Embed Size (px)
Citation preview

Theory of resonance Raman scattering and fluorescence from strongly vibronically coupled excited states of polyatomic molecules
G. Stock and W. Domcke Institut/ur Physikalische und Theoretische Chemie, Technische Universitiit Munchen, D-8046 Garching, Federal Republic a/Germany
(Received 29 May 1990; accepted 9 July 1990)
A theoretical description of secondary emission from complex absorption bands of isolated polyatomic molecules is developed. The strong non-Born-Oppenheimer coupling associated with conical intersections of the multidimensional excited-state potential-energy surfaces is included in a fully microscopic manner by solving the time-dependent Schrodinger equation for appropriate model systems incorporating the most relevant electronic states and vibrational modes. The effect of the large number of remaining vibrational modes and of the weaker coupling with additional electronic states is modeled by phenomenological relaxation terms (lifetime broadening and pure dephasing) in the framework of the density-matrix formalism. Explicit eigenstate-free expressions for absorption, resonance Raman, and fluorescence spectra are derived via density-matrix perturbation theory. The computational feasibility of the resulting mixed microscopic/phenomenological theory is demonstrated for a simple threemode model of the vibronic coupling of the SI (mr*) and S2 ( 1T1r*) states of pyrazine. The effect of excited-state vibronic coupling and ultrafast S2 ..... SI internal conversion on resonance Raman and fluorescence spectra is analyzed on the basis of these model calculations.
I. INTRODUCTION
As is well known, the secondary emission (SE) of polyatomic molecules following resonant excitation of an electronic transition generally consists of Raman and fluorescence emission, which are of different physical origin. 1-7 Resonance Raman (RR) scattering is a coherent two-photon process, whereas the fluorescence is a two-step process, where the emission occurs after phase relaxation (loss of coherence) in the excited state. This phase relaxation (dephasing) may be a consequence of the coupling of the molecule to the environment (pressure broadening in gases, solvation effects in solutions, or phonon coupling in solids) or may arise from intramolecular relaxation in isolated molecules, either on a single excited electronic potential surface [intrastate vibrational relaxation (IVR) ] or on vibronically coupled electronic potential surfaces [internal conversion (lC) orintersystem crossing (lSC)].
In this paper we shall be concerned with ultrafast and purely intramolecular electronic excited-state relaxation following resonant excitation of higher lying singlet states Sn (n)2) of polyatomic molecules. As a general rule, SE from Sn states of larger polyatomic molecules is strongly quenched or absent8 as a consequence of rapid IC of the Sn states into lower singlet states.
In contrast to the SI state, which typically exhibits well resolved spectra in absorption and emission, at least for excitation near the band origin under supersonic jet conditions, the higher singlet states typically exhibit largely featureless absorption spectra and fluorescence is difficult to detect. 8
Therefore very little is known about the dynamical processes occuring in these short-lived states. Only relatively recently, with the availability of tunable UV lasers, systematic RR
spectroscopy of higher singlet states has become possible.9 It has been demonstrated that RR and fluorescence measurements yield valuable information on the vibrational as well as relaxational dynamics of short-lived excited singlet states. 10-15
The present work is part of a series of investigations concerning the spectroscopic and dynamic effects of conical intersections 16 of adiabatic electronic potential-energy surfaces in polyatomic molecules. It has been shown on the basis of microscopic quantum calculations for a number offewstate few-mode vibronic coupling models that conical intersections are responsible for ultrafast (femtosecond) IC processes. 17-23 Spectroscopic effects of conical intersections have been discussed for photoe~~ctron spectra 17.19.20 and absorption spectra of neutral molecules. 19.21.24 A theoretical description of femtosecond real-time pump-probe spectroscopy of the dynamics on conically intersecting surfaces has been worked out recently.25-27 In the present work we address the problem of RR scattering and fluorescence from conically intersecting excited singlet states in larger polyatomic molecules. In extension of earlier work by Meyer and Koppel28 we adopt a Liouville-space density-matrix formulation which allows couplings with additional electronic states and additional vibrational modes to be included via phenomenological decay and dephasing constants. This is essential in order to account properly for the distinction between RR and redistributed fluorescence emission in complex systems. In contrast to Ref. 28, where the emphasis has been on the difficult problem of fluorescence emission within the strongly vibronically coupled manifold in cations, we shall be concerned with tpe somewhat simpler case of SE from strongly vibronically coupled excited states (SI ,S2 )
5496 J. Chern. Phys. 93 (8).15 October 1990 0021-9606/90/205496-14$03.00 © 1990 American Institute of Physics
Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

G. Stock and W. Domcke: Theory of resonance Raman scattering 5497
into the unperturbed ground state (So), Vibronic-coupling effects in RR spectra have been ex
tensively discussed within the framework of the classic Kramers-Heisenberg-Dirac (KHD) formula,29 see Refs. 30-33 and references given therein. More recently, the timedependent reformulation of the KHD formula34 has been extended to include vibronic coupling in the excited states. 35,36
In these works only the pure (coherent) Raman emission has been considered, which arises from SE emission processes which occur within the total dephasing time T2 of the system (typically less than 100 fs for larger polyatomic systems and molecules in solution). On the other hand, the simultaneous description of both RR and redistributed fluorescence, including the description of line shapes of dispersed SE spectra, has been a subject of considerable interest for more than two decades. In pioneering works of Huber l
and Hizhnyakov and Tehver2 collisional dephasing and dephasing due to phonon coupling in crystals have been treated, Subsequent investigations of the effect of dephasing on SE spectra have been based on the model of stochastic modulation of energy levels,1,37-42 on the density-matrix description with phenomenological damping terms,3,4,6,43,44 or on the so-called transform techniques. 2,45,46
All these formulations which include dephasing effects are restricted to the case of excited-state dynamics on uncoupled harmonic Born-Oppenheimer (BO) surfaces or to the case of uncoupled multilevel systems. The main motivation of the present work is to extend the theory towards a realistic description of strong non-BO coupling in excited electronic states. When dealing with femtosecond IC processes, one cannot a priori invoke perturbation theory for the non-BO coupling and the Markovian approximation, which would result in the usual exponential decay law.47 Our approach is rather to identify the strongest intramolecular interactions which are responsible for the fastest decay processes. These interactions are included in the model Hamiltonian for the system and the quantum dynamics is treated (numerically) exactly by solving the time-dependent Schrodinger equation. Additional relaxation effects (arising from the coupling of the electronic states and vibrational modes of the model system with the remaining states and modes of the molecule) are included in a phenomenological manner.
As a first application, we present the numerical realization of the theory for a simple three-mode model of the vibronic coupling of the SI and S2 states of pyrazine2
1,22 [see Ref. 21 for citations of the extensive literature on SI (mr* )-S2 ( 1T1T*) vibronic coupling in pyrazine]. While this simplified vibronic-coupling model is certainly an incomplete model of the photophysics of this moderately large molecule, its dynamics exhibits several nontrivial features which are presumably generic for ultrafast IC processes caused by conical intersections of potential-energy surfaces. 22 Apart from the demonstration of the technical feasibility of the computation ofRR and fluorescence spectra for strongly coupled model systems with 104 or more energy levels, we shall be particularly interested in elucidating the basic effects of conical intersections on RR and fluorescence spectra.
II. DENSITY-MATRIX DESCRIPTION OF RAMAN AND FLUORESCENCE SPECTROSCOPY OF VIBRONICALL Y COUPLED SYSTEMS
A. Hamiltonian and relaxation terms
We are concerned with molecular systems where the SE is affected by a strong vibronic interaction of the excited electronic states. As a simple but nontrivial model we consider an electronic three-state model system with a few vibrational degrees of freedom. Let IqJo) denote the electronic ground state and IqJI ), IqJ2) excited singlet electronic states of a polyatomic molecule. We assume that the excited electronic states IqJI ) and IqJ2) are close in energy and coupled by the strong intramolecular interaction V12 , whereas the intramolecular coupling of IqJI ) and IqJ2 ) with the well separated ground state IqJo) may be neglected to a first approximation. In a diabatic electronic representation,48 the model Hamiltonian H m reads
Hm = L IqJi)Hi(qJil + [lqJI)V12 (Q)(qJ21 +h.c.], ;=0,1,2
(2.1 )
where
H; = TN + V;(Q) (2.2)
denotes the multidimensional vibrational Hamiltonian in the diabatic state IqJ;), TN being the nuclear kinetic-energy operator. The symbol Q represents collectively the vibrational coordinates of the system.
Although being concerned with spontaneous emission, we may use the semiclassical approach, treating the material system quantum mechanically and the radiation field classically as an external field. I
,3 We assume that either one or both excited electronic states are coupled to the electronic ground state via the radiation field, whereas the IqJI )-lqJ2) transition is dipole forbidden. To simplify the analysis, we shall often specialize to the case where the lower lying electronic state IqJI ) can be considered as dark in absorption and emission, i.e., ~I = O. The model then describes the common situation of an optically bright excited state coupled to a dark background state.
The total Hamiltonian including the interaction of the model system with the field is thus written as
Hint(t) = - L IqJ;)lL.o°If(t)(qJol+h.c., ;= 1,2
(2.3a)
(2.3b)
where lL.o (i = 1,2) are the nonvanishing transition dipole moments and If (t) is the electric field. In the steady state the electric field is given by the superposition of the laser cw excitation and the spontaneous-emission field, i.e.,
If (t) = E[e - ;"'/' + Ese - ;"',1 + C.c., (2.4)
where WI' Ws are the frequencies and E[, Es are the polarization vectors of the laser field and the spontaneous-emission field, respectively.
The excited-state dynamics of larger polyatomic molecules can impossibly be treated in a fully microscopic manner. Thus the model Hamiltonian (2.1) is meant to include only the strongest interactions in the system which deter-
J. Chern. Phys., Vol. 93, No.8, 15 October 1990
Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

5498 G. Stock and W. Domcke: Theory of resonance Raman scattering
mine the dynamical evolution on the shortest time scales. We shall be particularly interested in situations where the adiabatic potential-energy surfaces of the excited electronic states exhibit a conical intersection. 16 The additional and presumably weaker intramolecular couplings due to the remaining vibrational degrees of freedom and additional electronic states will be taken into account here via phenomenological relaxation terms in the density-matrix formalism. 49
In the Schrodinger picture the equation of motion for the density matrix including relaxation terms is
ifz!....p(t) = [H,p(t)] - ifzr(t). at (2.5)
The physical effects to be described by the damping operator r ( t) are best characterized in the basis of eigenstates I "'v) of the molecular model Hamiltonian H m' First, the vibronic levels of the exited electronic singlet states IIPI ) and 11P2) are immersed in the dense manifold of vibrational levels of the electronic ground state and low-lying triplet states and can decay by Ie and ISC. In the Markovian approximation, these effects may be described by a population-decay constant lIT~V) for each vibronic level v of the excited-state manifold. For the two lowest excited singlet states of pyrazine, which will serve as an example below, it can be concluded from the experimentally observed relatively weak energy dependence of radiative lifetime and fluorescence quantum yield within the SI and the S2 bands that T I v) can be approximately taken as constant over the width of the SI and the S2 absorption band, respectively (see below). The phenomenological population decay can thus approximately be characterized by a single decay constant lIT[ within the energy range of either electronic state. The value of TI appropriate for the S2 absorption band is typically much shorter than the T[ for the S[ band.
