Embed Size (px)
Citation preview

Topics in Catalysis Vol. 19, No. 1, March 2002 125
Towards heterogeneous and green versions of Os dihydroxylationcatalysis
An Severeyns, Dirk E. De Vos and Pierre A. Jacobs ∗
Centre for Surface Chemistry and Catalysis, K.U. Leuven, Kasteelpark Arenberg 23, 3001 Leuven, BelgiumE-mail: [email protected]
Os-catalyzed dihydroxylation will find more widespread application if truly heterogeneous catalysts are available which use O2 orH2O2 as terminal oxidants. This paper discusses and critically evaluates the concepts by which Os can be anchored on solid supports. Newmethods are also available for reoxidation of hexavalent to octavalent Os.
KEY WORDS: osmium catalyst; dihydroxylation; catalysis
1. Introduction
Osmium tetroxide is an excellent catalyst for the cis-dihydroxylation (DH) of olefins [1]. This dihydroxylationefficiently produces cis-1,2-diols from olefins in only onestep. Obviously, this constitutes an advantage in compar-ison with the current industrial process, which comprisesthe epoxidation of an olefin followed by the hydrolysis ofthe formed epoxide. The DH can be catalyzed by osmium,ruthenium or manganese oxo species, osmium tetroxide be-ing by far the most versatile and selective catalyst for thisprocess. Thus, Mn catalysts also produce epoxide togetherwith the cis-diols [2]; with Ru and periodate, overoxidationis a major problem, unless the biphasic flash dihydroxyla-tion protocol is applied [3]. The wide range of applicationsof 1,2-diols and the unique properties of the osmium cata-lyst have stimulated a lot of research on the dihydroxylation,which has become even more useful with the discovery ofthe asymmetric variant by Sharpless et al. [4].
The Os-catalyzed dihydroxylation is a complex reactionsequence, consisting of (i) addition of OsO4 to the dou-ble bond (“osmylation”), with reduction of the Os, and(ii) reoxidation of OsVI to OsVIII and hydrolytic release ofthe diol. Several oxidants are used to effect the reoxida-tion. Nowadays, most synthetic organic chemists use N-methylmorpholine-N-oxide (NMO) or K3[Fe(CN)6] as ox-idant, which both allow the reaction to be performed withvery high chemoselectivity [5–7]. Although these optimizedmethods give excellent results on lab scale, industrial appli-cation remains impossible. Firstly, larger scale application isobstructed by the high toxicity and volatility of OsO4. Sec-ondly, the oxidants used to reoxidize OsVI are expensive anda large, stoichiometric amount of waste is produced. Thirdly,products may be contaminated with small amounts of non-hydrolyzed Os-diolate complexes.
Recently many efforts have been devoted to improvingthe applicability of the OsO4 catalyzed dihydroxylation, by
∗ To whom correspondence should be addressed.
dealing with aforementioned problems. Through heteroge-nization of the osmium catalyst, a non-volatile form of thecatalyst can be produced, which can be handled a lot moresafely and which allows an easy recycling. A few heterog-enization attempts have been reported and will be criticallydiscussed in this paper. By developing procedures whichuse “green” oxidants such as H2O2 and O2, the problemof the waste production can be solved and the process be-comes ecologically interesting. The examples demonstratethat all major improvements in the Os-catalyzed dihydrox-ylation critically rely on controlling the relative rates of thedifferent steps in the catalytic cycle, viz. osmylation, Os re-oxidation and hydrolytic diol release.
2. Heterogeneous Os catalysts
2.1. Amine-functionalized polymers as supports
Cainelli et al. and Herrmann et al. use polymers intheir attempts to immobilize an Os dihydroxylation cat-alyst [8–10]. Herrmann immobilizes OsO4 on commer-cial polyvinylpyridine (PVP), while Cainelli uses besidesPVP several amine-type polymers, prepared starting fromchloromethylated polystyrene:
The precise preparation procedure seems to influencethe state of the Os on the polymer before reaction. Thus,Cainelli’s material with immobilized diazabicyclooctane(DABCO) contains mostly OsVIII; in contrast, XPS andvibrational spectroscopy experiments demonstrate that af-ter exposure of the polyvinylpyridine to OsO4 in tetrahy-
1022-5528/02/0300-0125/0 2002 Plenum Publishing Corporation

126 A. Severeyns et al. / Heterogeneous Os dihydroxylation catalysis
Scheme 1.
drofuran, Os is predominantly available in the hexavalentstate [10]. XPS also evidences partial degradation of thePVP structure after exposure to the OsO4 solution in THF.However, it is uncertain whether the preparation mode influ-ences to any serious extent the eventual retention of Os onthese polymers in catalytic conditions.
