2.17 (s, 6H), 2.01 – 1.90 (m, 2H), 1.80 – 1.64 (m, 4H), 1.50 – 1.39 (m, 2H), 1.35 (s, 3H),
1.34 (s, 3H), 1.19 (s, 3H), 1.17 (s, 3H), 1.14 (s, 3H), 1.13 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 153.6, 153.5, 151.6, 139.6, 139.5, 126.9, 126.5, 117.4, 109.4,
109.3, 108.8, 108.7, 106.9, 106.8, 78.9, 78.6, 78.4, 78.1, 73.51, 73.47, 38.2, 37.9, 26.43, 26.38,
26.36, 26.31, 25.89, 25.87, 23.3, 23.2, 21.5.
HRMS (ESI): calculated for C17H25O4 293.1747 [M+H]+, found 297.1747.
H2O, reflux
O
OHO
1:1 d.r.
H
O
OHHO
OH
1:1 d.r.4.384.18 4.3: rhodonoid C
O
OOH
O
OHO
++
44% 4% 3%
4.35

290
To a solution of 4.18 (124 mg, 0.45 mmol) in DMF (5 mL) at room temperature was added p-TsOH
(86 mg, 0.45 mmol). The reaction was stirred at room temperature for 30 min, then diluted with
H2O (10 mL). The mixture was extracted with Et2O (4 × 10 mL). The combined organic extracts
were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (5:1 → 3:1 → 1:1, petrol/EtOAc gradient elution) to give 4.35 as
a white solid (3 mg, 2%). Further elution gave 4.39 as a colourless oil (40 mg, 28%). Data of 4.35
matched of previously obtained.
Data for 4.39:
Rf = 0.20 (2:1, petrol/EtOAc)
IR (neat): 3387, 2976, 2924, 1714, 1624, 1579, 1452, 1378, 1330, 1266 cm-1. 1H NMR (500 MHz, CDCl3): δ 8.18 (s, 1H), 8.18 (s, 1H), 6.64 (d, J = 10.0 Hz, 1H), 6.62 (d, J =
10.0 Hz, 1H), 6.21 (s, 1H), 6.20 (s, 1H), 6.12 (s, 2H), 5.43 (d, J = 10.0 Hz, 1H), 5.30 (d, J = 10.0
Hz, 1H), 4.89 – 4.87 (m, 2H), 2.19 (s, 6H), 1.90 – 1.62 (m, 8H), 1.23 (s, 6H), 1.21 (s, 6H), 1.20 (s,
3H), 1.19 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 161.5, 161.4, 153.8, 153.7, 151.31, 151.30, 139.7, 126.5, 126.2,
117.6, 117.4, 109.62, 109.56, 108.55, 108.51, 106.60, 106.57, 80.4, 80.2, 78.0, 77.7, 72.47, 72.46,
37.7, 37.5, 26.54, 26.47, 26.2, 24.9, 24.8, 24.3, 24.0, 21.5.
HRMS (ESI): calculated for C18H25O5 321.1697 [M+H]+, found 321.1699.
p-TsOH, DMF, rt
O
OHO
1:1 d.r.
H
O
OHO
OH
1:1 d.r.4.394.18
28%
O
OHO
4.35
+
2%
O H

291
To a solution of 4.18 (40 mg, 0.15 mmol) in EtOH (5 mL) was added 1 M HCl (5 mL) was stirred
at room temperature for 1 d. The mixture was extracted with Et2O (2 × 10 mL). The combined
organic extracts were dried over MgSO4, filtered and concentrated in vacuo. The residue was
purified by flash column chromatography on SiO2 (2:1, petrol/EtOAc) to give 4.36 as a colourless
oil (12 mg, 26%). Further elution gave 4.38 as a white solid (4 mg, 9%). Data of 4.38 matched
previously obtained.
Data of 4.36:
Rf = 0.45 (2:1, petrol/EtOAc)
IR (neat): 3342, 2974, 2922, 1624, 1579, 1511, 1452, 1370, 1330 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.64 (d, J = 10.1 Hz, 1H), 6.63 (d, J = 10.1 Hz, 1H), 6.23 (s, 2H),
6.12 (s, 2H), 6.48 (d, J = 10.0 Hz, 1H), 6.47 (d, J = 10.0 Hz, 1H), 4.97 (br s, 2H), 3.54 – 3.49 (m,
2H), 3.43 – 3.40 (m, 2H), 2.20 (s, 6H), 2.07 – 1.97 (m, 3H), 1.84 – 1.77 (m, 3H), 1.75 – 1.70 (m,
1H), 1.58 (s, 3H), 1.57 (s, 3H), 1.54 (s, 3H), 1.51 (s, 3H), 1.48 – 1.42 (m, 1H), 1.37 (s, 3H), 1.37 (s,
3H). 13C NMR (125 MHz, CDCl3): δ 153.9, 153.8, 151.2, 139.7, 139.6, 127.2, 126.7, 117.3, 117.2,
109.8, 109.7, 108.6, 108.5, 106.8, 106.7, 79.3, 79.2, 78.4, 78.0, 38.4, 37.9, 29.4, 29.2, 27.1, 26.9,
26.6, 26.4, 26.0, 25.9, 21.5, 16.1.
HRMS (ESI): calculated for C17H24ClO3 311.1408 [M+H]+, found 311.1410.
1 M HCl, EtOH, rt
O
OHO
1:1 d.r.
H
O
OHHO
1:1 d.r.4.364.18
Cl
26%
O
OHHO
1:1 d.r.4.38
OH
+
9%

292
To a solution of 4.18 (123 mg, 0.45 mmol) in CH2Cl2 (15 mL) at –78 ˚C was added BF3·OEt2 (0.05
mL, 0.45 mmol). The reaction was stirred at –78 ˚C for 30 min, then quenched with saturated
NaHCO3 solution (15 mL) and warmed to room temperature. The organic layer was separated and
the aqueous layer was extracted with CH2Cl2 (20 mL). The combined organic extracts were washed
with saturated NaHCO3 solution (3 × 20 mL), H2O (40 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (2:1,
petrol/EtOAc) to give 4.37 as a colourless oil (21 mg, 18%).
Data for 4.37:
Rf = 0.15 (2:1, petrol/EtOAc)
IR (neat): 3356, 2974, 2927, 1623, 1579, 1451, 1376, 1330, 1246 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.64 (d, J = 10.0 Hz, 2H), 6.22 (s, 1H), 6.12 (s, 1H), 5.46 (d, J =
10.0 Hz, 2H), 5.27 (br s, 2H), 3.58 (t, J = 11.0 Hz, 2H), 2.19 (s, 6H), 2.02 – 1.96 (m, 2H), 1.83 –
1.77 (m, 2H), 1.89 – 1.64 (m, 2H), 1.45 – 1.39 (m, 2H), 1.36 (s, 6H), 1.34 (s, 3H), 1.33 (s, 3H), 1.29
(s, 3H), 1.28 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 153.8, 151.3, 139.6, 126.6, 117.3, 109.6, 108.5, 106.7, 98.8, 97.5,
78.4, 77.2, 38.2, 26.6, 26.12, 26.08, 23.8, 23.6, 21.5, 21.2, 21.0. 19F NMR (470 MHz, CDCl3): δ –144.69 (hept, J = 22.3 Hz), –144.72 (hept, J = 22.3 Hz).
HRMS (ESI): calculated for C17H22FO3 293.1558 [M-H]-, found 293.1559.
BF3·OEt2, CH2Cl2, −78 °C
O
OHO
1:1 d.r.
H
O
OHHO
1:1 d.r.4.374.18
F
18%

293
To a solution of 4.18 (123 mg, 0.41 mmol) in CH2Cl2 (15 mL) at –78 ˚C was added FeCl3·6H2O
(110 mg, 0.41 mmol). The reaction was stirred at –78 ˚C for 1.5 h, then warmed to room
temperature for 20 min. The reaction was quenched with saturated NaHCO3 solution (15 mL). The
organic layer was separated and the aqueous layer was extracted with CH2Cl2 (20 mL). The
combined organic extracts were washed with saturated NaHCO3 solution (3 × 20 mL), H2O (40
mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (2:1, petrol/EtOAc) to give 4.35 as a white solid (45 mg, 40%).
Further elution gave rhodonoid C (4.3) as a white solid (26 mg, 23%). Further elution gave 4.36 as
a colourless oil (6 mg, 4%). Data of 4.3, 4.35 and 4.36 matched previously obtained.
To a solution of 4.18 (473 mg, 1.72 mmol) in CH2Cl2 (30 mL) at –78 ˚C was added FeCl3 (140 mg,
0.86 mmol). The reaction was stirred at –78 ˚C for 1.5 h. The reaction was quenched with saturated
NaHCO3 solution (30 mL), then warmed to room temperature. The organic layer was separated and
the aqueous layer was extracted with CH2Cl2 (30 mL). The combined organic extracts were washed
with saturated NaHCO3 solution (2 × 20 mL), H2O (40 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (2:1,
petrol/EtOAc) to give rhodonoid D (4.4) as a white solid (45 mg, 40%). Further elution gave 4.35 as
a white solid (165 mg, 35%). Further elution gave rhodonoid C (4.3) as a white solid (117 mg,
25%). Data of 4.3, 4.4 and 4.35 matched previously obtained.
FeCl3·6H2O, CH2Cl2, –78 °C to rt
O
OHO
1:1 d.r.
H
O
OHHO
1:1 d.r.4.364.18
Cl
4%23% 40%
O
OHO
4.3: rhodonoid C
O
OOH
4.35
+ +
FeCl3, CH2Cl2, –78 °C
25% 35%2%
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.35
O
OHO
1:1 d.r.
H
4.18

294
To a solution of 4.9 (1.0 g, 4.2 mmol) in CH2Cl2 (20 mL) was added m-CPBA (77%, 722 mg, 4.20
mmol). The reaction was stirred at room temperature for 30 min, followed by addition of p-TsOH
(80 mg, 0.42 mmol). The reaction was stirred at room temperature for 30 min, then quenched with
saturated NaHCO3 solution (30 mL). The organic layer was separated and the aqueous layer was
extracted with CH2Cl2 (2 × 30 mL). The combined organic extracts were washed with brine (100
mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
column chromatography on SiO2 (5:1 → 3:1, petrol/EtOAc gradient elution) to give ketone 4.35 as
a white solid (41 mg, 4%). Further elution gave rhodonoid D (4.4) as a white solid (25 mg, 2%).
Further elution gave rhodonoid C (4.3) as a white solid (62 mg, 5%). Data of 4.3, 4.4 and 4.35
matched previously obtained.
m-CPBA, CH2Cl2, rt;then p-TsOH
5% 4%2%
O
OHO
O
OH OH
H
H
4.4: rhodonoid D4.3: rhodonoid C
+ +O
OOH
4.34
OH
O
4.9

295
To a suspension of Mn (149 g, 2.71 mmol) and Cp2TiCl2 (168 mg, 0.68 mmol) in anhydrous THF
(5 mL) at room temperature was stirred until the solution changed from orange to dark green (1.5
h). 4.18 (93 mg, 0.34 mmol) in anhydrous THF (3 mL) was added and stirred for 2 h. The mixture
was filtered through Celite and washed with EtOAc (20 mL). The filtrate was concentrated in
vacuo. The residue was purified by flash column chromatography on SiO2 (3:1 → 2:1,
petrol/EtOAc) to give 4.40 (d.r. = 1:1) as an orange oil (48 mg, 51%).
Data for 4.40:
Rf = 0.45 (2:1, petrol/EtOAc)
IR (neat): 3342, 2971, 2929, 1623, 1578, 1510, 1450, 1421, 1366, 1329 cm-1. 1H NMR (500 MHz, CDCl3): δ 6.21 (s, 2H), 6.17 (s, 2H), 5.62 (br s, 2H), 3.51 (br s, 1H), 3.38 (dd,
J = 11.8, 3.7 Hz, 1H), 2.80 – 2.65 (m, 4H), 2.19 (s, 6H), 2.16 (ddd, J = 11.8, 4.1, 2.1 Hz, 1H), 2.08
– 2.03 (m, 2H), 1.99 (dd, J = 14.1, 4.3 Hz, 1H), 1.93 – 1.87 (m, 2H), 1.81 – 1.77 (m, 1H), 1.67 –
1.58 (m, 3H), 1.46 (d, J = 7.7 Hz, 1H), 1.21 (s, 3H), 1.17 (s, 3H), 1.08 (s, 3H), 1.05 (s, 3H), 0.69 (s,
3H), 0.64 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 155.03, 155.00, 153.38, 153.36, 136.8, 136.7, 110.09, 110.05,
107.4, 107.3, 106.5, 106.3, 78.4, 76.4, 74.9, 74.3, 43.5, 39.5, 37.90, 37.86, 37.3, 32.1, 27.3, 27.1,
26.44, 26.40, 26.38, 24.6, 21.8, 21.2, 17.8, 17.4, 14.0.
Mn, Cp2TiCl2, THF, rt
O
OHO
1:1 d.r.
H
4.401:1 d.r.
O
OH
4.18
51%H
OH

296
To a solution of 2-methyl-5-nitrophenol (4.41) (10.0 g, 65.3 mmol) and K2CO3 (13.5 g, 98.0 mmol)
in acetone (100 mL) was added BnBr (8.1 mL, 68.6 mmol). The mixture was heated at reflux or 3
h, then cooled to room temperature and diluted with CH2Cl2 (100 mL). The organic layer was
separated and washed with H2O (2 × 100 mL), brine (100 mL), dried over MgSO4, filtered through
a pad of SiO2 and washed with EtOAc (100 mL). The filtrate was concentrated in vacuo to give 4.42
as a colourless solid (15.9 g). 4.42 was used in next step without further purification. Data of 4.42
matched with literature.2
Data for 4.42:
Rf = 0.45 (5:1, petrol/EtOAc)
IR (neat): 1713, 1594, 1511, 1451, 1416, 1355, 1254, 1221 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.78 (d, J = 8.2 Hz, 1H), 7.75 (s, 1H), 7.46 (d, J = 7.0 Hz, 1H),
7.46 (s, 1H), 7.42 (t, J = 7.6 Hz, 2H), 7.37 – 7.35 (m, 1H), 7.29 (d, J = 8.2 Hz, 1H), 5.17 (s, 2H),
2.36 (s, 3H).
NO2
OH
4.41: 2-methyl-5-nitrophenol
BnBr, K2CO3acetone , reflux
NO2
OBn
4.42

297
To a solution of 4.42 (15.9 g, 65.3 mmol) in AcOH (200 mL) was adde Fe (36.5 g, 65.3 mmol). The
mixture was heated at 45 ºC for 14 h, then cooled to room temperature and diluted with EtOAc (200
mL). The mixture was filtered through Celite and washed with EtOAc (200 mL). The filtrate was
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (3:1,
petrol/EtOAc) to give 4.43 as a red oil (9.14 g, 66% over 2 step). Data for 4.43 matched from
literauture.2
Data for 4.43:
Rf = 0.45 (1:1, petrol/EtOAc)
IR (neat): 3448, 3366, 3072, 2924, 1709, 1616, 1588, 1512, 1454, 1436, 1361 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.44 (d, J = 7.6 Hz, 1H), 7.44 (s, 1H), 7.39 (t, J = 7.6 Hz, 2H),
7.32 (t, J = 7.3 Hz, 1H), 6.93 (d, J = 7.8 Hz, 1H), 6.29 (d, J = 2.1 Hz, 1H), 6.24 (dd, J = 7.9, 2.0 Hz,
1H), 5.03 (s, 2H), 3.58 (br s, 2H), 2.18 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 157.6, 145.4, 137.6, 131., 128.5, 127.7, 127.0, 117.0, 107.2,
100.0, 69.7, 15.5.
NO2
OBn
Fe, AcOH, 45 °C
NH2
OBn
4.434.42
66% over 2 steps

298
To a solution of 4.43 (9.14 g, 42.9 mmol), K2CO3 (5.90 g, 42.9 mmol), Cu(OAc)2 (15.6 g, 85.7
mmol), PhCO2H (5.93 g, 42.9 mmol) and PhB(OH)2 (13.1 g, 107 mmol) in toluene (200 mL) was
heated at reflux for 12 h. The reaction was cooled to room temperature and filtered through Celite,
then washed with EtOAc (200 mL). The filtrate was concentrated in vacuo. The residue was
purified by flash column chromatography on SiO2 (10:1 → 2:1, petrol/EtOAc) to give 4.44 as a
yellow solid (8.57 g, 69%). Data of 4.44 matched from literature.2
Data for 4.44:
Rf = 0.50 (5:1, petrol/EtOAc)
IR (neat): 3400, 3036, 2925, 2857, 1712, 1633, 1609, 1498, 1458, 1361, 1344, 1307 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.42 (d, J = 7.2 Hz, 1H), 7.42 (s, 1H), 7.39 (t, J = 7.5 Hz, 2H),
7.32 (t, J = 7.1 Hz, 1H), 7.22 (dd, J = 8.4, 7.5 Hz, 2H), 7.05 (d, J = 8.0 Hz, 1H), 6.95 (d, J = 7.7 Hz,
1H), 6.88 (t, J = 7.3 Hz, 1H), 6.67 (d, J = 2.0 Hz, 1H), 6.59 (dd, J = 7.9, 2.1 Hz, 1H), 5.61 (br s,
1H), 5.04 (s, 2H), 2.24 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 157.3, 143.7, 141.7, 137.4, 131.1, 129.3, 128.5, 127.7, 127.0,
120.3, 120.0, 117.0, 110.8, 103.1, 69.7, 15.8.
NH2
OBn K2CO3, Cu(OAc)2benzoic acid, PhB(OH)2
PhMe, reflux
HN
OBn
4.444.43
69%

299
To a solution of 4.44 (6.39 g, 22.0 mmol), K2CO3 (304 mg, 2.20 mmol) and Pd(OAc)2 (450 mg,
2.20 mmol) in pivalic acid (30 mL) was heated at 85 ºC under air for 20 h. The reaction was cooled
to room temperature, followed by addition of saturated K2CO3 solution (50 mL). The mixture was
filtered through SiO2 and washed with EtOAc (200 mL). The filtrated was washed with saturated
K2CO3 (100 mL), brine (100 mL), dried over MgSO4, filtered and concentrated in vacuo. The
residue was purified by flash column chromatography on SiO2 (10:1, petrol/EtOAc) to give 4.45 as
a white solid (4.0 g, 63%). Data of 4.45 matched from literature.2
Data for 4.45:
Rf = 0.35 (3:1, petrol/EtOAc)
IR (neat): 3381, 3032, 2924, 1710, 1615, 1560, 1510, 1496, 1454, 1411, 1362 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.95 (d, J = 7.7 Hz, 1H), 7.86 (br s, 1H), 7.82 (s, 1H), 7.50 (d, J =
7.4 Hz, 2H), 7.41 (t, J = 7.5 Hz, 2H), 7.37 – 7.30 (m, 3H), 7.21 – 7.17 (m, 1H), 6.91 (s, 1H), 5.17
(s, 2H), 2.44 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 156.4, 139.2, 139.0, 137.5, 128.5, 127.8, 127.1, 124.2, 123.5,
121.6, 119.7, 119.4, 119.3, 116.5, 110.2, 94.0, 70.2, 16.9.
HN
OBn
4.454.44
NH
OBn
K2CO3, Pd(OAc)2pivalic acid, 85 °C
63%

300
To a solution of 4.45 (4.39 g, 15.3 mmol) in MeOH/CH2Cl2 (3:1, 100 mL) at room temperature was
added Pd/C (5%, 800 mg). The mixture was stirred under H2 for 1.5 d, then filtered through SiO2
and washed with EtOAc (150 mL). The filtrated was concentrated in vacuo. The residue was
purified by flash column chromatography on SiO2 (4:1 → 2:1, petrol/EtOAc gradient elution) to
give 4.46 as a yellowish solid (2.65 g, 88%). Data of 4.46 matched from literature.2
Data of 4.46:
Rf = 0.15 (2:1, petrol/EtOAc)
IR (neat): 3639, 3532, 3403, 1699, 1637, 1613, 1459, 1437, 1416, 1309, 1248 cm-1. 1H NMR (500 MHz, CDCl3): δ 9.92 (br s, 1H), 8.23 (br s, 1H), 7.92 (d, J = 7.8 Hz, 1H), 7.77 (s,
1H), 7.37 (d, J = 8.0 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.96 (s, 1H), 2.34 (s,
3H).
4.46
NH
OH
4.45
NH
OBn
H2, Pd/C, CH2Cl2MeOH, rt
88%

301
To a solution of 4.46 (500 mg, 2.54 mmol) and citral (4.8) (443 mg, 5.08 mmol) in PhMe (10 mL)
at –78 ˚C was added Ti(Oi-Pr)4 (3.00 mL, 10.1 mmol). The solution was stirred at –78 ˚C for 15
min, then warmed to room temperature and stirred for 16 h. The reaction was quenched with
saturated hydroxylamine hydrochloride solution (10 mL). The mixture was filtered through Celite,
then washed with EtOAc (20 mL). The filtrate was washed sequentially with 1 M HCl solution (30
mL), brine (30 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was re-
dissolved in MeOH (10 mL), followed by addition of NaBH4 (182 mg, 5.08 mmol) to reduce
unreacted citral (4.8) that is difficult to separate from 4.5. The reaction was stirred at room
temperature for 30 min, then quenched with 1 M HCl solution (20 mL). The solution was extracted
with Et2O (2 × 30 mL). The combined organic extracts were washed with brine (30 mL), dried over
MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (4:1, petrol/EtOAc) to give mahanimbine (4.5) as a yellow oil (662 mg,
74%). Data for 4.5 matched that previously reported in the literature.2
Data for 4.5:
Rf = 0.45 (2:1, petrol/EtOAc)
IR (neat): 3423, 1968, 2920, 2853, 1645, 1610, 1491, 1458, 1440, 1307 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.89 (d, J = 7.7 Hz, 1H), 7.78 (br s, 1H), 7.64 (s, 1H), 7.31 – 7.27
(m, 2H), 7.16 (td, J = 6.5, 1.2 Hz, 1H), 6.54 (d, J = 9.8 Hz, 1H), 5.59 (d, J = 9.8 Hz, 1H), 5.11 (t, J
= 7.1 Hz, 1H), 2.32 (s, 3H), 2.20 – 2.11 (m, 2H), 1.75 (dd, J = 8.6, 8.1 Hz, 2H), 1.65 (s, 3H), 1.57
(s, 3H), 1.43 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 149.9, 139.2, 134.8, 131.6, 128.4, 124.18, 124.16, 123.9, 121.1,
119.4, 119.2, 118.3, 117.5, 116.6, 110.4, 104.2, 78.1, 40.8, 25.8, 25.6, 22.7, 17.6, 16.1.
HRMS (ESI): calculated for C23H26NO 332.2009 [M+H]+, found 332.2004.
O
NH
4.5: mahanimbine
4.8: citralTi(Oi-Pr)4, PhMe, −78 °C to rt;
then NaBH4, MeOH, rt
74%
O
H
NH
OH
4.46

302
To a solution of mahanimbine (4.5) (623 mg, 1.88 mmol) in CH2Cl2 (10 ml) at room temperature
was added m-CPBA (77%, 509 mg, 2.07 mmol). The reaction was stirred at room temperature for
10 min, then quenched with saturated Na2S2O3 solution (10 mL). The organic layer was separated,
then washed sequentially with saturated NaHCO3 solution (10 mL), brine (10 mL), dried over
MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (4:1, petrol/EtOAc) to give 4.22 (d.r. = 1:1) as a grey solid (400 mg,
61%).
Data for 4.22:
Rf = 0.20 (4:1, petrol/EtOAc)
IR (neat): 3333, 2972, 2925, 2853, 1646, 1612, 1459, 1441, 1405, 1323, 1214 cm-1. 1H NMR (500 MHz, CDCl3): δ 8.01 (s, 2H), 7.90 (d, J = 7.6 Hz, 2H), 7.65 (s, 1H), 7.64 (s, 1H),
7.34 (d, J = 8.1 Hz, 2H), 7.31 (t, J = 7.6 Hz, 2H), 7.16 (t, J = 7.4 Hz, 2H), 6.64 (d, J = 9.7 Hz, 2H),
5.61 (d, J = 9.8 Hz, 1H), 5.57 (d, J = 9.8 Hz, 1H), 2.78 (t, J = 6.2 Hz, 1H), 2.74 (t, J = 6.1 Hz, 1H),
2.31 (s, 3H), 2.30 (s, 3H), 1.92 – 1.85 (m, 4H), 1.79 – 1.71 (m, 4H), 1.44 (s, 3H), 1.43 (s, 3H), 1.28
(s, 3H), 1.27 (s, 3H), 1.23 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 149.6, 149.5, 139.5, 134.9, 128.1, 127.6, 124.25, 124.22, 123.8,
121.31, 121.30, 119.43, 119.41, 119.27, 119.26, 118.2, 118.14, 118.13, 117.9, 116.8, 116.7, 110.39,
110.37, 104.1, 103.9, 78.0, 77.7, 64.5, 64.3, 58.7, 58.6, 37.7, 37.1, 26.3, 25.6, 24.84, 24.83, 24.0,
23.7, 18.63, 18.57, 16.04, 16.03.
HRMS (ESI): calculated for C23H26NO2 348.1958 [M+H]+, found 348.1959.
m-CPBACH2Cl2, rt
61%O
NH
4.5: mahanimbine
1:1 d.r.
O
NHO
H
4.22

303
To a solution of 4.22 (d.r. = 1:1) (200 mg, 0.58 mmol) in CH2Cl2 (5 mL) at room temperature was
added p-TsOH⋅H2O (11 mg, 0.058 mmol). The reaction was stirred at room temperature for 1 h,
then quenched with H2O (5 mL). The organic layer was separated and the aqueous phase was
extracted with CH2Cl2 (5 mL). The combined organic extracts were washed with brine (10 mL),
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (5:1, petrol/EtOAc) to give 76 mg of a 7.4:1 mixture of murrayakonine D
(4.6) and ketone 4.47. This mixture was triturated with MeOH to give murrayakonine D (4.6) as a
white solid (65 mg, 33%). Trace quantities of ketone 4.47 were obtained for characterisation
purposes after repeated purification of the filtrate.
Data for murrayakonine D (4.6):
Rf = 0.45 (2:1, petrol/EtOAc)
IR (neat): 3280, 2964, 2931, 2853, 1633, 1613, 1460, 1313, 1215 cm-1.
MP: 244 ºC 1H NMR (500 MHz, CDCl3): δ 8.28 (br s, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.75 (s, 1H), 7.36 (d, J =
8.07 Hz, 1H), 7.29 (t, J = 7.3 Hz, 1H), 7.17 (t, J = 7.4 Hz, 1H), 5.29 (d, J = 4.2 Hz, 1H), 3.90 (d, J =
4.2 Hz, 1H), 2.32 (s, 3H), 1.97 (d, J = 3.9 Hz, 1H), 1.95 – 1.90 (m, 1H), 1.81 – 1.75 (m, 1H), 1.74
(s, 3H), 1.72 – 1.67 (m, 1H), 1.53 (dd, J = 14.5, 7.1 Hz, 1H), 1.38 (s, 3H), 1.32 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 149.0, 139.6, 138.4, 123.97, 123.95, 121.3, 119.3, 119.2, 118.5,
115.9, 110.6, 105.0, 82.1, 77.8, 70.4, 51.0, 42.7, 30.4, 28.1, 27.6, 27.1, 23.0, 16.3.
HRMS (ESI): calculated for C23H26NO2 348.1958 [M+H]+, found 348.1958.
Data for 4.47:
Rf = 0.45 (2:1, petrol/EtOAc)
IR (neat): 3369, 2971, 2926, 1703, 1646, 1611, 1459, 1443, 1405, 1310, 1265 cm-1. 1H NMR (500 MHz, CDCl3): δ 7.92 (br s, 1H), 7.91 (s, 1H), 7.67 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H),
7.31 (t, J = 7.6 Hz, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.68 (d, J = 9.8 Hz, 1H), 5.60 (d, J = 9.8 Hz, 1H),
2.73 (ddd, J = 17.4, 9.2, 6.3 Hz, 1H), 2.65 (ddd, J = 16.9, 8.6, 5.8 Hz, 1H), 2.59 (hept, J = 6.9 Hz,
1H), 2.32 (s, 3H), 2.08 – 2.04 (m, 2H), 1.43 (s, 3H), 1.07 (d, J = 6.9 Hz, 6H).
p-TsOH, CH2Cl2, rt
1:1 d.r.
O
NHO
H
4.6: murrayakonine D
O
ONH
33% O
NHO
+
4.22 4.47

304
13C NMR (125 MHz, CDCl3): δ 214.6, 149.6, 139.5, 134.8, 127.8, 124.3, 123.8, 121.4, 119.5,
119.3, 118.24, 118.21, 116.8, 110.4, 103.9, 77.9, 41.1, 35.3, 34.7, 26.2, 18.33, 18.27, 16.1.
HRMS (ESI): calculated for C23H26NO2 348.1958 [M+H]+, found 348.1954.

