Embed Size (px)
DESCRIPTION
.
Citation preview

The Role of Cytokines in Cancer Cachexia
Josep M. Argilés, Francisco J. López-Soriano
Departament de Bioquímica i Biologia Molecular, Facultat de Biologia,
Universitat de Barcelona, Barcelona, Spain
▼
Abstract: A large number of observations point towards cytokines, polypeptides released mainlyby immune cells, as the molecules responsible for the metabolic derangements associated withcancer-bearing states. Indeed, these alterations lead to a pathological state known as cancercachexia which is, unfortunately, one of the worst effects of malignancy, accounting for nearly athird of cancer deaths. It is characterized by weight loss together with anorexia, weakness, ane-mia, and asthenia. The complications associated with the appearance of the cachectic syndromeaffect both the physiological and biochemical balance of the patient and have effects on the effi-ciency of the anticancer treatment, resulting in a considerably decreased survival time. At the meta-bolic level, cachexia is associated with loss of skeletal muscle protein together with a depletion ofbody lipid stores. The cachectic patient, in addition to having practically no adipose tissue, is ba-sically subject to an important muscle wastage manifested as an excessive nitrogen loss. The meta-bolic changes are partially mediated by alterations in circulating hormone concentrations (insulin,glucagon, and glucocorticoids in particular) or in their effectiveness. The present study reviewsthe involvement of different cytokines in the metabolic and physiological alterations associatedwith tumor burden and cachexia. Among these cytokines, some can be considered as procachec-tic (such as tumor necrosis factor-a), while others having opposite effects can be named as anti-cachectic cytokines. It is the balance between these two cytokine types that finally seems to havea key role in cancer cachexia. © John Wiley & Sons, Inc. Med Res Rev, 19, No. 3, 223–248, 1999.
Key words: cancer cachexia; acute-phase; muscle wasting; metabolic changes; cytokines
1. I N T R O D U C T I O N
Approximately two thirds of patients who die with advanced cancer suffer from cancer cachexia.1
Indeed, the degree of cachexia is inversely correlated with the survival time of the patient and it al-
223
© 1999 John Wiley & Sons, Inc. CCC 0198-6325/99/030223-26
Correspondence to: Dr. Josep M. Argilés, Unitat de Bioquímica i Biologia Molecular B, Departament de Bio-química i Biologia Molecular, Facultat de Biologia, Universitat de Barcelona, Diagonal 645, 08071-Barcelona,Spain; e-mail: [email protected] grant sponsor: Fondo de Investigaciones Sanitarias de la Seguridad Social (F.I.S.) of the SpanishHealth Ministry; Contract grant number: 94/1048.Contract grant sponsor: DGICYT of the Spanish Ministry of Education and Science; Contract grant number:PB94-0938.Contract grant sponsor: Fundació Pi i Sunyer; Contract grant number: E00667.
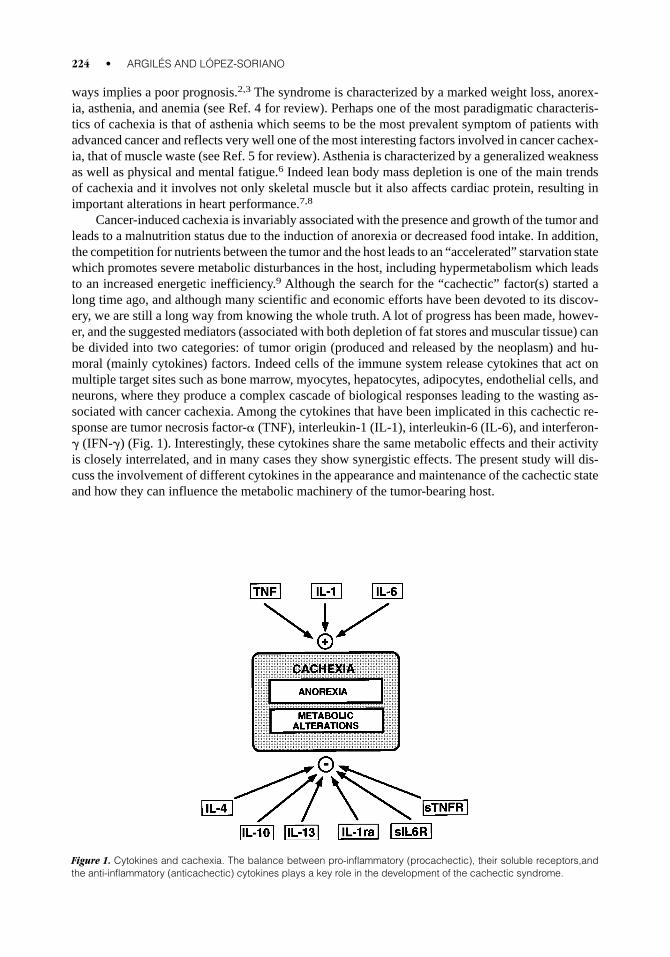
ways implies a poor prognosis.2,3 The syndrome is characterized by a marked weight loss, anorex-ia, asthenia, and anemia (see Ref. 4 for review). Perhaps one of the most paradigmatic characteris-tics of cachexia is that of asthenia which seems to be the most prevalent symptom of patients withadvanced cancer and reflects very well one of the most interesting factors involved in cancer cachex-ia, that of muscle waste (see Ref. 5 for review). Asthenia is characterized by a generalized weaknessas well as physical and mental fatigue.6 Indeed lean body mass depletion is one of the main trendsof cachexia and it involves not only skeletal muscle but it also affects cardiac protein, resulting inimportant alterations in heart performance.7,8
Cancer-induced cachexia is invariably associated with the presence and growth of the tumor andleads to a malnutrition status due to the induction of anorexia or decreased food intake. In addition,the competition for nutrients between the tumor and the host leads to an “accelerated” starvation statewhich promotes severe metabolic disturbances in the host, including hypermetabolism which leadsto an increased energetic inefficiency.9 Although the search for the “cachectic” factor(s) started along time ago, and although many scientific and economic efforts have been devoted to its discov-ery, we are still a long way from knowing the whole truth. A lot of progress has been made, howev-er, and the suggested mediators (associated with both depletion of fat stores and muscular tissue) canbe divided into two categories: of tumor origin (produced and released by the neoplasm) and hu-moral (mainly cytokines) factors. Indeed cells of the immune system release cytokines that act onmultiple target sites such as bone marrow, myocytes, hepatocytes, adipocytes, endothelial cells, andneurons, where they produce a complex cascade of biological responses leading to the wasting as-sociated with cancer cachexia. Among the cytokines that have been implicated in this cachectic re-sponse are tumor necrosis factor-a (TNF), interleukin-1 (IL-1), interleukin-6 (IL-6), and interferon-g (IFN-g) (Fig. 1). Interestingly, these cytokines share the same metabolic effects and their activityis closely interrelated, and in many cases they show synergistic effects. The present study will dis-cuss the involvement of different cytokines in the appearance and maintenance of the cachectic stateand how they can influence the metabolic machinery of the tumor-bearing host.
224 • ARGILÉS AND LÓPEZ-SORIANO
Figure 1. Cytokines and cachexia. The balance between pro-inflammatory (procachectic), their soluble receptors,andthe anti-inflammatory (anticachectic) cytokines plays a key role in the development of the cachectic syndrome.

2. M A L N U T R I T I O N
Malnutrition is indeed one of the hallmarks of cancer cachexia and it is basically associated withanorexia, or declining food intake. But are cytokines involved in the induction of anorexia? Feedingis a complex function resulting from the integration of peripheral and central nervous impulses inthe ventral hypothalamus.10,11 Stimulation of the medial hypothalamic nucleus inhibits feeding,while stimulation of the lateral nucleus promotes food intake. Of the peripheral impulses, oral stim-ulation by pleasant tastes elicits the feeding, while gastrointestinal distention prevents it. Concern-ing the possible involvement of cytokines in tumor-induced anorexia, although it is not preciselyconclusive, several points have to be taken into consideration. Cytokines such as IL-112,13 and TNF14
have been proposed as involved in cancer-related anorexia, possibly by increasing the levels of CRH,a central nervous system neurotransmitter that suppresses food intake and the firing of glucose-sensitive neurons, which would also decrease food intake.15 But cytokines do not seem to be the onlymediators of tumor-induced anorexia. Since multiple factors are involved in controlling food intake,it is therefore possible that there are also many factors contributing to the tumor-associated anorex-ia. Indeed, anorexigenic compounds are either released by the tumor into the host’s circulation or thepresence of the tumor may induce metabolic changes resulting in the release of such substances byhost tissues.4 Indeed, changes in the concentration of plasma amino acids associated with tumor bur-den16,17 may alter the bioavailability of some amino acids to the brain affecting neurotransmittersynthesis. This is the case for brain serotonin and its precursor, plasma tryptophan, which have beeninvolved in the pathogenic mechanisms leading to cancer-related anorexia. Increased brain avail-ability of tryptophan during cancer results in increased brain serotonin synthesis and therefore sero-tonergic activity, thus leading to reduced food intake.18 Other factors that could be involved in pro-moting the inhibitory impulses which are transmitted to the hypothalamus by stimulation ofserotoninergic and cathecolaminergic fibers are increased lactate and fatty acids blood levels,19,20
both associated with tumor burden. Fischer and colleagues have suggested that cancer anorexia isinduced through an elevation of circulating ammonia levels. Indeed, plasma ammonia concentrationsare elevated significantly before the onset of anorexia, and the intravenous infusion of ammoniumsalts into normal rats elicited anorexia at blood concentrations similar to those observed in anorec-tic tumor-bearing rats.21 Infusing these salts also produced nearly all the neurochemical aberrationsin neurotransmitters and brain amino acid profile observed in anorectic tumor-bearing rats.21 Inter-estingly, administration of methionine sulfoximine (an inhibitor of glutamine synthetase, one of theenzymes responsible for ammonia detoxification) intensifies cancer anorexia.22 Leptin seems to bethe main molecule that signals to the brain and controls food intake synthesized in adipose tissue, soit could well be a good candidate for mediating tumor-induced anorexia. Several studies have shownincreased leptin concentrations during sepsis and following cytokine administration.23,24 However,recent experiments carried out in our laboratory indicate that tumor-bearing rats underexpress theleptin gene in white adipose tissue, while the circulating concentrations of this compound also de-crease with tumor burden.25 In addition, plasma levels of leptin are not elevated in patients with lungcancer-associated cachexia.26 It can thus be concluded that the role of leptin in modulating food in-take during tumor growth is far from being understood. Chance et al. have shown that the hypothal-amic concentration and release of neuropeptide Y (which is decreased as leptin increases) is reducedin anorectic tumor-bearing rats.27 Interestingly, this neuropeptide blocks and reverses IL-1-inducedanorexia in rats.28
In any case, anorexia seems to be the effect rather than the cause of weight loss during cancer;cachexia perpetuates and worsens itself through a mechanism involving anorexia in a sort of posi-tive feedback which usually leads to death. There are pieces of evidence in favor of this argument.Firstly, total parenteral nutrition (which involves a high clinical cost) in patients with a high degreeof cachexia has led to controversial results, some being beneficial29 and some resulting in no im-provement in survival time.30 However, even in the cases where it promoted an increase in body
CYTOKINES AND CANCER CACHEXIA • 225