The resulting polymers have been tested with different Osreoxidizing agents. Cainelli et al. obtained better diol yieldsfrom most olefins with (CH3)3NO than with peroxides, e.g.,from ethyl oleate [8]:
The same material with Os immobilized on DABCOgroups has been used in combination with NaIO4. The pe-riodate reoxidizes the Os, but also cleaves the diols to twoaldehyde products [9]. This procedure with supported Osand NaIO4 is an alternative to ozonation. In contrast, H2O2seems the preferred oxidant for Herrmann’s material. Whileoveroxidation to the ketol is often a problem with Os andH2O2, the catalyst prepared by OsO4 adsorption on PVP inTHF leads to excellent diol yields, e.g., 95% from ethyl cro-tonate [10]. A disadvantage is that up to 40-fold excesses ofH2O2 are required, implying that the unproductive peroxidedecomposition is the major process.
In general, these approaches rely on the coordination ofnitrogen bases of the polymer to the Lewis acidic OsVI orOsVIII center. Such a coordination is at the basis of theconcept of “ligand accelerated catalysis”, which means thatat least in one step of the catalytic cycle, the ligand-boundmetal reacts much faster than the free metal complex [11].In the case of dihydroxylation, the addition of the cis-dioxoOs group to the double bond is accelerated by nitrogen basecoordination. For quinuclidine-coordinated OsO4, the os-mylation step is about 100 times faster than for free OsO4, ir-respective of the olefin substitution pattern [12]. Even largeracceleration effects are obtained with a chiral ligand such ascinchonidine. However, acceleration of one step does notimply that the overall reaction becomes faster. The over-all reaction rate is a complex result of several parameterssuch as steric hindrance and basicity of the nitrogen com-
pound, olefin substitution pattern, Os reoxidizing agent, andpresence of co-catalysts, particularly for accelerating the hy-drolysis of the osmate ester. Generally, hydrolysis of anosmate ester is much retarded by coordination of a nitro-gen base [13]. As a result, such base-Os-diolate adducts arestable and can easily be isolated. A base such as quinucli-dine binds strongly to the Os-diolate complex, and conse-quently, the overall reaction is strongly inhibited by quinu-clidine, even if the osmylation in se is accelerated [14,15](see scheme 1).
In contrast, in the case of cinchonidine, binding of thebase to the Os diolate complex is relatively weak; hence alarger fraction of the osmate ester is available in the freeform and is easily hydrolyzed.
In view of these considerations, catalytic turnover withthe Cainelli and Hermann catalysts is in our opinion onlypossible when the intermediate Os diolate complex is de-tached from the polymeric Lewis base, before the hydroly-sis. This implies an inherent liability to Os leaching. A clearmerit of the approach with polymeric amines is that the Os isintroduced into the reaction mixture as a non-volatile, harm-less compound. However, in no case has it been demon-strated that the filtrates of the reactions are free of catalyticactivity. Leaching tests in our laboratory with an OsO4/PVPmaterial have indicated that during catalytic turnover con-ditions, the filtrate contains active Os. If filtrate and solidare separated, the reaction progresses at least as fast in thefiltrate as in the presence of the polymeric support. Sim-ilar problems are encountered with heterogeneous versionsof the asymmetric dihydroxylation. Attention has focusedalmost exclusively on immobilizing the cinchona alkaloids,with various attachment points on the alkaloid structure,with varying spacer lengths between support and alkaloid,with inorganic or organic supports, with soluble or insol-uble polymers, or with different heteroaromatic groups atthe crucial C9-O position of the alkaloid [16–19]. Evenunusual supports such as monolayer-covered gold colloidshave been proposed [20]. Two examples are shown below,of a cinchona monomer and a silica-supported alkaloid, re-spectively [21,22] (see scheme 2).
In the best cases, the ee’s match those of the optimizedhomogeneous processes, and the ligand can effectively berecycled. However, even if the cost of the Os is comparableto that of, e.g., the phthalazine-substituted cinchona ligand,no attention is paid to recycling of the Os, and several groupsmention that Os leaching necessitates Os supplementation in

A. Severeyns et al. / Heterogeneous Os dihydroxylation catalysis 127
Scheme 2.
subsequent runs [18]. Again, Os-diolate hydrolysis seems torequire detachment from the polymeric ligand, and hencecauses leaching.
