Embed Size (px)
Citation preview

The Use of Density Functional Theory in the
Calculation of Thermodynamic Properties
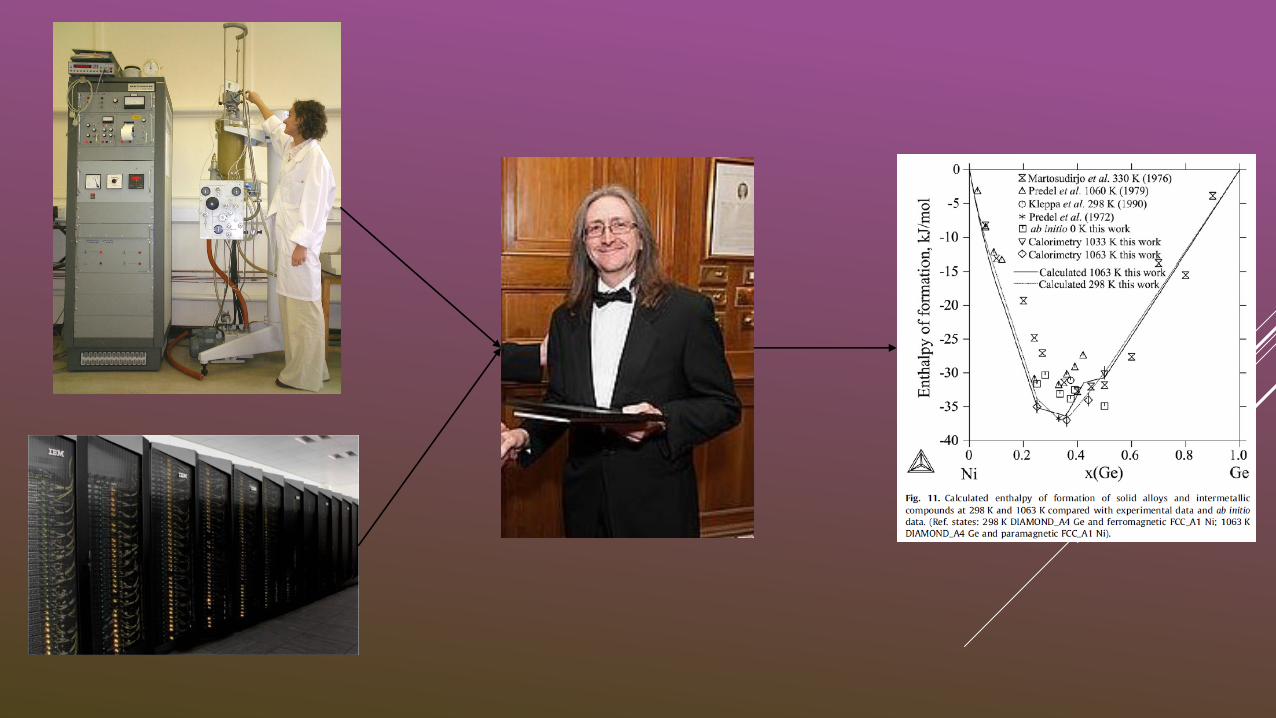
Dr Andrew Scott, University of Leeds, UK
Hume-Rothery Seminar, Derby, 2016
Materials Chemistry Committee

Contents
Introduction
Density functional theory
- background
- accuracy – exchange correlation functional
- precision – k-point mesh, basis set expansion
- geometry optimisation
Case studies
- Ni, V
- Ni3V
- Ni,V sigma phase
Computational cost
Summary

- Accurate thermodynamic data and phase diagrams are vital in materials research
- Data historically provided by experimental determination of properties
- Accurate data determined directly from theory without any empirical input – the so-called ab-
initio methods – can been used as an important additional source of information
- Density functional theory suitable for studying all elements and compounds and shown to
provide accurate property data for metals, alloys, ionic, covalent and molecular systems.
- Properties such as heats of formation, phase stabilities, surface energies and elastic constants
can be obtained from known stable or metastable structures
- Information added to experimental data to improve phase diagram assessments using the
CALPHAD method

Exact – all terms known except the exchange correlation potential, Vxc
Each electron is surrounded by its own mutual exclusion zone or hole.
The exact shape of this hole is only known for a free electron gas and this is the
basis of the first approximation to this term, the local density approximation
(LDA) (Kohn, 1965)
)()()]()([)(2
22
rErrVrVrm
xcH
Ab-initio methods are based on quantum theory
Schrodinger equation (1926) – exact, essentially impossible to solve for
systems with more than one electron
Density functional theory (1964) – Hohenberg and Kohn
‘simplified’ – now based on electron density
Walter Kohn
Nobel Prize, 1998
1887-1961

Exchange-correlation potential – gives the accuracy of the calculation
Local density approximation (LDA)
- assumed to have limited applicability – metallic bonding
- proved to be successful for a whole range of ionic, covalent and metallic materials
- limitations
- tends to ‘over-bind’ – lattice parameters/ bonds too short cf expt
- underestimates band gaps in semi-conductors and insulators
- incorrectly predicts the ground state of structure of iron to be non-magnetic and fcc
Generalised gradient approximations (GGA)
- ‘improved’ exchange-correlation functionals developed from early 1990s
Dispersion corrected functionals – molecular systems
Hybrid functionals
You should always review the literature, talk to colleagues and test the validity of the
approximation for your system of interest.
