Embed Size (px)
Citation preview

REVIEWEndocrine-Related Cancer (2011) 18 R53–R77
Tumor-induced osteomalacia
William H Chong1, Alfredo A Molinolo2, Clara C Chen3
and Michael T Collins1
1Skeletal Clinical Studies Unit, Craniofacial and Skeletal Diseases Branch, 2Oral Pharyngeal Cancer Branch, National Institute of
Dental and Craniofacial Research and 3Nuclear Medicine, Radiology and Imaging Sciences, Hatfield Clinical Research Center,
National Institutes of Health, Bethesda, Maryland 20892, USA
(Correspondence should be addressed to M T Collins; Email: [email protected])
Abstract
Tumor-induced osteomalacia (TIO) is a rare and fascinating paraneoplastic syndrome in whichpatients present with bone pain, fractures, and muscle weakness. The cause is high blood levelsof the recently identified phosphate and vitamin D-regulating hormone, fibroblast growth factor 23(FGF23). In TIO, FGF23 is secreted by mesenchymal tumors that are usually benign, but aretypically very small and difficult to locate. FGF23 acts primarily at the renal tubule and impairsphosphate reabsorption and 1a-hydroxylation of 25-hydroxyvitamin D, leading to hypophos-phatemia and low levels of 1,25-dihydroxy vitamin D. A step-wise approach utilizing functionalimaging (F-18 fluorodeoxyglucose positron emission tomography and octreotide scintigraphy)followed by anatomical imaging (computed tomography and/or magnetic resonance imaging),and, if needed, selective venous sampling with measurement of FGF23 is usually successful inlocating the tumors. For tumors that cannot be located, medical treatment with phosphatesupplements and active vitamin D (calcitriol or alphacalcidiol) is usually successful; however, themedical regimen can be cumbersome and associated with complications. This review summarizesthe current understanding of the pathophysiology of the disease and provides guidance inevaluating and treating these patients. Novel imaging modalities and medical treatments, whichhold promise for the future, are also reviewed.
Endocrine-Related Cancer (2011) 18 R53–R77
Introduction
Tumor-induced osteomalacia (TIO), also known as
oncogenic osteomalacia, is a rare paraneoplastic
syndrome of abnormal phosphate and vitamin D
metabolism caused by typically small endocrine
tumors that secrete the phosphaturic hormone, fibro-
blast growth factor 23 (FGF23; Drezner 2001, Folpe
et al. 2004, Jan de Beur 2005). Biochemical hallmarks
of the disorder are hypophosphatemia due to renal
phosphate wasting, inappropriately normal or low
1,25-dihydroxy vitamin D, and elevated or inappropri-
ately normal plasma FGF23. TIO is counted among the
ranks of endocrine neoplasms that have a striking
presentation and, when resected, a dramatic and
satisfying resolution. Due to a lack of knowledge of
the existence of the disease, the length of time from
onset of symptoms until diagnosis is often long. As a
result, patients frequently present with multiple
fractures, height loss, and generalized debilitated
status, reminiscent of how patients in the past would
Endocrine-Related Cancer (2011) 18 R53–R77
1351–0088/11/018–R53 q 2011 Society for Endocrinology Printed in Great
present with advanced primary hyperparathyroidism
(Fig. 1). If the condition develops before growth plate
closure, rickets is also present. There is also a group
of patients with a TIO-like syndrome in which a tumor
is never found. Whether or not this is due to the
inability to find the tumor or this represents a separate
syndrome is not known. In our series of 31 patients
with TIO syndrome in which genetic causes of
hypophosphatemia have been excluded, we have
been able to find the tumor in 19 (61%) of them.
Given that most of the patients referred to our center
have already failed tumor localization at the referring
institution at least once, the percent of patients in
whom we have not been able to find the tumor is
probably higher than is seen in patients being evaluated
for the first time.
A TIO-like syndrome can also be seen in association
with other diseases such as prostate cancer, oat cell
cancer, hematologic malignancies, neurofibromatosis,
Britain
DOI: 10.1530/ERC-11-0006
Online version via http://www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

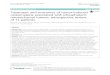
A B C D
Figure 1Clinical effects of advanced tumor-induced osteomalacia (TIO). The patient in the gown in panel A is depicted standing nextto his father. The patient was previously taller than his father, but this is no longer the case. Panel B demonstrates kyphosis andpectus carinatum, which resulted from multiple compression fractures due to osteomalacia. While these findings are the result ofadvanced osteomalacia, they are strikingly similar to those seen in advanced hyperparathyroidism, as demonstrated by the famouspatient reported by Fuller Albright, Captain Martell, shown in panels C and D, who suffered from years of untreatedhyperparathyroidism. (Photo of patient and father are reproduced with their permission. Material is reproduced with permission fromAlbright & Reifenstein (1948)).
W H Chong et al.: Tumor-induced osteomalacia
epidermal nevus syndrome, and polyostotic fibrous
dysplasia of bone (FD; Saville et al. 1955, Dent &
Gertner 1976, Taylor et al. 1984, Carey et al. 1986,
Rao et al. 1987, Konishi et al. 1991, Nakahama et al.
1995, Ivker et al. 1997, Reese & Rosen 1997, Collins
et al. 2001, Riminucci et al. 2003). In these cases, the
primary disease is usually obvious, and as such it may
be useful to refer to this as secondary TIO. In cases of
secondary TIO, the goal is treatment of the underlying
disease. However, when the underlying disease is not
amenable to cure or adequate treatment, as is the case
in FD, the medical treatment of the hypophosphatemic
syndrome is the same as in cases of primary TIO.
Robert McCance is often credited with the first
reported case of TIO. McCance (1947) reported a
patient with manifestations of what was clearly TIO.
The patient had pain, weakness, gait abnormalities, and
low phosphorus levels. She was treated with high doses
of vitamin D, but her symptoms did not completely
resolve until a tumor in her femur was resected.
Her cure, however, was attributed to the high-dose
vitamin D therapy. She was diagnosed with resistance
to vitamin D. During that period, vitamin D resistance
was believed to be the mechanism of what would
eventually come to be understood as FGF23-mediated
phosphate wasting disorders (Albright et al. 1937).
R54
The first person to clearly recognize that the disease
was the result of a ‘rachitogenic’ substance was Andrea
Prader. In 1959, he described an 11 1⁄2 -year-old girl
who developed severe rickets over the course of a year
(Prader et al. 1959). Evaluation showed decreased
tubular phosphate reabsorption but otherwise normal
studies of kidney function. A tumor, classified as a
giant cell granuloma, was identified in a rib and
resected with resultant healing of her rickets. Prader
highlighted the association between the resection of the
tumor and the cure of the rickets and posited that the
granuloma was secreting a rachitogenic substance.
Since this association was first made, w337 cases of
what may be referred to as primary TIO have been
reported in the literature (English) (Sharkis et al. 1997,
Yang et al. 1997, Malabanan et al. 1998, Baronofsky
et al. 1999, Drezner 1999, Fukumoto et al. 1999,
Gascon et al. 1999, Hasegawa et al. 1999, Heylen et al.
1999, Lamont et al. 1999, Nguyen & Wang 1999,
Ohashi et al. 1999, Zura et al. 1999, Clunie et al. 2000,
Nelson et al. 2001, 2003, Ogose et al. 2001, Park et al.
2001, Rhee et al. 2001, Sakamoto et al. 2001, Sato
et al. 2001, Seufert et al. 2001, Furco et al. 2002,
Garcia & Spencer 2002, Jan de Beur et al. 2002, Lui
et al. 2002, Moran & Paul 2002, Paglia et al. 2002,
Reis-Filho et al. 2002, 2004, Teasell & Shapiro 2002,
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
Yamazaki et al. 2002, Casari et al. 2003, Dissanayake
et al. 2003, Fuentealba et al. 2003, Kimizuka et al.
2004, Nayak et al. 2004, Takeuchi et al. 2004,
Toyosawa et al. 2004, Ungari et al. 2004, Ward et al.
2004, Auethavekiat et al. 2005, Colt et al. 2005,
Dupond et al. 2005a, Narvaez et al. 2005, Zimering
et al. 2005, Cheung et al. 2006, Dowman & Khattak
2006, Hodgson et al. 2006, Imel et al. 2006,
Inokuchi et al. 2006, Kaylie et al. 2006, Koriyama
et al. 2006, Ladha et al. 2006, Nguyen 2006, Sahnoune
et al. 2006, Tartaglia et al. 2006, Vandergheynst et al.
2006, Yoshioka et al. 2006, Ahn et al. 2007, Beech
et al. 2007, Elston et al. 2007, Geller et al. 2007,
Gershinsky et al. 2007, Halperin et al. 2007, Hesse
et al. 2007a,b, Jacob et al. 2007, Kaul et al. 2007,
Khosravi et al. 2007, Oka et al. 2007, Roarke &
Nguyen 2007, Umphrey et al. 2007, Williams et al.
2007, van Boekel et al. 2008, Duet et al. 2008, Endo
et al. 2008, von Falck et al. 2008, Habra et al. 2008,
Hannan et al. 2008, Harish et al. 2008, Kenealy et al.
2008, Lewiecki et al. 2008, Mannstadt et al. 2008,
Nasu et al. 2008, Ogura et al. 2008, Policarpio-Nicolas
et al. 2008, Ratanasuwan et al. 2008, Vollbrecht & Rao
2008, Westerberg et al. 2008, Woznowski et al. 2008,
Bahrami et al. 2009, Gore et al. 2009, Harbeck et al.
2009, Khadgawat et al. 2009, Mussig et al. 2009,
Nawrot-Wawrzyniak et al. 2009, Pirola et al. 2009,
Radaideh et al. 2009, Rendina et al. 2009, Romualdo-
Silva et al. 2009, Savage & Zimmer 2009, Sciubba
et al. 2009, Seijas et al. 2009, Szumera-Cieckiewicz
et al. 2009, Uramoto et al. 2009, Woo et al. 2009,
Yun et al. 2009, Chouhan et al. 2010, Dehghani et al.
2010, Haeusler et al. 2010, Ishii et al. 2010, Ito et al.
2010, Jagtap et al. 2010, Jung et al. 2010, Kobayashi
et al. 2010, Kurien et al. 2010, Marshall et al. 2010,
Mori et al. 2010, Pedrazzoli et al. 2010, Peters et al.
2010, Peterson et al. 2010, Xia et al. 2010). The fact
that over 200 of these cases have been reported in the
last 10 years indicates a growing recognition of this
disease. This growing recognition has paralleled the
identification of FGF23 as the phosphaturic agent
(ADHR Consortium 2000, White et al. 2001). The
discovery of FGF23 has not only paved the way toward
a better understanding of the pathophysiology and
treatment of TIO, but has also provided a window
into areas of mineral metabolism physiology that for
years had been unexplained. Hopefully, greater
recognition of TIO as a distinct disease entity, a better
understanding of the underlying pathophysiology,
improved tumor localization strategies, and better
medical treatment will make the sort of presentation
that is shown in Fig. 1, a historical footnote, as is
www.endocrinology-journals.org
now the case with the primary hyperparathyroidism
(Albright & Reifenstein 1948).
This review aims to provide insight into the
pathophysiology and mechanism of TIO as well as
guidance in evaluating and diagnosing this rare
disease. Clinical presentation, diagnostic testing and
therapeutic options are discussed with an emphasis on
recent advances. Future areas of interest for research
are also discussed.
Physiology and pathophysiology
Phosphate homeostasis
Phosphate is vital to normal physiologic functioning; it
plays a role in intracellular signaling, membrane
function, energy metabolism, and bone mineralization
(Sommer et al. 2007, Renkema et al. 2008).
Approximately, 65% of dietary phosphate is absorbed
in the duodenum and jejunum (Mount & Yu 2008).
Phosphate is predominantly stored in the skeleton with
a small amount available in the extracellular fluid.
This freely circulating phosphate is filtered by the
glomerulus, and under normal physiologic conditions,
85–95% of filtered phosphate is reabsorbed. As renal
phosphate load increases, phosphate reabsorption
increases until a threshold is reached, at which point
phosphate is excreted in the urine (Mount & Yu 2008).
Renal phosphate excretion is the primary mode of
phosphate clearance and regulation of phosphate
balance. The majority of phosphate reabsorption
takes place in the proximal renal tubule through type
2a and 2c Na-dependent phosphate cotransporters
(NaPi-2a and NaPi-2c; Mount & Yu 2008, Bergwitz
& Juppner 2010).
For years, it has been recognized that phosphate
concentrations are under the control of parathyroid
hormone (PTH), 1,25-vitamin D, and the so-called
‘phosphatonins’ (Mount & Yu 2008, Bergwitz &
Juppner 2010). While the primary role of PTH is
thought to be the maintenance of serum calcium levels,
it also plays an important role in phosphate regulation.
In the process of mobilizing calcium from bone, PTH
also mobilizes phosphate from bone. To help excrete
this increased blood phosphate, PTH acts to inhibit
renal phosphate reabsorption through endocytosis of
NaPi-2a, thus increasing renal phosphate excretion.
The overall effect is to lower blood phosphate levels
(Renkema et al. 2008). 1,25-vitamin D is thought to
play a role in phosphate regulation in both the
gastrointestinal tract and in the kidney, but the
mechanism is less understood. Increases in 1,25-
vitamin D lead to an increase in phosphate absorption
R55
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
from the gastrointestinal tract. In the kidney, the role
is more complex. With chronic administration of
vitamin D, there is reduction of NaPi-2a and sub-
sequent phosphaturia (Friedlaender et al. 2001). With
acute administration of vitamin D metabolites, there
is reduced renal phosphate excretion (Taketani et al.
1998). However, many of the effects of 1,25-vitamin D
on phosphate metabolism were posited before the
discovery of FGF23. It is now clear that FGF23 and
PTH play much more important roles in phosphate
homeostasis than 1,25-vitamin D.
Fibroblast growth factor 23
While Prader was the first to propose the idea of a
circulating factor that could cause phosphate wasting,
the first evidence that a circulating factor was
responsible for the hypophosphatemia of phosphaturic
disorders such as TIO was demonstrated in an elegant
set of experiments by Meyer et al. By performing
parabiosis experiments in hyp mice, the mouse model
for X-linked hypophosphatemic rickets, Meyer et al.
(1989) were able to demonstrate that a factor in the hyp
mouse’s circulation could induce hypophosphatemia in
wild-type (WT) mice. A similarly elegant set of
experiments by Nesbitt et al. (1992), in which the
transplantation of WT kidneys into hyp mice failed to
correct hypophosphatemia, confirmed the etiology as a
circulating factor and not a primary renal defect. The
first evidence to support this concept in humans was the
work by Miyauchi et al. (1988), in which a TIO tumor
resected from a patient and transplanted into nude mice
caused hypophosphatemia and the work that showed
that the supernatant from cultured tumor cells could
also cause hypophosphatemia in mice (Cai et al. 1994).
This phosphaturic substance was termed ‘phospha-
tonin’ by Econs & Drezner (1994) because of its ability
to lower blood phosphorus level.
The first identification of FGF23 as the putative
phosphatonin was when mutations in FGF23 were
identified by Econs and the autosomal-dominant
hypophosphatemic rickets (ADHR) consortium as the
cause of ADHR (ADHR Consortium 2000). FGF23 is a
member of the FGF ligand superfamily and functions
as an endocrine factor. It has a FGF-like amino
terminus and a unique carboxy-terminus domain
(ADHR Consortium 2000). Once identified as the
cause of ADHR, elevations in serum FGF23 were soon
found in TIO (White et al. 2001), X-linked hypopho-
sphatemia (XLH; Jonsson et al. 2003), FD (Riminucci
et al. 2003), ADHR (Imel et al. 2007) and autosomal-
recessive hypophosphatemic rickets (ARHR; Feng
et al. 2006). The first insight into the physiologic
R56
source of FGF23 was from the study of patients with
FD, wherein it was found that dysplastic osteogenic
cells are the source of FGF23 (Riminucci et al. 2003).