305
To a solution of 4.22 (d.r. = 1:1) (200 mg, 0.58 mmol) in CH2Cl2 (15 mL) at –78 ˚C was added
SnCl4 (0.06 mL, 0.58 mmol). The reaction was stirred at –78 ˚C for 15 min. The reaction was
quenched with saturated NaHCO3 solution (10 mL). The organic layer was separated and the
aqueous phase was extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were washed
with brine (2 × 10 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was
triturated with MeOH to give murrayakonine D (4.6) as a white solid (42 mg, 21%). Data of 4.6
matched previously obtained.
SnCl4, CH2Cl2, −78 °C
1:1 d.r.
O
NHO
H
4.6: murrayakonine D
O
ONH
21%4.22
306
4.5.3. NMR spectra
OH
O
4.91H NMR500 MHz
CDCl3
OH
O
4.913C NMR125 MHz
CDCl3
307
O
OHO
1:1 d.r.
H
4.18
1H NMR500 MHz
CDCl3
O
OHO
1:1 d.r.
H
4.18
13C NMR125 MHz
CDCl3
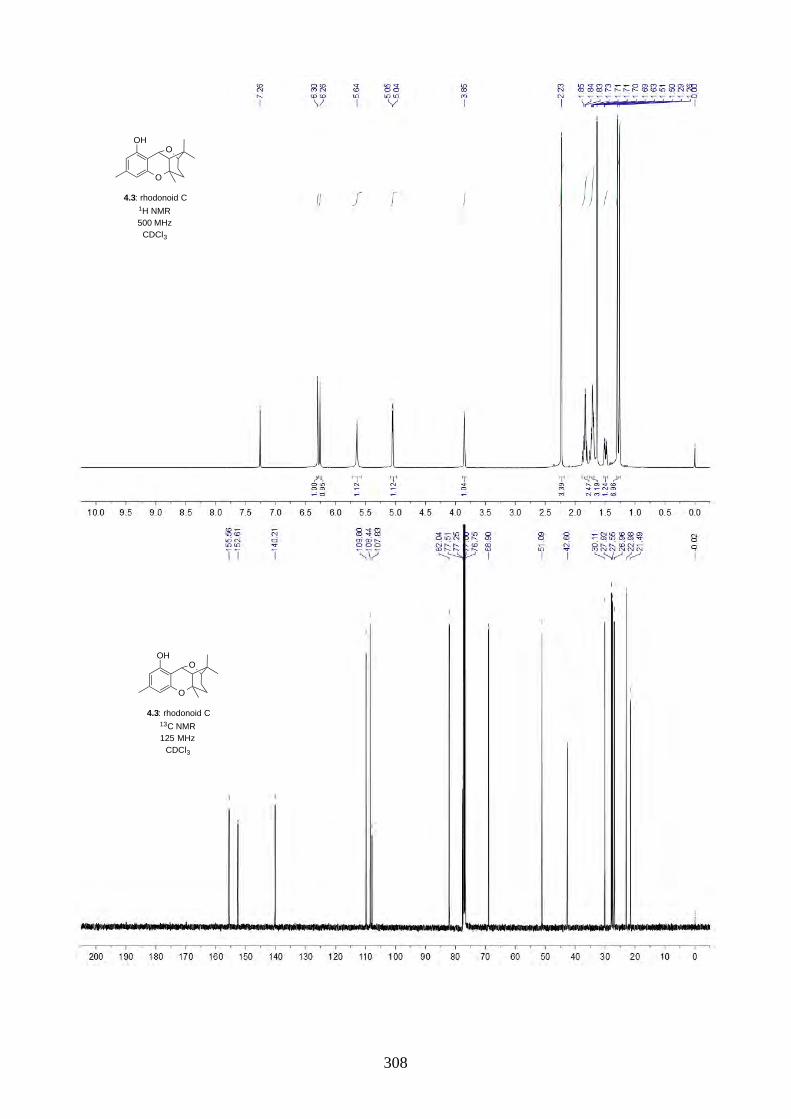
308
4.3: rhodonoid C
O
OOH
1H NMR500 MHz
CDCl3
4.3: rhodonoid C
O
OOH
13C NMR125 MHz
CDCl3
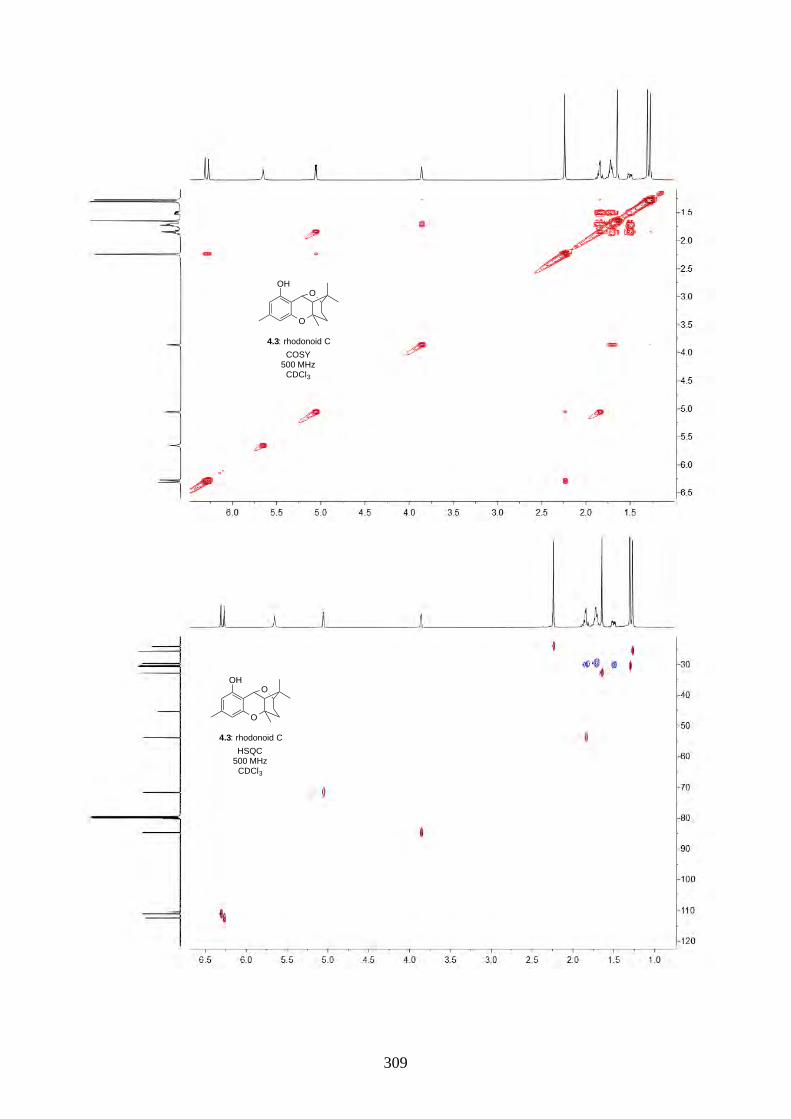
309
4.3: rhodonoid C
O
OOH
COSY500 MHz
CDCl3
4.3: rhodonoid C
O
OOH
HSQC500 MHz
CDCl3
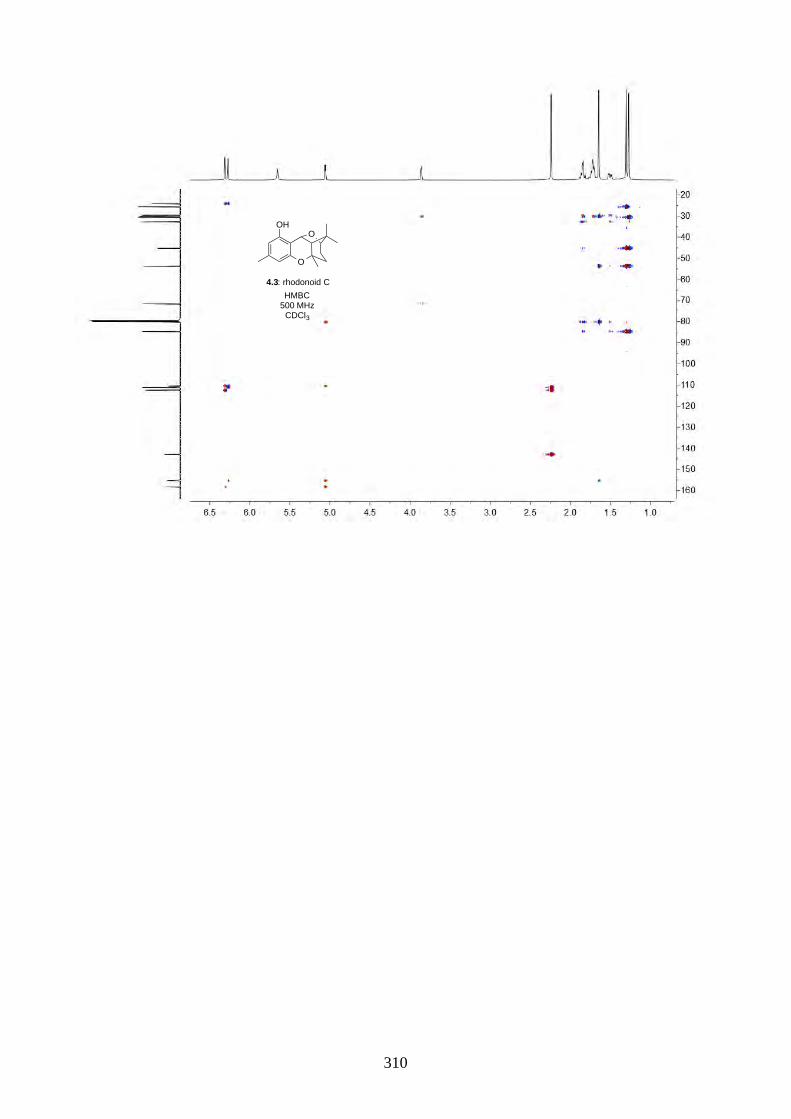
310
4.3: rhodonoid C
O
OOH
HMBC500 MHz
CDCl3
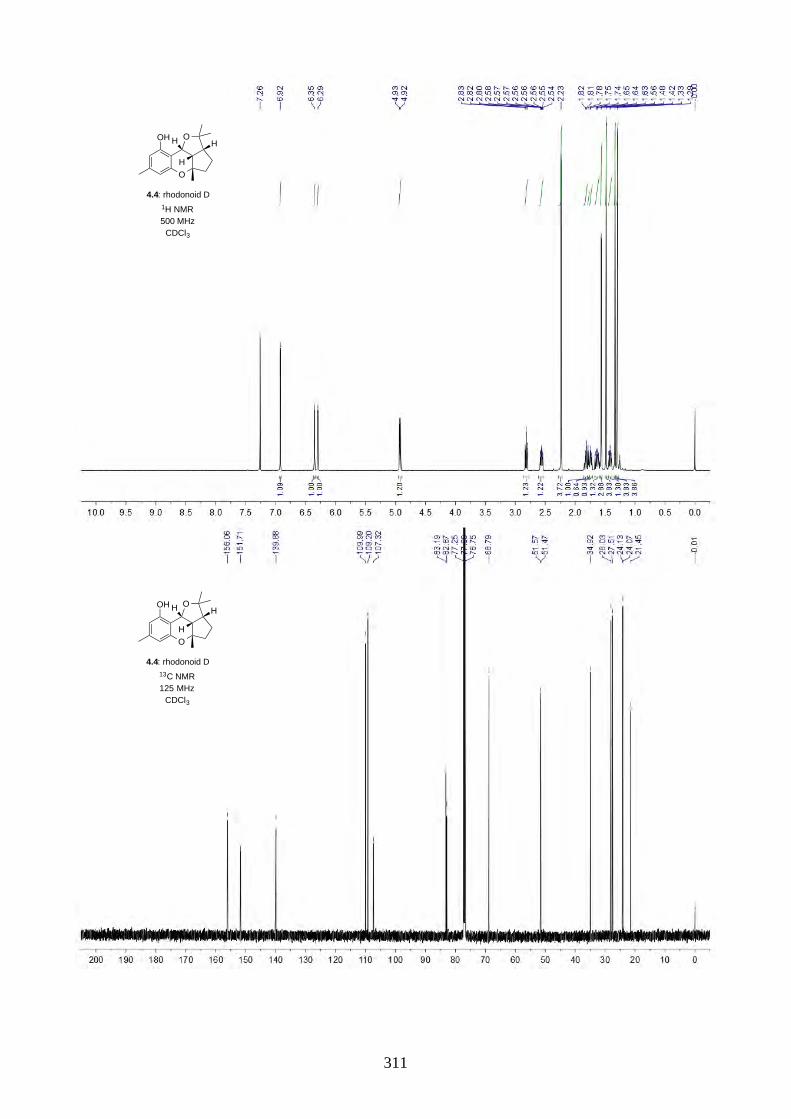
311
O
OH OH
H
H
4.4: rhodonoid D1H NMR500 MHz
CDCl3
O
OH OH
H
H
4.4: rhodonoid D13C NMR125 MHz
CDCl3
312
O
OH OH
H
H
4.4: rhodonoid D
COSY500 MHz
CDCl3
O
OH OH
H
H
4.4: rhodonoid D
HSQC500 MHz
CDCl3
313
O
OH OH
H
H
4.4: rhodonoid D
HMBC500 MHz
CDCl3
O
OH OH
H
H
4.4: rhodonoid D
NOESY500 MHz
CDCl3
314
O
OHO
4.351H NMR500 MHz
CDCl3
O
OHO
4.3513C NMR125 MHz
CDCl3
315
O
OHO
4.35COSY
500 MHzCDCl3
O
OHO
4.35HSQC
500 MHzCDCl3
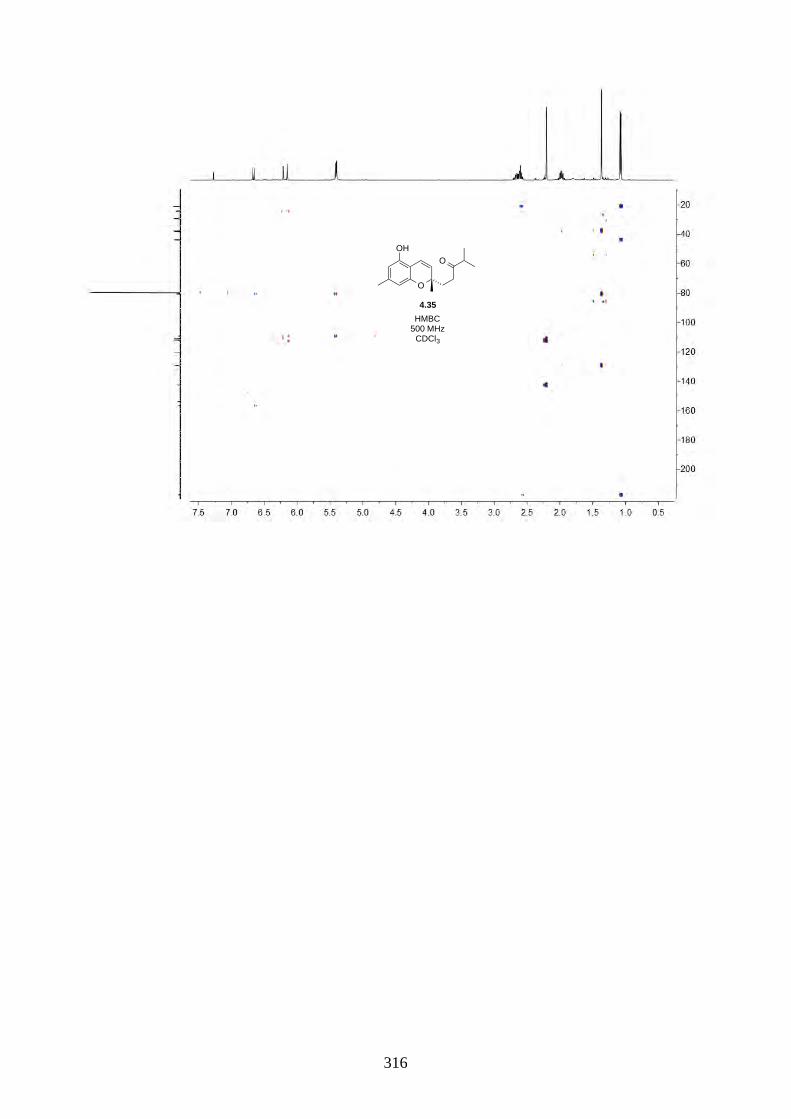
316
O
OHO
4.35HMBC
500 MHzCDCl3
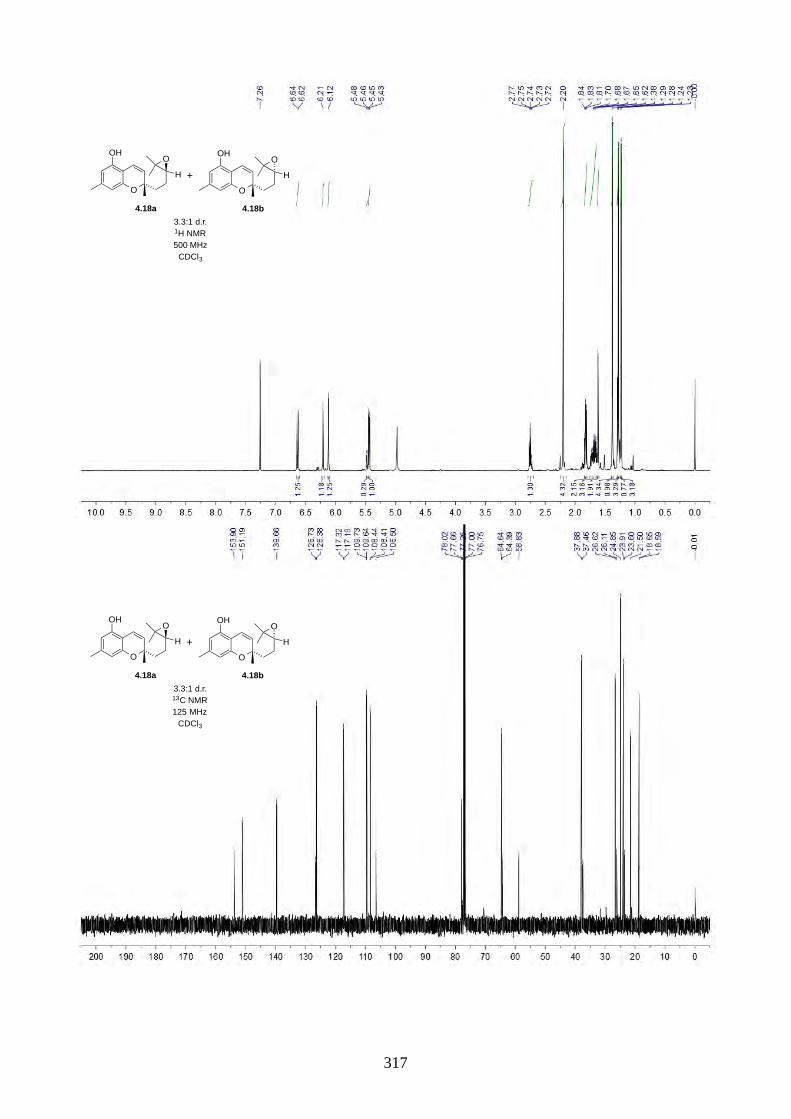
317
O
OHO
H
O
OHO
H +
4.18a3.3:1 d.r.1H NMR500 MHz
CDCl3
4.18b
O
OHO
H
O
OHO
H +
4.18a3.3:1 d.r.13C NMR125 MHz
CDCl3
4.18b
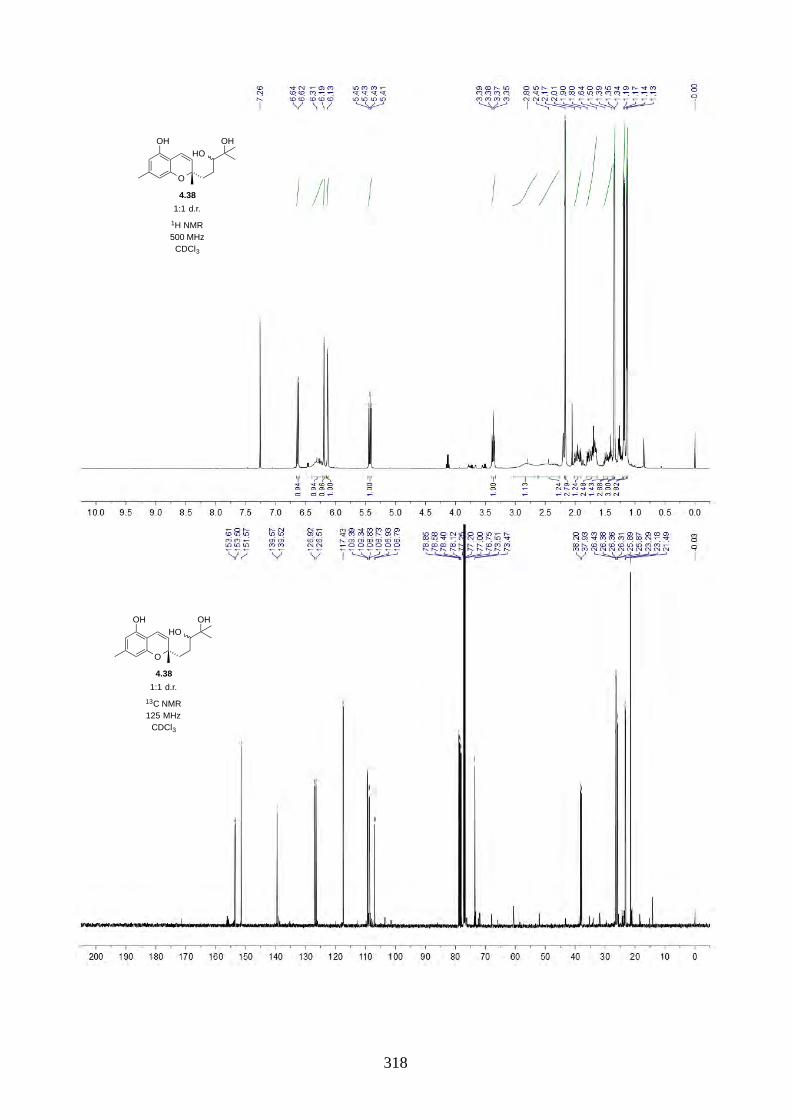
318
O
OHHO
OH
1:1 d.r.4.38
1H NMR500 MHz
CDCl3
O
OHHO
OH
1:1 d.r.4.38
13C NMR125 MHz
CDCl3
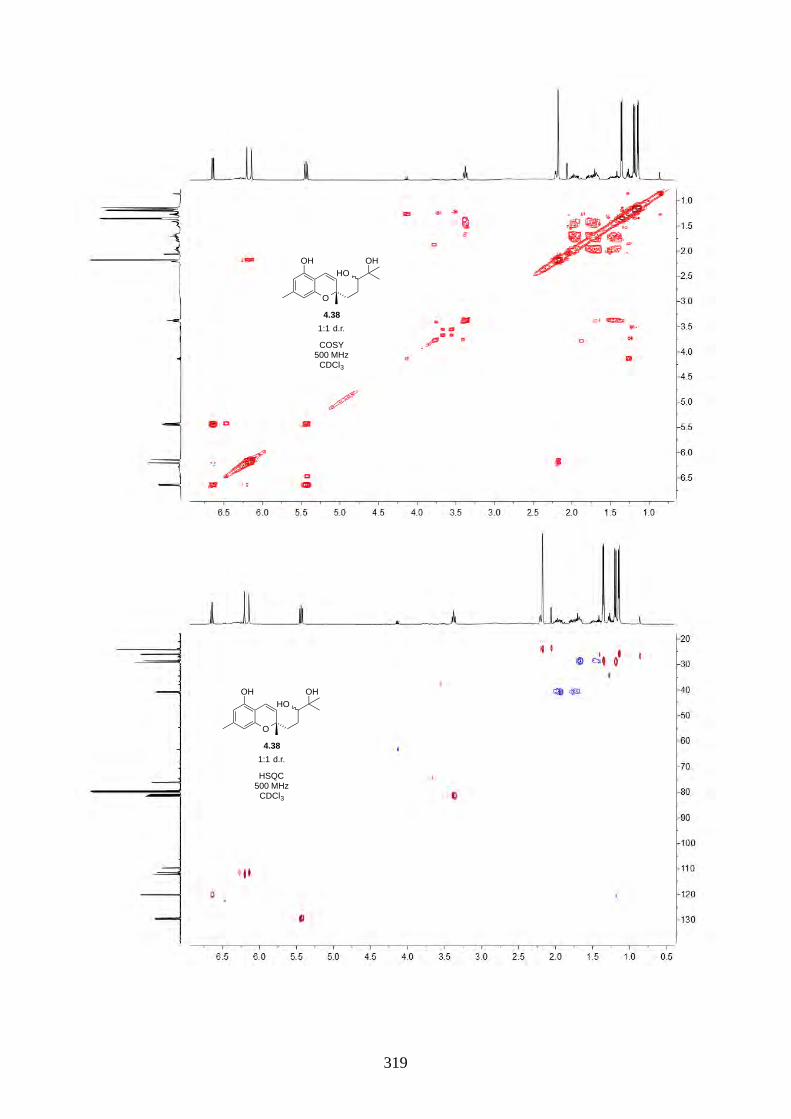
319
O
OHHO
OH
1:1 d.r.4.38
COSY500 MHz
CDCl3
O
OHHO
OH
1:1 d.r.4.38
HSQC500 MHz
CDCl3
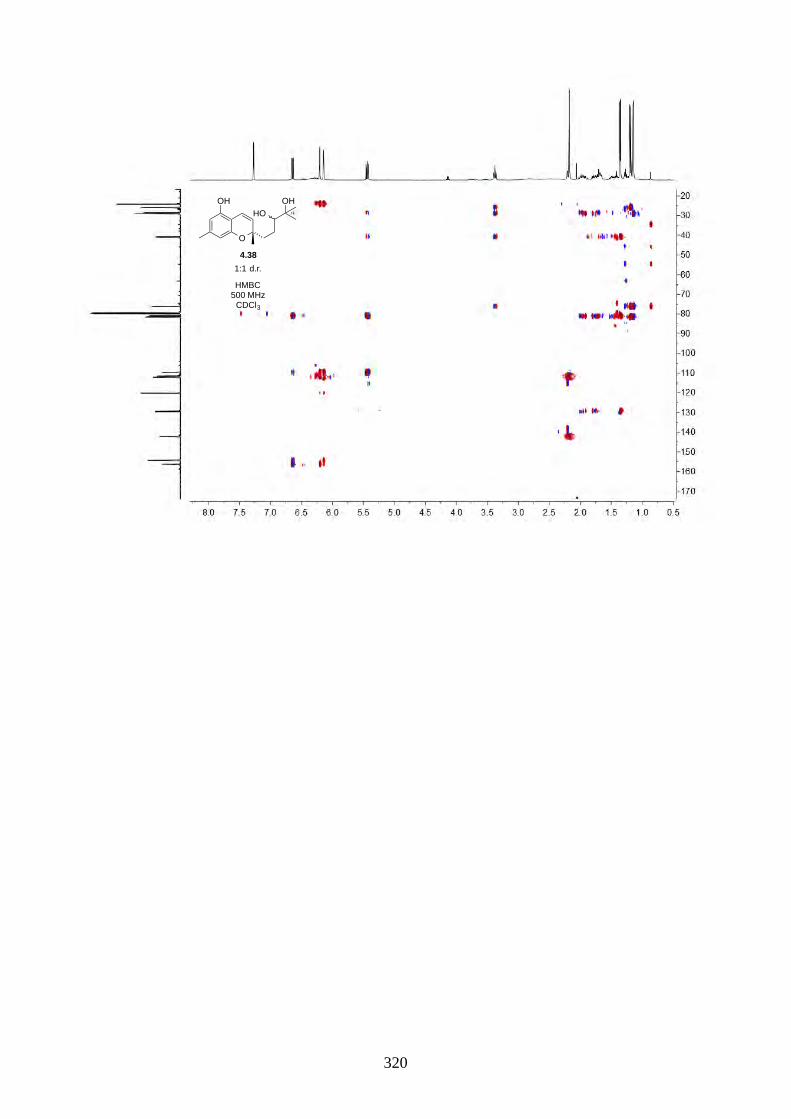
320
O
OHHO
OH
1:1 d.r.4.38
HMBC500 MHz
CDCl3
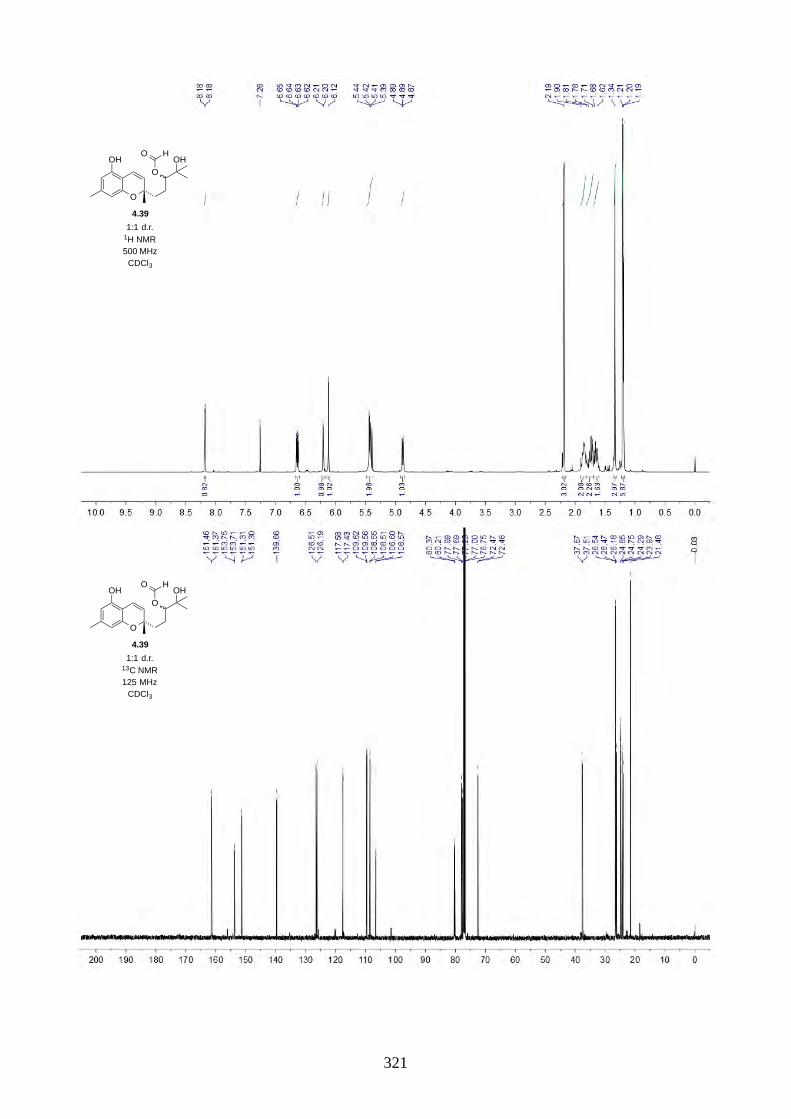
321
O
OHO
OH
1:1 d.r.4.39
O H
1H NMR500 MHz
CDCl3
O
OHO
OH
1:1 d.r.4.39
O H
13C NMR125 MHz
CDCl3
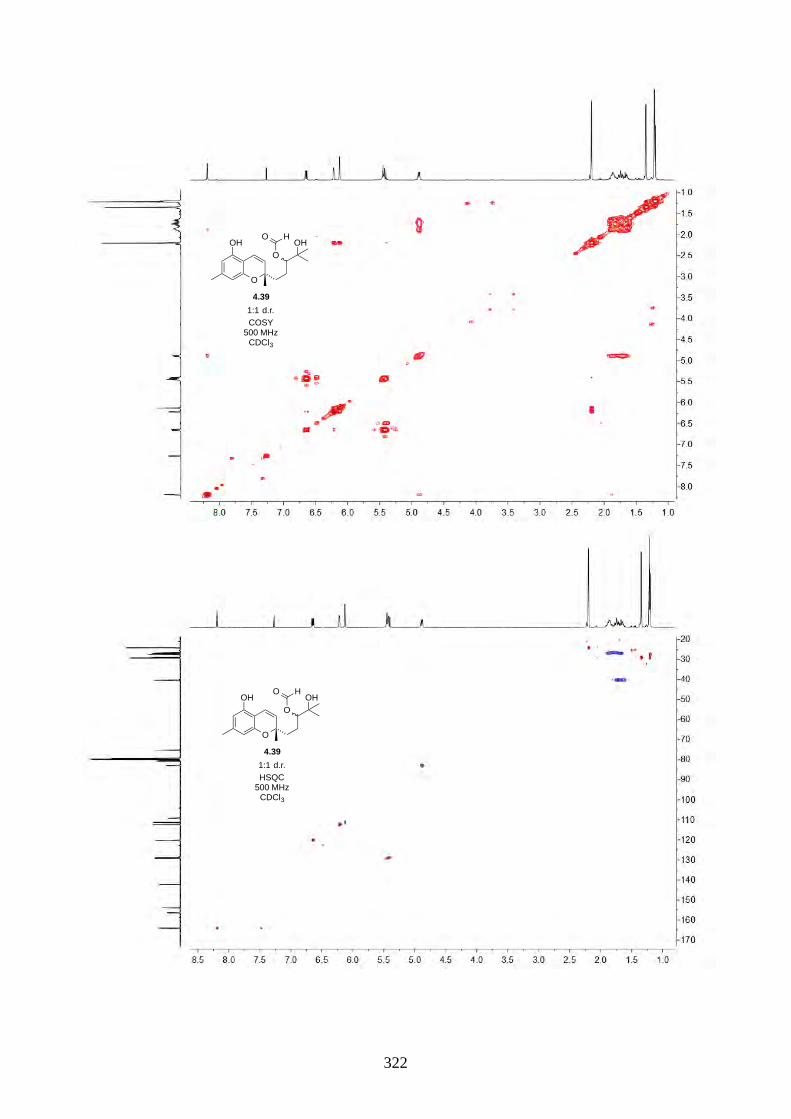
322
O
OHO
OH
1:1 d.r.4.39
O H
COSY500 MHz
CDCl3
O
OHO
OH
1:1 d.r.4.39
O H
HSQC500 MHz
CDCl3
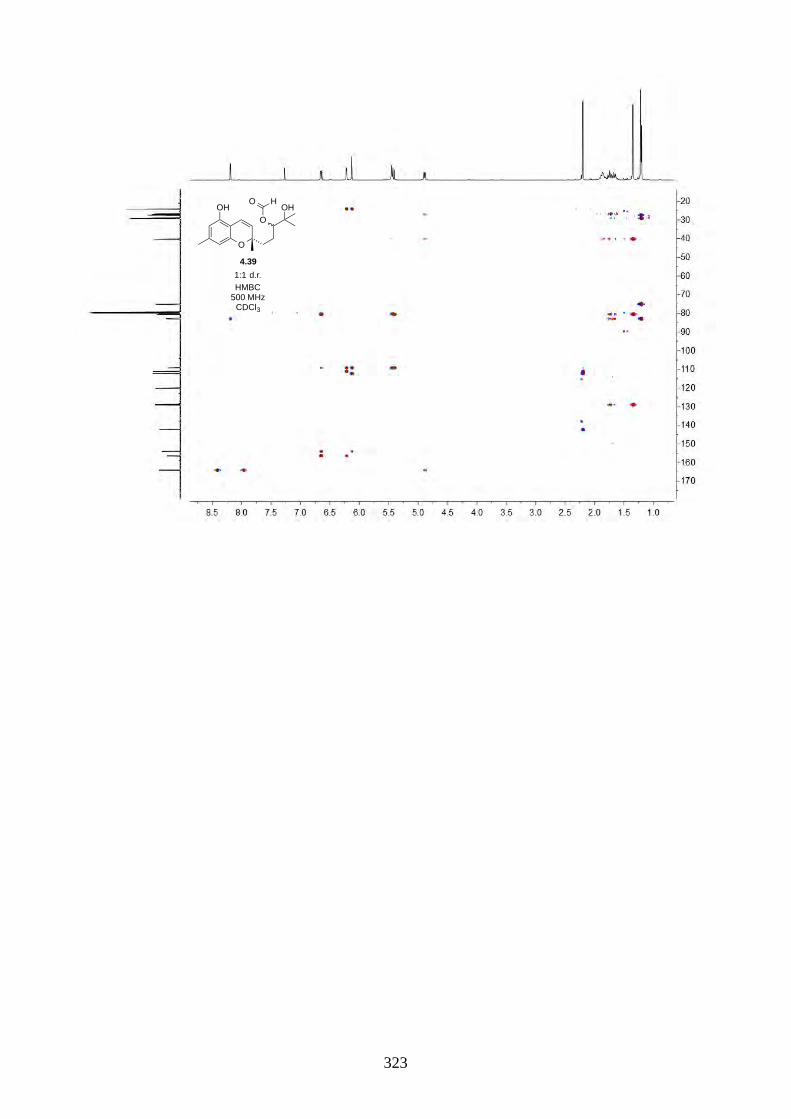
323
O
OHO
OH
1:1 d.r.4.39
O H
HMBC500 MHz
CDCl3
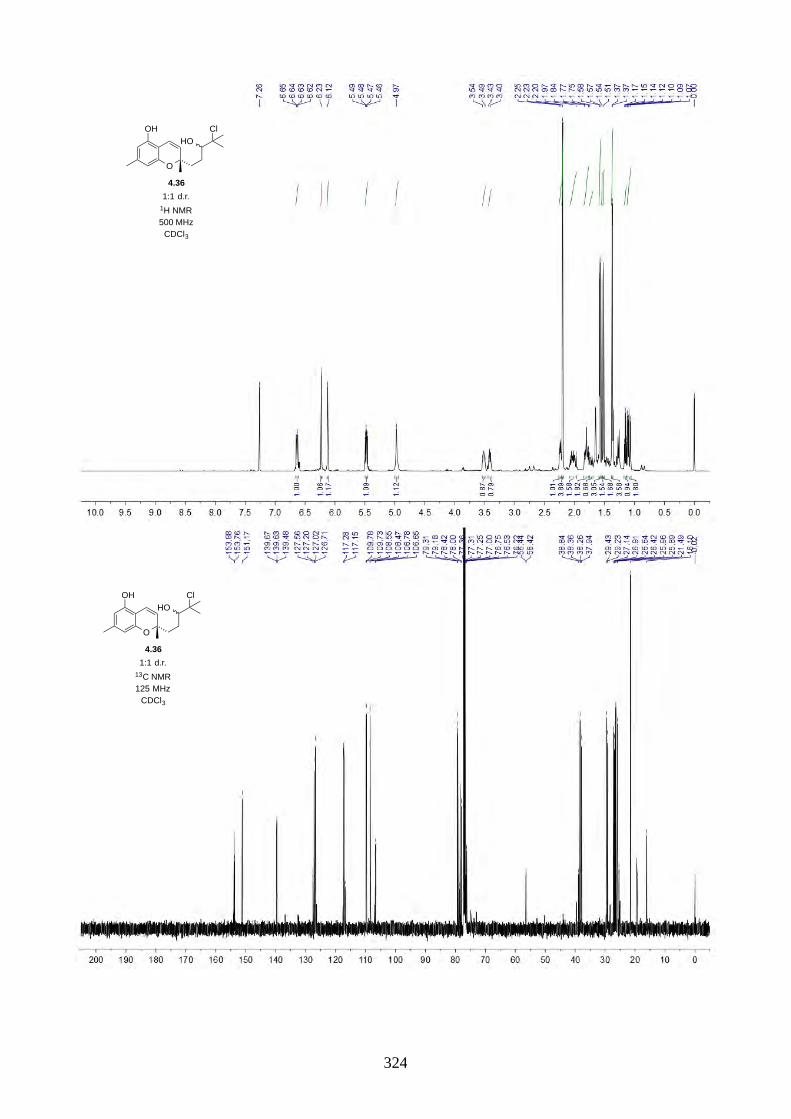
324
O
OHHO
1:1 d.r.4.36
Cl
1H NMR500 MHz
CDCl3
O
OHHO
1:1 d.r.4.36
Cl
13C NMR125 MHz
CDCl3
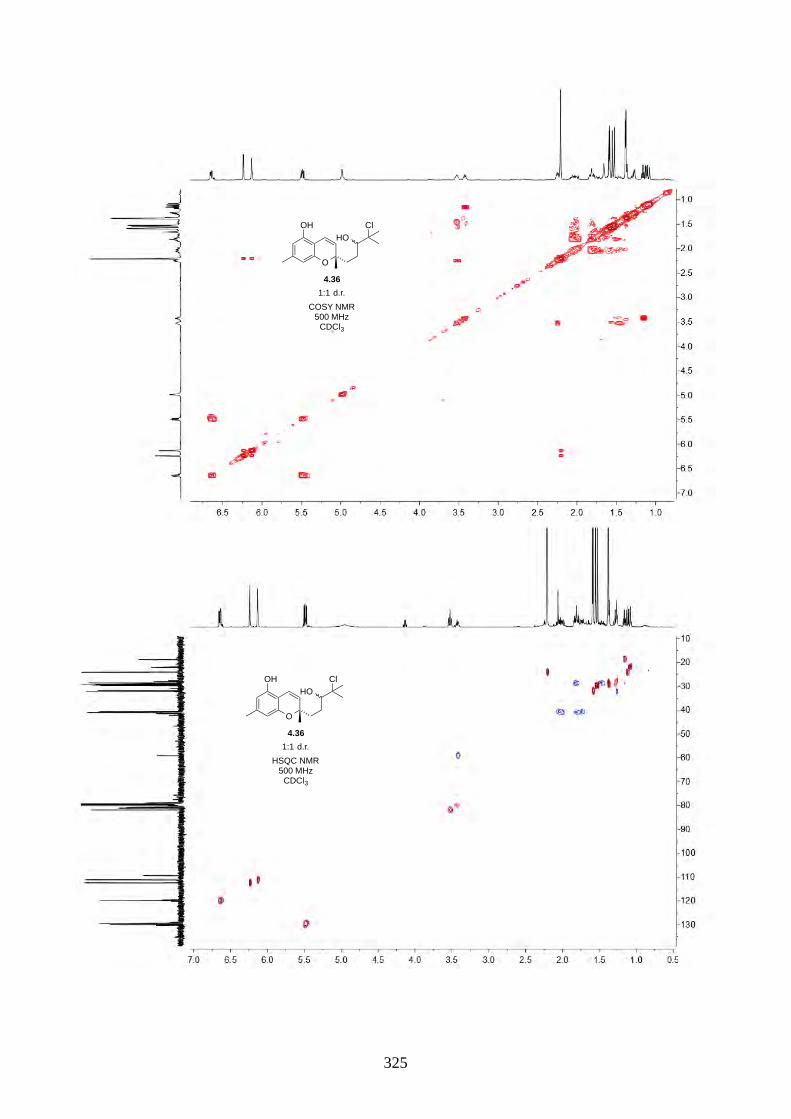
325
O
OHHO
1:1 d.r.4.36
Cl
COSY NMR500 MHz
CDCl3
O
OHHO
1:1 d.r.4.36
Cl
HSQC NMR500 MHz
CDCl3
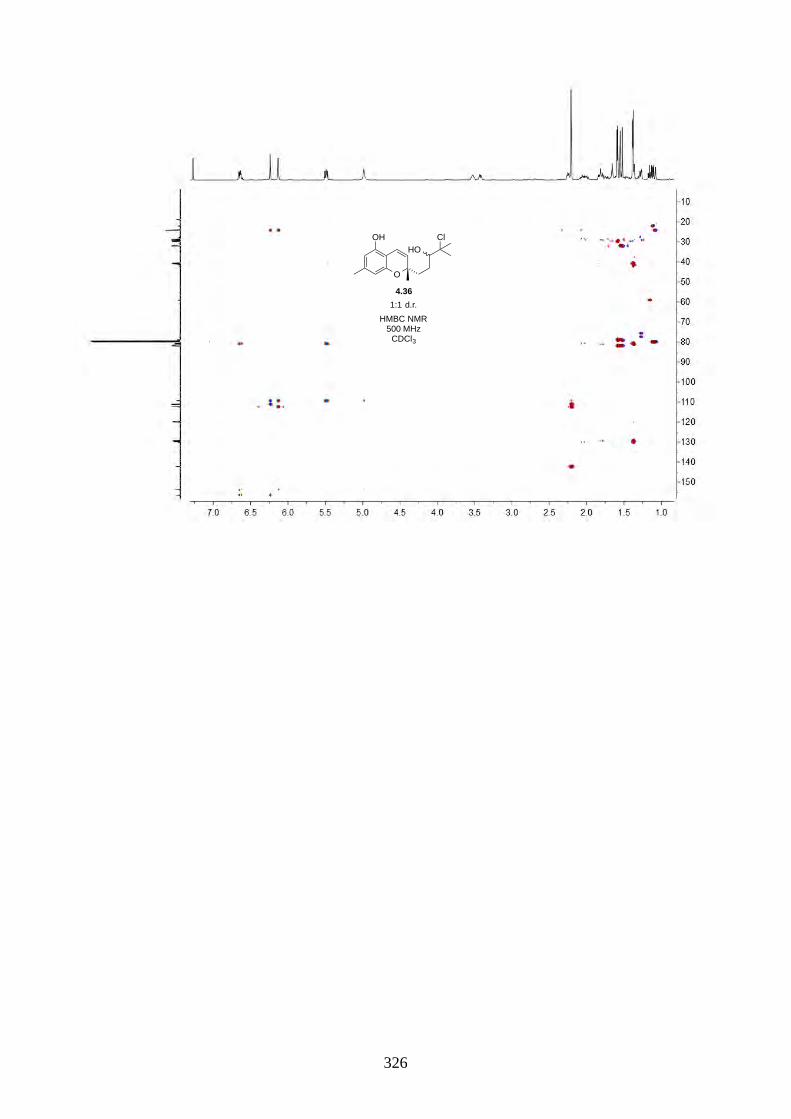
326
O
OHHO
1:1 d.r.4.36
Cl
HMBC NMR500 MHz
CDCl3
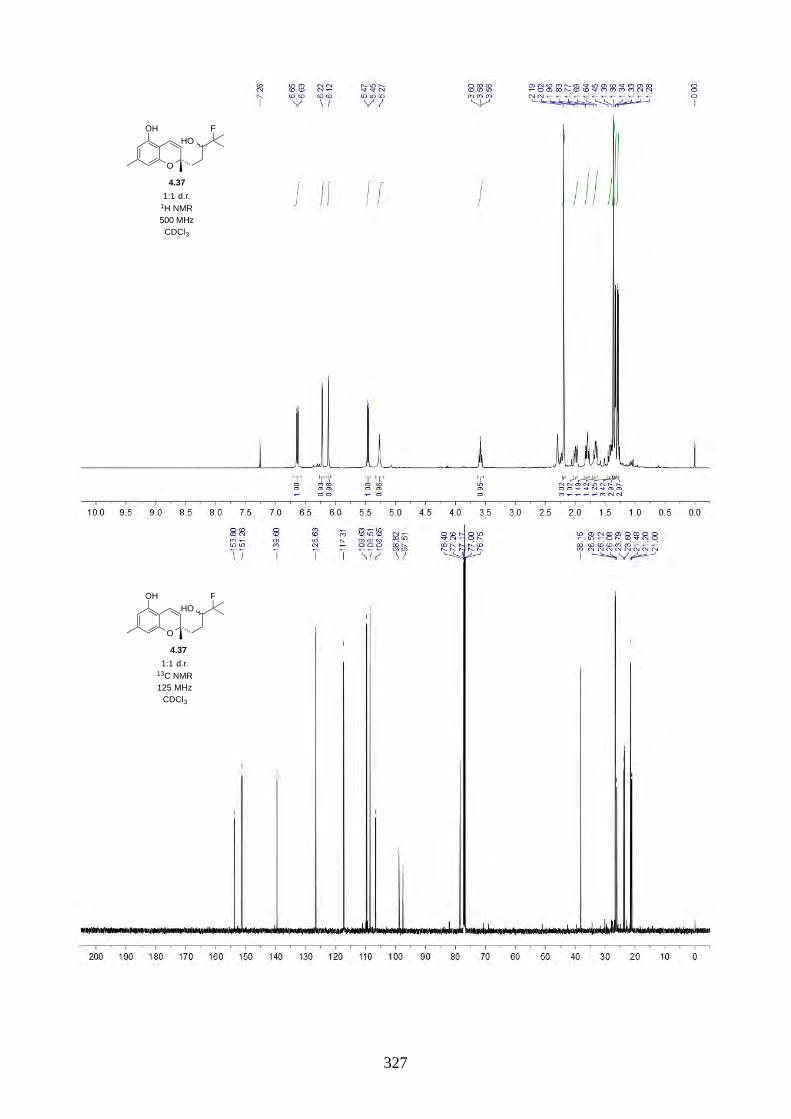
327
O
OHHO
F
1:1 d.r.4.37
1H NMR500 MHz
CDCl3
O
OHHO
F
1:1 d.r.4.37
13C NMR125 MHz
CDCl3
328
O
OHHO
F
1:1 d.r.4.37
COSY500 MHz
CDCl3
O
OHHO
OH
F
1:1 d.r.4.37
HSQC500 MHz
CDCl3
329
O
OHHO
F
1:1 d.r.4.37
HMBC500 MHz
CDCl3
O
OHHO
F
1:1 d.r.4.37
19F NMR470 MHz
CDCl3
330
4.401:1 d.r.
O
OH
HOH
1H NMR500 MHz
CDCl3
4.401:1 d.r.
O
OH
HOH
13C NMR125 MHz
CDCl3
331
4.401:1 d.r.
O
OH
HOH
COSY500 MHz
CDCl3
4.401:1 d.r.
O
OH
HOH
HSQC500 MHz
CDCl3
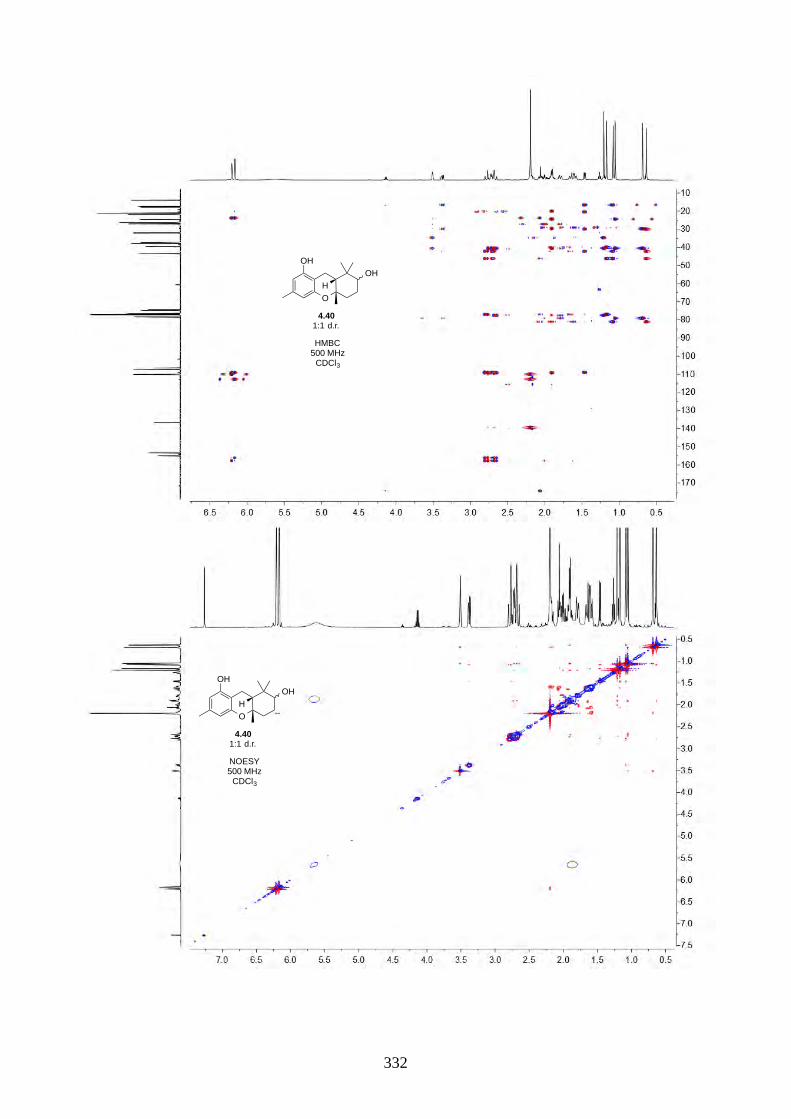
332
4.401:1 d.r.
O
OH
HOH
HMBC500 MHz
CDCl3
4.401:1 d.r.
O
OH
HOH
NOESY500 MHz
CDCl3
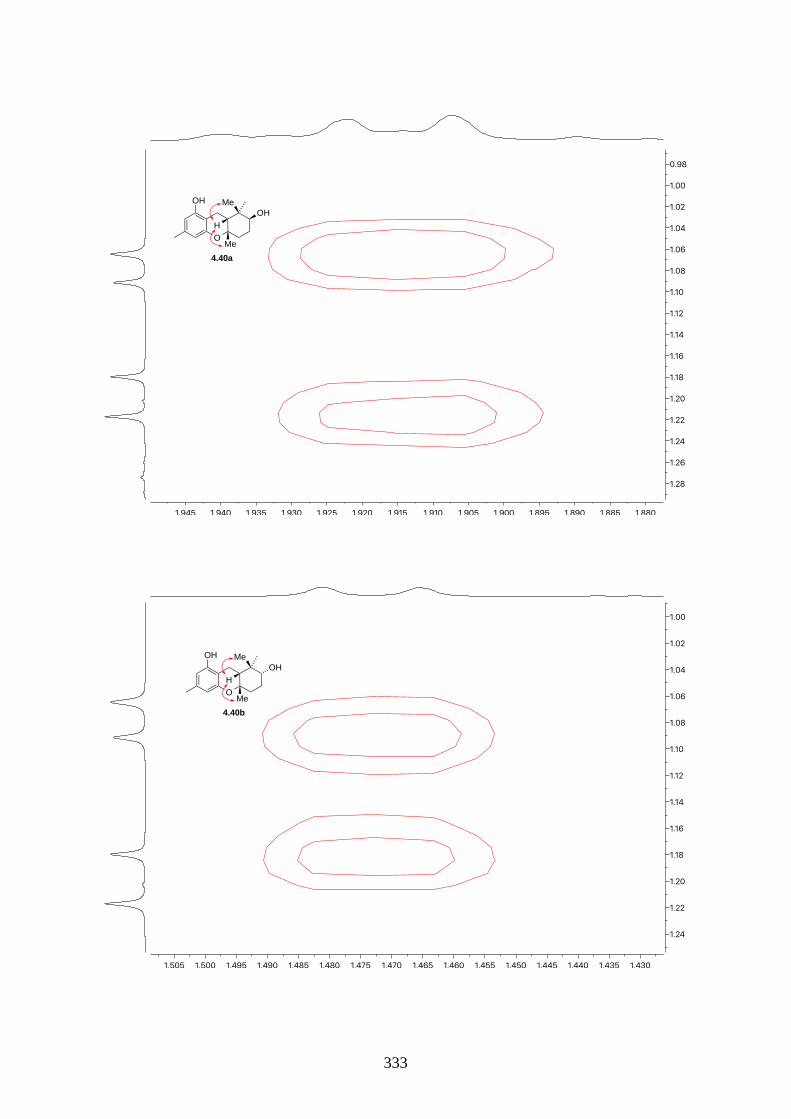
333
4.40a
O
OH MeOH
Me
H
4.40b
O
OH MeOH
Me
H
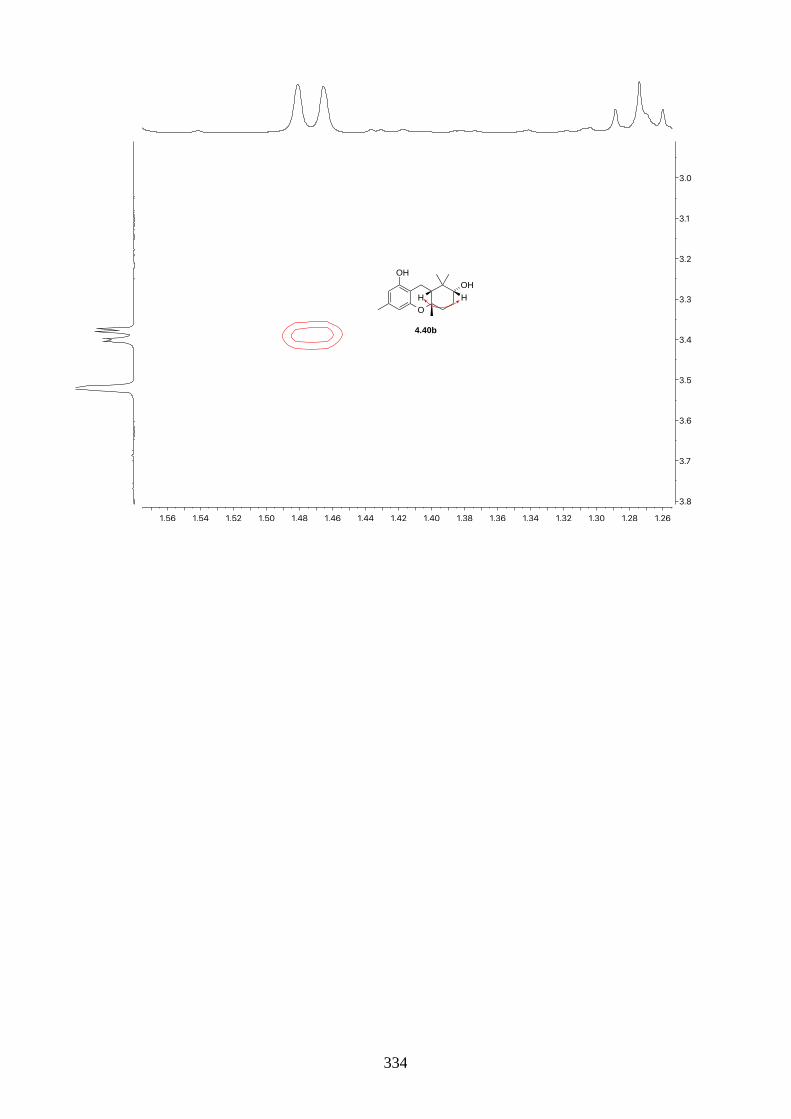
334
4.40b
O
OHOH
H H
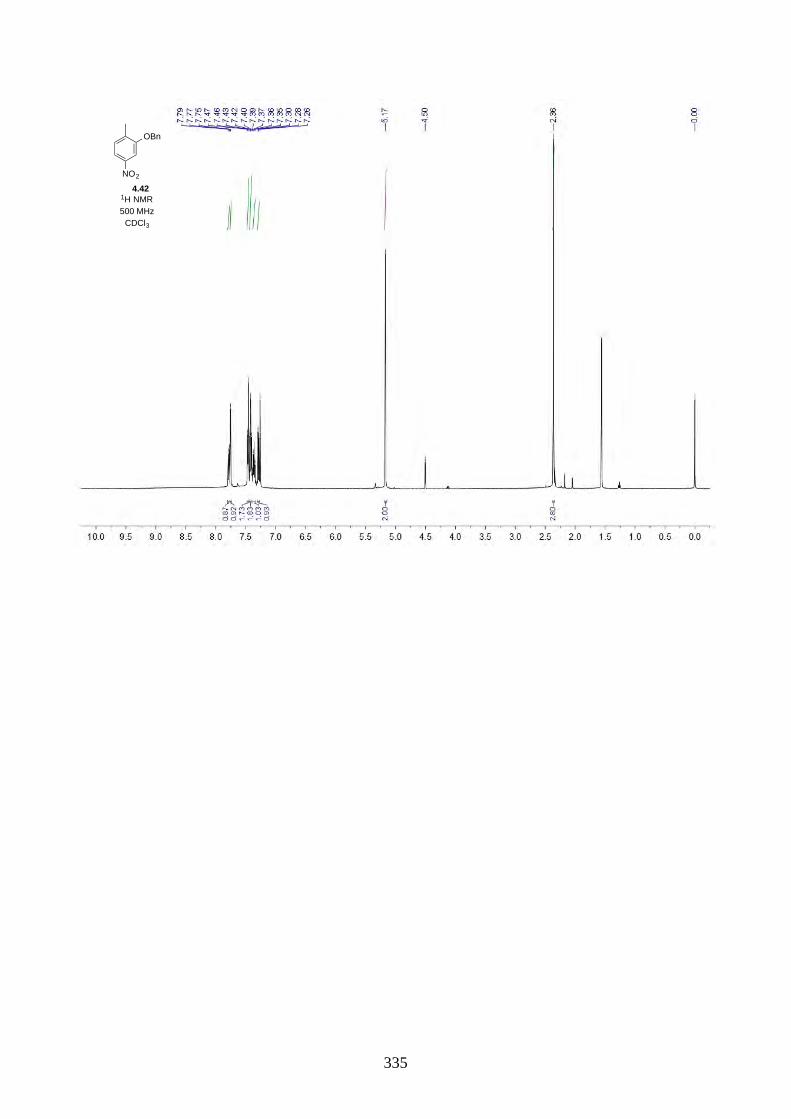
335
NO2
OBn
4.421H NMR500 MHz
CDCl3
336
NH2
OBn
4.431H NMR500 MHz
CDCl3
NH2
OBn
4.4313C NMR125 MHz
CDCl3
337
HN
OBn
4.441H NMR500 MHz
CDCl3
HN
OBn
4.4413C NMR125 MHz
CDCl3
338
4.45
NH
OBn
1H NMR500 MHz
CDCl3
4.45
NH
OBn
13C NMR125 MHz