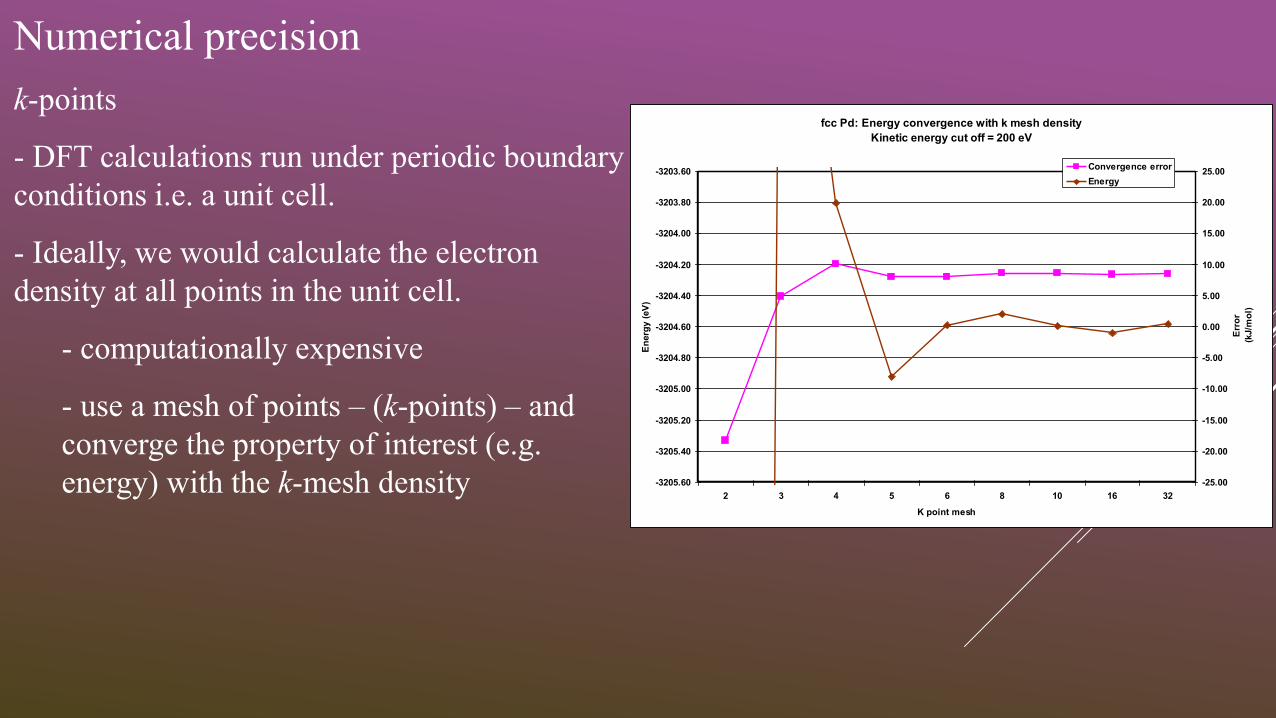
Numerical precision
k-points
- DFT calculations run under periodic boundary
conditions i.e. a unit cell.
- Ideally, we would calculate the electron
density at all points in the unit cell.
- computationally expensive
- use a mesh of points – (k-points) – and
converge the property of interest (e.g.