Reasoning that if dysplastic osteogenic cells are the
source of FGF23 in FD, we went on to show that
normal bone cells are the physiologic source of FGF23
(Riminucci et al. 2003). Physiologic regulation of
FGF23 secretion is still being defined, but probably
serum phosphorus (Larsson et al. 2003, Ferrari et al.
2005, Nishida et al. 2006, Ito et al. 2007) and/or serum
1,25-vitamin D (Collins et al. 2005) are important in
regulating the levels of FGF23.
FGF23 acts by binding to target cells via an FGF
receptor (probably FGFR1), but signaling requires the
co-receptor Klotho (Razzaque 2009). When FGFR is
activated, there is reduction of NaPi-2a transcription
and less NaPi-2a on the basal cell surface of proximal
tubule cells, which in turn leads to renal phosphate
excretion (Shimada et al. 2004). At this point, it is not
clear how this occurs, as Klotho expression has been
clearly reported only in distal tubule cells so far (Farrow
et al. 2009). There is a secreted form of Klotho, and it
is possible that this circulating Klotho may play a role
in FGF23 signaling (Kurosu et al. 2006).
There is evidence that the phosphaturic action of
FGF23 is to some extent PTH-dependent. Subjects
with hypoparathyroidism, who have very low or
undetectable PTH levels, have high serum phosphorus
in the setting of high serum FGF23, which is consis-
tent with the need for PTH for the full phosphaturic
effect of FGF23 (Gupta et al. 2004). This observation
is further supported by the fact that in patients with
TIO and XLH, medically induced hypoparathyroidism
by cinacalcet results in increased renal phosphate
reabsorption and an increase in serum phosphorus
(Geller et al. 2007, Alon et al. 2008).
In addition to its action on NaPi-2a and NaPi-2c,
FGF23 is also a regulatory hormone for 1,25-vitamin D
(Shimada et al. 2004). Through downregulation of
1a-hydroxylase and up-regulation of 24-hydroxylase,
it leads to a decrease in 1,25-dihydroxy vitamin.
Other compounds such as frizzled related protein-4,
matrix extracellular phosphoglycoprotein, and FGF7 have
also been suggested to be phosphatonins (De Beur et al.
2002, White et al. 2006). However, the preponderance of
data to date suggests that FGF23 is the primary, if not the
only, clinically relevant phosphatonin.
Histopathology
Tumors associated with TIO have included a wide
range of histopathological diagnoses, and despite the
description and classification scheme proposed by
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
Weidner (1991) and Folpe et al. (2004), many
clinicians and pathologists continue to be unaware of
these tumors as a distinct entity. The prototypical
phosphaturic mesenchymal tumor (mixed connective
tissue variant) (PMTMCT) contains neoplastic cells
that are spindled to stellate in shape, normochromatic
with small nuclei and indistinct nucleoli. A spectrum of
histopathological features is shown in Fig. 2A–F.
The nuclear grade is low, and mitotic activity is usually
absent or very low. The cells are typically embedded
A B
C D
E F
Figure 2 TIO tumor histopathology. (A) This tumor area showsimmature mesenchymal cells, with no particular differentiation.There are areas of edema and blood lacunae (arrow). (B) Thisproliferation is solid, with what seems to be abundantintercellular matrix (arrow). The nuclei are typical with somevariation in shape and size. (C) Numerous and irregularvascular structures (arrows), as well as areas of solidproliferation, are seen in this tumor area. (D) This tumor iscomposed mostly of irregular vascular structures (arrows)embedded in a relatively soft matrix. Variations in size andshapes of the nuclei are evident. (E) Lattice-like areas withossification (arrow). (F) This photomicrograph was taken from alung metastasis. The area shows numerous very largeosteoclastic-like giant cells (arrows). Note that even though theproliferation is biologically malignant, there are few or nohistological signs of malignancy.
www.endocrinology-journals.org
within a myxoid or myxochondroid matrix with
‘grungy’ calcification that can resemble chondroid or
osteoid. Numerous osteoclast-like giant cells are a
frequent finding, and mature fat and even lamellar bone
may also be seen. A prominent feature of these tumors
is an elaborate intrinsic microvasculature with an
admixture of vessel size and vascular pattern (Folpe
et al. 2004). The most common diagnosis for these
tumors has been hemangiopericytoma, but it has also
included hemangioma, sarcomas, ossifying fibromas,
granulomas, giant cell tumors, and osteoblastomas
(Weidner 1991, Drezner 2001, Folpe et al. 2004).
Weidner (1991) reviewed the literature of w60
cases of TIO that had been described at that time.
They were the first to propose a classification system
based on the histological findings of their 16 cases
of TIO, and designated the tumors as phosphaturic
mesenchymal tumors. These were then subdivided into
four categories; mixed connective tissue variant
(PMTMCT), osteoblastoma-like variant, non-ossifying
fibroma-like variant, and ossifying fibroma-like variant.
The first group, PMTMCT, comprised neoplasias
containing primitive stromal cells, prominent vessel,
and osteoclast-like giant cells (Fig. 2D and F). Osseous
metaplasia and poorly formed cartilage-like areas with
dystrophic calcification were also present (Fig. 2B and
E). They noted that these tumors usually occurred in soft
tissue and were typically benign in behavior. The
remaining three groups tended to occur in bone and
were also typically benign in behavior. Folpe et al.
(2004) reviewed the clinic-pathological features of 32
new cases and re-reviewed all of the previously
published cases, and made the assertion that virtually
all of the cases fell into the category of PMTMCT.
Antigen expression was first evaluated in two
immunohistochemical studies by Weidner et al. In the
first study, the immunostainings were negative for
FVIII-related antigen, S-100, and cytokeratin (Weidner
et al. 1985). The second study revealed only vimentin
immunoreactivity in some cases within the tumor cells.
All other antibodies (desmin, S-100 protein, leu-M 1,
chromogranin, cytokeratin, neuron-specific enolase,
leukocyte common antigen, and factor VIII-related
antigen) were negative (Weidner et al. 1985). In their
series, Folpe et al. (2004) performed a series of
immunohistochemical stainings, including pan-cyto-
keratin, desmin, S-100, smooth muscle actin, CD34,
and FGF23. With the exception of smooth muscle actin,
which they found reactive in three cases, and FGF23,
which was positive in about 70% of all the cases
studied, all other markers were negative. In terms of
FGF23 staining, it is the proliferating cells within the
tumor that usually stain positive for FGF23 (Fig. 3A).
R57
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
In terms of ultrastructural features, Stone et al.
(1992) described features consistent with a neuroendo-
crine tumor in a PMTMCT from a 33-year-old woman.
Neurosecretory granules were also found in the case by
Wilkins et al. (1995). However, immunostaining for
typical markers of neurosecretory tumors, such as
S-100, neuron-specific enolase, chromogranin and
synaptophysin, were negative, as was staining for
actin, cytokeratin, epithelial membrane antigen, and
FVIII. The only positive finding was vimentin,
confirming what had been already described by
Weidner. An additional case communicated by
Shelekhova et al. (2006) also showed similar neuro-
secretory granules.
While typically benign, malignant presentation and
metastases can occur (Wyman et al. 1977, Rico et al.
Lung parenchyma
Metastasis
A
B
Figure 3 (A) FGF23 immunohistochemistry of lung metastasisof a PMTMCT. (B) The tumor shown in this slide was relativelyhomogeneous and composed mainly of spindle-shaped cells.Note the relatively benign histological appearance of theneoplasia (inset). FGF23 reactivity was present in almost 100%of the tumor cells.
R58
1986, Harvey et al. 1992, Ogose et al. 2001, Uramoto
et al. 2009). Two of the 19 tumors in our series went on
to metastasize (Figs 2F and 3B), a feature that is hard
to predict from the benign histological appearance.
While metastases are rare, infiltration of surrounding
connective tissue is typically present, which has
significant implications for surgical management and
emphasizes the importance for wide surgical margins
to avoid persistence or recurrence – a point that cannot
be emphasized enough in the management of TIO.
It seems to be that PMTMCT constitute a single,
albeit morphologically heterogeneous, histopatho-
logical entity. Regardless of tumor morphology, the
hallmark of the diagnosis of a PMTMCT is the
association of the tumor with the clinical syndrome
of TIO, which includes an elevation in plasma FGF23
and its disappearance after tumor resection. It is this
heterogeneity that may account for their frequent
misdiagnosis (Folpe et al. 2004).
To date, the immunohistochemical and electron
microscopy findings have not shed light on the cell of
origin of these neoplasias, but only served to confirm
their mesenchymal origin. It is also quite possible that
all of the tumors share a common origin: a primitive
mesenchymal cell that itself has the ability to secrete
the hormone and which can differentiate into several
cell lineages.
To summarize, PMTMCT are a group of tumors
with a spectrum of histopathologic findings that
include a background of spindle/stellate cells with
low nuclear and mitotic activity. This is true even
in cases of metastatic disease. Prominent vascularity
is common and includes vessels of different sizes
and patterns, consistent with the fact that they are
most commonly classified as hemangiopericytomas.
Osteoclast-like giant cells are frequently seen in these
tumors and mature fat or lamellar bone can be present
as well. FGF23 staining is positive and appears in the
cytoplasm of the tumor cells. It is important to note that
histopathologic diagnosis of malignant disease is
difficult, as even in clinically proven metastatic disease
the cellular features appear benign.
Clinical evaluation
A summary of our approach to the evaluation of these
patients can be found in Fig. 4, and a summary of
medical treatment recommendations can be found
in Box 1. Patients with TIO often present with
many years of symptoms before they are diagnosed.
The symptoms, which lead to their evaluation, are
non-specific, and often progressive. Common com-
plaints are bone pain, muscle weakness, and multiple
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Diagnosis and treatment of tumor-induced osteomalacia
2. Confirm diagnosis
1. Signs and symptoms of tumor-induced osteomalacia
• ↓ Pi, ↓ %TRP, ↓ 1,25-D, ↑ FGF23• Exclude familial forms (if appropriate) Check: PHEX, FGF23, DMP-1, ENPP1
3. Tumor localization
• Functional imaging: FDG-PET/CT + Octreoscan/CT
• Anatomical imaging: MRI and/or CT
• Venous sampling and/or aspiration (if necessary)
Medical treatmentUnsuccessful
localized Repeat localization (1 year?)
4. Surgical excision(with wide margins)
Figure 4 Summary of the approach to diagnosis and treatment of patients with TIO. Pi, phosphate; %TRP, tubular reabsorption ofphosphate; 1,25-D, 1,25-dihydroxyvitamin D.
Endocrine-Related Cancer (2011) 18 R53–R77
fractures (Jan de Beur 2005). Pediatric patients can
develop rickets and growth retardation (Jan de Beur
2005, Haeusler et al. 2010). These patients are often
misdiagnosed with a variety of musculoskeletal
ailments, rheumatologic diseases, and sometimes
even psychiatric disorders (Teasell & Shapiro 2002,
Lewiecki et al. 2008). Hypophosphatemia caused by
impaired renal phosphate reabsorption is the bio-
chemical hallmark of the disease. In many institutions,
phosphate is no longer part of the routine chemistry
panels, thus hypophosphatemia can often go unrecog-
nized, further delaying diagnosis (Halperin et al. 2007).
Box 1 Medical treatment of tumor-induced osteomalacia
Goal of therapy
Medication
Phosphorus
Calcitriol or alphacalcidiol
Monitoring
Baseline renal ultrasound for evaluation of nephrolithiasis/nephro
† Repeat if concerning symptoms arise or there is persistent inc
Serum calcium, phosphorus and PTH every 3 months
† If phosphorus level is below target, increase phosphorus supp
† If calcium level is below target, add/increase calcium supplem
† If PTH elevated, increase calcitriol
Urinary calcium (UCa) and creatinine (UCr) every 3 months (2nd
† If UCa/UCrR0.2, check for urinary hemoglobin. Also check 24h
calcium. Decrease calcitriol dose if positive urinary hemoglobin o
† If UCa/UCr %0.2, and serum phosphorus and PTH are at targ
www.endocrinology-journals.org
Differential diagnosis
The differential diagnosis for hypophosphatemia
should first be separated into genetic versus acquired
causes. Genetic causes include XLH, ADHR, and
ARHR, which are essentially biochemical phenocopies
of TIO. In all of these genetic forms of hypopho-
sphatemia, the plasma FGF23 is either directly
elevated or inappropriately normal. XLH is almost
invariably present in early childhood, while ADHR can
present in either childhood or adulthood (Econs &
McEnery 1997). Therefore, a detailed personal history
to identify the age of onset, which can often be aided by
Low-end of normal for age-appropriate normal range
of phosphorus
Dosing
15–60 mg/kg per day (w1–3 g/day for adults)
Dose should be divided into 4–5 times/day to improve
tolerance
15–60 ng/kg per day (w0.75–3 mg/day for adults)
Start 1.5 mg/day and titrate. Dose can be divided into
BID and TID
calcinosis.
rease in urinary calcium
lements
ents
AM void)
urinary calcium and creatinine with goal of normal range urinary
r elevated 24h urinary calcium
et, continue current regimen
R59
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
review of the growth chart in the case of young
patients, and a detailed family history, looking for
family members with short stature and bowed legs, is
especially important in children and young adults.
Some genetic forms, especially XLH, are associated
with dental findings, including enamel hypoplasia,
dental abscesses, and caries; thus, a detailed dental
history is important (Baroncelli et al. 2006). Generally,
the younger the patient is at presentation, the more
likely is a genetic cause. Additional genetic disorders
that can present with hypophosphatemia and should
be considered in the hypophosphatemic patient are
hereditary hypophosphatemic rickets with hypercal-
ciuria (HHRH; Bergwitz et al. 2006), X-linked
recessive hypophosphatemia (XLRH)/Dent’s disease
(Scheinman 1998), and the inherited renal Fanconi
syndromes, Fanconi-Bickel syndrome (MIM ID
227810) (Santer et al. 1997) and Fanconi renal tubular
syndrome (MIM ID 134600) (Lichter-Konecki et al.
2001). HHRH and XLRH differ from the other genetic
causes of hypophosphatemia in that hypercalciuria due
to increased 1,25-vitamin D accompanied by nephro-
calcinosis and/or nephrolithiasis is a feature. Patients
with XLRH also have proteinuria. Patients with the
genetic Fanconi syndromes have a more generalized
renal tubulopathy. This can include any combination of
Table 1 Differential diagnosis of hypophosphatemia
Disease Gene FGF23 Other findings
Genetic causes
XLH PHEX Elevated Childhood onset,
ADHR FGF23 Elevated Variable age of o
remit and recur
ARHR DMP-1,
ENPP1
Elevated May have consan
HHRH SLC34A3 Low Increased 1,25-vi
calcium, low PT
XLRH/Dent’s CLCN5 Unknown Male predominan
nephrocalcinos
renal failure
Inherited Fanconi Various Low Glucosuria, amino
proximal renal t
Acquired causes
TIO NA Elevated Variable age of o
TmP/GFR
Acquired Fanconi NA Low Glucosuria, amino
tubular acidosis
heavy metals, c
etc. (see text)
XLH, X-linked hypophosphatemic rickets; ADHR, autosomal-dominhypophosphatemic rickets; XLRH, X-linked recessive hypophosphahypercalciuria; XLRH, X-linked recessive hypophosphatemic ricket
R60
aminoaciduria, low molecular weight proteinuria,
bicarbonaturia, calciuria, and others. However, the
most important fact that distinguishes these genetic
syndromes of hypophosphatemia from TIO is that
plasma FGF23 is high in TIO, but low in HHRH and
Fanconi syndrome. FGF23 levels in XLRH have not
been reported, but should be low as well. The genes
for the genetic forms of hypophosphatemia, as well as
various features, are detailed in Table 1.