CDCl3
339
4.46
NH
OH
1H NMR500 MHz
d6-acetone
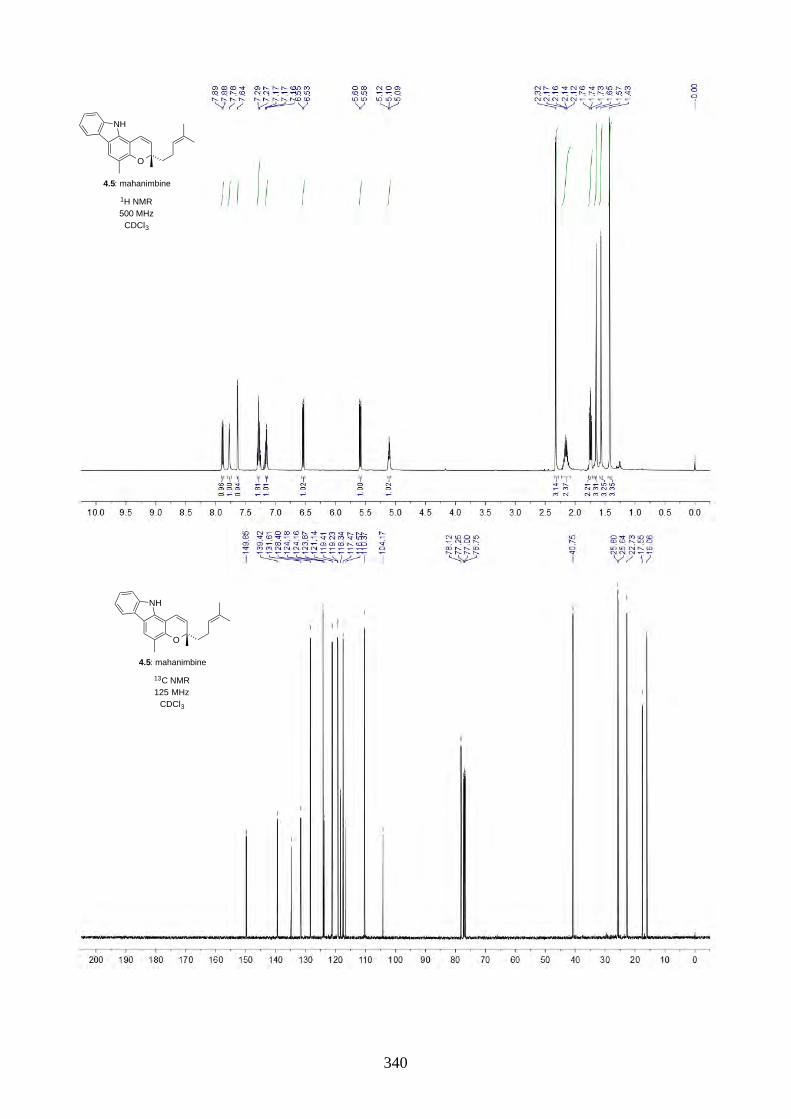
340
O
NH
4.5: mahanimbine
1H NMR500 MHz
CDCl3
O
NH
4.5: mahanimbine
13C NMR125 MHz
CDCl3
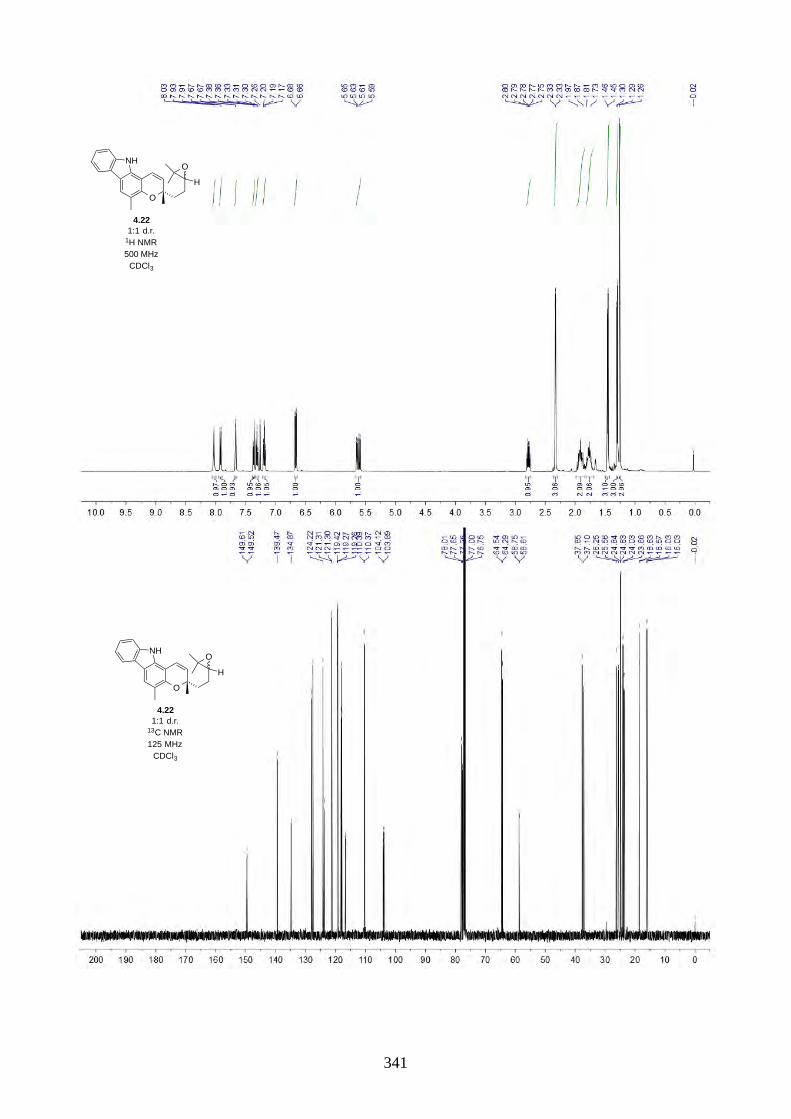
341
1:1 d.r.1H NMR500 MHz
CDCl3
O
NHO
H
4.22
1:1 d.r.13C NMR125 MHz
CDCl3
O
NHO
H
4.22
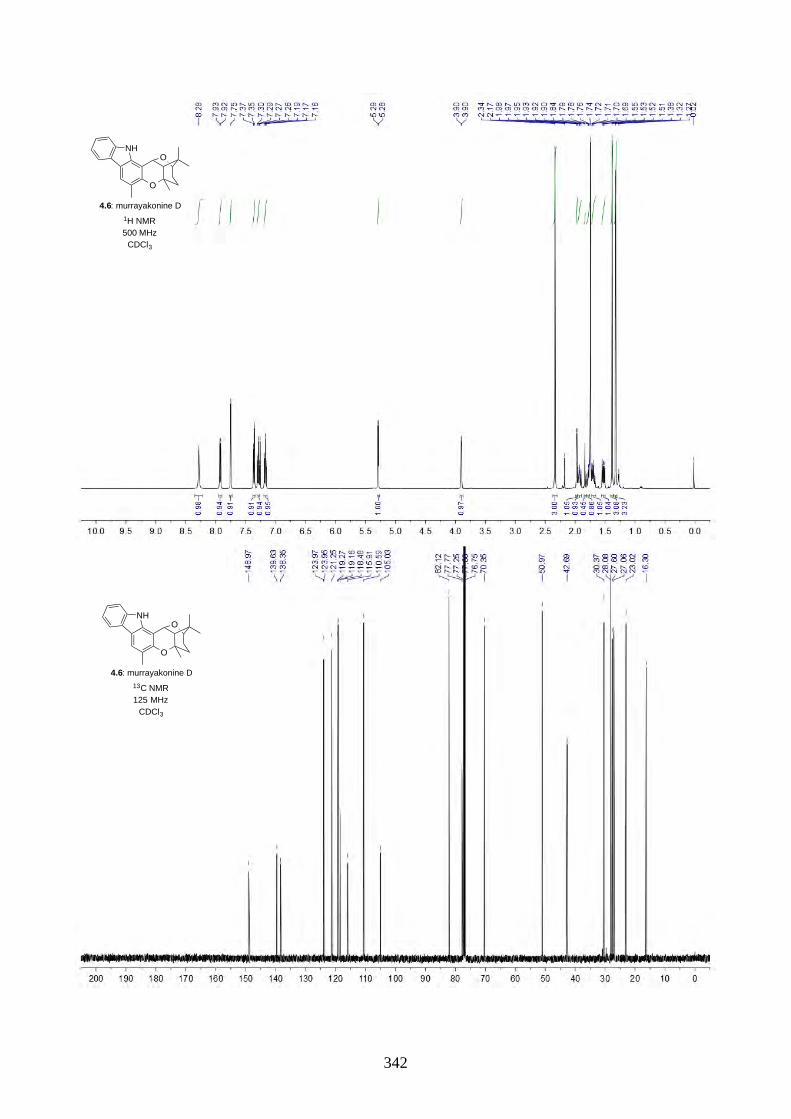
342
4.6: murrayakonine D
O
ONH
1H NMR500 MHz
CDCl3
4.6: murrayakonine D
O
ONH
13C NMR125 MHz
CDCl3
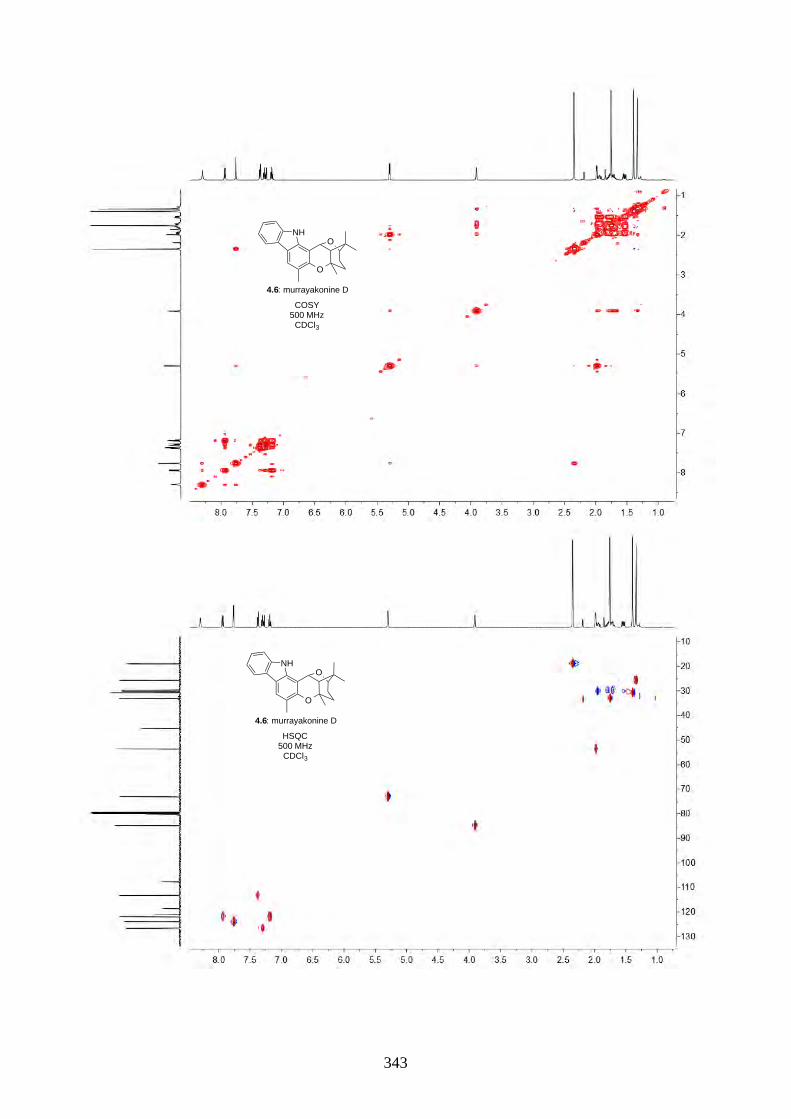
343
4.6: murrayakonine D
O
ONH
COSY500 MHz
CDCl3
4.6: murrayakonine D
O
ONH
HSQC500 MHz
CDCl3
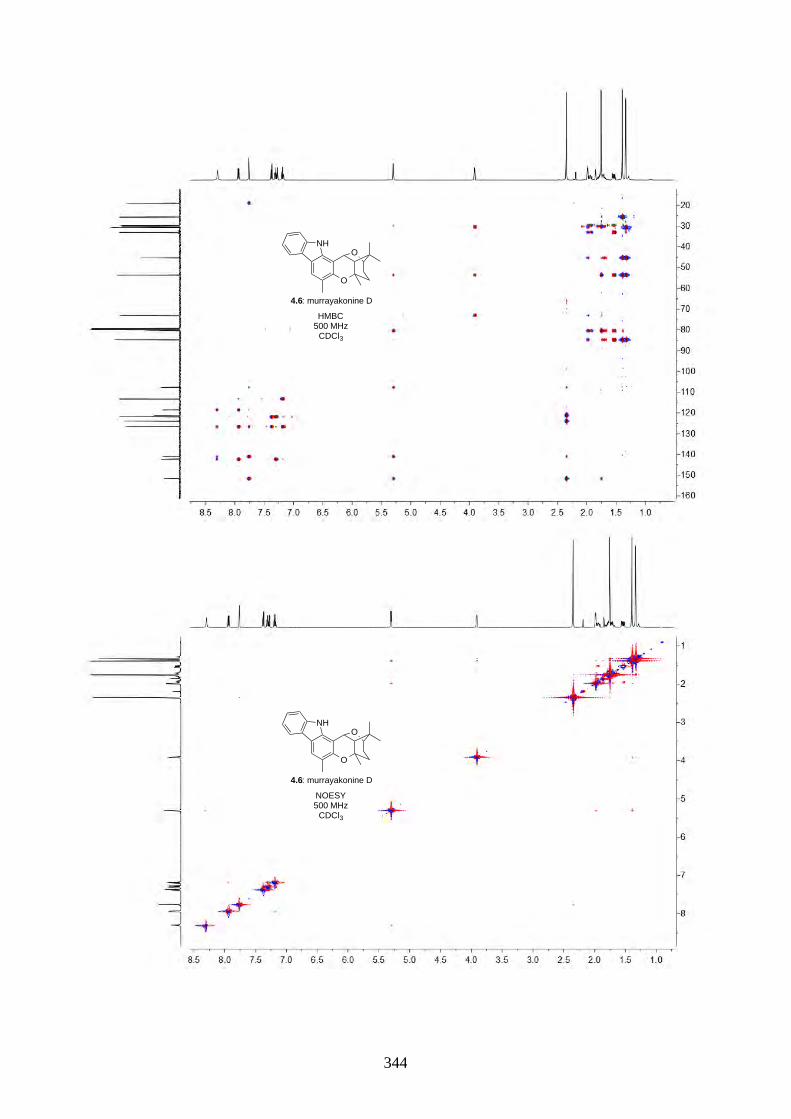
344
4.6: murrayakonine D
O
ONH
HMBC500 MHz
CDCl3
4.6: murrayakonine D
O
ONH
NOESY500 MHz
CDCl3
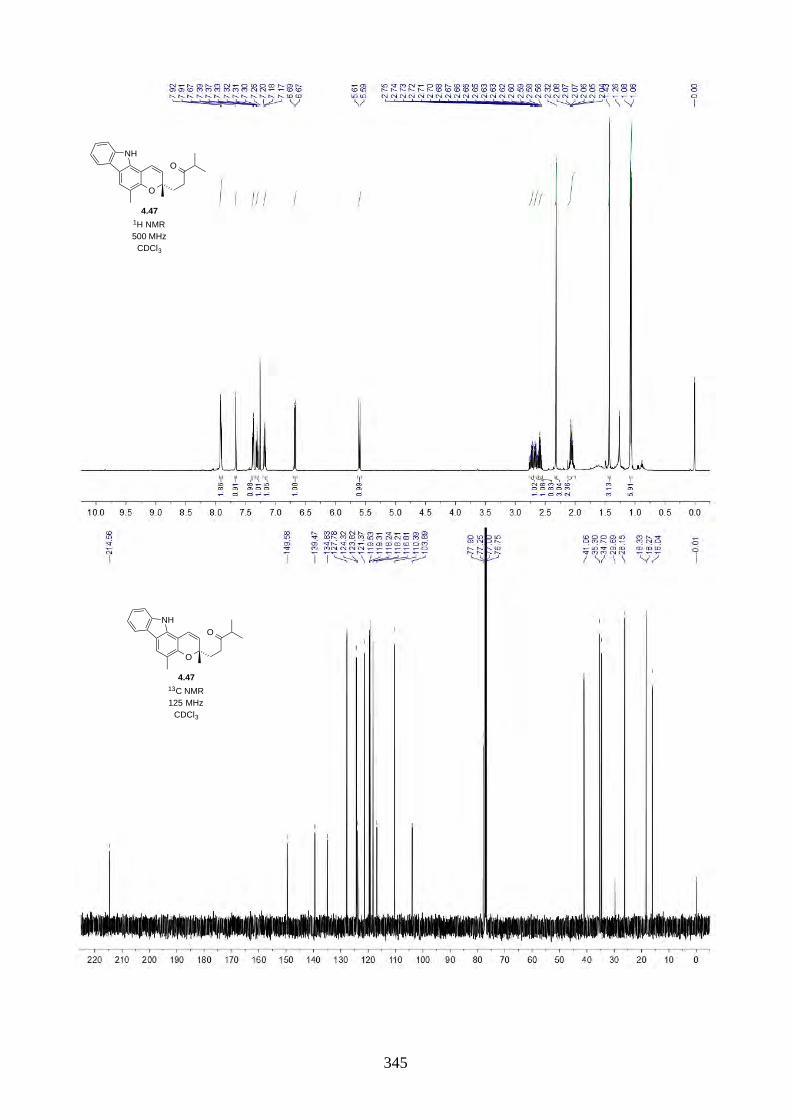
345
O
NHO
4.471H NMR500 MHz
CDCl3
O
NHO
4.4713C NMR125 MHz
CDCl3
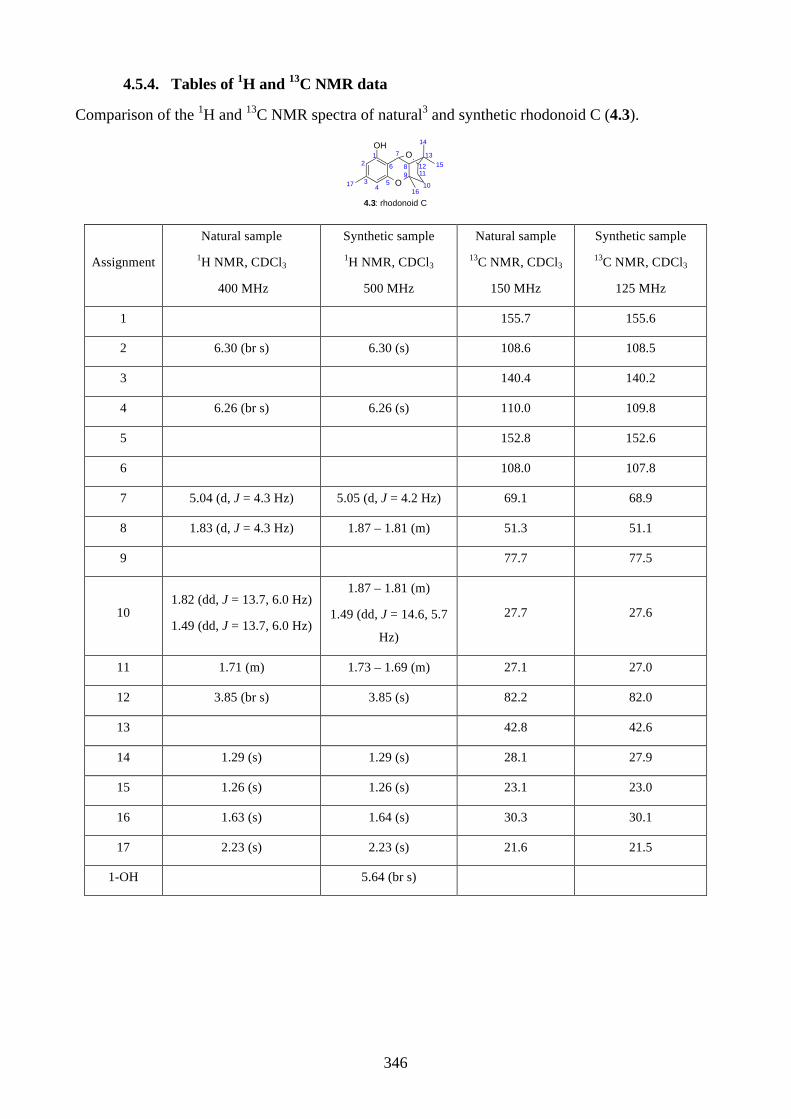
346
4.5.4. Tables of 1H and 13C NMR data
Comparison of the 1H and 13C NMR spectra of natural3 and synthetic rhodonoid C (4.3).
Assignment
Natural sample 1H NMR, CDCl3
400 MHz
Synthetic sample 1H NMR, CDCl3
500 MHz
Natural sample 13C NMR, CDCl3
150 MHz
Synthetic sample 13C NMR, CDCl3
125 MHz
1 155.7 155.6
2 6.30 (br s) 6.30 (s) 108.6 108.5
3 140.4 140.2
4 6.26 (br s) 6.26 (s) 110.0 109.8
5 152.8 152.6
6 108.0 107.8
7 5.04 (d, J = 4.3 Hz) 5.05 (d, J = 4.2 Hz) 69.1 68.9
8 1.83 (d, J = 4.3 Hz) 1.87 – 1.81 (m) 51.3 51.1
9 77.7 77.5
10 1.82 (dd, J = 13.7, 6.0 Hz)
1.49 (dd, J = 13.7, 6.0 Hz)
1.87 – 1.81 (m)
1.49 (dd, J = 14.6, 5.7
Hz)
27.7 27.6
11 1.71 (m) 1.73 – 1.69 (m) 27.1 27.0
12 3.85 (br s) 3.85 (s) 82.2 82.0
13 42.8 42.6
14 1.29 (s) 1.29 (s) 28.1 27.9
15 1.26 (s) 1.26 (s) 23.1 23.0
16 1.63 (s) 1.64 (s) 30.3 30.1
17 2.23 (s) 2.23 (s) 21.6 21.5
1-OH 5.64 (br s)
4.3: rhodonoid C
O
OOH1
3
13
1211
1098
7
6
5
2
4
14
17
15
16
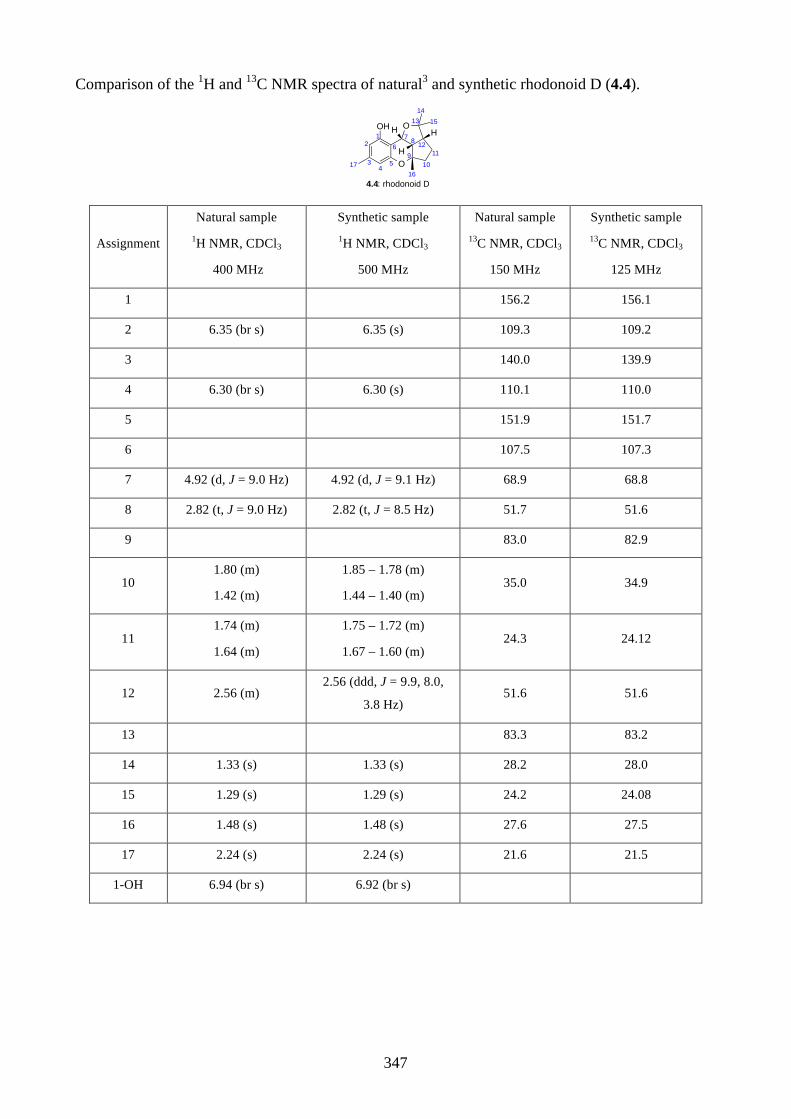
347
Comparison of the 1H and 13C NMR spectra of natural3 and synthetic rhodonoid D (4.4).
Assignment
Natural sample 1H NMR, CDCl3
400 MHz
Synthetic sample 1H NMR, CDCl3
500 MHz
Natural sample 13C NMR, CDCl3
150 MHz
Synthetic sample 13C NMR, CDCl3
125 MHz
1 156.2 156.1
2 6.35 (br s) 6.35 (s) 109.3 109.2
3 140.0 139.9
4 6.30 (br s) 6.30 (s) 110.1 110.0
5 151.9 151.7
6 107.5 107.3
7 4.92 (d, J = 9.0 Hz) 4.92 (d, J = 9.1 Hz) 68.9 68.8
8 2.82 (t, J = 9.0 Hz) 2.82 (t, J = 8.5 Hz) 51.7 51.6
9 83.0 82.9
10 1.80 (m)
1.42 (m)
1.85 – 1.78 (m)
1.44 – 1.40 (m) 35.0 34.9
11 1.74 (m)
1.64 (m)
1.75 – 1.72 (m)
1.67 – 1.60 (m) 24.3 24.12
12 2.56 (m) 2.56 (ddd, J = 9.9, 8.0,
3.8 Hz) 51.6 51.6
13 83.3 83.2
14 1.33 (s) 1.33 (s) 28.2 28.0
15 1.29 (s) 1.29 (s) 24.2 24.08
16 1.48 (s) 1.48 (s) 27.6 27.5
17 2.24 (s) 2.24 (s) 21.6 21.5
1-OH 6.94 (br s) 6.92 (br s)
O
OH OH
H
H
4.4: rhodonoid D
1
3
13
1211
109
876
5
2
4
14
17
15
16
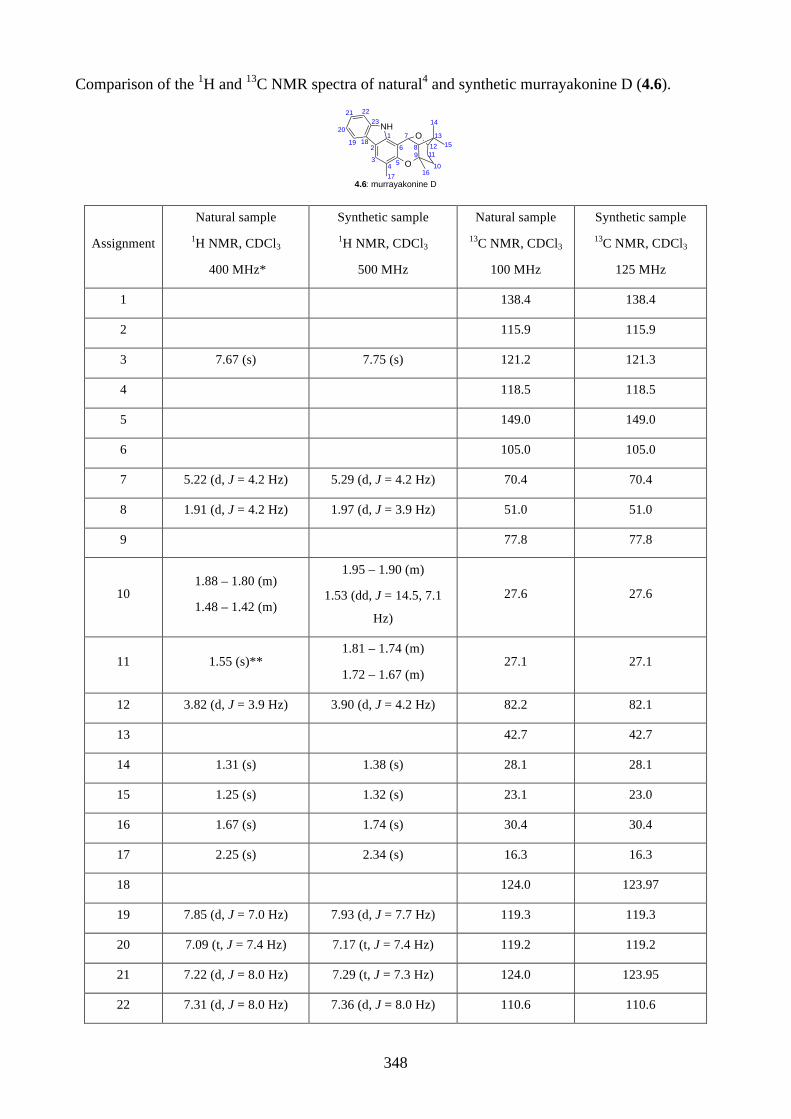
348
Comparison of the 1H and 13C NMR spectra of natural4 and synthetic murrayakonine D (4.6).
Assignment
Natural sample 1H NMR, CDCl3
400 MHz*
Synthetic sample 1H NMR, CDCl3
500 MHz
Natural sample 13C NMR, CDCl3
100 MHz
Synthetic sample 13C NMR, CDCl3
125 MHz
1 138.4 138.4
2 115.9 115.9
3 7.67 (s) 7.75 (s) 121.2 121.3
4 118.5 118.5
5 149.0 149.0
6 105.0 105.0
7 5.22 (d, J = 4.2 Hz) 5.29 (d, J = 4.2 Hz) 70.4 70.4
8 1.91 (d, J = 4.2 Hz) 1.97 (d, J = 3.9 Hz) 51.0 51.0
9 77.8 77.8
10 1.88 – 1.80 (m)
1.48 – 1.42 (m)
1.95 – 1.90 (m)
1.53 (dd, J = 14.5, 7.1
Hz)
27.6 27.6
11 1.55 (s)** 1.81 – 1.74 (m)
1.72 – 1.67 (m) 27.1 27.1
12 3.82 (d, J = 3.9 Hz) 3.90 (d, J = 4.2 Hz) 82.2 82.1
13 42.7 42.7
14 1.31 (s) 1.38 (s) 28.1 28.1
15 1.25 (s) 1.32 (s) 23.1 23.0
16 1.67 (s) 1.74 (s) 30.4 30.4
17 2.25 (s) 2.34 (s) 16.3 16.3
18 124.0 123.97
19 7.85 (d, J = 7.0 Hz) 7.93 (d, J = 7.7 Hz) 119.3 119.3
20 7.09 (t, J = 7.4 Hz) 7.17 (t, J = 7.4 Hz) 119.2 119.2
21 7.22 (d, J = 8.0 Hz) 7.29 (t, J = 7.3 Hz) 124.0 123.95
22 7.31 (d, J = 8.0 Hz) 7.36 (d, J = 8.0 Hz) 110.6 110.6
4.6: murrayakonine D
O
ONH
1
3
131211
10
98
7
6
5
2
4
14
17
15
16
1819
2320
21 22

