Embed Size (px)
Citation preview

Tissue Transglutaminase-Catalyzed Formation of High-Molecular-Weight Aggregates in Vitro Is Favored with LongPolyglutamine Domains: A Possible MechanismContributing to CAG-Triplet Diseases
Vittorio Gentile,*,1 Carlo Sepe,* Menotti Calvani,† Mariarosa A. B. Melone,‡ Roberto Cotrufo,‡Arthur J. L. Cooper,§,¶,\ John P. Blass,§,\,** and Gianfranco Peluso††*Dipartimento di Biochimica e Biofisica e ‡Istituto di Scienze Neurologiche, Seconda Universita di Napoli, 80138 Naples,Italy; †Direzione Scientifica Sigma Tau, Pomezia, Rome, Italy; ††Istituto di Biochimica delle Proteine ed Enzimologia,CNR, Naples, Italy; §Department of Neurology and Neuroscience, ¶Department of Biochemistry, and **Department ofMedicine, Cornell University Medical College, New York, New York 10021; and \Burke Medical Research Institute,Cornell University Medical College, White Plains, New York 10605
Received December 15, 1997
To investigate possible biochemical mechanisms un-derlying the ‘‘toxic gain of function’’ associated withpolyglutamine expansions, the ability of guinea pigliver tissue transglutaminase to catalyze covalent at-tachments of various polyamines to polyglutaminepeptides was examined. Of the polyamines tested,spermine is the most active substrate, followed byspermidine and putrescine. Formation of covalentcross links between polyglutamine peptides and poly-amines yields high-Mr aggregates—a process that isfavored with longer polyglutamines. In the presence oftissue transglutaminase, purified glyceraldehyde-3-phosphate dehydrogenase (a key glycolytic enzymethat binds tightly to the polyglutamine domains ofboth huntingtin and dentatorubral–pallidoluysian at-rophy proteins) is covalently attached to polyglu-tamine peptides in vitro, resulting in the formation ofhigh-Mr aggregates. In addition, endogenous glyceral-dehyde-3-phosphate dehydrogenase of a Balb-c 3T3 fi-broblast cell line overexpressing human tissue trans-glutaminase forms cross-links with a Q60 polypeptideadded to the cell homogenate. Possibly, expansion ofpolyglutamine domains (thus far known to occur inthe gene products associated with at least seven neu-rodegenerative diseases) leads to increased/aberranttissue transglutaminase-catalyzed cross-linking reac-
tions with both polyamines and susceptible proteins,such as glyceraldehyde-3-phosphate dehydrogenase.Formation of cross-linked heteropolymers may lead todeposition of high-Mr protein aggregates, thereby con-tributing to cell death. © 1998 Academic Press
Key Words: tissue transglutaminase; Huntington dis-ease; Qn domains; CAG expansions; glyceraldehyde-3-phosphate dehydrogenase; polyamines; neurodegen-erative diseases.
Expansions of polyglutamine (Qn)2 domains (encodedby CAG repeats) are now known to occur in the mu-tated gene products associated with an increasingnumber of neurodegenerative diseases. These diseasesinclude Huntington disease (HD), spinobulbar muscu-lar atrophy (SBMA), several spinocerebellar ataxias[types 1, 2, 3 (Machado–Joseph disease), and 7 (SCA-1,SCA-2, SCA-3, and SCA-7, respectively)], and denta-torubral–pallidoluysian atrophy (DRPLA) (1–4) (for alist of recent references see Ref. 5). Although verydifferent mutated proteins are specific for each disease,
1 To whom correspondence and reprint requests should be ad-dressed at Dipartimento di Biochimica e Biofisica, Seconda Univer-sita di Napoli, via Costantinopoli 16, 80138 Naples, Italy. Fax: 0039-81-5665863. E-mail: [email protected].
2 Abbreviations used: DRPLA, dentatorubral–pallidoluysian atro-phy; DTT, dithiothreitol; TCA, trichloroacetic acid; F-MOC, N-(9-fluoromethoxycarbonyl)-; GAPDH, glyceraldehyde-3-phosphate de-hydrogenase; HAP 1, huntingtin-associated protein 1; HD, Hunting-ton disease; Qn, polyglutamine; Pt, putrescine; SBMA, spinobulbarmuscular atrophy; SCA, spinocerebellar ataxia; Spd, spermidine;Spm, spermine; tTGase, tissue transglutaminase; DMC, N,N-di-methylcasein.
314 0003-9861/98 $25.00Copyright © 1998 by Academic Press
All rights of reproduction in any form reserved.
ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS
Vol. 352, No. 2, April 15, pp. 314–321, 1998Article No. BB980592

the underlying defect is remarkably similar. In everycase, disease occurs when n is $40. Moreover, gener-ally, the longer the repeat, the earlier the age of onsetand the more rapid the progression of disease (6, 7).The biological roles of Qn domains are not understood,but Qn stretches are known to bind to selected proteins(8–10) and to DNA (11). These findings, together withthe observation that mutated proteins are expressed invivo (12), have prompted several authors to suggestthat an expansion of Qn domains leads to a ‘‘toxic gainof function’’ (13–17). Neuropathology associated withmouse models of Machado–Joseph disease (17) and HD(18, 19) supports this hypothesis. Moreover, althoughhuntingtin (the effected protein in HD) is a large pro-tein (Mr ;350,000), all that is needed to produce theprogressive neurological phenotype in transgenic miceis exon 1 of the HD gene containing an expanded CAGrepeat (20).