In addition to the genetic causes of non-TIO hypo-
phosphatemia, there are acquired causes. Most of the
acquired forms of hypophosphatemia are the result of
direct renal tubular damage by a drug or a toxin. Tubular
damage usually results in a generalized tubulopathy,
similar to what is seen in the genetic Fanconi-type
tubulopathies mentioned above (Bonnardeaux & Bichet
2008). This type of tubulopathy can be seen as a result
of burns (Nordstrom et al. 1977), heavy metal exposure
(cadmium, lead and arsenic) (Omdahl & DeLuca
1971, Aranami et al. 2010), aminoglycoside antibiotics,
certain chemotherapeutic agents, especially cisplatin,
and the anti-retroviral drug, tenfovir, which is used in
the treatment of HIV and hepatitis B (Davis et al. 1980,
Izzedine et al. 2003, Earle et al. 2004). It can also occur
in association with multiple myeloma and other dyspro-
teinemias (Dash et al. 1997). 1,25-vitamin D levels are
References
rickets and dental caries The HYP Consortium (1995)
nset, may spontaneously White et al. (2000)
guinity in parents Feng et al. (2006)
Lorenz-Depiereux et al. (2006a)
Levy-Litan et al. (2010)
Lorenz-Depiereux et al. (2010)
tamin D, increased urinary
H, and nephrocalcinosis
Bergwitz et al. (2006)
Ichikawa et al. (2006)
Lorenz-Depiereux et al. (2006b)
ce, hypercalciuria,
is, kidney stones, and
Pook et al. (1993)
Lloyd et al. (1996)
Oudet et al. (1997)
aciduria, calciuria, and
ubular acidosis
Chadha & Alon (2009)
nset, low 1,25-D, low Drezner (2001)
Jan de Beur (2005)
aciduria, proximal renal
, history of exposure to
hemotherapeutic agents,
Izzedine et al. (2003)
ant hypophosphatemic rickets; ARHR, autosomal-recessivetemic rickets; HHRH, hereditary hypophosphatemic rickets withs; TIO, tumor-induced osteomalacia.
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
variable, and can be low, as seen in TIO. Unlike TIO,
Fanconi-type syndromes tend to be associated with more
severe proximal renal tubular defects and metabolic
acidosis. Again, the key factor in discriminating these
sorts of disorders from TIO is the plasma FGF23 level,
which is low in cases of tubular damage, and high in TIO.
Other disorders which can be associated with hypopho-
sphatemia are hematologic malignancies, total parenteral
nutrition, organ transplant, refeeding syndrome, correc-
tion of diabetic ketoacidosis, and dietary deficiency.
Approach to the diagnosis of TIO
Diagnosis confirmation
TIO should be suspected in patients who present with
consistent symptoms and with hypophosphatemia.
If hypophosphatemia is present, the presence of renal
phosphate wasting should be confirmed.
Two ways to evaluate urinary phosphate wasting are
the calculation of percent tubular reabsorption of
phosphate (%TRP) and tubular maximum for phos-
phate corrected for glomerular filtration rate (GFR)
(TmP/GFR). Open access programs that calculate
%TRP and/or TmP/GFR can be found on the Internet
(by searching under these terms), but caution should be
exercised to enter the proper units (traditional versus
SI), as indicated by the site. TmP/GFR can be
calculated only in the fasting state, but %TRP can be
calculated at any time using the following formula:
100!ð1Kððurine phosphate=urine creatinineÞ
!ðserum creatinine=serum phosphateÞÞÞ:
When phosphate is normal, the normal range is
between 85 and 95%.
The TmP/GFR is another measure of renal phos-
phate handling and is independent of plasma phosphate
and renal function. TmP/GFR can be determined using
a nomogram or algorithm (Kenny & Glen 1973,
Walton & Bijvoet 1975). Use of the algorithm is less
cumbersome and there is no clinically significant
difference between the values obtained, making it the
preferred method (Barth et al. 2000). The formula used
to calculate TmP/GFR is dependent on the value of
TRP and can be calculated using the formulas below:
If TRP is %0:86 ð86%Þ; then TmP=GFR
ZTRP!phosphate:
If TRP is O0:86 ð86%Þ; then TmP=GFR
Z 0:3!TRP=ð1K0:8!TRPÞ!phosphate:
www.endocrinology-journals.org
In using these algorithms, it is important that units
for urine creatinine, urine phosphate, serum creatinine,
and serum phosphate are consistent.
The normal reference ranges for TmP/GFR are age-
and gender-dependent, but have not been well defined
in large patient populations. Reasonable age- and
gender-dependent values are listed in Table 2, and are
derived from data compiled by Stark et al. (1986),
Alon & Hellerstein (1994) and Payne (1998).
TmP/GFR should be calculated from testing done in
the fasting state, typically from second morning-void
urine and blood samples taken at the same time. In the
non-disease state, the values for TRP and TmP/GFR
will be high when there is hypophosphatemia. In TIO,
these values are abnormally low. It is important to note
that the calculations of %TRP and TmP/GFR in
patients with suspected TIO need to be done off of
phosphate supplementation. Phosphate supple-
mentation will elevate urinary phosphate in both
subjects with and without TIO and can lead to falsely
low determinations of %TRP and TmP/GFR in patients
without the disease.
After confirming renal phosphate wasting as the
etiology for hypophosphatemia, additional lab tests
can be helpful in making the diagnosis of TIO. 1,25-
vitamin D can be low or inappropriately normal.
Calcium and PTH are usually normal, but PTH can
be high reflecting secondary hyperparathyroidism,
which is the normal response to low 1,25-vitamin D
caused by elevated FGF23. Measurement of blood
FGF23 is now commercially available and is essential
for the diagnosis. Currently, only the less-sensitive
C-terminus assay is widely available for commercial
testing (Imel et al. 2006). It is important to note that
this is only valid for plasma, not serum, samples. The
most sensitive and specific assay for use in the
diagnosis of TIO is the intact assay manufactured by
Kainos (Imel et al. 2006), which at present is typically
only used in research laboratories.
Once the diagnosis of an FGF23-dependent, phos-
phate wasting disorder is made, a thorough history can
aid in excluding the genetic causes, such as XLH,
ADHR, and ARHR. Genetic testing can also be done.
Having narrowed the diagnosis to TIO, a careful
physical examination should be performed, as the
tumors that cause TIO can sometimes be found in the
subcutaneous tissue (Fig. 5; Colt et al. 2005, Dewitt
et al. 2007, Ogura et al. 2008). It is also prudent to ask
the patient if she/he has noticed any new ‘lumps’ or
‘bumps’ as well as to examine the oral cavity, as
tumors have been reports in the jaws (Yun et al. 2009).
R61
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Table 2 Normal ranges for tubular maximum for phosphate
corrected for GFRa
Age Male mg/dl (mmol/l) Female mg/dl (mmol/l)
Newborn 5.7–8.1 (1.27–2.59) 5.7–8.1 (1.27–2.59)
1 month–2 years 3.6–5.4 (1.15–1.73) 3.6–5.4 (1.15–1.73)
2–12 years 3.8–5.0 (1.22–1.60) 3.8–5.0 (1.22–1.60)
12–16 years 3.4–4.6 (1.09–1.47) 3.4–4.6 (1.09–1.47)
16–25 years 3.33–5.9 (1.07–1.89) 3.18–6.41 (1.02–2.05)
25–45 years 3.09–4.18 (0.99–1.34) 2.97–4.45 (0.95–1.42)
45–65 years 2.78–4.18 (0.89–1.34) 2.72–4.39 (0.87–1.40)
65–75 years 2.47–4.18 (0.79–1.34) 2.47–4.18 (0.79–1.34)
aReferences (Stark et al. 1986, Alon & Hellerstein 1994,Payne 1998).
W H Chong et al.: Tumor-induced osteomalacia
Localizing studies: functional imaging
As tumors can arise in bone or soft tissue, occur from
head to toe, and are typically very small in size, locating
these tumors is often quite challenging. We have seen
in our cohort of patients that tumors are found more
commonly in bone than in soft tissue.
We advocate a stepwise approach, first performing
functional tests. In our hands, F-18 fluorodeoxyglucose
positron emission tomography, with computed tomo-
graphy (FDG-PET/CT), has proven to be most sensitive
for localizing TIO tumors (Dupond et al. 2005b,
Andreopoulou et al. 2010a, Jagtap et al. 2010). However,
while FDG-PET/CT may be very sensitive, it is also
non-specific and identifies areas of metabolic activity
A B
C
Figure 5 Subcutaneous TIO tumor detectable on physical examinatshown in Fig. 1) revealed a subcutaneous nodule (A). This nodule won functional imaging, FDG-PET (B). The lesion was visualized on(C, arrow). On the low-power view, it can be seen that the tumor wahigh-power view, it was revealed that the calcification in the lesion wTIO resolved after excision.
R62
that are not tumors. This is especially true in patients with
many areas of active fracture healing (Fig. 6).
Another important functional imaging modality is111Indium octreotide scintigraphy, ideally combined with
single photon emission CT and CT (SPECT/CT).
Somatostatin receptors have been found to be present
on many TIO tumors (Duet et al. 2008) and 111Indium-
octreotide has a high affinity for the somatostatin receptor,
especially subtype 2 and to a lesser extent subtype 5. As
with FDG PET/CT, emphasis should be placed on making
sure these imaging tests cover the entire body, from
head to toe, including the hands and feet. Standard
PET/CT and octreotide often exclude portions of the
extremities and may exclude portions of the head.
Addition of co-registered CT to both FDG-PET and
octreotide significantly increases the ability to localize
tumors, and whenever possible it should be performed.
More recently, 68Ga-DOTANOC PET/CT has been
explored as a means of finding TIO tumors (Hesse et al.
2007a, von Falck et al. 2008). This scan combines the
specificity of octreotide scanning with the sensitivity of
PET/CT. It utilizes a modified octreotide molecule
(DOTANOC) that has increased affinity for both
somatostatin receptor 2 and 5 (Wild et al. 2003,
2005). Labeling this compound with the positron
emitter 68Ga results in a PET compound that may be
more specific than FDG. However, studies comparing68Ga-DOTANOC PET/CT with standard octreotide
SPECT/CT in the diagnosis of TIO have not been done.
D
E
ion. Physical examination of a patient with TIO (the same patientas implicated as the culprit tumor by the fact that it was detectedCT scan, which also suggested intralesional calcifications completely contained in the subcutaneous tissue (D), and on aas actually ossification and contained areas of lamellar bone (E).
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

A B
C
D
E
Figure 6Multiple imaging modalities may be needed to localize TIO tumors. In this patient, FDG PET/CT revealed multiple areas ofincreased uptake (A). Octreotide scan only demonstrated a single lesion (B–D). MRI revealed a tumor in the area identified onfunctional imaging (E). TIO resolved after excision of the lesion.
Endocrine-Related Cancer (2011) 18 R53–R77
201Thallium and 99Technetium MIBI scintigraphy
have been used in TIO (Kimizuka et al. 2004, Hodgson
et al. 2006), but we have not found them to add
anything to FDG-PET or octreotide scanning.99Tc-MDP bone scintigraphy (bone scans) has not
proven to be a useful study in localization of TIO
tumors (Lee et al. 1995, Garcia & Spencer 2002). It
often reveals multiple foci of uptake at areas of fracture
and may actually misdirect the effort to localize the
tumor. Of interest, though, is that 99Tc-MDP bone
scans often show uptake at the costochondral junctions
(a sort of adult rachitic rosary) and areas of the bone in
skeletally mature adults where the growth plates were
previously located (Fig. 7). This finding should suggest
the diagnosis of TIO, and may represent a sort of
‘pseudo-reactivation’ of growth plates in the adult
skeleton. The pathophysiology underlying this inter-
esting and frequently observed phenomenon is unclear
at this time. Importantly, this finding should not be
misinterpreted as evidence of metastatic tumor, as it
sometimes is.
Localizing studies: anatomical imaging
Once suspicious lesions have been identified with
functional imaging, one should proceed to anatomical
imaging to confirm the location of the tumor. Anatomic
imaging studies include X-rays, CT, and/or magnetic
www.endocrinology-journals.org
resonance imaging (MRI) scans. Some investigators
advocate total body MRI as an initial imaging study.
However, we have not found this approach to be
fruitful. It should also be noted that there are ‘blind’
spots on both FDG-PET and octreoscan; brain uptake
on FDG-PET may obscure intracranial tumors, and
liver and spleen uptake of octreotide may obscure
potential lesions in these regions. Therefore, ana-
tomical imaging of these areas may be indicated if a
tumor has not been identified.
Venous sampling
Usually, the combination of functional and anatomical
imaging is successful in localizing the FGF23-
secreting tumor. However, there are certain circum-
stances in which more certainty and testing are
indicated. Often, more than one lesion is found on
functional imaging, particularly FGD-PET, each with a
reasonable degree of suspicion or the suspicious lesion
is located in an area where the indicated operation is
associated with a high level of potential morbidity. In
these cases, additional certainty and testing are
indicated. Of particular utility is selective venous
sampling with measurement of FGF23 (Andreopoulou
et al. 2010b). Examples of the utility of venous
sampling in distinguishing between multiple sites and
difficult sites are shown in Fig. 8.
R63
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

A
B
C
Figure 7 ‘Pseudo-reactivation’ of growth plate. X-ray (A) andbone scan (B) of a patient with TIO showing areas of intensetracer uptake and evidence of ‘growth plates’ in a 54-year-oldman who had senesced his growth plates decades before. Abone scan of another adult patient with TIO (C) revealed intensetracer uptake at multiple costochondral junctions, creating thenuclear medicine equivalent of an adult rachitic rosary.
W H Chong et al.: Tumor-induced osteomalacia
Venous sampling has been attempted in localizing
tumors in the absence of any suspicious lesions
identified on either functional or anatomical imaging,
the so called ‘blind sampling’. In a trial designed to test
this, we were unable to localize tumors by venous
sampling without a ‘target’ lesion suggested by
anatomical or functional imaging, and concluded this
was not a useful approach (Andreopoulou et al.
2010b). Van Boekel et al. (2008) advocated a two-
step approach to venous sampling. They suggested that
if a suspected tumor cannot be localized by imaging,
whole body venous sampling can be performed with
assessment of the average values for samples from
different anatomical regions. If the average values in
samples from a region appear to be higher, more
detailed sampling is performed in the smaller branches
of the veins in that region. In the one patient studied by
this approach, it appeared to suggest a particular
region. However, in retrospect, the tumor was evident
on an MRI that had been performed prior to venous
sampling, so the utility of this approach is not clear.
An additional approach that can be used for
confirmation that a suspicious lesion identified on
functional or anatomical imaging is the culprit tumor is
aspiration of the lesion. Elevated FGF23 in the aspirate
R64
is diagnostic of a culprit lesion. Inspection of cells in
the aspirate may reveal cellular morphology consistent
with that of a phosphaturic mesenchymal tumor,
further supporting that the aspirated lesion represents
the culprit lesion (Sciubba et al. 2009).
Despite all of the advances in imaging that are
available today, tumor localization may not be
successful. If this is the case, imaging studies should
be repeated in hopes that a tumor may be more evident
with time. This can be done every 1–2 years.
Treatment
Surgical resection
The treatment of choice for TIO is resection of the
tumor with a wide margin to insure complete resection.
Resection with a wide surgical margin is of utmost
importance, as recurrences of these tumors have been
reported (Clunie et al. 2000, Ogose et al. 2001,
Uramoto et al. 2009). Tumor resection is almost
always curative, and following complete resection of
the tumor, there is relatively rapid improvement.