A second important relaxation mechanism is the decay of coherences, that is, off-diagonal elements of the density matrix. As is well known,49 the interaction of the material system with a laser pulse leads, in first order in the field strength, to a coherence of the levels of the electronic ground state with the levels of dipole-allowed excited electronic states. The decay of this particular coherence is reflected by the homogeneous linewidth of the absorption spectrum.49 It can again be inferred from experimental data (absorption profiles of the SI and the S2 states of pyrazine, see below) that the coherence decay does not vary strongly over the width of the SI and S2 absorption bands and can thus approximately be characterized by a single electronic dephasing constant lIT2 within the energy range of either state. As is generally observed in larger polyatomic molecules, 8 the value of T2 appropriate for the S2 absorption band is much shorter than the T2 for the S[ band.
As is well known,49 the total dephasing rate lIT2 contains a contribution from the population decay of the excited-state manifold as well as from the so-called pure dephasing rate lIT* according to
lIT2 = 1I(2T[ ) + lIT*. (2.6)
A simple physical picture of the origin of the very fast pure dephasing after S2 excitation is provided by the model of stochastic modulation of the electronic excitation energy by
fluctuations in the large number of vibrational modes not included in Hm .37-42 It should be noted that after the initial ultrafast S2 -SI internal conversion process the microscopic model system possesses a large amount of vibrational energy (= 8000 cm - [ for the example of pyrazine to be discussed below) which will subsequently be distributed over the large number of remaining vibrational modes. The stochastic-modulation model, which corresponds to the hightemperature limit of the coupling of the system with a heat bath,37-42 thus appears appropriate.
To take account of these effects, we introduce the following damping operator in the basis of diabatic electronic states:
nt) = I lIT1 1IPi)Pij(t)(lPj l iJ= 1.2
+ I lIT2 [llPo)Poi(t)(lPil+ h.c.], (2.7) i= 1,2
with
Pij (t) = (lPi Ip(t) IlPj)' (2.8)
When Eqs. (2.5) and (2.7) are formally transformed into the basis of eigenstates of H m' it is seen that this ansatz precisely represents the above-described phenomenological relaxation effects, namely a population decay of all excitedstate vibronic levels with the rate lITI as well as a dephasing of the coherence between excited-state vibronic levels and electronic-ground-state vibrational levels with the rate lIT2 • It should be stressed that we do not assign different phenomenological relaxation rates to the unperturbed diabatic electronic states. The latter would be inconsistent with available spectroscopic data. For example, lines in the S[ (mr*) absorption spectrum which arise by intensity borrowing from the strongly allowed SO-S2 ( 1m*) transition would thus acquire the large homogeneous linewidth of the S2 absorption band.
It is clear that there may exist, in general, additional relaxation effects within the level structure of H m which are not included in the ansatz (2.7), e.g., pure dephasing of coherences within the manifold of excited-state vibronic levels. It should be stressed, however, that the model Hamiltonian H in itself describes relaxational behavior of observables which pertain to electronic or vibrational subsystems, in particular an ultrafast decay of the population of the higher excited electronic state and a dephasing of the vibrational motion (see Refs. 21 and 22 for a detailed discussion). We believe that the ansatz (2.7) provides a phenomenological description of the most essential relaxation effects which are not contained in the microscopic treatment of the problem and which are of relevance for the description of RR and fluorescence spectra.
B. Measurable quantities: Rates, cross sections, and quantum yield
It is well known from classical electrodynamics that the rate of absorption or emission of photons by a medium, characterized by its polarization P(t), is given by49
W = - l/cu( if (t). :t(t) ), (2.9)
J. Chern. Phys., Vol. 93, No.8, 15 October 1990
Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

G. Stock and W. Domcke: Theory of resonance Raman scattering 5499
where If (t) is the electric field and the brackets indicate time averaging over an optical period. The sign convention is such that W is negative in the case of absorption (disappearance of photons) and positive for emission (creation of photons). We prefer the concept of the rate of absorption or emission of photons, because this rate is directly related both to the .theoreticaltreatment (via the polarization) as well as to experimentally measurable cross sections.
In the CASe oflinear absorption, the electric cw laser field If 1 (t) and the linear polarization P( 1) (t) in the steady state are given by
,z; ( ) - iw,l (l.) I t = E1e + C.C.,
PO) () .. p( ) -/ru,t t = r- OJ/ e + c.C.,
resulting in the photon ab~orption rate
W(OJ/) = 2 1m l-L*oE/P*(OJ/).
(2.10)
(2.11 )
(2.12)
Including all prefactors, the linear-response absorption cross section at frequency OJ 1 is given byso
2 a uA(OJ/) =-1T-OJ/W(OJ/),
3 e2 (2.13)
where a is the fine structure constant. In the case of SE, we have to consider the total electric
field [Eq. (2.4)], consisting of the laser excitation field (2.10) 'and the spontaneous emission field, yielding the SE photon rate
W(OJs,OJ/) == W(I) (OJs,OJ/) + W(U) (OJs,OJ/),
W(I)(OJ OJ ) = 2 1m "*OE p*(I)(OJ OJ ) s' I, r- s s' I,'
(2.14a)
(2.14b)
W(II)(OJ.,OJ/) = 2 1m I-L*OE/P*(U) (OJs,OJ/). (2.14c)
P * (I,ll) (OJ s,OJ / ) are the time-independent parts of the nonlinear third-order polarization, depending on the frequency of the spontaneous emission OJs as well as on the frequency of the excitation OJ / [see Eq. (2.28) below] . The corresponding SE cross section is given by51
USE (OJs;OJ/) = !. ~ ...!!..-)2OJ/OJ: W(OJs,OJ/). 9 U\e2c
(2.15 )
When considering the total intensity emitted into all vibrational final states, i.e., the SE exCitation profile, we have to perform the integration over the photon frequencies OJ s'
yielding the total emission cross section
8 ~ a )2 3 UTE (OJ/) = - - OJ/ 2:(OJ/ - Ey) 1I(21T) Wy (OJ/), 9 ~c y
(2.16)
where Wy (OJ/) is the total photon rate scattered into a certain final state with the energy Ey •
Finally, we shall consider the excitation-energy dependent fluorescence quantum yield YF , defined as
YF(OJ/) = UTE (OJ/)/UA (OJ/). (2.17)
C. Calculation of the nonlinear polarization using density-matrix pertur:batlon·theory
In order to calculate the pOlarization of the medium, we have to solve the equation of motion (2.5) for the density operator P (t). Having in mind to evaluate the time evolution' of P ( t) using time-dependent perturbation theory, 52 it is
convenient to change to the interaction picture. The operator A '(t) in the interaction representation is related to the operator A in the SchrOdinger representation by the transformation (henceforth Ii = 1)
A '(t) = Ut(t)AU(t), (2.18)
where
U(t)=e- iHml (2.19a)
is the unitary time-evolution operator of the model system with electronic matrix elements
(2.19b)
The Liouville-von-Neumann equation. in the interaction representation reads
i i. P' (t) = [H int (t),p' (t)] - ir' (t), at where
(2.20)
Hint(t) = - 2: l~i)H~<~ol +h.c., (2,21a) i= 1,2
H ~I (t) = - 1f*(t)eiwIU~ (t) [I-LOloE*UI1 (t)
+ 1-Lo2°E *U21 (t)], (2.21b)
H ~2 (t) = - 1f*(t)eiwIU~ (t) [1-Lo1 0E*UI2 (t)
+ 1-L02°E *U22 (t)], (2.21c)
and
r'(t) = 2: lITII~i)pij(t)<~jl iJ= 1,2
+ 2: lITd I~O)P~i(t)(~il + h.c.]. i= 1,2
(2.22)
In the derivation ofEQ.s. (2.21b) and (2.21c) the standard rotating-wave approxi~ation (RWA)s3' has been employed. Assuming the molecular system to be initially in its electronic ground state I~o) and the vibrational 81:ound state
10) = 10,0 ... ),
the zero-order density matrix is given by
p'(O) (t) = P' (t = 0)
= 1~0)·10)(01(~01· (2.23)
The formal integration of Eq. (2.20) yields a system of coupled first-order integral equations
Poo(t) =Poo(O) -i fdt'(H~IP;o -p~lH;o
(2.24a)
P~·(t) = - il' dt' e-1/T,(t-I')(H" , H' ) II /OPOi - P/O Oi
o
(i=1,2), (2.24b)
I (t) . t d , -1/T2~I-t')(H' I , H' PDt· = - l Jo t e OlPII - Poo 01
+ H ~2pil ), (2.24c)
J. Chern. Phys" Vol. 93, No. 8,15 October 1990
Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

5500 G. Stock and W. Domcke: Theory of resonance Raman scattering
, ( ) 'lldt' -IIT2(t-I')(H" , H' P02 t = - I e 02Pn - Poo 02 o
(2.24d)
'lldt' -IIT,(t-I')(H" , H' ) - I e lOPOl - PlO 02'
o (2.24e)
pij(l) =p;/(I). (2.240
Using standard time-dependent density-matrix perturbation theory, 52 we shall solve the equations of motion successively up to third order, yielding the populations P;; (I) (i = 1,2) and the coherence P;2 (I) in even orders and the coherences Po; (I) (i = 1,2) in odd orders.
D. Linear absorption
In order to evaluate the absorption rate (2.12), we have to calculate the polarization
P(I) = Tr[WJ(I)]
where
= 2 Re{~ol Trv [POI (I)] + ~02 Trv [POl (I) p, (2.25)
(2.26a)
POI (I) = Uoo (I)POI (I) Uri (I) + Uoo (I)P02 (I) UTI (I),
(2.26b)
P02 (I) = Uoo (I)POI (I) Ur2 (I) + Uoo (I)P02 (I) UT2 (I). (2.26c)
In Eq. (2.25) the trace refers to a complete set of electronic and vibrational states (Tr) and to a complete set of vibrational states (Trv), respectively. Note that the vibronic coupling mixes the contributions of the two excited electronic states to the polarization when we change back to the Schr6-dinger representation.