Qn domains of both huntingtin and DRPLA proteinsbind tightly to glyceraldehyde-3-phosphate dehydroge-nase (GAPDH) (8, 9). Other proteins, such as hunting-tin-associated protein (HAP 1), also bind strongly tothe Qn domain of huntingtin (10). Thus, Qn domainscan apparently be components of potentially large pro-tein heterocomplexes. The structure and formation ofsuch complexes may be disturbed when the Qn domainis excessively large. Aberrant protein–protein interac-tions may represent, therefore, a key event in thepathogenesis of Qn repeat diseases. An intriguing hy-pothesis is that an expansion of Qn domains gives riseto new or more reactive substrates of tissue transglu-taminase (tTGase, type II transglutaminase) (13).
tTGase belongs to a family of Ca21-dependent en-zymes that are encoded by at least five unique genes inmammalian cells. These enzymes catalyze covalentposttranslational modifications of protein substratesby promoting the covalent formation of cross-links be-tween the g-carboxamido group of a glutamine (Q)endoresidue (electrophile) and the e-amino group of alysine (K) endoresidue (nucleophile) (21–24). The re-sulting isopeptide bond [Ne-(g-glutamyl)lysine] is resis-tant to proteolysis and its formation commonly resultsin the modified protein becoming much less soluble.Simple amines or polyamines, including polyaminesbound at one end to a protein Q residue, may substitutefor protein K residues as attacking nucleophile. tTGaseis the most widespread transglutaminase in mamma-lian organs. This enzyme participates in extracellularmatrix stabilization, receptor-mediated endocytosis,and cell signal transduction (24). tTGase, through itsability to catalyze intracellular protein cross links, hasbeen suggested to play a key role in apoptosis of manycell types—including neuroblastoma cell lines (25–28).
In previous work, Kahlem et al. (29) showed that (a)peptides containing non-pathological-length Qn do-mains (where n # 18) are excellent substrates of
tTGase ([14C]glycine ethyl ester as attacking nucleo-phile), (b) generally, the longer the Qn domain, thegreater the enzyme activity, and (c) R5Q18R5 formshighly insoluble aggregates in the presence of brainproteins and tTGase. Recently, we showed that gluta-thione S-transferase constructs containing either anon-pathological-length Qn domain (i.e., GSTQ20) or apathological-length Qn domain (i.e., GSTQ62) are excel-lent substrates of tissue transglutaminase ([1,4-14C]putrescine as attacking nucleophile) (5).
To examine further the mechanisms underlying ourprevious findings (5) and those of Kahlem et al. (29) wedetermined whether naked Qn domains (i.e., Qn do-mains that are not part of a larger protein or polypep-tide construct) are substrates of tTGase. Binding ofsuch constructs to the enzyme will be independent ofpossible secondary influences associated with sur-rounding non-Q protein/peptide residues. Here, we re-port that naked, extended Qn domains (Q20, Q40, andQ60) are indeed excellent substrates of tTGase with thenaturally occurring polyamines as attacking nucleo-phile. We also show that this capacity is enhanced withincreasing size of the Qn domain.
Previously, we provided several lines of evidencesuggesting that crystalline rabbit muscle GAPDH (ahomotetramer in its glycolytic form) possesses at leastone K residue/monomer that is a substrate of tTGase(30). Covalent attachment to a Q residue leads to lossof glycolytic function. This loss is much more pro-nounced with protein constructs containing pathologi-cal-length Qn domains (GSTQ62, GSTQ81) than withsmaller protein constructs containing non-pathologi-cal-length Q domains (GSTQ0, GSTQ10) (30). Here, weextend these findings to show that GAPDH monomerdoes indeed possess at least one substrate K residueper monomer and that high-Mr heteropolymers formedby covalent attachment of GAPDH to naked Qn do-mains are readily detected by Western blotting. Theevidence suggests that covalent attachment of Qn do-mains to GAPDH results in dramatic alterations of theproperties of the enzyme. Finally, we show thatGAPDH in a Balb-c 3T3 cell line overexpressing hu-man tTGase is covalently linked to Q60 when the cellcontents are exposed to the polymer.
MATERIALS AND METHODS
Materials. Qn peptides of different lengths [n 5 20 (Mr 2581), 40(Mr 5144), and 60 (Mr 7811)] were synthesized at Genemed Biotech-nologies, Inc. (San Francisco, CA) by using F-MOC-L-glutamine asprecursor. The Qn peptides were dissolved in water by sonication andstored at 220°C. Precise concentrations of stock solutions were de-termined by HPLC analysis to be 5.5, 6.3, and 30 mM for Q20, Q40,and Q60, respectively. Rabbit muscle GAPDH (106 U/mg of protein;Mr of monomer ;36,000), guinea pig liver tTGase (;2 units/mg ofprotein), Substance P (Mr 1348), and N,N-dimethylated bovine milkcasein (Mr ;35,000) were purchased from Sigma (St. Louis, MO).[1,4-14C]Putrescine dihydrochloride [H2N14CH2(CH2)2
14CH2NH2 z
315TISSUE TRANSGLUTAMINASE AND CAG-TRIPLET DISEASES

2HCl] (Pt; sp act 110 mCi/mmol) and [14C]spermine tetrahydrochlo-ride [(H2NCH2CH2CH2NH14CH2CH2)2 z 4HCl] (Spm; sp act 114 mCi/mmol) were purchased from Amersham (Little Chalfont, Bucking-hamshire, UK). [3H]Spermidine trihydrochloride [H2NC3H2(CH2)NH(CH2)3C3H2NH2 z 3HCl] (Spd; sp act 16.4 Ci/mmol) was pur-chased from NEN Du Pont (Dreiech, Germany).
Cell culture conditions. tTGase-transfected (clone 5) Balb-c 3T3fibroblasts were obtained and grown as previously described by Gen-tile et al. (26). Control cells (clone 1) were stably transfected withpSG5 expression vector (Stratagene, La Jolla, CA) alone and ex-pressed no detectable tTGase (see below).
Standard tTGase assay procedure. The reaction mixture (finalvolume 100 ml) contained 100 mM Tris–HCl buffer (pH 7.4), 10 mMdithiotreithol (DTT), 2 mg of purified guinea pig liver tTGase, 2.5 mMCaCl2, and 20 mM labeled polyamine (21). Measurements were madein triplicate, and the blank contained 5 mM EGTA. Either SubstanceP (0.1 mg) or Qn peptide (0.1 mg, except where stated) was used as Qdonor. Controls contained the complete assay mixture except thatthe Q donor substrate was omitted. The assay mixture was incubatedfor either 2 or 18 h at 37°C. To retain enzymatic activity over thelonger period, additional tTGase was added repeatedly during thereaction, as previously described by Kahlem et al. (29). At the end ofthe incubation, protein within the assay mixture was precipitatedwith 20% (w/v) trichloroacetic acid (TCA). The pellet was removed bycentrifugation for 10 min in a microfuge at 12,000g, washed twice,and then counted in a liquid scintillation counter. Alternatively, 50ml of the reaction mixture was subjected to SDS–PAGE (12.5% acryl-amide) (31) and the gel was analyzed by fluorography (32).