Similar problems may be encountered when it is at-tempted to immobilize other catalysts that are subject toligand-accelerated catalysis. A typical example is the epox-idation catalyst CH3ReO3, which becomes more active andstable by addition of nitrogen bases such as pyridine, pyra-zole or 3-cyanopyridine. Moreover, such bases prevent hy-drolysis of the epoxide by buffering of the Lewis acidity [23–25]. Recently, CH3ReO3 was supported on pyridine groups,covalently anchored to a siliceous matrix prepared via thesol–gel route [26]. With a ratio of anchored pyridine overRe of only 1, it was observed that the catalyst activity wasstable over several runs, even if the reactions needed a tem-perature of 80 ◦C and only proceeded easily with the veryreactive cyclooctene.
2.2. Microencapsulated osmium tetroxide
In another attempt to heterogenize the Os catalyst, OsO4
was entrapped in polystyrene microspheres. These micro-spheres have previously been applied for immobilizationof water-tolerant Lewis acids such as Sc triflate [27].The microspheres are prepared starting from a solution ofpolystyrene in cyclohexane, by addition of OsO4, coolingand addition of methanol to harden the capsule walls [28].In this method the OsO4 is distributed over the inner coreand the outer shell of the polystyrene particles; it remainsimmobilized in the polystyrene by physical envelopmentand by electronic interactions between the π electrons ofthe aromatic rings of the polymer and the Lewis acidicosmium center. The successful re-use of the polymer hasbeen demonstrated. A variant of this catalyst with anacrylonitrile–butadiene–styrene polymer as support can beused for asymmetric dihydroxylation, when chiral alkaloidligands are added [29]. With this simple and straightfor-ward approach, future successes in heterogeneous oxidationcatalysis can be expected.
2.3. Os deposition on inorganic oxides and on carbon
The series of patents by Michaelson and co-workers atExxon deserves attention for several reasons [30–32]. First,this team has done a serious attempt to use Os in large scaledihydroxylations, even in continuous reactor setups or withsmall volatile olefins such as ethylene. Secondly, they haveinvestigated in depth the conditions of facile Os dihydroxy-lations, often with O2 as the terminal oxidant. For reactionswith organic hydroperoxides or H2O2, several salts havebeen proposed as promotors, e.g., sodium acetate or NaI.These increase the olefin conversion or the diol selectivity.
In their approach to heterogeneously catalyzed oxida-tions, this group employs Os reduction catalysts, which areoften prepared by adsorption of Os3(CO)12 or OsCl3 on sup-ports such as MgO, Al2O3 or other oxides [30]. Moreover,co-catalysts such as NaI, NaOH were used in combinationwith t-BuOOH as the oxidant and t-BuOH as the solvent.In the presence of NaOH, Os is easily detected in the liquidphase of the reaction mixture. However, no Os is found byelemental analysis of the filtrate if NaI is added as a pro-moter. The examples give further experimental evidence forthe strong association of Os with the surface in the presenceof NaI: (i) an Os3(CO)12/Al2O3 catalyst can be reused upto six times without activity loss; (ii) the filtrate from anOs3(CO)12/Al2O3/NaI reaction does not produce additionaldiol when fresh oxidant and olefin are added. The latter ex-periment proves that in the presence of NaI, all the Os isassociated with the support at least at the end of the reaction.Most of the NaI, however, is simply dissolved:

128 A. Severeyns et al. / Heterogeneous Os dihydroxylation catalysis
Remark that even very small amounts of iodide are effec-tive. As the precise speciation of the Os in these conditionsis not known, it is unclear by which mechanism the Os iswithheld by the alumina support.
In a further development, the iodide promotor was incor-porated in the catalyst by use of an Amberlyst IRA-400 an-ion exchange polymer [30]. This resin was converted to theiodide form by reaction with HI and subsequently exposedto OsO4. The resulting resin can be reused at least 3 times inthe formation of isobutylene glycol, with unchanged activity.However, no specific leaching data are provided.