weight, this was not associated with an increase in lean body mass but it seemed rather to increasewater retention, which was manifested as a peripheral edema together with a decreased hematocrit.31
Secondly, in different experimental models, pair-feeding does not lead either to the same extent ofweight loss or metabolic abnormalities as found in the tumor-bearing animals. In fact, tumor burdeninduces metabolic changes which do not resemble those of caloric restriction or starvation but ratherthose found in infection or injury.32 Thirdly, sometimes tumors involve the alimentary tract and re-sult in diminished food intake as a consequence of mechanical obstruction, pain or early satiety.33
In addition, anticancer treatment (such as chemotherapy, radiotherapy or immunotherapy) may beassociated with varying degrees of nausea, vomiting, and, consequently, anorexia. Furthermore, al-terations in food perception (taste and smell) together with psychological causes (such as depres-sion) may contribute to the reduced food intake.33
3. B O D Y W E I G H T L O S S A N D C I R C U L A T I N GC Y T O K I N E S
Perhaps weight loss, the most paradigmatic trend of cancer cachexia, can be mimicked by cytokinesunder some experimental conditions. Nevertheless, episodic TNF administration has proved unsuc-cessful at inducing cachexia in experimental animals.34 – 37 Indeed, repetitive TNF administrationsinitially induce a cachectic effect although tolerance to the cytokine soon develops and food intakeand body weight return to normal. Others have shown that escalating doses of TNF are necessary tomaintain the cachectic effects.38 However, a very elegant approach involving the implantation ofCHO cells which were transfected with the human TNF gene in nude mice39 seems to indicate thatTNF may have a clear and important role in the induction of cachexia.
Raised concentrations of TNF have been detected in the serum of about 70% of patients withparasitic infections such as leishmaniasis and malaria40,41 and in patients with septicemia,42 all ofwhich are pathological states where a high degree of cachexia is achieved. The increase in plasmaTNF in septicemia is likely to be due to increased concentrations of endotoxin which can elicit a tran-sitory rise in plasma TNF when administered to healthy control subjects.43 – 45 In contrast, evidencefor increased TNF in the plasma of cancer patients is controversial. Balkwill et al. found that 50%of 226 fresh serum samples from cancer patients had a positive response for TNF with an enzyme-linked immunosorbent assay,46 and more recently the presence of this cytokine has been observedin the serum of children with acute lymphoblastic leukemia.47 By contrast, other studies have reported no increase,42,48,49 even in patients with small-cell lung cancer.50 Similarly, in tumor-bearing mice, no TNF could be detected in plasma with bioassays.48 In contrast, other studies havefound considerable amounts of TNF in the blood of tumor-bearing rats.51,52 These divergent find-ings may be due to different sensitivities or specificities of the assay methods, stability of TNF onstorage, short half-life of TNF in vivo or localized paracrine production of TNF.53
Strassman et al., also using a murine colon adenocarcinoma, have shown that treatment with ananti-mouse IL-6 antibody was successful in reversing the key parameters of cachexia in tumor-bearing mice.54 These results seem to indicate that, at least in certain types of tumors, IL-6 couldhave a more direct involvement than TNF in the cachectic state of the animals. Conversely, otherstudies have revealed that IL-6 is not involved in cachexia in a very similar mouse tumor model.55,56
In addition, in vitro studies using incubated rat skeletal muscle have clearly shown that IL-6 had nodirect effect on muscle proteolysis.57
Another interesting candidate for cachexia is IFN-g. This molecule, which is produced by acti-vated T and NK cells and possesses biological activities that overlap with those of TNF, is best knownfor its ability to activate mononuclear phagocytes and to enhance expression of class II MHC anti-gens. Matthys et al., using a monoclonal antibody against IFN-g, were able to reverse the wastingsyndrome associated with the growth of the Lewis lung carcinoma in mice,58 thus indicating that en-
226 • ARGILÉS AND LÓPEZ-SORIANO

dogenous production of IFN-g occurs and subsists in the tumor-bearing mice, and is instrumental inbringing about some of the generalized metabolic changes characteristic of cancer cachexia. Thesame group has also demonstrated that severe cachexia also develops rapidly in nude mice inocu-lated with CHO cells constitutively producing IFN-g, as a result of the transfection of the corre-sponding gene.59
Other cytokines, such as the leukemia inhibitory factor (LIF),60 transforming growth factor-b(TGF-b)61 or IL-162 have also been suggested as mediators of cachexia. Thus, mice engrafted withtumors secreting LIF (like the human melanoma line SEKI) develop severe cachexia.63 ConcerningIL-1, although its anorectic64 and pyrogenic65 effects are well-known, administration of IL-1 recep-tor antagonist to tumor-bearing rats did not result in any improvement in the degree of cachexia,66
thus suggesting that its role in cancer cachexia (if any) may be secondary to the actions of other me-diators. Interestingly the levels of both IL-6 and LIF have been shown to be increased in patientswith different types of malignancies.67,68
Cyliary neurotrophic factor (CNTF) is a member of the family of cytokines which include IL-6and LIF and which is produced predominantly by glial cells of the peripheral nervous system.69
However this cytokine also seems to be expressed in skeletal muscle.69 Henderson et al. have demon-strated that this cytokine induced potent cachectic effects and acute-phase proteins (independent ofthe induction of other cytokine family members) in mice implanted with C6 glioma cells, genetical-ly modified to secrete CNTF.70
Bearing all this information in mind, it may be concluded that, although TNF has a very im-portant role in the induction of cachexia, the metabolic derangement leading to this pathological statecan also be influenced by other cytokines produced by immune cells in response to invasive stimuli.
4. H Y P E R M E T A B O L I S M
If anorexia is not the only factor involved in cancer cachexia it becomes clear that metabolic abnor-malities must have a very important role. Basically, due to hypermetabolic state, the tumor-bearinghost is energetically more inefficient than in the normal nontumor-bearing state, and this leads to anincreased energy expenditure that, together with the decreased food intake, has a key role in the de-velopment of cancer cachexia. Indeed, body weight maintenance requires energy intake to equal en-ergy expenditure. In fact, these two variables are normally interconnected since when energy intakeincreases, so does expenditure and vice versa. For instance, starvation is characterized by an impor-tant drop in oxygen consumption71 while carbohydrate overeating is associated with an increase inthermogenesis.72 This relation between caloric intake and energy expenditure can be viewed as amechanism to save calories when intake is low and to prevent obesity when excess food is eaten. Incancer cachexia, however, the often decreased caloric intake is not accompanied by a drop in ener-gy expenditure. Hyltander et al., in a wide population study, have showed that cancer patients havea higher resting energy expenditure as compared with normal controls.73 Different mechanisms canbe involved in the increase in energy expenditure. Thus, the activity of futile cycles, such as the Coricycle (glucose to lactate to glucose) or lactate recycling that takes place between the tumor and thehost4 is certainly involved in generating energetic inefficiency. Indeed the gluconeogenic utilizationof the tumor-derived lactate is a very inefficient metabolic process consuming 6 molecules of ATPper cycle but it is essential for compensating tumor acidosis. IL-6 seems to be clearly involved instimulating gluconeogenesis in cultured hepatocytes.74 Yasmineh and Theologides, however, haveshown that TNF does not seem responsible for the increase in gluconeogenesis observed in the tu-mor-bearing host.75 In addition, Christ and Nath have shown that both TNF and IL-1 impair theglucagon-mediated increase in phosphoenolpyruvate carboxykinase (one of the rate limiting en-zymes in gluconeogenesis) in cultured rat hepatocytes.76 Conversely, Zentella et al. have clearlyshown that TNF action in cultured myocytes is linked with an important activation of a futile cy-
CYTOKINES AND CANCER CACHEXIA • 227

cle.77 Thus, the cytokine stimulates glucose utilization and lactate formation activating the substratecycle between phosphofructokinase and fructose-1,6-bisphosphatase, therefore increasing glucoseand glycogen utilization with a subsequent increase in lactate production (Fig. 2).77 Protein synthe-sis and degradation (protein turnover) constitutes another wasteful metabolic process which is acti-vated by TNF (Fig. 2).78 Another futile cycle is that which involves an abnormal function of the Na1-K1-ATPase. The enzyme complex works by pumping Na1 out of the cells at a certain energetic costin order to fulfil the demands of the active transport. Alterations in the stoichiometry (more ATP perNa1 translocated) have been observed in Ehrlich ascites tumor cells.79
Nonshivering thermogenesis takes place in brown adipose tissue (BAT). Brown adipocytes con-tain numerous mitochondria and are characterized by the presence of a 32 kDa protein, thermogeninor uncoupling protein (UCP1),80 which uncouples oxidative phosphorylation in the mitochondrialcompartment. Consequently, the energy associated with substrate oxidation is not employed for ATPsynthesis and thus is released as heat. Injection of low doses of TNF either peripherally or into thebrain of laboratory animals elicits rapid increases in metabolic rate which are not associated with in-creased metabolic activity but rather with an increase in blood flow and thermogenic activity (asmeasured by GDP binding) of BAT.81 Interestingly, during cachectic states there is an increase inBAT thermogenesis both in humans82 and experimental animals.83 In addition, TNF stimulates li-pogenesis in BAT.84 Consequently, nonshivering thermogenesis due to BAT activity may be a veryimportant factor contributing to the decreased energy efficiency found in the cachectic state.
Until very recently, the UCP1 protein (present only in brown adipose tissue) was considered tobe the only mitochondrial protein carrier that stimulated heat production by dissipating the proton
228 • ARGILÉS AND LÓPEZ-SORIANO
Figure 2. Cytokines and futile cycling. Changes in futile substrate cycling have been suggested as participating in bodyweight regulation by increasing energy expenditure. Cytokines may participate in the activity of some of these cycles.

gradient generated during respiration across the inner mitochondrial membrane and therefore un-coupling respiration from ATP synthesis. Recently, two new proteins sharing the same function,UCP2 and UCP3, have been described. While UCP2 is expressed ubiquitously,85,86 UCP3 is ex-pressed abundantly and specifically in skeletal muscle in humans87– 89 and also in brown adipose tis-sue of rodents.90 Concerning the regulation of their expression, UCP2 mRNA levels are upregulat-ed in WAT and skeletal muscle in obesity-resistant mice but not in an obesity-prone strain,91 thereforesuggesting a possible role as a defense mechanism against high-fat induced obesity. Fasting increasesUCP2 mRNAin skeletal muscle92 while it does not seem to affect the expression of the gene in brownadipose tissue. In skeletal muscle UCP3 gene expression is also modulated by food intake: its ex-pression is increased by fasting.93 Very recently, Faggioni et al. have reported that UCP2 gene ex-pression is induced by LPS in several cell types (the effect also being observed following IL-1 orTNF administration),94 and have therefore suggested that it could represent a mechanism for the in-creased thermogenesis during infection. Our research group has demonstrated that both UCP2 andUCP3 mRNAs are elevated in skeletal muscle during tumor growth95 and that TNF is able to mim-ic it.96
5. H Y P E R L I P E M I A A N D F A T M E T A B O L I S M
During cachexia there is a dramatic loss of white adipose tissue, basically due a fall in LPL activityand an increase in the activity of hormone-sensitive lipase (the rate-limiting enzyme of the lipolyticpathway). In addition to these metabolic events associated with cachectic states, there is an inhibi-tion of glucose transport and de novo lipogenesis in the tissue. TNF has been shown to decrease LPLactivity in 3T3-L1 cells,97 associated with a decrease in LPL mRNA.98 Fried and Zechner reportedthat TNF produced a dose-dependent marked suppression of LPL activity in human adipose tissuemaintained in organ culture.99 In vivo administration of TNF results in a decrease of adipose tissueLPL activity in rat, mouse, and guinea pig.98,100 This decreased activity has been shown to depressthe uptake of exogenous [14C]lipid by adipose tissue and to increase circulating triacylglycerols inthe rat.101 Such elevation may, in part, be the result of stimulation of lipolysis in adipose tissue withsubsequent increased secretion of very low density lipoproteins (VLDL) from the liver.102,103 In con-trast to these observations, in human primary cultures of isolated adipocytes the cytokine was un-able to decrease LPL.104 The addition of TNF to 3T3-L1 cells increased lipolysis,105 which has beenconfirmed by others using fully differentiated adipocytes.106 TNF and IL-1 have both been shownto inhibit glucose transport in adipocytes107 and consequently decrease the availability of substratesfor lipogenesis. Conversely, no direct action of TNF has been shown on de novo lipogenesis in adi-pose tissue of starved rats.102 However, TNF decreased acetyl-CoA carboxylase (a key lipogenic en-zyme) during preadipocyte differentiation by a decrease in its mRNA; this did not occur in fully dif-ferentiated adipocytes.108
IFN-g, like TNF, can inhibit LPL activity in 3T3L1 adipocytes and can diminish the rate of syn-thesis of long-chain lipids from smaller chain fatty acids.109 This effect is similar to that of the inhi-bition of lipogenesis and LPL seen with TNF. With this ability of IFN-g to mimic the effects of TNFon fat metabolism, and with its apparent synergy with TNF, IFN-g may have a prominent role in can-cer cachexia. In cultured adipocytes, IL-1, TNF-b (lymphotoxin), IFN-g, and LIF were all shown todecrease LPL activity.109 –111 Similarly, IL-1 and IFN a, b, and g increased lipolysis in adipocytesin culture.106,109,112
Elevation of circulating lipid seems to be a hallmark of cancer-bearing states to the extent thatsome authors have suggested that plasma levels may be used to screen patients for cancer.113 Hy-perlipemia in cancer-bearing states seems to be the result of an elevation of both triacylglycerols andcholesterol. Hypertriglyceridemia is the consequence of the decreased LPL activity which results ina decrease in the plasma clearance of both endogenous (transported as VLDL) and exogenous (trans-
CYTOKINES AND CANCER CACHEXIA • 229