Determination of GAPDH as a substrate of tTGase. To determinewhether GAPDH is a K donor in the tTGase reaction, GAPDH (50 mg,;1.4 nmol of monomer) was incubated with a Q donor substrate(Q60) (0.017, 0.033, 0.067, or 0.135 nmol) or with N,N-dimethylcasein(;0.17, 0.35, 0.7, and 1.4 nmol) under conditions described above forthe tTGase assay, except that polyamines were omitted. At the endof the incubation time (18 h at 37°C), aliquots (5–20 ml) were sub-jected to SDS–PAGE (10% acrylamide) and blotted to nitrocellulose(Bio-Rad, 0.45 mm). To obtain effective transfer of high-Mr aggre-gates from the upper part of the polyacrylamide gel to the nitrocel-lulose, the time of transfer was 6 h rather than the more customary2 h (0.7 mA/cm2). For Western blot analysis, the primary antibodywas a monoclonal IgG to human GAPDH (Biogenesis, Bournemouth,UK) (1:5000 dilution). The secondary antibody (1:3000 dilution) wasa rabbit anti-mouse IgG conjugated to peroxidase. Staining wascarried out with an AEC chromogen kit (Sigma) according to themanufacturer’s instructions.
To determine whether GAPDH can act as a Q-donor substrate oftTGase, GAPDH [;0.17, 0.35, 0.7, and 1.4 nmol (calculation based onthe size of the monomer)] was incubated for 2 h with labeled Spm (2.0nmol) as described above for the tTGase assay. At the end of theincubation, protein in the assay mixture was precipitated with 1 mlof 20% (w/v) TCA and washed twice, and the pellet was counted in aliquid scintillation counter.
Cross-linking of GAPDH in Balb-c 3T3 cells. To determinewhether cross-linking of GAPDH to Qn stretches can occur in acellular environment, Balb-c 3T3 fibroblasts transfected with thehuman tTGase gene (clone 5) were compared to their nontransfectedcounterpart (clone 1). Cells at 80% confluency were suspended inhomogenization buffer [Tris–HCl (pH 7.40) containing 2 mM EGTA]and sonicated at 4°C. This sonicated suspension (protein content ;4mg/ml) was used as a source of both GAPDH and tTGase. The assaymixture (100 ml) contained cell homogenate (80 mg of protein), 1 mgof Q60, 100 mM Tris–HCl buffer (pH 7.4), 10 mM DTT, and 2.5 mMCaCl2. Blanks were identical except that they contained 5 mM EGTAto inhibit the Ca21-dependent tTGase. At the end of the incubation(18 h at 37°C), analysis of the monomeric form of GAPDH wasperformed by Western blotting as described above.
Stability of GAPDH in postmortem rat brain. Adult male Dawleyrats were killed by decapitation. In one group of animals the brainwas immediately removed, weighed, and homogenized in a 10-foldvolume of ice-cold 10 mM potassium phosphate buffer (pH 7.2) bymeans of a glass-to-glass homogenizer. An aliquot of the suspensionwas then assayed for GAPDH activity as described above. In anothergroup each brain was bisected down the midline. One-half of thebrain was immediately frozen in liquid nitrogen. The other half wasfrozen after incubation at 21°C for 20 h. The two halves were thenthawed and homogenized. An aliquot of the homogenate was assayedfor GAPDH activity as described by Cooper et al. (30) and comparedto the activity in brains immediately homogenized after decapitationof the rats.
Statistical analyses. The data are reported as means 6 SE ob-tained from three experiments, each performed in triplicate. Themeans were compared using analysis of variance (ANOVA) plusBonferroni’s t test. A P value of less than 0.05 was taken as signifi-cant.
RESULTS
Analysis of Different Polyamines as Amine DonorSubstrates for Attachment to Q20 and Comparisonwith Substance P Peptide
Initially, Q20 was chosen as a Q-donor substrate toprobe the efficacy with which tTGase catalyzes cova-lent attachment of polyamines to Qn stretches. Amongthe three polyamines tested, Spm was the most potentsubstrate under the conditions used, whereas Spd andPt were less effective (Table I).
The size of the Q20 peptide (Mr 5 2581) used in theexperiment summarized in Table I is of about the samemagnitude as that of Substance P (Mr 5 1348), a bio-logically important peptide (containing two adjacent Qresidues) that is a good amine-acceptor (Q-donor) sub-strate of tTGase (33, 34). Thus, it was instructive tocompare the relative effectiveness of Q20 and Sub-stance P as Q donors in the presence of Spm. Thefluorograph depicted in Fig. 1 shows that, in the pres-ence of labeled Spm, both Substance P and Q20 aresubstrates of tTGase. However, Substance P and Q20exhibit very different behavior with respect to theircapacity to form high-Mr aggregates. Covalent attach-ment of Substance P to Spm yields an intense band atthe bottom of lane 2 (Fig. 1). This band is due either to
TABLE I
Relative Amounts of Labeled Polyamines Covalently Boundto Q20 in the Presence of tTGase
mol of polyamine incorporated/mol of peptide
Putrescine 0.091 6 0.004Spermidine 0.103 6 0.006Spermine 0.134 6 0.007
Note. Values are means 6 SE. n 5 3 different experiments. Eachvalue is significantly different from the other two values with P ,0.05 by ANOVA plus Bonferroni’s t test. Incubation time 5 2 hours.