An Os on carbon catalyst has been proposed for an un-usual reaction in the domain of alkane hydroxylation [33].Os is indeed capable of catalytically consuming organic hy-droperoxides, even in the absence of olefins. Exemplaryperoxides comprise secondary and tertiary hydroperoxides,such as ethylbenzene hydroperoxide, cumyl or t-butyl hy-droperoxide and cyclohexyl hydroperoxide. When alkanesare present during this decomposition, they are oxidized toan alcohol/ketone mixture, presumably in a radical process.A typical reported reaction is as follows:
The high yield of C8-oxygenates, based on the peroxideconsumed, is remarkable. Moreover, the selectivity for thealcohol is very high (over 90%), particularly when t-BuOHis used as a co-solvent. A plausible explanation for this prod-uct distribution is that cumyloxy radicals react largely withthe alkane, producing a cyclooctyl radical which recombinesalmost completely with hydroxyl radicals to produce the al-cohol. The overall reaction differs from a classical perox-ide induced alkane autoxidation, partly because it is con-ducted under Ar. Trapping of cyclooctyl radicals by adven-titious O2, or by O2 formed in peroxide disproportionation,seems insignificant; such trapping would lead to much morecyclooctyl hydroperoxide or cyclooctanone. Moreover, β-scission of the cumyloxy radical to form acetophenone isalso less important. A similar high alcohol selectivity is ob-served in the decomposition of cyclohexyl hydroperoxide incyclohexane to cyclohexanol, which is formed in a 132%yield based on the consumed peroxide. In most of these re-actions, OsO4 is the catalyst, but in some examples, the useof a carbon-supported Os is proposed. However, no data areprovided on Os leaching or catalyst reuse.
2.4. A heterogeneous cis-dihydroxylation catalyst withstable osmium-diolate reaction centers
We recently reported a new heterogeneous cis-dihydro-xylation catalyst with a persistent bond between Os and aninorganic support [34]. The immobilization is based on twoparticular properties of the Os-diolate species involved in aclassical dihydroxylation cycle:
(1) First, if the hydrolytic conditions are not too drastic,cis-diols are not formed out of tetrasubstituted olefins.In fact, the osmylation step is taking place, formingthe osmateVI ester, but depending on the conditions inwhich the reaction is performed, the sterically hinderedglycolate esters are inert to the subsequent oxidativehydrolysis. Literature contains ample information onOs reactions of tetrasubstituted olefins. Sharpless etal. report that the Upjohn method, with NMO as oxi-dant in water/acetone/t-butanol, fails with 2,3-dimethyl-2-octene as a substrate [35]; later, the same group devel-oped a cis-dihydroxylation procedure using t-BuOOHunder alkaline conditions. Reactions were performedwith t-BuOOH in t-butanol in the presence of the or-ganic base tetraethylammonium hydroxide, or with t-BuOOH in acetone, in the presence of tetraethylam-monium acetate [35,36]. The alkaline conditions favorrapid hydrolytic removal of the diol from the coordi-nation sphere of the osmium center, in this way sup-pressing overoxidation. With the Et4NOAc/t-BuOOHmethod, no diol is formed at all out of 2,3-dimethyl-2-octene, while this olefin is readily converted by theEt4NOH/t-BuOOH method (69% diol yield). The moredrastic hydrolytic conditions explain the success of thesecond method for the cis-dihydroxylation of tetrasub-stituted olefins.
(2) A second important element for the development ofour catalyst is the possibility of reoxidizing an OsVI
monodiolate complex to cis-dioxo OsVIII, without re-lease of the diol. A second olefin can add to this OsVIII
complex, resulting in an OsVI bisdiolate complex. Inthis way, two catalytic cycles are operative for the dihy-droxylation reaction (figure 1) [4,37]. The key interme-diate is the OsVIII-trioxoglycolate complex 3, which hasa junction position between the two cycles. Evidencefor the existence of the second cycle has been found byreplicating the steps shown in figure 1 under stoichio-metric conditions [37]. Thus, complex 1 was allowedto react with an olefin to form the monoglycolate es-ter 2. Then a different olefin was added together withone equivalent of the oxidant, leading to the putativebisglycolate ester 4. Via a stoichiometric reductive hy-drolysis, one equivalent of each diol was released. Thisexperiment proves that both catalytic cycles form diolsfrom olefins. Complexes 1, 2 and 4 can be isolated andcharacterized.
