Embed Size (px)
Citation preview

1
TP53INP1 down-regulation activates a p73-dependent DUSP10/ERK signaling pathway to
promote metastasis of hepatocellular carcinoma
Kai-Yu Ng1, Lok-Hei Chan1, Stella Chai1, Man Tong1, Xin-Yuan Guan2,4, Nikki P Lee3, Yunfei Yuan5,
Dan Xie5, Terence K Lee6, Nelson J Dusetti7, Alice Carrier7, Stephanie Ma1,4
1School of Biomedical Sciences, Departments of 2Clincial Oncology and 3Surgery, 4State Key
Laboratory for Liver Research, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong
Kong; 5State Key Laboratory of Oncology in Southern China, Sun Yat-Sen University Cancer Center,
Guangzhou, China; 6Department of Applied Biology and Chemical Technology, The Hong Kong
Polytechnic University, Hong Kong; 7Aix Marseille University, CNRS, INSERM, Institut Paoli-
Calmettes, CRCM, Marseille, France
Corresponding author: Stephanie Ma, PhD, School of Biomedical Sciences, Li Ka Shing Faculty of
Medicine, The University of Hong Kong, 1/F, Laboratory Block, 21 Sassoon Road, Pok Fu Lam,
Hong Kong. E-mail: [email protected]; Tel: 852-3917-9238; Fax: 852-2817-0857
Short title: TP53INP1 in HCC metastasis
Keywords: metastasis; TP53INP1; p73; DUSP10; ERK; liver cancer
Abbreviations: DUSP, dual-specificity MAP kinase phosphatases; EV, empty vector; HCC,
hepatocellular carcinoma; IHC, immunohistochemistry; KD, knockdown; MKP, MAP kinase
phosphatases; NTC, non-target control; OE, overexpression; qRT-PCR, quantitative real time
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

2
polymerase chain reaction; shRNA, short hairpin RNA; TP53INP1, tumor protein 53 inducible
nuclear 1
Conflicts of interest: The authors disclose no conflicts.
Funding: This work was supported in part by grants from Research Grants Council – General
Research Fund (HKU_773412M), – Collaborative Research Fund (C7027-14G) and the Croucher
Foundation Innovation Award to S. Ma.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

3
Abstract
Identifying critical factors involved in the metastatic progression of hepatocellular carcinoma
(HCC) may offer important therapeutic opportunities. Here we report that the pro-apoptotic
stress response factor TP53INP1 is often selectively down-regulated in advanced stage IV and
metastatic human HCC tumors. Mechanistic investigations revealed that TP53INP1 down-
regulation in early stage HCC cells promoted metastasis via DUSP10 phosphatase-mediated
activation of the ERK pathway. The DUSP10 promoter included putative binding sites for p73
directly implicated in modulation by TP53INP1. Overall, our findings showed how TP53INP1
plays a critical in limiting progression of early stage HCC, with implications for developing new
therapeutic strategies to attack metastatic HCC.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

4
Introduction
Liver cancer remains one of the most prevalent and deadliest cancer types worldwide.
Hepatocellular carcinoma (HCC) accounts for over 75% of all liver cancer cases. Metastasis and
post-surgical recurrence are common and represent major obstacles to the improvement of
patient survival. HCC patients are often diagnosed at an advanced stage when curative therapy is
no longer available and even after surgery, the prognosis of HCC remains unsatisfactory, with a
five year post-recurrence rate at >70%. Metastasis is a complex multistep process involving
alterations in the dissemination, invasion, survival and growth of new cancer cell colonies, which
are regulated by a complex network of intra- and inter-cellular signal transduction cascades (1).
However, metastasis remains the most poorly understood component of cancer pathogenesis (2).
Elucidation of the mechanisms underlying metastasis is fundamental for the development of new
therapeutic treatments for advanced metastatic HCC.
Extracellular signal-regulated kinases (ERKs) have been shown to play critical roles in malignant
transformation and cancer metastasis (3). Oncogenic activation of ERKs can be induced by various
mechanisms including transcriptional overexpression, mutations in upstream components of the
MAP kinase pathway, such as RAS and BRAF, and down-regulation of negative regulator dual-
specificity MAP kinase phosphatases (DUSPs) (4). ERK plays a major role in invasion by inducing
proteases that degrade the basement membrane, enhances cell migration, and increases cell
survival. Activated ERK pathway has been shown to correlate with the expression of epithelial-
mesenchymal transition (EMT) markers, a hallmark of metastasis. These findings collectively
suggest that ERK plays a major role in tumor progression and metastasis. However, our
knowledge of endogenous regulators of DUSP/ERK remains limited and how they work to
promote HCC metastasis is also not known.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

5
TP53INP1 is a stress-induced tumor suppressor gene with anti-proliferative and pro-apoptotic
activities (5-6). It is an alternatively spliced gene encoding two protein isoforms (α and β), and
when overexpressed, both isoforms exert a tumor suppressor function, mainly by inducing the
transcription of target genes involved in cell-cycle arrest and p53-mediated apoptosis as part of
the cell responses to genotoxic stress. Significant reduction or loss of TP53INP1 expression has
been shown in a number of cancer types including those of the stomach (7), breast (8), pancreas
(9), esophagus (10), lung (11), melanocyte (12), colon (13) and blood (14). In relation to
metastasis, TP53INP1 has only been implicated in a handful of studies including one report where
they found transcriptional levels of TP53INP1 to be down-regulated in metastatic lung of brain
cancers (15). A more recent study led by our collaborator Dusetti et al. found TP53INP1 to reduce
pancreatic cancer cell migration by regulating SPARC expression (16). TP53INP1 is a target gene
of the transcription factor p53. Conversely, TP53INP1 has also been shown to play a role in
cellular homeostasis through p53-dependent and p53-independent manners (5-6). In addition to
p53, TP53INP1, which is also a p73 target gene, can enhance transcriptional activity of p73 to
induce cell cycle arrest and cell death (17). Thus, TP53INP1 can exert its tumor suppressor
function by inducing the transcription of both p53 and p73 target genes.
In our previous studies, we found that the initiation, growth and self-renewal of CD133+ liver
tumors to be fine-tuned by a balance of miR-130b overexpression and TP53INP1 down-regulation
(18). This result suggests that TP53INP1 is a critical effector driving hepatocarcinogenesis.
Nevertheless, to date, no studies have determined the function of TP53INP1 in HCC metastasis or
the molecular mechanism by which TP53INP1 regulates invasion and metastasis in HCC. Here, we
demonstrate that TP53INP1 is frequently down-regulated in advanced stage and metastatic
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

6
human HCC tumors and that down-regulation of TP53INP1 in HCC functionally promotes
metastasis through ERK activation via a p73-dependent DUSP10 regulation. Findings from our
study not only provides new insight into how HCC metastasis is regulated but also provides a new
layer of mechanism by which DUSP10/ERK signaling is regulated by p73/TP53INP1 and also
identifies DUSP10 as a new transcriptional effector of p73.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

7
Materials and Methods
Gene expression profiling and patient samples. Gene expression profiling studies involving
multiple clinical samples were performed analyzing the expression of specific transcripts in two
datasets available through Gene Expression Omnibus (GSE25097 and GSE40367) (19-20). In
addition, human primary and matched metastatic HCC tissue samples were obtained from 37
patients undergoing hepatectomy at the Sun Yat‐Sen University Cancer Centre in Guangzhou,
China. Tissue samples were collected from patients who had not received any previous local or
systemic treatment prior to operation. Use of human samples was approved by the committee for
ethical review of research involving human subjects at the Sun Yat‐Sen University Cancer Centre.
Cell lines. Human HCC cell lines Hep3B, SNU182, SK-Hep-1, and human hepatoblastoma cell line
HepG2 were purchased from American Type Culture Collection. Human liver cell line LO2 and HCC
cell lines PLC8024, QSG-7701, QGY-7703 were obtained from the Institute of Virology, Chinese
Academy of Medical Sciences, Beijing, China. Human HCC cell line HLE, was obtained from
Japanese Collection of Research Bioresources Cell Bank. Immortalized normal liver cell line, MIHA,
was provided by Dr. J. R. Chowdhury, Albert Einstein College of Medicine, New York (21).
MHCC97L cells were obtained from Liver Cancer Institute, Fudan University, China (22). 293FT
cells were purchased from Invitrogen. All cell lines used in this study were obtained between
2013 to 2016, regularly authenticated by morphologic observation and AuthentiFiler STR
(Invitrogen) and tested for absence of mycoplasma contamination (MycoAlert, Lonza). Cells were
used within 20 passages after thawing.
Reagents. U0126 was purchased from Cell Signaling Technologies. Mitomycin C was purchased
from Calbiochem.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

8
Phospho-kinase array profiling. Proteome Profiler Human Phospho-kinase Array Kit was
purchased from R&D Systems (ARY003B).
Quantitative real‐time PCR (qRT-PCR). Total RNA was extracted using RNAisoPlus (Takara).
For qRT-PCR of mRNA targets, cDNA was synthesized by PrimeScript RT Master Mix (Takara) and
amplified with EvaGreen qPCR MasterMix-R (ABM) and primers listed in Supplementary Table 1.
β-actin was amplified as an internal control. Reactions were performed on an ABI Prism 7900
System (Applied Biosystems) with data analyzed using the ABI SDS v2.3 software (Applied
Biosystems). Relative expression differences were calculated using the 2‐ΔΔCt method.
Western blot. Protein lysates were quantified and resolved on a SDS-PAGE gel, transferred onto
PVDF membrane (Millipore) and immunoblotted with a primary antibody, followed by incubation
with a secondary antibody. Antibody signal was detected using an enhanced chemiluminescence
system (GE Healthcare). The following antibodies were used: TP53INP1 (1:250, Genway Biotech,
GWB-61D856), p-ERK1/2 (1:1000, Cell Signaling Technology, 9101), total ERK (1:1000, Cell
Signaling Technology, 9102), DUSP10 (1:500, Cell Signaling Technology, 3483), p73 (1:1000,
Novus Biologicals, NB100-56674), BAX (1:1000, Cell Signaling Technology, 2772), MDM2 (1:500,
Santa Cruz, sc-965) and β‐actin (1:5000, Sigma‐Aldrich, A5316).
Expression plasmids and lentiviral transduction. Expression plasmids for shRNAs were made
in a pLKO.1-puro vector (Sigma-Aldrich). The targeted sequences were: human TP53INP1 (464,
5’- CCGGCATAGATACTTGCACTGGTTTCTCGAGAAACCAGTGCAAGTATCTATGTTTTTTG -3’) and
(3834, 5’- CCGGGCGCCATGTTTCTCAAAGTTTCTCGAGAAACTTTGAGAAACATGGCGCTTTTTTG -3’);
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