ported as chylomicra) triacylglycerols. Muscaritoli et al. have clearly demonstrated that both thefractional removal rate and the maximum clearing capacity (calculated at high infusion rates whenLPL activity is saturated) are significantly decreased after the administration of an exogenous tria-cylglycerol load to cancer patients.114 In tumor-bearing animals with a high degree of cachexia thereis also an important association between decreased LPL activity and hypertriglyceridemia.20,115 An-other factor that could contribute to the elevation of circulating triacylglycerols is an increase in liv-er lipogenesis.116 Hypercholesterolemia is often seen in both tumor-bearing animals and humanswith cancer.117–119 Interestingly, most cancer cells show an altered regulation in cholesterol biosyn-thesis, showing a lack of feedback control on HMG-CoA reductase, the key enzyme in the regula-tion of cholesterol biosynthesis. Cholesterol perturbations during cancer include changes in lipopro-tein profiles, in particular an important decrease in the amount of cholesterol transported in the highdensity lipoproteins (HDL) fraction. This finding has been observed in both experimental animalsand human subjects.117–119 HDLl plays an important role in the transport of excess cholesterol fromextrahepatic tissues to the liver for reutilization or excretion into bile (reverse cholesterol transport).It is thus conceivable that the observed low levels of HDL-cholesterol may be related, at least in part,to a decreased cholesterol efflux to HDL as a consequence of increased utilization and/or storage inproliferating tissues, such as neoplasms. However, since precursor particles of HDL are thought toderive from lipolysis of triacylglycerol-rich lipoproteins such as VLDL and chylomicra,120 and sincea significant positive correlation between plasma HDL cholesterol and LPL activity in adipose tis-sue has also been reported,120 the possibility that the low HDL cholesterol concentrations observedduring tumor growth may be secondary to the decreased triacylglycerol clearance from plasma, as aresult of LPL inhibition, must also be considered. As described above, in lipid metabolism TNF canaffect different sites which can produce the serum elevations in lipid levels: adipose tissue LPL andlipolysis. Another important site that can account for hyperlipemia is the de novo fatty acid synthe-sis that takes place in the liver. Indeed, TNF has been shown to increase hepatic lipogenesis in vivoand subsequent VLDL production.121 In vivo administration of IL-1, IL-6, and IFN-a to mice alsoproduces a rapid increase in hepatic lipogenesis.122,123 IL-4 is a cytokine that has marked inhibito-ry properties in regulating the immune response. By itself, IL-4 has no effect on hepatic fatty acidsynthesis, but it inhibits the stimulation of hepatic lipogenesis induced by TNF, IL-1, and IL-6.124
Using a polyclonal rat anti-TNF antibody, we have demonstrated that TNF is involved in the abnor-malities in lipid metabolism found in tumor-bearing rodents.125 In addition, studies involving TNFreceptor 1-deficient mice have shown that the cytokine via receptor-1 is involved in the alterationsin lipid metabolism associated with the implantation of a cachexia-induced tumor.20 In conclusion,it may be suggested that TNF, together with perturbations in the hormonal homeostasis, is likely toplay an important role in forcing the metabolic balance of the adipocyte towards the catabolic side(Table I).
6. M U S C L E W A S T I N G
Asthenia or lack of strength is one of the main characteristics of cancer cachexia and it is directly re-lated to the muscle waste observed in cachectic states. During fasting, muscle proteins are degradedto provide amino acids which are used for gluconeogenesis; however, during longer starvation peri-ods, protein breakdown is decreased in order to conserve nitrogen and maintain lean body mass. Thisability, absolutely essential for conserving nitrogen when the intake is reduced, seems to be absentin cancer-bearing states, leading to a depletion of vital host protein both skeletal and functional. Theskeletal muscle, which accounts for almost half of the whole body protein mass, is severely affect-ed in cancer cachexia126 –128 and evidence has been provided for muscle protein waste as being as-sociated with enhanced turnover rates.129 –134 Since cachexia tends to develop at a rather advanced
230 • ARGILÉS AND LÓPEZ-SORIANO

stage of the neoplastic growth, preventing muscle waste in cancer patients is of great potential clin-ical interest. Whether the negative protein balance results from altered rates of synthesis or break-down, or from changes on both sides of muscle protein turnover is still debated.131,135 –137 Rennieet al. have suggested that, during cancer cachexia, the muscle mass is decreased as a result of a low-er rate of protein synthesis, while changes in protein degradation are secondary.138 Conversely, stud-ies involving the release of 3-methylhistidine (a marker of myofibrillar protein degradation) fromperipheral muscle in cancer patients suggest that protein degradation is clearly increased.139 Ourgroup has clearly demonstrated, using several experimental models, that protein synthesis is hardlyaltered in skeletal muscle during tumor growth and that there is a great increase in protein degrada-tion both studied in vivo51,125,131 and in vitro.140 In addition, we have identified the proteolytic mech-anism which is involved in skeletal muscle during cancer cachexia.141–143 A nonlysosomal, ATP andubiquitin-dependent proteolytic system is activated in skeletal muscle of tumor-bearing animals.
A large body of evidence suggests that TNF participates in the protein wasting and loss of ni-trogen associated with cachectic situations. Chronic treatment of rats with recombinant TNF result-ed in a depletion of body protein compared with pair-fed control animals.144 Indeed, chronic treat-ment with either recombinant TNF or IL-1b resulted in a body protein redistribution and a significantdecrease in muscle protein content, associated with coordinate decreases in muscle mRNA levels formyofibrillar proteins.144 Studies involving administration of recombinant TNF in vivo have shownan increase in nitrogen efflux from skeletal muscle of nonweight-losing humans with disseminatedcancer.145 Flores et al, by infusing 14C-leucine to rats, showed that chronic recombinant TNF ad-ministration significantly enhanced muscle protein breakdown.146 Goodman, measuring both tyro-sine and 3-methylhistidine release by incubated rat muscles of animals acutely treated with the cy-tokine, concluded that TNF was involved in activating muscle proteolysis.147 The mechanismsunderlying such actions still remain obscure. Our research group have clearly demonstrated that TNF
CYTOKINES AND CANCER CACHEXIA • 231
Table I. Metabolic Alterations Present in the Host That Can Be Mimicked by Cytokines
Metabolic Alteration Cytokines Involved
Carbohydrate metabolismIncreased hepatic gluconeogenesis IL-6Increased Cori cycle activity TNFIncreased glucose turnover TNFDecreased muscle insulin-estimated glucose uptake TNFLipid metabolismHyperlipidemia TNF, IL-1, LIF, IFN-gDecreased WAT LPL activity TNF, IL-1, IFN-g, LIFIncreased WAT lipolysis TNF, IL-1, IFN-a,b,gIncreased BAT thermogenesis TNFProtein metabolismIncreased whole body protein turnover TNFIncreased hepatic protein synthesis TNF, IL-1, LIF, IL-6Changes in circulating amino acid pattern TNFIncreased muscle protein degradation TNFDecreased muscle amino acid uptake TNFIncreased hepatic protein synthesis TNF, IL-1, LIF, IFN-gIncreased BCAA turnover TNF, IL-1Hormonal changesInsulin resistance TNFIncreased counter-regulatory hormones TNF, IL-1

treatment enhances protein degradation measured in vivo in rat skeletal muscle.78,148 In addition, wehave described that, at least during tumor growth, muscle wasting is associated with the activationof nonlysosomal ubiquitin-dependent proteases,141,142 and that this activation seems to be mediatedvia TNF.149 –151 Ubiquitin, an 8.6 kDa peptide, is involved in the targeting of proteins undergoingcytosolic ATP-dependent proteolysis. In the cell, ubiquitin can be found free or conjugated in anisopeptide linkage to other cellular proteins. Proteins with multiple ubiquitins are the ones targetedfor degradation by an ATP-dependent protease.152–156 However, it has been suggested that the ac-tivity of this system, which is integrated in a supramolecular structure called the proteasome,157 canalso be related to the turnover of long-lived proteins, such as those found in skeletal muscle.158 Wehave also reported that in vivo administration of TNF to rats results in an increased skeletal muscleproteolysis associated with an increase in both gene expression and higher levels of free and conju-gated ubiquitin.150,151 In addition, the in vivo action of TNF during cancer cachexia does not seemto be mediated by IL-166 or glucocorticoids.159 Concerning a possible direct action of TNF on mus-cle proteolysis, the presence of both p55 and p75 receptors has been described160 and we have re-cently demonstrated that the action of the cytokine on the induction of ubiquitin-dependent proteol-ysis can be direct.161 Other cytokines, such as IL-1 or IFN-g are also able to activate ubiquitin geneexpression.162 Therefore, TNF, alone or in combination with other cytokines, seems to mediate mostof the changes concerning nitrogen metabolism associated with cachectic states (Table I). In con-clusion, muscle protein degradation is perhaps the most important metabolic feature of the cachec-tic cancer-bearing host, and future studies will no doubt concentrate on the discovery of compounds(to abrogate cytokine action) which are able to block the activation of the proteolytic systems re-sponsible for the enhanced degradation.
In studying the mechanism which leads to protein waste, particular interest has been given tothe metabolism of branched-chain amino acids (BCAA: leucine, isoleucine, and valine)163 which areessential nutrients for both humans and animals, making up to 40% of the minimal daily require-ments of indispensable amino acids in man,164 leucine alone representing 8% of body protein in hu-mans.165 The carbon skeletons arising from transamination of BCAA provide a major source of metabolic fuel for skeletal muscle. In wasting disorders (including cancer and injury) plasma con-centrations of BCAA are often increased and their turnover rates altered.166 It has been previouslydemonstrated that in vivo leucine oxidation to CO2 is enhanced in sepsis167 and in tumor-bearing an-imals,166,168,169 related to an increased turnover of the amino acid.166 The trigger for the increasedleucine oxidation remains unknown but both TNF and IL-1 have been proposed as mediators.167,170
The growing tumor has a considerable demand for essential amino acids. Attempts have been madeto inhibit the development of cancer by altering the diet composition of ingested amino acids, in-ducing a state of amino acid imbalance.171,172 In particular, it has been shown that tumor growth isdelayed in rats maintained on a valine-depleted diet; however, this had a negative impact on the nu-tritional state of the host.173 According to Lazo,174 a tumor can considerably increase the daily needfor leucine in humans, this increase having a better correlation with the clinical deterioration of thepatient than the modification of carbohydrate and energy metabolism. As a result of the demand forleucine, there is an amino acid flux from muscle to the tumor associated with muscle waste in thehost. In general, however, the leucine taken up by the tumor only represents a minor part of the aminoacid demand in the tumor-bearing animal.166 It can thus be proposed that the major contribution toBCAA oxidation in cancer-bearing states is made by skeletal muscle.166 Indeed, BCAA are the onlyamino acids that are extensively degraded in skeletal muscle and have been shown to stimulate pro-tein synthesis and inhibit degradation in skeletal muscle in vitro175 –178 and in vivo.179,180 Pushingspeculation further, it could be suggested that during cancer cachexia the response of muscle proteinturnover to BCAA is altered. This is at present being investigated in our laboratory. Interestingly, theactivation of muscle proteolysis in skeletal muscle always parallels that of BCAAoxidation. We havedescribed that b2-agonists (clenbuterol in particular) are able to suppress the activation of the ubiq-
232 • ARGILÉS AND LÓPEZ-SORIANO