In the case of linear absorption we have to calculate p(l) (I). Insertion of p(l) (I) into Eq. (2.9) yields the absorption signal
W(aJ/) = - 2 Re .. 2: ~.o·E*~ft.E roo dt e - IIT2 /.1=1,2 Jo
x e;(w/ Holt (01 (IP; Ie - ;Hml IlPj) 10), (2.27)
where €o is the energy of the electronic and vibrational ground state,
In the derivation of the absorption signal we have employed the Condon approximation, i.e., we have assumed the transition dipole matrix element ~Oi (i = 1,2) to be independent of the coordinate Q in the adopted diabatic electronic representation.
E. Secondary emission in the case of a single bright excited electronic state
In this section we are concerned with the special case that only the 11P0 )-11P2) transition couples to the radiation field, the 11P0 )-IIPI) transition being dipole forbidden or only weakly allowed. This assumption leaves us with the commonly encountered situation of SE from a bright electronic state coupled to a dark background state, which is a physically more transparent problem. In this case no coherence PI2 (I) is induced by the laser field and no interference effects will arise in the emission. Furthermore, the calculation simplifies considerably, because the number of terms contributing to the SE signal is significantly reduced.
In order to calculate the SE signal, we have to evaluate Eq. (2.9) using the nonlinear polarization p(3) (I). Restricting ourselves to contributions to p(3) (I) which are resonant in all optical transitions (R W A), the nonlinear polarization is obtained in the usual manner by solving successively the equations of motion (2.24) up to third order and transforming the resulting coherences PoP) (I) back to the Schr6dinger representation [Eq. (2,26) ]. Due to the special choice of the damping matrix r(1) in Eq. (2.7), as discussed and motivated above, it is possible to employ the closure relation of the electronic states (~; lIP; ) (IP; I = 1) and contract the terms arising from the different electronic potential surfaces, yielding for the SE rates54
W(aJs,aJ/) = 2: W~I)(aJs,aJ/) + W~II)(aJs,aJ/), (2.28a) v
W (I)( )-2R I 121 121' dt dt dt -(t-13+ 12- ll)IT2 -(t3- 12)IT, -;(W,+Ev )(I-13) II ll3 ll2 v aJs,aJ/ - e ~20·Es ~20·E/ 1m 3 2 lee e
1_ 00 0 0 0
(2.28b)
(2.28c)
where
ct>yO (I) = (vi (1P21e - iHml 11P2) 10) (2.29)
are correlation functions representing the intramolecular excited-state dynamics and €y denotes the energies of the final vibrational levels Iv) in the electronic ground state. As has been discussed by many authors for three-level systems and multilevel systems,I-7,41-44 the SE signal consists of two physically distinct components. The first term W (I) (aJ"aJ/) is often referred to as fluorescence-like emission, as it is directly proportional to the (second-order) excited-state excess population, while the second term W(I1)(w s 'w/) is referred to as coherent Raman-like emission. It is seen that the proposed model [i.e.,
J. Chem. Phys., Vol. 93, No.8, 15 October 1990
Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

G. Stock and W. Domcke: Theory of resonance Raman scattering 5501
the Hamiltonian (2.1) and the damping matrix (2.7) ] yields formally the same SE signal as is obtained for simple uncoupled multilevel systems, although we are dealing here with a vibronic coupling problem. The whole information on the complex dynamics of strongly vibronically coupled excited states is incorporated in the intramolecular correlation functions <llvO (t) .
Although our actual computations are based on the "eigenstate-free" correlation functions <llvO (f), it is instructive to introduce for interpretative purposes the exact eigenstates of H m belonging to the vibronically coupled manifold 1q:>1 ), 1q:>2),
Hm ItPv) = E'vltPv). (2.30)
It should be stressed that it is not possible to compute these eigenstates even for the simple three-mode model to be discussed below, as this would require the diagonalization of Hamiltonian matrices of the order of 104
•
Inserting Eq. (2.30) into Eq. (2.28), the time integrations are easily performed, yielding the well-known result for the SE photon rate scattered into a certain final statel
-7
•3
8.-43
W ( ) _ 21 ° 121 12 ~ (0 I (q:>21 tP a ) ( tP a I q:>2 ) Iv) (vi (q:>zl tP p ) ( tP p I q:>2 ) 10) y It)s,lt)/ - 1-L20 Es 1-L20 °E/ ~ . .
a.p (E'O + It)/ - E'a - I/T2 )(E'O + It)/ - E'p + zlTz )
X [11'6(lt)/ + E'O -It)s - E'.) + lIT* ( 1 + 1 )] . (2.31) (E'a - E'p + i/TI ) E'y + It)s - E'a - ilT2 E'y + It)s - E'p + ilT2
In this frequency-domain expression the two emission types mentioned above are clearly separated. The first term is recognized as the well-known KHD expression,29 describing the infinitely sharp Raman peaks that occur in the emission spectrum according to the conservation of energy. The second term accounts for the redistributed fluorescence that disappears in the absence ofpUfe dephasing (lIT* = 0).
Equations (2.28) and (2.31) completely describe the SE in our theoretical framework for the case of a single bright excited state. A considerable simplification arises if we are only interested in the total emission intensity scattered into a certain final state, i.e., the total emission excitation profile, that is obtained by performing the integration over the photon energies3•7•55
= 211'1 l-L2o °Es 1211-L~ooEd2 Re 1"" dt21t, dtle - IIT-(t, - t,le - (t,+ tl)/2T,i(""+ €O)(t2 - t\)<llvO (t2 )<ll:«, (tl ). (2.32)
The corresponding expression in the frequency domain reads
W(TE)( )-2 I ° 121. 0'12~ (OI(q:>2ItPa)(tPalq:>z)lv)(vl(q:>zltPp)(tPplq:>2)IO) [1+ 2i1T* ] y It)/ - 11' 1-L20 Es I-Lzo E/ ~ a.p (E'o +It)/ - E'a - i1T2) (E'o + It)/ - E'p + i1T2) (E'a - E'p + i/TI )
= W~R)(lt)/) + W~F)(lt)/),
where W~R) (It)/) is the total Raman emission into a certain final state for an excitation frequency It)/ and W~F) (It)/) is the corresponding total redistributed fluorescence emission.
Transforming W~R)(lt)/) back into the time-dependent picture, we finally obtain the time-dependent form of the (It).-integrated) KHD expression, which has been made popular by Heller et al.:34
W~R)(lt)/) = 211'11-L2ooEsI21I-LzooE/12
X 11"" dte- tIT2i(OJI+€O,t<llvO(t) IZ. (2.34)
It is noteworthy that in the derivation ofEq. (2.34) we did not assume the absence of pure dephasing. Thus the correlation function <llvO (t) is damped with the total dephasing rate IITz, arising from the population decay as well as from pure dephasing [Eq. (2.6) ].55 As Eq. (2.34l can also be derived in the Schrodinger wave-function formalism, the damping of the correlation function has often been misinterpreted as being due to pure lifetime effects. This explains inconsistencies in the literature, arising from the interpretation of dephasing times of 10 fs or less as excited-state lifetimes. to
(2.33) I F. Secondary emission In the case of two coupled bright excited electronIC states
So far we have studied in some detail the simplified situation of a single bright excited state coupled to dark background state. Consiciering the more general case where both the transition Iq:>o )-[q:>1 ) as well as Iq:>o )-1q:>2 ) couples to the radiation field, we have to solve the equation of motion (2.20) with the full interaction operator (2.21) (I-LO! #0). For the sake of brevity, we restrict ourselves here to the calculation of the total (It)s-integrated) emission rates. A lengthy calculation yields for the total emission excitation spectrum
W~TE)(lt)/) = 411'Re L l-LoioE=-l-LpoErl-LokoEsl-LlOoE/ iJ.k./= 1.2
(2.35)
where
<ll~)(t) = (vi (q:>;\e- iHmt 19'})IO). (2.36)
J. Chern. Phys .• Vol. 93. No.8. 15 October 1990
Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

5502 G. Stock and W. Domcke: Theory of resonance Raman scattering
Analogously, the Raman excitation profile [cf. Eq. (2.34)] is given by
W~R)(ml) = 21T.I I foo dte- tIT2/(OJI+€o)t
1= 1,2 Jo
X [ .... 0;·Es .... 20·Ei"<I>~~)(t)
+ .... o;.Es .... IO.Ei"<I>~~l)(t)] /2 (2.37)
As it is seen from Eqs. (2.35) and (2.37), we may expect interference effects due to the interference of the emission of the two bright electronic states.
III. MODEL SYSTEM AND COMPUTATIONAL METHODS
Recently a conical intersection of the SI and S2 surfaces of pyrazine has been identified on the basis of semiempirical calculations and the analysis of experimental spectra. 2 I It has been shown that this intersection is responsible for an ultrafas't (=20 fs) S2 -+SI internal conversion process and causes a dephasing of the vibrational motion on a time scale of a few hundred fs.22 We adopt here this model system as a representative of strong vibronic coupling of excited electronic states in polyatomic molecules. For a discussion of the extensive literature on the SI [IB3u (n1T*)]S2 [ I B2u ( 1T1T*)] vibronic coupling problem we refer to Ref. 21 and the references given therein. The H; and V12 in the molecular Hamiltonian (2.1) are specified as21 .22
Ho = I( - J..- mj a2
2 +J..-mjQJ) (j= 1,6a,lOa),
j 2 aQj 2 (3.1)
H; =Ho +E; + I KY)Qj (i= 1,2,j= 1,6a), (3.2) j
VI2 = AQlOa' (3.3)
where the Q 's are dimensionless normal coordinates and the m's the associated harmonic vibrational frequencies. QlOa is the (single) normal coordinate of pyrazine of B Ig symmetry which can couple SI (IB3u) and S2 (IB2U) in first order, A being the vibronic coupling constant. The Qj,j = 1, 6a, are the normal coordinates of the totally symmetric modes VI and V 6a of pyrazine. EI and E2 are the vertical excitation energies and KY), i = I, 2,j = I, 6a, represent the gradients of the excited-state potentials with respect to the A Ig coordinates and account for the shift of the eqUilibrium geometry of the excited states in these coordinates. The parameters of the model as determined in Ref. 21 for pyrazine are collected in Table I.
In pyrazine, the SO-S2 (1T1T*) optical transition is strongly allowed, the SO-SI transition is weakly allowed, and the SI -S2 transition is dipole forbidden. The oscillator strength of the SO-SI transition is about a factor of 10 smaller than the SO-S2 oscillator strength. 56
The computational methods have been described in detail previously,22,26 and are only briefly sketched. It has been pointed out in Sec. II that the microscopic model Hamiltonian H m enters the calculations only via the correlation functions <I> ~g) (t), which carry the complete information on the intramolecular dynamics of the vibronically coupled ex-
TABLE I. Vertical electronic excitation energies, vibrational (v, 'V6a ) and vibronic (v lOa ) coupling constants, and vibrational frequencies of the S, -S2 vibronic coupling model for pyrazine. All quantities are in electron volts.