9
human p73 (753, 5’-
CCGGATCCGCGTGGAAGGCAATAATCTCGAGATTATTGCCTTCCACGCGGATTTTTTG -3’) and (1643,
5’- CCGGCCAAGGGTTACAGAGCATTTACTCGAGTAAATGCTCTGTAACCCTTGGTTTTTG -3’); human
ERK1 (5’- CCGGCTATACCAAGTCCATCGACATCTCGAGATGTCGATGGACTTGGTATAGTTTTTG -3’)
and ERK2 (5’- CCGGGACATTATTCGAGCACCAACCCTCGAGGGTTGGTGCTCGAATAATGTCTTTTTG -
3’) and scrambled shRNA non-target control (NTC) (5’-
CCGGCAACAAGATGAAGAGCACAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT -3’). Sequences
were transfected into 293FT cells, packaged using MISSION Lentiviral Packaging Mix (Sigma-
Aldrich). The full-length complementary DNA of human DUSP10 was amplified in cDNA of human
adult normal liver tissue RNA (BioChain) as a template using the following primers (forward 5’-
GGGGACAAGTTTGTACAAAAAAGCAGGCTGCCACCATGCCTCCGTCTCCTTTAGAC -3’; reverse 5’-
GGGGACCACTTTGTACAAGAAAGCTGGGTCACACAACCGTCTCCACG -3’); and then cloned into the
Gateway entry vector pDONR201. DUSP10 was then shuttled into the Gateway destination vector
pEZ-Lv199 (GeneCopoeia). Sequences were transfected into 293FN cells, packaged using Lenti-
Pac HIV expression packaging mix (GeneCopoeia). Stable clones were selected with puromycin.
The full-length complementary DNA of human TP53INP1 was amplified in cDNA of human adult
normal liver tissue RNA (BioChain) as a template using the following primers (forward 5’-
GGGGACAAGTTTGTACAAAAAAGCAGGCTGCCACCATGTTCCAGAGGCTGAATAAAATGT -3’; reverse
5’-GGGGACCACTTTGTACAAGAAAGCTGGGTTAGTAATTGTACTGACGCGGG -3’); and then cloned
into the Gateway entry vector pDONR201. TP53INP1 was then shuttled into the Gateway
destination vector pLenti CMV Blast DEST (706-1) (Addgene plasmid #17451). Sequences were
transfected into 293FN cells, packaged using LentiPac HIV expression packaging mix
(GeneCopoeia). Stable clones were selected with blasticidin.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

10
Cell motility and invasion assays. Migration and invasion assays were conducted in 24‐well
Millicell hanging inserts (Millipore) and 24‐well BioCoat Matrigel Invasion Chambers (BD
Biosciences), respectively. Cells re‐suspended in serum free DMEM were added to the top
chamber and medium supplemented with 10% FBS was added to the bottom chamber as a
chemoattractant. After 48 hrs of incubation at 37°C, cells that migrated or invaded through the
membrane (migration) or Matrigel (invasion) were fixed and stained with crystal violet
(Sigma‐Aldrich). The number of cells was counted in 3 random fields under 20x objective lens and
imaged using SPOT imaging software (Nikon).
Immunohistochemistry. Immunohistochemical staining of paraffin sections was carried out
using a two-step protocol. After antigen retrieval, sections were incubated with the following
antibodies against anti-human TP53INP1 (clone A25-E12; 6µg/ml) (9), anti-human p73 (1:500,
Novus Biologicals, NB100-56674), anti-human DUSP10 (1:50, Cell Signaling Technology, 3483)
and anti-human p-ERK1/2 (1:500, abcam; ab50011). Anti-mouse, -rabbit and -rat HRP-labeled
polymer (DAKO) was used as secondary antibodies. Color detection was performed by liquid
DAB+ substrate chromogen system (DAKO). Slides were counterstained with Mayer’s
hematoxylin. According to the intensity and total area of the staining, the expression of TP53INP1
was scored as either low (< 30%), medium (30 to 60%) or high (> 60%) expression.
Luciferase reporter assay. Both fragments of the DUSP10 promoter regions S1 (-4400 to -
2201bp, carrying predicted site sequences ATTAAGTTTCAACATGTA and
ATCATGTTACAACATCCA) and S2 (-2200 to -1bp, carrying predicted site sequences
GGTATGTGCCTGCATGTA and GGCAAGGGGCGGCTTGCC) were amplified and cloned into the XhoI
and HindIII sites of a pGL3 basic vector (Promega) for luciferase reporter assay. All PCR products
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

11
cloned into the plasmid were verified by DNA sequencing to ensure that they were free of
mutations and in the correct cloning direction. Primer sequences used listed in Supplementary
Table 2.
Chromatin immunoprecipitation (ChIP) assay. ChIP assay was performed using the
MagnaChIP™ A kit (Millipore). Briefly, cells were sonicated and lysed after cross-link treatment by
1% formaldehyde for 10 min. The crosslinked protein / DNA complex was immunoprecipitated by
anti-p73 antibody or normal IgG bound to protein A magnetic beads. After overnight incubation at
4oC, the complex was eluted and DNA was purified. The immunoprecipitated DNA was quantified
by qPCR using primer sequences designed to detect specific regulatory regions listed in
Supplementary Table 3.
Animal studies. The study protocol was approved by and performed in accordance with the
Committee of the Use of Live Animals in Teaching and Research at The University of Hong Kong.
Metastasis was assessed by orthotopically injecting into the liver to observe for extrahepatic
metastasis to the lung. Luciferase‐labeled cells were injected into the left lobes of the livers of 6-
weeks old BALB/c nude mice (n = 6-10/group). Six to eight weeks after implantation, mice were
administered with 100mg/kg D-luciferin (Gold Biotechnology) via peritoneal injection 5 mins
before bioluminescent imaging (IVISTM 100 Imaging System, Xenogen). Livers and lungs were
harvested for ex vivo imaging and histological analysis. Metastatic nodules in the lungs were
counted.
Statistical analysis. Data were analyzed by SPSS 21.0 or GraphPad Prism 6.0 and shown as mean
± standard deviations, unless otherwise specified. Differences between groups were analyzed by
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