uitin-dependent proteolytic system during tumor growth.181 In a similar manner, b2-agonists are alsoable to suppress the increase in BCAA oxidation in skeletal muscle during cancer cachexia.169
7. A C U T E - P H A S E R E S P O N S E
The result of the enhanced muscle proteolysis is a large release of amino acids from skeletal musclewhich takes place specially as alanine and glutamine (Fig. 3). The release of amino acids is also po-tentiated by an inhibition of amino acid transport into skeletal muscle.140 While glutamine is basi-cally taken up by the tumor to sustain both the energy and nitrogen demands of the growing mass,182
alanine is mainly channeled to the liver for both gluconeogenesis (as previously commented) andprotein synthesis. Interestingly, liver fractional rates of protein synthesis are increased in tumor-bearing animals, accounting for the production of the so-called acute phase proteins, while there isa decrease in albumin synthesis, leading to hypoalbuminemia.183
The acute phase response is a systemic reaction to tissue injury, typically observed during in-fection or trauma, characterized by a series of hepatocyte-derived plasma proteins known as acute-phase reactants [including C-reactive protein (CRP), serum amyloid A (SAA), a1-antitrypsin, fibrinogen, complement factors B and C3] and by reduced synthesis of albumin and transferrin.184
In cancer patients, an acute-phase response is observed.185 –187 For many years investigators havebeen searching for mediators involved in the regulation of acute-phase protein synthesis. Interest-ingly the cytokines IL-6, IL-1, and TNF are now regarded as the major mediators of acute-phase pro-tein induction in the liver.188 In fact, acute-phase proteins can be divided into two groups: type I andtype II acute phase proteins.189,190 Type I proteins include SAA, CRP, C3, haptoglobin (rat), and a1-acid glycoprotein, and are induced by IL-1 and TNF. Type II proteins include fibrinogen, haptoglo-bin (human), a1-antichymotrypsin, and a2-macroglobulin (rat). These proteins are induced by IL-6,LIF, OSM (oncostatin M), CNTF, and CT-1 (cardiotrophin-1). Unfortunately, the role of acute-phasereactants during cancer growth is still far from understood.
CYTOKINES AND CANCER CACHEXIA • 233
Figure 3. Muscle waste and acute-phase response. Perhaps the most paradigmatic metabolic derrangement inducedby the tumor is the activation of proteolysis in skeletal muscle and the redistribution of protein synthesis in the liver. Thesealterations can be mimicked by different cytokines.

8. I N S U L I N R E S I S T A N C E A N D O T H E RH O R M O N A L A L T E R A T I O N S
In addition to having increased glucose production191 and glucose intolerance,192 cancer patientsshow a clear insulin resistance status that involves adipose tissue, skeletal muscle, and liver. The in-creased hepatic glucose production is partially the result of a lack of inhibition of gluconeogenesisby insulin due to a certain degree of liver insulin resistance. Similarly, glucose utilization by skele-tal muscle is reduced both in experimental animals and cancer patients, this being the result of clearinsulin resistance. In addition, increases in counter-regulatory hormones, such as glucocorticoids193
or glucagon,194 also seem to be involved. As Tayek points out: “the cancer patient behaves like thetype II diabetic who is unable to maximize glucose uptake in the skeletal muscle in the presence oflarge amounts of insulin”.195 The decreased stimulation of glucose uptake does not seem to be theconsequence of a defect in insulin-binding but rather a postreceptor defect. The insulin resistanceobserved in skeletal muscle also affects glycogen synthesis, which is clearly reduced in cancer pa-tients.196 Insulin resistance in the tumor-bearing host may be overcome by the use of exogenous in-sulin, which in animal studies has improved the degree of cachexia197,198 as well as the response toanticancer therapy, including surgery and chemotherapy.199
Pathological situations associated with a high TNF production show a state of peripheral insulinresistance. Among them, endotoxemia, cancer, and trauma are normally associated with increases incirculating TNF and peripheral insulin resistance. Thus, the general clinical impression is that the in-sulin dose for diabetic patients must be increased if infection is present, thus suggesting an additionalimpairment to insulin resistance.200 Lang and Dobrescu, using an euglycemic clamp in septic rats(induced by injection of live E. coli), found that the septic state induced peripheral insulin resis-tance.201 It is well known that during infection there is a stimulation of macrophage TNF productionby means of either LPS or other endotoxins released by the infecting agent. In addition, chronic ad-ministration of TNF to rats induces systemic insulin resistance.202 Clinical administration of TNF tohealthy humans has been reported to reduce insulin sensitivity, by inducing hyperglycemia withoutlowering insulin levels.203
Thus, since NIDDM is characterized by profound insulin resistance, TNF could also be re-sponsible for this pathological state. Measurement of gene expression and protein levels for the cy-tokine in type II diabetic patients has confirmed this hypothesis.204 In addition, elevated expressionof TNF was implicated in animal models of obesity and in the insulin-resistant diabetes mellituswhich accompanies the disorder. In four models of obesity and insulin-resistant diabetes (the fa/faobese Zucker rat, the ob/ob obese mice, the tub/tub obese mice, and the db/db obese diabetic mice),expression of TNF mRNA in adipose tissue was induced, with corresponding elevation of TNF pro-tein both locally and systemically.205 Interestingly, in vitro studies using fully differentiated 3T3-L1adipocytes have shown that when the cells are exposed to TNF they became insulin-resistant sinceinsulin is not able to stimulate hexose transport.206 This appears to be a consequence of a down reg-ulation in GLUT4 expression, the insulin-stimulated glucose transporter. This observation may ex-plain why in the same kind of cells incorporation of glucose into lipids is diminished after incuba-tion with supernatants of activated macrophages.207 The same authors reported that long-termtreatment of 3T3-F442A adipocytes with TNF led to down regulation of the GLUT4 mRNA, and,although no similar results have yet been reported in humans, their results suggest that the cytokinecould be a key mediator of abnormal gene expression in obesity-diabetes syndromes and could af-fect glucose homeostasis. In the same study, in vivo administration of the human TNF soluble re-ceptor partially reversed the resistance to insulin-stimulated glucose uptake, thus suggesting thatTNF could be one of the mediators of insulin resistance in several experimental obesity models.208
Very interestingly, Saghizadeh et al. have found that the TNF gene is also overexpressed in skeletalmuscle in diabetic subjects,209 these results further supporting the role of the cytokine in the induc-tion of a generalized state of insulin resistance.
234 • ARGILÉS AND LÓPEZ-SORIANO

Considerable attention is now being focused on the mechanism by which TNF induces resis-tance in the cascade of insulin signal transduction, the mechanism for tissue specific overexpressionin the obese adipocyte, and the possibility that interference with the pathway could be a new thera-peutic approach to abrogate insulin resistance and thereby obesity-induced diabetes. Hofmann et al.have shown an induction of TNF mRNA and TNF receptors mRNA in adipose tissue and musclefrom a mouse model of noninsulin-dependent diabetes mellitus, the obese KKAy mice.210 They alsoobserved that the elevated expression of TNF mRNA can be partially reduced by oral administrationof an insulin-sensitizing drug. Hotamisligil et al. have shown that TNF has the ability to decrease thetyrosine kinase activity of the insulin receptor.211 Treatment of cultured murine adipocytes with TNFwas shown to induce serine phosphorylation of insulin receptor substrate-1 (IRS-1) and convert IRS-1 into an inhibitor of the insulin receptor tyrosine kinase in vitro, thus indicating that TNF inducesinsulin resistance through an unexpected action of IRS-1 to attenuate insulin receptor signaling. Itmay thus be suggested that TNF expression is involved in a physiological loop, perhaps related to alimitation of obesity at the expense of promoting insulin resistance. For this reason, experiments arebeing undertaken in our laboratory involving obese mice that are transgenic for the soluble TNF receptor-1. Future exciting research will, no doubt, point in this direction.
9. U S I N G C Y T O K I N E S I N T H E R A P Y
The therapeutic strategies that can be used are based on either blocking cytokine synthesis or ac-tion.
A. Blocking Synthesis
Pentoxifylline, a methylxantine derivative, is a phosphodiesterase inhibitor that inhibits TNF syn-thesis by decreasing gene transcription. This drug was originally used for the treatment of varioustypes of vascular insufficiency because of its hemorheological activity, thought to be based on itsability to reduce blood viscosity and increase the filterability of blood cells. While several studiesusing animal models suggest that pentoxifylline is able to decrease the cytokine-induced toxicity ofantineoplasic agents while preserving antitumor treatment efficacy,212 clinical studies have shownthat the drug failed to improve the appetite or to increase the weight of cachectic patients.213 In ad-dition, pentoxifylline has also been used in the treatment of cachectic AIDS patients with very poorresults since it did not influence the weight, temperature, well-being or tiredness of the subjects. Infact, the patients frequently reported gastrointestinal side-effects. However, the reported clinical trials have been relatively limited and, therefore, larger randomized studies are necessary to assessthe efficacy of pentoxifylline in the treatment of cancer cachexia.
Rolipram is another type IV phosphodiesterase inhibitor that has been shown to decrease TNFproduction by LPS-stimulated human monocytes.214 This compound has previously been used in thetreatment of endogenous depression in both animals and man, and it may have therapeutic activityin disease states where TNF seems to play a role in the pathogenesis, such as in endotoxic shock.215
Thalidomide (a-N-phthalimidoglutaramide) is a drug unfortunately associated with tragedy. In-deed, its use as a sedative in pregnant women caused over 10,000 victims of severe malformationsin newborn children. Recently, however, a certain revival has affected the drug since it has beendemonstrated to suppress TNF production in monocytes in vitro216 and to normalize elevated TNFlevels in vivo.217 Apparently thalidomide activity is due to a selective desestabilization of the TNFmRNA.218 The drug has certainly been used to counteract high cytokine levels in tuberculosis pa-tients.219 Its use in cancer cachexia remains to be established but it could potentially have a certainrole in counteracting TNF-mediated metabolic changes.
CYTOKINES AND CANCER CACHEXIA • 235

