Embed Size (px)
Citation preview

Thyroid Hormone Resistance SyndromeInhibition of Normal Receptor Function by Mutant Thyroid Hormone Receptors
V. Krishna K. Chatterjee, Takashi Nagaya, Laird D. Madison, Shoumen Datta, Anne Rentoumis, and J. Larry JamesonThyroid Unit, Massachusetts General Hospital, Boston, Massachusetts 02114; and Harvard Medical School, Boston, Massachusetts 02114
Introduction
Thyroid hormone (T3) resistance is inherited in most cases in anautosomal dominant manner. The disorder is characterized byelevated free thyroid hormone levels and partial resistance tothyroid hormone at the cellular level. Distinct single amino acidsubstitutions in the ligand binding domain of the ft form of thethyroid hormone receptor have been described in two kindredswith this disorder. Weused transient expression assays to char-acterize the functional properties of these receptor mutants,one containing a Gly to Arg change at amino acid 340 (G340R)and the other a Pro to His change at amino acid 448 (P448H).A nine amino acid carboxy terminal deletion (A448456), analo-gous to an alteration that occurs in v-erbA, was also studied forcomparison with the mutations that occur in the T3 resistancesyndrome. None of the receptor mutants were able to mediatethyroid hormone dependent activation (TreTKCAT) or repres-sion (TSHaCAT) of reporter genes when compared with thewild type receptor. In addition, the mutants inhibited the activ-ity of normal a and (3 receptor isoforms when examined in coex-pression assays. This activity, referred to as dominant negativeinhibition, was manifest with respect to both the positively andnegatively regulated reporter genes. Although mutant receptorbinding to DNAwas unaffected, ligand binding studies showedthat the G340R and A448456 mutants failed to bind T3,whereas the P448H mutant bound hormone with reduced affin-ity (- 10%of normal) compared to the wild type receptor. Con-sistent with this finding, the P448H mutant receptor was par-tially active at higher T3 concentrations. Furthermore, the domi-nant negative inhibition elicited by the P448H receptor mutantat higher T3 concentrations was reversed in the presence of highdoses of T3. These findings indicate that mutant (3 receptors inpatients with thyroid hormone resistance have reduced affinityfor T3 and are functionally deficient, but impair the activity ofnormal receptors, thereby providing a mechanism for the domi-nant mode of inheritance in this disorder. (J. Clin. Invest. 1991.87:1977-1984.) Key words: transcriptional regulation - erbA -
thyroid-stimulating hormone - thyroid hormone resistance * thy-roid hormone
V. K. K. Chatterjee and T. Nagaya contributed equally to this work.Address reprint requests to Dr. J. Larry Jameson, Thyroid Unit,
Jackson 1021, Massachusetts General Hospital, Boston, MA02114.Dr. Chatterjee's present address is Department of Medicine, Universityof Cambridge, Addenbrooke's Hospital, Hills Road, Cambridge,United Kingdom.
Receivedfor publication 8 August 1990 and in revised form 18 De-cember 1990.
The syndrome of generalized thyroid hormone resistance(GTHR)' was first described by Refetoff et al. (1), and is char-acterized by elevated circulating levels of free thyroxine (T4)and triiodothyronine (T3) and inappropriately normal or in-creased levels of thyroid-stimulating hormone (TSH). Al-though patients with GTHRare relatively refractory to themetabolic effects of increased thyroid hormone, there is vari-ability in the degree of resistance in different target tissueswithin an individual and the clinical phenotype varies in differ-ent kindreds (2). An autosomal recessive mode of inheritancewas suggested in original GTHRkindred, but the disorder hasbeen shown to segregate in an autosomal dominant manner inmost families (1, 3).
Although both quantitative (4) and qualitative defects (5) inthyroid hormone binding to fibroblast nuclear receptors fromGTHRpatients have been described, these studies did notfirmly establish a defect at the level of the thyroid hormonereceptor. Recently, two thyroid hormone receptor genes, desig-nated hTRa and hTR#, have been identified, encoding highlyhomologous proteins with different tissue distributions (6, 7).Genetic analysis has now shown linkage between the GTHRsyndrome and the (3 receptor gene locus in three kindreds (8,9).These observations have been extended by sequencing (3 recep-tor cDNAs from two separate families with the disorder. Inboth cases, affected individuals have a point mutation in oneallele of the (3 receptor gene together with a second normalallele, whereas their unaffected relatives have two normal al-leles. In each kindred, the point mutation alters a single aminoacid codon in the carboxy-terminal ligand binding domain ofthe receptor. In one kindred, the mutation is a Glycine to Argi-nine substitution at amino acid 340 (G340R) (10). In the otherkindred, a Proline to Histidine substitution was identified atamino acid 448 (P448H) (9).
Although ligand binding studies have shown that these mu-tant proteins either fail to bind T3 (10) or bind hormone withreduced affinity (11), their ability to modulate target gene ex-pression has yet to be defined. Moreover, because the affectedindividuals also possess a second normal (3 receptor allele andpresumably two normal a receptor alleles, the mechanism bywhich the mutant receptor causes resistance to thyroid hor-mone action is unclear. It has been hypothesized that the mu-tant receptor might block the activity of normal receptors (9,10). In support of this concept, receptor homologues (v-erbA;splicing variant a2) that are unable to bind thyroid hormone
1. Abbreviations used in this paper: ABCD, avidin-biotin complexDNA; CAT, chloramphenicol acetyltransferase; GTHR, generalizedthyroid hormone resistance; K,, receptor affinity constant; RSV, Roussarcoma virus; TRE, thyroid response element; nTRE, negative TRE;TREp, positive TRE.
Dominant Negative Regulation by Thyroid Hormone Receptor Mutants 1977
Abstract
J. Clin. Invest.© The American Society for Clinical Investigation, Inc.0021-9738/91/06/1977/08 $2.00Volume 87, June 1991, 1977-1984

have been shown to inhibit the action of the wild type receptors(12-14).
Transient expression assays provide a convenient model forexamining the functional properties of thyroid hormone recep-tors. Wehave previously used a receptor deficient human cellline (JEG-3) for studies of the functional properties of human aand ,3 receptor isoforms (15, 16). Both types of thyroid hor-mone receptor are capable of activating expression of a re-porter gene (TreTKCAT) that contains a palindromic -positiveTRE (TREp) and repressing the activity of a reporter gene(TSHaCAT) that contains a negative thyroid response element(nTRE) (15, 16). Transcriptional stimulation and repressioneach require receptor activation by thyroid hormone.
In this study, we used this transient assay system to charac-terize the functional properties of the mutant receptors fromtwo GTHRkindreds (G340R and P448H) as well as a carboxy-terminal truncated form of the 1B receptor (A448-456) thatcorresponds to the deletion in v-erbA (13). Wedemonstratethat in addition to being nonfunctional, the receptor mutantsinhibit the action of their wild type receptor counterparts. Thisdominant negative action by the mutant receptors mayexplainthe dominant mode of inheritance as well as resistance to hor-mone action.