12
unpaired Student’s t test for continuous variables. Correlation between expressions was analyzed
by Chi-square test. Statistical significance was defined as p ≤ 0.05.
Results
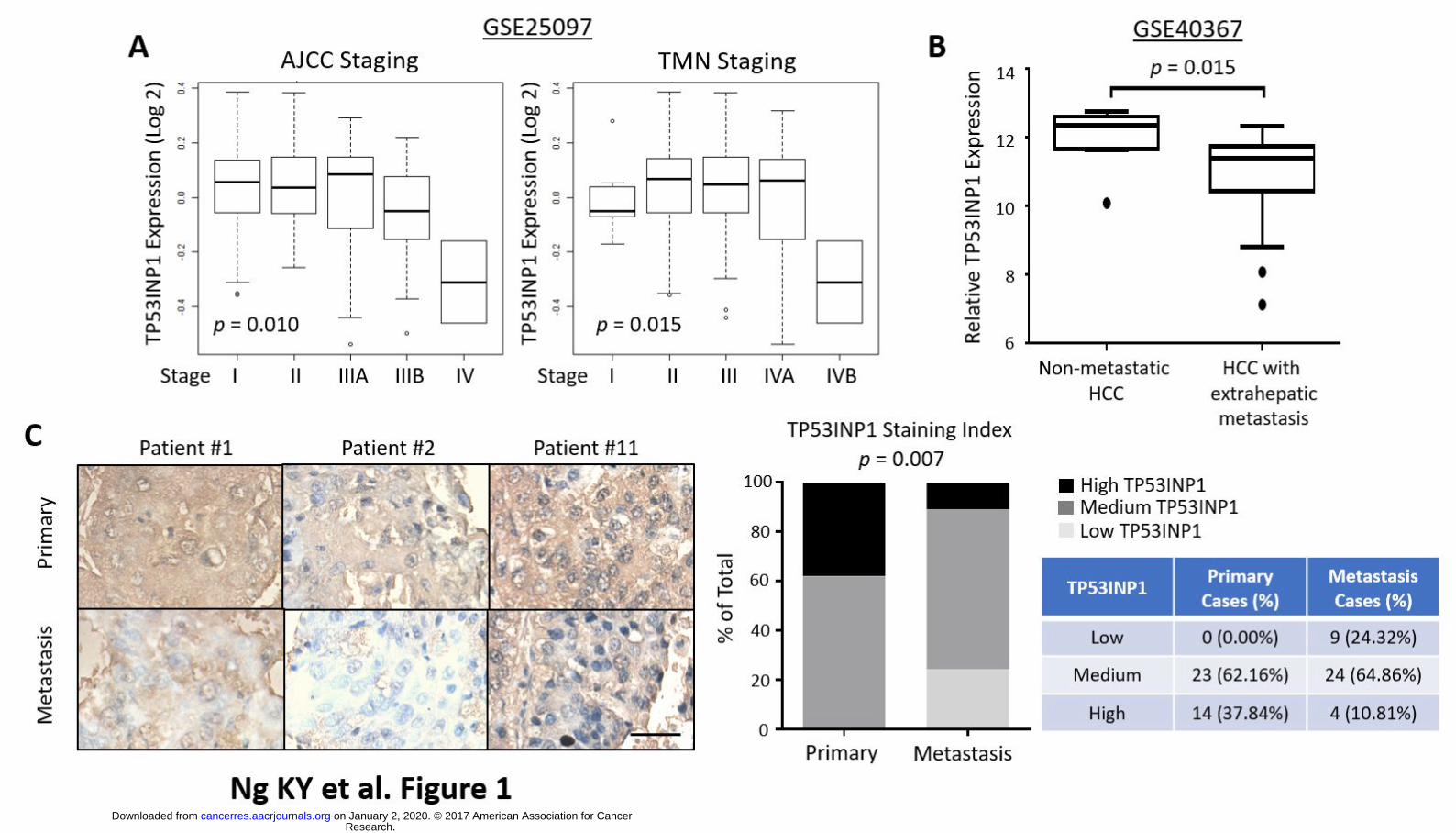
TP53INP1 is down-regulated in advanced stage and metastatic HCC tumors. As an initial
attempt to explore whether TP53INP1 expression is associated with metastasis, we evaluated the
expression of TP53INP1 transcripts in two public gene expression databases. We found that in
advanced stage HCC samples (Stage IV of AJCC and TNM) that is more likely associated with
recurrence and metastasis , the expression of TP53INP1 was significantly lower than that in early
stage samples (Stages I, II and III) (GSE25097) (19) (Fig. 1A). In a second, independent data set
(GSE40367) (20) that compares metastatic free HCCs and HCCs with extrahepatic metastases, we
also observed significantly lower expression of TP53INP1 in HCC samples with extrahepatic
metastases (Fig. 1B). To confirm these observations experimentally, we carried out
immunohistochemical analyses in 37 pairs of matched primary and metastatic HCC tissue
samples. Consistently, TP53INP1 was found to be down-regulated in metastatic HCC. Only 28 of
the 37 metastatic HCC samples were strong or moderately positive for TP53INP1 and 9 were
weak or negative. In contrast, moderate or strong immune positivity for TP53INP1 was present in
all 37 out of 37 primary HCC cases, suggesting that a down-regulation of TP53INP1 expression is
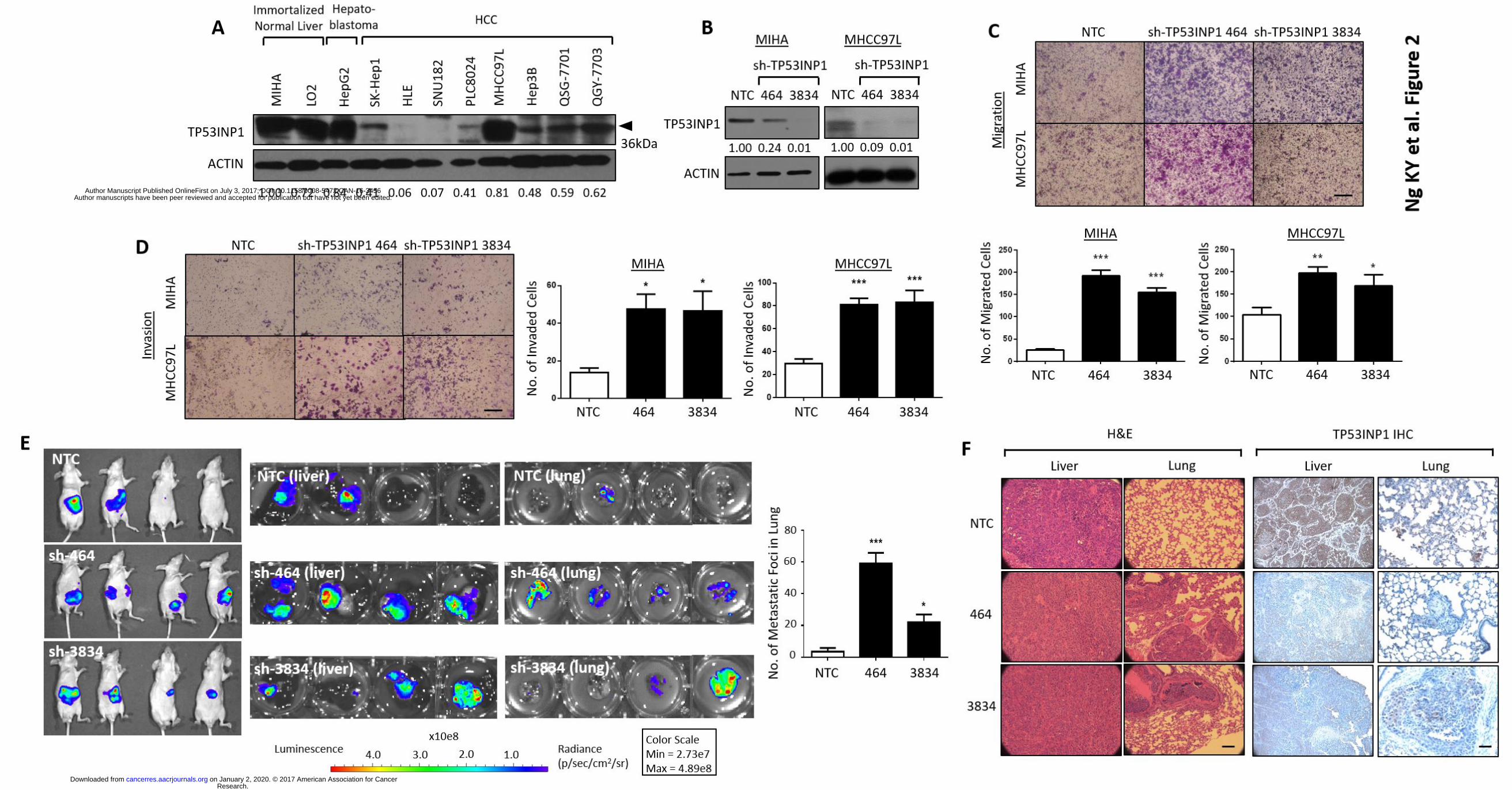
involved in HCC metastasis (Fig. 1C). We then carried out Western Blot analyses in a panel of
immortalized normal liver (MIHA and LO2), hepatoblastoma (HepG2) and HCC cell lines (SK-
Hep1, HLE, SNU182, PLC8024, MHCC97L, Hep3B, QSG-7701 and QGY-7703). The expression of
TP53INP1 was high in the immortalized normal liver and hepatoblastoma cells, while 7 of the 8
HCC cell lines examined displayed significantly lower or undetectable levels of TP53INP1
expression (Fig. 2A).
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

13
TP53INP1 knockdown promotes HCC metastasis. To assess the functional role of TP53INP1 in
cancer cells, we knocked down expression of TP53INP1 in immortalized normal liver cells MIHA
and HCC cells MHCC97L using two TP53INP1-specific shRNA lentiviruses (sh-TP53INP1 464 and
3834). As controls, we used lentiviruses expressing non-specific shRNA (non-target control, NTC).
Efficient TP53INP1 knockdown was confirmed by Western Blot (Fig. 2B). We found that
TP53INP1 shRNA-expressing cells had a significantly enhanced ability to migrate and invade
compared with control cells (Figs. 2C-D). Similar results were also obtained when the same
experiment was performed in the presence of mitomycin C, where cells were inhibited to
proliferate, suggesting that TP53INP1-mediated migration and invasion is not a misinterpretation
of the cells’ altered ability to proliferate (Supplementary Fig. 1). To confirm these findings, we
further examined the effects of TP53INP1 expression in an in vivo experimental metastasis model
where cells were orthotopically injected into the liver for observation of metastasis to the lung.
TP53INP1 suppression induced a potent increase in the ability of MHCC97L cells to not only form
tumors in the liver, but also metastasize to the lung (sh-464: 7 of 10 tumors formed in the liver
with 6 developing extrahepatic metastasis in the lung; sh-3834: 8 of 10 tumors formed in the liver
with 4 developing extrahepatic metastasis in the lung). In contrast, MHCC97L control cells only
resulted in tumor growth in the liver in 6 of 10 mice injected, with only 2 mice going on to develop
lung metastasis (Fig. 2E; only 4 representative mice shown). Mice were sacrificed after 8 weeks
and both livers and lungs were removed for histological analyses. H&E staining of the tumors
confirmed the bioluminescence signals observed to indeed represent tumor cells and that there is
altered ability of the cells to metastasize to the lung as evident by increased number of tumor
nodules present there (Figs. 2E-F). Immunohistochemical analysis also found TP53INP1
expression to be preferentially expressed in the livers and lungs of the non-target control
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