energy) with the k-mesh density
fcc Pd: Energy convergence with k mesh density
Kinetic energy cut off = 200 eV
-3205.60
-3205.40
-3205.20
-3205.00
-3204.80
-3204.60
-3204.40
-3204.20
-3204.00
-3203.80
-3203.60
2 3 4 5 6 8 10 16 32
K point mesh
En
erg
y (
eV
)
-25.00
-20.00
-15.00
-10.00
-5.00
0.00
5.00
10.00
15.00
20.00
25.00
Err
or
(kJ/m
ol)
Convergence error
Energy
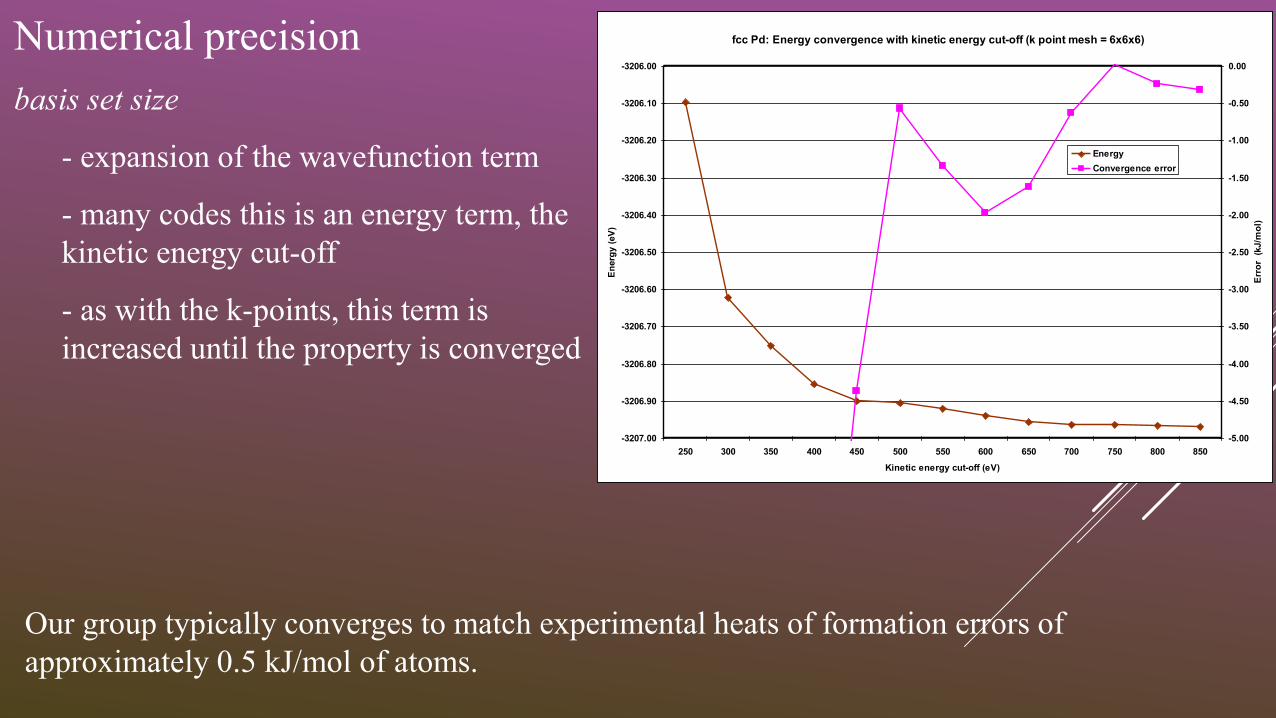
Numerical precision
basis set size
- expansion of the wavefunction term
- many codes this is an energy term, the
kinetic energy cut-off
- as with the k-points, this term is
increased until the property is converged
fcc Pd: Energy convergence with kinetic energy cut-off (k point mesh = 6x6x6)
-3207.00
-3206.90
-3206.80
-3206.70
-3206.60
-3206.50
-3206.40
-3206.30
-3206.20
-3206.10
-3206.00
250 300 350 400 450 500 550 600 650 700 750 800 850
Kinetic energy cut-off (eV)
En
erg
y (
eV
)
-5.00
-4.50
-4.00
-3.50
-3.00
-2.50
-2.00
-1.50
-1.00
-0.50
0.00
Err
or
(kJ/m
ol)
Energy
Convergence error
Our group typically converges to match experimental heats of formation errors of
approximately 0.5 kJ/mol of atoms.