Methods
Plasmid constructions and receptor mutagenesis. TSHaCATcontains846 bp, of 5' flanking sequence and 44 bp of exon I from the humanTSH a subunit gene, linked to the gene encoding chloramphenicolacetyltransferase (CAT) (15). TreTKCAT (provided by G. Brent andD. Moore, Massachusetts General Hospital, Boston, MA) contains twocopies of a palindromic TRE (5' gatc TCAGGTCATGACCTGAgatc-3'; linker sequences are shown in small case letters) inserted upstreamof the thymidine kinase (TK) promoter and CATgene (17).
For site directed mutagenesis (18), the human thyroid hormone ftreceptor cDNAwas subcloned into Ml3mpl8. The following oligonu-cleotides were used to direct second strand synthesis: G340R, 5' CAG-CTGAAAAATGGGCGTCTTGGGGTGGTG3', P448H, 5' ACA-GAACTCCTCCCCCATTTGTTCCTGGAA3', ,# A448-456, 5'-TGCCCCACAGAACTCCTCCCCATAGACTGACTGGATTCCT3'. Recombinant phage containing the desired mutations were identi-fied by sequence analysis. Both mutant and wild type receptor cDNAswere then subcloned into a vector in which expression is driven by theRous sarcoma virus (RSV) promoter (14). The mutations are illus-trated schematically in Fig. 1 A.
Cell culture and transient expression assays. JEG-3 cells (HTB 36,American Type Culture Collection, Rockville, MD) were grown inOptimem (BRL-Gibco Laboratories, Grand Island, NY) containing2%(vol/vol) FCS, penicillin (100 U/ml) and streptomycin (100 ug/ml).The cells were trypsinized 18 h before transfection and plated intomedium containing 2%FCSdepleted of T3 by treatment with activatedcharcoal. Triplicate plates of cells were transfected with 5 Mg reporterplasmid (TreTKCAT or TSHaCAT) together with 0.2-4 ug of wildtype or mutant receptor expression vector with the addition ofRSVLUCplasmid as necessary to maintain a constant amount ofRSV-containing plasmid. Following a 16-h exposure to a calciumphosphate-DNA precipitate (19), fresh serum-stripped media wereadded with or without T3. After an additional 48 h, the cells wereharvested and CATenzyme activity was measured by quantitating theacetylation of ["Clchloramphenicol (20). The intraassay variation inCATactivity in triplicate transfections was typically < 10%. AbsoluteCATassay values varied two to threefold in separate experiments, pre-sumably reflecting differences in transfection efficiency. Representa-tive experiments from a single group of transfections are therefore pre-sented, but similar results were obtained in independent experiments.
Name Receptor Strulcture410
13 1 1,,>X1 1T33 456
G3(34(R DAle1-
G 340 R456
1P4481i T3iL~~IP 448 1H
447
A 448-456 T
A 44M-451
+RinCi|1
+
B
TreTKCAT in Ad- mTATAX 3TREp TREp
TSHaCATuf cRECREoTATALnIRE
Figure 1. Structures of wild type and mutant T3 receptors and reportergenes. (A) Schematic representation of human a and j3 receptorisoforms and ft receptor mutants. Putative DNA(open boxes) and T3binding domains are shown schematically. The positions of aminoacid substitutions as well as a nine amino acid deletion in the i re-ceptor are indicated. The T3 binding properties of these receptors aresummarized at the right of the figure (+, normal binding; -, absentbinding; ±, reduced binding affinity). (B) Reporter gene constructsused in transient expression assays. In TreTKCAT, the location oftwo copies of a positive TRE (TREp) is indicated by arrows. InTSHaCAT, the positions of two cAMPresponse elements (CREs)and the putative negative TRE(nTRE, hatched box) are diagrammed.The locations of the TATA box, transcriptional start site, and theCATgene are also indicated for both constructs.
Receptor binding studies. Avidin-biotin complex DNA(ABCD)binding assays were used to assess receptor binding to DNAas de-scribed previously (15). Both wild type and mutant ft receptor cDNAswere subcloned into pGEM7 (Promega Biotec, Madison, WI) for invitro transcription and translation. The transcribed and cappedmRNAswere used to program translation in a rabbit reticulocyte lysatesystem in the presence of [35S]radiolabelled methionine according tothe protocol of the supplier (Promega). Aliquots of labeled protein wereanalyzed using SDSpolyacrylamide gel electrophoresis to confirm re-ceptor integrity and to demonstrate that comparable amounts of thedifferent mutants were synthesized (data not shown). Double strandedTREp (16) or a control oligonucleotide (-72 to -32 bp in the humanTSHa promoter) that does not bind to the thyroid receptor (15) wereannealed, and the 3' recessed ends were filled in with biotin- I l-dUTP(Enzo Biochem, Inc., NewYork, NY), dATP, dCTP, and dGTPusingKlenow polymerase. Biotinylated DNA(500 fmol) was incubated with[35S]methionine labelled receptor (100,000 cpm of TCA precipitableprotein) for 40 min at 240C. The receptor-DNA complexes were precip-itated with streptavidin-agarose beads (Bethesda Research Laborato-ries, Gaithersburg, MD) and quantitated by scintillation counting.
Receptor interactions with DNAwere also analyzed using gel mobil-ity shift assays in which nuclear receptor extracts were prepared fromCOS-I cells (16). COS-l cells (CRL 1650, American Type Culture Col-lection) were transferred to Optimem with 2%charcoal stripped fetalbovine serum before transfection. Cells were transfected with CDM8expression vectors ( 16) encoding human a I, P, ordifferent site-directedmutants of the ,B receptor. After 36 h, cells were harvested and extracts
1978 V. K K Chatterjee, T. Nagaya, L. D. Madison, S. Datta, A. Rentoumis, and J. L. Jameson

were prepared as described by Dammet al. (13). Binding reactions (50Al) contained 32P-labelled TREp(50 fmol) or a control oligonucleotide(-72 to -32 bp in the human TSHa promoter) incubated with trans-fected COScell nuclear extract (7.5 gg protein) in binding buffer (20mMHepes pH 7.9, 120 mMKCI, 2 mMdithiothreitol, 0.1% NP-40,100 ,g/ml poly [d(I-C)J, and 100 nMT3). The DNA-protein mixturewas incubated at room temperature for 60 min before electrophoresisthrough a 5% nondenaturing acrylamide gel.
For hormone binding assays, unlabelled in vitro translation prod-ucts (5 gl) were incubated with 0.01 nM ['25I]T3 and increasingamounts (0.001-0.4 nM) of T3 (21). The reactions were carried out at4°C for 16 h in a final vol of 200 Ml of binding buffer (10% (vol/vol)glycerol, 15 mMTris pH 7.6, 50 mMNaCl, 2 mMEDTA, 1 mMdithiothreitol). Nonsaturable binding was measured in the presence ofa 1,000-fold excess of unlabeled T3. Bound and free hormone was
separated by filtration (HAWP2540; Millipore/Continental WaterSystems, Bedford, MA) (22). Receptor affinity constants (K.) were cal-culated using Scatchard plot analyses.