The stability of the tetrasubstituted glycolate esters inspecific conditions and the possibility to perform a second
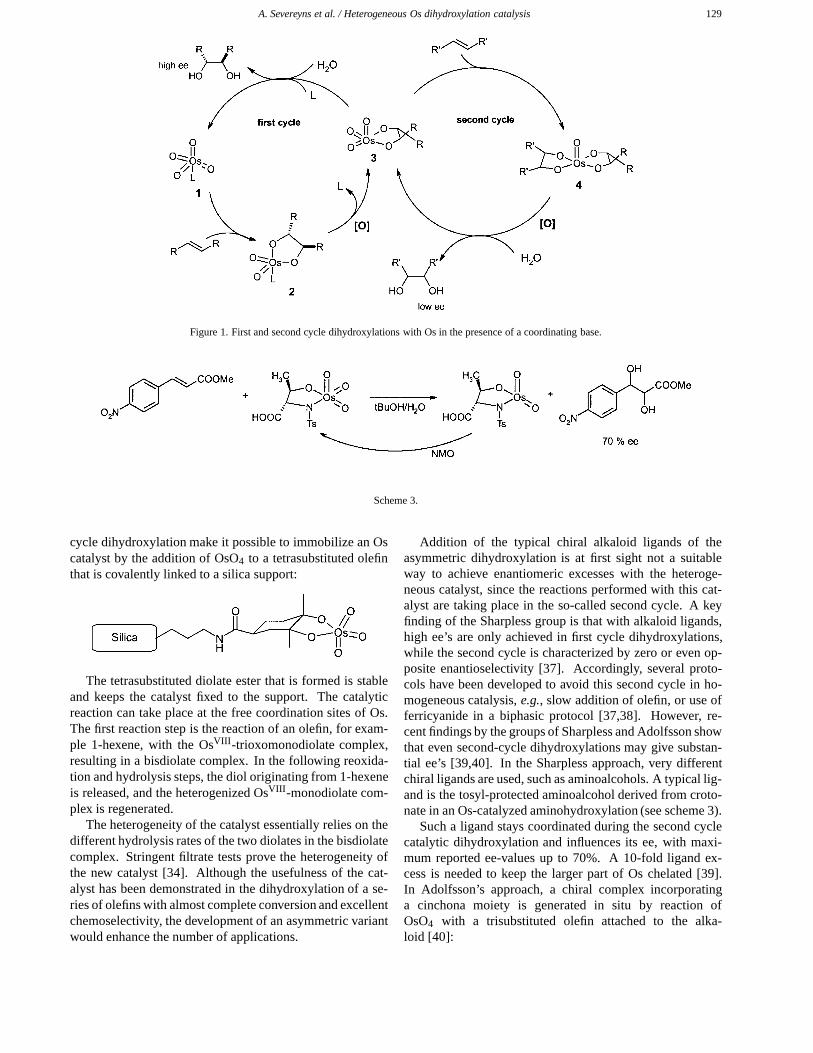
A. Severeyns et al. / Heterogeneous Os dihydroxylation catalysis 129
Figure 1. First and second cycle dihydroxylations with Os in the presence of a coordinating base.
Scheme 3.
cycle dihydroxylation make it possible to immobilize an Oscatalyst by the addition of OsO4 to a tetrasubstituted olefinthat is covalently linked to a silica support:
The tetrasubstituted diolate ester that is formed is stableand keeps the catalyst fixed to the support. The catalyticreaction can take place at the free coordination sites of Os.The first reaction step is the reaction of an olefin, for exam-ple 1-hexene, with the OsVIII-trioxomonodiolate complex,resulting in a bisdiolate complex. In the following reoxida-tion and hydrolysis steps, the diol originating from 1-hexeneis released, and the heterogenized OsVIII-monodiolate com-plex is regenerated.
The heterogeneity of the catalyst essentially relies on thedifferent hydrolysis rates of the two diolates in the bisdiolatecomplex. Stringent filtrate tests prove the heterogeneity ofthe new catalyst [34]. Although the usefulness of the cat-alyst has been demonstrated in the dihydroxylation of a se-ries of olefins with almost complete conversion and excellentchemoselectivity, the development of an asymmetric variantwould enhance the number of applications.
Addition of the typical chiral alkaloid ligands of theasymmetric dihydroxylation is at first sight not a suitableway to achieve enantiomeric excesses with the heteroge-neous catalyst, since the reactions performed with this cat-alyst are taking place in the so-called second cycle. A keyfinding of the Sharpless group is that with alkaloid ligands,high ee’s are only achieved in first cycle dihydroxylations,while the second cycle is characterized by zero or even op-posite enantioselectivity [37]. Accordingly, several proto-cols have been developed to avoid this second cycle in ho-mogeneous catalysis, e.g., slow addition of olefin, or use offerricyanide in a biphasic protocol [37,38]. However, re-cent findings by the groups of Sharpless and Adolfsson showthat even second-cycle dihydroxylations may give substan-tial ee’s [39,40]. In the Sharpless approach, very differentchiral ligands are used, such as aminoalcohols. A typical lig-and is the tosyl-protected aminoalcohol derived from croto-nate in an Os-catalyzed aminohydroxylation (see scheme 3).