316 GENTILE ET AL.
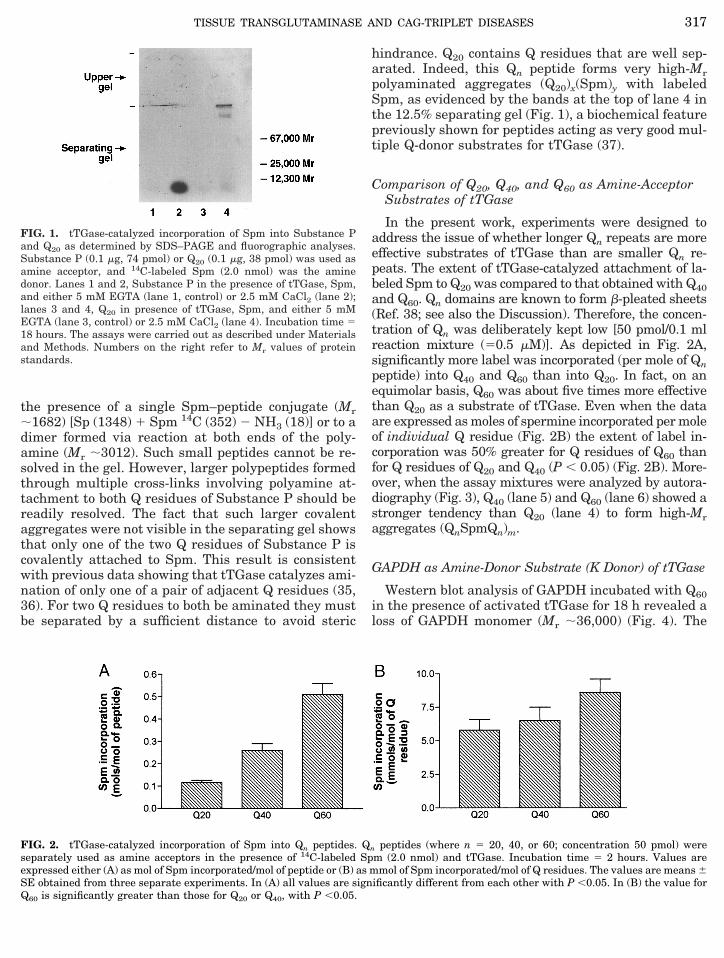
the presence of a single Spm–peptide conjugate (Mr;1682) [Sp (1348) 1 Spm 14C (352) 2 NH3 (18)] or to adimer formed via reaction at both ends of the poly-amine (Mr ;3012). Such small peptides cannot be re-solved in the gel. However, larger polypeptides formedthrough multiple cross-links involving polyamine at-tachment to both Q residues of Substance P should bereadily resolved. The fact that such larger covalentaggregates were not visible in the separating gel showsthat only one of the two Q residues of Substance P iscovalently attached to Spm. This result is consistentwith previous data showing that tTGase catalyzes ami-nation of only one of a pair of adjacent Q residues (35,36). For two Q residues to both be aminated they mustbe separated by a sufficient distance to avoid steric
hindrance. Q20 contains Q residues that are well sep-arated. Indeed, this Qn peptide forms very high-Mrpolyaminated aggregates (Q20)x(Spm)y with labeledSpm, as evidenced by the bands at the top of lane 4 inthe 12.5% separating gel (Fig. 1), a biochemical featurepreviously shown for peptides acting as very good mul-tiple Q-donor substrates for tTGase (37).
Comparison of Q20, Q40, and Q60 as Amine-AcceptorSubstrates of tTGase
In the present work, experiments were designed toaddress the issue of whether longer Qn repeats are moreeffective substrates of tTGase than are smaller Qn re-peats. The extent of tTGase-catalyzed attachment of la-beled Spm to Q20 was compared to that obtained with Q40and Q60. Qn domains are known to form b-pleated sheets(Ref. 38; see also the Discussion). Therefore, the concen-tration of Qn was deliberately kept low [50 pmol/0.1 mlreaction mixture (50.5 mM)]. As depicted in Fig. 2A,significantly more label was incorporated (per mole of Qnpeptide) into Q40 and Q60 than into Q20. In fact, on anequimolar basis, Q60 was about five times more effectivethan Q20 as a substrate of tTGase. Even when the dataare expressed as moles of spermine incorporated per moleof individual Q residue (Fig. 2B) the extent of label in-corporation was 50% greater for Q residues of Q60 thanfor Q residues of Q20 and Q40 (P , 0.05) (Fig. 2B). More-over, when the assay mixtures were analyzed by autora-diography (Fig. 3), Q40 (lane 5) and Q60 (lane 6) showed astronger tendency than Q20 (lane 4) to form high-Mraggregates (QnSpmQn)m.
GAPDH as Amine-Donor Substrate (K Donor) of tTGase
Western blot analysis of GAPDH incubated with Q60in the presence of activated tTGase for 18 h revealed aloss of GAPDH monomer (Mr ;36,000) (Fig. 4). The
FIG. 1. tTGase-catalyzed incorporation of Spm into Substance Pand Q20 as determined by SDS–PAGE and fluorographic analyses.Substance P (0.1 mg, 74 pmol) or Q20 (0.1 mg, 38 pmol) was used asamine acceptor, and 14C-labeled Spm (2.0 nmol) was the aminedonor. Lanes 1 and 2, Substance P in the presence of tTGase, Spm,and either 5 mM EGTA (lane 1, control) or 2.5 mM CaCl2 (lane 2);lanes 3 and 4, Q20 in presence of tTGase, Spm, and either 5 mMEGTA (lane 3, control) or 2.5 mM CaCl2 (lane 4). Incubation time 518 hours. The assays were carried out as described under Materialsand Methods. Numbers on the right refer to Mr values of proteinstandards.
FIG. 2. tTGase-catalyzed incorporation of Spm into Qn peptides. Qn peptides (where n 5 20, 40, or 60; concentration 50 pmol) wereseparately used as amine acceptors in the presence of 14C-labeled Spm (2.0 nmol) and tTGase. Incubation time 5 2 hours. Values areexpressed either (A) as mol of Spm incorporated/mol of peptide or (B) as mmol of Spm incorporated/mol of Q residues. The values are means 6SE obtained from three separate experiments. In (A) all values are significantly different from each other with P ,0.05. In (B) the value forQ60 is significantly greater than those for Q20 or Q40, with P ,0.05.
317TISSUE TRANSGLUTAMINASE AND CAG-TRIPLET DISEASES

loss was progressive with increasing amounts of Q60(Fig. 4B). As determined by densitometric scanning(Gel-Pro Analyzer; Media Cybernetics, version 1.0.1) adramatic decrease in the monomeric form of GAPDH(to $75%) was observed when the enzyme was incu-bated in the presence of Q60 and activated tTGase,while increasing amounts of high-Mr aggregates couldbe detected at the top of each corresponding lane in the5% stacking gel (Fig. 4A).