Results
Functional properties of mutant receptors. To assess the abilityof wild type and mutant receptors to activate or repress tran-scription, TreTKCAT and TSHaCAT(Fig. 1 B) were used asreporter genes to measure transcriptional activation and re-
pression, respectively, in thyroid hormone receptor deficientJEG-3 choriocarcinoma cells. Receptors and mutants were
subcloned into an RSVexpression vector. Using this system,maximal receptor activity is elicited using 100 ng of the recep-tor expression vector and 1 nMT3 (16). In these experiments,supramaximal doses (200 ng) of wild type or mutant receptorand T3 (5 nM) were used to ensure maximal receptor activity.
In the absence of cotransfected receptor, T3 treatment re-pressed TSHaCATactivity (20-50% suppression in differentexperiments) (Fig. 2 B). In other experiments, we have alsoobserved minimal induction of TreTKCAT expression in theabsence of transfected receptor (one to threefold, data notshown). These T3-mediated effects may reflect the presence of
A
1.0*
0.8
0.6
0.4
0.2
0
fl-T3 BTreTKCAT * ;T3
Control G340R P448H A 448-4 56
Receptors
E i-3ISHCATLU Q-3
low amounts of endogenous receptor in these cells. Coexpres-sion of wild type ,3 receptor conferred a marked increase in T3responsiveness. TreTKCAT expression was induced 18-foldand TSHaCATwas suppressed by 79% (Fig. 2 A). Under thesame conditions, the mutant receptors exhibited markedly di-minished activity. The ,B A448-456 mutant failed to mediateactivation of TreTKCAT or repression of TSHaCAT. TheGTHRreceptor mutants (G340R and P448H) were also func-tionally deficient, but differed in their activities. WhereasG340R was essentially inactive (0.9-fold induction of TreTK-CATand 17% repression of TSHaCAT), P448H exhibited par-tial T3 responsiveness (4.2-fold induction of TreTKCAT and35% repression of TSHaCAT).
Dominant negative action of mutant receptors. Having es-tablished the functional capabilities of the individual receptormutants, their ability to modulate the activity of wild type a
and ,3 receptors was examined. Wild type ,B receptor (200 ng)was cotransfected with or without a 10-fold excess (2 ,g) ofeach of the mutant receptors. Under these conditions, the re-ceptor mutants markedly inhibited both positive and negativetranscriptional responses mediated by the wild type receptor(Fig. 3). For example, T3-dependent activation of TreTKCATwas reduced from 73-fold with ,B receptor alone to 21-fold and24-fold in the presence of coexpressed G340R or P448H, re-spectively. The A448-456 mutant was more potent and re-duced activation to sixfold. A similar pattern of results wasobtained with respect to negative regulation using TSHaCAT.Repression of TSHaCATactivity in the presence of wild typereceptor alone (80%) was blunted to 16% by G340R, 50% byP448H, and repression was eliminated using A448-456.
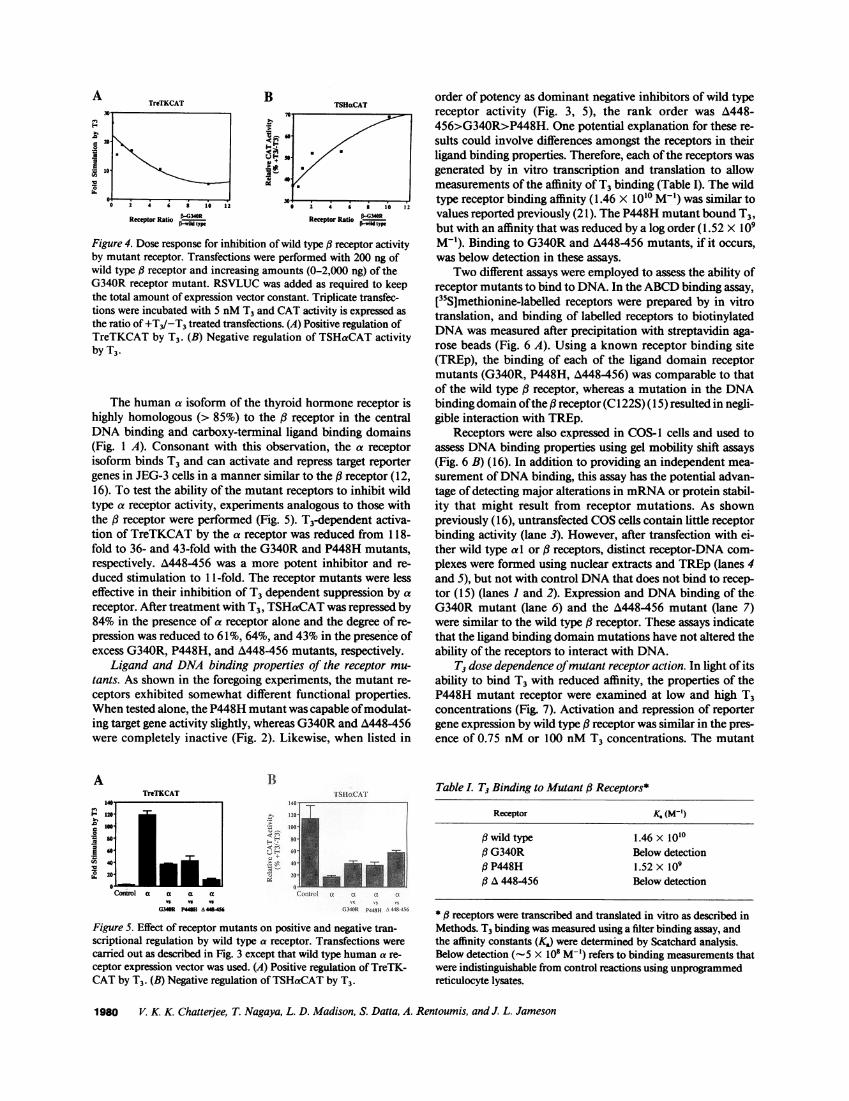
Dose response studies were carried out to determine theamount of mutant receptor expression vector required to in-hibit wild type receptor activity. Increasing concentrations ofthe G340R mutant receptor progressively reduced transcrip-tional activation of TreTKCAT from 22- to 7-fold (Fig. 4 A).Similarly, the degree of repression of TSHaCATwas reducedfrom 62%with wild type receptor alone to 30%with a 10-foldexcess of G340R(Fig. 4 B). In both cases, half-maximal inhibi-tion of wild type receptor activity occurred using a four tofivefold excess of mutant receptor.
A B
40'4 40-
20-
_ I0
__0.53xlIM20 Cuntru 0.3(3 0 .644H5x 04#98X
Recepturs~~O.