14
xenografts (Fig. 2F). In addition, to rule out any potential off-target effects of our knockdown
shRNAs, we performed experiments to rescue the effects of TP53INP1 shRNAs on migration and
invasion by overexpressing TP53INP1 in the same cells. Overexpression of TP53INP1 in MHCC97L
cells with TP53INP1 stably repressed rescued the ability of the cells to attenuate migration and
invasion in both knockdown clones, further demonstrating the importance of TP53INP1 in
regulating metastasis in HCC (Supplementary Fig. 2).
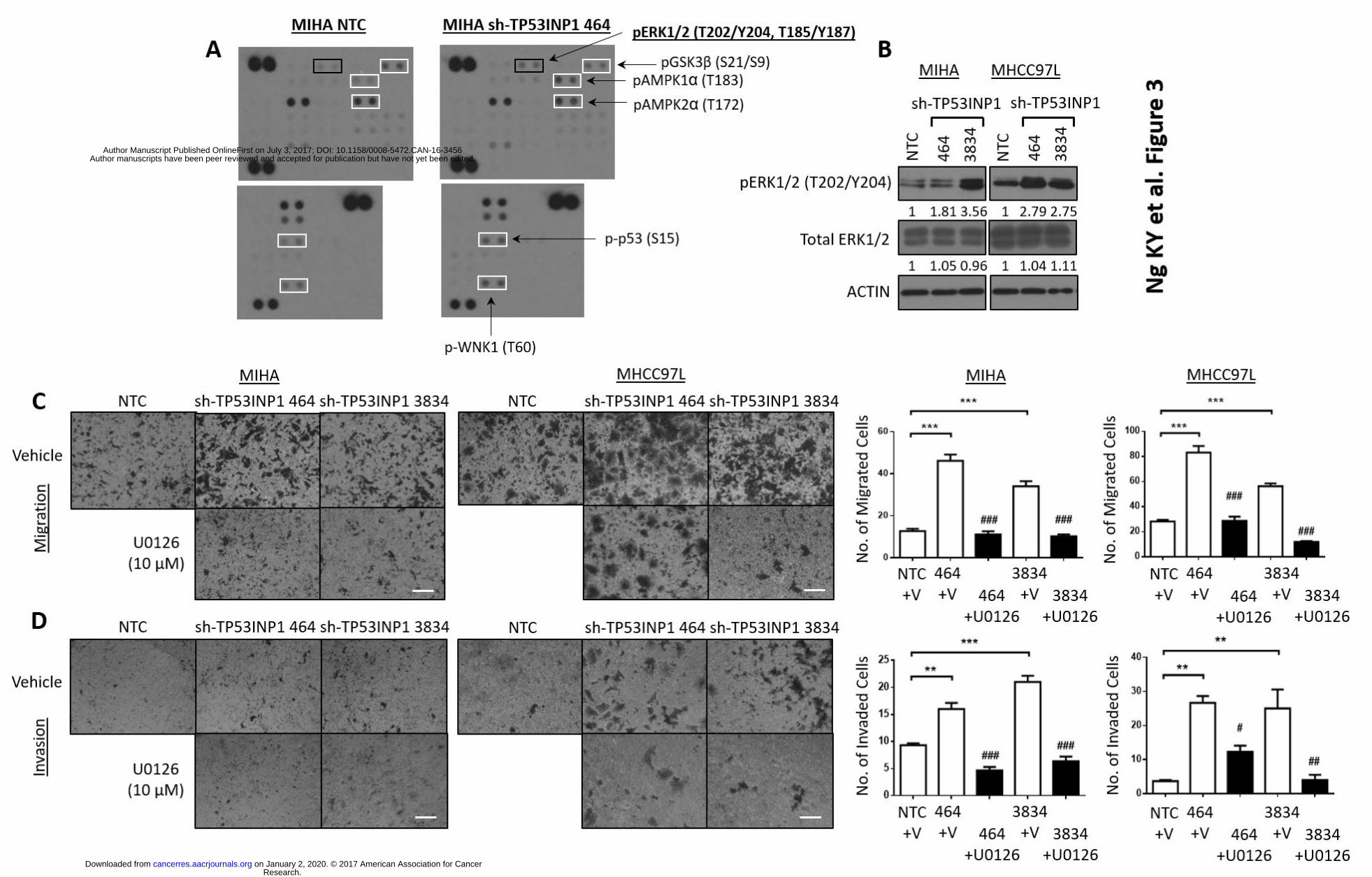
Phopsho-kinase array profiling analysis identifies activation of ERK to be involved in
TP53INP1-mediated HCC metastasis. To elucidate the molecular mechanism of TP53INP1 in
regulating HCC metastasis, a Proteome Profiler Human Phospho-Kinase Array Kit was utilized to
compare the relative levels of 43 human protein kinase phosphorylation between HCC cells with
or without TP53INP1 knocked down. Intensity of the spots on the array was quantified by ImageJ
analyses and those spots that displayed > 1.5 fold change between control and TP53INP1 knocked
down cells were selected for further validation by Western Blot analyses. Altogether, 6 phospho-
kinases were found altered, including pERK1/2 (T202/Y204, T185/Y187), pGSK3β (S21/S9),
pAMPK1α (T183), pAMPK2α (T172), p-p53 (S15) and p-WNK1 (T60) (Fig. 3A), of which only p-
ERK1/2 could be validated to be commonly increased in both MIHA and MHCC97L cells (Fig 3B).
To further validate the role of pERK1/2 signaling in TP53INP1 regulated metastasis, we analyzed
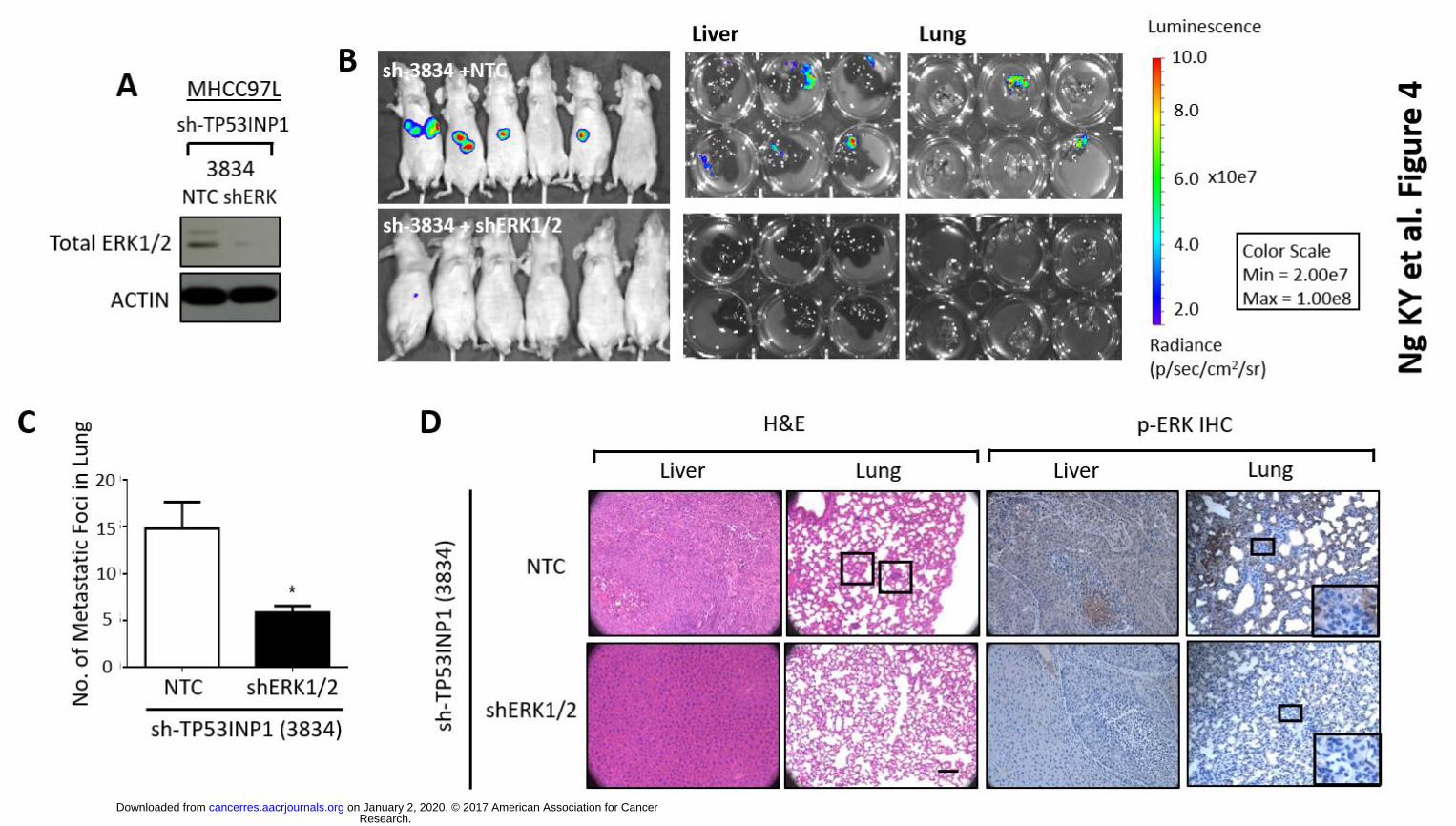
the impact of introducing an ERK inhibitor (U0126) or stable shRNA against ERK1/2 into HCC
cells with TP53INP1 stably repressed on these altered metastatic phenotype. Introduction of
U0126 or sh-ERK1/2 in TP53INP1 suppressed HCC cells attenuated in vitro cell migration and
invasion abilities (Figs. 3C-D, Fig. 4A, Supplementary Fig. 3), as well as lung metastasis in vivo
(Figs. 4B-D), suggesting that ERK signaling is needed to drive metastasis in TP53INP1 deficient
HCC. Immunohistochemical analysis also found p-ERK expression to be preferentially expressed
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