FGF23 has a halflife of w45 min and disappears
rapidly from the circulation (Khosravi et al. 2007). The
majority of patients demonstrate surgical cure, as
evidenced by the return of serum phosphate to normal,
by post-operative day 5. Some patients may take as
long as 10 days and, in children, we have seen
phosphate return to normal in as few as 2 days. In fact,
it is return of serum phosphorus to normal after tumor
resection that confirms the diagnosis of TIO. Most
patients feel better within days to weeks of tumor
removal. Bone healing starts immediately, but depend-
ing on the severity of the disease, it may take up to a
year or more for significant clinical improvement.
Late recurrence due to metastatic disease is rare but
possible. This probably occurs in !5% of the patients
with TIO (Ogose et al. 2001, Folpe et al. 2004). Lung is
a common site for metastasis (Fig. 3), and as such
should be closely evaluated when there is a late
recurrence without evidence of local disease. The
lesions can be quite small and difficult to visualize.
Therefore, high-resolution CT is recommended as the
imaging modality of choice. More advanced disease
may present with a miliary pattern. The course after
metastasis is quite variable, and survival of up to 30
years has been reported (Harvey et al. 1992). In our
series of 31 cases of TIO, we have seen two cases of
recurrence due to tumor metastasis in the lung (Fig. 3).
There is no chemotherapeutic regimen with any
demonstrated efficacy in treating metastatic TIO.
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

600
700C
L acetabular lesion
A
400
500
R patellar lesion
200
300
FG
F23
(pg
/ml)
0
100B
D E F
90100G H
4050607080
0102030
FG
F23
(pg
/ml)
Catheter
Catheter tip
Venous sampling
r pop
abo
ve p
atell
a
Periph
eral
Right a
nter
ior fa
cial
Right s
inus
Right ju
gular
(low
)
Super
ior sa
gitta
l sinu
s (m
id)
Super
ior sa
gitta
l sinu
s (m
id)
Super
ior sa
gitta
l sinu
s (po
sterio
r)
Super
ior sa
gitta
l sinu
s (an
terio
r)
Right p
etro
sal
r sfv
high
r dee
p fe
m lo
w
r com
mon
iliac
r sfv
lower
r int
erna
l iliac
r hep
atic
r low
er p
op ve
ry lo
w
r pop
abo
ve kn
ee
r pop
very
high
R pop
low
I ver
tebr
al
I sfv
prox
I sfv
very
low d
istal
I circ
umfle
x pro
x
I com
mon
iliac
I com
mon
fem
I int il
iac is
ch b
orde
r
I int il
iac lo
w
I inte
rnal
iliac s
mall
I inte
rnal
iliac l
arge
bra
nch.
..
I inte
rnal
iliac l
arge
bra
nch
low
I inte
rnal
iliac l
arge
bra
nch.
.
I int il
iac m
id
Figure 8 Utility of selective venous sampling in TIO. Selective venous sampling is useful in distinguishing between multiple suspectlesions, as in this patient who had uptake at both the region of the acetabulum (A) and patella (B). Elevated FGF23 in the veinsdraining the acetabular region (C) identified the lesion in this region as the causative lesion. Selective venous sampling is also usefulin identifying lesions in places difficult to image or to approach surgically. The brain shows generalized increased uptake on FDGPET/CT (D) making identification of a lesion in this area difficult. A lesion was seen on octreotide scan (E) and MRI (F). It was felt thatthis could be a TIO tumor or a meningioma, which are also octreotide positive. Venous sampling (G) demonstrated elevated FGF23(H), confirming that the lesion was the FGF23-secreting tumor. This material is reproduced from Andreopoulou et al. 2010b withpermission of John Wiley & Sons, Inc. r, right; l, left; pop, popliteal; sfv, superficial femoral vein; fem, femoral; prox, proximal; comm,common; int, internal; isch, ischial; mid, middle.
Endocrine-Related Cancer (2011) 18 R53–R77
Radiofrequency ablation (RFA) has also been
reported as a possible treatment modality (Hesse et al.
2007b). Hesse et al. reported use of RFA in a 40-year-old
woman with TIO, in whom the tumor was located in
the femoral head. In order to preserve the hip joint,
CT-guided RFA was performed in two rounds of
www.endocrinology-journals.org
treatment. The patient showed complete biochemical
and symptomatic recovery within weeks, and had
unremarkable follow-up at 1 year. While this is
promising, long-term follow-up and effectiveness in
other cases remains to be seen. Again, the need for a wide
margin is advocated to avoid recurrence or metastasis.
R65
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
Medical treatment
When the tumor cannot be localized or is not surgically
resectable, medical therapy with phosphate supple-
mentation and calcitriol or alfacalcidiol is used. The
treatment regimen that follows is essentially the same
as that used in non-TIO causes of hypophosphatemia.
When initiating treatment, it is prudent to check
weekly labs to guide titration of medications until
treatment targets are reached.
Phosphorus supplementation is the mainstay of
treatment. However, since phosphorus is rapidly
absorbed and cleared, multiple doses throughout the
day are necessary (at least 3–4 times per day) in an
attempt to try and get the serum phosphorus to the
lower end of the age-appropriate normal range.
Treatment to the lower end of the age-appropriate
range has been shown by bone biopsy to improve bone
disease. Whether treatment to targets below the normal
range is effective remains unclear. GI upset and
diarrhea may develop as a result of phosphate
supplementation. GI side effects can sometimes be
alleviated with divided dosing and administration with
food; however, they should not be provided with
calcium-rich foods. GI side effects can also sometimes
be avoided by using concentrated oral preparations that
were developed for treatment of constipation or for
bowel preparation prior to endoscopy. Various
phosphorus supplement preparations are listed in
Table 3. Secondary hyperparathyroidism can be seen
on presentation, due to suppression of 1,25-vitamin D
by FGF23, or it can develop as a result of phosphorus
supplementation. Active vitamin D (calcitriol or
alfacalcidiol) is used to prevent or treat secondary
hyperparathyroidism. The dose is titrated to keep the
PTH in the normal range. In the early phases of the
treatment of severe bone disease, additional calcium
supplementation may be necessary to provide mineral
ion substrate to heal the bone. Addition of calcium
supplements or increases in calcitriol and/or alfacalci-
diol are indicated for difficult to suppress PTH, very
Table 3 Phosphorus supplements
Phosphorus source Amount of elemental phosphorus
Neutra-Phos 8 mmol (248 mg) per capsule/packet
Neutro-Phos K 8 mmol (248 mg) per capsule/packet
K-Phos Neutral 8 mmol (248 mg) per tablet
K Phos Original 3.68 mmol (114 mg) per tablet
Fleet’s Phospho Soda 4.15 mmol (128.65 mg) per ml
Joulies Solution Varies depending on compounding
pharmacy
Adapted fromhttp://www.globalrph.com/phosphate_supplements.htm; http://www.newbornnetworks.org.uk/staffs/Nutrition.pdf.
R66
low urinary calcium, or hypocalcemia. As bone healing
progresses, the regimen needs to be modified. Calcium
supplementation and/or active vitamin D treatment
usually need to be decreased. One consequence of
over-treatment with active vitamin D is the develop-
ment of hypercalciuria and the risk for nephrocalci-
nosis/nephrolithiasis. While on chronic treatment,
periodic measurement of urine calcium should be
performed. Prolonged phosphorus supplementation can
lead to the development of tertiary hyperparathy-
roidism. This may require partial parathyroidectomy,
or treatment with the calcium-sensing receptor
agonist, cinacalcet.
The treatment regimen is to give 15–60 mg/kg per
day of elemental phosphorus (typically 1–3 g/day)
divided into 4–6 doses. Various formulations with
varying amounts of phosphorus are available (Table 3).
Calcitriol or alfacalcidiol is given at 15–60 ng/kg
per day, with a typical starting dose of 1.5 mg/day in
an adult.
A new treatment approach that holds promise, but
needs additional study for confirmation of efficacy and
establishment of safety, is cinacalcet, an agonist of the
calcium-sensing receptor that lowers blood PTH levels
(Geller et al. 2007). The use of cinacalcet was
advocated on the basis of evidence that FGF23 action
was PTH-dependent. Gupta et al. (2004) found that
both FGF23 and serum phosphorus were high in the
blood of patients with hypoparathyroidism, indicating
that in the absence of PTH, FGF23 was unable to
adequately lower blood phosphorus level. This led to
the notion that medically induced hypoparathyroidism
may be a potential treatment for TIO (Geller et al.
2007). In this paper, we were able to show that
cinacalcet increased %TRP and serum phosphorus,
allowed for a decrease in phosphate supplementation,
and led to bone healing, as assessed by iliac crest bone
biopsy. However, hypercalciuria developed, necessi-
tating the addition of a thiazide diuretic to lower
urinary calcium. Similarly, cinacalcet has shown
promise in treating patients with XLH, which is
also a disorder of excess FGF23 (Alon et al. 2008,
Yavropoulou et al. 2010). In patients with TIO
treated with cinacalcet, urinary calcium must be
monitored carefully to avoid nephrocalcinosis and/or
nephrolithiasis.
A previously advocated treatment for TIO was the
somatostatin analog, octreotide (Seufert et al. 2001).
This appeared to be a logical treatment, given the
presence of somatostatin receptors on the cell surface
of TIO tumors and the ability to detect the tumors with
radiolabeled octreotide. However, most clinicians have
not been able to reproduce the success that Seufert
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
et al. had in the single patient they reported. Paglia
et al. (2002) reported their lack of success in a single
patient, and we saw no effect in five patients and have
abandoned further attempts (unpublished data).
Few reports are available on the role of external
beam radiation or chemotherapy in the treatment of
these patients. In the few reports that mention radiation
therapy, there does not appear to be any clear benefit
(Fuentealba et al. 2003, Uramoto et al. 2009).
Chemotherapy regimens that have been reported
include the combination of cisplatin, doxorubicin,
and methotrexate (Terek & Nielsen 2001) as well as
the use of dasatinib (Peters et al. 2010). In the first case,
chemotherapy was pursued as the first option as the
patient was felt to have an osteosarcoma. Following
treatment, the patient’s phosphate normalized (Terek
& Nielsen 2001). In the second case, the patient had
recurrent disease that could not be completely resected.
Due to strongly positive immunohistochemical stain-
ing of platelet-derived growth factor receptor, the
patient was started on dasatinib and has been stable on
this therapy (Peters et al. 2010). Given the limited
evidence for either radiation or chemotherapy, their
roles in treating these patients are not known at this
time. Given the slowly proliferating nature of these
tumors, we would expect that these treatments would
have little efficacy.
Medical treatment: monitoring
A baseline ultrasound should be obtained, and blood
and urine studies should be monitored approximately
every 3 months. For urine tests, checking the second
morning void for calcium and creatinine is suggested to
assess for hypercalciuria. If the calcium/creatinine is
R0.2, urinalysis should be done to check for the
presence of blood in the urine. If this is present,
calcitriol should be decreased and a 24 h urine for
calcium and creatinine should be checked with a goal
of obtaining normal urinary calcium/creatinine ratio. If
this remains elevated, calcitriol should be decreased
further. If calcium/creatinine is !0.2 and the serum
phosphorus and PTH are within targets, the current
regimen can be maintained. A summary of medical
therapy and monitoring is provided in Box 1.
Future directions
Treatment with calcitonin has also been explored as a
method to suppress FGF23 (van Boekel et al. 2008). A
single s.c. injection of calcitonin was able to suppress
FGF23 levels by 44.6% at 9 h post-injection. Long-
term treatment, however, was not pursued as focus was
www.endocrinology-journals.org
given to localization of the tumor. We were unable to
replicate these findings in a single patient in whom we
attempted this treatment (unpublished data). Whether
calcitonin is truly an effective treatment awaits further
investigation.
Future treatment will likely be guided by a better
understanding of the biology of FGF23 and the nature
of these tumors. Recent investigations have led to a
rudimentary understanding of FGF23 synthesis, post-
translational modification, and signaling. While eluci-
dation of the transcriptional and translational
regulation of FGF23 is still lacking, it has become
evident that posttranslational glycosylation by UDP-N-
acetyl-alpha-D-galactosamine:polypeptide N-acetylga-
lactosaminyltransferase 3 (GalNAc-T3) is essential for
the secretion of biologically active FGF23 (Topaz et al.
2004, Benet-Pages et al. 2005, Frishberg et al. 2005,
Dumitrescu et al. 2009). The fact that patients null for
GalNAc-T3 have a phenotype that is completely
confined to abnormalities in mineral metabolism
suggests that GalNAc-T3 may eventually be a
therapeutic target for diseases of FGF23 excess, such
as TIO. A promising therapeutic currently in clinical
trials are monoclonal antibodies that target the FGF23–
FGFR1 interaction (Aono et al. 2009, 2010). A
monoclonal antibody that disrupts the interaction of
FGF23 with the FGFR appears to be the mechanism of
action with this approach.
Conclusion
TIO is a fascinating paraneoplastic syndrome caused
by unregulated over-secretion of the recently identified
phosphate and vitamin D regulating hormone, FGF23.
It is a debilitating disease that is cured with excision of
the tumors. The benign-appearing histopathology
typically seen in these tumors can be misleading, as
even clinically proven metastatic disease can have
benign cellular features. While the tumors can be
difficult to locate, a step-wise approach that involves
functional imaging, followed by anatomical imaging,
and, if necessary, selective venous sampling or
aspiration for confirmation is usually successful.
Excision with wide margins is important to avoid late
recurrence. When tumors cannot be identified, medical
treatment can be successful though periodic surveil-
lance is necessary.
Declaration of interest
The authors declare that there is no conflict of interest that
could be perceived as prejudicing the impartiality of the
research reported.
R67
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
Funding
This work was supported by the funding from the Division of
Intramural Research, National Institutes of Dental and
Craniofacial Research, National Institutes of Health,
Bethesda, Maryland, USA.
References
ADHR Consortium 2000 Autosomal dominant hypopho-
sphataemic rickets is associated with mutations in FGF23.
The ADHR Consortium. Nature Genetics 26 345–348.
(doi:10.1038/81664)
Ahn JM, Kim HJ, Cha CM, Kim J & Yim SG 2007
Oncogenic osteomalacia: induced by tumor, cured by
surgery. Oral Surgery, Oral Medicine, Oral Pathology,
Oral Radiology, and Endodontics 103 636–641. (doi:10.
1016/j.tripleo.2005.12.027)
Albright F & Reifenstein E 1948 Clinical hyperparathyroid-
ism. In Parathyroid Glands & Metabolic Bone Disease,
ch 3, p 56. Baltimore, MD, USA: The Williams & Wilkins
Company.
Albright F, Butler AM & Bloomber E 1937 Rickets resistant
to vitamin D therapy. American Journal of Diseases of
Children 54 529–547.
Alon U & Hellerstein S 1994 Assessment and interpretation
of the tubular threshold for phosphate in infants and
children. Pediatric Nephrology 8 250–251. (doi:10.1007/
BF00865491)
Alon US, Levy-Olomucki R, Moore WV, Stubbs J, Liu S &
Quarles LD 2008 Calcimimetics as an adjuvant treatment
for familial hypophosphatemic rickets. Clinical
Journal of the American Society of Nephrology 3
658–664. (doi:10.2215/CJN.04981107)
Andreopoulou P, Dumitrescu CE, Kelly MH, Brillante B,
Cutler CM, Wodajo FM, Chang R & Collins MT 2010a
Selective venous catheterization for the localization of
phosphaturic mesenchymal tumors. Journal of Bone and
Mineral Research [in press]. (doi:10.1002/jbmr.316)
Andreopoulou P, Millo C, Reynolds J, Kelly M, Brillante B,
Wodajo FM, Chang R, Chen CC & Collins MT 2010b
Multimodality Diagnosis and Treatment of Tumor
Induced Osteomalacia. Endocrine Reviews
31 (Supplement 1) OR08–6 S49.
Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H,
Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S
et al. 2009 Therapeutic effects of anti-FGF23 antibodies
in hypophosphatemic rickets/osteomalacia. Journal of
Bone and Mineral Research 24 1879–1888. (doi:10.1359/
jbmr.090509)
Aono Y, Hasegawa H, Yamazaki Y, Shimada T, Fujita T,
Yamashita T & Fukumoto S 2010 Anti-FGF23
neutralizing antibodies ameliorate muscle weakness and
decreased spontaneous movement of Hyp mice.
Journal of Bone and Mineral Research 26 803–810.
(doi:10.1002/jbmr.275)
Aranami F, Segawa H, Furutani J, Kuwahara S, Tominaga R,
Hanabusa E, Tatsumi S, Kido S, Ito M & Miyamoto K
R68
2010 Fibroblast growth factor 23 mediates the phos-
phaturic actions of cadmium. Journal of Medical
Investigation 57 95–108. (doi:10.2152/jmi.57.95)
Auethavekiat P, Roberts JR, Biega TJ, Toney MO,
Christensen RS, Belnap CM & Berenberg JL 2005 Case 3.
Oncogenic osteomalacia associated with hemangioper-
icytoma localized by octreotide scan. Journal of Clinical
Oncology 23 3626–3628. (doi:10.1200/JCO.2005.05.043)
Bahrami A, Weiss SW, Montgomery E, Horvai AE, Jin L,
Inwards CY & Folpe AL 2009 RT-PCR analysis for
FGF23 using paraffin sections in the diagnosis of
phosphaturic mesenchymal tumors with and without
known tumor induced osteomalacia. American Journal of
Surgical Pathology 33 1348–1354. (doi:10.1097/PAS.
0b013e3181aa2311)
Baroncelli GI, Angiolini M, Ninni E, Galli V, Saggese R &
Giuca MR 2006 Prevalence and pathogenesis of dental
and periodontal lesions in children with X-linked
hypophosphatemic rickets. European Journal of
Paediatric Dentistry 7 61–66.
Baronofsky SI, Kalbhen CL, Demos TC & Sizemore GW
1999 Oncogenic osteomalacia secondary to a hemangio-
pericytoma of the hip: case report. Canadian Association
of Radiologists Journal 50 26–28.
Barth JH, Jones RG & Payne RB 2000 Calculation of renal
tubular reabsorption of phosphate: the algorithm performs
better than the nomogram. Annals of Clinical Biochemistry
37 79–81. (doi:10.1258/0004563001901371)
Beech TJ, Rokade A, Gittoes N & Johnson AP 2007 A
haemangiopericytoma of the ethmoid sinus causing
oncogenic osteomalacia: a case report and review of the
literature. International Journal of Oral and Maxillofacial
Surgery 36 956–958. (doi:10.1016/j.ijom.2007.03.005)
Benet-Pages A, Orlik P, Strom TM & Lorenz-Depiereux B
2005 An FGF23 missense mutation causes familial
tumoral calcinosis with hyperphosphatemia. Human
Molecular Genetics 14 385–390. (doi:10.1093/hmg/
ddi034)
Bergwitz C & Juppner H 2010 Regulation of phosphate
homeostasis by PTH, vitamin D, and FGF23. Annual
Review of Medicine 61 91–104. (doi:10.1146/annurev.
med.051308.111339)
Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe
M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO,
Anderson D et al. 2006 SLC34A3 mutations in patients
with hereditary hypophosphatemic rickets with hyper-
calciuria predict a key role for the sodium-phosphate
cotransporter NaPi-IIc in maintaining phosphate homeo-
stasis. American Journal of Human Genetics 78 179–192.
(doi:10.1086/499409)
van Boekel G, Ruinemans-Koerts J, Joosten F, Dijkhuizen P,
van Sorge A & de Boer H 2008 Tumor producing
fibroblast growth factor 23 localized by two-staged
venous sampling. European Journal of Endocrinology
158 431–437. (doi:10.1530/EJE-07-0779)
Bonnardeaux A & Bichet D 2008 Inherited disorders
associated with generalized dysfunction of the proximal
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
tubule (renal Fanconi syndrome). In Brenner & Rectors:
The Kidney, 8 edn, pp 1390–1399. Ed B Brenner.
Philadelphia, PA: Saunders Elsevier.
Cai Q, Hodgson SF, Kao PC, Lennon VA, Klee GG,
Zinsmiester AR & Kumar R 1994 Brief report: inhibition
of renal phosphate transport by a tumor product in a
patient with oncogenic osteomalacia. New England
Journal of Medicine 330 1645–1649. (doi:10.1056/
NEJM199406093302304)
Carey DE, Drezner MK, Hamdan JA, Mange M, Ahmad MS,
Mubarak S & Nyhan WL 1986 Hypophosphatemic
rickets/osteomalacia in linear sebaceous nevus syndrome:
a variant of tumor-induced osteomalacia. Journal of
Pediatrics 109 994–1000. (doi:10.1016/S0022-3476(86)
80283-9)
Casari S, Rossi V, Varenna M, Gasparini M, Parafioriti A,
Failoni S & Sinigaglia L 2003 A case of oncogenic
osteomalacia detected by 111In-pentetreotide total body
scan. Clinical and Experimental Rheumatology 21
493–496.
Chadha V & Alon US 2009 Hereditary renal tubular
disorders. Seminars in Nephrology 29 399–411. (doi:10.
1016/j.semnephrol.2009.03.013)
Cheung FM, Ma L, Wu WC, Siu TH, Choi PT & Tai YP 2006
Oncogenic osteomalacia associated with an occult
phosphaturic mesenchymal tumour: clinico-radiologico-
pathological correlation and ultrastructural studies. Hong
Kong Medical Journal 12 319–321.
Chouhan V, Agrawal K, Vinothkumar TK & Mathesul A
2010 Bilateral insufficiency fracture of the femoral head
and neck in a case of oncogenic osteomalacia. Journal of
Bone and Joint Surgery. British Volume 92 1028–1031.
(doi:10.1302/0301-620X.92B7.24526)
Clunie GP, Fox PE & Stamp TC 2000 Four cases of acquired
hypophosphataemic (‘oncogenic’) osteomalacia.
Problems of diagnosis, treatment and long-term management.
Rheumatology 39 1415–1421. (doi:10.1093/rheumatology/
39.12.1415)
Collins MT, Chebli C, Jones J, Kushner H, Consugar M,
Rinaldo P, Wientroub S, Bianco P & Robey PG 2001
Renal phosphate wasting in fibrous dysplasia of bone is
part of a generalized renal tubular dysfunction similar to
that seen in tumor-induced osteomalacia. Journal of Bone
and Mineral Research 16 806–813. (doi:10.1359/jbmr.
2001.16.5.806)
Collins MT, Lindsay JR, Jain A, Kelly MH, Cutler CM,
Weinstein LS, Liu J, Fedarko NS & Winer KK 2005
Fibroblast growth factor-23 is regulated by
1alpha,25-dihydroxyvitamin D. Journal of Bone
and Mineral Research 20 1944–1950. (doi:10.1359/
JBMR.050718)
Colt E, Gopan T & Chong HS 2005 Oncogenic osteomalacia
cured by removal of an organized hematoma. Endocrine
Practice 11 190–193.
Dash T, Parker MG & Lafayette RA 1997 Profound
hypophosphatemia and isolated hyperphosphaturia in
www.endocrinology-journals.org
two cases of multiple myeloma. American Journal of
Kidney Diseases 29 445–448. (doi:10.1016/S0272-6386
(97)90207-9)
Davis S, Kessler W, Haddad BM & Maesaka JK 1980 Acute
renal tubular dysfunction following cis-dichlorodiammine
platinum therapy. Journal of Medicine 11 133–141.
De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D,
Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY
et al. 2002 Tumors associated with oncogenic osteomalacia
express genes important in bone and mineral metabolism.
Journal of Bone and Mineral Research 17 1102–1110.
(doi:10.1359/jbmr.2002.17.6.1102)
Dehghani M, Dabaghmanesh MH & Omrani GH 2010
Photoclinic. Oncogenic osteomalacia. Archives of Iranian
Medicine 13 253–254.
Dent CE & Gertner JM 1976 Hypophosphataemic osteoma-
lacia in fibrous dysplasia. Quarterly Journal of Medicine
45 411–420.
Dewitt CA, Collins MT & Cowen EW 2007 Diffuse pain,
hypophosphatemia, and a subcutaneous nodule.
Journal of the American Academy of Dermatology 57
509–512. (doi:10.1016/j.jaad.2007.05.010)
Dissanayake AM, Wilson JL, Holdaway IM & Reid IR 2003
Oncogenic osteomalacia: culprit tumour detection whole
body magnetic resonance imaging. Internal Medicine
Journal 33 615–616. (doi:10.1111/j.1445-5994.2003.
00488.x)
Dowman JK & Khattak FH 2006 Oncogenic hypopho-
sphataemic osteomalacia mimicking bone metastases on
isotope bone scan. Annals of Rheumatic Disease 65 1664.
(doi:10.1136/ard.2006.057943)
Drezner M 1999 Tumor-induced osteomalacia. In Primer on
the Metabolic Bone Diseases and Disorders of Mineral
Metabolism, 4 edn, Ed M Favus. Philadelphia, PA:
Lippincott Williams & Wilkins
Drezner MK 2001 Tumor-induced osteomalacia. Reviews in
Endocrine & Metabolic Disorders 2 175–186. (doi:10.
1023/A:1010006811394)
Duet M, Kerkeni S, Sfar R, Bazille C, Liote F & Orcel P 2008
Clinical impact of somatostatin receptor scintigraphy in
the management of tumor-induced osteomalacia. Clinical
Nuclear Medicine 33 752–756. (doi:10.1097/RLU.
0b013e31818866bf)
Dumitrescu CE, Kelly MH, Khosravi A, Hart TC, Brahim J,
White KE, Farrow EG, Nathan MH, Murphey MD &
Collins MT 2009 A case of familial tumoral calcino-
sis/hyperostosis-hyperphosphatemia syndrome due to
a compound heterozygous mutation in GALNT3
demonstrating new phenotypic features. Osteoporosis
International 20 1273–1278. (doi:10.1007/s00198-008-
0775-z)
Dupond JL, Mahammedi H, Magy N, Blagosklonov O,
Meaux-Ruault N & Kantelip B 2005a Detection of a
mesenchymal tumor responsible for hypophosphatemic
osteomalacia using FDG-PET. European Journal of
Internal Medicine 16 445–446. (doi:10.1016/j.ejim.2005.
07.003)
R69
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
Dupond JL, Mahammedi H, Prie D, Collin F, Gil H,
Blagosklonov O, Ricbourg B, Meaux-Ruault N &
Kantelip B 2005b Oncogenic osteomalacia: diagnostic
importance of fibroblast growth factor 23 and F-18
fluorodeoxyglucose PET/CT scan for the diagnosis and
follow-up in one case. Bone 36 375–378. (doi:10.1016/j.
bone.2005.01.001)
Earle KE, Seneviratne T, Shaker J & Shoback D 2004
Fanconi’s syndrome in HIVC adults: report of three cases
and literature review. Journal of Bone and Mineral
Research 19 714–721. (doi:10.1359/jbmr.2004.19.5.714)
Econs MJ & Drezner MK 1994 Tumor-induced osteomalacia
– unveiling a new hormone. New England Journal of
Medicine 330 1679–1681. (doi:10.1056/NEJM19940609
3302310)
Econs MJ & McEnery PT 1997 Autosomal dominant
hypophosphatemic rickets/osteomalacia: clinical
characterization of a novel renal phosphate-wasting
disorder. Journal of Clinical Endocrinology and
Metabolism 82 674–681. (doi:10.1210/jc.82.2.674)
Elston MS, Stewart IJ, Clifton-Bligh R & Conaglen JV 2007
A case of oncogenic osteomalacia with preoperative
secondary hyperparathyroidism: description of the
biochemical response of FGF23 to octreotide therapy
and surgery. Bone 40 236–241. (doi:10.1016/j.bone.2006.
07.027)
Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D,
Minagawa M, Sugimoto T, Yamauchi M, Michigami T
et al. 2008 Clinical usefulness of measurement of
fibroblast growth factor 23 (FGF23) in hypophosphatemic
patients: proposal of diagnostic criteria using FGF23
measurement. Bone 42 1235–1239. (doi:10.1016/j.bone.
2008.02.014)
von Falck C, Rodt T, Rosenthal H, Langer F, Goesling T,
Knapp WH & Galanski M 2008 (68)Ga-DOTANOC
PET/CT for the detection of a mesenchymal tumor
causing oncogenic osteomalacia. European Journal of
Nuclear Medicine and Molecular Imaging 35 1034.
(doi:10.1007/s00259-008-0755-8)
Farrow EG, Davis SI, Summers LJ & White KE 2009
Initial FGF23-mediated signaling occurs in the distal
convoluted tubule. Journal of the American Society of
Nephrology 20 955–960. (doi:10.1681/ASN.
2008070783)
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X,
Rauch F, Davis SI, Zhang S et al. 2006 Loss of DMP1
causes rickets and osteomalacia and identifies a role for
osteocytes in mineral metabolism. Nature Genetics 38
1310–1315. (doi:10.1038/ng1905)
Ferrari SL, Bonjour JP & Rizzoli R 2005 Fibroblast growth
factor-23 relationship to dietary phosphate and renal
phosphate handling in healthy young men. Journal of
Clinical Endocrinology and Metabolism 90 1519–1524.
(doi:10.1210/jc.2004-1039)
Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M,
Bertoni F, Cho JY, Econs MJ, Inwards CY, Jan de Beur
SM, Mentzel T et al. 2004 Most osteomalacia-associated
R70
mesenchymal tumors are a single histopathologic entity:
an analysis of 32 cases and a comprehensive review of the
literature. American Journal of Surgical Pathology 28
1–30. (doi:10.1097/00000478-200401000-00001)
Friedlaender MM, Wald H, Dranitzki-Elhalel M, Zajicek HK,
Levi M & Popovtzer MM 2001 Vitamin D reduces renal
NaPi-2 in PTH-infused rats: complexity of vitamin D action
on renal P(i) handling. American Journal of Physiology.
Renal Physiology 281 F428–F433.
Frishberg Y, Topaz O, Bergman R, Behar D, Fisher D,
Gordon D, Richard G & Sprecher E 2005 Identification of
a recurrent mutation in GALNT3 demonstrates that
hyperostosis-hyperphosphatemia syndrome and familial
tumoral calcinosis are allelic disorders. Journal of
Molecular Medicine 83 33–38. (doi:10.1007/s00109-004-
0610-8)
Fuentealba C, Pinto D, Ballesteros F, Pacheco D, Boettiger O,
Soto N, Fernandez W, Gabler F, Gonzales G & Reginato AJ
2003 Oncogenic hypophosphatemic osteomalacia associ-
ated with a nasal hemangiopericytoma. Journal of Clinical
Rheumatology 9 373–379. (doi:10.1097/01.rhu.00001019
06.15276.ed)
Fukumoto S, Takeuchi Y, Nagano A & Fujita T 1999
Diagnostic utility of magnetic resonance imaging skeletal
survey in a patient with oncogenic osteomalacia. Bone 25
375–377. (doi:10.1016/S8756-3282(99)00170-2)
Furco A, Roger M, Mouchet B, Richard O, Martinache X &
Fur A 2002 Osteomalacia cured by surgery. European
Journal of Internal Medicine 13 67–69. (doi:10.1016/
S0953-6205(01)00196-0)
Garcia CA & Spencer RP 2002 Bone and In-111 octreotide
imaging in oncogenic osteomalacia: a case report.