Figure 2. Functional properties of the wild type and mutant ,B recep-tors. Control plasmid (RSVLUC) or RSVexpression vector (200 ng)encoding either wild type or mutant fl receptors were transfected intoJEG-3 cells with 5 ug of the indicated TreTKCAT or TSHaCATreporter genes. Triplicate transfections were incubated in either theabsence (dark bars) or presence (hatched bars) of 5 nM T3. CATactivity is expressed as percent chloramphenicol acetylation per mil-liliter of cell extract per hour. The data shown is the mean±SDoftriplicate transfections. Relative stimulation or repression in the pres-ence of T3 treatment is indicated above the bars. (A) Positive regula-tion of TreTKCAT by T3. (B) Negative regulation of TSHaCATby T3.
TreTKCAT TSHcZCAT
Control 0Vs Vs AsG;34OR P448H A 448456
s vs * s
C;340R P44RH A 449-456
Figure 3. Effect of receptor mutants on positive and negative tran-scriptional regulation by wild type 6 receptor. JEG-3 cells were trans-fected with 5 gg of TreTKCAT or TSHaCATtogether with either2,200 ng of RSVLUC(Control), or 200 ng of ,B receptor and a 10-foldexcess (2,000 ng) of RSVLUCor mutant # receptors. Triplicatetransfections were incubated in either the absence or presence of 5nMT3 and the results are presented as the relative activity of +T3/-T3 treated plates. (A) Positive regulation of TreTKCAT by T3. (B)Negative regulation of TSHaCATby T3.
Dominant Negative Regulation by Thyroid Hormone Receptor Mutants 1979
17.9x
1L. 0.9x 4.2x lOx
TreTKCAT B
7'.
S +
A:i i 6 8 10 12
Receptor Ratio P-G3 aR
TSHaCAT
*/
0 2 4 8 i 10
Receptor Ratio P-G34uRA-wlldtype
12
Figure 4. Dose response for inhibition of wild type ,3 receptor activityby mutant receptor. Transfections were performed with 200 ng ofwild type # receptor and increasing amounts (0-2,000 ng) of theG340R receptor mutant. RSVLUCwas added as required to keepthe total amount of expression vector constant. Triplicate transfec-tions were incubated with 5 nMT3 and CATactivity is expressed asthe ratio of +T3/-T3 treated transfections. (A) Positive regulation ofTreTKCAT by T3. (B) Negative regulation of TSHaCATactivityby T3.
The human a isoform of the thyroid hormone receptor ishighly homologous (> 85%) to the ,3 receptor in the centralDNAbinding and carboxy-terminal ligand binding domains(Fig. 1 A). Consonant with this observation, the a receptorisoform binds T3 and can activate and repress target reportergenes in JEG-3 cells in a manner similar to the ft receptor (12,16). To test the ability of the mutant receptors to inhibit wildtype a receptor activity, experiments analogous to those withthe f3 receptor were performed (Fig. 5). T3-dependent activa-tion of TreTKCAT by the a receptor was reduced from 118-fold to 36- and 43-fold with the G340R and P448H mutants,respectively. A448-456 was a more potent inhibitor and re-duced stimulation to 11-fold. The receptor mutants were lesseffective in their inhibition of T3 dependent suppression by areceptor. After treatment with T3, TSHaCATwas repressed by84% in the presence of a receptor alone and the degree of re-pression was reduced to 61%, 64%, and 43% in the presence ofexcess G340R, P448H, and A448-456 mutants, respectively.
Ligand and DNA binding properties of the receptor mu-tants. As shown in the foregoing experiments, the mutant re-ceptors exhibited somewhat different functional properties.Whentested alone, the P448H mutant was capable of modulat-ing target gene activity slightly, whereas G340Rand A448-456were completely inactive (Fig. 2). Likewise, when listed in
order of potency as dominant negative inhibitors of wild typereceptor activity (Fig. 3, 5), the rank order was A448-456>G340R>P448H. One potential explanation for these re-sults could involve differences amongst the receptors in theirligand binding properties. Therefore, each of the receptors wasgenerated by in vitro transcription and translation to allowmeasurements of the affinity of T3 binding (Table I). The wildtype receptor binding affinity (1.46 X 10" M-') was similar tovalues reported previously (21). The P448H mutant bound T3,but with an affinity that was reduced by a log order (1.52 X 109M-l). Binding to G340R and A448-456 mutants, if it occurs,was below detection in these assays.
Two different assays were employed to assess the ability ofreceptor mutants to bind to DNA. In the ABCDbinding assay,[35S]methionine-labelled receptors were prepared by in vitrotranslation, and binding of labelled receptors to biotinylatedDNAwas measured after precipitation with streptavidin aga-rose beads (Fig. 6 A). Using a known receptor binding site(TREp), the binding of each of the ligand domain receptormutants (G340R, P448H, A448-456) was comparable to thatof the wild type A receptor, whereas a mutation in the DNAbinding domain of the # receptor (C 1 22S) (15) resulted in negli-gible interaction with TREp.
Receptors were also expressed in COS-1 cells and used toassess DNAbinding properties using gel mobility shift assays(Fig. 6 B) (16). In addition to providing an independent mea-surement of DNAbinding, this assay has the potential advan-tage of detecting major alterations in mRNAor protein stabil-ity that might result from receptor mutations. As shownpreviously (16), untransfected COScells contain little receptorbinding activity (lane 3). However, after transfection with ei-ther wild type a 1 or is receptors, distinct receptor-DNA com-plexes were formed using nuclear extracts and TREp (lanes 4and 5), but not with control DNAthat does not bind to recep-tor (15) (lanes I and 2). Expression and DNAbinding of theG340R mutant (lane 6) and the A448-456 mutant (lane 7)were similar to the wild type # receptor. These assays indicatethat the ligand binding domain mutations have not altered theability of the receptors to interact with DNA.
T3 dose dependence of mutant receptor action. In light of itsability to bind T3 with reduced affinity, the properties of theP448H mutant receptor were examined at low and high T3concentrations (Fig. 7). Activation and repression of reportergene expression by wild type ,3 receptor was similar in the pres-ence of 0.75 nM or 100 nM T3 concentrations. The mutant
TreTKCATB
TSHaCAT
a a a a
G340R P44H Abe84
Control at a to
3400R 1'4481[ A448-456
Figure 5. Effect of receptor mutants on positive and negative tran-scriptional regulation by wild type a receptor. Transfections werecarried out as described in Fig. 3 except that wild type human a re-
ceptor expression vector was used. (A) Positive regulation of TreTK-CATby T3. (B) Negative regulation of TSHaCATby T3.
Table I. T3 Binding to Mutant # Receptors*
Receptor K. (M-')
iBwild type 1.46 x 1010, G340R Below detection,BP448H 1.52 x 109AB/ 448-456 Below detection
* is receptors were transcribed and translated in vitro as described inMethods. T3 binding was measured using a filter binding assay, andthe affinity constants (K.) were determined by Scatchard analysis.Below detection (- 5 x 10 M-') refers to binding measurements thatwere indistinguishable from control reactions using unprogrammedreticulocyte lysates.