349
23 139.7 139.6
1-NH 8.21 (s) 8.28 (s)
* 1H spectrum is incorrectly referenced (our chemical shift values for synthetic 4.6 are 0.07-0.09
ppm higher than for natural 4.6).
** Misassigned water peak.

350
4.5.5. Single crystal X-ray data
A single crystal was mounted in paratone-N oil on a plastic loop and X-ray diffraction data were
collected at 150(2) K on an Oxford X-Calibur single crystal diffractometer (λ = 0.71073 Å). Data
was corrected for absorption using a multi-scan method, and the structure solved by direct methods
using SHELXS-975 and refined by full-matrix least squares on F2 by SHELXL-2014,6 interfaced
through the program X-Seed.7 All non-hydrogen atoms were refined anisotropically and hydrogen
atoms were included as invariants at geometrically estimated positions. X-ray experimental data is
given below. CIF data have been deposited with the Cambridge Crystallographic Data Centre,
CCDC reference number CCDC 1538308 (murrayakonine D).
X-Ray experimental data for murrayakonine D (4.6): C23H25NO2, Fw 347.44, monoclinic, I2/a, a
20.969(3), b 10.2106(14), c 17.083(3) Å, β 99.423(16)º, Vol. 3608.3(9) Å3, Z = 8, density (calc.)
1.279 Mg/m3, abs. coefficient 0.081 mm-1, F(000) 1488, crystal size 0.34×0.18×0.11 mm3, θ range
3.36 to 28.47°, reflns collected 12832, Obs. reflns 1910 [R(int) = 0.1658], GoF2 0.974, R1 [I>2σ(I)]
0.0927, wR2 (all data) 0.3054, Largest diff. peak and hole 0.339 & -0.459 e.Å-3.
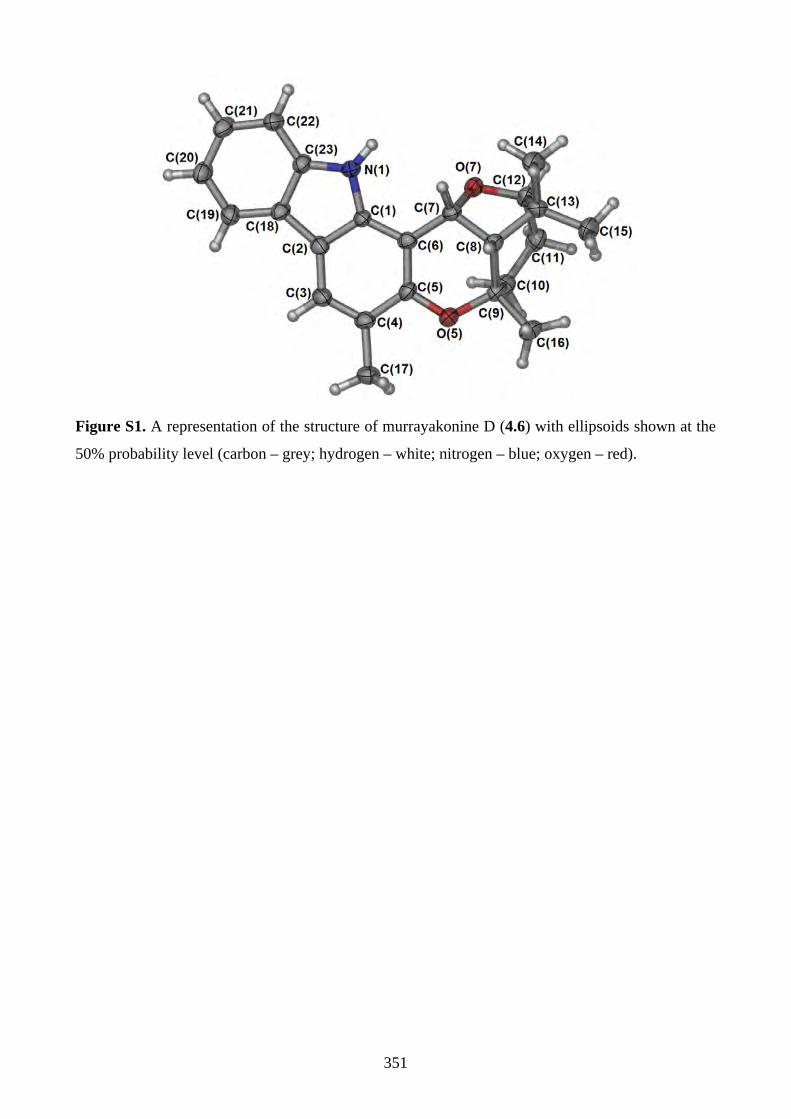
351
Figure S1. A representation of the structure of murrayakonine D (4.6) with ellipsoids shown at the
50% probability level (carbon – grey; hydrogen – white; nitrogen – blue; oxygen – red).