15
in the livers and lungs of the non-target control of sh-TP53INP1 xenografts (Fig. 4D). Note that
1µM and 10µM of ERK inhibitor U0126 was initially used to test which concentration was most
appropriate for experimental use. At the end, 10µM concentration was chosen as it resulted in
complete abolishment of ERK expression as evident by Western Blot analysis, with no sign of
toxicity to the cells (data not shown).
TP53INP1 inhibits HCC metastasis through DUSP10-dependent modulation of ERK. Dual-
specificity MAP kinase (MAPK) phosphatases (MKPs or DUSPs) are well-established negative
regulators of MAPK/ERK signaling in mammalian cells and tissues. By virtue of their differential
subcellular localization and ability to specifically recognize, dephosphorylate and inactivate
different MAPK isoforms, they are key spatiotemporal regulators of pathway activity. The MKPs
constitute a distinct subgroup of eleven catalytically active enzymes within the larger family of
DUSPs, which all share a conserved cluster of basic amino acid residues involved in MAPK
recognition (23-25). A screen of these DUSP members at the genomic level by qRT-PCR in HCC
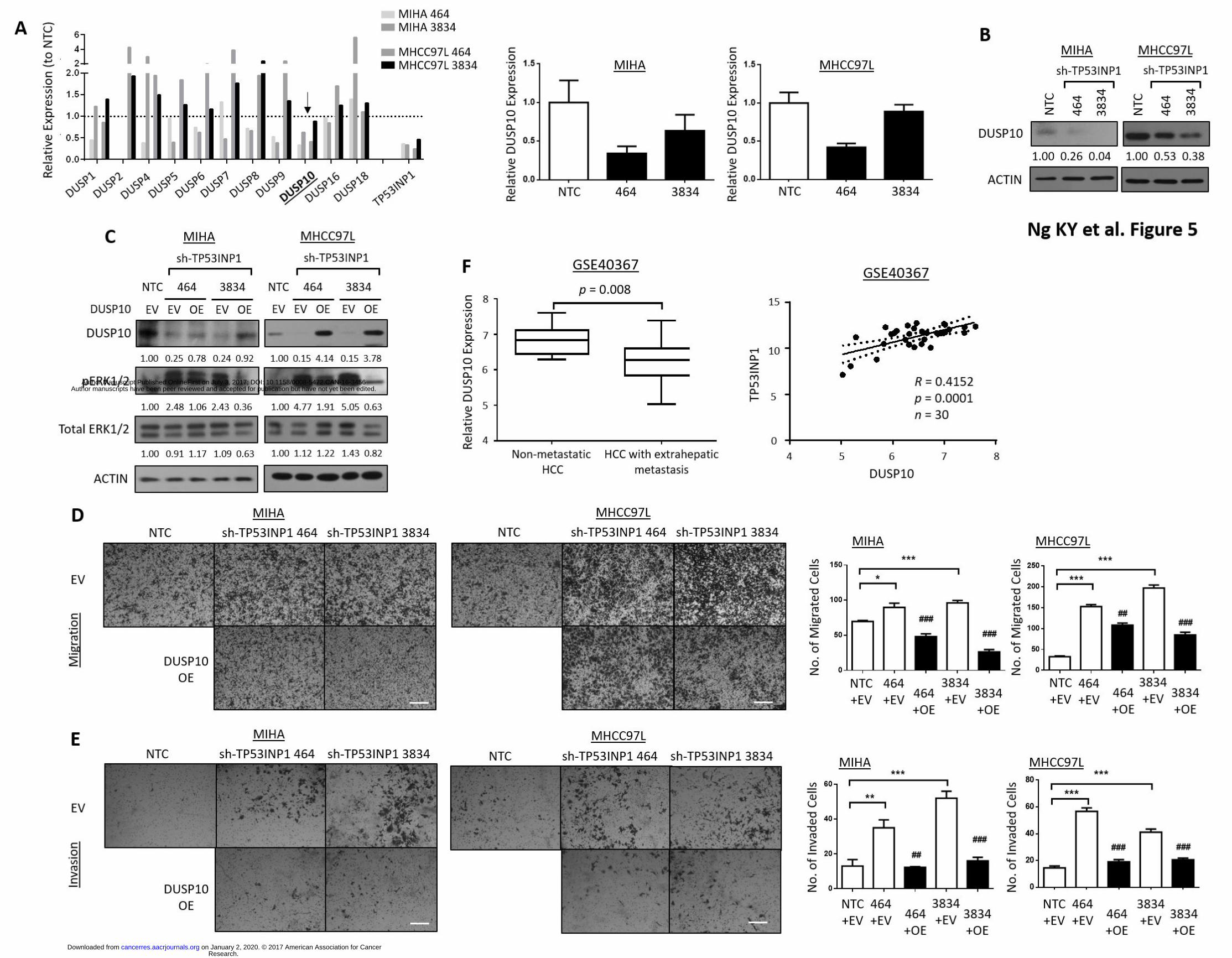
cells with or without TP53INP1 suppressed identified DUSP10/MKP-5 to be consistently down-
regulated in both MIHA and MHCC97L cells following TP53INP1 knock down (Fig. 5A). This
observation was further validated at the proteomic level by Western Blot where DUSP10 was
found to be significantly down-regulated (Fig. 5B), concomitant with p-ERK1/2 activation in
TP53INP1 shRNA-expressing cells as compared with control cells (Fig. 3B). To further validate the
role of DUSP10-mediated pERK1/2 signaling in TP53INP1 regulated metastasis, rescue
experiments where DUSP10 was re-introduced into HCC cells with TP53INP1 stably repressed
was carried out (Fig. 5C). Introduction of DUSP10 in TP53INP1 suppressed HCC cells resulted in a
marked decrease in phosphorylated ERK (Fig. 5C) concomitant with attenuated abilities of HCC
cells to migrate and invade in vitro (Figs. 5D-E), suggesting that DUSP10-mediated alteration of p-
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

16
ERK in TP53INP1 low/absent HCC cells can indeed promote metastasis. Consistently, we also
observed a significantly lower expression of DUSP10 in human HCC samples with extrahepatic
metastases as compared to metastatic free HCC samples in the GSE40367 dataset (20). A positive
correlation between TP53INP1 and DUSP10 expression was also found in the same sample cohort
(R = 0.4152; p = 0.0001) (Fig. 5F).
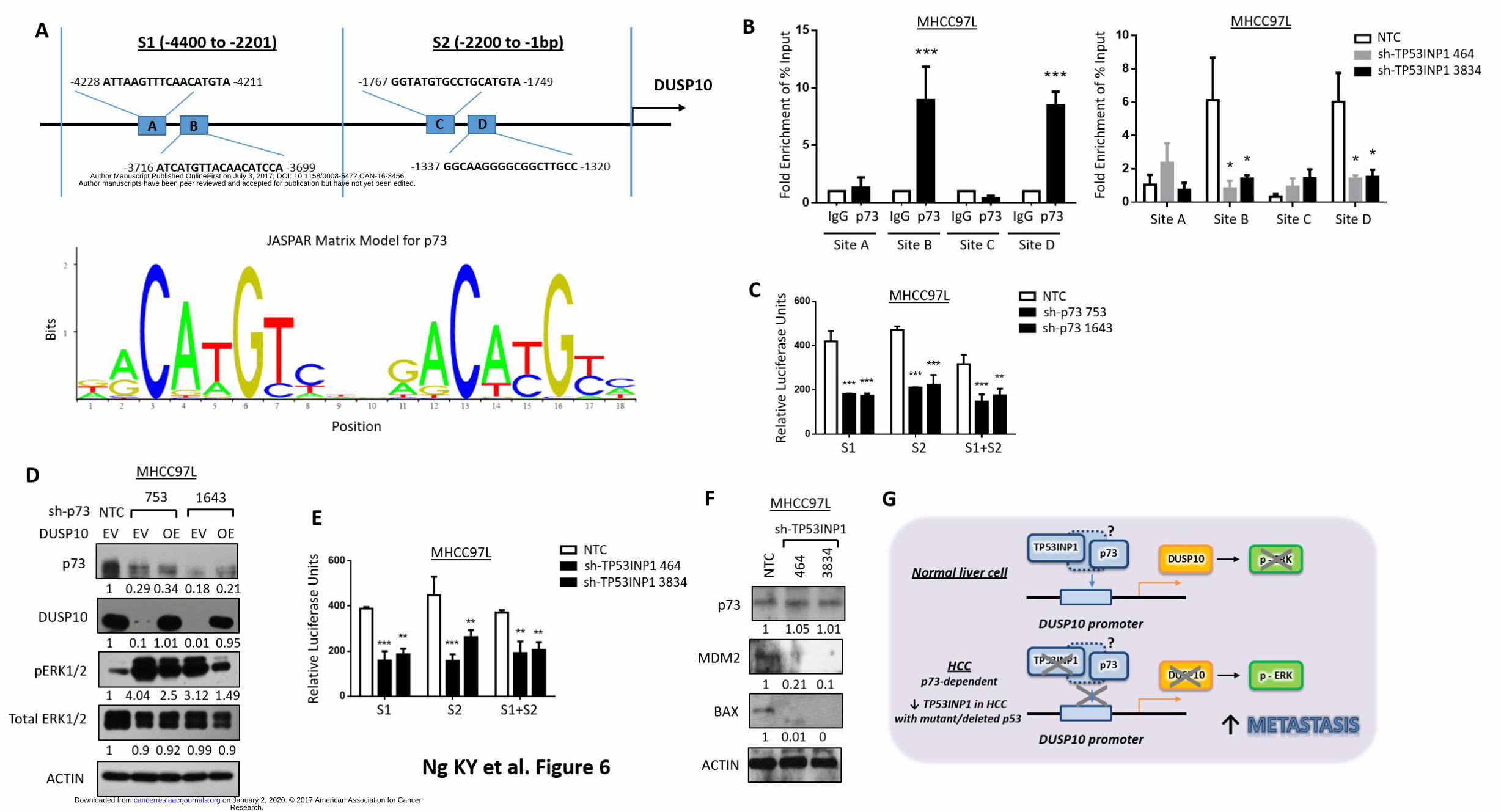
p73, which transcriptional activity is known to be modulated by TP53INP1, binds and
regulates DUSP10 via promoter binding and cooperatively drive ERK activation in HCC. To
determine the link between TP53INP1 and DUSP10 mediated ERK signaling in regulating HCC
metastasis, the upstream region of DUSP10 (-1 to -4400) was analyzed using JASPAR
(http://jaspar.genereg.net). Four predicted binding sites of p73, which activity is known to be
modified by TP53INP1 (17), was found in the upstream region of DUSP10 [two sites in S1 (A and
B); and two sites in S2 (C and D)], with a high relative score of >0.75 (Figs. 6A). ChIP assays
showed high physical binding affinity of endogenous p73 to DUSP10 in MHCC97L cells in two of
the four predicted sites, namely site B (at -3716 to -3699) and site D (at -1337 to -1320) (Fig. 6B,
left). To delineate the involvement of TP53INP1 in the regulation of p73 activity and its
subsequent binding to the promoter of DUSP10, we knocked down TP53INP1 in MHCC97L and
repeated the ChIP assay again. Silencing of TP53INP1 attenuated binding of p73 to DUSP10
promoter in the same two binding sites (B and D) (Fig. 6B, right), suggesting that TP53INP1 does
indeed play a role in modulating the binding affinity of p73 to the DUSP10 promoter. Note that it
has previously been reported that TP53INP1 can also alter the transactivation capacity of p73 on a
number of genes, demonstrating a functional association between p73 and TP53INP1 (17).
Notably, both MIHA liver and MHCC97L HCC cell lines that were used for functional experiments
in our current study are either p53 absent (MIHA) or mutant (MHCC97L). Both cell types are
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