1980 V. K K. Chatterjee, T. Nagaya, L. D. Madison, S. Datta, A. Rentoumis, and J. L. Jameson
A
I-
0 24-
ca,q 10.
W
I:-o
A
Pto1..au-
A
1,t:
-
.-, n6
;;i
W +
el.
7;:x

A_, 2000- 001
00-
LULControl T~
B
:7 0-~ ~~~00 $
$10 $,oss 9
lRECEPTOR al 1t 1 10 o
and mutant receptors were transfected inllevels of expression. Nuclear extracts contreceptors were analyzed in gel mobility shilabeled TREp (lanes 3-7) or a control DI}TSHa promoter (-72 to -32 bp) that doeI and 2). Expressed receptors (al, fl, G34(at the top of the gel. An extract from untrnas none (lane 3). Because it had not beevector, mutant P448H was omitted fromtions of free DNAand receptor-bound DPwith arrows.
P448H was only weakly active (fourfCAT, 40% repression of TSHaCAT)tion, but became fully functional at ition. The apparent increase in the actant relative to the wild type (3 receptor
A * Low T3TreTKCA r * High T3
60-
Control P445H
P44811
B
tl
u
<Rpi.r .1
lu .-
'Z e,2z
-
Figure 7. Effect of T3 concentration on mudominant negative activity. Transfections i
scribed in previous figures except that the c
ther low (0.75 nM) or high (100 nM) T3 ccregulation of TreTKCAT by T3. (B) NegTSHaCATby T3.
Figure 6. Expressionand DNAbinding prop-erties of mutant ft re-ceptors. (A) ABCDre-ceptor-DNA binding as-says. In vitro translated,3S methionine-labeledwild type (3 or (3 receptormutants (G340R,P448H, A448-456,C122S) were bound tobiotinylated TREp and
j the receptor-DNA com-plexes precipitated withstreptavidin agarosebeads. Results are themean±SD of triplicateassays. Backgroundbinding in the absenceof added DNAwas sub
observed in other experiments. Thus, although P448H bindsT3 with reduced affinity, it appears to function normally in thepresence of saturating doses of hormone. As expected basedupon the T3 binding data, the reduced activity of mutantsG340Rand A448-456 was unaffected by increasing the dose ofT3 (data not shown).
The dominant negative effects of the P448H receptor mu-tant were also dependent on T3 concentration (Fig. 7). TreTK-CATactivation by wild type (3 receptor was reduced from 25-fold to 4-fold by mutant P448H at 0.75 nM T3, but TreTK-CAT activation was not inhibited at 100 nM T3. Similarly,repression of TSHaCAT activity was effectively blocked bymutant P448H at a low T3 concentration, but full repressionwas retained at the high T3 concentration.
Discussion
%J1Q%%A%%AA-11I-1'"a au_ In this study, we have characterized the functional properties of
binctdigBidn toail peiimutant thyroid hormone receptors (G340R and P448H) that
control. BidinA frgetoa
were identified previously in two separate kindreds with(human TSHa pro- GTHRsyndrome (9, 10). Several main conclusions can bemoter -72 to -32 bp) drawn from these studies. First, the mutant receptors bind T3that does not bind re- with greatly reduced affinity and their ability to transcription-ceptor with high affinity ally activate or repress target genes in response to T3 is thereforeis indicated as control impaired. This observation is in keeping with previous studiesDNA. (B) Gel mobility showing T3 receptor isoforms (e.g., v-erbA, a2) that cannotshift assays. Wild ty'le bind T3 are transcriptionally inactive (1 2, 13, 16) and empha-
to COS-lI cells to attain high siethfattatrncitoargutonsa gndeenaining transiently expressed siethfattatrncitoareutonsalgndep-ift assays using either 32p_ dent process. Second, the different receptor mutants have dis-'IA fr-agment derived human tinct functional properties. Whereas G340Rand A448-456 ex-cs not bind receptors (lanes hibit no T3 binding and are completely inactive in transfectionOR, A448-456) are indicated assays, P448H binds T3 with reduced affinity, and conse-ansfected cells is denoted quently is partially active in response to T3. Third, we find thatnsublonedinto CDM8 the mutant receptors inhibit the transcriptional activity of the
this experiment. The loca- wild type thyroid hormone receptor. Notably, the inhibition byNIA complexes are indicated mutant receptors applies to the activity of both (3 and a recep-
tors and is manifest using both positively and negatively regu-lated target genes.
Two major mechanisms might be considered to explain theold activation of TreTK- fact that GTHRcan be inherited in an autosomal dominantat the low T3 concentra- manner. Given that the mutant receptors are transcriptionallythe hger T3 concentra- inactive, one possibility is that the amount of functional recep-,tivity of the P448H mu- tor is reduced as a consequence of having the mutation occur inr at high T3 doses, was not one of two autosomal alleles. Alternatively, it is possible that
the mutant receptor in some manner interferes with the func-tion of normal receptors, thereby accounting for its phenotypic
TSHOOCAT of gT3 expression in the heterozygous state. The receptor deficiency80 ~~~~~mechanism assumes that the quantity of functional receptor60 ~~~~~~within the cell is a limiting step for hormone action and that the
normal allele is not upregulated to compensate for the mutant40 ~~~~~~allele. There is currently no evidence that excludes this mecha-20 ~~~~~nism. However, the observation that mutant receptors can
block the transcriptional regulation of two different reporterControl 3P448H genes supports an interference mechanism as the basis for resis-
P44~8H tance to thyroid hormone action in heterozygotes.itant receptor function and Given the apparent complexity of transcriptional regula-were carried out as de- tion by thyroid hormone receptors, it is useful to consider po-cells were treated with ei- tential cellular mechanisms by which mutant receptors couldncentrations. (A) Positive inhibit wild type receptors. One mechanism could involve theative regulation of formation of nonfunctional heterodimers between wild type
and mutant receptor proteins (Fig. 8, model 1). This type of
Dominant Negative Regulation by Thyroid Hormone Receptor Mutants 1981

Figure 8. Potential mecha-nisms for dominant negativeinhibition. Three potentialmodels for inhibition by ligandbinding domain (LBD) mu-tants of T3 receptor action are
presented. Wild type receptors(WT) can bind T3, whereasmutant receptors (Mut) areunable to bind T3. The forma-tion of homodimers and het-erodimers of wild type andmutant receptors is hypotheti-cal. Model 1 suggests that mu-tant receptor could form non-functional heterodimers withthe wild type receptor to de-
plete the amount of residualactive receptor. Model 2 sug-gests that mutant receptorscould block wild type activityby competing for binding toa common target TREse-
quence. Model 3 indicates amechanism in which function-
ally inactive receptors interactwith and deplete a limitingcellular factor (X) that is re-
quired for the function of normal receptors. The formation of dimersis shown, but is not required to invoke mechanisms 2 or 3. Thesemodels are not mutually exclusive and it is possible that dominantnegative inhibition involves several different mechanisms.