Such a ligand stays coordinated during the second cyclecatalytic dihydroxylation and influences its ee, with maxi-mum reported ee-values up to 70%. A 10-fold ligand ex-cess is needed to keep the larger part of Os chelated [39].In Adolfsson’s approach, a chiral complex incorporatinga cinchona moiety is generated in situ by reaction ofOsO4 with a trisubstituted olefin attached to the alka-loid [40]:

130 A. Severeyns et al. / Heterogeneous Os dihydroxylation catalysis
Particularly when alternative Os sources such as imidocomplexes are used, higher ee’s are obtained than in thenormal first cycle asymmetric dihydroxylation; this suggeststhat second cycle dihydroxylation can indeed induce highee’s. Although this process must probably be optimized,further development of the concept of an enantioselectivesecond-cycle process offers a perspective for a future het-erogeneous asymmetric catalyst.
3. Osmium-catalyzed dihydroxylation withenvironmentally friendly oxidants
3.1. With hydrogen peroxide
It has been known for a long time that H2O2 can be usedfor the reoxidation of OsVI to OsVIII [41]. Unfortunately,this Milas chemistry results in nonselective reactions, due tooveroxidations. Bäckvall et al. designed a coupled catalyticsystem, using H2O2 as the terminal oxidant [42,43]. In thissystem OsVI is reoxidized to OsVIII by a coupled electron–transfer–mediator system based on NMO and a flavin. Thisbiomimetic catalytic system combines the chemoselectivityof NMO with the ecologically favorable properties of H2O2.Moreover, even asymmetric dihydroxylations can be per-formed with this coupled system. Because the flavin is ratherunstable, a more robust mediator was looked for, to partici-pate in the triple catalytic dihydroxylation system. Vanadylacetylacetonate is an appropriate peroxide activator [44]:
3.2. With oxygen
An even more obvious way to oxidize hexavalent os-mate to OsVIII is using O2 in aqueous alkaline conditions.However, this property has only recently become useful forpreparative dihydroxylations. Early attempts to use O2 re-sulted in very poor selectivity because reactions were per-formed in water with water-soluble reactants such as allyl
alcohol [45]. In alkaline water, the OsVI-glycolates are read-ily oxidized by O2, and the coordinated diols are further ox-idized, eventually to form oxalate. This deep oxidation canbe avoided in the biphasic conditions elaborated by Bellerand his group, particularly for dihydroxylation of aromaticolefins [46,47]. The proposed catalytic cycle is similar tothe one presented by Sharpless for the osmium-catalyzed di-hydroxylation with K3[Fe(CN)6]. In the biphasic system,the addition of the OsVIII species to an olefin such as α-methylstyrene proceeds in the organic layer. As long as theOsVI-glycolate stays within the organic t-BuOH layer, it isnot susceptible to reoxidation. The next step, hydrolysis ofthe Os-glycolate, is relatively fast at pH 10.4, but for ster-ically hindered olefins, a pH up to 12 may be required toachieve sufficient rates. After hydrolysis, the diol, e.g., 2-phenyl-1,2-propanediol stays in the organic layer, while os-mate returns to the aqueous phase and is reoxidized. Clearly,the two-phase operation has a highly beneficial effect on theselectivity; moreover, since there is almost no contributionof the second cycle, the asymmetric dihydroxylation in pres-ence of O2 and chiral alkaloids shows only marginally lowerenantioselectivities than with K3[Fe(CN)6]. It is unclearwhether the procedure works equally well for small, watersoluble olefins such as allylic alcohols. The Os adducts ofthese molecules will likely reside in the aqueous layer whereundesired overoxidation may take place. Rather high tem-peratures (50 ◦C) and pressures (between 3 and 10 bar O2)are useful to increase the rate of Os oxidation and thus thewhole process. Even if air can be used instead of O2, work-ing with free OsO4 at high temperature and pressure remainstroublesome.
Two alternative systems have been proposed in which O2
is the eventual oxidant. First Michaelson et al. used copperbromide as a co-catalyst, possibly in combination with pyri-dine [48]. Instead of working at alkaline pH, reactions wereconducted at pH 4.5 or even in more strongly acid media, ina sulfolane/water mixture. In these conditions, OsVI is notspontaneously oxidized by air, and acceptable diol selectiv-ities are obtained. Thus propanediol is formed with 85%selectivity, using 1 mol% of Os. Obviously, the CuI/CuII
couple mediates the electron transfer from OsVI in a Wackertype transfer chain; CuI is itself easily reoxidized by O2.