'B3U (mr*) 'B2u (1T1r*) (J)
E 3.94 4.84 v, 0.037 - 0.254 0.126 V 6a - 0.105 0.149 0.074
VIDa 0.262 0.118
cited electronic states. In order to compute these correlation functions, we represent the model Hamiltonian in a directproduct basis which is built from the electronic states 19'1), 19'2) and harmonic oscillator states Iv) (j= I, 6a, lOa). Converged results are obtained with a Hamiltonian matrix of dimension 14 112 (see Ref. 26 for details). The field-free time-dependent Schrodinger equation with the appropriate initial condition is solved with a fourth-order predictor-corrector finite-differences method using a time step of 0.04 fs. 22,26 The resulting vector of time-dependent coefficients represents directly the required correlation functions <I>~g) (t).
The time-dependent propagation of the state vectors of dimension = 104 and the subsequent multiple time integrations have been performed with a Silicon Graphics Iris 4D/20 workstation. The time-dependent state vectors have been propagated up to 3 ps (75000 time steps) which is necessary to obtain converged fluorescence intensities for an excited-state lifetime of TI = 500 fs. The propagation requires about 5 CPU hours per symmetry (B3u or B 2u ' respectively). The first 40 correlation functions of each symmetry were stored for subsequent evaluation. The absorption.spectrum [Eq. (2.27)] and the pure Raman signals [Eqs. (2.34) and (2.37) ] are easily calculated using a standard FFT routine in a few CPU seconds. In this case there is only a single integral to be performed which converges quickly (the integral is damped with l/T2 ). The double time integrations required for the evaluation of the total emission intensities [Eqs. (2.32) and (2.35)] can be performed in a single loop, using the trapezoidal rule with a time step of 0.32 fs, requiring = 1 CPU minute per final state. The (synthesized) dispersed fluorescence spectra and the fluorescence excitation spectra (see below) have been computed by wave function propagation with simultaneous integrations. As we could not find a trick to eliminate the triple time integration in the dispersed fluorescence photon rate [Eq. (2.28b)], we solved the problem numerically by brute force, which required about 3 CPU hours per final vibrational state.
IV. COMPUTATIONAL RESULTS
A. Absorption spectrum and determination of phenomenological relaxation rates
To get an impression of how the model is related to reality, we first consider the simplest response of the model system, that is, the (linear) absorption spectrum. Figure 1 (a) shows the experimental gas-phase absorption spectrum
J. Chem. Phys., Vol. 93, No. 8,15 October 1990
Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs
G. Stock and W. Domcke: Theory of resonance Raman scattering 5503
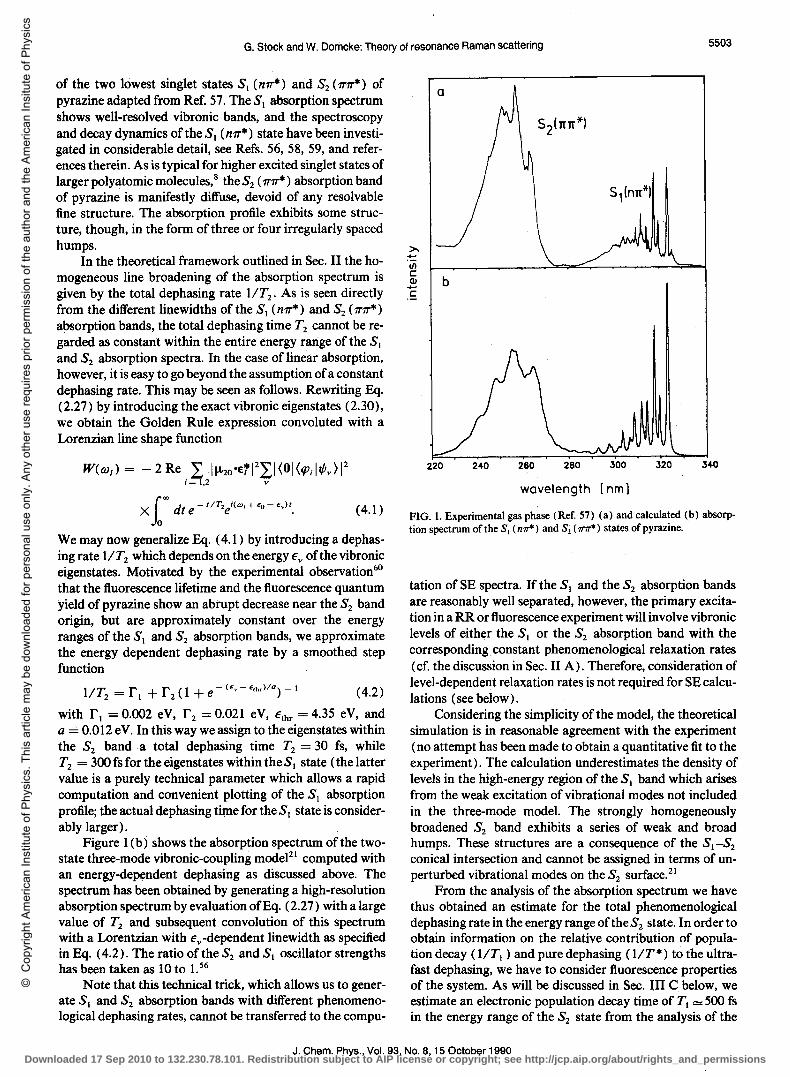
of the two lowest singlet states SI (mT*) and S2 (-17"1T*) of pyrazine adapted from Ref. 57. The SI absorption spectrum shows well-resolved vibronic bands, and the spectroscopy and decay dynamics of the SI (m1"*) state have been investigated in considerable detail, see Refs. 56, 58, 59, and references therein. As is typical for higher excited singlet states of larger polyatomic molecules,8 the S2 ( 11"11"*) absorption band of pyrazine is manifestly diffuse, devoid of any resolvable fine structure. The absorption profile exhibits some structure, though, in the form of three or four irregularly spaced humps.
In the theoretical framework outlined in Sec. II the homogeneous line broadening of the absorption spectrum is given by the total dephasing rate lIT2. As is seen directly from the different linewidths of the SI (m1"*) and S2 ( 11"11"*)
absorption bands, the total dephasing time T2 cannot be regarded as constant within the entire energy range of the SI and S2 absorption spectra. In the case of linear absorption, however, it is easy to go beyond the assumption of a constant dephasing rate. This may be seen as follows. Rewriting Eq. (2.27) by introducing the exact vibronic eigenstates (2.30), we obtain the Golden Rule expression convoluted with a Lorenzian line shape function
W(w/) = - 2 Re L 1~20·ErI2LI (01 (/Pi ItPv) 12 i= 1.2 v
(4.1 )
We may now generalize Eq. (4.1) by introducing a dephasing rate lIT2 which depends on the energy €v of the vibronic eigenstates. Motivated by the experimental observation60
that the fluorescence lifetime and the fluorescence quantum yield of pyrazine show an abrupt decrease near the S2 band origin, but are approximately constant over the energy ranges of the SI and S2 absorption bands, we approximate the energy dependent dephasing rate by a smoothed step function
lIT2 =rl +r2(1+e-(E,,-E'h,)IQ)-1 (4.2)
with r l = 0.002 eV, r 2 = 0.021 eV, €thr = 4.35 eV, and a == 0.012 eV. In this way we assign to the eigenstates within the S2 banda total dephasing time T2 = 30 fs, while T2 = 300 fs for the eigenstates within the S 1 state (the latter value is a purely technical parameter which allows a rapid computation and convenient plotting of the SI absorption profile; the actual dephasing time for the SI state is considerably larger).
Figure 1 (b) shows the absorption spectrum of the twostate three-mode vibronic-coupling model21 computed with an energy-dependent dephasing as discussed above. The spectrum has been obtained by generating a high-resolution absorption spectrum by evaluation ofEq. (2.27) with a large value of T2 and subsequent convolution of this spectrum with a Lorentzian with €v-dependent linewidth as specified in Eq. (4.2). The ratio of the S2 and S) oscillator strengths has been taken as 10 to 1.56
Note that this technical trick, which allows us to generate SI and S2 absorption bands with different phenomenological dephasing rates, cannot be transferred to the compu-
a
S1 Inrr*)
>--III ~ b -c
220 240 340
wavelength [nm 1
FIG. 1. Experimental gas phase (Ref. 57) (a) and calculated (b) absorption spectrum of the S, (mr*) and Si ( 1T1r*) states of pyrazine.
tation of SE spectra. If the SI and the S2 absorption bands are reasonably well separated, however, the primary excitation in a RR or fluorescence experiment will involve vibronic levels of either the SI or the S2 absorption band with the corresponding constant phenomenological relaxation rates (cf. the discussion in Sec. II A). Therefore, consideration of level-dependent relaxation rates is not required for SE calculations (see below) .
Considering the simplicity of the model, the theoretical simulation is in reasonable agreement with the experiment (no attempt has been made to obtain a quantitative fit to the experiment). The calculation underestimates the density of levels in the high-energy region of the SI band which arises from the weak excitation of vibrational modes not included in the three-mode model. The strongly homogeneously broadened S2 band exhibits a series of weak and broad humps. These structures are a consequence of the S) -S2 conical intersection and cannot be assigned in terms of unperturbed vibrational modes on the S2 surface. 2
1
From the analysis of the absorption spectrum we have thus obtained an estimate for the total phenomenological dephasing rate in the energy range of the S2 state. In order to obtain information on the relative contribution of populationdecay (liT) andpuredephasing (lIT*) to the ultrafast dephasing, we have to consider fluorescence properties of the system. As will be discussed in Sec. III C below, we estimate an electronic population decay time of TI = 500 fs in the energy range of the S2 state from the analysis of the
J. Chern. Phys., Vol. 93, No.8, 15 October 1990 Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs
5504 G. Stock and W. Domcke: Theory of resonance Raman scattering
fluorescence quantum yield. The result that TI is considerably larger than T2 (500 vs 30 fs) implies that the homogeneous broadening of the S2 absorption spectrum arises almost completely from pure dephasing, i.e., T2 = T * = 30 fs.