mechanism has been documented with other DNAbindingtranscription factors. For example, in the CREB/ATF familyof transcription factors, homodimers and heterodimers formvia a conserved leucine zipper motif (23) and mutant CREBproteins function in a dominant negative manner by dimeriz-ing with the wild type protein to inhibit transcriptional capabil-ity (24). Another such example is provided by the Id proteinwhich inhibits the transcriptional activity of the muscle differ-entiation factor MyoDby forming heterodimers via a sharedhelix-loop-helix (25). In the case of thyroid hormone receptors,deletion mutants of the chicken a receptor repress wild typereceptor function and this inhibitory property has been local-ized to the carboxy-terminal domain that may contain a dimer-ization interface (14, 26). In support of this concept, the thy-roid hormone receptor has recently been shown to form heter-odimers with the. retinoic acid receptor via a shared structuralmotif within the carboxy-terminal region (27). Both of theGTHRreceptor mutations occur outside of this putative di-merization domain, raising the possibility that they could stillform dimers. Further structure-function analyses within thisregion will be required to precisely delineate the receptor se-
quences that are required for receptor-receptor interactions.A second mechanism for dominant negative activity would
be for the transcriptionally defective mutants to compete withthe wild type receptor for binding to a commonDNAtargetsequence, which in this case is the TRE (Fig. 8, model 2). Thefinding that a mutation in the DNAbinding domain of theviral oncogene, v-erbA, abolishes its dominant negative proper-ties, lends support to this type of mechanism (13). The intro-duction of DNAbinding domain mutations into the GTHR
receptors may provide further insight regarding the contribu-tion of this mechanism to their dominant negative activity. It isinteresting to note that in another receptor resistance syn-drome (hereditary resistance to vitamin D), the disorder is in-herited in an autosomal recessive manner and heterozygotesare phenotypically normal (28). In this instance, the mutationsoccur within the DNAbinding domain of the vitamin D recep-tor, consistent with the hypothesis that binding to DNAmayberequired for receptors to exert inhibitory activity in the hetero-zygous state.
The mutant receptors could also inhibit the action of nor-mal receptors by sequestering a shared accessory factor that isnecessary for wild type receptor modulation of gene transcrip-tion (Fig. 8, model 3). This property, sometimes referred to as
"squelching" (29), has been postulated to explain transcrip-tional interference between coexpressed estrogen and proges-terone receptors (30), and recent analyses of transcriptionalregulation by the adenovirus Ela protein also support this con-cept (31). Because inhibition by the GTHRand A448-456 mu-
tants occurs with relatively low doses of coexpressed mutantreceptors, we do not favor such a mechanism for these mu-
tants, but it cannot be excluded at this point. These three po-tential pathways for transcriptional interference by the mutantreceptors are not mutually exclusive and inhibition may wellresult from a combination of several different mechanisms.
In many respects, the dominant negative properties that wehave observed with the GTHRreceptor mutants are analogousto the findings with other transcriptionally inactive thyroidhormone receptor variants (12, 13, 16). For example, both thev-erbA oncogene and the a2 isoform have been shown to act as
dominant negative inhibitors when coexpressed with the wildtype receptor in transient expression assays. The GTHRmu-
tants, v-erbA, and a2 receptors share in commonthe fact thatthey contain sequence alterations in the extreme carboxy ter-minus of the receptor. Although these alterations eliminate orgreatly reduce hormone binding in each of these mutants, itmay be premature to conclude that this is the only modifica-tion in receptor function that is required for dominant negativeactivity. For example, the carboxy-terminal nine amino aciddeletion that occurs in v-erbA has been proposed to serve as a
transactivation domain (32). Thus, its deletion could poten-tially eliminate both T3 binding and the potential for transacti-vation. As suggested in Fig. 8, the activity of heterodimersformed between v-erbA and a normal cellular receptor couldpotentially be influenced by the presence or absence of a trans-activation domain within v-erbA. This feature might accountfor the somewhat greater inhibition that is observed with thev-erbA homologue (A448-456) than with the GTHRmutants(G340R, P448H) (Figs. 3 and 5), or with the a2 isoform (16).
If an interference mechanism is the basis for thyroid hor-mone resistance in the heterozygous state, then in the simplestcase it might be proposed that the normal and mutant alleleswould be expressed equally resulting in a 1 :1 ratio of normaland mutant receptor. The fact that mutant and wild type se-
quences comprise a similar number of isolated cDNA clones(10) provides some data to support this assumption. However,several lines of evidence suggest that the situation in vivo maybe complex. For example, T3 can regulate the expression of itsown receptors (33, 34) and the impact of thyroid hormoneresistance on this process is currently unknown. Furthermore,the levels of expressed a and ,B receptors varies widely in differ-ent tissues (34, 35) and a number of receptor splicing variants
1982 V. K. K Chatterjee, T. Nagaya, L. D. Madison, S. Datta, A. Rentoumis, and J. L. Jameson
Potential NMechlanismis for IB Nititantsto inhihit T3 RLectjeptor Actiiti
Moldel IInhib itioi Sit frt il'i- in iSnolt o' i litatI i WIele. t-od im' 's:
d ep ili 'r or I-cce ')t 9
Aloded21ufbliirth ,1miill of, i nadc 's ti tan~ ceptons:orn peli ti on Forricc crplor-1 5i ndr~ig Io 1)NA
j.
W Il~ts_..
Model .1ni itio b (11intio roh-inirrd Itransactinl Iactoi-s
'1
IX ,itIINIXI

have been recognized for both forms of receptor (36-38). Thus,if the mutation occurs in one of the ( receptor genes, the degreeof resistance may reflect the composition of receptor isoformswithin a given cell as well as compensatory responses of thevarious receptor isoforms to the T3 resistant state. Future stud-ies of receptor expression and regulation in vivo may help toclarify the relative proportions of different receptor isoforms.