Clinical Nuclear Medicine 27 582–583. (doi:10.1097/
00003072-200208000-00007)
Gascon A, Cobeta-Garcia JC, Iglesias E, Lazaro JM &
Muniesa JA 1999 Oncogenic osteomalacia in a patient
with a fibrocystic nodule of the breast. Nephrology,
Dialysis, Transplantation 14 1561–1563. (doi:10.1093/
ndt/14.6.1561)
Geller JL, Khosravi A, Kelly MH, Riminucci M, Adams JS &
Collins MT 2007 Cinacalcet in the management of tumor-
induced osteomalacia. Journal of Bone and Mineral
Research 22 931–937. (doi:10.1359/jbmr.070304)
Gershinsky M, Croitoru S, Dickstein G, Bardicef O, Gelman R
& Barmeir E 2007 Imaging of oncogenic osteomalacia.
Israel Medical Association Journal 9 566–567.
Gore MO, Welch BJ, Geng W, Kabbani W, Maalouf NM,
Zerwekh JE, Moe OW & Sakhaee K 2009 Renal
phosphate wasting due to tumor-induced osteomalacia:
a frequently delayed diagnosis. Kidney International 76
342–347. (doi:10.1038/ki.2008.355)
Gupta A, Winer K, Econs MJ, Marx SJ & Collins MT 2004
FGF-23 is elevated by chronic hyperphosphatemia.
Journal of Clinical Endocrinology and Metabolism 89
4489–4492. (doi:10.1210/jc.2004-0724)
Habra MA, Jimenez C, Huang SC, Cote GJ, Murphy WA Jr,
Gagel RF & Hoff AO 2008 Expression analysis of
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
fibroblast growth factor-23, matrix extracellular phos-
phoglycoprotein, secreted frizzled-related protein-4, and
fibroblast growth factor-7: identification of fibroblast
growth factor-23 and matrix extracellular phosphoglyco-
protein as major factors involved in tumor-induced
osteomalacia. Endocrine Practice 14 1108–1114.
Haeusler G, Freilinger M, Dominkus M, Egerbacher M,
Amann G, Kolb A, Schlegel W, Raimann A &
Staudenherz A 2010 Tumor-induced hypophosphatemic
rickets in an adolescent boy – clinical presentation,
diagnosis, and histological findings in growth plate and
muscle tissue. Journal of Clinical Endocrinology and
Metabolism 95 4511–4517. (doi:10.1210/jc.2010-0543)
Halperin F, Anderson RJ & Mulder JE 2007 Tumor-induced
osteomalacia: the importance of measuring serum
phosphorus levels. Nature Clinical Practice.
Endocrinology & Metabolism 3 721–725. (doi:10.1038/
ncpendmet0639)
Hannan FM, Athanasou NA, Teh J, Gibbons CL, Shine B &
Thakker RV 2008 Oncogenic hypophosphataemic
osteomalacia: biomarker roles of fibroblast growth
factor 23, 1,25-dihydroxyvitamin D3 and lymphatic
vessel endothelial hyaluronan receptor 1. European
Journal of Endocrinology 158 265–271. (doi:10.1530/
EJE-07-0485)
Harbeck B, Schocklmann H, Seekamp A, Czech N &
Monig H 2009 Tumor-induced osteomalacia: successful
treatment by radio-guided tumor surgery. Journal of
Clinical Rheumatology 15 31–34. (doi:10.1097/RHU.
0b013e3181960483)
Harish S, Jurriaans E, Jan E, Sur M & Colterjohn N 2008
Giant cell tumour of soft tissue causing oncogenic
osteomalacia: report demonstrating the use of octreotide
scintigraphy in tumour localization. Clinical Radiology
63 101–107. (doi:10.1016/j.crad.2007.05.017)
Harvey JN, Gray C & Belchetz PE 1992 Oncogenous
osteomalacia and malignancy. Clinical Endocrinology 37
379–382. (doi:10.1111/j.1365-2265.1992.tb02342.x)
Hasegawa T, Shimoda T, Yokoyama R, Beppu Y, Hirohashi
S & Maeda S 1999 Intracortical osteoblastic osteosar-
coma with oncogenic rickets. Skeletal Radiology 28
41–45. (doi:10.1007/s002560050470)
Hesse E, Moessinger E, Rosenthal H, Laenger F, Brabant G,
Petrich T, Gratz KF & Bastian L 2007a Oncogenic
osteomalacia: exact tumor localization by co-registration
of positron emission and computed tomography.
Journal of Bone and Mineral Research 22 158–162.
(doi:10.1359/jbmr.060909)
Hesse E, Rosenthal H & Bastian L 2007b Radiofrequency
ablation of a tumor causing oncogenic osteomalacia. New
England Journal of Medicine 357 422–424. (doi:10.1056/
NEJMc070347)
Heylen A, Dasnoy D, Hustin J & Pochet JM 1999 Tumor-
related osteomalacia followed after treatment by
hyperparathyroidism. Revue du Rhumatisme (English ed.)
66 53–57.
www.endocrinology-journals.org
Hodgson SF, Clarke BL, Tebben PJ, Mullan BP, Cooney WP III
& Shives TC 2006 Oncogenic osteomalacia: localization
of underlying peripheral mesenchymal tumors with use
of Tc 99m sestamibi scintigraphy. Endocrine Practice 12
35–42.
Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner
JM & Econs MJ 2006 Intronic deletions in the SLC34A3
gene cause hereditary hypophosphatemic rickets
with hypercalciuria. Journal of Clinical
Endocrinology and Metabolism 91 4022–4027. (doi:10.
1210/jc.2005-2840)
Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM,
Shulman D, Kassem M, Rackoff P, Zimering M, Dalkin A
et al. 2006 Sensitivity of fibroblast growth factor 23
measurements in tumor-induced osteomalacia. Journal of
Clinical Endocrinology and Metabolism 91 2055–2061.
(doi:10.1210/jc.2005-2105)
Imel EA, Hui SL & Econs MJ 2007 FGF23 concentrations
vary with disease status in autosomal dominant hypo-
phosphatemic rickets. Journal of Bone and Mineral
Research 22 520–526. (doi:10.1359/jbmr.070107)
Inokuchi G, Tanimoto H, Ishida H, Sugimoto T, Yamauchi M,
Miyauchi A & Nibu K 2006 A paranasal tumor associated
with tumor-induced osteomalacia. Laryngoscope 116
1930–1933. (doi:10.1097/01.mlg.0000231295.67060.89)
Ishii A, Imanishi Y, Kobayashi K, Hashimoto J, Ueda T,
Miyauchi A, Koyano HM, Kaji H, Saito T, Oba K et al.
2010 The levels of somatostatin receptors in causative
tumors of oncogenic osteomalacia are insufficient for their
agonist to normalize serum phosphate levels. Calcified
Tissue International 86 455–462. (doi:10.1007/s00223-
010-9369-9)
Ito N, Fukumoto S, Takeuchi Y, Takeda S, Suzuki H,
Yamashita T & Fujita T 2007 Effect of acute changes of
serum phosphate on fibroblast growth factor (FGF) 23
levels in humans. Journal of Bone and Mineral
Metabolism 25 419–422. (doi:10.1007/s00774-007-
0779-3)
Ito N, Shimizu Y, Suzuki H, Saito T, Okamoto T, Hori M,
Akahane M, Fukumoto S & Fujita T 2010 Clinical utility
of systemic venous sampling of FGF23 for identifying
tumours responsible for tumour-induced osteomalacia.
Journal of Internal Medicine 268 390–394. (doi:10.1111/
j.1365-2796.2010.02262.x)
Ivker R, Resnick SD & Skidmore RA 1997 Hypopho-
sphatemic vitamin D-resistant rickets, precocious pub-
erty, and the epidermal nevus syndrome. Archives of
Dermatology 133 1557–1561. (doi:10.1001/archderm.
133.12.1557)
Izzedine H, Launay-Vacher V, Isnard-Bagnis C & Deray G
2003 Drug-induced Fanconi’s syndrome. American
Journal of Kidney Diseases 41 292–309. (doi:10.1053/
ajkd.2003.50037)
Jacob JJ, Finny P, Thomas M, Thomas N & John M 2007
Oncogenic osteomalacia. Journal of Association of
Physicians of India 55 231–233.
R71
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
Jagtap VS, Sarathi V, Lila AR, Malhotra G, Sankhe SS,
Bandgar T, Menon P & Shah NS 2010 Tumor induced
osteomalacia: a single center experience. Endocrine
Practice 16 1–19. (doi:10.4158/10024.GL)
Jan de Beur SM 2005 Tumor-induced osteomalacia.
Journal of the American Medical Association 294
1260–1267. (doi:10.1001/jama.294.10.1260)
Jan de Beur SM, Streeten EA, Civelek AC, McCarthy EF,
Uribe L, Marx SJ, Onobrakpeya O, Raisz LG, Watts NB,
Sharon M et al. 2002 Localisation of mesenchymal
tumours by somatostatin receptor imaging. Lancet 359
761–763. (doi:10.1016/S0140-6736(02)07846-7)
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T,
Imanishi Y, Yamamoto T, Hampson G, Koshiyama H,
Ljunggren O et al. 2003 Fibroblast growth factor 23 in
oncogenic osteomalacia and X-linked hypophosphatemia.
New England Journal of Medicine 348 1656–1663.
(doi:10.1056/NEJMoa020881)
Jung GH, Kim JD, Cho Y, Chung SH, Lee JH & Sohn KR
2010 A 9-month-old phosphaturic mesenchymal tumor
mimicking the intractable rickets. Journal of Pediatric
Orthopaedics. Part B/ European Paediatric Orthopaedic
Society, Pediatric Orthopaedic Society of North America
19 127–132. (doi:10.1097/BPB.0b013e32832f59cb)
Kaul M, Silverberg M, Dicarlo EF, Schneider R, Bass AR &
Erkan D 2007 Tumor-induced osteomalacia. Clinical
Rheumatology 26 1575–1579. (doi:10.1007/s10067-006-
0468-y)
Kaylie DM, Jackson CG & Gardner EK 2006 Oncogenic
osteomalacia caused by phosphaturic mesenchymal tumor
of the temporal bone. Otolaryngology – Head and Neck
Surgery 135 653–654. (doi:10.1016/j.otohns.2005.03.086)
Kenealy H, Holdaway I & Grey A 2008 Occult nasal sinus
tumours causing oncogenic osteomalacia. European
Journal of Internal Medicine 19 516–519. (doi:10.1016/j.
ejim.2008.01.011)
Kenny AP & Glen AC 1973 Tests of phosphate reabsorption.
Lancet 2 158. (doi:10.1016/S0140-6736(73)93112-7)
Khadgawat R, Singh Y, Kansara S, Tandon N, Bal C, Seith A
& Kotwal P 2009 PET/CT localisation of a scapular
haemangiopericytoma with tumour-induced osteomala-
cia. Singapore Medical Journal 50 e55–e57.
Khosravi A, Cutler CM, Kelly MH, Chang R, Royal RE,
Sherry RM, Wodajo FM, Fedarko NS & Collins MT 2007
Determination of the elimination half-life of fibroblast
growth factor-23. Journal of Clinical Endocrinology and
Metabolism 92 2374–2377. (doi:10.1210/jc.2006-2865)
Kimizuka T, Ozaki Y & Sumi Y 2004 Usefulness of 201Tl
and 99mTc MIBI scintigraphy in a case of oncogenic
osteomalacia. Annals of Nuclear Medicine 18 63–67.
(doi:10.1007/BF02985616)
Kobayashi K, Nakao K, Kawai K, Ito K, Hukumoto S,
Asakage T, Oota S & Motoi R 2010 Tumor-induced
osteomalacia originating from the temporal bone: a case
report. Head & Neck [in press]. (doi:10.1002/hed.21355)
Konishi K, Nakamura M, Yamakawa H, Suzuki H, Saruta T,
Hanaoka H & Davatchi F 1991 Hypophosphatemic
R72
osteomalacia in von Recklinghausen neurofibromatosis.
American Journal of the Medical Sciences 301 322–328.
(doi:10.1097/00000441-199105000-00006)
Koriyama N, Nishimoto K, Kodama T, Nakazaki M, Kurono
Y, Yoshida H & Tei C 2006 Oncogenic osteomalacia in a
case with a maxillary sinus mesenchymal tumor.
American Journal of the Medical Sciences 332 142–147.
(doi:10.1097/00000441-200609000-00010)
Kurien R, Manipadam MT & Rupa V 2010 Oncogenic
osteomalacia in a patient with an ethmoid sinus tumour.
Journal of Laryngology and Otology 124 799–803.
(doi:10.1017/S0022215109992313)
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A,
Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW
et al. 2006 Regulation of fibroblast growth factor-23
signaling by klotho. Journal of Biological Chemistry 281
6120–6123. (doi:10.1074/jbc.C500457200)
Ladha SS, Whitaker MD & Bosch EP 2006 Oncogenic
osteomalacia: muscular weakness and multiple fractures.
Neurology 67 364–365. (doi:10.1212/01.wnl.
0000225057.56863.33)
Lamont EB, Cavaghan MK & Brockstein BE 1999
Oncogenic osteomalacia as a harbinger of recurrent
osteosarcoma. Sarcoma 3 95–99. (doi:10.1080/
13577149977712)
Larsson T, Nisbeth U, Ljunggren O, Juppner H & Jonsson KB
2003 Circulating concentration of FGF-23 increases as
renal function declines in patients with chronic kidney
disease, but does not change in response to variation
in phosphate intake in healthy volunteers. Kidney
International 64 2272–2279. (doi:10.1046/j.1523-1755.
2003.00328.x)
Lee HK, Sung WW, Solodnik P & Shimshi M 1995 Bone
scan in tumor-induced osteomalacia. Journal of Nuclear
Medicine 36 247–249.
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N,
Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S,
Hadad Y, Goding J et al. 2010 Autosomal-recessive
hypophosphatemic rickets is associated with an inacti-
vation mutation in the ENPP1 gene. American Journal of
Human Genetics 86 273–278. (doi:10.1016/j.ajhg.2010.
01.010)
Lewiecki EM, Urig EJ Jr & Williams RC Jr 2008 Tumor-
induced osteomalacia: lessons learned. Arthritis and
Rheumatism 58 773–777. (doi:10.1002/art.23278)
Lichter-Konecki U, Broman KW, Blau EB & Konecki DS
2001 Genetic and physical mapping of the locus for
autosomal dominant renal Fanconi syndrome, on
chromosome 15q15.3. American Journal of Human
Genetics 68 264–268. (doi:10.1086/316923)
Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach
B, Scheinman SJ, Harding B, Bolino A, Devoto M,
Goodyer P et al. 1996 A common molecular basis for
three inherited kidney stone diseases. Nature 379
445–449. (doi:10.1038/379445a0)
Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M,
Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
SM, Rittmaster RS, Shlossberg AH et al. 2006a DMP1
mutations in autosomal recessive hypophosphatemia
implicate a bone matrix protein in the regulation of
phosphate homeostasis. Nature Genetics 38 1248–1250.
(doi:10.1038/ng1868)
Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-
Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R,
Albers N, Lichtner P, Schnabel D et al. 2006b Hereditary
hypophosphatemic rickets with hypercalciuria is caused
by mutations in the sodium-phosphate cotransporter gene
SLC34A3. American Journal of Human Genetics 78
193–201. (doi:10.1086/499410)
Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G &
Strom TM 2010 Loss-of-function ENPP1 mutations
cause both generalized arterial calcification of infancy
and autosomal-recessive hypophosphatemic rickets.
American Journal of Human Genetics 86 267–272.
(doi:10.1016/j.ajhg.2010.01.006)
Lui CY, Khoo R, Law TC & Chong SF 2002 Case report.
Tumour-induced osteomalacia in a patient with osseous
haemangioma. Clinical Radiology 57 1125–1127.