17
however p73 wild type. Luciferase reporter assays showed high transcriptional activity of
endogenous p73 to DUSP10 in MHCC97L cells in both sites 1 and 2, as knockdown of p73 would
decrease the activation of DUSP10 promoter by two folds (Fig. 6C). Stable knockdown of p73 in
MHCC97L cells led to a marked decrease in DUSP10 and concomitant increase in pERK1/2
expression; while overexpression of DUSP10 in cells with p73 stably suppressed can cancel this
effect (Fig. 6D). Further, we found stable TP53INP1 knockdown in MHCC97L cell to also result in a
similar decrease in DUSP10 promoter activation (Fig. 6E). Immunohistochemical staining of
xenograft tumors generated from HCC cells with and without TP53INP1 knockdown further
validated these observations as TP53INP1 repressed tumors displayed elevated pERK1/2
concomitant with a decrease in DUSP10 (Supplementary Fig. 4). Note p73 expression levels
remain unchanged in TP53INP1 repressed HCC cells, as evidenced by both Western Blot and IHC
analyses (Fig. 6F and Supplementary Fig. 4). In addition, we also noted that in addition to DUSP10,
knockdown of TP53INP1 would similarly lead to a marked down-regulation of other known p73
targets, including MDM2 and BAX2 (17) (Fig. 6F). Taken together, TP53INP1 can enhance p73
ability to drive DUSP10 transcription, thereby altering downstream ERK signaling to drive HCC
metastasis. In HCC tumors where p73 mutations are rarely observed, TP53INP1 down-regulation
promotes HCC metastasis through DUSP10 inactivation via p73-dependent DUSP10 promoter
binding and regulation, resulting in activation of the ERK signaling pathway (Fig. 6G).
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

18
Discussion
Metastasis is a major hallmark of cancer and yet remains the most poorly understood component
of cancer pathogenesis (2). It is a complex multistep process involving alterations in the
dissemination, invasion, survival and growth of new cancer cell colonies, which are regulated by a
complex network of intra- and inter-cellular signal transduction cascades (1). In this study, we
demonstrate that TP53INP1 is frequently down-regulated in advanced stage and metastatic
human HCC tumors and that down-regulation of TP53INP1 in HCC promotes metastasis through
DUSP10 inactivation via p73-dependent DUSP10 promoter binding and regulation, resulting in
activation of the ERK signaling pathway. Findings from our study not only provides new insight
into how HCC metastasis is regulated but also provides a new layer of mechanism by which
DUSP10/ERK signaling is regulated by p73/TP53INP1. Note that since TP53INP1-mediated
ERK1/2 activation can also lead to increased cell proliferation, we must take caution when we
interpret our metastasis findings, such that we must ensure that the metastasis effect is not a by-
product of the cells’ altered proliferation potential. To address this, we repeated our migration
and invasion assays again, in the presence of mitomycin C, a drug used to inhibit cell proliferation.
TP53INP1 is a stress-induced p53-target gene whose expression is modulated by transcription
factors such as p53, p73 and E2F1 (6, 17, 26). It encodes two protein isoforms, TP53INP1α and
TP53INP1β (5), which have similar functions and can induce cell cycle arrest and apoptosis when
overexpressed (6). In association with homeodomain-interacting protein kinase-2 (HIPK2),
TP53INP1 phosphorylates p53 protein at serine 46, thereby enhancing p53 protein stability and
its transcriptional activity, leading to transcriptional activation of p53-target genes, cell growth
arrest and apoptosis upon DNA damage stress (27). The anti-proliferative and pro-apoptotic
activities of TP53INP1 indicate that TP53INP1 has an important role in cellular homeostasis and
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

19
DNA damage response. TP53INP1 can be subcellularly localized in the nucleus or cytoplasm
depending on the context. In addition to its role in the nucleus where it stimulates the
transcriptional activity of p53 and p73 (17, 27), it also contributes to autophagy and regulation of
energetic metabolism and reactive oxygen species (28-31).
Deficiency in TP53INP1 expression results in increased tumorigenesis. A number of studies have
demonstrated a significant reduction of TP53INP1 expression during cancer formation of the
stomach (7), breast (8), pancreas (9), esophagus (10), lung (11), melanocyte (12), colon (13) and T-
cell leukemia (14); and that down-regulation of TP53INP1 correlated with more aggressive
clinico-pathological behaviors in several human cancer types (7-9, 11). TP53INP1-deficient mice
exhibited exacerbated colitis-associated carcinogenesis (32), while TP53INP1 expression was
found to be lost in rat pre-neoplastic liver lesions (33). In contrast to this, two recent studies
published by the same group in 2012 have found TP53INP1 to be frequently overexpressed in
prostate cancer and castration-resistant prostate cancer, and that its overexpression correlated
with poor prognostic factors and is predictive of tumor relapse (34-35), suggesting that TP53INP1
appears to play a dual role as both a tumor-suppressing and tumor-promoting gene and that its
expression trend is cancer type specific. TP53INP1 down-regulation in cancers is regulated at
multiple levels by DNA methylation (10), the transcription factors c-myc (10) and n-myc (36),
histone deacetylase 2 (36) as well as a plethora of miRNAs including miR-569 (37), miR-155 (9,
38-40), miR-182 (41), miR-93, miR-130b (14, 18), miR-30a, miR-205 (42-43) and miR-125b (11).
Studies have not only demonstrated a functional tumor suppressive role of TP53INP1 but also a
role in modulating cancer stem cell phenotypes (38), cisplatin and gemcitabine resistance (41, 44),
as well as oxidative stress (45). In our previous studies, we found that the initiation, growth and
self-renewal of CD133+ liver tumors are regulated by a balance of miR-130b overexpression and
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

20
TP53INP1 down-regulation (18), yet to date, the role of TP53INP1 in HCC metastasis or the
molecular mechanism by which TP53INP1 regulates migration and invasion in HCC has not been
explored. Prior to findings presented in this study, only three reports have linked TP53INP1 to
metastasis where they found TP53INP1 to reduce pancreatic cancer cell migration by regulating
SPARC expression (16); that TP53INP1 is down-regulated in distant lung metastasis of brain
cancer (15); and TP53INP1 3’UTR to function as a competitive endogenous RNA (ceRNA) in
repressing the metastasis of glioma cells by regulating miRNA activity (46). Specifically, using a
mouse model of skin wound healing in TP53INP1 wild-type and deficient mice, our collaborators
elegantly showed TP53INP1 to suppress cell migration in vivo. Similar observations were also
noted in vitro in TP53INP1 wild-type and deficient mouse embryonic fibroblasts (MEFs). Above
studies collectively support a role of TP53INP1 in regulating metastasis.
As mentioned above, TP53INP1 encodes two protein isoforms (α and β) (5). This current study
did not examine these two isoforms separately, but just looked at the role of both isoforms
collectively in HCC. The antibody used for Western Blot analysis binds to the N-terminus of
TP53INP1, which detects both protein isoforms. However, it should be noted that a predominant
36kDa band was observed in the Western Blot, which accordingly to our previous experience and
studies would represent the α isoform. We did observe a much weaker band at 55 kDa band,
which in our experience would correspond to the β isoform of TP53INP1. However, this band was
only detected upon extensive exposure. RT-PCR analysis on HCC cell lines, clinical samples and sh-
TP53INP1 HCC cells using primers specific to just α isoform, β isoform or both α and β isoforms,
revealed that expression levels were similarly expressed or unexpressed (data not shown).
Whether the two isoforms are differentially expressed at the mRNA and protein level or would
exert different functional roles in HCC would need to be further studied.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

21
There is ample evidence to show that TP53INP1 can alter the transactivation capacity of a number
of genes through both p53 and p73-dependent manners (17). P53 is mutated in ~30% of all liver
cancers (47). But unlike p53, mutation of p73 is not a common event in HCC nor other human
tumors. P73 was also not found to be differentially expressed in non-tumor vs. HCC (GSE25097)
not metastatic-free HCC vs. HCC with extrahepatic metastasis (GSE40367) (data not shown). Here,
we have uncovered a novel mechanism by which TP53INP1 down-regulation contributes to HCC
metastasis, through a p73-dependent DUSP10/ERK signaling pathway. The immortalized normal
liver and HCC cell lines that were used in this current study, namely MIHA and MHCC97L,
respectively, were either p53 deleted or mutated, but were both p73 intact. Whether down-
regulation of TP53INP1 promotes HCC metastasis through a similar DUSP10/ERK mechanism in a
p53-dependent manner needs to be further studied using appropriate cell lines that harbors wild-
type p53. It is interesting to note that we were also able to predict five p53 putative binding sites
on the DUSP10 promoter with a relative score higher than 0.75 (which is the same setting used for
prediction of p73 binding sites on DUSP10), suggesting that TP53INP1 may also indeed control
DUSP10/ERK pathway via a p53-dependent manner. If experimentally proven, TP53INP1 down-
regulation would be able to regulate DUSP10/ERK via both p53 and p73 means, uncovering a new
mechanism for all p53 wild-type, mutated/deleted HCC tumors.
p63 also exhibits significant structural homology to p53 and p73, has been reported to bind to the
same responsive element as p73 and plays a role in cancer metastasis (48). It would also be
intriguing to study the possible involvement of p63 and TP53INP1-mediated suppression of
metastasis. Towards this end, we went back to examine the p63 status in HCC tissues and found
that p63 expression is largely absent in HCC (49-50). With this, we cannot conclude that p63 has
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