To obtain more information on the ability of K resi-due(s) of GAPDH monomer to form tTGase-catalyzedcovalent linkages to protein Q residues, we chose to useDMC as Q donor. [DMC contains only Q-residue sub-strates of tTGase (the potential K-residue substratesare blocked) and therefore does not form covalentlylinked homopolymers in the presence of the cross-link-
ing enzyme.] A strong reduction (to $65%, as deter-mined by densitometric scanning) of the monomericform of GAPDH (Mr ;36,000) was observed when itwas incubated in the presence of DMC and activatedtTGase (Fig. 5), whereas no such effect was observedwhen GAPDH was incubated in the presence of bovineserum albumin (a protein that is not a substrate oftTGase). We previously noted that tTGase does notcatalyze the covalent attachment of [1,4-14C]Pt toGAPDH (30). We have now found that tTGase simi-larly does not catalyze incorporation of labeled Spminto GAPDH (data not shown). The results stronglyindicate that GAPDH acts as an amine (K)-donor sub-strate, but not as a Q donor for tTGase-catalyzed reac-tions. Finally, the formation of high-Mr aggregates(Fig. 4A) argues that more than one K residue may becovalently attached to a single Qn molecule (see Dis-cussion).
Cross-Linking of GAPDH in Cell Homogenates
To determine whether it is likely that GAPDH can becross-linked to Qn domains by the action of tTGase intissues or cells, a Balb-c 3T3 fibroblast cell line stablytransfected with a eukaryotic expression vector (pSG5–tTGase; clone 5) containing high levels of human tTGase(sp. act. 107 pmol/30 min/mg of protein) (26) was used asan experimental model. Clone 1 (control), the nontrans-fected counterpart of clone 5, has no detectable tTGase(by activity, Western blot, and Northern blot analyses)(26). These two clones, therefore, represent appropriatecellular models in which the biological effects of tTGasecan be easily determined and analyzed.
Total cell homogenates obtained from clone 5 andfrom clone 1 (control) were incubated in the presence ofQ60. A significant decrease (about 50%, as determinedby densitometric scanning) in the monomeric form ofGAPDH (Mr 36,000) occurred only when homogenatesobtained from clone 5 cells were incubated in the pres-ence of both Q60 and 2.5 mM free Ca21 (lane 2 versus
FIG. 3. tTGase-catalyzed incorporation of Spm into Qn peptides asassessed by SDS–PAGE and fluorographic analyses. Qn peptides(where n 5 20, 40, or 60; concentration 50 pmol) were separatelyused as amine acceptors in the presence of 14C-labeled Spm (2.0nmol) and tTGase. Lanes 1, 2, and 3 were controls which contained2.5 mM EGTA and Q20 (lane 1), Q40 (lane 2), or Q60 (lane 3). Lanes4, 5, and 6 contained 2.5 mM CaCl2 and Q20 (lane 4), Q40 (lane 5), andQ60 (lane 6). Incubation time 5 18 hours. The assays were carried outas described under Materials and Methods. Numbers on the rightrefer to Mr values of marker proteins.
FIG. 4. Western blot analysis of GAPDH after incubation withtTGase and Q60. GAPDH (50 mg, 1.35 nmol of monomer) was incu-bated with various amounts of Q60 and tTGase. Lane 1 (control),GAPDH incubated with tTGase and Q60 in the presence of 5 mMEGTA; lanes 2, 3, 4, and 5, GAPDH incubated with tTGase and 2.5mM Ca21 in the presence of increasing amounts of Q60 (0.125, 0.250,0.5, and 1 mg, equivalent to 16, 32, 64, and 128 pmol, respectively).(A) Upper gel (stacking gel). (B) Lower gel (separating gel). Incuba-tion time 5 18 hours. For further details see Materials and Methods.
FIG. 5. Western blot analysis of GAPDH after incubation withtTGase and N,N-dimethylcasein. GAPDH (50 mg, 1.35 nmol of mono-mer) was incubated with various amounts of N,N-dimethylcaseinand tTGase. Lane 1 (control), GAPDH incubated with bovine serumalbumin (92 mg; 1.4 nmol) and Ca21-activated tTGase; lanes 2, 3, 4,and 5. GAPDH incubated with Ca21-activated tTGase in the pres-ence of increasing amounts of N,N-dimethylcasein (6, 12, 25, and 50mg corresponding to ;0.17, 0.35, 0.7, and 1.4 nmol, respectively).Incubation time 5 18 hours. For further details see Materials andMethods.
318 GENTILE ET AL.

lane 1, Fig. 6). This finding shows that GAPDH deriveddirectly from a cell homogenate can be covalently crosslinked to a Qn domain by endogenous tTGase.
DISCUSSION
Longer Qn Domains Are More Potent Q-DonorSubstrates of tTGase Than Are SmallerQn Domains
We previously showed that equimolar GSTQ20 andGSTQ62 are about equally effective as substrates oftTGase (with [1,4-14C]Pt as attacking nucleophile) (5).However, the concentration of the GSTQn constructsused in that study (;16–18 mM) was relatively high,and noncovalent intermolecular bonding may havebeen a problem. In the present study we used a nakedQn in place of the GSTQn construct. This arrangementminimizes the possibility of noncovalent interactionbetween Qn and hydrophobic sites of proteins withinthe assay mixture (see below). Moreover, in the presentstudy, we used a dilute concentration of Qn domain (0.5mM), so that the chances of forming b-pleated sheetsbetween adjacent Qn molecules in the assay mixturewere also minimized. Thus, by utilizing conditions thatminimized noncovalent interactions and permittedmaximal ‘‘exposure’’ of Q residues, we showed for thefirst time that a pathological-length Qn domain (i.e.,Q60) is a much more effective Q substrate of tTGasethan is a non-pathological-length Qn domain (i.e., Q20)or borderline-pathological-length Qn domain (i.e., Q40).Additionally, we found that, under our experimentalconditions and in agreement with findings previouslyreported by others (39), Spm is the best polyamine(nucleophile) substrate for tTGase-catalyzed reactionsin vitro. Moreover, we found that Q40 and Q60 exhibit astronger tendency than Q20 to form insoluble high-Mrprotein aggregates in the presence of Spm and tTGase.This last finding is of particular interest, because it isthe first demonstration of the formation of high-Mraggregates of Qn peptides linked via polyaminebridges. The present findings therefore are entirelyconsistent with the hypothesis (see the introduction)
that the toxic gain of function associated with expres-sion of proteins containing expanded Qn domains canbe due at least in part to aberrant or increased tTGaseactivity.