B. Raman spectra
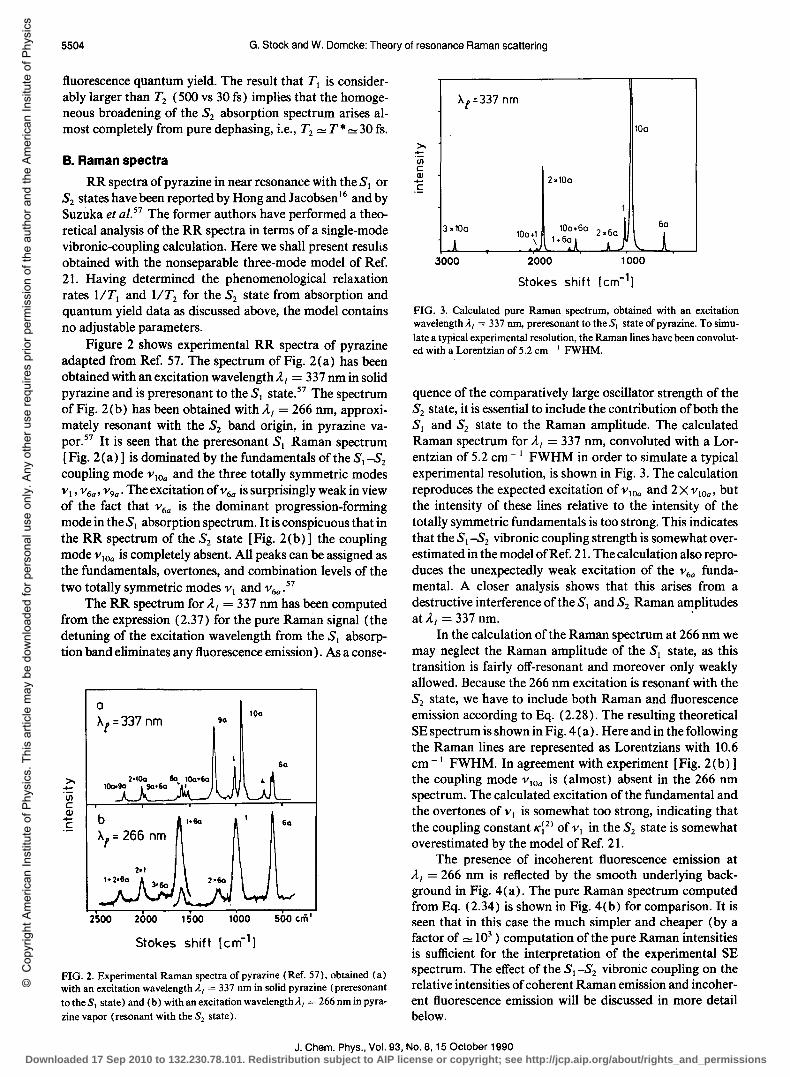
RR spectra of pyrazine in near resonance with the SI or S2 states have been reported by Hong and Jacobsen 16 and by Suzuka et al. 57 The former authors have performed a theoretical analysis of the RR spectra in terms of a single-mode vibronic-coupling calculation. Here we shall present results obtained with the nonseparable three-mode model of Ref. 2!. Having determined the phenomenological relaxation rates lITI and lIT2 for the S2 state from absorption and quantum yield data as discussed above, the model contains no adjustable parameters.
Figure 2 shows experimental RR spectra of pyrazine adapted from Ref. 57. The spectrum of Fig. 2(a) has been obtained with an excitation wavelength AI = 337 nm in solid pyrazine and is preresonant to the SI state. 57 The spectrum of Fig. 2(b) has been obtained with AI = 266 nm, approximately resonant with the S2 band origin, in pyrazine vapor.57 It is seen that the preresonant SI Raman spectrum [Fig. 2(a)] is dominated by the fundamentals of the SI-S2
coupling mode VlOa and the three totally symmetric modes VI' V 6a , V9a' The excitation ofv6a is surprisingly weak in view of the fact that V6a is the dominant progression-forming mode in the S I absorption spectrum. It is conspicuous that in the RR spectrum of the S2 state [Fig. 2(b)] the coupling mode V lOa is completely absent. All peaks can be assigned as the fundamentals, overtones, and combination levels of the two totally symmetric modes VI and V 6a • 57
The RR spectrum for AI = 337 nm has been computed from the expression (2.37) for the pure Raman signal (the detuning of the excitation wavelength from the SI absorption band eliminates any fluorescence emission). As a conse-
>.-(/) C <II .-C
a 'At =337 nm 9a
b 1.60 6a
'At = 266 nm
2·1
Stokes shift [cm-1)
FIG. 2. Experimental Raman spectra ofpyrazine (Ref. 57), obtained (a) with an excitation wavelength AI = 337 nm in solid pyrazine (preresonant totheS, state) and (b) with an excitation wavelength AI = 266 nmin pyra
zine vapor (resonant with the S2 state).
Af=337 nm
100
>-.... (/) c (II
2.100 .-C
1
3.100 100.60 2.:0)
60 100+1
A A \ 1 +60 A
3000 2000 1000
Stokes shift [cm-1j
FIG. 3. Calculated pure Raman spectrum, obtained with an excitation wavelength AI = 337 nm, preresonant to the S, state of pyrazine. To simu
late a typical experimental resolution, the Raman lines have been convoluted with a Lorentzian of 5.2 cm ~, FWHM.
quence of the comparatively large oscillator strength of the S2 state, it is essential to include the contribution of both the SI and S2 state to the Raman amplitude. The calculated Raman spectrum for AI = 337 nm, convoluted with a Lorentzian of 5.2 cm -I FWHM in order to simulate a typical experimental resolution, is shown in Fig. 3. The calculation reproduces the expected excitation ofvlOa and 2XvlOa , but the intensity of these lines relative to the intensity of the totally symmetric fundamentals is too strong. This indicates that the SI-S2 vibronic coupling strength is somewhat overestimated in the model of Ref. 21. The calculation also reproduces the unexpectedly weak excitation of the V6a fundamental. A closer analysis shows that this arises from a destructive interference of the SI and S2 Raman amplitudes at AI = 337 nm. .
In the calculation of the Raman spectrum at 266 nm we may neglect the Raman amplitude of the SI state, as this transition is fairly off-resonant and moreover only weakly allowed. Because the 266 nm excitation is resonant with the S2 state, we have to include both Raman and fluorescence emission according to Eq. (2.28). The resulting theoretical SE spectrum is shown in Fig. 4 (a). Here and in the following the Raman lines are represented as Lorentzians with 10.6 cm -I FWHM. In agreement with experiment [Fig. 2(b)] the coupling mode V lOa is (almost) absent in the 266 nm spectrum. The calculated excitation of the fundamental and the overtones of VI is somewhat too strong, indicating that the coupling constant Kj2) of VI in the S2 state is somewhat overestimated by the model of Ref. 21.
The presence of incoherent fluorescence emission at AI = 266 nm is reflected by the smooth underlying background in Fig. 4(a). The pure Raman spectrum computed from Eq. (2.34) is shown in Fig. 4(b) for comparison. It is seen that in this case the much simpler and cheaper (by a factor of = 103
) computation of the pure Raman intensities is sufficient for the interpretation of the experimental SE spectrum. The effect of the SI -S2 vibronic coupling on the relative intensities of coherent Raman emission and incoherent fluorescence emission will be discussed in more detail below.
J. Chem. Phys., Vol. 93, No.8, 15 October 1990 Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs
G. Stock and W. Domcke: Theory of resonance Raman scattering 5505
If) C Q.o ..... .!:
a
Xt = 266 nm
b
2.1+ 60
3000
60
2.1
2.60
2000 1000
Stokes shift [cm-')
FIG. 4. Calculated SE spectrum (a) and pure Raman spectrum (b) for approximately resonant excitation of the S, state ofpyrazine. The Raman lines are represented as Lorentzians with 10.6 cm - I FWHM.
In summary, the two-state three-mode vibronic coupling model of Ref. 21, augmented by phenomenological relaxation terms, yields an acceptable qualitative description of the Raman spectra for resonant or near-resonant excitation of the SI and S2 states of pyrazine, considering that no parameters have been adjusted. Improvements of the model towards a more quantitative description of the RR spectra, preferably in conjunction with accurate ab initio calculations ofthe SI and S2 potential-energy surfaces, will be the subject of future work.
c. Analysis of the effect of vlbronic coupling on Raman and fluorescence spectra
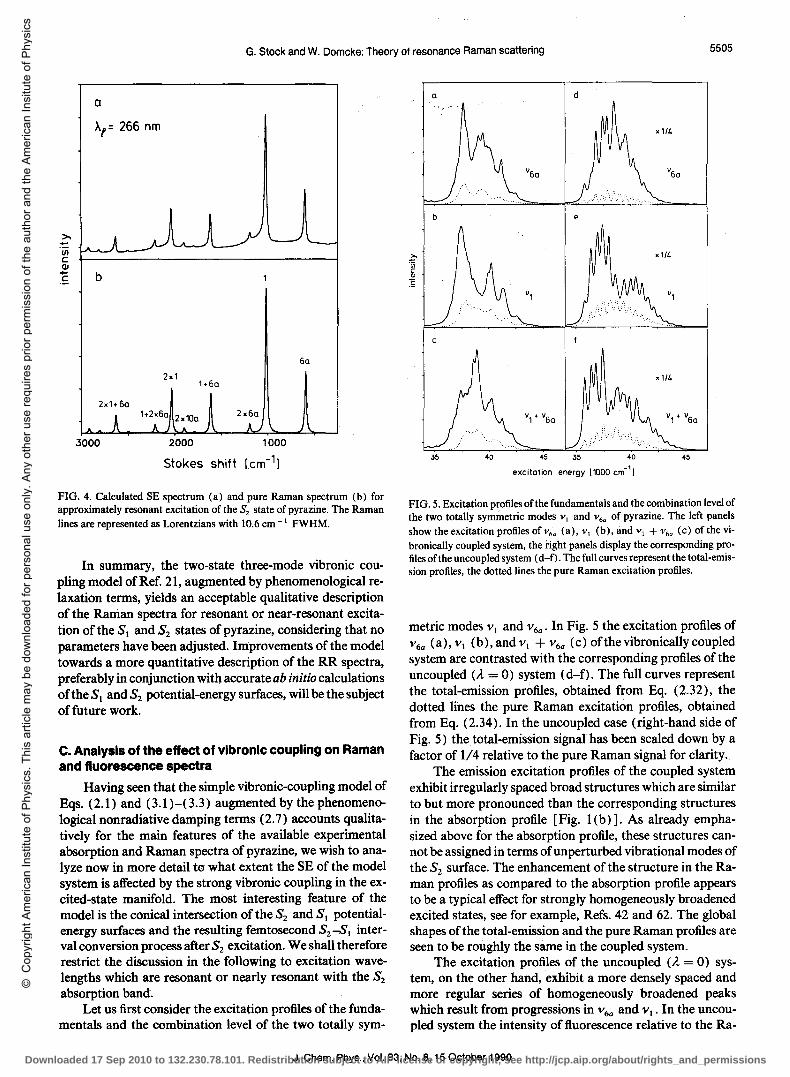
Having seen that the simple vibronic-coupling model of Eqs. (2.1) and (3.1)-(3.3) augmentedbythephenomenological nonradiative damping terms (2.7) accounts qualitatively for the main features of the available experimental absorption and Raman spectra of pyrazine, we wish to analyze now in more detail to what extent the SE of the model system is affected by the strong vibronic coupling in the excited-state manifold. The most interesting feature of the model is the conical intersection of the S2 and SI potentialenergy surfaces and the resulting femtosecond S2-81 interval conversion process after S2 excitation. We shall therefore restrict the discussion in the following to excitation wavelengths which are resonant or nearly resonant with the S2 absorption band.