22
no role in TP53INP1-mediated DUSP10/ERK signaling, but at least in the context of HCC, where
p63 expression is absent, chances are low.
Our study benefitted from the fast growing publicly available transcriptome datasets deposited in
NCBI Gene Expression Omnibus. The two datasets used, namely GSE40367 (20) and GSE25097
(19), were chosen in particular as the clinical samples profiled are all representative of Asian
ethnicity and are thus more relevant to the disease in our locality. In particular, the GSE40367
dataset was sampled from laser capture microdissected tissue of pure tumor cells of HCCs with
extrahepatic metastases and metastasis-free HCCs. Samples of these are rare and of particular
importance to studies like this that focuses specifically on HCC metastasis.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

23
Acknowledgements
We thank the Faculty Core Facility at the LKS Faculty of Medicine, The University of Hong Kong for
providing and maintaining the equipment needed for animal imaging.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

24
References
1. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011;331:1559-64.
2. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific
colonization. Nat Rev Cancer 2009;9:274-84.
3. Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumor progression and invasion.
Cancer Metastasis Rev 2003;22:395-403.
4. Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome
of MAP kinase signalling. FEBS J 2013;280:489-504.
5. Tomasini R, Samir AA, Vaccaro MI, Pebusque MJ, Dagorn JC, Iovanna JL, et al. Molecular and
functional characterization of the stress-induced protein (SIP) gene and its two transcripts
generated by alternative splicing. SIP induced by stress and promotes cell death. J Biol Chem
2001;276:44185-92.
6. Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, et al. p53DINP1, a p53-
inducible gene, regulates p53-dependent apoptosis. Mol Cell 2001;8:85-94.
7. Jiang PH, Motoo Y, Garcia S, Iovanna JL, Pebusque MJ, Sawabu N. Down-expression of tumor
protein 53-induced nuclear protein 1 in human gastric cancer. World J Gastroenterol
2006;12:691-6.
8. Ito Y, Motoo Y, Yoshida H, Iovanna JL, Takamura Y, Miya A, et al. Decreased expression of
tumor protein p53-induced nuclear protein 1 (TP53INP1) in breast carcinoma. Anticancer
Res 2006;26:4391-5.
9. Gironella M, Seux M, Xie MJ, Cano C, Tomasini R, Gommeaux J, et al. Tumor protein 53-induced
nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic
tumor development. Proc Natl Acad Sci USA 2007;104:16170-5.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

25
10. Weng W, Yang Q, Huang M, Qiao Y, Xie Y, Yu Y, et al. c-Myc inhibits TP53INP1 expression via
promoter methylation in esophageal carcinoma. Biochem Biophys Res Commun
2011;405:278-84.
11. Li Q, Han Y, Wang C, Shan S, Wang Y, Zhang J, et al. MicroRNA-125b promotes tumor
metastasis through targeting tumor protein 53-induced nuclear protein 1 in patients with
non-small-cell lung cancer. Cancer Cell Int 2015;15:84.
12. Bonazzi VF, Irwin D, Hayward NK. Identification of candidate tumor suppressor genes
inactivated by promoter methylation in melanoma. Genes Chromosomes Cancer 2009;48:10-
21.
13. Shibuya H, Iinuma H, Shimada R, Horiuchi A, Watanabe T. Clinicopathological and prognostic
value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology 2010;79:313-20.
14. Yeung ML, Yasunaga J, Bennasser Y, Dusetti N, Harris D, Ahmad N, et al. Roles for microRNAs,
miR-93 and miR-130b, and tumor protein 53-induced nuclear protein 1 tumor suppressor in
cell growth dysregulation by human T-cell lymphotrophic virus 1. Cancer Res 2008;68:8976-
85.
15. Zohrabian VM, Nandu H, Gulati N, Khitrov G, Zhao C, Mohan A, et al. Gene expression profiling
of metastatic brain cancer. Oncol Rep 2007;18:321-8.
16. Seux M, Peuget S, Montero MP, Siret C, Rigot V, Clerc P, et al. TP53INP1 decreases pancreatic
cancer cell migration by regulating SPARC expression. Oncogene 2011;30:3049-61.
17. Tomasini R, Seux M, Nowak J, Bontemps C, Carrier A, Dagorn JC, et al. TP53INP1 is a novel p73
target gene that induces cell cycle arrest and cell death by modulating p73 transcriptional
activity. Oncogene 2005;24:8093-104.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

26
18. Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, et al. miR-130b promotes CD133(+)
liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear
protein 1. Cell Stem Cell 2010;7:694-707.
19. Lamb JR, Zhang C, Xie T, Wang K, Zhang B, Hao K, et al. Predictive genes in adjacent normal
tissue are preferentially altered by sCNV during tumorigenesis in liver cancer and may rate
limiting. PLoS One 2011;6:e20090.
20. Ye QH, Zhu WW, Zhang JB, Qin Y, Lu M, Lin GL, et al. GOLM1 modulates EGFR/RTK cell-surface
recycling to drive hepatocellular carcinoma metastasis. Cancer Cell 2016;30:444-58.
21. Brown JJ, Parashar B, Moshage H, Tanaka KE, Engelhardt D, Rabbani E, et al. A long-term
hepatitis B viremia model generated by transplanting nontumorigenic immortalized human
hepatocytes in Rag-2-deficient mice. Hepatology 2000;31:173-81.
22. Li Y, Tang ZY, Ye SL, Liu Y, Chen J, Xue Q, et al. Establishment of cell clones with different
metastatic potential from the metastatic hepatocellular carcinoma cell line MHCC97. World J
Gastroenterol 2001;6:630-6.
23. Kidger AM, Keyse SM. The regulation of oncogenic Ras/ERK signalling by dual-specificity
mitogen activated protein kinase phosphatases (MKPs). Semin Cell Dev Biol 2016;50:125-32.
24. Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis
Rev 2008;27:253-61.
25. Nomura M, Shiba K, Katagiri C, Kasugai I, Masuda K, Sato I, et al. Novel function of MKP-
5/DUSP10, a phosphatase of stress-activated kinases, on ERK-dependent gene expression,
and upregulation of its gene expression in colon carcinomas. Oncol Rep 2012;28:931-6.
26. Tomasini R, Samir AA, Pebusque MJ, Calvo EJ, Totaro S, Dagorn JC, et al. P53-dependent
expression of the stress-induced protein (SIP). Eur J Cell Biol 2002;81:294-301.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

27
27. Tomasini R, Samir AA, Carrier A, Isnardon D, Cecchinelli B, Soddu S, et al. TP53INP1s and
homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity. J
Biol Chem 2003;278:37722-9.
28. Seillier M, Pouyet L, N’Guessan P, Nollet M, Capo F, Guillaumond F, et al. Defects in mitophagy
promotes redox-driven metabolic syndrome in the absence of TP53INP1. EMBO Mol Med
2015;7:802-18.
29. Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, et al. Tumor protein 53-induced
nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res 2009;69:219-
26.
30. Seillier M, Peuget S, Gayet O, Gauthier C, N’Guessan P, Monte M, et al. TP53INP1, a tumor
suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region
(LIR) and promotes autophagy-dependent cell death. Cell Death Differ 2012;19:1525-35.
31. Saadi H, Seillier M, Carrier A. The stress protein TP53INP1 plays a tumor suppressive role by
regulating metabolic homeostasis. Biochimie 2015;118:44-50.
32. Gommeaux J, Cano C, Garcia S, Gironella M, Pietri S, Culcasi M, et al. Colitis and colitis-
associated cancer are exacerbated in mice deficient for tumor protein 53-induced nuclear
protein 1. Mol Cell Biol 2007;27:2215-28.
33. Ogawa K, Asamoto M, Suzuki S, Tsujimura K, Shirai T. Downregulation of apoptosis revealed
by laser microdissection and cDNA microarray analysis of related genes in rat liver
preneoplastic lesions. Med Mol Morphol 2005;38:23-9.
34. Giusiano S, Baylot V, Andrieu C, Fazli L, Gleave M, Iovanna JL, et al. TP53INP1 as new
therapeutic target in castration-resistant prostate cancer. Prostate 2012;72:1286-94.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

28
35. Giusiano S, Garcia S, Andrieu C, Dusetti NJ, Bastide C, Gleave M, et al. TP53INP1
overexpression in prostate cancer correlates with poor prognostic factors and is predictive of
biological cancer relapse. Prostate 2012;72:117-28.
36. Shahbazi J, Scarlett CJ, Norris MD, Liu B, Haber M, Tee AE, et al. Histone deacetylase 2 and N-
Myc reduce p53 protein phosphorylation at serine 46 by repressing gene transcription of
tumor protein 53-induced nuclear protein 1. Oncotarget 2014;5:4257-68.
37. Chaluvally-Raghavan P, Zhang F, Pradeep S, Hamilton MP, Zhao X, Rupaimoole R, et al. Copy
number gain of hsa-miR-569 at 3q26.2 leads to loss of TP53INP1 and aggressiveness of
epithelial cells. Cancer Cell 2014;26:863-79.
38. Liu F, Kong X, Lv L, Gao J. TGF-β1 acts through miR-155 to down-regulate TP53INP1 in
promoting epithelial-mesenchymal transition and cancer stem cell phenotypes. Cancer Lett
2015;359:288-98
39. Zhang J, Cheng C, Yuan X, He JT, Pan QH, Sun FY. microRNA-155 acts as an oncogene by
targeting the tumor protein 53-induced nuclear protein 1 in esophageal squamous cell
carcinoma. Int J Clin Exp Pathol 2014;7:602-10.
40. Zhang CM, Zhao J, Deng HY. MiR-155 promotes proliferation of human breast cancer MCF-7
cells through targeting tumor protein 53-induced nuclear protein 1. J Biomed Sci 2013;20:79.
41. Qin J, Luo M, Qian H, Chen W. Upregulated miR-182 increases drug resistance in cisplatin-
treated HCC cell by regulating TP53INP1. Gene 2014;538:342-7.
42. Xu CG, Yang MF, Fan JX, Wang W. MiR-30a and miR-205 are downregulated in hypoxia and
modulate radiosensitivity of prostate cancer cells by inhibiting autophagy via TP53INP1. Eur
Rev Med Pharmacol Sci 2016;20:1501-8.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