B. Blocking Action
The use of anticytokine antibodies (either mono- or polyclonal) and cytokine receptor antagonists orsoluble receptors has led to very interesting results. Thus, in rats bearing the Yoshida AH-130 asciteshepatoma (a highly cachectic tumor) anti-TNF therapy resulted in a partial reversal of the abnor-malities associated with both lipid125 and protein metabolism.51 In humans, however, clinical trialsusing anti-TNF treatment have led to poor results in reverting the protein waste associated with sep-sis.220 Concerning IL-6, experimental models have proved that the use of antibodies is very effec-tive in preventing tumor-induced waste.221 Strassman et al. have demonstrated that the experimen-tal drug suramine (which prevents the binding of IL-6 to its cell surface receptor, as demonstratedby radioreceptor binding assay and affinity binding experiments) partially blocks (up to 60%) thecatabolic effects associated with the growth of the colon-26 adenocarcinoma in mice.54 In humans,administration of an anti-IL-6 monoclonal antibody to patients with AIDS and suffering from an im-munoblastic or a polymorphic large-cell cell lymphoma had a very positive effect on fever andcachexia.222 Concerning other cytokines, anti-IFN-g therapy has also been effective in revertingcachexia in mice bearing the Lewis lung carcinoma58 but there is a lack of clinical data. On the oth-er hand, blocking IL-1 action by means of the IL-1 receptor antagonist (IL-1ra) in tumor-bearing ratshad no effects on either body weight or reversal of metabolic changes.66 It has to be pointed out herethat the routine use of anticytokine antibodies is, at present, too expensive due to the fact that re-quires a very large number of antibody molecules in order to block cytokine action completely.
The appearance of the cachectic syndrome is dependent not only on the production of the above-mentioned cytokines, known as catabolic proinflammatory cytokines, but also on the so-called anti-inflammatory cytokines such as interleukin-4 (IL-4) or interleukin-10 (IL-10) (Fig. 4). Mori et al.have demonstrated that the administration of interleukin-12 (IL-12) to mice bearing the colon-26 car-cinoma alleviates the body weight loss and other abnormalities associated with cachexia, such as adi-pose tissue wasting and hypoglycemia.223 The anticachectic properties are seen at low doses of IL-12, insufficient to inhibit tumor growth. The effects of IL-12 seem to be dependent on a considerabledecrease of IL-6,223 cytokine which has been responsible for the cachexia associated with this tu-mor model.224,225 A similar action has been described for INF-a. Administration of this cytokinepromoted a decrease in both IL-6 mRNA expression in the tumor and serum IL-6 levels, resulting inan amelioration of the cachexia in a murine model of malignant mesothelioma.226
Insulin-like growth factor-I (IGF-I) is able to ameliorate the catabolic effects of TNF227 andcould therefore be used as a potential therapeutic tool in cancer cachexia. Indeed, administration ofIGF-I to tumor-bearing animals results in an amelioration of their cachectic state.228 Interleukin-15
236 • ARGILÉS AND LÓPEZ-SORIANO
Figure 4. Anti-inflammatory cytokines in cachexia. Anti-inflammatory (anticachectic) cytokines, such as IL-4 or IL-10, actby both inhibiting pro-inflammatory (procachectic) cytokine release and by increasing the production of cytokine-antagonist molecules such as IL-1ra. In addition, they block the production of NO and of ROS.

(IL-15) has been reported to be an anabolic factor for skeletal muscle229 and very recent experimentscarried out in our laboratory clearly demonstrate that the cytokine is able to reverse most of the ab-normalities associated with cancer cachexia in a rat tumor model.230
10. C O N C L U D I N G R E M A R K S A N D F U T U R ER E S E A R C H
Under normal circumstances maintenance of homeostasis in mammals, including humans, is guar-anteed by a number of mechanisms. Deviations from this stable situation which impose a seriousthreat to the health of the organism may occur. The organism responds to these challenges, such astissue injury or infection, by a coordinated sequence of systemic and metabolic changes or by localchanges such as inflammatory reactions. The macrophage-derived proinflammatory cytokines IL-1,IL-6, and TNF have key roles in inducing these changes and are therefore involved in many patho-physiological conditions, not only immune and inflammatory reactions but also in the developmentof cachexia. In fact, the balance between these and the anti-inflammatory cytokines such as inter-leukin-1 receptor antagonist (IL-1ra), interleukin-10 (IL-10), and transforming growth factor-b(TGF-b) is pivotal for the fine tuning of many biochemical processes.
Acomplex interaction of procachectic and anticachectic cytokines or cytokine-neutralizing mol-ecules probably determines the critical presentation and course of cachexia (Table II). Intervening inthis sequence of events to modify the host responses may prove to be a beneficial treatment strate-gy for cachexia. Currently tested anti-proinflammatory cytokines have produced interesting results.Among the cytokines that can protect against TNF-mediated cachexia, IL-4 seems to be a good can-didate since Th2-response deficient IL-4 (2/2) mice were very susceptible during acute schistoso-miasis, exhibiting severe acute cachexia followed by death.231 Similarly monocyte chemoattractantprotein-1 (MCP-1) seems to protect mice against endotoxin-induced mortality, possible by increas-ing IL-10 (an anti-inflammatory cytokine) and decreasing TNF. Indeed, administration of recombi-nant-derived MCP-1 to LPS-challenged mice supports this view.232 Similarly administration of an-other cytokine related with Th2 response, IL-13, also reduces TNF and increases survival againstLPS challenge.233 Fujiki et al. have shown that while mice inoculated with an adenocarcinoma cellline expressing IL-6 developed major wasting and cachexia, the inoculation of IL-10 producing-transfectant cells to mice did not cause anorexia or progressive weight loss, suggesting that the cy-tokine was able to counteract cachexia by inhibition of IL-6 production by the tumor cells.234
Hyperalimentation (either enteral or parenteral) of cancer patients has unfortunately proved tobe an inefficient tool for fighting cachexia. It either leads to an increased accumulation of body flu-ids or to an acceleration of tumor growth without any increases in lean body mass. Consequently, fu-ture clinical and research efforts will be devoted to investigating the metabolic alterations inducedin the host by the presence of the tumor and how this knowledge may be used in the clinical man-agement of cancer patients more deeply. Particularly interesting is the idea of controlling protein me-
CYTOKINES AND CANCER CACHEXIA • 237
Table II. Procachectic and Anticachectic Cytokines
Procachectic Anticachectic
IL-1 sTNFrIL-6 IL-1raTNF IL-4IFN-g IL-10LIF IL-13CNTF IL-15

tabolism in the patients essentially by means of blocking protein degradation in skeletal muscle. Thiswould result both in preservation of the muscle mass and, to some extent, in a depletion of aminoacids for tumor growth. Investigations are currently being undertaken in our laboratory to understandthe regulation of the ATP-dependent proteolysis, the main proteolytic pathway activated during can-cer cachexia in skeletal muscle.
Bearing in mind all the information presented here, it can indeed be concluded that no definitemediator of cancer cachexia has been yet identified. However, among all the possible mediators con-sidered here, TNF (one of the earliest molecules related to cachexia) is one of the most relevant can-didates. Indeed, TNF can mimic most of the abnormalities found during cancer cachexia: weight loss,anorexia, increased thermogenesis, alterations in lipid metabolism and adipose tissue dissolution, in-sulin resistance and muscle waste including activation of protein breakdown and increased BCAAmetabolism. However, TNF alone cannot explain all the cachectic metabolic alterations present indifferent types of human cancers and experimental tumors. Another important drawback is the factthat TNF circulating concentrations are not always elevated in cancer-bearing states, and, althoughit may be argued that in those cases local tissue production of the cytokine may be high, cachexiadoes not seem to be a local tumor effect. Consequently, both tumor-produced and humoral factorsmust collaborate in the full induction of the cachectic state.
In conclusion, and because metabolic alterations often appear early after the onset of tumorgrowth, the scope of appropriate treatment, although not aimed at achieving immediate eradicationof the tumor mass, could influence the course of the patient’s clinical state or, at least, prevent thesteady erosion of dignity that the patient may feel in association with the syndrome. This would nodoubt contribute to improving the patient’s quality of life and, possibly, prolong survival. Althoughexploration of the role that cytokines play in the host response to invasive stimuli is an endeavor thathas been underway for many years, considerable controversy still exists over the mechanisms of leantissue and body fat dissolution that occur in the patient with either cancer or inflammation andwhether humoral factors regulate this process. A better understanding of the role of cytokines inter-fering with the molecular mechanisms accounting for protein wasting in skeletal muscle is essentialfor the design of future effective therapeutic strategies. In any case, understanding the humoral re-sponse to inflammation and modifying cytokine actions pharmacologically may prove very effec-tive and no doubt future research will concentrate on this interesting field.
R E F E R E N C E S
1. Warren, S. The immediate cause of death in cancer. Am J Med Sci 1932;184:610–613.2. Nixon DW, Heymsfield SB, Kutner MM, Ansley J, Lawson, DH. Am J Med Protein-caloric undernutri-
tion in hospitalized cancer patients. 1980;68:491–497.3. De Wys, WD. Management of cancer cachexia. Semin Oncol 1985;12:452–460.4. Argilés JM, Alvarez B, López-Soriano FJ. The metabolic basis of cancer cachexia. Med Res Rev
1997;17:477–498.5. Argilés, JM, García-Martínez, C, Llovera M, López-Soriano FJ. The role of cyotkines in muscle wasting:
its relation with cancer cachexia. Med Res Rev 1992;12:637–652.6. Adams R, Victor, M. In: Adams R, Victor M editors. Principles of neurology. New York: McGraw-Hill;
1981. pp. 341–345.7. Houten L, Reilley AA. An investigation of the cause of death from cancer. J Surg Oncol 1980;13,111–
116.8. McBride W, Jackman JD, Grayburn PA. Prevalence and clinical characteristics of a high cardiac output
states in patients with multiple myeloma. Am J Med 1990;89:21–24.9. Argilés JM, Azcón-Bieto The metabolic environment of cancer. Mol Cell Biochem 1988;81:3–17.
10. Davis JD, Levine MW. A model for the control of ingestion. Psychol Rev 1977;84:379–412.11. De Wys WD. Anorexia as a general effect of cancer. Cancer 1979;43:2013–2019.
238 • ARGILÉS AND LÓPEZ-SORIANO

12. Opara EI, Laviano A, Meguid MM, Yang Z. Correlation between food intake and CSF IL-1a in anorectictumor bearing rats. Neuroreport 1995;6:750–752.
13. Plata-Salamán CR. Anorexia during acute and chronic disease. Nutrition 1996;12:69–78.14. Stovroff MC, Fraker, DL, Swedenborg JA, Norton JA. Cachectin/tumor necrosis factor, a possible medi-
ator of cancer anorexia in the rat. Cancer Res 1988;48:920–925.15. Plata-Salamán C, Borkoski JP. Chemokines/intercrines and central regulation of feeding. Am J Physiol
1994;266:R1711–R1515.16. Rivera S, López-Soriano FJ, Azcón-Bieto J, Argilés JM. Blood amino acid compartmentation in mice bear-
ing Lewis lung carcinomes. Cancer Res 1987;47:5644–5646.17. García-Martínez C, López-Soriano FJ, Argilés JM. Alanine metabolism in rats bearing the Yoshida AH-
130 ascites hepatoma. Cancer Lett 1994;87:123–130.18. Rossi-Fanelli F, Cangiano C. Increased availability of tryptophan in brain as a common pathogenic mech-
anism for anorexia associated with different diseases. Nutrition 1991;7:364–367.19. Argilés JM, López-Soriano FJ. The energy stae of tumor-bearing rats. J Biol Chem 1991;266:2978–
2982.20. López-Soriano J, Argilés JM, López-Soriano FJ, Lipid metabolism in rats bearing the Yoshida AH-130 as-
cites hepatoma. Mol Cell Biochem 1996;165:17–23.21. Chance WT, Zhang FS, Fischer JE. Hyperammonemia and anorexia in Morris hepatoma-bearing rats.
Physiol Behav 1991;50: 397–401.22. Chance WT, Zhang FS, Fischer JE. Methionine sulfoximine intensifies cancer anorexia. Pharmacol Phys-
iol Behav 1991;39:115–118.23. Grunfeld C, Zhao C, Fuller J, Pollock A, Moser A, Friedman J, Feingold KR. Endotoxin and cytokines in-
duce expression of leptin, the ob gene product, in hamsters. A role for leptin in the anorexia or infection.J Clin Invest 1996;97:2152–2157.
24. Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet III DJ, Flier JS, Lowell BB, Fraker DL,Alexander HR. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in in-flammatory anorexia. J Exp Med 1997;185:171–175.
25. López-Soriano J, Carbó N, Tessitore L, López-Soriano FJ, Argilés JM. Leptin and tumour growth in therat. Int J Cancer. In press.
26. Simons JP, Schols AM, Campfield LA, Wouters EF, Saris WH. Plasma concentration of total leptin andhuman lung-cancer-associated cachexia. Clin Sci 1997;93:273–277.
27. Chance WT, Balasubraniam A, Sheriff S, Fischer JE. Possible role of neuropeptide Y in experimental can-cer anorexia. Adv Exp Med Biol 1994;354:185–201.
28. Sonti G, Ilyin SE, Plata-Salamán CR. Neuropeptide Y blocks and reverses interleukin-1b-induced anorex-ia in rats. Peptides 1996;17: 517–520.
29. Copeland EM, Macfayder BV, Dudrick SJ. Effect of hyperalimentation in established delayed hypersen-sibility in the cancer patient. Ann Surg 1976;184:60–64.
30. Heber D, Byerley LO, Chi J, Grosvenor M, Bergman RN, Coleman N, Chlebowski RT. Pathophysiologyof malnutrition in the adult cancer patient. Cancer 1988;58:1867–1873.
31. Evans WK, Makuch R, Clamon GH, Feld R, Weiner RS, Moran E, Blum R, Shepherd FA, Jeejeebhoy KN,Dewys WD. Limited impact of total parenteral nutrition on nutritional status during treatment for smallcell lung cancer. Cancer Res 1985;45:3347–3353.
32. Lowry SF. Cancer cachexia revisited: old problems and new perspectives. Eur J Cancer 1991; 27:1–3.33. Balducci L, Hardy C. Cancer and malnutrition: a critical interaction. A review. Am J Hematol 1985;18:91–
103.34. Mahony SM, Tisdale MJ. Induction of weight loss and metabolic alterations by human recombinant tu-
mour necrosis factor. Br J Cancer 1988;58:345–349.35. Socher SH, Friedman A, Martinez D. Recombinant human tumour necrosis factor induces acute reduc-
tions in food intake and body weight in mice. J Exp Med 1988;167:1211–1227.36. Stovroff MC, Fraker DL, Swedenborg JA, Norton JA. Cachectin/tumor necrosis factor: a possible medi-
ator of cancer anorexia in the rat. Cancer Res 1988;48:4567–4572.37. Mullen BJ, Harris RBS, Patton JS, Martin RJ. Recombinant tumor necrosis factor-α chronically admin-
istered in rats: lack of cachectic effect. Proc Soc Exp Biol Med 1990;193:318–325.38. Tracey KL, Wei H, Manogue KR, Fong Y, Hesse DG, Nguyen HT, Kuo GC, Beutler B, Cotran RS, Cera-
CYTOKINES AND CANCER CACHEXIA • 239