Secondly, a multicatalytic system was proposed for Osreoxidation, containing a sensitizer and selenides in a bipha-sic t-BuOH/water system buffered by K2CO3 [49,50]. First,singlet dioxygen is produced by the action of light on thewater-soluble dye Rose Bengal. Next, 1O2 oxidizes e.g.,benzylphenylselenide to the corresponding oxide, which re-oxidizes osmate even at 12 ◦C. Note that in aqueous con-ditions, the lifetime of 1O2 is small; excessive irradiationmay damage the sensitizer or may cause decomposition ofthe selenide to elemental Se. It should be remarked that ex-cept for the rather low reaction temperature, all other con-ditions are optimal to allow air reoxidation of Os; hence abackground contribution of reoxidation with O2 cannot beexcluded.

A. Severeyns et al. / Heterogeneous Os dihydroxylation catalysis 131
4. Summary and outlook
Even if the reaction of OsO4 with unsaturated substrateshas been known for almost a century [1], a lot of recentprogress improves the chances for large scale applicationof Os-catalyzed dihydroxylations. A particularly impor-tant evolution is the use of O2 or H2O2 as an Os reoxi-dizing agent [44,46]. The most crucial hurdle in Os catal-ysis remains a reliable heterogenization, preferably of anenantioselective catalyst. Several new heterogenization ap-proaches have been brought forward recently, and time willtell which concepts really do work well [10,28,34]. A partic-ularly pleasing point is that important new insights are beinggained in the mechanisms of Os dihydroxylations. As an ex-ample, the concept of an asymmetric second-cycle dihydrox-ylation may eventually allow to design solid asymmetric Oscatalysts [39,40]. Finally, extension of Os catalysis to otherreaction types, such as aminohydroxylations, deserves con-tinuous attention, in view of their relevance for biologicallyimportant molecules.
Acknowledgement
We are grateful to F.W.O. (Flanders, Belgium) for a re-search grant (PAJ, DDV) and for a fellowship (AS). Ourwork in catalyst heterogenization is sponsored by the Bel-gian Federal Government in the frame of an IAP program onSupramolecular Chemistry and Catalysis.
References
[1] M. Schröder, Chem. Rev. 80 (1980) 187.[2] D.E. De Vos, S. De Wildeman, B. Sels, P.J. Grobet and P.A. Jacobs,
Angew. Chem. 111 (1999) 1033.[3] T.K.M. Shing, V.W.F. Tai and E.K.W. Tam, Angew. Chem. 106 (1994)
2408.[4] H.C. Kolb, M.S. VanNieuwenhze and K.B. Sharpless, Chem. Rev. 94
(1994) 2483.[5] V. VanRheenen, R.C. Kelly and D.Y. Cha, Tetrahedron Lett. 23 (1976)
1973.[6] J.S. Mayell, Ind. Eng. Chem. Prod. Res. Dev. 7 (1968) 130.[7] M. Minamoto, K. Yamamoto and J. Tsuji, J. Org. Chem. 55 (1990)
766.[8] G. Cainelli, M. Contento, F. Manescalchi and L. Plessi, Synthesis
(1989) 45.[9] G. Cainelli, M. Contento, F. Manescalchi and L. Plessi, Synthesis
(1989) 47.[10] W.A. Herrmann, R.M. Kratzer, J. Blümel, H.B. Friedrich, R.W.
Fisher, D.C. Apperley, J. Mink and O. Berkesi, J. Mol. Catal. A 120(1997) 197.
[11] D.J. Berrisford, C. Bolm and K.B. Sharpless, Angew. Chem. 107(1995) 1159.
[12] P.G. Andersson and K.B. Sharpless, J. Am. Chem. Soc. 115 (1993)7047.
[13] E. Erdik and D. Kahya, Int. J. Chem. Kinet. 29 (1997) 359.[14] E.N. Jacobsen, I. Markó, W.S. Mungall, G. Schröder and K.B.
Sharpless, J. Am. Chem. Soc. 110 (1988) 1968.[15] E.N. Jacobsen, I. Markó, M.B. France, J.S. Svendsen and K.B.
Sharpless, J. Am. Chem. Soc. 111 (1989) 737.[16] D. Pini, A. Petri, A. Nardi, C. Rosini and P. Salvadori, Tetrahedron
Lett. 32 (1991) 5175.[17] C. Bolm and A. Gerlach, Angew. Chem. Int. Ed. Engl. 36 (1997) 741.[18] C. Bolm, A. Maischak and A. Gerlach, Chem. Commun. (1997) 2353.[19] P. Salvadori, D. Pini, A. Petri and A. Mandoli, in: Chiral Catalyst
Immobilization and Recycling, eds. D.E. De Vos, I.F.J. Vankelecomand P.A. Jacobs (Wiley–VCH, 2000) p. 235.