29
43. Nie G, Duan H, Li X, Yu Z, Luo L, Lu R, et al. MicroRNA-205 promotes the tumorigenesis of
nasopharyngeal carcinoma through targeting tumor protein p53-inducible nuclear protein 1.
Mol Med Rep 2015;12:5715-22.
44. Nakaya N, Ishigaki Y, Nakajima H, Murakami M, Shimasaki T, Takata T, et al. Meaning of tumor
protein 53-induced nuclear protein 1 in the molecular mechanism of gemcitabine sensitivity.
Mol Clin Oncol 2013;1:100-104.
45. Al Saati T, Clerc P, Hanoun N, Peuget S, Lulka H, Gigous V, et al. Oxidative stress induced by
inactivation of TP53INP1 cooperates with KrasG12D to initiate and promote pancreatic
carcinogenesis in the murine pancreas. Am J Pathol 2013;182:1996-2004.
46. Wang Y, Lin G. TP53INP1 3’-UTR functions as a ceRNA in repressing the metastasis of glioma
cells by regulating miRNA activity. Biotechnol Lett 2016;38:1699-707.
47. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences,
and clinical use. Cold Spring Harb Perspect Biol 2010;2:a001008.
48. DeYoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene
2007;26:5169-83.
49. Ramalho FS, Ramalho LN, Della Porta L, Zucoloto S. Comparative immunohistochemical
expression of p63 in human cholangiocarcinoma and hepatocellular carcinoma. J
Gastroenterol Hepatol 2006;21:1276-80.
50. Tannapfel A, Wittekind C. Genes involved in hepatocellular carcinoma: deregulation in cell
cycling and apoptosis. Virchows Arch 2002;440:345-52.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

30
Figure Legends
Figure 1. TP53INP1 is down-regulated in advanced stage and metastatic HCC tumors. (A) Gene
expression levels of human TP53INP1 mRNA (NM_033285) in HCC tumors categorized by both
AJCC (Stages I, II, IIIA, IIIB and IV) (n = 219) and TNM (Stages I, II, III, IVA and IVB) (n = 229)
staging systems (GSE25097). Open circles represent outliers. (B) Box and whisker plot analysis of
TP53INP1 mRNA levels in metastasis-free HCCs (n = 10) and HCCs with extrahepatic metastasis (n
= 20) (GSE40367). The horizontal lines indicate data within median ± 1.5 inter-quartile range
(IQR). Closed circles represent outliers. (C) TP53INP1 immunostaining of tissue microarray
comprising of 37 paired human primary and metastatic HCC tissue samples. Shown are
representative images of the immunostaining. Scale bar, 50µm. p = 0.0007. Graph indicates the
percentage of cases displaying low, medium and high staining intensity of TP53INP1.
Figure 2. TP53INP1 knockdown promotes HCC metastasis. (A) Western Blot analysis of
TP53INP1 expression in a panel of immortalized normal liver (MIHA and LO2), hepatoblastoma
(HepG2) and HCC (SK-Hep1, HLE, SNU182, PLC8024, MHCC97L, Hep3B, QSG-7701 and QGY-7703)
cell lines. (B) Validation of TP53INP1 knockdown in MIHA and MHCC97L cells by Western Blot.
NTC, non target control. sh-TP53INP1 clones 464 and 3834. Representative images and
quantification of number of cells that (C) migrated or (D) invaded in MIHA and MHCC97L cells
with or without TP53INP1 suppressed. Scale bar, 50µm. * p < 0.05, ** p < 0.01 and *** p < 0.001
compared to NTC control. (E) Bioluminescence imaging of 4 representative nude mice injected
intrahepatically with luciferase labeled MHCC97L cells with or without TP53INP1 suppressed. Ex
vivo imaging of the livers and lungs harvested from nude mice that received orthotopic liver
injections. n = 10 mice per group. Bar chart summary of number of metastatic foci observed in
lung. * p < 0.05 and *** p < 0.001 compared to NTC control. (F) Representative H&E and
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

31
immunohistochemistry staining of TP53INP1 images of liver and lung tissues harvested. Scale bar,
50µm. n = 10. NTC, non target control. sh-TP53INP1 clones 464 and 3834.
Figure 3. Phopsho-kinase array profiling analysis identifies activation of ERK to be involved in
TP53INP1-mediated HCC metastasis. (A) Western blot images of deregulated phospho-kinases
spotted on the Proteome Profiler Human Phospho-kinase Array, comparing MIHA cells transduced
with non target control (NTC) or sh-TP53INP1 clone 464. (B) Western Blot analysis for levels of
phosphorylated and total ERK1/2 in HCC cells expressing NTC or sh-TP53INP1 clones (464 and
3834). Representative images and quantification of number of cells that (C) migrated or (D)
invaded in MIHA and MHCC97L cells expressing NTC or sh-TP53INP1 clones (464 and 3834) that
are treated with DMSO vehicle control (V) or ERK inhibitor U0126 (10µm). Scale bar, 50µm. ** p <
0.01 and *** p < 0.001 compared to NTC/vehicle control. # p < 0.05, ## p < 0.01 and ### p < 0.001
compared to vehicle.
Figure 4. TP53IN1 inhibits HCC metastasis via modulation of ERK signaling. (A) Western Blot
analysis for levels of total ERK1/2 in MHCC97L cells co-expressing sh-TP53INP1 clones and NTC
or sh-ERK1/2. (B) Bioluminescence imaging of nude mice injected intrahepatically with luciferase
labeled MHCC97L cells co-expressing sh-TP53INP1 clones and NTC or sh-ERK1/2. Ex vivo imaging
of the livers and lungs harvested from nude mice that received orthotopic liver injections. (C) Bar
chart summary of number of metastatic foci observed in lung. * p < 0.05 compared to NTC control.
(D) Representative H&E and immunohistochemistry staining of p-ERK images of liver and lung
tissues harvested.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

32
Figure 5. TP53INP1 inhibits HCC metastasis through DUSP10-dependent modulation of ERK. (A,
left) Relative expression of selected DUSP/MKP family members and TP53INP1 in MIHA and
MHCC97L cells expressing NTC or sh-TP53INP1 clones (464 and 3834) by qRT-PCR. (A, right)
Validation of down-regulated DUSP10 expression following TP53INP1 knockdown in MIHA and
MHCC97L cells by qRT-PCR. (B) Western Blot analysis for levels of DUSP10 in HCC cells
expressing NTC or sh-TP53INP1 clones (464 and 3834). (C) Western Blot analysis for levels of
DUSP10, phosphorylated and total ERK1/2 in HCC cells co-expressing NTC or sh-TP53INP1 clones
and empty vector or DUSP10 overexpression. Representative images and quantification of
number of cells that (D) migrated or (E) invaded in MIHA and MHCC97L cells co-expressing NTC
or sh-TP53INP1 clones and empty vector or DUSP10 overexpression. Scale bar, 50µm. * p < 0.05,
** p < 0.01 and *** p < 0.001 compared to NTC/EV control. ## p < 0.01 and ### p < 0.001
compared to EV control. (F, left) Box and whisker plot analysis of DUSP10 mRNA levels in
metastasis-free HCCs (n = 10) and HCCs with extrahepatic metastasis (n = 20) (GSE40367). The
horizontal lines indicate data within median ± 1.5 inter-quartile range (IQR). (F, right) Pearson
correlation analysis of TP53INP1 and DUSP10 mRNA levels in human HCC samples (n = 30)
(GSE40367).
Figure 6. p73, which transcriptional activity is modulated by TP53INP1, binds and regulates
DUSP10 via promoter binding and cooperatively drive ERK activation in HCC. (A) Computational
prediction of p73 binding sites (S1 at -4400 to -2201bp and S2 at -2200 to 1bp) on DUSP10
promoter region by JASPAR matrix model. (B) Confirmation of p73 binding to candidate DUSP10
sites by ChIP-qPCR analysis in MHCC97L cells with or without TP53INP1 suppressed. Chromatins
were immunoprecipitated by anti-p73 antibody, and the enrichment of predicted p73 binding
sites on DUSP10 (sites A, B, C and D) relative to IgG control were confirmed by qPCR. (C)
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

33
Luciferase reporter assays in MHCC97L cells expressing NTC or sh-p73 clones (753 and 1643) to
validate the interaction between p73 and DUSP10 at both predicted regions. pRL-TK renilla
luciferase plasmid co-transfected for normalization. ** p < 0.01 and *** p < 0.001 compared to NTC
control. (D) Western Blot analysis for levels of p73, DUSP10, phosphorylated and total ERK1/2 in
HCC cells expressing NTC or sh-p73 clones (753 and 1643), with empty vector (EV) or DUSP10
overexpressed. (E) Luciferase reporter assays in MIHA and MHCC97L cells expressing NTC or sh-
TP53INP1 clones (464 and 3834) to validate the interaction between p73 and DUSP10 at both
predicted regions. pRL-TK renilla luciferase plasmid co-transfected for normalization. ** p < 0.01
and *** p < 0.001 compared to NTC control. (F) Western Blot analysis for levels of p73, MDM2 and
BAX in HCC cells expressing NTC or sh-TP53INP1 clones (464 and 3834). (G) Proposed model
illustrates how TP53INP1 down-regulation promotes HCC metastasis through a p73-dependent
DUSP10/ERK signaling pathway. Dotted box with question mark indicates how TP53INP1
interacts with p73 is still unknown.
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456

Published OnlineFirst July 3, 2017.Cancer Res Kai-Yu Ng, Lok-Hei Chan, Stella Chai, et al. hepatocellular carcinomaDUSP10/ERK signaling pathway to promote metastasis of TP53INP1 down-regulation activates a p73-dependent
Updated version
10.1158/0008-5472.CAN-16-3456doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2017/07/01/0008-5472.CAN-16-3456.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/early/2017/07/01/0008-5472.CAN-16-3456To request permission to re-use all or part of this article, use this link
Research. on January 2, 2020. © 2017 American Association for Cancercancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 3, 2017; DOI: 10.1158/0008-5472.CAN-16-3456





