mi A, Lowry SF. Cachectin/tumor necrosis factor induces cachexia, anemia and inflammation. J Exp Med1988;167:1211–1227.
39. Oliff A, Defeo-Jones D, Boyer M, Martinez D, Kiefer D, Vuocolo G, Wolfe A, Socher SH. Tumors se-creting human TNF/cachectin induce cachexia in mice. Cell 1987;50:555–563.
40. Scuderi, P, Lam KS, Ryan KJ, Petersen E, Sterling KE, Finley PR, Ray CG, Slymen DJ, Salmon SE. Raisedserum levels of tumour necrosis factor in parasitic infections. Lancet 1986;2:1364–1365.
41. Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P. Tumor necrosis factor (cachectin) as anessential mediator in murine cerebral malaria. Science 1987;237:1210–1212.
42. Waage A , Espevik T, Lamvik J. Detection of tumor necrosis factor-like cytotocity in serum from patientswith septicaemia but not from untreated cancer patients. Scan J Immunol 1986;24:739–743.
43. Beutler B, Milsark IV, Cerami A. Passive immunization against cachectin/tumor necrosis factor protectsmice from lethal effect of endotoxin. Science 1985;229:869–871.
44. Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anticachectin/TNFmonoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987;330:662–664.
45. Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S, Dinarello CA, Cerami, A, Wolff SM,Wilmore DW. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med1988;318:1481–1486.
46. Balkwill F, Burke F, Talbot D, Tavernier J, Osborne R, Naylor S, Durbin H, Fiers W. Evidence for tumournecrosis factor/cachectin production in cancer. Lancet 1987;2:1229–1232.
47. Saarinen UM, Kosfelo EK, Teppo AM, Simes MA. Tumor necrosis factor in children with malignancies.Cancer Res 1990;50: 592–595.
48. Moldawer LL, Drott C, Lundholm K. Monocytic production and plasma bioactivities of interleukin-1 andtumour necrosis factor in human cancer. Eur J Clin Invest 1988;18:486–492.
49. Socher SM, Martinez D, Craig JB, Kuhn JG, Oliff A. Tumour necrosis factor nondetectable in patientswith cancer cachexia. J Natl Cancer Inst 1988;80:595–598.
50. Selby PJ, Hobbs S, Niner C, Jackson, E, Smith IE, McElwain TJ. Endogenesis tumour necrosis factor incancer patients. Lancet 1988;1:483.
51. Costelli P, Carbó N, Tessitore L, Bagby GJ, López-Soriano FJ, Argilés JM, Baccino FM. Tumour necro-sis factor-a mediates changes in muscle protein turnover in a cachectic rat tumour model. J Clin Invest1993;92:2783–2789.
52. Stovroff MC, Fraker DL, Norton JA. Cachectin activity in the serum of cachectic, tumor-bearing rats. ArchSurg 1988;124:94–99.
53. Aderka D, Fischer D, Levo Y, Holtmann H, Hahn T, Wallach D. Cachectin/tumour necrosis factor pro-duction by cancer patients. Lancet 1985;2:1190.
54. Strassmann G, Fong M, Freter CE, Windsor S, D’Alessandro F, Nordan RP. Suramin interferes with in-terleukin-6 receptor binding in vitro and inhibits colon-26-mediated experimental cancer cachexia in vivo.J Clin Invest 1993;92:2152–2159.
55. Soda K, Kawakami M, Kashii A, Miyata M. Characterization of mice bearing subclones of colon 26 ade-nocarcinoma disqualifies interleukin-6 as the sole inducer of cachexia. Jpn J Cancer Res 1994;85:1124–1130.
56. Soda K, Kawakami M, Kashii A, Miyata M, Manifestations of cancer cachexia induced by colon 26 adenocarcinoma are not fully ascribable to interleukin-6. Int J Cancer 1995;62:332–336.
57. García-Martínez C, López-Soriano FJ, Argilés JM. Interleukin-6 does not activate protein breakdown inrat skeletal muscle. Cancer Lett 1994;76:1–4.
58. Matthys P, Heremans H, Opdenakker G, Billiau A. Anti-interferon-g antibody treatment, growth of Lewislung tumours in mice and tumour-associated cachexia. Eur J Cancer 1991;27:182–187.
59. Matthys P, Dukmans R, Proost P, Van Damme J, Heremans H, Sobis H, Billiau A. Severe cachexia in miceinoculated with interferon-g-producing tumor cells. Int J Cancer 1991;49:77–82.
60. Mori M, Yamaguchi K, Honda S, Nagasaki K, Ueda M, Abe O, Abe K. Cancer Res 1991;51:6656–6659.61. Zugmaier G, Paik S, Wilding G, Knabbe K, Bano M, Lupu R, Deschauer B, Simpson S, Dickson RB,
Lippman M. Transforming growth factor b1 induces cachexia and systemic fibrosis without an antitumoreffect in nude mice. Cancer Res 1991;51:3590–3594.
62. Moldawer LL, Georgieff M, Lundholm K. Interleukin-1, tumour necrosis factor-a/cachexin and thepathogenesis of cancer cachexia. Clin Physiol 1987;7:263–274.
240 • ARGILÉS AND LÓPEZ-SORIANO

63. Mori M, Yamaguchi K, Abe K. Purification of a lipoprotein lipase-inhibiting protein produced by a melanomacell line associated with cancer cachexia. Biochem Biophys Res Commun 1989;160:1085–1092.
64. Mrosovsky N, Molony LA, Conn CA, Kluger MJ. Anorexic effects of interleukin-1 in the rat. Am J Phys-iol 1989;257:R1315–R1321.
65. Hashimoto M. Characterization and mechanism of fever induction by interleukin-1-beta. Pflügers Arch1991;419:616–621.
66. Costelli P, Llovera M, Carbó N, García-Martínez C, López-Soriano FJ, Argilés JM. Interleukin-1 recep-tor antagonist (IL-Ira) is unable to reverse cachexia in rats bearing an ascites hepatoma. (Yoshida AH-130). Cancer Lett 1995;95:33–38.
67. Vanderschueren B, Dumon JC, Oleffe V, Heymans C, Gérain J, Body JJ. Circulating concentrations of in-terleukin-6 in cancer patients and their pathogenic role in tumor-induced hypercalcemia. Cancer ImmunolImmunother 1994;39:286–290.
68. Lorgeot V, Praloran V, Turlure P, Denizot Y. Concentrations of serum leukemia inhibitory factor (LIF) inpatients with hematologic malignancies. Leukemia 1997;11:311–312.
69. Ip NY, McClain J, Barrezueta NX, Aldrich TH, Pan L, Li Y, Wiegand SJ, Friedman B, Davis S, Yan-copoulos GD. The alpha component of the CNTF receptor is required for signalling and defines potentialCNTF targets in the adult and during development. Neuron 1993;10: 89–102.
70. Henderson JT, Mullen BJ, Roder JC. Physiological effects of CNTF-induced wasting. Cytokine1996;8:784–793.
71. P. Felig. Endocrinology. New York: Grune & Stratton; 1979. Vol. 3, pp. 1927–1940.72. Stock MJ, Rothwell NJ. Brown adipose tissue and the response to overfeeding. Biochem Soc Trans
1986;14:239–240.73. Hyltander A, Drott C, Korner V. Elevated energy expenditure in cancer patients with solid tumors. Eur J
Cancer 1991;27:9–15.74. Blumberg D, Hochwald S, Brennan MF, Burt M, Interleukin-6 stimulates gluconeogenesis in primary cul-
tures of rat hepatocytes. Metabolism 1995;44:145–146.75. Yasmineh WG, Theologides A, Effect of tumor necrosis factor on enzymes of gluconeogenesis in the rat.
Proc Soc Exptl Biol Med 1992;199:97–103.76. Christ B, Nath A. Impairment by interleukin-1b and tumour necrosis factor-a of the glucagon-induced in-
crease in phosphoenolpyruvate carboxykinase gene expression and gluconeogenesis in cultures rat hepa-tocytes. Biochem J 1996;320:161–166.
77. Zentella A, Manogue K, Cerami A. Cachectin/TNF-mediated lactate production in cultured myocytes islinked to activation of a futile substate cycle. Cytokine 1993;5:436–447.
78. Llovera M, López-Soriano FJ, Argilés JM. Effects of tumor necrosis factor-a on muscle protein turnoverin female Wistar rats. J Natl Cancer Inst 1993;85:1334–1339.
79. Racker E. Why do tumor cells have a high aerobic glycolysis? J Cell Physiol 1976;89:697–700.80. Nicholls DG. The thermogenic mechanism of brown adipose tissue. Biosci Rep 1983;3:431–441.81. Rothwell NJ. Cytokines and thermogenesis. Int J Obesity 1993;17:S98–S101.82. Bianchi A, Bruce J, Cooper AL, Childs C, Kohli M, Morris ID, Morris-Jones P, Rothwell NJ. Increased
brown adipose tissue activity in children with malignant disease. Horm Metab Res 1989;21:640–641.83. Oudart H, Calgari C, Andriamampandry M, LeMaho Y, Malan A. Stimulation of brown adipose tissue ac-
tivity in tumor-bearing rats. Can J Physiol Pharmacol 1995;73:1625–1631.84. López-Soriano J, Argilés JM, López-Soriano FJ. Metabolic effects of tumour necrosis factor-a on rat
brown adipose tissue. Mol Cell Biochem 1995;143:113–118.85. Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, Bouillaud F, Seldin MF,
Surwit RS, Ricquier D, Warden CH. Uncoupling protein-2: a novel gene linked to obesity and hyperin-sulinemia. Nature-Genet S, 1997;15:269–272.
86. Gimeno RE, Dembski M, Weng X, Deng N, Shyjan AW, Gimeno C, Iris F, Ellis SJ, Woolf EA, TartagliaLA. Cloning and characterization of an uncoupling protein homolog. Diabetes 1997;46:900–906.
87. Vidal-Puig A, Solanes G, Grujic D, Flier JS, Lobell BB. UCP3: an uncoupling protein homologue ex-pressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys ResCommun 1997;235:79–82.
88. Solanes G, Vidal-Puig A, Grujic D, Flier JS, Lobell BB. The human uncoupling protein-3 gene. J BiolChem 1997;272:26433–26436.
CYTOKINES AND CANCER CACHEXIA • 241