352
4.5.6. References
1. Luo, G. Y.; Wu, H.; Tang, Y.; Li, H.; Yeom, H. S.; Yang, K.; Hsung, R. P. Synthesis, 2015, 47, 2713.
2. Hesse, R.; Gruner, K. K.; Kataeva, O.; Schmidt, A. W.; Knolker, H. J. Chemistry, 2013, 19, 14098.
3. Liao, H. B.; Huang, G. H.; Yu, M. H.; Lei, C.; Hou, A. J. J. Org. Chem., 2017, 82, 1632. 4. Nalli, Y.; Khajuria, V.; Gupta, S.; Arora, P.; Riyaz-Ul-Hassan, S.; Ahmed, Z.; Ali, A. Org.
Biomol. Chem., 2016, 14, 3322. 5. Sheldrick, G. M. Acta Crystallogr A, 1990, 46, 467. 6. Sheldrick, G. M. Acta Crystallographica a-Foundation and Advances, 2015, 71, 3. 7. Barbour, L. J. J. Supramol. Chem., 2003, 1, 189.

353
Chapter 5
Biomimetic Total Synthesis of Yezo’otogirin C
5.1. Introduction
5.1.1. Reductive radical cyclization
Historically, most radical cyclizations in organic chemistry were conducted under reductive
conditions. For example, starting from an alkyl halide (e.g. 5.1), reductive radical initiation
using Bu3Sn· could generate the radical 5.2. 5.2 could undergo 5-exo-trig or 6-endo-trig
cyclization to give 5.3 and 5.4 respectively (Scheme 5.1). Although both reactions are not
forbidden under Baldwin’s rule,1 5-exo-trig cyclization is generally favoured from the kinetic
perspective.2
Scheme 5.1: An illustration of reductive radical cyclization.
The transition state of 5-exo-trig cyclization was proposed by Beckwith2,3,4 and Houk,5 their
calculations and experiments concluded that the chair-like transition state 5.2a is more stable
than the boat-like transition state 5.2b (Figure 5.1).
Figure 5.1: Transition states of 5-exo-trig cyclization.
5.1.2. Oxidative radical cyclization
In contrast to a reductive radical cyclization, oxidative radical cyclization does not require a
halide but often a dicarbonyl group.6,7 The mechanism is shown in Scheme 5.2, the
dicarbonyl 5.7 first binds to a metal ion (e.g. Mn3+), then the enolate 5.8 oxidizes to a radical
intermediate 5.9. The radical is stabilized by resonance effects on both carbonyl groups
(Scheme 5.2). Radical 5.9 then undergoes 5-exo-trig cyclization to give 5.10. Oxidation of
BrBu3Sn
single electronreduction
6-endo-trig
5-exo-trig
single electronreduction
single electronreduction
5.65.35.5
5.45.25.1
chair-like transition state
boat-like transition state
5.2a 5.2b

354
radical 5.10 to the carbocation 5.11, followed by deprotonation to afford 5.12 and thus
terminate the radical cyclization.
Scheme 5.2: Mechanism of oxidative radical cyclization.7
An example of the oxidative radical cyclization is the biomimetic total synthesis of
garcibracteatone (5.18) by the George group (Scheme 5.3).7 The radical cyclization cascade
started from the oxidation of the Δ2,3-enol of weddellianone A (5.13) by Mn(OAc)3 which
gave radical 5.14, then subsequently underwent 7-endo-trig cyclization to the lavandulyl
alkene to give the tertiary radical 5.15. 5-exo-trig cyclization of 5.15 onto the Δ7,8-enol gave
5.16, which then underwent a 5-exo-trig cyclization onto the Δ17,18-alkene to give the tertiary
radical 5.17. 5.17 could either undergo an intramolecular aromatic radical substitution, or the
radical was oxidized to a carbocation which subsequently underwent a Friedel-Crafts reaction
to give gracibracteatone (5.18).8 This biomimetic synthesis of garcibracteatone showcases the
predisposed reactivity of weddellianone A (5.13), where 4 rings, 4 C-C bonds, and 4
stereocenters were formed in 1 step.
Scheme 5.3: Biomimetic total synthesis of garcibracteatone (5.18) by the George group.8
O O
R
O O
R
Mn3+ O O
RMn3+
single electronoxidation
5-exo-trig
O O
R
5.8 5.9 5.9
5.11 5.10 5.9
O O
R
O O
Re.g. Mn3+, Cu2+
deprotonation
5.12
O O
R
single electronoxidation
O O
R
5.7
O
OH
HO
Ph O
OHO O
OH
H
5.18:(±)-garcibracteatone14%
O
O
HO
Ph O
H
O
O
HO
Ph O
H
OPh
O
OH
HOOHO O
OH
H
5.13: weddellianone A
Mn(OAc)3, Cu(OAc)2AcOH, rt, 3 h
single electron oxidation
7-endo-trig
5-exo-trig
5-exo-trig
Intramolecular aromaticradical substitution
orsingle electron
oxidation followed byintramolecularFriedel-Crafts
5.17 5.16
5.14 5.15
2
37
8
18

355
5.1.3. Isolation of yezo’otogirins A-C
Figure 5.2: Yezo’otogirins A-C.9
Yezo’otogirins A-C were isolated from a flowering plant Hypericum yezoense, alongside an
unnamed natural product 5.22 which was proposed to be the biosynthetic precursor of
yezo’otogirin A (5.19) (we later named 5.22 as pre-yezo’otogirin A).9 5.22 was previously
co-isolated with hyperforin (5.23) from Hypericum perforatum (St. John’s Wort).10
Hyperforin (5.23) is the active ingredient in St. John’s Wort which is used as an anti-
depressant.11 We propose hyperforin (5.23), pre-yezo’otogirin A (5.22) and yezo’otogirin A
(5.19) are all biosynthetically linked (Figure 5.2).
O
H
O
H
5.21: yezo'otogirin C
O
H
O
H
5.19: yezo'otogirin A
O
H
O
H
5.20: yezo'otogirin B
O
OH
5.22: pre-yezo'otogirin A
O
O
5.23: hyperforin
O
OH

356
5.1.4. Proposed biosynthesis of yezo’otogirn A (5.19)
Scheme 5.4: Proposed biosynthesis of yezo’otogirin A (5.19).12
The detailed proposed biosynthesis is shown in Scheme 5.4, which begins from the
fragmentation of hyperforin (5.23) to give pre-yezo’otogirin A (5.22). We then propose pre-
yezo’otogirin A (5.22) can be converted into yezo’otogirin A (5.19) via an oxidative radical
cyclization. The cascade reaction could start from a single electron oxidation of the enolate of
pre-yezo’otogirin A (5.22) at C-2 to give radical 5.24. 5-exo-trig cyclization of 5.24 onto the
Δ14,15-alkene could give radical 5.25. For the 5-exo-trig cyclization to occur in radical 5.24, it
must adopt a boat-like transition state to avoid the steric clash between the Δ14,15-alkene and
the isopropyl group. After the cyclization, radical 5.25 could be oxidized to carbocation 5.26,
followed by ring closure by the carbonyl group at C-1 to give yezo’otogirin A (5.19) (Scheme
5.4).
O
OH
5.22: pre-yezo'otogirin A
O
O
5.23: hyperforin
O
OHfragmentation
OO
Beckwith-Houk boat-like
transition state
single electronoxidation
5.19: yezo'otogirin A
ring closuresingle electron
oxidation
5-exo-trig
O
O
HH
O
O
HH
O
O
HH
5.24
5.255.26
1415
1
2