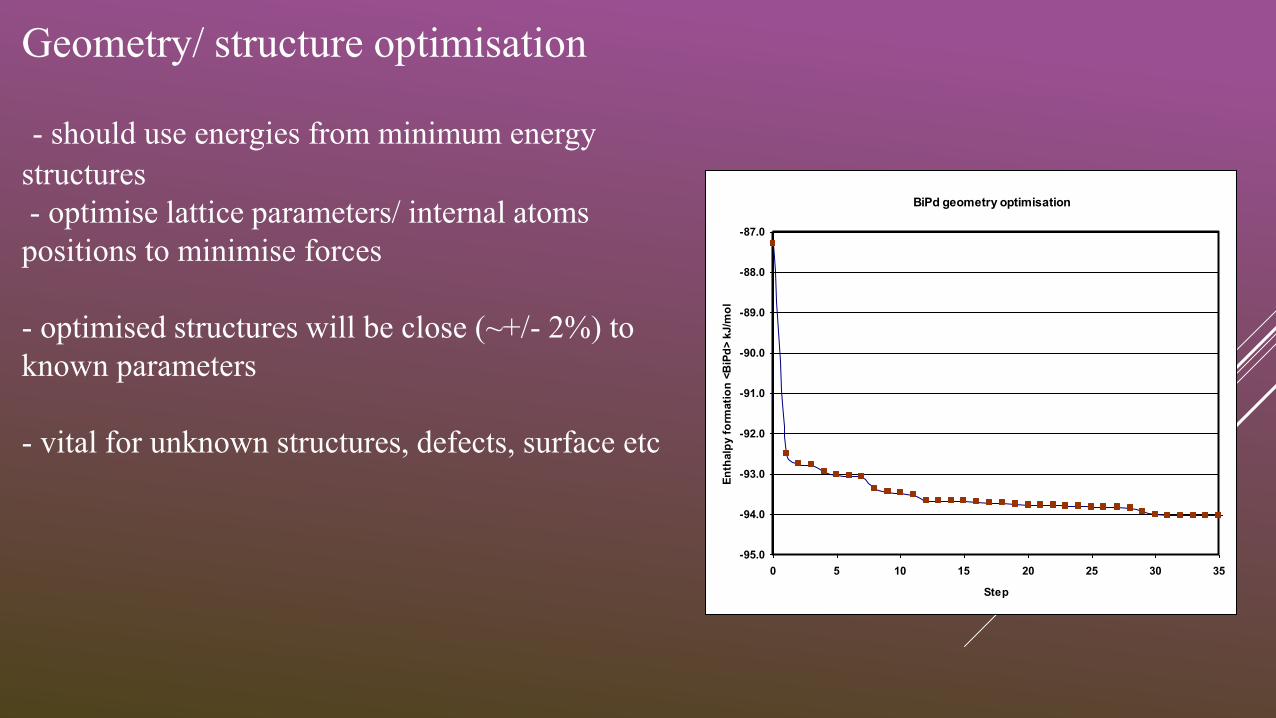
BiPd geometry optimisation
-95.0
-94.0
-93.0
-92.0
-91.0
-90.0
-89.0
-88.0
-87.0
0 5 10 15 20 25 30 35
Step
En
thalp
y f
orm
ati
on
<B
iPd
> k
J/m
ol
Geometry/ structure optimisation
- should use energies from minimum energy
structures
- optimise lattice parameters/ internal atoms
positions to minimise forces
- optimised structures will be close (~+/- 2%) to
known parameters
- vital for unknown structures, defects, surface etc
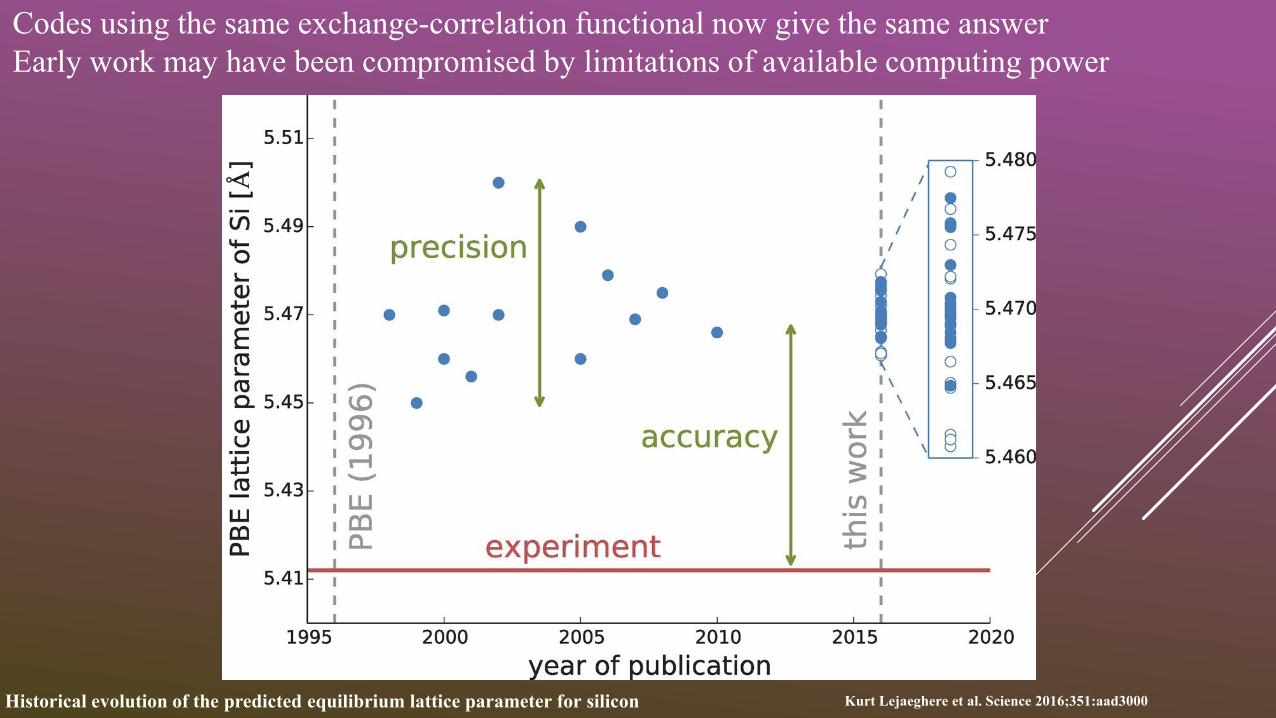
Historical evolution of the predicted equilibrium lattice parameter for silicon Kurt Lejaeghere et al. Science 2016;351:aad3000
Codes using the same exchange-correlation functional now give the same answer
Early work may have been compromised by limitations of available computing power

Case studies
Elements – phase/ lattice stabilities
- vanadium
- nickel
Simple compound –
- Ni3V – heat of formation
Sigma phase, Ni,V
- 3 sub-lattice model
- 5 sub-lattice model
Structure databases
e.g. http://icsd.cds.rsc.org/

DFT code, parameters
- calculations were performed using the pseudopotential code, Castep