Weexamined the issue of the stoichiometry of mutant re-ceptor inhibition in vitro by altering the ratio of mutant to wildtype receptors in transient expression assays (Fig. 4). Althoughsome degree of dominant negative activity was apparent at lowmutant/wild type receptor ratios (1: 1 or 2:1), it was somewhatsurprising that 50% inhibition required at least a fourfold ex-cess of mutant receptor. The basis for the requirement for ex-cess mutant receptor and its relevance to clinical thyroid hor-mone resistance are unclear. It is possible that clinical measure-ments of resistance (e.g., elevated thyroid hormone levels) aremore sensitive indicators of receptor inhibition than our invitro assays of transient gene expression. A more likely basis forthe requirement for excess mutant receptor may involve theartificial nature of the transient expression assay. Although thepredicted ratios of expressed receptor are likely to correlatewith the amounts of transfected plasmid, we do not have aquantitative measure of the expressed receptor proteins. Basedupon their levels of expression in transfected COScells (Fig. 6B), there is some evidence that the stabilities of the mutant andwild type receptor mRNAsand proteins are similar in trans-fected cells. It is possible that quantitative differences in domi-nant negative activity would be found under different condi-tions such as performing the experiments at a lower or higherlevel of expressed reporter gene or using a different level of wildtype receptor. In addition, the current studies have been per-formed in only a single receptor deficient cell line (JEG-3 cells).A number of recent reports indicate that T3 receptors interactwith other cellular proteins (27, 39-41) and the levels and com-positions of such factors may alter dominant negative regula-tion in a cell type specific manner. One must also consider thatthe inhibitory effects of the mutant receptors may be depen-dent upon the target gene for thyroid hormone receptor action.For example, coexpressed thyroid hormone and retinoic acidreceptors act cooperatively to induce the expression of a targetgene containing a palindromic TRE, but inhibit the expressionof a gene containing TREs from the myosin heavy chain gene(27). Because of these issues, it is apparent that one shouldinterpret transient receptor assays of receptor functioncautiously, particularly with respect to quantitative effects. De-spite these reservations, this technique appears to provide avaluable in vitro assay for the transcriptional activity of recep-tor mutants and to serve as an index of dominant negativeactivity.
The observation that different kindreds with GTHRdis-play distinct clinical features is a particularly intriguing aspectof this disorder (2, 3, 9). Wehave shown that the receptormutants from two kindreds differ in their functional proper-ties. At a low T3 concentration, the properties of P448H andG340Rare essentially indistinguishable in vitro. However, at aT3 concentration which saturates P448H, this mutant is tran-scriptional active and loses its dominant negative activity. How-ever, we note that individuals with the P448H mutation (9) andthe G340Rmutation (10) have similar circulating thyroid hor-mone levels and the clinical features of resistance are different,but not necessarily less severe, in individuals with the P448H
mutation. Thus, it is unclear at this stage whether differences inligand affinity or some other aspect of mutant receptor func-tion account for phenotypic differences in this disorder. Tobetter understand the clinical features that result from thyroidhormone resistance, it will be necessary to acquire more infor-mation about the in vivo distribution, regulation, and mecha-nisms of action of normal and mutant receptors. Recent stud-ies (9, 42) suggest that a number of different mutations mayexist in other kindreds with GTHR. Analyses of their func-tional properties will be of interest to further elucidate thepathophysiology of this disorder.
Acknowledgments
Wethank G. Brent and D. Moore for providing the plasmid TreTK-CAT, R. Evans for the human # receptor cDNA, and L. J. DeGroot forthe a receptor cDNA.
This work was supported in part by Public Health Service grantDK42144 to J. L. Jameson. V. K. K. Chatterdee is a Wellcome TrustSenior Clinical Fellow.
References
1. Refetoff, S., L. T. DeWind, and L. J. DeGroot. 1967. Familial syndromecombining deaf-mutism, stippled epiphyses, goiter and abnormally high PBI:possible target organ refractoriness to thyroid hormone. J. Clin. Endocrinol. &Metab. 27:279-294.
2. Magner, J. A., P. Petrick, M. M. Menezes-Ferreira, M. Stelling, and B. D.Weintraub. 1986. Familial generalized resistance to thyroid hormones: report ofthree kindreds and correlation of affected tissues with the binding of '25I triiodo-thyronine to fibroblast nuclei. J. Endocrinol. Invest. 9:459-470.
3. Refetoff, S. 1982. Syndromes of thyroid hormone resistance. Am. J. Phys-iol. 243:E88-E98.
4. Ichikawa, K., I. A. Hughes, A. L. Horwitz, and L. J. DeGroot. 1987. Charac-terization of nuclear thyroid hormone receptors of cultured skin fibroblasts frompatients with resistance to thyroid hormone. Metab. Clin. Exp. 36:392-399.
5. Menezes-Ferreira, M. M., C. Eil, J. Wortsman, and B. D. Weintraub. 1984.Decreased uptake of '25I triiodo-l-thyronine in fibroblasts from patients withperipheral thyroid hormone resistance. J. Clin. Endocrinol. & Metab. 59:1081-1087.
6. Weinberger, C., C. C. Thompson, E. S. Ong, R. Lebo, D. J. Gruol, andR. M. Evans. 1986. The c-erbA gene encodes a thyroid hormone receptor. Nature(Lond.). 324:641-646.
7. Nakai, A., A. Sakurai, G. I. Bell, and L. J. DeGroot. 1988. Characterizationof a third human thyroid hormone receptor co-expressed with other thyroidhormone receptors in several tissues. Mol. Endocrinol. 2:1087-1092.
8. Usala, S. J., A. E. Bale, N. Gesundheit, C. Weinberger, R. W. Lash, F. E.Wondisford, 0. W. McBride, and B. D. Weintraub. 1988. Tight linkage betweenthe syndrome of generalized thyroid hormone resistance and the human c-erbA,3gene. Mol. Endocrinol. 2:1217-1220.
9. Usala, S. J., G. E. Tennyson, A. E. Bale, R. W. Lash, N. Gesundheit, F. E.Wondisford, D. Accili, P. Hauser, and B. D. Weintraub. 1990. A base mutation ofthe c-erbAft thyroid hormone receptor in a kindred with generalized thyroidhormone resistance: molecular heterogeneity in two other kindreds. J. Clin. In-vest. 85:93-100.
10. Sakurai, A., K. Takeda, K. Ain, P. Ceccarelli, A. Nakai, S. Seino, G. I. Bell,S. Refetoff, and L. J. DeGroot. 1989. Generalized resistance to thyroid hormoneassociated with a mutation in the ligand-binding domain of the human thyroidhormone receptor ,B. Proc. Nat/. Acad. Sci. USA. 86:8977-8981.
11. Usala, S. J., F. E. Wondisford, T. L. Watson, J. B. Menke, and B. D.Weintraub. 1990. Thyroid hormone and DNAbinding properties of a mutantc-erbA# receptor associated with generalized thyroid hormone resistance. Bio-chem. Biophys. Res. Commun. 171:575-580.
12. Koenig, R. J., M. A. Lazar, R. A. Hodin, G. A. Brent, P. R. Larsen, W. W.Chin, and D. D. Moore. 1989. Inhibition of thyroid hormone action by a non-
hormone binding c-erbA protein generated by alternative mRNAsplicing. Na-ture (Lond.). 337:659-661.
13. Damm,K., C. C. Thompson, and R. M. Evans. 1989. Protein encoded byv-erbA functions as a thyroid hormone receptor antagonist. Nature (Lond.).339:593-596.
14. Forman, B. M., C. Yang, M. Au, J. Casanova, J. Ghysdael, and H. H.Samuels. 1989. A domain containing a leucine-zipper like motif mediates novelin vivo interactions between the thyroid hormone and retinoic acid receptors.Mol. Endocrino/. 3:1610-1626.