357
5.1.5. Previous biomimetic total synthesis of yezo’otogirin A (5.19)
Scheme 5.5: Biomimetic total synthesis of yezo’otogirin A.12
During my research studies as an honour student, we achieved a biomimetic total synthesis of
yezo’otogirin A (5.19). We started with 5.28, which was prepared from 3-ethoxy-2-
cyclohexenone (5.27) in 2 steps.13 Prenylation of 5.28 gave 5.29 where the prenyl group at C-
6 was in the unnatural relative stereochemistry. Conjugate addition of 5.29 with (4-
methylpent-3-enyl)magnesium bromide followed by aldol reaction with isobutyraldehyde
gave β-hydroxyketone 5.30. Oxidation of 5.30 gave 6-epi-pre-yezo’otogirin A (5.31). With
numerous conditions trialled, 6-epi-pre-yezo’otogirin A (5.31) failed to give yezo’otogirin A
(5.19) by oxidative radical cyclization. Epimerisation of 6-epi-pre-yezo’otogirin A (5.31) to
pre-yezo’otogirin A (5.22) was also unsuccessful.
We then attempted the epimerisation of β-hydroxyketone 5.30. We first protected the β-
hydroxyketone 5.30 using TMSCl, followed by epimerisation by LDA and subsequent
deprotection by TBAF to give 5.32.14 Oxidation of 5.32 using Dess-Martin periodinane gave
pre-yezo’otogirin A (5.22). Pre-yezo’otogirin A (5.22) was then converted to yezo’otogirin A
O
80%
prenyl bromideLDA, THF,–78 °C to rt
O
O
OHH
CuBr, Me2S, THF, 0 °C; then LDA; i-PrCHO
MgBr
5.28 5.29
5.30
O
OH
5.31: 6-epi-pre-yezo'otogirin A
6
O
OHH
5.32
19% over4 steps
Dess-Martin peridinaneNaHCO3, CH2Cl2, rt
36%over 2 steps
O
OH
5.22: pre-yezo'otogirin A
6 Dess-Martin peridinaneNaHCO3, CH2Cl2, rt
67%
5.19: yezo'otogirin A
O
O
HH
Mn(OAc)3Cu(OTf)2
DMF, 150 °C29%
oxidative radical cyclization
cascade
6
6
O
OEt
5.27: 3-ethoxy-2-cyclohexenone
1. LDA, prenyl bromideTHF, –78 °C to rt2. MeLi, THF, –78 °C to rt
82% over 2 steps
1. TMSCl, imidazole, DMF, 0 °C to rt2. LDA, THF, –78 °C to –40 °C3. TBAF, THF, 0 °C

358
(5.19) using Mn(OAc)3 and Cu(OTf)2 in DMF at 150 °C, presumably proceeding via the
oxidative radical cyclization cascade (Scheme 5.5).
5.1.6. Previous bioinspired total synthesis of yezo’otogirin C (5.21) by Lee
Scheme 5.6: Bioinspired total synthesis of yezo’otogirin X (5.21) by Lee.15
While our manuscript on the total synthesis of yezo’otogirin A was in preparation, a
bioinspired total synthesis of yezo’otogirin C (5.21) was reported by Lee.15 Starting from
5.28, methylation of 5.28 gave 5.33. Conjugate addition of 5.33 with (4-methylpent-3-
enyl)magnesium bromide followed by trapping of the enolate with CNCO2Me gave 5.34.
5.34 was then oxidized by Mn(OAc)2 with catalytic co-oxidant Mn(OAc)3 under O2 to give
endoperoide 5.35 in 55% yield. Note this reaction was conducted at relatively mild conditions
(room temperature) compared to our system (150 °C) and the yield of 5.35 was quite high.
This suggests the Lee’s synthetic conditions are potentially more biomimetic than ours.
Reduction and ring closure of 5.35 by thiourea gave 5.36, which contains the skeleton of
yezo’otogirin C (5.19). Functional group conversion of the ester 5.36 to the isobutyryl group
in 4 steps then gave yezo’otogirin C (5.19) (Scheme 5.6).15
O
90%
LDA, MeI, THF,–78 °C to rt
OO
O
OH
CuBr, Me2S, THF, reflux; then CNCO2Me
MgBr
5.28 5.33 5.34
OO
OO
HO
Mn(OAc)2, Mn(OAc)3EtOH, air, rt
5.35
O
H
O
H
5.21: yezo'otogirin C
O
H
O O
H
5.36
55%
thiourea, MeOHreflux
92%60%over 4 steps

359
5.1.7. Aims of this project
Figure 5.3: Aims of this study.
Our aims were to synthesize 6-epi-pre-yezo’otogirin C (5.37) and pre-yezo’otogirin C (5.38)
(a proposed “undiscovered” natural product), and to convert 5.37 and 5.38 into yezo’otogirin
C (5.21) via the oxidative radical cyclization cascade12 or the stepwise cyclization developed
by Lee (Figure 5.3).15
O
OH
5.37: 6-epi-pre-yezo'otogirin C
6
O
OH
6
5.38: pre-yezo'otogirin C
O
H
O
H
5.21: yezo'otogirin C
oxidativeradical cyclization
cascade
orstepwise
cyclizationby Lee
undiscovered natural product?

360
5.2. Results and discussion
5.2.1. Synthesis of 6-epi-pre-yezo’otogirin C (5.37)
Scheme 5.7: Synthesis of ketone 5.28.13
The synthesis started from prenylation of 3-ethoxy-2-cyclohexenone (5.27) which gave 5.39
in quantitative yield, followed by nucleophilic attack of the ketone by MeLi and elimination
to give 5.28 in 82% yield (Scheme 5.7).13 We discovered that it was crucial to use MeLi⋅LiBr
as the reagent for this reaction, whereas using MeLi⋅LiCl would decrease the yield to 50%.
Scheme 5.8: Synthesis of β-hydroxyketone 5.42.
Methylation of 5.28 gave 5.33 as an inseparable mixture of two diastereoisomers in a ratio of
7:1, unfortunately, favouring the isomer with the unnatural relative stereochemistry at C-6
which was also observed by Lee.15 We attempted to epimerise 5.33 at C-6 with various bases
but no reaction was observed. Despite having two diastereoisomers in 5.33, we always
obtained only a single diastereoisomer in the next reaction. Therefore, 5.33 would always be
used as a mixture in the following step.
Conjugate addition of 5.33 with (4-methylpent-3-enyl)magnesium bromide followed by an
aldol reaction of the enolate intermediate 5.40 with isobutyraldehyde (5.41) (Scheme 5.8)
O
OEt
O
OEtLDA, prenyl bromide, THF–78 °C to rt
99%
OMeLi·LiBr, THF–78 °C to rt
82%
5.27: 3-ethoxy-2-cyclohexenone 5.39 5.28
O
71%
LDA, MeI, THF,–78 °C to rt
O
O
OHH
CuBr, Me2S, THF, 0 °C; then LDA; i-PrCHO
MgBr
29%
5.28 5.33
5.421 stereoisomer
d.r. 7:1
6
O
O
H
5.41: isobutyraldehyde
5.40

361
afforded β-hydroxyketone 5.42. We discovered the addition of LDA before isobutyraldehyde
(5.41) would improve the yield of 5.42 to 29%. 5.42 was assigned as an anti-aldol product
because the enolate intermediate 5.40 could only adopt the Z-configuration due to the
restriction of the ring.
Scheme 5.9: Oxidation of 5.42 to 6-epi-yezo’otogirin C (5.37).
The oxidation of 5.42 with Dess-Martin periodinane was a clean reaction and gave 6-epi-
yezo’otogirin C (5.37) in excellent yield (Scheme 5.9). Unfortunately, epimerisation of 5.37
at C-6 to pre-yezo’otogirin C (5.38) was unsuccessful. We were aware the proton H-2 is most
likely to be deprotonated by base, and we attempted to use excess of LDA or n-BuLi to
achieve a secondary deprotonation at H-6 but this led to the decomposition of 5.37.16
5.2.2. Synthesis of yezo’otogirin C (5.21) from 6-epi-pre-yezo’otogirin C (5.37)
Scheme 5.10: Synthesis of yezo’otogirin C (5.21).
Despite the previous failure in the radical cyclization of 6-epi-pre-yezo’otogirin A (5.31) to
yezo’otogirin A (5.19), we were optimistic that the cyclization might occur with 6-epi-pre-
yezo’otogirin C (5.37). As the methyl group at C-6 is less bulky than a prenyl group, it is
more likely for the radical cyclization to overcome the interference from a methyl group in
the transition state 5.44 (Scheme 5.11). Indeed, when 6-epi-pre-yezo’otogirin C (5.37) was
heated with Mn(OAc)3 and Cu(OTf)2 in DMF, we observed a 5% yield of yezo’otogirin C
(5.21) (Scheme 5.10). The NMR of yezo’otogirin C matched the isolation data.9 We also
observed 5% yield of 5.43, presumably formed from the oxidative radical cyclization to give
intermediate 5.45. However, the cyclization of 5.46 did not take place from the enol at C-1 to
O
OH
Dess-Martin periodinane,NaHCO3, CH2Cl2, rt
O
OHH
92%
5.42
O
OH
5.38: pre-yezo'otogirin C
base
epimersation
5.37: 6-epi-pre-yezo'otogirin C
662
O
H
O
H
O
OH Mn(OAc)3·2H2O, Cu(OTf)2,
DMF, 150 °C
5% 5%
5.21: yezo'otogirin C 5.43
+
5.37: 6-epi-pre-yezo'otogirin C
O6
14
1 O7
2
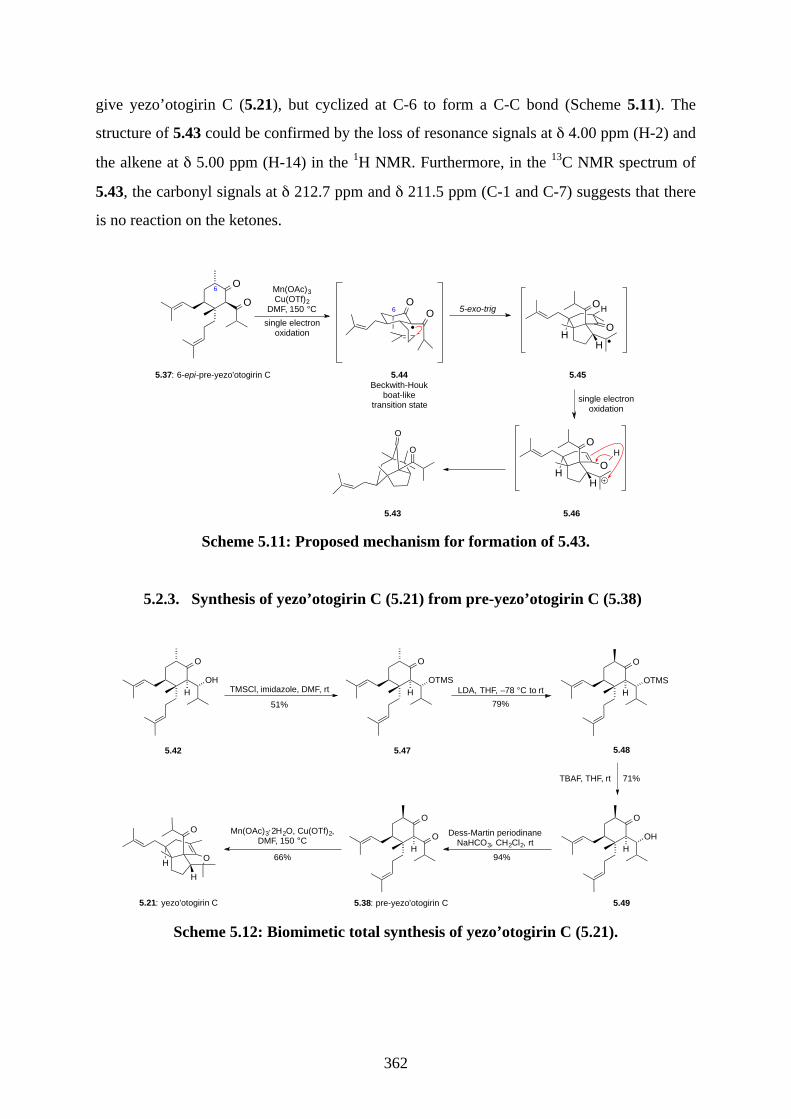
362
give yezo’otogirin C (5.21), but cyclized at C-6 to form a C-C bond (Scheme 5.11). The
structure of 5.43 could be confirmed by the loss of resonance signals at δ 4.00 ppm (H-2) and
the alkene at δ 5.00 ppm (H-14) in the 1H NMR. Furthermore, in the 13C NMR spectrum of
5.43, the carbonyl signals at δ 212.7 ppm and δ 211.5 ppm (C-1 and C-7) suggests that there
is no reaction on the ketones.
Scheme 5.11: Proposed mechanism for formation of 5.43.
5.2.3. Synthesis of yezo’otogirin C (5.21) from pre-yezo’otogirin C (5.38)
Scheme 5.12: Biomimetic total synthesis of yezo’otogirin C (5.21).
O
O
5.37: 6-epi-pre-yezo'otogirin C
O
O
HH
5.465.43
6
O
O
OO6
Beckwith-Houk boat-like
transition state
5.44
single electronoxidation
O
O
HH
H
5.45
5-exo-trig
H
single electronoxidation
Mn(OAc)3Cu(OTf)2
DMF, 150 °C
O
OHH TMSCl, imidazole, DMF, rt
O
OTMSH
51%
LDA, THF, –78 °C to rt
O
OTMSH
79%
TBAF, THF, rt
O
OHH
71%
Dess-Martin periodinaneNaHCO3, CH2Cl2, rt
O
OH
94%O
H
O
H
Mn(OAc)3·2H2O, Cu(OTf)2, DMF, 150 °C
66%
5.42 5.47 5.48
5.495.21: yezo'otogirin C 5.38: pre-yezo'otogirin C
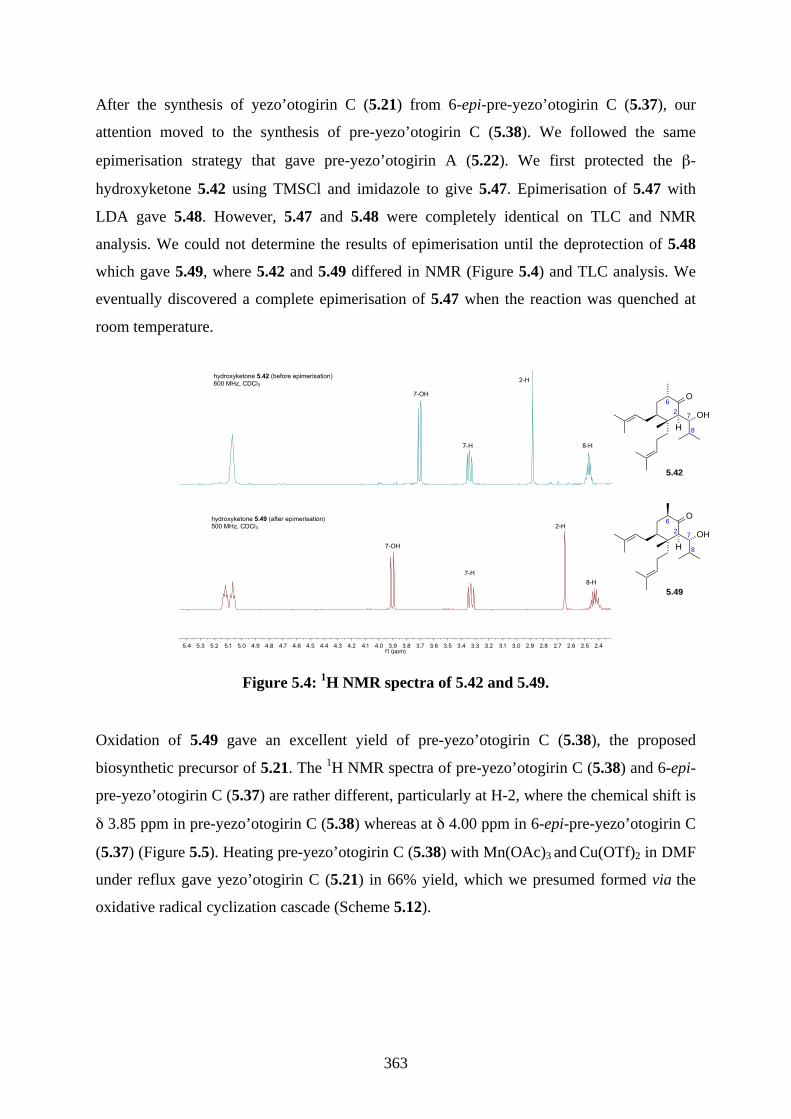
363
After the synthesis of yezo’otogirin C (5.21) from 6-epi-pre-yezo’otogirin C (5.37), our
attention moved to the synthesis of pre-yezo’otogirin C (5.38). We followed the same
epimerisation strategy that gave pre-yezo’otogirin A (5.22). We first protected the β-
hydroxyketone 5.42 using TMSCl and imidazole to give 5.47. Epimerisation of 5.47 with
LDA gave 5.48. However, 5.47 and 5.48 were completely identical on TLC and NMR
analysis. We could not determine the results of epimerisation until the deprotection of 5.48
which gave 5.49, where 5.42 and 5.49 differed in NMR (Figure 5.4) and TLC analysis. We
eventually discovered a complete epimerisation of 5.47 when the reaction was quenched at
room temperature.
Figure 5.4: 1H NMR spectra of 5.42 and 5.49.
Oxidation of 5.49 gave an excellent yield of pre-yezo’otogirin C (5.38), the proposed
biosynthetic precursor of 5.21. The 1H NMR spectra of pre-yezo’otogirin C (5.38) and 6-epi-
pre-yezo’otogirin C (5.37) are rather different, particularly at H-2, where the chemical shift is
δ 3.85 ppm in pre-yezo’otogirin C (5.38) whereas at δ 4.00 ppm in 6-epi-pre-yezo’otogirin C
(5.37) (Figure 5.5). Heating pre-yezo’otogirin C (5.38) with Mn(OAc)3 and Cu(OTf)2 in DMF
under reflux gave yezo’otogirin C (5.21) in 66% yield, which we presumed formed via the
oxidative radical cyclization cascade (Scheme 5.12).
O
OHH
5.49
62 7
8
O
OHH
5.42
62 7
8
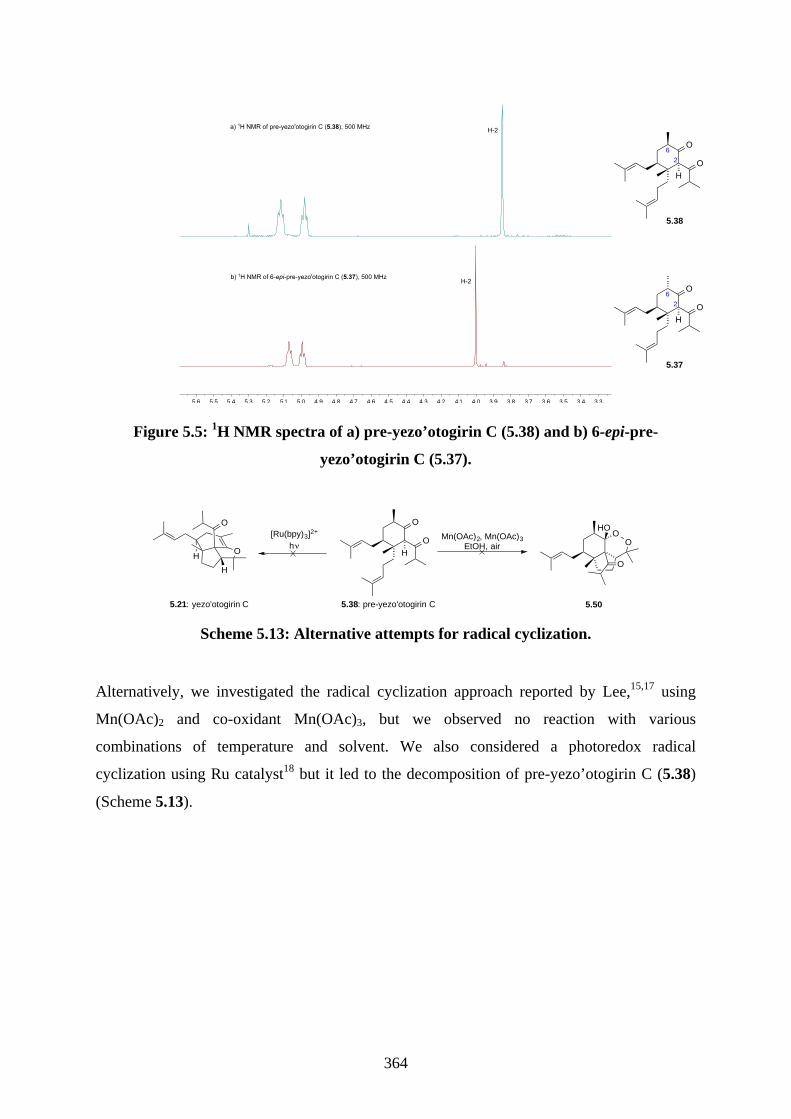
364
Figure 5.5: 1H NMR spectra of a) pre-yezo’otogirin C (5.38) and b) 6-epi-pre-
yezo’otogirin C (5.37).
Scheme 5.13: Alternative attempts for radical cyclization.
Alternatively, we investigated the radical cyclization approach reported by Lee,15,17 using
Mn(OAc)2 and co-oxidant Mn(OAc)3, but we observed no reaction with various
combinations of temperature and solvent. We also considered a photoredox radical
cyclization using Ru catalyst18 but it led to the decomposition of pre-yezo’otogirin C (5.38)
(Scheme 5.13).
O
OH
5.38: pre-yezo'otogirin C
Mn(OAc)2, Mn(OAc)3EtOH, air
5.50
O
H
O
H
5.21: yezo'otogirin C
[Ru(bpy)3]2+
hν OO
O
HO
O
OH
5.38
62
O
OH
5.37
62

365
5.2.4. Improved total synthesis of yezo’otogirin C (5.21) reported by Lee.
Scheme 5.14: Improved synthesis of yezo’otogirin C reported by Lee.17
After the completion of synthesis of yezo’otogirin C (5.21), Lee reported an improved
synthesis of yezo’otogirin C (5.21).17 Epimerisation and radical cyclization of 6-epi-pre-
yezo’otogirin C (5.37) was achieved in one pot by i-Pr2NH and Mn(OAc)3, the reaction gave
5.21 in 52% yield which is comparable to the yield of our synthesis from pre-yezo’otogirin C
(5.38). However, Lee and coworkers were not able to isolate 5.38.
O
H
O
H
O
OH Mn(OAc)3·2H2O, i-Pr2NH
EtOH, 90 °C
5.21: yezo'otogirin C5.37
52%

366
5.3. Summary
After the previous biomimetic synthesis of yezo’otogirin A, we have completed a total
synthesis of yezo’otogirin C using a similar strategy. We believe the transformation from pre-
yezo’otogirin C to yezo’otogirin C via an oxidative radical cyclization cascade is biomimetic,
in which a C=C bond, a C-C bond, a C-O bond, 2-rings and 2 stereocenters are formed in 1
step. We have also managed to convert 6-epi-pre-yezo’otogirin C to yezo’otogirin C,
although the yield of this transformation was 5%, the overall synthesis was protecting group
free with good step economy. We have sent our synthetic pre-yezo’otogirin C sample to the
isolation chemists, which may help to investigate whether this proposed biosynthetic
precursor could be found in the plant extracts.
Figure 5.6: Summary of the total synthesis of yezo’otogirin C.
O
OHH
5.42
3 stepsepimersation
6O
OHH
5.49
6
O
OH
5.37: 6-epi-pre-yezo'otogirin C
6O
OH
6
5.38: pre-yezo'otogirin C
O
H
O
H
5.21: yezo'otogirin C
oxidative radicalcyclisation
O
OEt
5.27: 3-ethoxy-2-cyclohexenone
oxidation oxidation
4 steps

367
5.4. References
1. Baldwin, J. E. J. Chem. Soc. Chem. Comm., 1976, 734. 2. Beckwith, A. L. J. Tetrahedron, 1981, 37, 3073. 3. Beckwith, A. L. J.; Schiesser, C. H. Tetrahedron Lett., 1985, 26, 373. 4. Beckwith, A. L. J.; Zimmerman, J. J. Org. Chem., 1991, 56, 5791. 5. Spellmeyer, D. C.; Houk, K. N. J. Org. Chem., 1987, 52, 959. 6. Julia, M. Acc. Chem. Res., 1971, 4, 386. 7. Snider, B. B. Chem. Rev. (Washington, D. C.), 1996, 96, 339. 8. Pepper, H. P.; Lam, H. C.; Bloch, W. M.; George, J. H. Org. Lett., 2012, 14, 5162. 9. Tanaka, N.; Kakuguchi, Y.; Ishiyama, H.; Kubota, T.; Kobayashi, J. i. Tetrahedron
Lett., 2009, 50, 4747. 10. Shan, M. D.; Hu, L. H.; Chen, Z. L. J. Nat. Prod., 2001, 64, 127. 11. Vollmer, J. J.; Rosenson, J. J. Chem. Educ., 2004, 81, 1450. 12. Lam, H. C.; Kuan, K. K.; George, J. H. Org. Biomol. Chem., 2014, 12, 2519. 13. Kuramochi, A.; Usuda, H.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. J. Am. Chem.
Soc., 2005, 127, 14200. 14. Shimizu, Y.; Shi, S.-L.; Usuda, H.; Kanai, M.; Shibasaki, M. Angew. Chem., Int. Ed.,
2010, 49, 1103. 15. He, S. Z.; Yang, W.; Zhu, L. Z.; Du, G. Y.; Lee, C. S. Org. Lett., 2014, 16, 496. 16. Norris, M. D.; Perkins, M. V.; Sorensen, E. J. Org. Lett., 2015, 17, 668. 17. Yang, W.; Cao, J.; Zhang, M.; Lan, R.; Zhu, L.; Du, G.; He, S.; Lee, C. S. J. Org.
Chem., 2015, 80, 836. 18. Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev., 2013, 113, 5322.