89. Boss O, Samec S, Paoloni-Giacobino A, Rossier C, Dulloo A, Seydoux J, Muzzin P, Giacobino JP. Un-coupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression.FEBS Lett 1997;408:39–42.
90. Matsuda J, Hosoda K, Itoh H, Son C, Doi K, Tanaka T, Fukunaga Y, Inoue G, Nishimura H, YoshimasaY, Yamori Y, Nakao K. Cloning of rat uncoupling protein-3 and uncoupling protein-2 sDNAs: their geneexpression in rats fed high-fat diet. FEBS Lett 1997;418:200–204.
91. Surwit RS, Wang S, Petro AE, Sanchís D, Raimbault S, Ricquier D, Collins S. Diet-induced changes inuncoupling protein in obesity-prone and obesity-resistant strains of mice. Proc Natl Acad Sci USA1998;95:4061–4065.
92. Boss O, Samec S, Dulloo A, Seydoux J, Muzzin P, Giacobino JP. Tissue-dependent upregulation of rat un-coupling protein-2 expression in response to fasting or cold. FEBS Lett 1997;412:111–114.
93. Boss O, Samec S, Kúhne F, Bijlenga P, Assimacopoulos-Jeannet F, Seydoux J, Giacobino JP, Muzzin P.Uncoupling protein-3 expression in rodent skeletal muscle is modulated by food intake but not by changesin environmental temperature. J Biol Chem 1998;273: 5–8.
94. Faggioni R, Shigenaga J, Moser A, Feingold KR, Grunfeld C. Induction of UCP2 gene expression by LPS,a potential mechanism for increased thermogenesis during infection. Biochem Biophys Res Commun1998;244:75–78.
95. Sanchís D, Busquets S, Alvarez B, Ricquier D, López-Soriano FJ, Argilés JM. Skeletal muscle UCP2 andUCP3 gene expression in a rat cancer cachexia model. FEBS Lett 1998;436:415–418.
96. Busquets S, Sanchís D, Alvarez B, Ricquier D, López-Soriano FJ, Argilés In the rat, TNF-a administra-tion results in an increase in both UCP2 and UCP3 mRNAs in skeletal muscle: a possible mechanism forcytokine-induced thermogenesis? FEBS Lett 1998;440:348–350.
97. Price SR, Olivecrona T, Pekala PH. Regulation of lipoprotein lipase synthesis by recombinant tumornecrosis factor: the primary regulatory role of the hormone in 3T3-L1 adipocytes. Arch Biochem Biophys1986;251:738–746.
98. Cornelius P, Enerback S, Bjursell G, Olivecrona T, Pekala PH. Regulation of lipoprotein lipase mRNAcontent in 3T3-L1 cells by tumour necrosis factor. Biochem J 1988;249:765–769.
99. Fried SK, Zechner R. Cachectin/tumor necrosis factor decreases human adipose tissue lipoprotein lipasemRNA levels, synthesis, and activity. J Lipid Res 1989;30:1917–1923.
100. Semb H, Peterson J, Tavernier J, Olivecrona T. Multiple effects of tumor necrosis factor on lipoprotein lipase in vivo. J Biol Chem 1987;262: 8390–8394.
101. Evans RD, Williamson DH. Tumour necrosis factor-α (cachectin) mimics some of the effects of tumourgrowth on the disposal of a [14C]lipid load in virgin, lactating and litter-removed rats. Biochem J1988;256:1055–1058.
102. Feingold KR, Grunfeld C. Tumor necrosis factor-a stimulates hepatic lipogenesis in the rat in vivo. J ClinInvest 1987;80:1384–1389.
103. Krauss RM, Grunfeld C, Doerrler WT, Feingold KR. Tumor necrosis factor acutely increases plasma lev-els of very low density lipoproteins of normal size and composition. Endocrinology 1990;127:1016–1021.
104. Kern PA. Recombinant human tumor necrosis factor does not inhibit lipoprotein lipase in primary culturesof isolated human adipocytes. J Lipid Res 1988;29:909–914.
105. Kawakami M, Murase T, Ogawa H, Ishibashi S, Mori N, Takaku F, Shibata S. Human recombinant TNFsuppresses lipoprotein lipase activity and stimulates lipolysis in 3T3-L1 cells. J Biochem 1987;101:331–338.
106. Feingold KR, Doerrler W, Dinarello CA, Fiers W, Grunfeld C. Stimulaton of lipolysis in cultured fat cellsby tumor necrosis factor, interleukin-1, and interferons is blocked by inhibition of prostaglandins synthe-sis. Endocrinology 1992;130:10–16.
107. Hauner H, Petruschke T, Russ M, Röhrig K, Eckel J. Effects of tumour necrosis factor-alpha (TNFa) onglucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetolo-gia 1995;38:764–771.
108. Pape ME, Kim KH. Effect of tumor necrosis factor on acetyl-coenzyme A carboxylase gene expressionand pre-adipocyte differentiation. Mol Endocrinol 1988;2:395–403.
109. Patton JS, Shepard HM, Wilking H, Lewis G, Aggarwal BB, Eessalu TE, Gavin LA, Grunfeld C. Inter-ferons and tumor necrosis factor have similar catabolic effects on 3T3L1 cells. Proc Natl Acad Sci USA1986;83:8313–8318.
242 • ARGILÉS AND LÓPEZ-SORIANO

110. Beutler BA, Cerami A. Recombinant interleukin-1 suppresses lipoprotein lipase activity in 3T3-L1 cells.J Immunol 1985;135: 3969–3971.
111. Marshall MK, Doerrler W, Feingold KR, Grunfeld C. Leukemia inhibitory factor induces changes in lipidmetabolism in cultures adipocytes. Endocrinology 1994;135:141–147.
112. Price SR, Mizel SB, Pekala PH. Regulation of lipoprotein lipase synthesis and 3T3-L1 adipocyte metab-olism by recombinant interleukin-1. Biochim Biophys Acta 1986;889:374–381.
113. Rossi-Fanelli F, Cangiano C, Muscaritoli M, Conversano L, Torelli GF, Cascino A. Nutrition 1995;11:595–600.
114. Muscaritoli M, Cangiano C, Cascino A, Ceci F, Giacomelli L, Cardellicangiano P, Mulieri M, Rossi-Fanelli F. Plasma clearance of exogenous lipids in patients with malignant disease. Nutrition 1990;6:147–151.
115. Evans RD, Williamson DH. Tissue-specific effects of rapid tumour growth on lipid metabolism in the ratduring lactation and on litter removal. Biochem J 1988;252:65–72.
116. Mulligan HD, Tisdale MJ. Lipogenesis in tumour and host tissues in mice bearing colonic adenocarcino-mas. Br J Cancer 1991;63:719–722.
117. Dessí S, Batetta B, Pulisci D, Accogli P, Pani P, Broccia G. Total and HDL cholesterol in human and hema-tologic neoplasms. Int J Hematol 1991;54:483–486.
118. Dessí S, Batetta B, Anchisi C, Pani P, Costelli P, Tessitore L, Baccino FM. Cholesterol metabolism dur-ing the growth of a rat ascites hepatoma (Yoshida AH-130). Br J Cancer 1992;66:787–793.
119. Dessí S, Batetta B, Spano O, Bagby GJ, Tessitore L, Costelli P, Baccino FM, Pani P, Argilés JM. Pertur-bations of trigylcerides but not of cholesterol metabolism are prevented by anti-tumour necrosis factor-atreatment in rats bearing an ascites hepatoma (Yoshida AH-130). Br J Cancer 1995;72:1138–1143.
120. Eisenberg S. High density lipoprotein metabolism. J Lipid Res 1984;25:1017–1058.121. Feingold KR, Serio MJ, Adi S, Moser AH, Grunfeld C. Tumor necrosis factor stimulates hepatic lipid syn-
thesis and secretion. Endocrinology 1989;124:2336–2342.122. Feingold KR, Mounzer S, Serio MK, Moser AH, Dinarello CA, Grunfeld C. Multiple cytokines stimulate
hepatic lipid synthesis in vivo. Endocrinology 1989;125:267–274.123. Grunfeld C, Adi S, Soued M, Moser AH, Fiers W, Feingold KR. The search for mediators of the lipogenic
effects of tumor necrosis factor: potential role for interleukin-6. Cancer Res 1990;50:4233–4238.124. Grunfeld C, Soued M, Adi S, Soued M, Moser AH, Fiers W, Dinarello CA, Feingold KR. Interleukin-4
inhibits stimulation of hepatic lipogenesis by tumor necrosis factor, interleukin-1 and interleukin-6 but notby interferon-alpha. Cancer Res 1991;51:2803–2807.
125. Carbó N, Costelli P, Tessitore L, Bagby GJ, López-Soriano FJ, Baccino FM, Argilés JM. Anti-tumournecrosis factor-a treatment interferes with changes in lipid metabolism in a cachectic tumour model. ClinSci 1994;87:349–355.
126. Lawson DH, Richmond A, Nixon DW, Rudman D. Metabolic approaches to cancer cachexia. Annu RevNutr 1982;2:277–301.
127. Morrison SD. In: Liotta AL, editor. Influence of tumor development on the host. The Netherlands: Klu-wer Acad. Publ.; 1989, pp. 176–213.
128. Tisdale MJ. Cancer cachexia. Br J Cancer 1992;63:337–342.129. Kien CL, Camitta BM. Increased whole-body protein turnover in children with newly diagnosed leukemia
or lymphoma. Cancer Res 1983;43:5592–5596.130. Kien CL, Camitta BM. Close association of accelerated rates of whole body protein turnover (synthesis
and breakdown) and energy expenditure in children with newly diagnosed acute lymphocytic leukemia. J Parent Enteral Nutr 1987;11:129–134.
131. Tessitore L, Bonelli G, Baccino FM. Early development of protein metabolic perturbation in the liver andskeletal muscle of tumour-bearing rats. Biochem J 1987;241:153–159.
132. Beck SA, Tisdale MJ. Nitrogen excretion in cancer cachexia and its modification by a high fat diet in mice.Cancer Res 1989;49:3800–3804.
133. Melville S, McNurlan MA, Graham Calder A, Garlick PJ. Increased protein turnover despite normal en-ergy metabolism and responses to feeding in patients with lung cancer. Cancer Res 1990;50:1125–1131.
134. Beck SA, Smith KL, Tisdale MJ. Anticachetic and antitumour effect of eicosapentaenoic acid and its ef-fect on protein turnover. Cancer Res 1991;51:6089–6093.
135. Lundholm K, Ekman D, Edstrom S, Karlberg I, Jagenberg R, Scherstén T. Protein synthesis in liver tis-
CYTOKINES AND CANCER CACHEXIA • 243