- GGA-PBE exchange-correlation functional
- k-point density 0.04 Å-1 and kinetic energy cut-off of 440 eV giving good numerical
precision
- ‘on the fly’ ultrasoft pseudopotentials were used for all the elements
- calculations were spin polarized and all structures were geometry optimized

Lattice
parameter
Energy Lattice
stability
(DFT)
Lattice
stability
(SGTE)
Å eV/atom kJ/(mole
atoms)
kJ/(mole
atoms)
bcc a=3.0016
(-0.9%)
-1951.77778 0
fcc a=3.8206 -1951.51756 +25.1 7.5
hcp a=2.8125
c=4.0737
-1951.44057 +32.5 4.0
DFT predicts vanadium to be body centred cubic and non-magnetic bcc V

fcc Ni
Lattice
parameter
Energy Lattice
stability
(DFT)
Lattice
stability
(SGTE)
Å eV kJ/(mole
atoms)
kJ/(mole
atoms)
fcc a=3.520
(-0.1%)
-1375.96867 0 0
hcp a=2.4893 -1375.94478 +2.3 +2.9
c=4.0823
bcc a=2.8020 -1375.87072 +9.5 +8.0
DFT predicts nickel to be face centred cubic and magnetic
Turchi, 2007, addresses the discrepancies between ab-initio and CALPHAD lattice
stabilities, with the conclusion that ab-initio data from dynamically unstable structures are
not valid. (can discuss further at coffee)
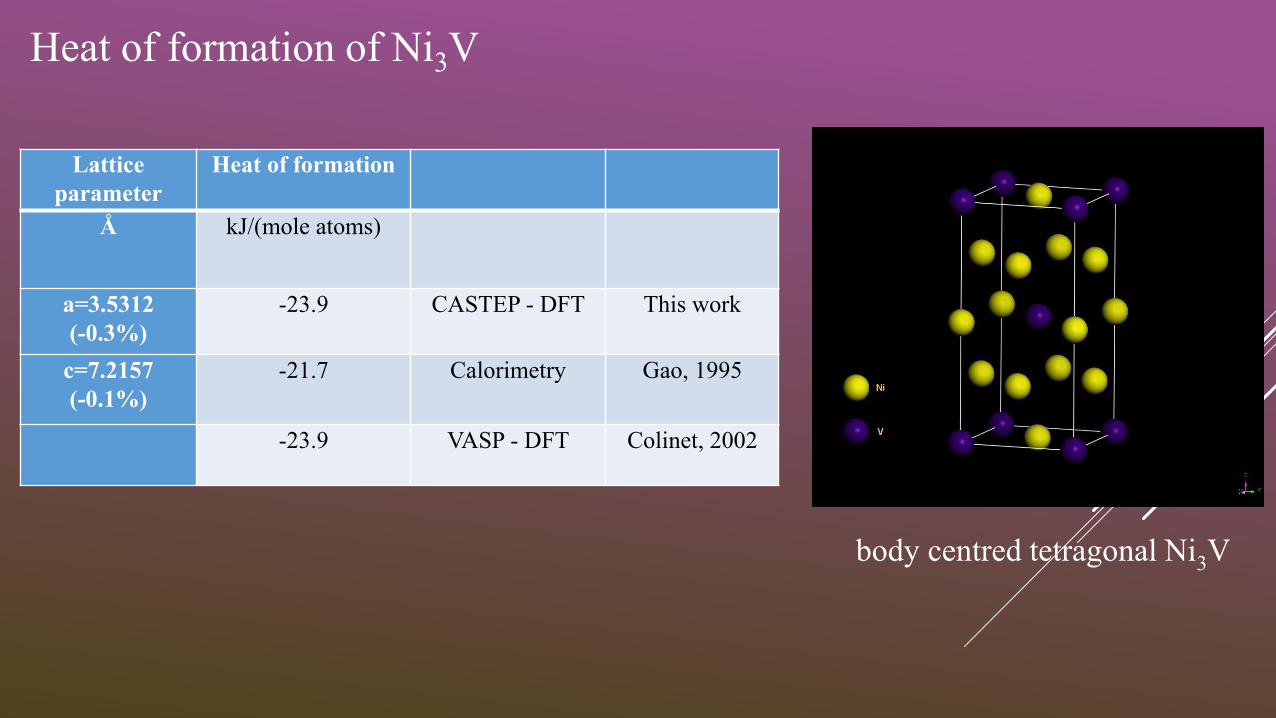