368
5.5. Experimental
5.5.1. General methods
All chemicals used were purchased from commercial suppliers and used as received. All reactions were
performed under an inert atmosphere of N2. All organic extracts were dried over anhydrous magnesium
sulfate. Thin layer chromatography was performed using aluminium sheets coated with silica gel F254.
Visualization was aided by viewing under a UV lamp and staining with ceric ammonium molybdate or
KMnO4 stain followed by heating. All Rf values were measured to the nearest 0.05. Flash column
chromatography was performed using 40-63 micron grade silica gel. Melting points were recorded on a
digital melting point apparatus and are uncorrected. Infrared spectra were recorded using an FT-IR
spectrometer as the neat compounds. High field NMR spectra were recorded using a 600 MHz or 500
MHz spectrometer (1H at 600/500 MHz, 13C at 150/125 MHz). Solvent used for spectra were CDCl3
unless otherwise specified. 1H chemical shifts are reported in ppm on the δ-scale relative to TMS (δ 0.0)
or CDCl3 (δ 7.26), and 13C NMR are reported in ppm relative to CDCl3 (δ 77.00). Multiplicities are
reported as (br) broad, (s) singlet, (d) doublet, (t) triplet, (q) quartet, (quin) quintet, (sext) sextet, (hept)
heptet and (m) multiplet. All J-values were rounded to the nearest 0.1 Hz. ESI high resolution mass
spectra were recorded on a ESI-TOF mass spectrometer.

369
5.5.2. Experimental procedures
To a solution of 3-ethoxy-2-cyclohexenoe (5.27) (1.00 g, 7.13 mmol) in anhydrous THF (3 mL) at –78
°C was added LDA (2 M in THF, 4.28 mL, 8.56 mmol) dropwise and stirred for 30 min. Prenyl
bromide (0.90 mL, 7.84 mmol) was added at –78 °C and stirred for 1 h before warming the reaction to
room temperature and stirred for 2 h. The mixture was quenched with saturated NH4Cl solution (10
mL). The organic layer was separated and aqueous layer was extracted with Et2O (2 × 10 mL). The
combined organic extracts were washed with saturated NH4Cl solution (30 mL) and brine (30 mL),
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (10:1, petrol/EtOAc) to give 5.39 as a yellowish oil (1.49 g, 99%). Data of
5.39 matched from literature.1
Data for 5.39:
Rf = 0.48 (2:1, petrol/EtOAc)
IR (neat): 2930, 1655, 1607, 1452, 1379, 1189 cm-1. 1H NMR (600 MHz, CDCl3): δ 5.33 (s, 1H), 5.13 – 5.10 (m, 1H), 3.92 – 3.87 (m, 2H), 2.56 – 2.52 (m,
1H), 2.42 – 2.40 (m, 2H), 2.22 – 2.18 (m, 2H), 2.13 – 2.07 (m, 1H), 2.03 (dd, J = 13.3, 4.9 Hz, 1H),
1.71 (s, 3H), 1.62 (s, 3H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (CDCl3, 150 MHz): δ 201.3, 176.9, 133.3, 122.0, 102.4, 64.2, 45.6, 28.2, 28.1, 25.9, 25.8,
17.8, 14.2.
O
OEt
O
OEtLDA, prenyl bromide, THF–78 °C to rt
99%
5.27: 3-ethoxy-2-cyclohexenone 5.39

370
To a solution of 5.39 (1.49 g, 7.15 mmol) in anhydrous THF (10 mL) at –78 °C was added MeLi·LiBr
(1.5 M in Et2O, 7.1 mL, 10.7 mmol) dropwise and stirred for 2 h. The reaction mixture was warmed to
room temperature and stirred for 2 h, then acidified by 1 M HCl (10 mL). The organic layer was
separated and aqueous layer was extracted with Et2O (2 × 20 mL). The combined organic layer was
washed with saturated NaHCO3 solution (30 mL) and brine (30 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (10:1,
petrol/EtOAc) to give 5.28 as a colourless oil (1.05g, 82%).1
Data for 5.28:
Rf = 0.53 (2:1, petrol/EtOAc)
IR (neat): 2917, 1669, 1625, 1439, 1378 cm-1 1H NMR (CDCl3, 600 MHz): δ 5.85 (s, 1H), 5.11 (ddd, J = 6.5, 4.6, 3.2 Hz, 1H), 2.44 (ddd, J = 17.2,
10.6, 5.2 Hz, 1H), 2.32 − 2.26 (m, 3H), 2.19 – 2.15 (m, 1H), 2.04 − 2.00 (m, 1H), 1.98 (s, 3H), 1.90 −
1.85 (m, 1H), 1.73 (s, 3H), 1.63 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ 199.6, 165.6, 133.9, 126.9, 121.8, 40.0, 34.0, 29.8, 26.5, 25.8, 23.0,
17.9.
O
OEt OMeLi·LiBr, THF–78 °C to rt
82%
5.39 5.28

371
To a solution of 5.28 (5.00 g, 28.0 mmol) in anhydrous THF (20 mL) at –78 °C was added LDA (2.0 M
in THF, 16.9 mL, 33.7 mmol) and stirred for 30 min. MeI (1.92 mL, 30.8 mmol) was added at –78 °C
and stirred for 30 min. The reaction mixture was warmed to room temperature over 30 min and stirred
for 4 h. The reaction was quenched with saturated NH4Cl solution (20 mL). The organic layer was
separated and the aqueous layer was extracted with Et2O (2 × 20mL). The combined organic extracts
were washed with saturated NH4Cl solution (40 mL) and brine (40 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (10:1 → 4:1,
petrol/EtOAc gradient elution) to give a 7:1 mixture of 5.33 as a yellow oil (3.84 g, 71%). Data of 5.33
matched with literature.2
Data for 5.33:
Rf = 0.39 (4:1, petrol/EtOAc)
IR (neat): 2966, 2914, 2971, 1670, 1444, 1377 cm-1
Data for major diastereoisomer: 1H NMR (600 MHz, CDCl3): δ 5.80 (s, 1H), 5.17 – 5.14 (m, 1H), 2.49 – 2.43 (m, 1H), 2.23 – 2.18 (m,
4H), 1.96 (d, J = 1.2 Hz, 3H), 1.95 – 1.94 (m, 1H), 1.74 (s, 3H), 1.63 (s, 3H), 1.11 (d, J = 6.8 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 201.9, 164.6, 133.8, 126.2, 122.3, 40.1, 36.1, 34.8, 29.7, 25.8, 22.9,
17.8, 15.4.
Data for minor diastereoisomer: 1H NMR (600 MHz, CDCl3): δ 5.89 (s, 1H), 5.05 – 5.03 (m, 1H), 2.49 – 2.43 (m, 1H), 2.23 – 2.18 (m,
4H), 1.97 (d, J = 2.8 Hz, 3H), 1.95 – 1.94 (m, 1H), 1.72 (s, 3H), 1.62 (s, 3H), 1.11 (d, J = 6.6 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 202.0, 163.6, 134.0, 127.8, 120.8, 41.2, 40.6, 36.8, 30.7, 25.9, 22.0,
18.0, 14.9.
HRMS (ESI): calculated for C13H20O 193.1592 [M+H]+, found 193.1588.
O
71%
LDA, MeI, THF−78 °C to rt
O
5.28 5.33d.r. 7:1

372
To a solution of Mg (316 mg, 13.4 mmol) in anhydrous THF (5 mL) was added 5-bromo-2-methyl-2-
pentene (0.70 mL, 5.20 mmol) at room temperature and the resultant mixture was stirred for 30 min.
The mixture was added to a suspension of CuBr (750 mg, 5.20 mmol), Me2S (0.40 mL, 5.20 mmol)
and 5.33 (500 mg, 2.60 mmol) in anhydrous THF (5 mL) at 0 °C and stirred for 2 h. LDA (2.0 M in
THF, 0.26 mL, 0.52 mmol) was added and stirred at 0 °C for 15 min. Isobutyraldehyde (0.34 mL, 3.70
mmol) was then added and stirred at 0 °C for 45 min. The mixture was quenched with saturated NH4Cl
solution (10 mL). The organic layer was separated and the aqueous layer was extracted with Et2O (2 ×
20 mL). The combined organic extracts were washed with saturated NaHCO3 solution (30 mL), brine
(30 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash
chromatography on SiO2 (100:1 → 30:1, petrol/EtOAc gradient elution) to give 5.42 (265 mg, 29%) as
a colourless oil. Data of 5.42 matched from literature.2
Data for 5.42:
Rf = 0.35 (petrol/EtOAc, 10:1)
IR (neat): 3520, 2966, 2928, 2869, 1693, 1455, 1377, 1265 cm-1 1H NMR (600 MHz, CDCl3): δ 5.07 (m, 2H), 3.70 (d, J = 3.7 Hz, 1H), 3.34 (dd, J = 9.0, 11.2 Hz, 1H),
2.88 (s, 1H), 2.47 (hept, J = 7.2 Hz, 1H), 2.13 – 2.16 (m, 1H), 1.95 – 2.06 (m, 2H), 1.74 – 1.83 (m, 4H),
1.72 (s, 3H), 1.69 (s, 3H), 1.62 (s, 3H), 1.60 (s, 3H), 1.55 – 1.58 (m, 3H), 1.23 (d, J = 7.3 Hz, 3H), 1.01
(d, J = 6.6 Hz, 3H), 0.97 (s, 3H), 0.85 (d, J = 6.7 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 214.6, 132.4, 131.5, 124.1, 123.7, 75.8, 60.1, 58.7, 41.5, 41.1, 40.4,
39.8, 35.1, 34.0, 27.7, 25.8, 25.7, 21.7, 20.2, 18.1, 18.0, 17.6, 14.8, 1.0, 0.9.
HRMS (ESI): calculated for C23H41O2 349.3107 [M+H]+, found 349.3113.
O
O
OHH
CuBr, Me2S, THF, 0 °C; then LDA; i-PrCHO
MgBr
29%
5.33 5.42d.r. 7:1

373
To a solution of hydroxyketone 5.42 (260 mg, 0.75 mmol) and NaHCO3 (81 mg, 0.97 mmol) in CH2Cl2
(5 mL) was added Dess-Martin periodinane (411 mg, 0.97 mmol) at room temperature. The mixture
was stirred for 30 min, then quenched with saturated NaHCO3 solution (5 mL). The organic layer was
separated and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic
extracts were washed with saturated NaHCO3 solution (2 × 10 mL), brine (10 mL), dried over MgSO4,
filtered and concentrated in vacuo. The residue was purified by flash chromatography on SiO2 (10:1,
petrol/EtOAc) to give 6-epi-pre-yezo’otogirin C 5.37 (235 mg, 92%) as a colourless oil. Data for 5.37
matched from literature.2
Data for 6-epi-pre-yezo’otogirin C (5.37):
Rf = 0.50 (10:1, petrol/EtOAc)
IR (neat): 2968, 2928, 1726, 1701, 1457, 1382 cm-1 1H NMR (600 MHz, CDCl3): δ 5.07 (t, J = 7.6 Hz, 1H), 4.99 (t, J = 6.7 Hz, 1H), 4.00 (s, 1H), 2.62 –
2.67 (m, 1H), 2.51 (hept, J = 6.9 Hz, 1H), 2.10 – 2.13 (m, 1H), 2.01 – 2.07 (m, 1H), 1.80 – 1.90 (m,
3H), 1.70 – 1.78 (overlapped m, 2H), 1.72 (s, 3H), 1.67 (s, 3H), 1.62 (s, 3H), 1.59 (s, 3H), 1.42 – 1.45
(m, 2H), 1.24 (d, J = 7.1 Hz, 3H), 1.07 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 6.8 Hz, 3H), 1.05 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 212.9, 211.3, 132.9, 131.8, 123.6, 123.2, 64.2, 45.3, 43.3, 43.1, 38.0,
37.9, 33.9, 26.9, 25.9, 25.7, 22.0, 18.1, 18.0, 18.0, 17.9, 17.7, 17.6.
HRMS (ESI): calculated for C23H39O2 347.2950 [M+H]+, found 347.2948.
O
OH
Dess-Martin periodinaneNaHCO3, CH2Cl2, rt
O
OHH
92%
5.42 5.37

374
To a solution of 5.37 (117 mg, 0.34 mmol) in degassed DMF (3 mL) at room temperature was added
Mn(OAc)3·2H2O (358 mg, 1.36 mmol) and Cu(OTf)2 (246 mg, 0.68 mmol). The mixture was heated at
150 °C for 1 h. The reaction mixture was cooled to room temperature and quenched with water. The
aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic extracts were washed with
H2O (2 × 20 mL) and brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo. The
residue was purified by flash column chromatography on SiO2 (100:1→20:1, petrol/EtOAc gradient
elution) to give yezo’otogirin C 5.21 (6 mg, 5%) as a colourless oil and rearranged product 5.43 (6 mg,
5%) as a colourless oil.
Data for yezo’otogirin C 5.21:
Rf = 0.65 (5:1, petrol/EtOAc)
IR (neat): 2971, 2937, 2877, 1694, 1449, 1382, 1259 cm-1. 1H NMR (600 MHz, CDCl3): δ 5.13 – 5.10 (m, 1H), 3.19 (dd, J = 10.2, 9.0 Hz, 1H), 2.96 (hept, J =
6.6 Hz, 1H), 1.96 (dd, J =14.8, 11.6 Hz, 1H), 1.94 (dd, J = 11.1, 1.0 Hz, 1H), 1.86 (dd, J = 3.3, 15.5
Hz, 1H), 1.84 – 1.80 (m, 1H), 1.76 – 1.74 (m, 1H), 1.73 (s, 3H), 1.71 (s, 3H), 1.61 (s, 3H), 1.56 – 1.52
(m, 2H), 1.39 – 1.32 (m, 1H), 1.23 (tt, J = 11.4, 3.0 Hz, 1H), 1.18 (s, 3H), 1.15 (s, 3H), 1.02 (d, J = 3.4
Hz, 3H), 1.01 (d, J = 3.7 Hz, 3H), 0.75 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 217.4, 149.2, 132.4, 124.2, 107.5, 83.4, 73.6, 54.9, 48.5, 47.0, 41.5,
37.9, 32.7, 29.6, 29.5, 25.9, 25.40, 25.37, 21.5, 19.6, 18.3, 17.9, 16.3.
HRMS (ESI): calculated for C23H35O2 343.2637 [M−H]−, found 343.2641.
Data for rearranged compound 5.43:
Rf = 0.60 (Petrol/EtOAc, 10:1)
IR (neat): 2971, 2937, 2877, 1695, 1449, 1381, 1259, 1104 cm-1 1H NMR (600 MHz, CDCl3): δ 5.07 – 5.05 (m, 1H), 3.52 (d, J = 10.0 Hz, 1H), 3.47 (hept, J = 6.6 Hz,
1H), 2.22 – 2.16 (m,1H), 2.02 – 2.00 (m, 3H), 1.84 – 1.82 (m, 3H), 1.68 (s, 3H), 1.57 (s, 3H), 1.44 (dd,
J = 7.7, 9.3 Hz, 2H), 1.26 (s, 3H), 1.21 (s, 3H), 1.12 (d, J = 6.5 Hz, 3H), 1.07 (d, J = 6.7 Hz, 3H), 0.94
(s, 6H).
O
H
O
H
O
OH Mn(OAc)3·2H2O, Cu(OTf)2
DMF, 150 °C
5% 5%
5.21: yezo'otogirin C 5.43
+
5.37
O
O

375
13C NMR (150 MHz, CDCl3): δ 211.7, 211.5, 132.2, 123.7, 73.5, 58.6, 49.4, 42.7, 42.3, 39.5, 35.2,
30.0, 28.9, 27.2, 25.6, 23.6, 23.5, 23.4, 21.0, 19.1, 17.6.
HRMS (ESI): calculated for C23H37O2 345.2794 [M+H]+, found 345.2795.

376
To a solution of 5.42 (530 mg, 1.52 mmol) in DMF (20 mL) at room temperature was added TMSCl
(0.58 mL, 4.56 mmol) and stirred for 2 hr. The reaction was quenched with saturated NaHCO3 solution
(15 mL). The organic layer was separated and the aqueous layer was extracted with Et2O (2 × 20 mL).
The combined organic extracts were washed with H2O (2 × 30 mL), saturated NaHCO3 solution (30
mL), brine (30 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by
flash column chromatography on SiO2 (50:1, petrol/EtOAc) to give 5.47 as a colourless oil (326 mg,
51%).
Data for 5.47:
Rf = 0.60 (5:1, petrol/EtOAc)
IR (neat): 2965, 2929, 2873, 1708, 1452, 1376, 1252, 1055 cm-1 1H NMR (500MHz, CDCl3): δ 5.08 (t, J = 6.9 Hz, 1H), 5.00 (t, J = 6.8 Hz, 1H), 3.93 (t, J = 3.6 Hz,
1H), 2.81 – 2.72 (m, 1H), 2.34 (d, J = 3.0 Hz, 1H), 2.25 – 2.21 (m, 1H), 2.08 – 2.02 (m, 1H), 1.89 –
1.76 (m, 5H), 1.72 (s, 3H), 1.65 (s, 3H), 1.63 (s, 3H), 1.57 (s, 3H), 1.53 (s, 1H), 1.50 – 1.37 (m, 2H),
1.06 (s, 3H), 0.98 (d, J = 6.5 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H), 0.11 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 214.6, 132.4, 131.5, 124.1, 123.7, 75.8, 60.1, 58.7, 41.5, 41.1, 40.4,
39.8, 35.1, 34.0, 27.7, 25.8, 25.7, 21.7, 20.2, 18.1, 18.0, 17.6, 14.8, 1.0, 0.9.
HRMS (ESI): calculated for C26H49O2Si 421.3502 [M+H]+, found 421.3508.
O
OHH
TMSCl, imidazole, DMF, rt
O
OTMSH
51%
5.42 5.47

377
To a solution of 5.47 (316 mg, 0.75 mmol) in anhydrous THF (10 mL) at -78 °C was added LDA (2.0
M in THF, 1.1 mL, 2.20 mmol). The mixture was stirred for 3 hours and warmed to room temperature
over 30 min. The reaction was quenched with saturated NH4Cl solution (10 mL). The organic layer was
separated and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic extracts
were washed with saturated NH4Cl solution (20 mL) and brine (20 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (50:1,
petrol/EtOAc) to give 5.48 as a colourless oil (249 mg, 79%).
Data for 5.48:
Rf = 0.60 (5:1, petrol/EtOAc)
IR (neat): 2964, 2925, 2873, 1707, 1451, 1378, 1265, 1252 cm-1 1H NMR (500 MHz, CDCl3): δ 5.08 (t, J = 7.2 Hz, 1H), 5.00 (t, J = 7.0 Hz, 1H), 3.93 (t, J = 3.7Hz,
1H), 2.80 – 2.75 (m, 1H), 2.34 (d, J = 3.2 Hz, 1H), 2.25 – 2.21 (m, 1H), 2.08 – 2.02 (m, 1H), 1.89 –
1.77 (m, 5H), 1.72 (s, 3H), 1.65 (s, 3H), 1.64 (s, 3H), 1.57 (s, 3H), 1.53 (s, 1H), 1.50 – 1.39 (m, 2H),
1.06 (s, 3H), 0.98 (d, J = 6.5 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 6.7 Hz, 3H), 0.12 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 214.7, 132.4, 131.5, 124.1, 123.8, 75.8, 41.6, 41.1, 40.4, 39.9, 35.1,
34.1, 27.7, 25.9, 25.7, 21.8, 20.3, 18.1, 18.0, 17.7, 14.9, 0.95.
HRMS (ESI): calculated for C26H49O2Si 421.3502 [M+H]+, found 421.3508.
LDA, THF, −78 °C to rt
O
OTMSH
O
OTMSH
79%
5.47 5.48

378
To a solution of 5.48 (240 mg, 0.57 mmol) in anhydrous THF (5 mL) at room temperature was added
TBAF (1.0 M in THF, 0.86 mL, 0.86 mmol). The mixture was stirred for 30 min. The reaction was
quenched with saturated NH4Cl solution (5 mL). The organic layer was separated and the aqueous layer
was extracted with Et2O (2 × 5 mL). The combined organic extracts were washed with brine (10 mL),
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on SiO2 (30:1, petrol/EtOAc) to give 5.49 in a colourless oil (141 mg, 71%).
Data for 5.49:
Rf = 0.55 (5:1, petrol/EtOAc)
IR (neat): 3514, 2965, 2929, 2872, 1695, 1451, 1377, 1248 cm-1 1H NMR (500MHz, CDCl3): δ 5.12 (t, J = 7.2 Hz, 1H), 5.07 (t, J = 7.0 Hz, 1H), 3.90 (d, J = 11.4 Hz,
1H), 3.33 (dd, J = 9.1, 11.0 Hz, 1H), 2.65 (s, 1H), 2.47 – 2.39 (m, 2H), 2.16 – 2.09 (m, 2H), 2.05 – 1.98
(m, 1H), 1.91 – 1.85 (m, 1H), 1.82 – 1.75 (m, 2H), 1.72 (s, 3H), 1.69 (s, 3H), 1.61 (s, 6H), 1.59 – 1.54
(m, 2H), 1.22 (q, J = 12.7 Hz, 1H), 1.00 (d, J = 6.6 Hz, 3H), 0.97 (d, J = 6.3 Hz, 3H), 0.93 (s, 3H), 0.84
(d, J = 6.7 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 219.3, 132.7, 131.7, 123.8, 123.3, 75.9, 55.5, 46.6, 46.3, 42.9, 38.2,
36.5, 34.4, 27.5, 25.9, 25.7, 21.1, 20.5, 19.9, 18.5, 17.9, 17.7, 14.1.
HRMS (ESI): calculated for C23H41O2 349.3107 [M+H]+, found 349.3113.
TBAF, THF, rt
O
OHH
O
OTMSH
71%
5.48 5.49

379
To a solution of 5.49 (130 mg, 0.37 mmol) in CH2Cl2 (5 mL) at room temperature was added Dess-
Martin periodinane (316 mg, 0.75 mmol) and NaHCO3 (63 mg, 0.75 mmol). The mixture was stirred
for 1.5 h, then quenched with saturated NaHCO3 solution (5 mL). The organic layer was separated and
the aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic extracts were washed
with saturated NaHCO3 solution (10 mL) and brine (10 mL), dried over MgSO4, filtered and
concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (30:1,
petrol/EtOAc) to give pre-yezo’otogirin C (5.38) as a colourless oil (120 mg, 94%).
Data for pre-yezo’otogirin C (5.38):
Rf = 0.55 (5:1, petrol/EtOAc)
IR (neat): 2968, 2928, 2872, 1723, 1703, 1453, 1378, 1280, 1252 cm-1 1H NMR (500 MHz, CDCl3): δ 5.12 (t, J = 7.0 Hz, 1H), 4.98 (t, J = 6.7 Hz, 1H), 3.85 (s, 1H), 2.50 –
2.43 (m, 2H), 2.13 – 2.01 (m, 3H), 1.86 – 1.75 (m, 2H), 1.73 (s, 3H), 1.71 – 1.68 (m, 1H), 1.66 (s, 3H),
1.61 (s, 3H), 1.58 (s, 3H), 1.50 – 1.46 (m, 2H), 1.26 – 1.18 (m, 1H), 1.06 (d, J = 6.8 Hz, 3H), 1.04 (d, J
= 7.0 Hz, 3H), 1.01 (d, J = 8.0 Hz, 3H), 1.00 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 211.0, 210.6, 132.9, 131.7, 123.7, 123.0, 66.8, 45.5, 42.7, 42.5, 37.0,
36.6, 26.8, 25.9, 25.7, 21.9, 18.5, 18.0, 17.7, 17.7, 17.3, 14.3.
HRMS (ESI): calculated for C23H39O2 347.2950 [M+H]+, found 347.2948.
Dess-Martin periodinanNaHCO3, CH2Cl2, rt
O
OH
O
OHH
94%
5.49 5.38