Let us first consider the excitation profiles of the fundamentals and the combination level of the two totally sym-
a d
b e
35 40 45 35 40 45
excitation energy [1000 cm-')
FIG. 5. Excitation profiles ofthe fundamentals and the combination level of the two totally symmetric modes VI and V 6a of pyrazine. The left panels show the excitation profilesofv6a (a), VI (b), and VI + V 6a (c) ofthe vibronically coupled system, the right panels display the corresponding profiles of the uncoupled system (d-f). The full curves represent the total-emission profiles, the dotted lines the pure Raman excitation profiles.
metric modes VI and V6a' In Fig. 5 the excitation profiles of V 6a (a),vI (b),andv i +v6a (c)ofthevibronicallycoupled system are contrasted with the corresponding profiles of the uncoupled (A = 0) system (d-f). The full curves represent the total-emission profiles, obtained from Eq. (2.32), the dotted lines the pure Raman excitation profiles, obtained from Eq. (2.34). In the uncoupled case (right-hand side of Fig. 5) the total-emission signal has been scaled down by a factor of 1/4 relative to the pure Raman signal for clarity.
The emission excitation profiles of the coupled system exhibit irregularly spaced broad structures which are similar to but more pronounced than the corresponding structures in the absorption profile [Fig. I (b) ]. As already emphasized above for the absorption profile, these structures cannot be assigned in terms of unperturbed vibrational modes of the S2 surface. The enhancement of the structure in the Raman profiles as compared to the absorption profile appears to be a typical effect for strongly homogeneously broadened excited states, see for example, Refs. 42 and 62. The global shapes of the total-emission and the pure Raman profiles are seen to be roughly the same in the coupled system.
The excitation profiles of the uncoupled (A = 0) system, on the other hand; exhibit a more densely spaced and more regular series of homogeneously broadened peaks which result from progressions in V 6a and VI' In the uncoupled system the intensity of fluorescence relative to the Ra-
J. Chem. Phys., Vol. 93, No.8, 15 October 1990 Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs
5506 G. Stock and W. Domcke: Theory of resonance Raman scattering
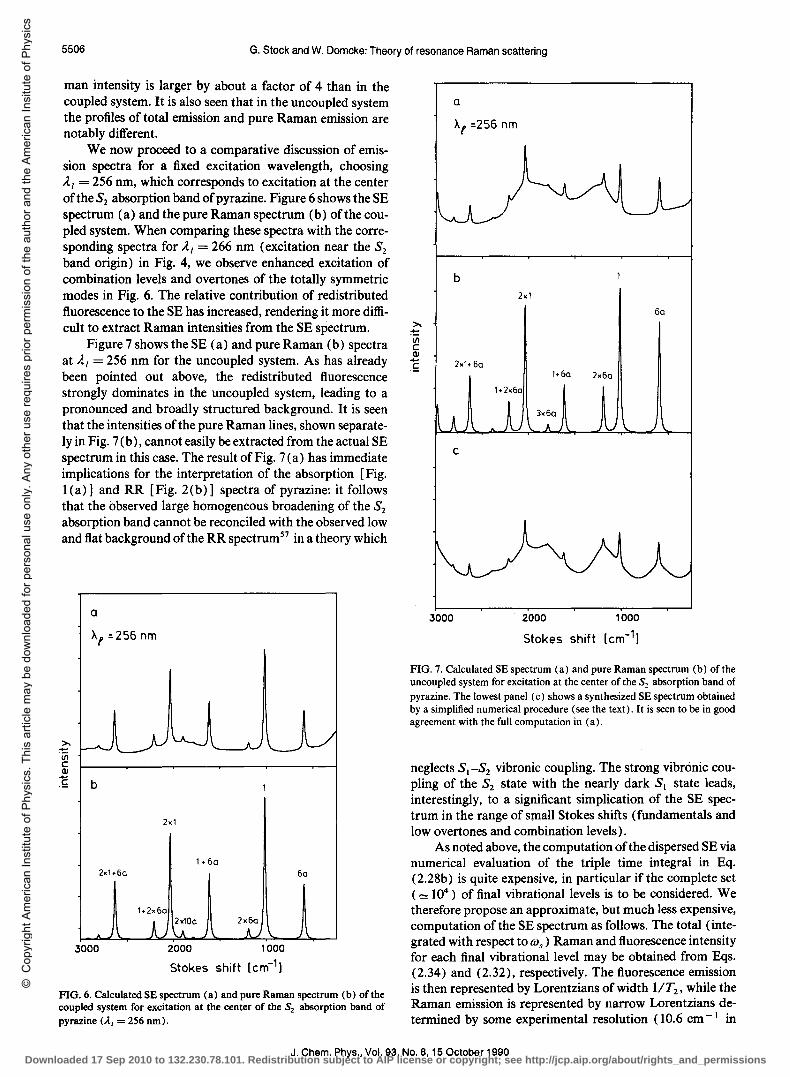
man intensity is larger by about a factor of 4 than in the coupled system. It is also seen that in the uncoupled system the profiles of total emission and pure Raman emission are notably different.
We now proceed to a comparative discussion of emission spectra for a fixed excitation wavelength, choosing Al = 256 nm, which corresponds to excitation at the center of the S2 absorption band of pyrazine. Figure 6 shows the SE spectrum (a) and the pure Raman spectrum (b) of the coupled system. When comparing these spectra with the corresponding spectra for Al = 266 nm (excitation near the S2 band origin) in Fig. 4, we observe enhanced excitation of combination levels and overtones of the totally symmetric modes in Fig. 6. The relative contribution of redistributed fluorescence to the SE has increased, rendering it more difficult to extract Raman intensities from the SE spectrum.
Figure 7 shows the SE (a) and pure Raman (b) spectra at Al = 256 nm for the uncoupled system. As has already been pointed out above, the redistributed fluorescence strongly dominates in the uncoupled system, leading to a pronounced and broadly structured background. It is seen that the intensities of the pure Raman lines, shown separately in Fig. 7 (b), cannot easily be extracted from the actual SE spectrum in this case. The result of Fig. 7 (a) has immediate implications for the interpretation of the absorption [Fig. l(a)] and RR [Fig. 2(b)] spectra of pyrazine: it follows that the observed large homogeneous broadening of the S2 absorption band cannot be reconciled with the observed low and flat background of the RR spectrumS7 in a theory which
a
At =256 nm
>. .... 'iii c CII
~ b
3000
2.1+60
2.1
1.2.60
1.60
2000 1000
Stokes shift [cm-1)
60
FIG. 6. Calculated SE spectrum (a) and pure Raman spectrum (b) of the coupled system for excitation at the center of the S2 absorption band of pyrazine (A.I = 256 nm).
>. .... If) c CII .... c
a
At =256 nm
b
2.1.60
c
3000
2.1
1.60 2.60
1+2.60
2000 1000
Stokes shift [cm-')
60
FIG. 7. Calculated SE spectrum (a) and pure Raman spectrum (b) of the uncoupled system for excitation at the center of the S2 absorption band of pyrazine. The lowest panel (c) shows a synthesized SE spectrum obtained by a simplified numerical procedure (see the text). It is seen to be in good agreement with the full computation in (a).
neglects S) -S2 vibronic coupling. The strong vibronic coupling of the S2 state with the nearly dark S) state leads, interestingly, to a significant simplication of the SE spectrum in the range of small Stokes shifts (fundamentals and low overtones and combination levels).
As noted above, the computation of the dispersed SE via numerical evaluation of the triple time integral in Eq. (2.28b) is quite expensive, in particular if the complete set (= 104
) of final vibrational levels is to be considered. We therefore propose an approximate, but much less expensive, computation of the SE spectrum as follows. The total (integrated with respect to (i)s) Raman and fluorescence intensity for each final vibrational level may be obtained from Eqs. (2.34) and (2.32), respectively. The fluorescence emission is then represented by Lorentzians of width lIT2 , while the Raman emission is represented by narrow Lorentzians determined by some experimental resolution (10.6 cm -) in
J. Chem. Phys., Vol. 93, No.8, 15 October 1990 Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs
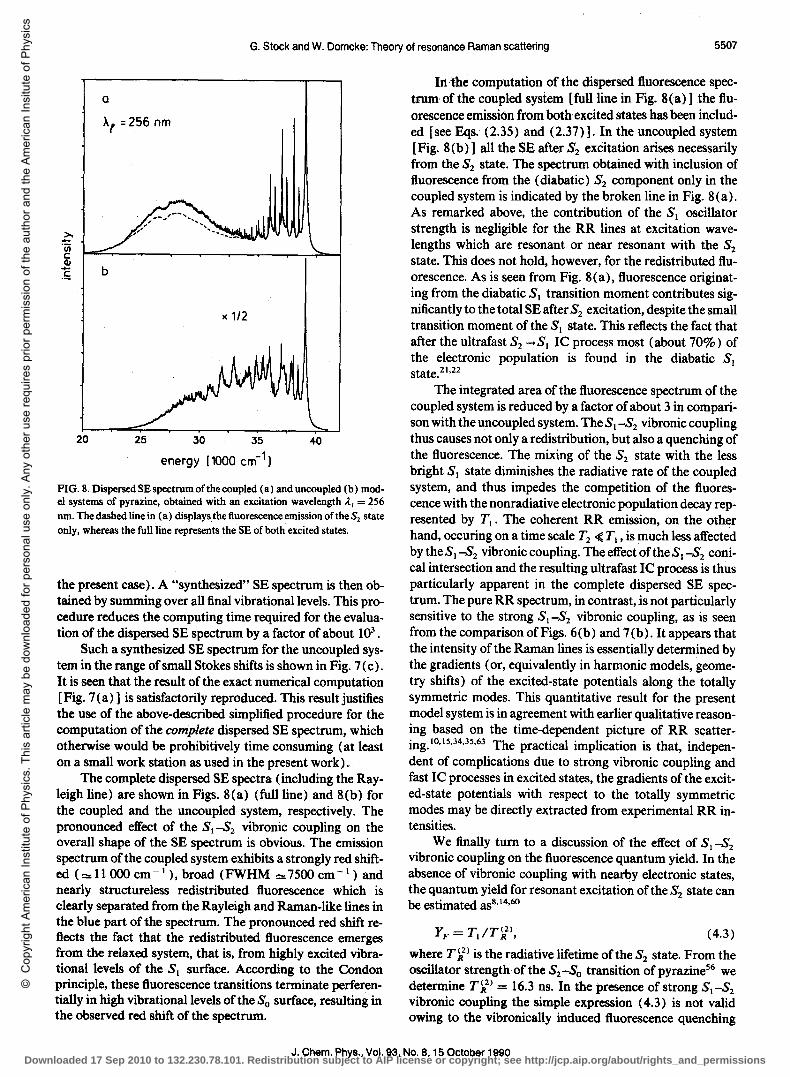
G. Stock and W. Domcke: Theory of resonance Raman scattering 5507
a
Af = 256 nm
)( 1/2
20 25 30 35 40
energy [1000 em-1)
FIG. 8. Dispersed SE spectrum of the coupled (a) and uncoupled (b) model systems of pyrazine, obtained with an excitation wavelength .1/ = 256 nm. The dashed line in (a) displaysthe fluorescence emission of the S2 state only, whereas the full line represents the SE of both excited states.