(doi:10.1053/crad.2002.1091)
Malabanan AO, Turner AK, Rosenberg IN & Holick MF 1998
Oncogenic osteomalacia: clinical presentation, densito-
metric findings, and response to therapy. Journal of
Clinical Densitometry 1 77–80. (doi:10.1385/JCD:1:1:77)
Mannstadt M, Lorente C & Juppner H 2008 Rapid detection
of intact FGF-23 in tumor tissue from patients with
oncogenic osteomalacia. Clinical Chemistry 54
1252–1254. (doi:10.1373/clinchem.2007.102418)
Marshall AE, Martin SE, Agaram NP, Chen JH, Horn EM,
Douglas-Akinwande AC & Hattab EM 2010 A 61-year-
old woman with osteomalacia and a thoracic spine lesion.
Brain Pathology 20 499–502. (doi:10.1111/j.1750-3639.
2009.00360.x)
McCance R 1947 Osteomalacia with Looser’s nodes
(Milkman’s syndrome) due to a raised resistance to
vitamin D acquired about the age of 15 years. Quarterly
Journal of Medicine 16 33–46.
Meyer RA Jr, Meyer MH & Gray RW 1989 Parabiosis
suggests a humoral factor is involved in X-linked
hypophosphatemia in mice. Journal of Bone and Mineral
Research 4 493–500. (doi:10.1002/jbmr.5650040407)
Miyauchi A, Fukase M, Tsutsumi M & Fujita T 1988
Hemangiopericytoma-induced osteomalacia: tumor
transplantation in nude mice causes hypophosphatemia
and tumor extracts inhibit renal 25-hydroxyvitamin D
1-hydroxylase activity. Journal of Clinical Endocrinology
and Metabolism 67 46–53. (doi:10.1210/jcem-67-1-46)
Moran M & Paul A 2002 Octreotide scanning in the
detection of a mesenchymal tumour in the pubic
symphysis causing hypophosphataemic osteomalacia.
International Orthopaedics 26 61–62. (doi:10.1007/
s00264-001-0318-0)
Mori Y, Ogasawara T, Motoi T, Shimizu Y, Chikazu D,
Tamura K, Fukumoto S & Takato T 2010 Tumor-induced
osteomalacia associated with a maxillofacial tumor
www.endocrinology-journals.org
producing fibroblast growth factor 23: report of a case and
review of the literature. Oral Surgery, Oral Medicine,
Oral Pathology, Oral Radiology, and Endodontics 109
e57–e63. (doi:10.1016/j.tripleo.2009.10.052)
Mount D &Yu A 2008 Phosphate transport. In Brenner &
Rectors: The Kidney, 8 edn, pp 196–204. Ed B Brenner.
Philadelphia, PA: Saunders Elsevier
Mussig K, Oksuz MO, Pfannenberg C, Adam P, Zustin J,
Beckert S & Petersenn S 2009 Somatostatin receptor
expression in an epitheloid hemangioma causing
oncogenic osteomalacia. Journal of Clinical
Endocrinology and Metabolism 94 4123–4124. (doi:10.
1210/jc.2009-0927)
Nakahama H, Nakanishi T, Uno H, Takaoka T, Taji N,
Uyama O, Kitada O, Sugita M, Miyauchi A, Sugishita T
et al. 1995 Prostate cancer-induced oncogenic hypopho-
sphatemic osteomalacia. Urologia Internationalis 55
38–40. (doi:10.1159/000282746)
Narvaez J, Domingo-Domenech E, Narvaez JA, Nolla JM &
Valverde J 2005 Acquired hypophosphatemic osteoma-
lacia associated with multiple myeloma. Joint, Bone,
Spine 72 424–426. (doi:10.1016/j.jbspin.2004.10.012)
Nasu T, Kurisu S, Matsuno S, Tatsumi K, Kakimoto T,
Kobayashi M, Nakano Y, Wakasaki H, Furuta H, Nishi M
et al. 2008 Tumor-induced hypophosphatemic osteoma-
lacia diagnosed by the combinatory procedures of
magnetic resonance imaging and venous sampling for
FGF23. Internal Medicine 47 957–961. (doi:10.2169/
internalmedicine.47.0745)
Nawrot-Wawrzyniak K, Varga F, Nader A, Roschger P,
Sieghart S, Zwettler E, Roetzer KM, Lang S, Weinkamer
R, Klaushofer K et al. 2009 Effects of tumor-induced
osteomalacia on the bone mineralization process.
Calcified Tissue International 84 313–323. (doi:10.1007/
s00223-009-9216-z)
Nayak A, Sharma SG, Tandon N & Ray R 2004 Phosphaturic
mesenchymal tumor: a case report. Indian Journal of
Pathology & Microbiology 47 530–533.
Nelson AE, Mason RS, Robinson BG, Hogan JJ, Martin EA,
Ahlstrom H, Astrom G, Larsson T, Jonsson K, Wibell L
et al. 2001 Diagnosis of a patient with oncogenic
osteomalacia using a phosphate uptake bioassay of serum
and magnetic resonance imaging. European Journal of
Endocrinology 145 469–476. (doi:10.1530/eje.0.
1450469)
Nelson AE, Bligh RC, Mirams M, Gill A, Au A, Clarkson A,
Juppner H, Ruff S, Stalley P, Scolyer RA et al. 2003
Clinical case seminar: fibroblast growth factor 23: a new
clinical marker for oncogenic osteomalacia. Journal of
Clinical Endocrinology and Metabolism 88 4088–4094.
(doi:10.1210/jc.2002-021919)
Nesbitt T, Coffman TM, Griffiths R & Drezner MK 1992
Crosstransplantation of kidneys in normal and Hyp mice.
Evidence that the Hyp mouse phenotype is unrelated to an
intrinsic renal defect. Journal of Clinical Investigation 89
1453–1459. (doi:10.1172/JCI115735)
R73
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
Nguyen BD 2006 Coexisting hyperparathyroidism
and oncogenic osteomalacia: sestamibi and
somatostatin receptor scintigraphy. Clinical Nuclear
Medicine 31 648–651. (doi:10.1097/01.rlu.0000238192.
37389.28)
Nguyen BD & Wang EA 1999 Indium-111 pentetreotide
scintigraphy of mesenchymal tumor with oncogenic
osteomalacia. Clinical Nuclear Medicine 24 130–131.
(doi:10.1097/00003072-199902000-00016)
Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F,
Taniguchi A, Sato T, Shuto E, Nashiki K, Arai H,
Yamamoto H et al. 2006 Acute effect of oral phosphate
loading on serum fibroblast growth factor 23 levels in
healthy men. Kidney International 70 2141–2147.
(doi:10.1038/sj.ki.5002000)
Nordstrom H, Lennquist S, Lindell B & Sjoberg HE 1977
Hypophosphataemia in severe burns. Acta Chirurgica
Scandinavica 143 395–399.
Ogose A, Hotta T, Emura I, Hatano H, Inoue Y, Umezu H &
Endo N 2001 Recurrent malignant variant of phosphaturic
mesenchymal tumor with oncogenic osteomalacia.
Skeletal Radiology 30 99–103. (doi:10.1007/
s002560000306)
Ogura E, Kageyama K, Fukumoto S, Yagihashi N, Fukuda Y,
Kikuchi T, Masuda M & Suda T 2008 Development of
tumor-induced osteomalacia in a subcutaneous tumor,
defined by venous blood sampling of fibroblast growth
factor-23. Internal Medicine 47 637–641. (doi:10.2169/
internalmedicine.47.0761)
Ohashi K, Ohnishi T, Ishikawa T, Tani H, Uesugi K &
Takagi M 1999 Oncogenic osteomalacia presenting as
bilateral stress fractures of the tibia. Skeletal Radiology 28
46–48. (doi:10.1007/s002560050471)
Oka M, Kamo T, Sasaki E, Kaji H, Nishizawa H, Imanishi Y
& Nishigori C 2007 A case of phosphaturic mesenchymal
tumour (mixed connective tissue variant) that developed
in the subcutaneous tissue of a patient with oncogenic
osteomalacia and produced fibroblast growth factor 23.
British Journal of Dermatology 157 198–200. (doi:10.
1111/j.1365-2133.2007.07940.x)
Omdahl JL & DeLuca HF 1971 Strontium induced rickets:
metabolic basis. Science 174 949–951. (doi:10.1126/
science.174.4012.949)
Oudet C, Martin-Coignard D, Pannetier S, Praud E,
Champion G & Hanauer A 1997 A second family with
XLRH displays the mutation S244L in the CLCN5
gene. Human Genetics 99 781–784. (doi:10.1007/
s004390050448)
Paglia F, Dionisi S & Minisola S 2002 Octreotide for tumor-
induced osteomalacia. New England Journal of Medicine
346 1748–1749 (author reply 1748–1749). (doi:10.1056/
NEJM200205303462215)
Park JM, Woo YK, Kang MI, Kang CS & Hahn ST 2001
Oncogenic osteomalacia associated with soft tissue
chondromyxoid fibroma. European Journal of Radiology
39 69–72. (doi:10.1016/S0720-048X(00)00279-5)
R74
Payne RB 1998 Renal tubular reabsorption of phosphate
(TmP/GFR): indications and interpretation. Annals of
Clinical Biochemistry 35 201–206.
Pedrazzoli M, Colletti G, Ferrari M, Rossetti G, Moneghini L
& Autelitano L 2010 Mesenchymal phosphaturic neo-
plasm in the maxillary sinus: a case report. International
Journal of Oral and Maxillofacial Surgery 39 1027–1032.
(doi:10.1016/j.ijom.2010.04.039)
Peters KB, McLendon R, Morse MA & Vredenburgh JJ 2010
Treatment of recurrent intracranial hemangiopericytoma
with SRC-related tyrosine kinase targeted therapy: a case
report. Case Reports in Oncology 3 93–97. (doi:10.1159/
000307468)
Peterson NR, Summerlin DJ & Cordes SR 2010 Multiple
phosphaturic mesenchymal tumors associated with
oncogenic osteomalacia: case report and review of the
literature. Ear, Nose, & Throat Journal 89 E11–E15.
Pirola E, Vergani F, Casiraghi P, Leone EB, Guerra P &
Sganzerla EP 2009 Oncogenic osteomalacia caused by a
phosphaturic mesenchymal tumor of the thoracic spine.
Journal of Neurosurgery. Spine 10 329–333. (doi:10.
3171/2009.1.SPINE08351)
Policarpio-Nicolas ML, Abbott TE, Dalkin AC, Bennett-
Wick J & Frierson HF Jr 2008 Phosphaturic mesenchymal
tumor diagnosed by fine-needle aspiration and core
biopsy: a case report and review of literature. Diagnostic
Cytopathology 36 115–119. (doi:10.1002/dc.20772)
Pook MA, Wrong O, Wooding C, Norden AG, Feest TG &
Thakker RV 1993 Dent’s disease, a renal Fanconi
syndrome with nephrocalcinosis and kidney stones, is
associated with a microdeletion involving DXS255 and
maps to Xp11.22. Human Molecular Genetics 2
2129–2134. (doi:10.1093/hmg/2.12.2129)
Prader A, Illig R, Uehlinger E & Stalder G 1959 Rickets
following bone tumor. Helvetica Paediatrica Acta 14
554–565.
Radaideh AR, Jaradat D, Abu-Kalaf MM & Nusier MK 2009
Resolution of severe oncogenic hypophosphatemic
osteomalacia after resection of a deeply located soft-tissue
tumour. Current Oncology 16 87–90.
Rao DS, Parfitt AM, Villanueva AR, Dorman PJ &
Kleerekoper M 1987 Hypophosphatemic osteomalacia
and adult Fanconi syndrome due to light-chain nephro-
pathy. Another form of oncogenous osteomalacia.
American Journal of Medicine 82 333–338. (doi:10.1016/
0002-9343(87)90081-7)
Ratanasuwan T, Chetsurakarn S, Ongphiphadhanakul B &
Damrongkitchaiporn S 2008 A case report of tumor-
induced osteomalacia: eight year followed-up. Journal of
the Medical Association of Thailand 91 1900–1902.
Razzaque MS 2009 The FGF23-Klotho axis: endocrine
regulation of phosphate homeostasis. Nature Reviews.
Endocrinology5 611–619. (doi:10.1038/nrendo.2009.196)
Reese DM & Rosen PJ 1997 Oncogenic osteomalacia
associated with prostate cancer. Journal of Urology 158
887. (doi:10.1016/S0022-5347(01)64351-9)
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
Reis-Filho JS, Paiva ME & Lopes JM 2002 Pathologic quiz
case. A 36-year-old woman with muscle pain and
weakness. Phosphaturic mesenchymal tumor (mixed
connective tissue variant)/oncogenic osteomalacia.
Archives of Pathology & Laboratory Medicine 126
1245–1246.
Reis-Filho JS, Paiva ME & Lopes JM 2004 August 2003:
47-year-old female with a 7-year history of osteomalacia
and hypophosphatemia. Brain Pathology 14 111–112,
115.
Rendina D, De Filippo G, Tauchmanova L, Insabato L,
Muscariello R, Gianfrancesco F, Esposito T, Cioffi M,
Colao A, Strazzullo P et al. 2009 Bone turnover and the
osteoprotegerin-RANKL pathway in tumor-induced
osteomalacia: a longitudinal study of five cases. Calcified
Tissue International 85 293–300. (doi:10.1007/s00223-
009-9275-1)
Renkema KY, Alexander RT, Bindels RJ & Hoenderop JG
2008 Calcium and phosphate homeostasis: concerted
interplay of new regulators. Annals of Medicine 40 82–91.
(doi:10.1080/07853890701689645)
Rhee Y, Lee JD, Shin KH, Lee HC, Huh KB & Lim SK 2001
Oncogenic osteomalacia associated with mesenchymal
tumour detected by indium-111 octreotide scintigraphy.
Clinical Endocrinology 54 551–554. (doi:10.1046/j.1365-
2265.2001.01056.x)
Rico H, Fernandez-Miranda E, Sanz J, Gomez-Castresana F,
Escriba A, Hernandez ER & Krsnik I 1986 Oncogenous
osteomalacia: a new case secondary to a malignant tumor.
Bone 7 325–329. (doi:10.1016/8756-3282(86)90251-6)
Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A,
White KE, Waguespack S, Gupta A, Hannon T, Econs MJ
et al. 2003 FGF-23 in fibrous dysplasia of bone and its
relationship to renal phosphate wasting. Journal of
Clinical Investigation 112 683–692. (doi:10.1172/
JCI18399)
Roarke MC & Nguyen BD 2007 PET/CT localization of
phosphaturic mesenchymal neoplasm causing tumor-
induced osteomalacia. Clinical Nuclear Medicine 32
300–301. (doi:10.1097/01.rlu.0000257180.03964.51)
Romualdo-Silva DD, Silva BC, Caetano CV, Tiburcio AM,
Nunes MB, Chagas SA, Polito ET, Ferreira AR & Purisch S
2009 Tumor-induced osteomalacia: a case report. Arquivos
Brasileiros de Endocrinologia e Metabologia 53 378–382.
(doi:10.1590/S0004-27302009000300014)
Sahnoune I, Tazi-Mezalek Z, Essaadouni L, Harmouche H,
Ismael F, Adnaoui M, Aouni M, Kettani F & Maaouni A
2006 Oncogenic osteomalacia in a patient with heman-
gioma: a clinical diagnosis. Joint, Bone, Spine 73
115–118. (doi:10.1016/j.jbspin.2005.06.001)
Sakamoto A, Oda Y, Nagayoshi Y, Iwakiri K, Tamiya S,
Iwamoto Y & Tsuneyoshi M 2001 Glomangiopericytoma
causing oncogenic osteomalacia. A case report with
immunohistochemical analysis. Archives of Orthopaedic
and Trauma Surgery 121 104–108. (doi:10.1007/
s004020000187)
www.endocrinology-journals.org
Santer R, Schneppenheim R, Dombrowski A, Gotze H,
Steinmann B & Schaub J 1997 Mutations in GLUT2, the
gene for the liver-type glucose transporter, in patients
with Fanconi-Bickel syndrome. Nature Genetics 17
324–326. (doi:10.1038/ng1197-324)
Sato K, Obara T, Yamazaki K, Kanbe M, Nakajima K,
Yamada A, Yanagisawa T, Kato Y, Nishikawa T &
Takano K 2001 Somatic mutations of the MEN1 gene and
microsatellite instability in a case of tertiary hyperpara-
thyroidism occurring during high phosphate therapy for
acquired, hypophosphatemic osteomalacia. Journal of
Clinical Endocrinology and Metabolism 86 5564–5571.