[20] H. Li, Y.-Y. Luk and M. Mrksich, Langmuir 15 (1999) 4957.[21] C. Bolm and A. Maischak, Synlett (2001) 93.[22] P. Salvadori, D. Pini and A. Petri, J. Am. Chem. Soc. 119 (1997) 6929.[23] W.A. Herrmann, R.M. Kratzer, H. Ding, W.R. Thiel and H. Glas,
J. Organometal. Chem. 555 (1998) 293.[24] H. Adolfsson, A. Converso and K.B. Sharpless, Tetrahedron Lett. 40
(1999) 3991.[25] H. Adolfsson, C. Copéret, J.P. Chiang and A.K. Yudin, J. Org. Chem.
65 (2000) 8651.[26] K. Dallmann and R. Buffon, Catal. Commun. 1 (2000) 9.[27] S. Kobayashi and S. Nagayama, J. Am. Chem. Soc. 120 (1998) 2985.[28] S. Nagayama, M. Endo and S. Kobayashi, J. Org. Chem. 63 (1998)
6094.[29] S. Kobayashi, M. Endo and S. Nagayama, J. Am. Chem. Soc. 121
(1999) 11229.[30] R.C. Michaelson, R.G. Austin and D.A. White, US Patent 4413151
(1983).[31] R.C. Michaelson, R.G. Austin and D.A. White, US Patent 4486613
(1984).[32] R.C. Michaelson and R.G. Austin, US Patent 4533773 (1985).[33] M. Costantini and J.-P. Lecomte, US Patent 4918238 (1990).[34] A. Severeyns, D.E. De Vos, L. Fiermans, F. Verpoort, P.J. Grobet and
P.A. Jacobs, Angew. Chem. 113 (2001) 606.[35] K. Akashi, R.E. Palermo and K.B. Sharpless, J. Org. Chem. 43 (1978)
2063.[36] K.B. Sharpless and K. Akashi, J. Am. Chem. Soc. 98 (1976) 1986.[37] J.S.M. Wai, I. Markó, J.S. Svendsen, M.G. Finn, E.N. Jacobsen and
K.B. Sharpless, J. Am. Chem. Soc. 111 (1989) 1123.[38] H.L. Kwong, C. Sorato, Y. Ogino, H. Chen and K.B. Sharpless, Tetra-
hedron Lett. 31 (1990) 2999.[39] M.A. Andersson, V.V. Fokin and K.B. Sharpless, presented at the
221st National Am. Chem. Soc. Meeting (2001).[40] H. Adolfsson and F. Stålfors, presented at the 221st National Am.
Chem. Soc. Meeting (2001).[41] N.A. Milas and S. Sussman, J. Am. Chem. Soc. 58 (1936) 1302.[42] K. Bergstad, S.Y. Jonsson and J.-E. Bäckvall, J. Am. Chem. Soc. 121
(1999) 10424.[43] S.Y. Jonsson, K. Färnegardh and J.-E. Bäckvall, J. Am. Chem. Soc.
123 (2001) 1365.[44] A.H. Éll, S.Y. Jansson, A. Börje, H. Adolfsson and J.-E. Bäckvall,
Tetrahedron Lett. 42 (2001) 2569.[45] J.F. Cairns and H.L. Roberts, J. Chem. Soc. C (1968) 640.[46] C. Döbler, G.M. Mehltretter, U. Sundermeier and M. Beller, J. Am.
Chem. Soc. 122 (2000) 10289.[47] C. Döbler, G.M. Mehltretter, U. Sundermeier and M. Beller,
J. Organomet. Chem. 621 (2001) 70.[48] R.C. Michaelson and R.G. Austin, US patent 4533772 (1985).[49] A. Krief and C. Colaux-Castillo, Tetrahedron Lett. 40 (1999) 4189.[50] L. Hevesi and A. Krief, Angew. Chem. 88 (1976) 413.






















![[Charles N. Satterfield] Heterogeneous Catalysis](https://img.pdfslide.us/doc/110x75/577cc1a51a28aba71193945e/charles-n-satterfield-heterogeneous-catalysis.jpg)