the present case). A "synthesized" SE spectrum is then obtained by summing over all final vibrational levels. This procedure reduces the computing time required for the evaluation of the dispersed SE spectrum by a factor of about 103 .
Such a synthesized SE spectrum for the uncoupled system in the range of small Stokes shifts is shown in Fig. 7 ( c ) . It is seen that the result of the exact numerical computation [Fig. 7 (a) ] is satisfactorily reproduced. This result justifies the use of the above-described simplified procedure for the computation of the complete dispersed SE spectrum, which otherwise would be prohibitively time consuming (at least on a small work station as used in the present work).
The complete dispersed SE spectra (including the Rayleigh line) are shown in Figs. 8(a) (full line) and 8(b) for the coupled and the uncoupled system, respectively. The pronounced effect of the SI-S2 vibronic coupling on the overall shape of the SE spectrum is obvious. The emission spectrum of the coupled system exhibits a strongly red shifted ("'" 11000 cm- I ), broad (FWHM "",7500 cm -1) and nearly structureless redistributed fluorescence which is clearly separated from the Rayleigh and Raman-like lines in the blue part of the spectrum. The pronounced red shift reflects the fact that the redistributed fluorescence emerges from the relaxed system, that is, from highly excited vibrationallevels of the SI surface. According to the Condon principle, these fluorescence transitions terminate perferentially in high vibrational levels of the So surface, resulting in the observed red shift of the spectrum.
In, the computation of the dispersed ft.uorescence spectrum of the coupled system [full line in Fig. 8(a)] the ft.uorescence emission from both excited states has been included [see Eqs. (2.35) and (2.37)]. In the uncoupled system [Fig. 8(b)] all the SE after S2 excitation arises necessarily from the S2 state. The spectrum obtained with inclusion of fluorescence from the (diabatic) S2 component only in the coupled system is indicated by the broken line in Fig. 8 (a) . As remarked above, the contribution of the SI oscillator strength is negligible for the RR lines at excitation wavelengths which are resonant or near resonant with the S2 state. This does not hold, however, for the redistributed fluorescence. As is seen from Fig. 8(a), fluorescence originating from the diabatic SI transition moment contributes significantly to the total SE after S2 excitation, despite the small transition moment of the S) state. This reflects the fact that after the ultrafast S2 .... S) IC process most (about 70%) of the electronic population is found in the diabatic SI state.2l ,22
The integrated area of the fluorescence spectrum of the coupled system is reduced by a factor of about 3 in comparison with the uncoupled system. The SI -S2 vibronic coupling thus causes not only a redistribution, but also a quenching of the fluorescence. The mixing of the S2 state with the less bright SI state diminishes the radiative rate of the coupled system, and thus impedes the competition of the fluorescence with the nonradiative electronic population decay represented by T1 • The coherent RR emission, on the other hand, occuring on a time scale T2 4: TI , is much less affected bytheSI-S2 vibroniccoupling. TheeffectoftheSI-S2 conical intersection and the resulting ultrafast IC process is thus particularly apparent in the complete dispersed SE spectrum. The pure RR spectrum, in contrast, is not particularly sensitive to the strong SI -S2 vibronic coupling, as is seen from the comparison of Figs. 6 (b) and 7 (b). It appears that the intensity of the Raman lines is essentially determined by the gradients (or, equivalently in harmonic models, geometry shifts) of the excited-state potentials along the totally symmetric modes. This quantitative result for the present model system is in agreement with earlier qUalitative reasoning based on the time-dependent picture of RR scattering. IO,15,34,35,63 The practical implication is that, indepen-dent of complications due to strong vibronic coupling and fast IC processes in excited states, the gradients ofthe excited-state potentials with respect to the totally symmetric modes may be directly extracted from experimental RR intensities.
We finally turn to a discussion of the effect of S 1 -S2 vibronic coupling on the fluorescence quantum yield. In the absence of vibronic coupling with nearby electronic states, the quantum yield for. resonant excitation of the S2 state can be estimated as8,I4,6O
YF = TIIT}fl, (4.3)
where Tk2) is the radiative lifetime of the S2 state. From the
oscillator strength of the S2 -So transition of pyrazine56 we determine T}fl = 16.3 ns. In the presence of strong S)-S2 vibronic coupling the simple expression (4.3) is not valid owing to the vibronically induced fluorescence quenching
J. Chern. Phys., Vol. 93, No.8, 15 October 1990 Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

5508 G. Stock and W. Domcke: Theory of resonance Raman scattering
mentioned above. We have therefore computed the fluorescence quantum yield of the coupled system from the definition (2.17), that is, the ratio of the total SE cross section to the absorption cross section. Comparison with the experimental result YF ::::: 10 - 5 at the center of the S2 absorption band of pyrazine60 allows us to determine the phenomenological model parameter TI for the S2 absorption band of pyrazine ( YF would be strictly unity for TI = T2 = 00 ). We thus estimate, as anticipated in Sec. IV A, TI "'" 500 fs. Application of Eq. (4.3), that is, neglecting the strong SI -S2 vibronic mixing, would have resulted in a much shorter population-decay lifetime, namely TI "'" 160 fs. This result illustrates that it is essential to take account of excited-state vibronic mixing in the estimation of phenomenological non-radiative decay rates. This point has also been emphasized recently by Nickel and Hertzberg for a different molecular system. 14
v. DISCUSSION AND CONCLUSIONS
A theoretical framework for the analysis of Raman and fluorescence emission from strongly vibronically coupled excited electronic states of isolated polyatomic molecules has been outlined. The main emphasis of our work is on a truly microscopic description of the ultrafast and strongly non-Markovian IC dynamics of conically intersecting higher excited singlet states of polyatomic systems, based on the (numerically) exact solution of the time-dependent Schr6-dinger equation for suitable multimode vibronic-coupling models. 18-23 Since any such microscopic model must necessarily be incomplete for larger polyatomic molecules, in particular with respect to the dynamical evolution at longer time scales, it is necessary to include additional relaxation effects in a phenomenological manner. Adopting the density-matrix formulation, which allows pure dephasing effects to be incorporated in addition to lifetime broadening effects,49,52 and assuming phenomenological relaxation rates which are approximately constant (that is, energy independent) within a given electronic absorption band, we have formulated a computationally manageable mixed microscopic/phenomenological theory, which should provide a powerful tool for the description of complex relaxation phenomena in excited electronic states. The present formulation extends existing eigenstate-free density-matrix formulations of RR scattering and fluorescence42.44 by including explicitly and nonperturbatively the strong non-BO phenomena associated with conical intersections in excited electronic states.
In order to illustrate the general ideas, we have performed numerical calculations of absorption, RR, and dispersed fluorescence spectra for a simple three-mode model of the vibronic coupling of the SI (mr*) and S2 ( 1T1r*) states ofpyrazine which has been proposed earlier. 21 As has been shown in detail elsewhere,22 this simple model describes an ultrafast (femtosecond) S2 --+SI IC process. The additional phenomenological total dephasing (lIT2 ) and lifetime broadening (lITI ) rates for the S2 state have been determined by comparison with the experimental S2 absorption profile and fluorescence quantum yield, respectively. It has been shown that the resulting model accounts on a qualita-
tive level for the main features of the available absorption and RR spectra of pyrazine.
The essential effects of excited-state vibronic coupling on the RR and fluorescence spectra found for the present model system can be summarized as follows: (i) The coupling mode v lOa' which is the only vibrational mode of pyrazine which can couple SI (mr*) and S2 emr*) in first order, is strongly active in the RR spectrum oflower (SI ) state, but is virtually absent in the RR spectrum of the higher (S2) state. (ii) The shape of the diffuse absorption profile and RR excitation profiles of the S2 state is strongly affected by the vibronic coupling: the SI -S2 conical intersection results in the appearance of widely and irregularly spaced broad structures which cannot be assigned in terms of unperturbed vibrational modes on the S2 surface. (iii) The vibronic coupling causes a suppression of the broad redistributed fluorescence background in the Raman region of the SE spectrum observed after S2 excitation. The main part of the fluorescence from S2 emerges strongly red shifted ( "'" 11 000 cm - 1 ). (iv) The mixing of S2 with the less efficiently fluorescing SI state is reflected by a significant decrease of the fluorescence quantum yield.
Most of these effects are presumably rather general features of SE from optically allowed excited singlet states which are coupled via a conical intersection to a lower lying dark (or only weakly allowed) singlet state in larger polyatomic molecules. The relative intensitivity of the pure RR signal to the ultrafast IC process of the higher excited state reflects the fact that the coherent Raman emission is restricted to ultrashort times by the very fast total dephasing of the S2 -So coherence. The Raman signal carries, therefore, relatively little information on the complex excited-state dynamics. It provides a simple means, however, to determine the gradients (or shifts of the equilibrium geometry) of the potential energy surface of short-lived excited states with respect to totally symmetric normal coordinates. 10,15,34,35,63 The redistributed fluorescence emission, on the other hand, arises from the dephased and electronically relaxed system and is therefore significantly affected by the non-BO dynamics in the excited states. The dramatic red shift of the center of the fluorescence band predicted by the present quantum dynamical calculations, for example, reflects most directly the existence ofa S2 --+SI IC process. We have also seen that consideration of the excited-state vibronic coupling is essential when estimating electronic population decay times from experimentally observed quantum yields.