body centred tetragonal Ni3V
Heat of formation of Ni3V
Lattice
parameter
Heat of formation
Å kJ/(mole atoms)
a=3.5312
(-0.3%)
-23.9 CASTEP - DFT This work
c=7.2157
(-0.1%)
-21.7 Calorimetry Gao, 1995
-23.9 VASP - DFT Colinet, 2002
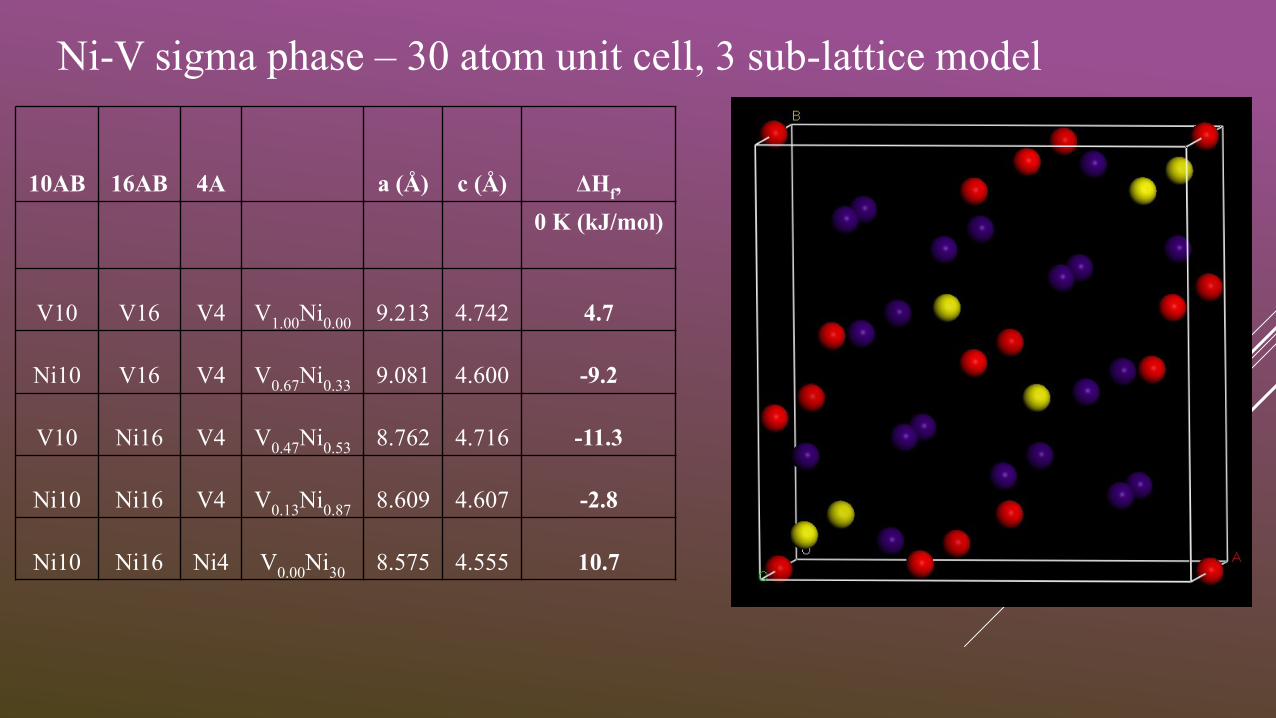
Ni-V sigma phase – 30 atom unit cell, 3 sub-lattice model
10AB 16AB 4A a (Å) c (Å) ΔHf,
0 K (kJ/mol)
V10 V16 V4 V1.00
Ni0.00
9.213 4.742 4.7
Ni10 V16 V4 V0.67
Ni0.33
9.081 4.600 -9.2
V10 Ni16 V4 V0.47
Ni0.53
8.762 4.716 -11.3
Ni10 Ni16 V4 V0.13
Ni0.87
8.609 4.607 -2.8
Ni10 Ni16 Ni4 V0.00
Ni30
8.575 4.555 10.7
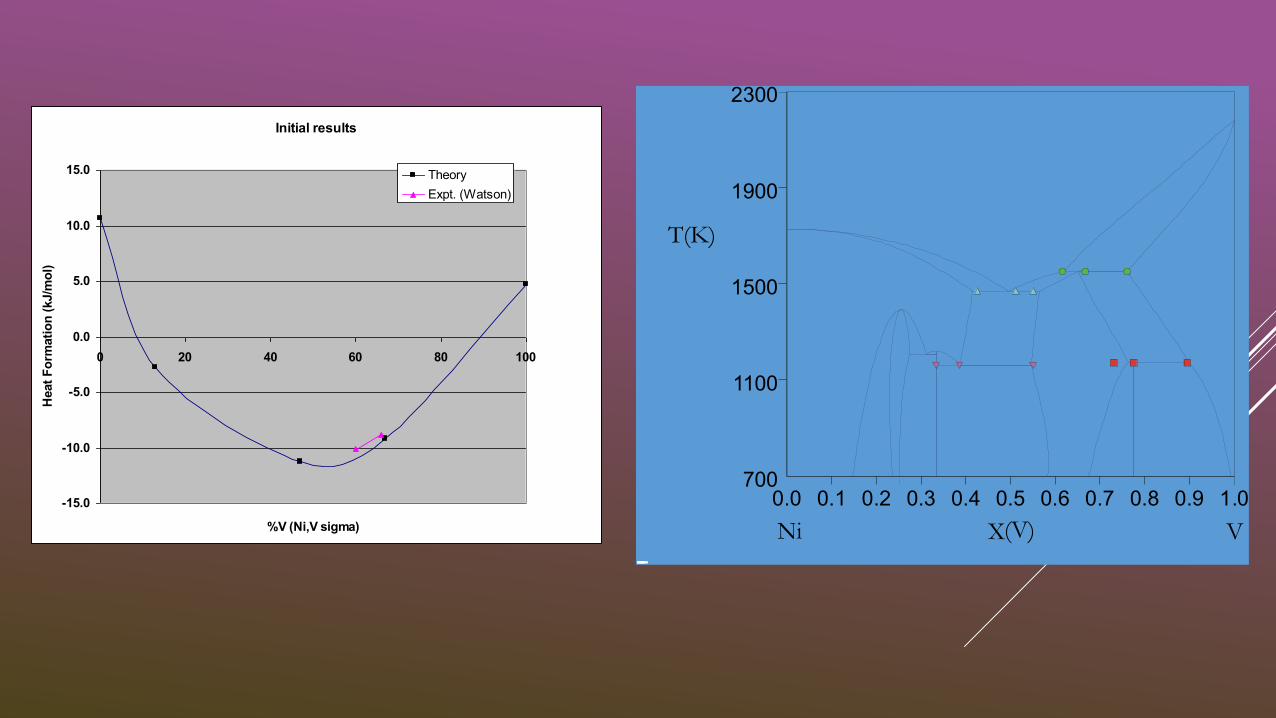
Initial results
-15.0
-10.0
-5.0
0.0
5.0
10.0
15.0
0 20 40 60 80 100
%V (Ni,V sigma)
Heat
Fo
rmati
on
(kJ/m
ol)
Theory
Expt. (Watson)
T(K)
X(V)
700
1100
1500
1900
2300
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Ni V
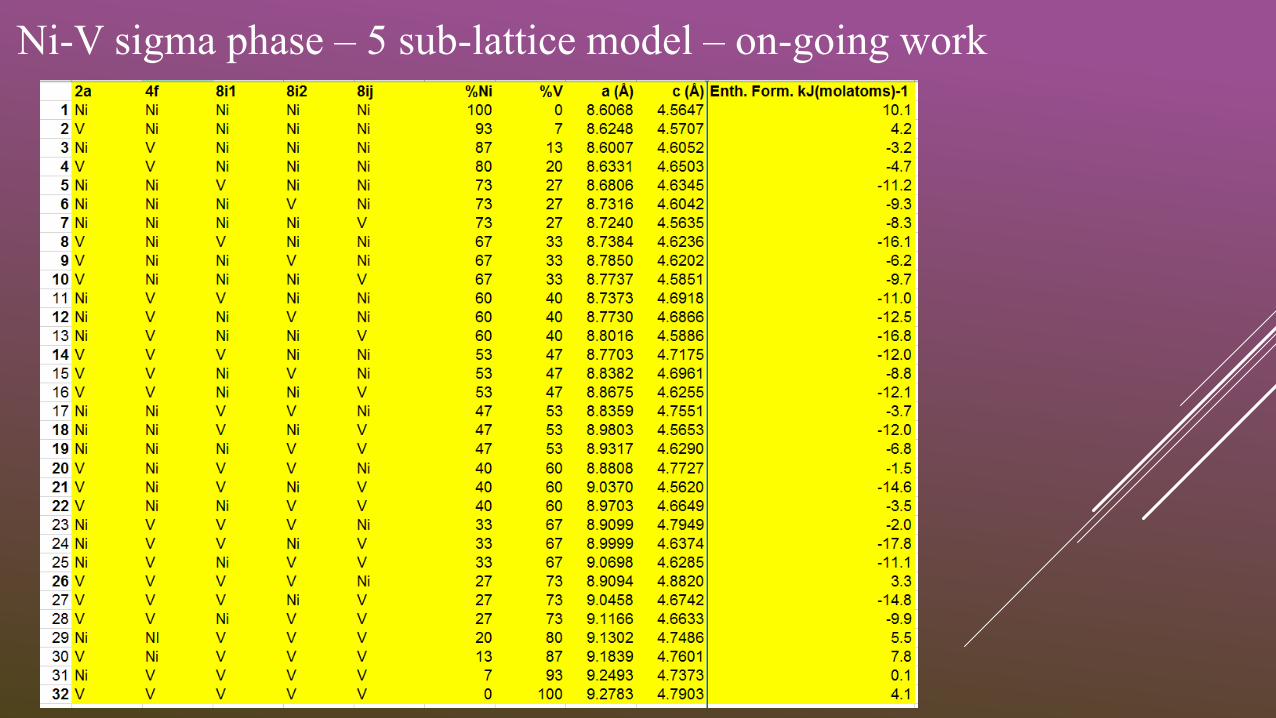
Ni-V sigma phase – 5 sub-lattice model – on-going work
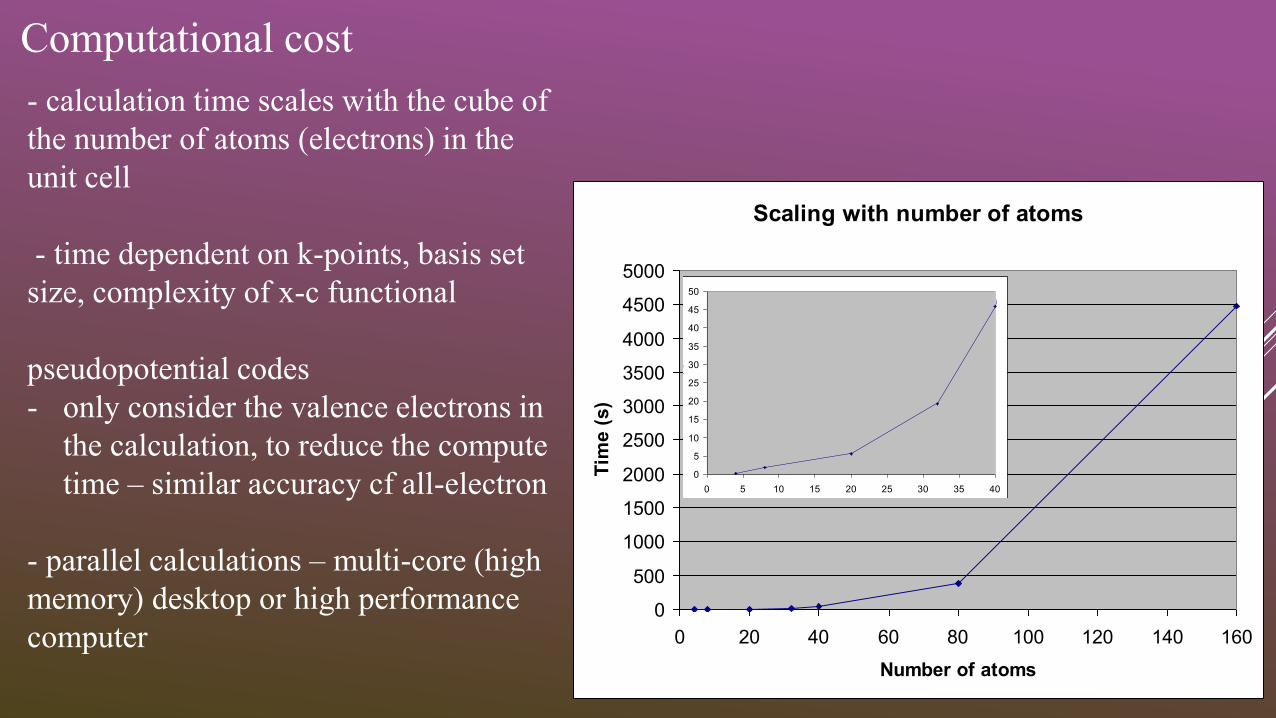
Computational cost
- calculation time scales with the cube of
the number of atoms (electrons) in the
unit cell