Dominant Negative Regulation by Thyroid Hormone Receptor Mutants 1983

15. Chatterjee, V. K. K, J-K. Lee, A. Rentoumis, and J. L. Jameson. 1989.Negative regulation of the thyroid-stimulating hormone a gene by thyroid hor-mone: receptor interaction adjacent to the TATA box. Proc. Nati. Acad. Sci.USA. 86:9114-9118.
16. Rentoumis, A., V. K. K. Chattedjee, L. D. Madison, S. Datta, G. D.Gallagher, L. J. DeGroot, and J. L. Jameson. 1990. Negative and positive tran-scriptional regulation by thyroid hormone receptor isoforms. Mol. Endocrinol.4:1522-1531.
17. Brent, G. A., J. W. Harney, Y. Chen, R. L. Warne, D. D. Moore, and P. R.Larsen. 1989. Mutations of the rat growth hormone promoter which increase anddecrease response to thyroid hormone define a consensus thyroid response ele-ment. Mol. Endocrinol. 3:1996-2004.
18. Kunkel, T. A. 1985. Rapid and efficient site-specific mutagenesis withoutphenotypic selection. Proc. Nati. Acad. Sci. USA. 82:488-492.
19. Graham, B., and A. van der Eb. 1973. A new technique for the assay ofinfectivity of human adenovirus DNA. Virology. 52:456-467.
20. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinantgenomes which express chloramphenicol acetyltransferase in mammalian cells.Mol. Cell. Biol. 2:1044-1051.
21. Scheuler, P. A., H. L. Schwartz, K. A. Strait, C. N. Mariash, and J. H.Oppenheimer. 1990. Binding of 3,5,3'-triiodothyronine (T3) and its analogs tothe in vitro translational products of the c-erbA protooncogenes: differences inthe affinity of the a and j5 forms for the acetic acid analog and failure of thehuman testis and kidney a-2 products to bind T3. Mol. Endocrinol. 4:227-234.
22. Inoue, A., J. Yamakawa, M. Yukioka, and S. Morisawa. 1983. Filter-binding assay procedure for thyroid hormone receptors. Anal. Biochem.134:176-183.
23. Abel, T., and T. Maniatis. 1989. Action of leucine zippers. Nature (Lond.).341:24-25.
24. Dwarki, V. J., M. Montminy, and I. M. Verma. 1990. Both the basicregion and the leucine zipper domain of CREBprotein are essential for transcrip-tional activation. EMBO(Eur. Mol. Biol. Organ.) J. 9:225-232.
25. Benezra, R., R. L. Davis, D. Lockshon, D. L. Turner, and H. Weintraub.1990. The protein Id: a negative regulator of helix-loop-helix DNAbinding pro-teins. Cell. 61:49-59.
26. Forman, B. M., and H. H. Samuels. 1990. Minireview: interactionsamong a subfamily of nuclear hormone receptors: the regulatory zipper model.Mol. Endocrinol. 4:1293-1301.
27. Glass, C. K., S. M. Lipkin, 0. V. Devary, and M. G. Rosenfeld. 1989.Positive and negative regulation of gene transcription by a retinoic acid-thyroidhormone receptor heterodimer. Cell. 59:697-708.
28. Hughes, M. R., P. J. Malloy, D. G. Kieback, R. A. Kesterson, J. W. Pike,D. Feldman, and B. W. O'Malley. 1988. Point mutationsin the human vitamin D
receptor gene associated with hypocalcemic rickets. Science (Wash. DC).242: 1702-1705.
29. Ptashne, M. 1988. Howeukaryotic transcriptional activators work. Na-ture (Lond.). 335:683-689.
30. Meyer, M., H. Gronemeyer, B. Turcotte, M. Bocquel, D. Tasset, and P.Chambon. 1989. Steroid hormone receptors compete for factors that mediatetheir enhancer function. Cell. 57:433-442.
31. Martin, K. J., J. W. Lillie, and M. R. Green. 1990. Evidence for interac-tion of different eukaryotic transcriptional activators with distinct cellulartargets.Nature (Lond.). 346:147-152.
32. Zenke, M., A. Munoz, J. Sap, B. Vennstrom, and H. Beug. 1990. v-erbAoncogene activation entails the loss of hormone-dependent regulator activity ofc-erbA. Cell. 61:1035-1049.
33. Lazar, M. A., and W. W. Chin. 1988. Regulation of two c-erbA messengerribonucleic acids in rat GH3cells by thyroid hormone. Mol. Endocrinol. 2:479-484.
34. Hodin, R. A., M. A. Lazar, B. J. Wintman, D. S. Darling, R. J. Koenig,P. R. Larsen, D. D. Moore, and W. W. Chin. 1989. Identification of a thyroidhormone receptor that is pituitary specific. Science (Wash. DC). 244:76-79.
35. Sakurai, A., A. Nakai, and L. J. DeGroot. 1989. Expression of three formsof thyroid hormone receptor in human tissues. Mol. Endocrinol. 3:392-399.
36. Mitsuhashi, T., G. E. Tennyson, and V. M. Nikodem. 1988. Alternativesplicing generates messages encoding rat c-erbA proteins that do not bind thyroidhormone. Proc. NatL Acad. Sci. USA. 85:2781-2785.
37. Izumo, S., and V. Mahdavi. 1988. Thyroid hormone receptor a isoformsgenerated by alternative splicing differentially activate myosin HCgene transcrip-tion. Nature (Lond.). 334:539-542.
38. Yaoita, Y., Y. Shi, and D. D. Brown. 1990. Xenopus laevis a and P thyroidhormone receptors. Proc. Natl. Acad. Sci. USA. 87:7090-7094.
39. Murray, M. B., and H. C. Towle. 1989. Identification of nuclear factorsthat enhance binding of the thyroid hormone receptor to a thyroid hormoneresponse element. Mol. Endocrinol. 3:1434-1442.
40. Burnside, J., D. S. Darling, and W. W. Chin. 1990. A nuclear factor thatenhances binding of thyroid hormone receptors to thyroid hormone responseelements. J. Biot. Chem. 265:2500-2504.
41. Lazar, M. A., and T. J. Berrodin. 1990. Thyroid hormone receptors formdistinct nuclear protein-dependent and independent complexes with a thyroidhormone response element. Mol. Endocrinol. 4:1627-1635.
42. Takeda, K., S. Balzano, A. Sakurai, L. J. DeGroot, and S. Refetoff. 1990.Screening of 19 unrelated families with generalized resistance to thyroid hormone(GRTH) for known point mutations in the thyroid receptor (TR) P5 and thedetection of a new mutation. The 72nd Annual Meeting of The Endocrine Soci-ety, 20-23 June 1990, Atlanta, GA. (3):25 (Abstr.)
1984 V. K K Chatterjee, T. Nagaya, L. D. Madison, S. Datta, A. Rentoumis, and J. L. Jameson