RR scattering and fluorescence emission thus provide largely complementary information on the excited-state dynamics. The consistent prediction of both RR scattering and fluorescence emission from short-lived excited states of polyatomic molecules on an ab initio basis is still a major challenge for the theory. The aim of such a theory should be to include as much of the dynamics as possible in the microscopic model Hamiltonian H m' with electronic energies and coupling constants derived from ab initio computations. We believe that a rigorous treatment of the ultrafast non-BO dynamics associated with conical intersections of excited state surfaces, though computationally laborious, is essentially for the understanding of the spectroscopy of short-
J. Chem. Phys., Vol. 93, No.8, 15 October 1990 Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs

G. Stock and W. Domcke: Theory of resonance Raman scattering 5509
lived excited states in polyatomic molecules. Present-day computer technology allows the treatment of the time-dependent quantum dynamics of vibronic-coupling models with up to about ten nonseparable vibrational degrees of freedom (see Ref. 20 for an example). Once the correlation functions ofEq. (2.29) have been obtained for the appropriate final vibrational levels, the expressions given in Sec. II provide a comparatively simple means to evaluate the RR and fluorescence signals, involving only two phenomenological parameters (T\ and T2 ). While the ansatz (2.7) for the relaxation terms, in particular the assumption of energy-independent T\ and T2 within a given absorption band, is certainly an oversimplification, it may serve as a useful reference for future more ambitious treatments of dephasing and lifetime broadening effects in photon scattering from polyatomic molecules.
ACKNOWLEDGMENTS
The authors would like to thank Rudolf Schneider for stimulating discussions and help with the numerical calculations. This work has been supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
I D. L. Huber, Phys. Rev. 158, 843 (1967); 170, 418 (1968); 178, 93 (1969); Phys. Rev. B I, 3409 (1970).
2V. Hizhnyakov and I. Tehver, Phys. Status Solidi 21, 755 (1967). 3y. R. Shen, Phys. Rev. B 9, 622 (1974). 4Y.Toyozawa, J. Phys. Soc. Jpn. 41, 400 (1976). 5D. L. Rousseau and P. F. Williams,J. Chem. Phys. 64, 3519 (1976); J. M.
Friedman, ibid. 71, 3147 (1979). 6y. Fujimura, H. Kono, T. Nakajima, and S. H. Lin, J. Chem. Phys. 75, 99
(1981). 7S. Mukamel, J. Chem. Phys. 82, 5398 (1985). 8M. Kasha, Discuss. Faraday Soc. 9,14 (1950);J. B. Birks, Photo physics of Aromatic Molecules (Wiley, New York, 1970), Chap. 5.
9S. A. Asher, Annu. Rev. Phys. Chem. 39,537 (1988). 10 A. B. Myers, R. A. Mathies, I}; J. Tannor, and E. J. Heller,J. Chem. Phys.
77,3857 (1982). liD. P. Gerrity, L. D. Ziegler, P. B. Kelly, R. A. Desiderio, and B. Hudson,
J. Chem. Phys. 83,3209 (1985). 12C. M. Jones and S. A. Asher, J. Chern. Phys. 89, 2649 (1988). 13 R. J. Sension and B. S. Hudson, J. Chem. Phys. 90, 1377 (1989). 14B. Nickel and J. Hertzberg, Chem. Phys. 132,219 (1989). ISM. O. Trulson, G. D. Dollinger, and R. A. Mathies, J. Chem. Phys. 90,
4274 (1989); P. J. Reid, S. J. Doig, and R. A. Mathies, Chem. Phys. Lett. ·156, 163 (1989).
16G. Herzberg and H. C. Longuet-Higgins, Discuss. Faraday Soc. 35, 77 (1963).
17H. Koppel, L. S. Cederbaum, and w. Domcke, J. Chem. Phys. 77, 2014 ( 1982).
18H. Koppel, Chem. Phys. 77, 359 (1983). 19H. Koppel, W. Domcke, and L. S. Cederbaum, Adv. Chem. Phys. 57, 59
(1984). 2°H. Koppel, L. S. Cederbaum, and W. Domcke, J. Chem. Phys. 89, 2023
(1988). 21 R. Schneider and W. Domcke, Chem. Phys. Lett. ISO, 235 (1988). 22R. Schneider and W. Domcke, Chern. Phys. Lett. 159,61 (1989); R.
Schneider, W. Domcke, and H. Koppel.J. Chern. Phys. 92,1045 (1990). 23U. Manthe and H. Koppel, J. Chem. Phys. 93, 345, 1658 (1990). 24E. Haller, H. Koppel, and L. S. Cederbaum, J. Mol. Spectrosc.l11, 377
( 1985). 25W. Domckeand H. KOppel, Chern. Phys. Lett. 140, 133 (1987). 26 G. Stock, R. Schneider, and W. Domcke, J. Chern. Phys. 90, 7184 ( 1989). 27 G. Stock and W. Domcke, J. Opt. Soc. Am. B (in press). 28 H.-D. MeyerandH. KOppel,J. Chem. Phys. 81, 2605 (1984); H. Koppel
andH.-D. Meyer, Chern. Phys. Lett. 107, 149 (1984). 29H. A. Kramers and W. Heisenberg, Z. Phys. 31, 681 (1925); P. A. M.
Dirac, Proc. R. Soc. London 114, 710 (1927). lOW. H. Henneker, W. Siebrand, and M. Z. Zgierski, Chem. Phys. Lett. 43,
11 (1976); W. H. Henneker, A. P. Penner, W. Siebrand, and M. Z. Zgierski, ibid. 48, 197 (1977); M. Z. Zgierski and M. Pawlikowski, ibid. 63,221 (1979).
31 H. Hong, J. Chem. Phys. 67, 801, 813 (1977); 68, 1253 (1978). 32 J. A. Shelnutt and D. C. O'Shea, J. Chem. Phys. 69, 5361 (1978). 33W. H. Henneker, W. Siebrand, and M. Z. Zgierski, J. Chern. Phys. 74,
6560 (1981). 34S._Y. LeeandE. J. Heller,J. Chem. Phys. 71,4777 (1979);E.J. Heller, R.
L. Sundberg, and D. J. Tannor, J. Phys. Chem. 86, 1822 (1982); D. J. Tannor and E. J. Heller, J. Chem. Phys. 77, 202 (1982).
35R. D. Coalson and J. L. ~insey, J. Chem. Phys. 85, 4322 (1986). 36R. Heather and H. Metiu, J. Chem. Phys. 90, 6903 (1989). 37 R. Kubo, in Fluctuation. Relaxation. and Resonance in Magnetic Sys
tems, edited by D. ter Haar (Oliver and Boyd, Edinburgh, 1962), pp. 23-68.
38 R. M. Hochstrasser and F. A. Novak, Chem. Phys. Lett. 48, 1 (1977). 39T. Takagahara, E. Hanamura, and R. Kubo, J. Phys. Soc. Jpn. 43,802
(1977); 43,811 (1977). 4OZ. Deng and S. Mukamel, J. Chem. Phys. 85,1738 (1986). 41R. Kubo, Pure Appl. Chem. 57, 201 (1985). 42S. Mukamel, Adv. Chem. Phys. 70, 165.(1988), and references therein. 43R. M. Hochstrasser and F. A. Novak, Chem. Phys. Lett. 53, 3 (1978). 44K. Shan, Y. Yan, and S. Mukamel, J. Chern. Phys. 87, 2021 (1987). 45D. L. Tonks and J. B. Page, J. Chem. Phys. 75,5694 (1981). 46p. M. Champion and A. C. Albrecht, Annu. Rev. Phys. Chem. 33,353
(1982). 47 See, for example, F. Haake in Quantum Statistics in Optics and Solid-State
Physics, edited by G. Hohler, Vol. 66 of Springer Tracts in Modem Physics (Springer, Berlin, 1973), p. 98.
48 H. C. Longuet-Higgins, in Advances in Spectroscopy, edited by H. W. Thompson (Interscience, New York, 1961), Vol. 2, p. 429; F. T. Smith, Phys. Rev. 179, III (1969).
49 Y. R. Shen, The Principles of Nonlinear Optics (Wiley, New York, 1984). so R. H. Pantell and H. E. Puthoff, Fundamentals of Quantum Electronics
(Wiley, New York, 1969). 51 J. Tang and A. C. Albrecht, in Raman Spectroscopy, edited by H. A. Szy
manski (Plenum, New York, 1970), Vol. 2, p. 33. 52 W. H. Louisell, Quantum Statistical Properties of Radiation (Wiley, New
York,1973). 53F. Bloch and A. Siegert, Phys. Rev. 57, 522 (1940); M. P. Silverman and
F. M. Pipkin, J. Phys. B 5, 1844 (1972). 54G. Stock, thesis, Technical University of Munich, 1990 (unpublished). 55 R. A. Harris, R. A. Mathies, and W. T. Pollard, J. Chem. Phys. 85,3744
(1986). 56K. K. Innes, J. G. Ross, and W. R. Moomaw, J. Mol. Spectrosc.132, 492
(1988). 57 I. Suzuka, Y. Udagawa, and M. Ito, Chem. Phys. Lett. 64, 333 (1979). 58 J. Kommandeur, W. A. Majewski, w. L. Meerts, and D. W. Pratt, Annu.
Rev. Phys. Chem. 38, 433 (1987). 59 A. Amirav, Chern. Phys. 126, 327 (1988). 601. Yamazaki, T. Murao, T. Yamanaka, and K. Yoshihara, Faraday Dis-
cuss. Chern. Soc. 75, 395 (1983). 61 H.-K. Hong and C. W. Jacobsen, J. Chem. Phys. 68,1170 (1978). 62 C. K. Chan and J. B. Page, J. Chern. Phys. 79, 5234 (1983). 63 A. B. Myers, R. A. Harris, and R. A. Mathies, J. Chern. Phys. 79, 603
( 1983); A. B. Myers and R. A. Mathies, ibid. 81, 1552 (1984).
J. Chem. Phys., Vol. 93, No.8, 15 October 1990 Downloaded 17 Sep 2010 to 132.230.78.101. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
© C
opyr
ight
Am
eric
an In
stitu
te o
f Phy
sics
. Thi
s ar
ticle
may
be
dow
nloa
ded
for p
erso
nal u
se o
nly.
Any
oth
er u
se re
quire
s pr
ior p
erm
issi
on o
f the
aut
hor a
nd th
e A
mer
ican
Insi
tute
of P
hysi
cs





