(doi:10.1210/jc.86.11.5564)
Savage CR & Zimmer LA 2009 Oncogenic osteomalacia
from pterygopalatine fossa mass. Journal of Laryngology
and Otology 123 1052–1054. (doi:10.1017/S0022215
109004927)
Saville PD, Nassim JR, Stevenson FH, Mulligan L & Carey M
1955 Osteomalacia in Von Recklinghausen’s neuro-
fibromatosis; metabolic study of a case. BMJ 1 1311–1313.
(doi:10.1136/bmj.1.4925.1311)
Scheinman SJ 1998 X-linked hypercalciuric nephrolithiasis:
clinical syndromes and chloride channel mutations.
Kidney International 53 3–17. (doi:10.1046/j.1523-1755.
1998.00718.x)
Sciubba DM, Petteys RJ, Shakur SF, Gokaslan ZL, McCarthy
EF, Collins MT, McGirt MJ, Hsieh PC, Nelson CS &
Wolinsky JP 2009 En bloc spondylectomy for treatment
of tumor-induced osteomalacia. Journal of Neurosurgery.
Spine 11 600–604. (doi:10.3171/2009.6.SPINE08120)
Seijas R, Ares O, Sierra J & Perez-Dominguez M 2009
Oncogenic osteomalacia: two case reports with surprisingly
different outcomes. Archives of Orthopaedic and Trauma
Surgery 129 533–539. (doi:10.1007/s00402-008-0808-2)
Seufert J, Ebert K, Muller J, Eulert J, Hendrich C, Werner E,
Schuuze N, Schulz G, Kenn W, Richtmann H et al. 2001
Octreotide therapy for tumor-induced osteomalacia. New
England Journal of Medicine 345 1883–1888. (doi:10.
1056/NEJMoa010839)
Sharkis DH, Devereux JP, Chako AC & Estep HL 1997
Reversible avascular necrosis of the femoral head in
tumor-induced osteomalacia. Endocrine Practice 3
137–139.
Shelekhova KV, Kazakov DV, Hes O, Treska V & Michal M
2006 Phosphaturic mesenchymal tumor (mixed connec-
tive tissue variant): a case report with spectral analysis.
Virchows Archiv 448 232–235. (doi:10.1007/s00428-005-
0149-2)
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R,
Takeuchi Y, Fujita T, Nakahara K, Fukumoto S &
Yamashita T 2004 FGF-23 is a potent regulator of vitamin
D metabolism and phosphate homeostasis. Journal of
Bone and Mineral Research 19 429–435. (doi:10.1359/
JBMR.0301264)
Sommer S, Berndt T, Craig T & Kumar R 2007 The
phosphatonins and the regulation of phosphate transport
R75
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

W H Chong et al.: Tumor-induced osteomalacia
and vitamin D metabolism. Journal of Steroid Biochem-
istry and Molecular Biology 103 497–503. (doi:10.1016/j.
jsbmb.2006.11.010)
Stark H, Eisenstein B, Tieder M, Rachmel A & Alpert G 1986
Direct measurement of TP/GFR: a simple and reliable
parameter of renal phosphate handling. Nephron 44
125–128. (doi:10.1159/000184216)
Stone MD, Quincey C & Hosking DJ 1992 A neuroendocrine
cause of oncogenic osteomalacia. Journal of Pathology
167 181–185. (doi:10.1002/path.1711670204)
Szumera-Cieckiewicz A, Ptaszynski K, Pawelas A &
Rutkowski P 2009 Oncogenic osteomalacia associated
with phosphaturic mesenchymal tumour, mixed connec-
tive tissue type of the knee. Polish Journal of Pathology
60 193–198.
Taketani Y, Segawa H, Chikamori M, Morita K, Tanaka K,
Kido S, Yamamoto H, Iemori Y, Tatsumi S, Tsugawa N
et al. 1998 Regulation of type II renal NaC-dependent
inorganic phosphate transporters by 1,25-dihydroxyvita-
min D3. Identification of a vitamin D-responsive element
in the human NAPi-3 gene. Journal of Biological
Chemistry 273 14575–14581. (doi:10.1074/jbc.273.23.
14575)
Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y,
Yamashita T, Miyamoto Y, Okazaki H, Nakamura K,
Nakahara K et al. 2004 Venous sampling for fibroblast
growth factor-23 confirms preoperative diagnosis
of tumor-induced osteomalacia. Journal of Clinical
Endocrinology and Metabolism 89 3979–3982. (doi:10.
1210/jc.2004-0406)
Tartaglia F, Minisola S, Sgueglia M, Blasi S, Brunelli D,
Degli Effetti E, Maturo A, Cola A, Custureri F &
Campana FP 2006 Tumor-induced hypophosphatemic
osteomalacia associated with tertiary hyperparathyroid-
ism: a case report. Il Giornale di Chirurgia 27 9–13.
Taylor HC, Fallon MD & Velasco ME 1984 Oncogenic
osteomalacia and inappropriate antidiuretic hormone
secretion due to oat-cell carcinoma. Annals of Internal
Medicine 101 786–788.
Teasell RW & Shapiro AP 2002 Misdiagnosis of conversion
disorders. American Journal of Physical Medicine &
Rehabilitation 81 236–240. (doi:10.1097/00002060-
200203000-00015)
Terek R & Nielsen G 2001 Case records of the Massachusetts
General Hospital. Weekly clinicopathological exercises.
Case 29-2001. A 14-year-old with abnormal bones and a
sacral mass. New England Journal of Medicine 345
903–908. (doi:10.1056/NEJMcpc010029)
The HYP Consortium 1995 A gene (PEX) with homologies
to endopeptidases is mutated in patients with
X-linked hypophosphatemic rickets. The HYP
Consortium. Nature Genetics 11 130–136. (doi:10.1038/
ng1095-130)
Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak
P, Mizrachi M, Khamaysi Z, Behar D, Petronius D,
Friedman V et al. 2004 Mutations in GALNT3, encoding
R76
a protein involved in O-linked glycosylation, cause
familial tumoral calcinosis. Nature Genetics 36 579–581.
(doi:10.1038/ng1358)
Toyosawa S, Tomita Y, Kishino M, Hashimoto J, Ueda T,
Tsujimura T, Aozasa K, Ijuhin N & Komori T 2004
Expression of dentin matrix protein 1 in tumors causing
oncogenic osteomalacia. Modern Pathology 17 573–578.
(doi:10.1038/modpathol.3800084)
Umphrey LG, Whitaker MD, Bosch EP & Cook CB 2007
Clinical and bone density outcomes of tumor-induced
osteomalacia after treatment. Endocrine Practice 13
458–462.
Ungari C, Rocchi G, Rinna C, Agrillo A, Lattanzi A &
Pagnoni M 2004 Hypophosphaturic mesenchymal tumor
of the ethmoid associated with oncogenic osteomalacia.
Journal of Craniofacial Surgery 15 523–527. (doi:10.
1097/00001665-200405000-00036)
Uramoto N, Furukawa M & Yoshizaki T 2009 Malignant
phosphaturic mesenchymal tumor, mixed connective
tissue variant of the tongue. Auris, Nasus, Larynx 36
104–105. (doi:10.1016/j.anl.2008.01.003)
Vandergheynst F, Van Dorpe J, Goldman S & Decaux G
2006 Increased 18F fluorodeoxyglucose uptake of a
vertebral hemangioma responsible for oncogenic osteo-
malacia. European Journal of Internal Medicine 17 223.
(doi:10.1016/j.ejim.2005.11.008)
Vollbrecht JE & Rao DS 2008 Images in clinical
medicine. Tumor-induced osteomalacia. New England
Journal of Medicine 358 1282. (doi:10.1056/
NEJMicm066066)
Walton RJ & Bijvoet OL 1975 Nomogram for derivation of
renal threshold phosphate concentration. Lancet 2
309–310. (doi:10.1016/S0140-6736(75)92736-1)
Ward LM, Rauch F, White KE, Filler G, Matzinger MA,
Letts M, Travers R, Econs MJ & Glorieux FH 2004
Resolution of severe, adolescent-onset hypophosphatemic
rickets following resection of an FGF-23-producing
tumour of the distal ulna. Bone 34 905–911. (doi:10.1016/
j.bone.2003.12.025)
Weidner N 1991 Review and update: oncogenic osteomalacia-
rickets. Ultrastructural Pathology 15 317–333. (doi:10.
3109/01913129109016242)
Weidner N, Bar RS, Weiss D & Strottmann MP 1985
Neoplastic pathology of oncogenic osteomalacia/rickets.
Cancer 55 1691–1705. (doi:10.1002/1097-0142(19850415)
55:8!1691::AID-CNCR2820550814O3.0.CO;2-S)
Westerberg PA, Olauson H, Toss G, Wikstrom B, Morales O,
Linde T, Jonsson K, Ljunggren O & Larsson TE 2008
Preoperative tumor localization by means of venous
sampling for fibroblast growth factor-23 in a patient with
tumor-induced osteomalacia. Endocrine Practice 14
362–367.
White K, Evans W, O’Riordan JL, Speer M, Econs MJ,
Lorenz-Depiereux B, Grabowski M, Meitinger T &
Strom T 2000 Autosomal dominant hypophosphataemic
rickets is associated with mutations in FGF23. Nature
Genetics 26 345–348. (doi:10.1038/81664)
www.endocrinology-journals.org
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access

Endocrine-Related Cancer (2011) 18 R53–R77
White KE, Jonsson KB, Carn G, Hampson G, Spector TD,
Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang
IM, Ljunggren O et al. 2001 The autosomal dominant
hypophosphatemic rickets (ADHR) gene is a secreted
polypeptide overexpressed by tumors that cause phos-
phate wasting. Journal of Clinical Endocrinology and
Metabolism 86 497–500. (doi:10.1210/jc.86.2.497)
White KE, Larsson TE & Econs MJ 2006 The roles of
specific genes implicated as circulating factors involved
in normal and disordered phosphate homeostasis: frizzled
related protein-4, matrix extracellular phosphoglycopro-
tein, and fibroblast growth factor 23. Endocrine Reviews
27 221–241. (doi:10.1210/er.2005-0019)
Wild D, Schmitt J, Ginj M, Macke H, Bernard B, Krenning E,
De Jong M, Wenger S & Reubi J 2003 DOTA-NOC, a
high-affinity ligand of somatostatin receptor subtypes 2, 3
and 5 fo labelling with various radiometals. European
Journal of Nuclear Medicine and Molecular Imaging 30
1338–1347. (doi:10.1007/s00259-003-1255-5)
Wild D, Macke HR, Waser B, Reubi JC, Ginj M, Rasch H,
Muller-Brand J & Hofmann M 2005 68Ga-DOTANOC: a
first compound for PET imaging with high affinity for
somatostatin receptor subtypes 2 and 5. European
Journal of Nuclear Medicine and Molecular Imaging 32
724. (doi:10.1007/s00259-004-1697-4)
Wilkins GE, Granleese S, Hegele RG, Holden J, Anderson DW
& Bondy GP 1995 Oncogenic osteomalacia: evidence for
a humoral phosphaturic factor. Journal of Clinical
Endocrinology and Metabolism 80 1628–1634. (doi:10.
1210/jc.80.5.1628)
Williams K, Flanagan A, Folpe A, Thakker R & Athanasou
NA 2007 Lymphatic vessels are present in phosphaturic
mesenchymal tumours. Virchows Archiv 451 871–875.
(doi:10.1007/s00428-007-0471-y)
Woo VL, Landesberg R, Imel EA, Singer SR, Folpe AL,
Econs MJ, Kim T, Harik LR & Jacobs TP 2009
Phosphaturic mesenchymal tumor, mixed connective
tissue variant, of the mandible: report of a case and review
of the literature. Oral Surgery, Oral Medicine, Oral
Pathology, Oral Radiology, and Endodontics 108
925–932. (doi:10.1016/j.tripleo.2009.07.005)
Woznowski M, Quack I, Stegbauer J, Buchner N, Rump LC
& Schieren G 2008 Oncogenic osteomalacia, a rare
paraneoplastic syndrome due to phosphate wasting – a
case report and review of the literature. Clinical
Nephrology 70 431–438.
Wyman AL, Paradinas FJ & Daly JR 1977 Hypophosphataemic
osteomalacia associated with a malignant tumour of the
tibia: report of a case. Journal of Clinical Pathology 30
328–335. (doi:10.1136/jcp.30.4.328)
www.endocrinology-journals.org
Xia WB, Jiang Y, Li M, Xing XP, Wang O, Hu YY, Zhang HB,
Liu HC, Meng XW & Zhou XY 2010 Levels and dynamic
changes of serum fibroblast growth factor 23 in hypo-
phosphatemic rickets/osteomalacia. Chinese Medical
Journal 123 1158–1162.
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K,
Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita
T et al. 2002 Increased circulatory level of biologically
active full-length FGF-23 in patients with hypopho-
sphatemic rickets/osteomalacia. Journal of Clinical
Endocrinology and Metabolism 87 4957–4960. (doi:10.
1210/jc.2002-021105)
Yang IM, Park YK, Hyun YJ, Kim DY, Woo JT, Kim SW,
Kim JW, Kim YS & Choi YK 1997 Oncogenic
osteomalacia caused by a phosphaturic mesenchymal
tumor of the oral cavity: a case report. Korean Journal of
Internal Medicine 12 89–95.
Yavropoulou MP, Kotsa K, Gotzamani Psarrakou A, Papazisi
A, Tranga T, Ventis S & Yovos JG 2010 Cinacalcet in
hyperparathyroidism secondary to X-linked hypopho-
sphatemic rickets: case report and brief literature review.
Hormones 9 274–278.
Yoshioka K, Nagata R, Ueda M, Yamaguchi T, Konishi Y,
Hosoi M, Inoue T, Yamanaka K, Iwai Y & Sato T 2006
Phosphaturic mesenchymal tumor with symptoms related
to osteomalacia that appeared one year after tumorect-
omy. Internal Medicine 45 1157–1160. (doi:10.2169/
internalmedicine.45.1797)
Yun KI, Kim DH & Pyo SW 2009 A phosphaturic
mesenchymal tumor of the floor of the mouth with
oncogenic osteomalacia: report of a case. Journal of Oral
and Maxillofacial Surgery 67 402–405. (doi:10.1016/
j.joms.2008.01.007)
Zimering MB, Caldarella FA, White KE & Econs MJ 2005
Persistent tumor-induced osteomalacia confirmed by
elevated postoperative levels of serum fibroblast growth
factor-23 and 5-year follow-up of bone density changes.
Endocrine Practice 11 108–114.
Zura RD, Minasi JS & Kahler DM 1999 Tumor-induced
osteomalacia and symptomatic looser zones secondary
to mesenchymal chondrosarcoma. Journal of Surgical
Oncology 71 58–62. (doi:10.1002/(SICI)1096-9098
(199905)71:1!58::AID-JSO12O3.0.CO;2-F)
Received in final form 11 March 2011Accepted 12 April 2011Made available online as an Accepted Preprint13 April 2011
R77
Downloaded from Bioscientifica.com at 02/10/2020 05:19:30AMvia free access