Possible Implications of Covalent Binding of GAPDHto Pathological-Length Qn Domains
In confirmation of our earlier findings (30), thepresent results show that GAPDH possesses at leastone K-residue (but no Q-residue) substrate of tTGasethat can form an isopeptide bond with a suitable Qsubstrate. In a previous study (30), the highly insolublenature of the covalent adducts investigated (i.e.,GAPDH 3 GSTQ60/82, GAPDH 3 casein) preventeddirect detection of high-Mr bands of GAPDH hetero-polymers on Western blots. In the present study, use ofan extended transfer time to the nitrocellulose filterand use of the Q substrate Q60 (in place of GSTQn)permitted detection for the first time of tTGase-cata-lyzed formation of high-Mr GAPDH 3 Qn heteropoly-mers (Fig. 4). Moreover, the present results show thatwhen a minimally diluted extract of a Balb-c 3T3 fibro-blast cell line expressing GAPDH and human tTGaseis incubated with Q60 (Fig. 6) and subjected to Westernblot analysis, the intensity of the GAPDH monomerprotein band is decreased (lane 4) relative to that incontrols (lanes 1–3). This finding strongly suggeststhat if tTGase, GAPDH, and Qn domains are juxta-posed in the same compartment in the cell, they canpotentially interact (see below).
GAPDH is an interesting protein. As noted above, inits tetrameric form, it is an important enzyme in theglycolytic pathway. In its monomeric form, GAPDH isa uracil DNA glycosylase and binds to RNA, ATP,calcylin, actin, tubulin, and the amyloid precursor pro-tein (8, 40). As also pointed out by Sawa et al. (40),expression of GAPDH is dynamic, changing with hyp-oxia, calcium influx, oncogene transformations, andgrowth-accelerated states and as a function of cell cy-cle. Recent publications by Chuang, Snyder, and theircolleagues suggest an important role for GAPDH inapoptosis of neuronal (40–42) and nonneuronal cells(40). Thus, apoptosis is accompanied by a remarkableinflux of glycolytically inactive and solubilization-resis-tant GAPDH into the nuclear compartment. Alter-ations in any of the multiple roles of GAPDH by cova-lent attachment of Qn domains may seriously compro-mise cell viability.
A tight, but noncovalent binding between immobi-lized GAPDH and several huntingtin fragments as wellas with androgen receptor (the gene product mutatedin SBMA) or ataxin 1 (the gene product mutated inSCA-1) has previously been demonstrated (8, 9). In allthese cases, it is thought that Qn domains bind toproteins by hydrogen bonding in b sheets as polar
FIG. 6. Western blot analysis of GAPDH in homogenates of Balb-c3T3 fibroblasts expressing human tTGase and exposed to Q60. Cellhomogenates (80 mg of protein) derived from clone 5 (expressinghuman tTGase) (lanes 1 and 2) and clone 1 (control) (lanes 3 and 4)were incubated in the presence of Q60 (1 mg, 740 pmol) and 2.5 mMCa21 (lanes 2 and 4) or 5 mM EDTA (lanes 1 and 3, controls).Incubation time 5 18 hours. For further details see Materials andMethods.
319TISSUE TRANSGLUTAMINASE AND CAG-TRIPLET DISEASES

zippers. Moreover, chymotrypsin inhibitor 2 containingan artificial Qn domain oligomerizes by formingb-pleated sheets (36, 43). Curiously, despite tight non-covalent interactions, GAPDH activity is not inhibitedby Qn constructs in the absence of tTGase (30). How-ever, as noted above, incubation of GAPDH withGSTQ62 (or GSTQ81) in the presence of tTGase leads torelatively rapid loss of enzyme activity (30). GAPDH isa very abundant cellular constituent and it is likely tobe present in a large excess over Qn domains in vivo.The experiment depicted in Fig. 4 was carried out byincubating Qn domains with a large molar excess ofGAPDH (.10-fold) in the presence of tTGase. Underthese conditions, GAPDH forms very high-Mr aggre-gates. Despite the very large molar excess of GAPDHover Q60, the intensity of the GAPDH band was con-siderably decreased (lanes 2–5) when incubated withtTGase compared to a control band (lane 1) (Fig. 4).Therefore, it is likely that in the presence of a largeexcess of GAPDH over Qn (Fig. 4), a ‘‘zipper’’ effectoccurs such that attachment of one GAPDH subunitvia a K 3 Q linkage is followed by attachment ofanother subunit via a K 3 Q linkage to the same Qnmolecule (Kx3 Qn). However, the possibility cannot beruled out that GAPDH subunit possesses more thanone K-residue substrate of tTGase, permitting singleGAPDH subunits to form bridges between individualQn strands (Qn 4 K–K9 3 Qn9) or loops to the samestrand (Qn 4 K–K9 3 Qn). Possibly, all three types oflinkages occur.
GAPDH and huntingtin are colocalized in the cell.Both GAPDH (references summarized in Ref. 8) andhuntingtin (44, 45) are predominantly cytosolic and inpart associated with cytoskeletal proteins and mem-branes. Huntingtin is also associated to a lesser extentwith synaptic vesicles (46) and possesses a nuclear-targeting motif (47). tTGase is present largely in thecytosol, but some is also present in the extracellularspace (26) and in the nucleus (48). The presence oftTGase, huntingtin, and GAPDH in the cytosolic com-partment suggests that the potential exists for inter-actions among the three proteins in vivo. Possibly,tTGase-mediated formation of intracellular aggregatesbetween Qn stretches and GAPDH could produce anabnormal and progressive stiffness of the cellular scaf-fold. Induction of rigidity in the cellular scaffold is amajor factor associated with apoptosis (26, 49).