380
To a solution of pre-yezo’otogirin C (5.38) (60 mg, 0.17 mmol) in degassed DMF (5 mL) at room
temperature was added Mn(OAc)3·2H2O (92 mg, 0.35 mmol) and Cu(OTf)2 (61 mg, 0.17 mmol). The
mixture was warmed to 150 °C for 2 hours, then cooled to room temperature. The reaction was diluted
with EtOAc (10 mL) and H2O (5 mL). The organic layer was separated and the aqueous layer was
extracted with EtOAc (2 × 10 mL). The combined organic extracts were washed with H2O (2 × 20 mL)
and brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by
flash column chromatography on SiO2 (50:1, petrol/EtOAc) to give yezo’otogirin C (5.21) as a
colourless oil (40 mg, 66%). Data for yezo’otogirin C (5.21) matched from previously obtained.
O
H
O
H
O
OH Mn(OAc)3·2H2O, Cu(OTf)2
DMF, 150 °C
66%
5.38: pre-yezo'otogirin C 4.21: yezo'otogirin C
381
5.5.3. NMR spectra
O
OEt
5.391H NMR600 MHz
CDCl3
O
OEt
5.3913C NMR150 MHz
CDCl3
382
O
5.281H NMR600 MHz
CDCl3
O
5.2813C NMR150 MHz
CDCl3
383
O
5.3313C NMR150 MHz
CDCl3
O
5.331H NMR600 MHz
CDCl3
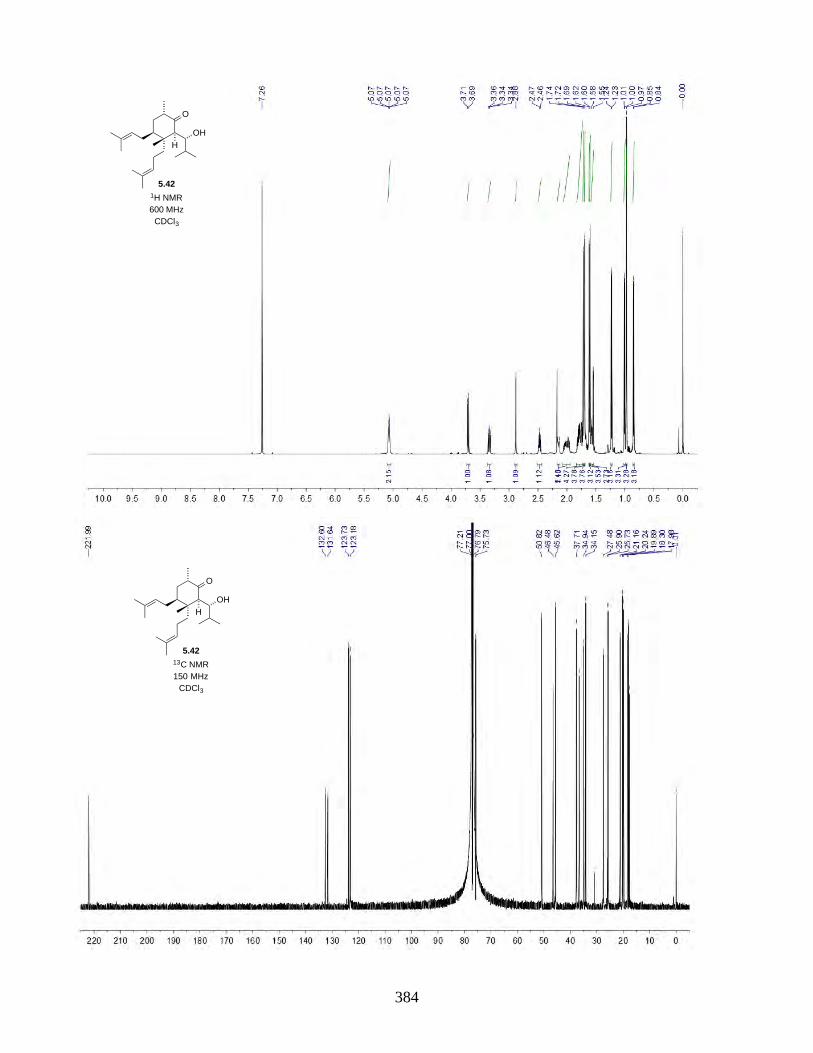
384
O
OHH
5.421H NMR600 MHz
CDCl3
O
OHH
5.4213C NMR150 MHz
CDCl3
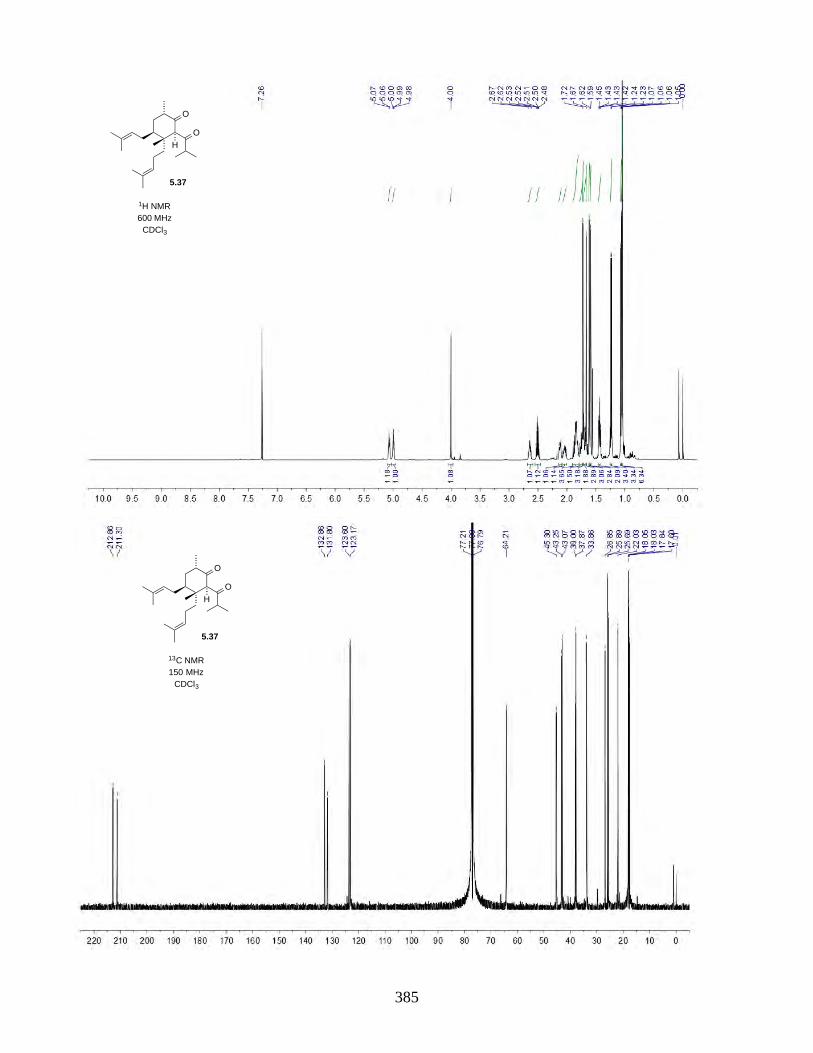
385
O
OH
5.37
1H NMR600 MHz
CDCl3
O
OH
5.37
13C NMR150 MHz
CDCl3
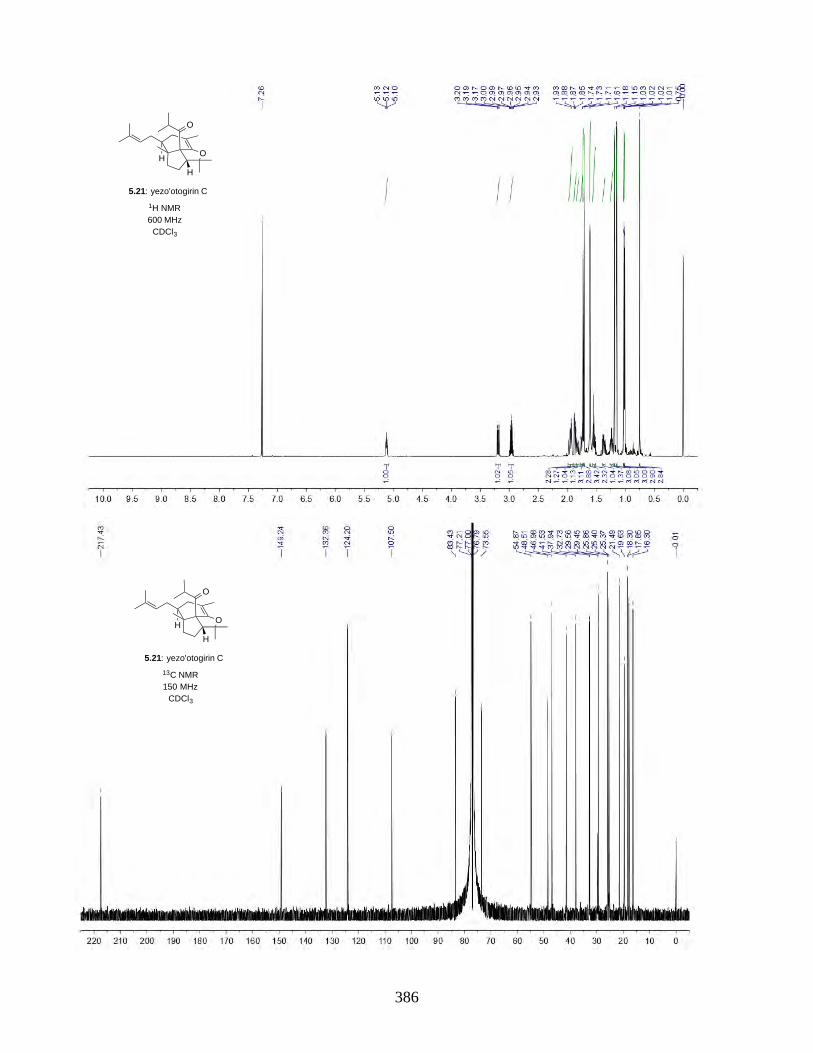
386
O
H
O
H
5.21: yezo'otogirin C1H NMR600 MHz
CDCl3
O
H
O
H
5.21: yezo'otogirin C13C NMR150 MHz
CDCl3
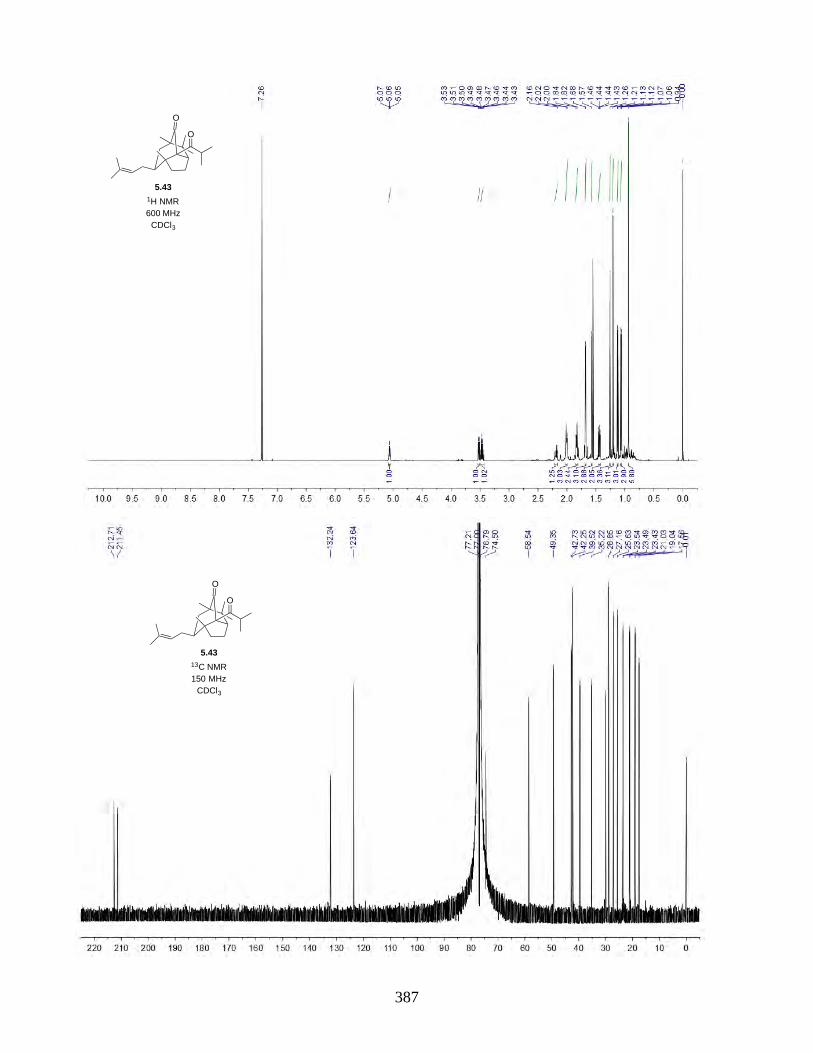
387
5.431H NMR600 MHz
CDCl3
O
O
5.4313C NMR150 MHz
CDCl3
O
O
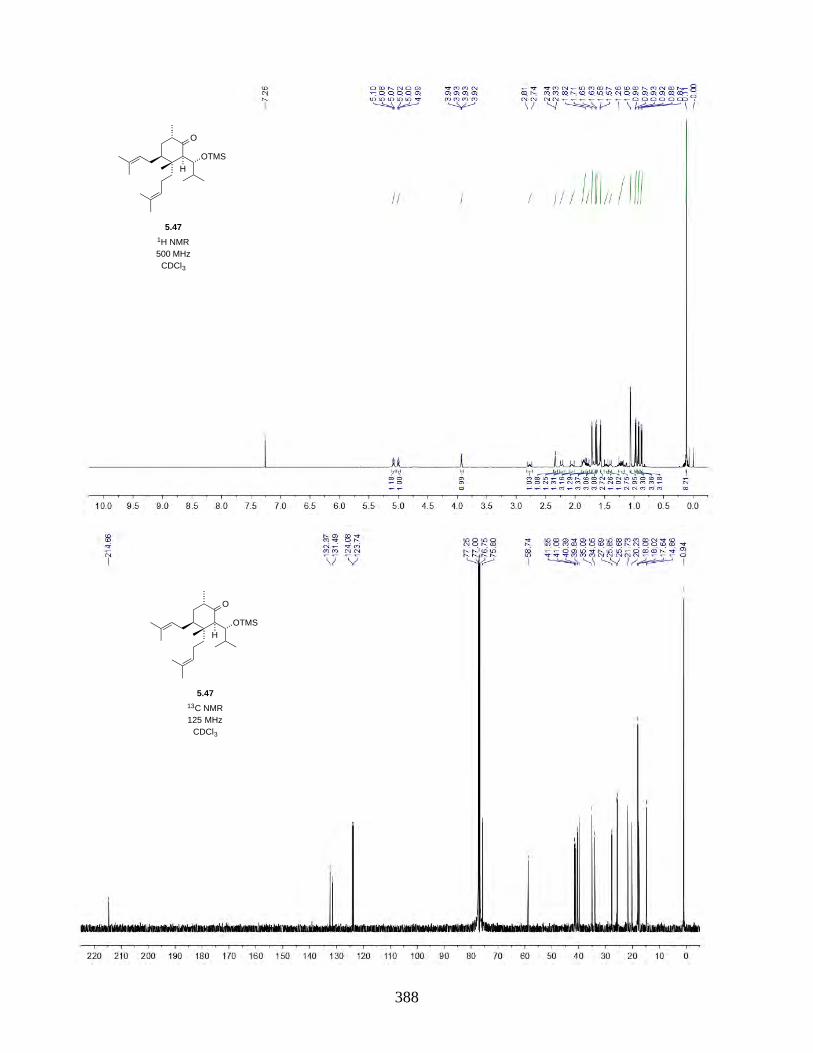
388
O
OTMSH
5.471H NMR500 MHz
CDCl3
O
OTMSH
5.4713C NMR125 MHz
CDCl3
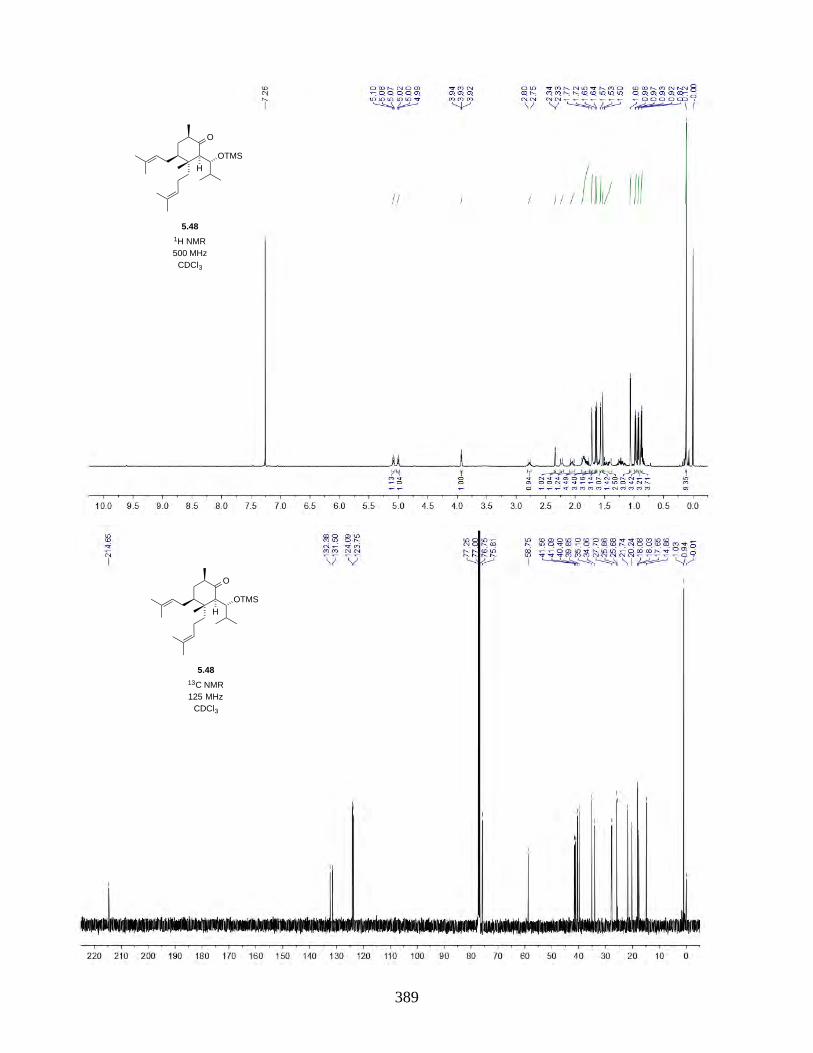
389
O
OTMSH
5.481H NMR500 MHz
CDCl3
O
OTMSH
5.4813C NMR125 MHz
CDCl3
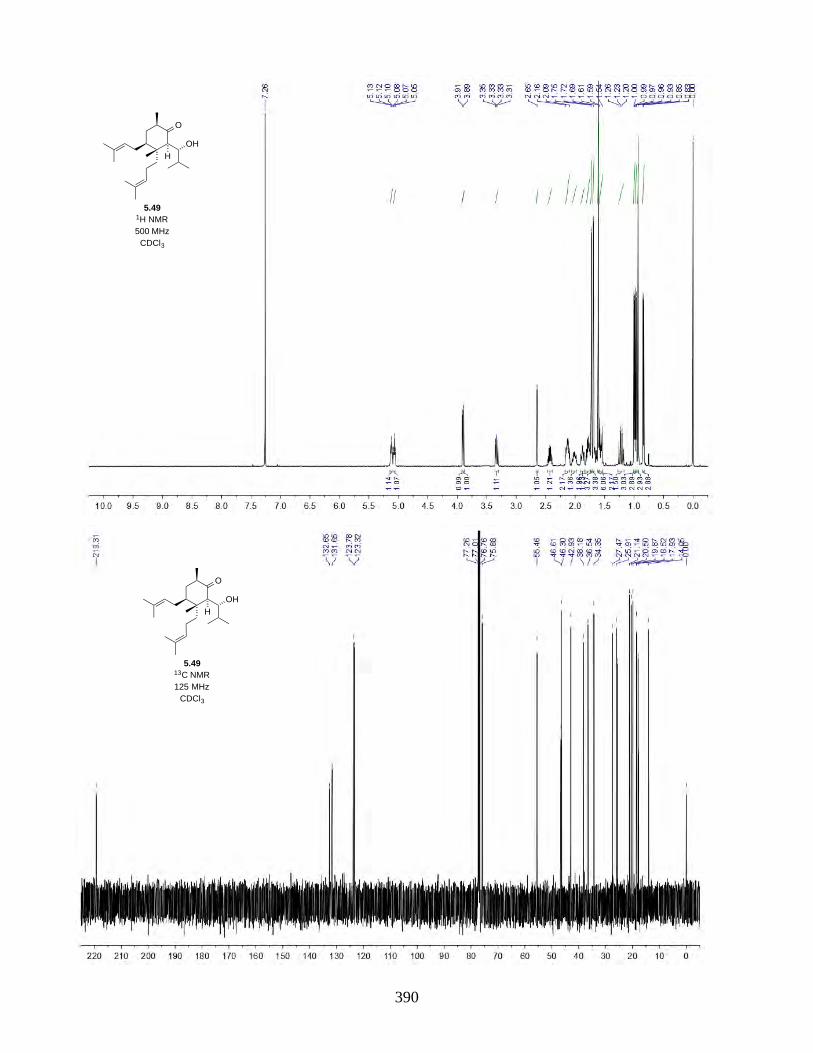
390
O
OHH
5.491H NMR500 MHz
CDCl3
O
OHH
5.4913C NMR125 MHz
CDCl3
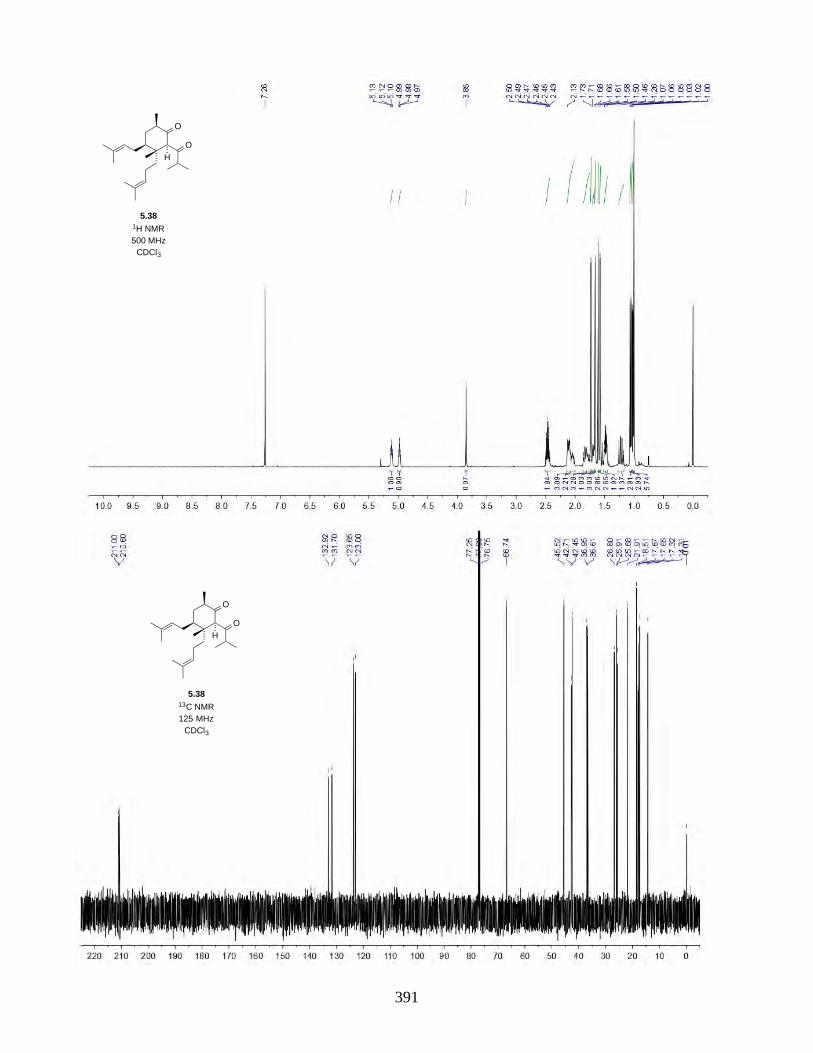
391
O
OH
5.381H NMR500 MHz
CDCl3
O
OH
5.3813C NMR125 MHz
CDCl3
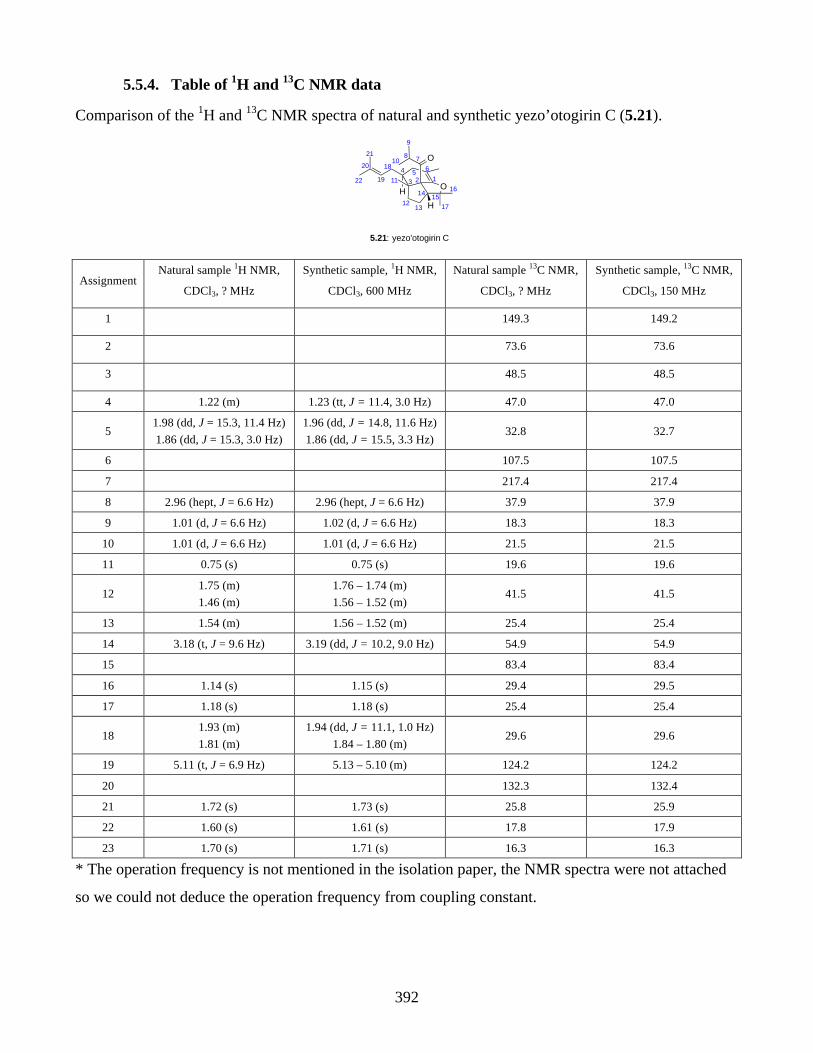
392
5.5.4. Table of 1H and 13C NMR data
Comparison of the 1H and 13C NMR spectra of natural and synthetic yezo’otogirin C (5.21).
Assignment Natural sample 1H NMR,
CDCl3, ? MHz
Synthetic sample, 1H NMR,
CDCl3, 600 MHz
Natural sample 13C NMR,
CDCl3, ? MHz
Synthetic sample, 13C NMR,
CDCl3, 150 MHz
1 149.3 149.2
2 73.6 73.6
3 48.5 48.5
4 1.22 (m) 1.23 (tt, J = 11.4, 3.0 Hz) 47.0 47.0
5 1.98 (dd, J = 15.3, 11.4 Hz) 1.86 (dd, J = 15.3, 3.0 Hz)
1.96 (dd, J = 14.8, 11.6 Hz) 1.86 (dd, J = 15.5, 3.3 Hz)
32.8 32.7
6 107.5 107.5
7 217.4 217.4
8 2.96 (hept, J = 6.6 Hz) 2.96 (hept, J = 6.6 Hz) 37.9 37.9
9 1.01 (d, J = 6.6 Hz) 1.02 (d, J = 6.6 Hz) 18.3 18.3
10 1.01 (d, J = 6.6 Hz) 1.01 (d, J = 6.6 Hz) 21.5 21.5
11 0.75 (s) 0.75 (s) 19.6 19.6
12 1.75 (m) 1.46 (m)
1.76 – 1.74 (m) 1.56 – 1.52 (m)
41.5 41.5
13 1.54 (m) 1.56 – 1.52 (m) 25.4 25.4
14 3.18 (t, J = 9.6 Hz) 3.19 (dd, J = 10.2, 9.0 Hz) 54.9 54.9
15 83.4 83.4
16 1.14 (s) 1.15 (s) 29.4 29.5
17 1.18 (s) 1.18 (s) 25.4 25.4
18 1.93 (m) 1.81 (m)
1.94 (dd, J = 11.1, 1.0 Hz) 1.84 – 1.80 (m)
29.6 29.6
19 5.11 (t, J = 6.9 Hz) 5.13 – 5.10 (m) 124.2 124.2
20 132.3 132.4
21 1.72 (s) 1.73 (s) 25.8 25.9
22 1.60 (s) 1.61 (s) 17.8 17.9
23 1.70 (s) 1.71 (s) 16.3 16.3
* The operation frequency is not mentioned in the isolation paper, the NMR spectra were not attached
so we could not deduce the operation frequency from coupling constant.
O
H
O
H
5.21: yezo'otogirin C
2
17
161
1514
1312
11
10
9
8 764 5
3
18
19
20
21
22

393
5.5.5. References
1. Kuramochi, A.; Usuda, H.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc., 2005, 127, 14200.
2. He, S.; Yang, W.; Zhu, L.; Du, G.; Lee, C.-S. Org. Lett., 2013, 16, 496.





