- time dependent on k-points, basis set
size, complexity of x-c functional
pseudopotential codes
- only consider the valence electrons in
the calculation, to reduce the compute
time – similar accuracy cf all-electron
- parallel calculations – multi-core (high
memory) desktop or high performance
computer
Scaling with number of atoms
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
0 20 40 60 80 100 120 140 160
Number of atoms
Tim
e (
s)
0
5
10
15
20
25
30
35
40
45
50
0 5 10 15 20 25 30 35 40
Number of atoms
Tim
e (
s)

1997 Silicon Graphics O2 (muffin)
1 core, memory 64Mb (£7,550)
2002 Leeds Grid Node 1 (maxima)
60 cores, total memory 168GB
2002 Leeds Grid Node 2 (Snowdon)
282 cores, total memory 282Gb, Myrinet B
2005 Leeds Grid Node 3 (Everest)
444 cores, total memory 1008Gb, Myrinet F
2008 National Grid Service (Leeds)
160 cores (Gb) total memory ~224Gb
2010- Leeds ARC1
2592 cores (1.5-2Gb), Infiniband,
2013- N8 (Leeds) Polaris
5056 cores (4Gb), 256 cores (16Gb),InfiniBand
2013 iDataplex - Blue Wonder
8192 cores; 64 TB memory, 158.7 TeraFlop/s
2014 - ? ARC2, ARC3, N8-2? + multicore, large memory pc

There are many DFT codes available for the researcher. What factors should be considered
when deciding on the code to use?
i) cost – free academic codes to commercial codes
ii) support – large user base (listserver)/ workshops
iii) ongoing development – latest X-C functionals, optimized to run in parallel on latest
computing architecture
iv) good interface to aid setting up the calculation and analyzing the results
Recommended: the all-electron code, WIEN2k (Blaha, 2001) and the pseudopotential codes,
Castep (Clark, 2005) and VASP (Kresse, 1996)

Summary
DFT can give ground state (0 K) properties:
- lattice parameters
- bulk energies
- lattice/phase stabilities
- elastic properties
- surface energies
Temperature dependent calculations possible but at computational cost
Excellent accuracy in most cases for stable or metastable structures. Systems of 10s of atoms
can be studied on a desktop pc and 100s of atoms on a parallel high performance computer.
Thermodynamic data used by CALPHAD community to improve existing phase diagram
assessments
All work in collaboration Dr Andy Watson





