A recent report suggests that the activity of GAPDHis unaltered in HD brain compared with that of controlhuman brain (50). The postmortem interval beforefreezing of the tissue was about 15 h and the stabilityof the enzyme in the human brain postmortem was notdetermined. Nevertheless, it is possible that the glyco-lytic activity of GAPDH in the human brains was un-altered during the postmortem interval in that study.In this regard, we have found that the activity of the
rat brain enzyme postmortem is surprisingly stable.Specific activities of GAPDH in rat brain frozen at 0and 20 h postmortem (21°C) were 170 6 13 and 161 612 mmol/min/g wet wt (n 5 3), respectively. The specificactivity of GAPDH in fresh, unfrozen rat brain was175 6 5 mmol/min/g wet wt (n 5 3). However, in lightof the findings discussed above that a glycolyticallyinactive form of GAPDH accumulates in the nucleus ofcells undergoing apoptosis, a more detailed analysis ofGAPDH protein in subcellular compartments of HDbrain is warranted. Inasmuch as tTGase and Qn do-mains can accumulate in the nucleus, it is possible thatcross-linking reactions in that compartment involvingGAPDH and Qn domains may contribute to the apopto-tic mechanisms. Certainly, our present results withfibroblasts stably transfected with the gene for humantTGase suggest that endogenous GAPDH in the pres-ence of Ca21-activated tTGase and a Qn stretch canbecome cross-linked and lose activity (30). This finding,described above, is the first demonstration of an effectof tTGase on Qn and GAPDH in the presence of allcellular components, including substrates and coen-zymes, that might theoretically protect againsttTGase-mediated GAPDH inactivation.
Slow neuronal death may occur in HD and otherCAG-triplet diseases via an increase in cytosolic freeCa21, subsequent activation of tTGase, and accumula-tion of Ne-[g-glutamyl]lysine cross-links on GAPDHand other substrate proteins. Loss of a critical form ofGAPDH or of other metabolically important enzymesmay lead to a defect in energy metabolism as is thoughtto occur in HD (50–52). Moreover, promotion ofGAPDH cross-links in the nuclear compartment maylead to increased apoptosis.
Finally, nuclei from dying neurons in the basal gan-glia and the brain cortex of animals transgenic for themutated huntingtin gene have been shown to containlarge, insoluble high-Mr protein aggregates (18, 19).This finding is consistent with a role of mutated hun-tingtin and other extended Qn proteins in the inductionof apoptotic processes (17). However, it is not clear howthese protein aggregates arise. As noted above, hun-tingtin has a nuclear-targeting motif. Moreover,tTGase is known to utilize histones as Q substratesand the nucleus contains large amounts of potentialpolyamine cross linkers (53). Whether nuclear tTGasecan catalyze cross-linking reactions in this compart-ment (especially in cells that may already be compro-mised) involving GAPDH, histones, polyamines, andQn domains is not yet known.
In conclusion, there are now several tantalizingclues suggesting the involvement of tTGase in theetiology and the molecular pathology of CAG-expan-sion diseases and possibly of other diseases. Theinvolvement of tTGase in such diseases deserves fur-ther study.
320 GENTILE ET AL.

ACKNOWLEDGMENTS
This work was supported in part by Telethon Grant number E. 217to V.G.; the Progetto Legge 41, Finanziamento C. E. E. RegioneCampania to G.P.; and the Will Rogers Foundation, the WinifredMasterson Burke Relief Foundation, and National Institute of Aging(AG 09014) to A.J.L.C. and J.P.B.
REFERENCES
1. The Huntington’s Disease Collaborative Research Group (1993)Cell 72, 971–983.
2. Mandel, J.-L. (1994) Nature Genet. 7, 453–455.3. Albin, R. L., and Tagle, D. A. (1995) Trends Neurosci. 18, 11–14.4. Roses, A. D. (1996) Nature Med. 2, 267–269.5. Cooper, A. J. L., Sheu, K.-F. R., Burke, J. R., Onodera, O.,
Strittmatter, W. J., Roses, A. D., and Blass, J. P. (1997) J. Neu-rochem. 69, 431–434.
6. Penney, J. B., Jr., Vonsattel, J.-P., MacDonald, M. E., Gusella,J. F., and Myers, R. H. (1997) Ann. Neurol. 41, 689–692.
7. Housman, D. E. (1995) Nature Genet. 10, 3–4.8. Burke, J. R., Enghild, J. J., Martin, M. E., Jou, Y.-S., Myers,
R. M., Roses, A. D., Vance, J. M., and Strittmatter, W. J. (1996)Nature Med. 2, 347–350.
9. Koshy, B., Matilla, T., Burright, E. N., Merry, D. E., Fischbeck,K. H., Orr, H. T., and Zoghbi, H. Y. (1996) Hum. Mol. Genet. 5,1311–1318.
10. Li, X.-J., Sharp, A. H., Li, S.-H., Dawson, T. M., Snyder, S. H.,and Ross, C. A. (1996) Proc. Natl. Acad. Sci. USA 93, 4839–4844.
11. Lescure, A., Lutz, Y., Eberhard, D., Jacq, X., Krol, A., Grummt,I., Davidson, I., Chambon, P., and Tora, L. (1994) EMBO J. 13,1166–1175.
12. Jou, Y.-S., and Myers, R. M. (1995) Hum. Mol. Genet. 4, 465–469.13. Green, H. (1993) Cell 74, 955–956.14. Portera-Cailliau, C., Hedreen, J. C., Price, D. L., and Koliatsos,
V. E. (1995) J. Neurosci. 15, 3775–3787.15. Thomas, L. B., Gates, D. J., Richfield, E. K., O’Brien, T. F.,
Schweitzer, J. B., and Steindler, D. A. (1995) Exp. Neurol. 133,265–272.
16. Zeitlin, S., Liu, J. P., Chapman, D. L., Papaioannou, V. E., andEfstratiadis, A. (1995) Nature Genet. 11, 155–163.
17. Ikeda, H., Yamaguchi, M., Sugai, S., Aze, Y., Narumiya, S., andKakizuka, A. (1996) Nature Genet. 13, 196–202.
18. Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp,A. H., Ross, C. A., Scherzinger, E., Wanker, E. E., Mangiarini, L.,and Bates, G. P. (1997) Cell 90, 537–548.
19. Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollen-bach, B., Hasenbank, R., Bates, G. P., Davies, S. W., Lehrach, H.,and Wanker, E. E. (1997) Cell 90, 549–558.
20. Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper,A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Da-vies, S. W., and Bates, G. P. (1996) Cell 87, 493–506.
21. Lorand, L. (1972) Ann. N. Y. Acad. Sci. 202, 6–30.22. Folk, J. E. (1983) Adv. Enzymol. 54, 1–56.23. Greenberg, C. S., Birckbichler, P. J., and Rice, R. H. (1991)
FASEB J. 5, 3071–3077.24. Aeschlimann, D., and Paulsson, M. (1994) Thromb. Haemostasis
71, 402–415.25. Fesus, L., Thomazy, V., Autuori, F., Ceru, M. P., Tarcsa, E., and
Piacentini, M. (1989) FEBS Lett. 245, 150–154.26. Gentile, V., Thomazy, V., Piacentini, M., Fesus, L., and Davies,
P. J. A. (1991) J. Cell Biol. 119, 463–474.
27. Piacentini, M., Davies, P. J. A., and Fesus, L. (1994) in ApoptosisII: The Molecular Basis of Apoptosis in Disease (Tomei, L. D.,and Cope, F. O., Eds.), pp. 143–165, Cold Spring Harbor Labo-ratory Press, Cold Spring Harbor, NY.
28. Melino, G., Annichiarico-Petruzzelli, M., Piredda, L., Candi, E.,Gentile, V., Davies, P. J. A., and Piacentini, M. (1994) Mol. Cell.Biol. 14, 6584–6596.
29. Kahlem, P., Terre, C., Green, H., and Djian, P. (1996) Proc. Natl.Acad. Sci. USA 93, 14580–14585.
30. Cooper, A. J. L., Sheu, K.-F. R., Burke, J. R., Onodera, O.,Strittmatter, W. J., Roses, A. D., and Blass, J. P. (1997) Proc.Natl. Acad. Sci. USA 94, 12604–12609.
31. Laemmli, U. K. (1970) Nature 277, 680–685.32. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular
Cloning: A Laboratory Manual, Cold Spring Harbor LaboratoryPress, Cold Spring Harbor, NY.
33. Porta, R., Gentile, V., Esposito, C., Fusco, A., Peluso, G., andMetafora, S. (1988) Neuropeptides 11, 89–92.
34. Esposito, C., Mancuso, F., Calignano, A., Di Pierro, P., Pucci, P.,and Porta, R. (1995) J. Neurochem. 65, 420–426.
35. Hohenadl, C., Mann, K., Mayer, U., Timpl, R., Paulsson, M., andAeschlimann, D. (1995) J. Biol. Chem. 270, 23415–23420.
36. Grootjans, J. J., Groenen, P. J. T. A., and de Jong, W. W. (1995)J. Biol. Chem. 270, 22855–22858.
37. Porta, R., Gentile, V., Esposito, C., Mariniello, L., and Auricchio,S. (1990) Phytochemistry 29, 2801–2804.
38. Stott, K., Blackburn, J. M., Butler, P. J. G., and Perutz, M. F.(1995) Proc. Natl. Acad. Sci. USA 92, 6509–6513.
39. Porta, R., Esposito, C., Metafora, S., Pucci, P., Malorni, A., andMarino, G. (1988) Anal. Biochem. 172, 499–503.
40. Sawa, A., Khan, A. A., Hester, L. D., and Snyder, S. H. (1997)Proc. Natl. Acad. Sci. USA 94, 11669–11674.
41. Ishitani, R., Sunaga, K., Hirano, A., Saunders, P., Katsube, N.,and Chuang, D.-M. (1996) J. Neurochem. 66, 928–935.
42. Saunders, P. A., Chaleka-Franaszek, E., and Chuang, D.-M.(1997) J. Neurochem. 69, 1820–1828.
43. Perutz, M. F., Johnson, T., Suzuki, M., and Finch, J. T. (1994)Proc. Natl. Acad. Sci. USA 91, 5355–5358.
44. DiFiglia, M., Sapp, E., Chase, K., Schwarz, C., Meloni, A., Young,C., Martin, E., Vonsattel, J.-P., Carraway, R., Reeves, S. A.,Boyce, F. M., and Aronin, N. (1995) Neuron 14, 1075–1081.
45. Trottier, Y., Devys, D., Imbert, G., Saudou, F., An, I., Lutz, Y.,Weber, C., Agid, Y., Hirsch, E. C., and Mandel, J.-L. (1995)Nature Genet. 10, 104–110.
46. Gutekuns, C. A., Levey, A. I., Heilman, C. J., Whaley, W. L., Yi,H., Nash, N. R., Rees, H. D., Madden, J. J., and Hersch, S. M.(1995) Proc. Natl. Acad. Sci. USA 92, 8710–8714.
47. Bessert, D. A., Gutridge, K. L., Dunbar, J. C., and Carlock, L. R.(1995) Mol. Brain Res. 33, 165–173.
48. Singh, U. S., Erickson, J. W., and Cerione, R. A. (1995) Biochem-istry 34, 15863–15871.
49. Arends, M. J., and Wyllie, A. H. (1991) Int. Rev. Exp. Pathol. 32,223–254.
50. Browne, S. E., Bowling, A. C., MacGarvey, U., Baik, M. J.,Berger, S. C., Muqit, M. M. K., Bird, E. D., and Beal, M. F. (1997)Ann. Neurol. 41, 646–653.
51. Beal, M. F. (1992) Ann. Neurol. 31, 119–130.52. Jenkins, B. G., Koroshetz, W. J., Beal, M. F., and Rosen, B. R.
(1993) Neurology 43, 2689–2695.53. Ballestar, E., Abad, C., and Franco, L. (1996) J. Biol. Chem. 271,
18817–18824.
321TISSUE TRANSGLUTAMINASE AND CAG-TRIPLET DISEASES





