sue under the influence of a methyl-cholanthrene-induced sarcoma in mice. Cancer Res 1979;39:4657–4661.
136. Emery PW, Neville AM, Edwards RHT, Rennie MJ. Increased myofibrillar degradation and decreasedprotein synthesis in tumor-bearing mice. Eur J Clin Invest 1982;12:10.
137. Pain VM, Randall DP, Garlick PJ. Protein synthesis in liver and skeletal muscle of mice bearing an ascitestumor. Cancer Res 1984;44:1054–1057.
138. Rennie MJ, Edwards RHT, Emery PW, Halliday D, Lundholm K, Millward DJ. Depressed protein syn-thesis is the dominant characteristic of wasting and cachexia. Clin Physiol 1983;3:387–398.
139. Lundholm K, Bennegard K, Eden E, Svaninger G, Emery PW, Rennie MJ. Efflux of 3-methylhistidinefrom the leg in cancer patients who experience weight loss. Cancer Res 1982;.42:4807–4811.
140. García-Martínez C, López-Soriano FJ, Argilés JM. Amino acid uptake in skeletal muscle of rats bearingthe Yoshida AH-130 ascites hepatoma. Mol Cell Biochem 1995;148:17–23.
141. Llovera M, García-Martínez C, Agell N, Marzábal M, López-Soriano FJ, Argilés JM. Ubiquitin gene ex-pression is increased in skeletal muscle of tumour-bearing rats. FEBS Lett 1994;338:311–318.
142. Llovera M, García-Martínez C, Agell N, López-Soriano FJ, Argilés JM. Muscle wasting associated withcancer cachexia is linked to an important activation of the ATP-dependent ubiquitin-mediated proteoly-sis. Int J Cancer 1995;61:138–141.
143. Argilés JM, López-Soriano FJ. The ubiquitin-dependent proteolytic pathway in skeletal muscle: its rolein pathological states. Trends Pharmacol Sci 1996;17:223–226.
144. Fong Y, Moldawer LL, Morano M, Wei H, Barber A, Manogue K, Tracey KJ, Kuo G, Fischman DA, Ce-rami A, Lowry SF. Cachectin/TNF or IL-1a induces cachexia with redistribution of body proteins. Am JPhysiol 1989;256:R659–R665.
145. Warren RS, Starnes Jr HF, Gabrilove JL, Oettgen HF, Brennan MF. The acute metabolic effects of tumornecrosis factor administration in humans. Arch Surg 1987;122:1396–1400.
146. Flores EA, Bistrian BR, Pomposelli JJ, Dinarello CA, Blackburn GL, Istfan NW. J Clin Invest1989;83:1614–1622.
147. Goodman MN. Tumor necrosis factor induces skeletal muscle protein breakdown in rats. Am J Physiol1991;260:E727–E730.
148. Llovera M, López-Soriano FJ, Argilés JM. Chronic tumour necrosis factor-α treatment modifies proteinturnover in rat tissues. Biochem Mol Biol Int 1993;30:29–36.
149. García-Martínez C, López-Soriano, FJ, Argilés JM. Acute treatment with tumour necrosis factor-a induceschanges in protein metabolism in rat skeletal muscle. Mol Cell Biochem 1993;125:11–18.
150. García-Martínez C, Agell N, Llovera M, López-Soriano FJ, Argilés JM. Tumour necrosis factor-a in-creases the ubiquitinization of rat skeletal muscle proteins. FEBS Lett 1993;323:211–214.
151. García-Martínez C, Llovera M, Agell N, López-Soriano FJ, Argilés JM. Ubiquitin gene expression inskeletal muscle is incresed by tumour necrosis factor-a. Biochem Biophys Res Commun 1994;201:682–686.
152. Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: con-jugation of proteins with multiple chains of polypeptide of ATP-dependent proteolysis. Proc Natl AcadSci USA 1980;77:1783–1786.
153. Ciechanover A, Finley D, Varshavsky A. The ubiquitin-dependent pathway and mechanims of energy-dependent intracellular protein degradation. J Cell Biochem 1984;24:27–53.
154. Finley D, Varshavsky A. The ubiquitin system: functions and mechanisms. Trends Biochem Sci1985;10:343–347.
155. Finley D, Chan V. Ubiquitination. Annu Rev Cell Biol 1991;7:25–69.156. Rechsteiner JM. Natural substrates of the ubiquitin proteolytic pathway. Cell 1991;66:615–618.157. Peters JM. Proteasomes: protein degradation machines of the cell. Trends Biochem Sci 1994;19:377–382.158. Hilenski LL, Terracio L, Haas AL, Borg TK. Immunolocalization of ubiquitin conjugates at Z-bands and
intercalates discs of rat cardiomyocytes in vitro and in vivo. J Histochem Cytochem 1992;40:1037–1042.159. Llovera M, Garcia-Martínez C, Costelli P, Agell N, Carbó N, López-Soriano FJ, Argilés JM. Muscle hy-
percatabolism during cancer cachexia is not reversed by the glucocorticoid receptor antagonist RU 38486.Cancer Lett 1996;99:7–14.
160. Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today 1992;13:151–153.161. Llovera M, García-Martínez C, Agell N, López-Soriano FJ, Argilés JM. TNF can directly induce the ex-
244 • ARGILÉS AND LÓPEZ-SORIANO

pression of ubiquitin-dependent proteolytic system in rat soleus muscles. Biochem Biophys Res Commun1997;230:238–241.
162. Llovera M, Carbó N, López-Soriano FJ, García-Martínez C, Busquets S, Alvarez B, Agell N, Costelli P,López-Soriano FJ, Celada A, Argilés JM. Different cytokines modulate ubiqutin-gene expression in ratskeletal muscle. Cancer Lett 1998;133:83–87.
163. Nair KS, Schwartz RG, Welle S. Leucine as a regulator of whole body and skeletal muscle protein me-tabolism in humans. Am J Physiol 1992;263:E928–E934.
164. Adibi SA. Metabolism of branched-chain amino acids in altered nutrition. Metabolism 1976;25:1287–1302.
165. Block RJ. Amino acid handbook. Springfield, IL: 1956, p. 344.166. Argilés JM, López-Soriano FJ. The oxidation of leucine in tumour-bearing rats. Biochem J 1990;268:241–
244.167. García-Martínez C, Llovera M, López-Soriano FJ, Del Santo B, Argilés JM. Lipopolysacharide (LPS) in-
creases the in vivo oxidation of branched-chain amino acids in the rat: a cytokine-mediated effect. MolCell Biochem 1995;148:9–15.
168. Goodlad GAJ, Tee MK, Clark CM. Leucine oxidation and protein degradation in the extensor digitorumlongus and soleus of the tumor-bearing host. Biochem Med 1981;26:143–147.
169. Costelli P, Llovera M, García-Martínez C, Carbó N, López-Soriano FJ, Argilés JM, Enhanced leucine ox-idation in rats bearing an ascites hepatoma (Yoshida AH-130) and its reversal by clenbuterol. Cancer Lett1995;91:73–78.
170. Nawabi MD, Block KP, Chakrabarti MC, Buse MG. Administration of endotoxin, tumor necrosis factor,or interleukin-1 to rats activates skeletal muscle branched-chain a-keto acid dehydrogenase. J Clin Invest1990; 85:256–263.
171. Brennan MF. Total parenteral nutrition in the cancer patient. N Engl J Med 1981;305:375–382.172. Nishihira T, Takagi T, Kawarabayashi Y, Izumi U, Ohkuma S, Koike N, Toyoda T, Mori S. Anticancer
therapy with valine-depleted amino acid imbalance solution. J Exp Med 1988;156:259–270.173. Nishihira T, Takagi T, Mori S. Leucine and manifestation of antitumor activiy by valine-depleted amino
acid imbalance. Nutrition 1993;9:146–152.174. Lazo PA. Tumour-host metabolic interaction and cachexia. FEBS Lett 1985;187:189-192.175. Buse MG, Reid SS. Leucine: a possible regulator of protein turnover in muscle. J Clin Invest
1975;56:1250–1261.176. Fulks RM, Li JB, Goldberg AL. Effects of insulin, glucose and amino acids on protein turnover in rat di-
aphragm. J Biol Chem 1975;250:290–298.177. Tischler ME, Desautels M, Goldberg AL. Does leucine, leucyl-tRNA, or some metabolite of leucine reg-
ulate protein synthesis and degradation in skeletal and cardiac muscle? J Biol Chem 1982;257:1613–1621.178. Mitch WE, Clark AS. Specificity of the effects of leucine and its metabolites on protein degradation in
skeletal muscle. Biochem J 1984;222:579–586.179. Blomstrand E, Hassmén P, Ekblom B, Newsholme EA. Administration of branched-chain amino acids
during sustained exercise: effects on performance and on plasma concentration of some amino acids. EurJ Appl Physiol 1991;63:83–88.
180. Smith K, Barua JM, Watt PW, Scrimgeour CM, Rennie MJ. Flooding with L-[113C]leucine stimulates hu-man muscle protein incorporation of continuously infused [1-13C]valine. Am J Physiol 1992;262:E372–E376.
181. Costelli P, García-Martínez C, Llovera M, Carbó N, López-Soriano FJ, Agell N, Tessitore L, Baccino FM,Argilés JM. Mucle protein waste in tumor-bearing rats is effectively antagonized by a b2-adrenergic ag-onist (clenbuterol). J Clin Invest 1995;95:2367–2372.
182. Medina MA, Sánchez-Jiménez F, Márquez J, Rodriguez-Quesada A, Nuñez de Castro I. Relevance of glu-tamine metabolism in tumor cell growth. Mol Cell Biochem 1992;113:1–15.
183. Kern KA, Norton JA. Cancer cachexia. J Parent Enteral Nutr 1988;12:286–298.184. Koj A. In: Gordon AH, Koj A, editors. The acute-phase response to injury and infection. Amsterdam: El-
sevier; 1985; p. 139.185. Fearon KC, Falconer JS, Slater C, McMillan DC, Ross JA, Preston T. Albumin synthesis rates are not de-
creased in hypoalbuminemic cachectic cancer patients with an ongoing acute-phase protein response. AnnSurg 1998;227:249–254.
CYTOKINES AND CANCER CACHEXIA • 245

186. Croce MV, Segal-Eiras A. Identification of acute-phase proteins (APP) in circulating immune complexes(CIC) in esophagal patients’ sera. Cancer Invest 1996;14:421–426.
187. Weinstein PS, Skinner M, Sipe JD, Lokich JJ, Zamcheck N, Cohen AS. Acute-phase proteins or tumormarkers: the role of SAA, SAP, CRP and CEA as indicators of metastasis in a broad spectrum of neoplasticdiseases. Scand J Immunol 1984;19:193–198.
188. Moshage H. Cytokines and the hepatic acute phase response. J Pathol 1997;181:257–266.189. Baumann H, Gauldie J. The acute phase response. Immunol Today 1994;15:74–80.190. Koj A. Biological functions of acute-phase proteins. In: Gordon AH, Koj A, editors. The acute-phase re-
sponse to injury and infection. Amsterdam: Elsevier; 1985. p. 145–160.191. Waterhouse C, Jeanpetre N, Keilson J. Glucogenesis from alanine in patients with progressive malignant
disease. Cancer Res 1979;39:1968–1972.192. Glicksman AS, Rawson RW. Diabetes and altered carbohydrate metabolism in patients with cancer. Can-
cer 1956;9;1127–1134.193. Werk EE, Sholiton LJ, Marnell RT. Altered cortisol metabolism in advanced cancer and other terminal
diseases. Metabolism 1964;13:1425–1438.194. Inculet RI, Peacock JL, Gorschboth CM, Norton JA. Gluconeogenesis in the tumor-influenced rat hepa-
tocyte: importance of tumor burden, lactate, insulin and glucagon. J Natl Cancer Inst 1987;79:1039 –1046.
195. Tayek JA. A review of cancer cachexia and abnormal glucose metabolism in humans with cancer. J AmColl Nutr 1992;11: 455–456.
196. Lundholm K, Holm G, Scherstén T. Insulin resistance in patients with cancer. Cancer 1978;38:4665–4670.197. Moley JF, Morrison FD, Norton JA. Preoperative insulin reverses cachexia and decreases mortality in tu-
mor-bearing rats. J Surg Res 1987;43:21–28.198. Tessitore L, Costelli P, Baccino FM. Humoral mediation for cachexia in tumor-bearing rats. Br J Cancer
1993;67:15–23.199. Peacock JL, Gorschboth CM, Norton JA. Impact of insulin on doxorubicin-induced host toxicity and tu-
mor regression. Cancer Res 1987;47:4318–4322.200. Rayfield EJ, Ault MJ, Keusch GT, Brothers MJ, Nechemias C, Smith H. Infection and diabetes: the case
for glucose control. Am J Med1982; 72:439–450.201. Lang CH, Dobrescu C. Sepsis-induced changes in in vivo insulin action in diabetic rats. Am J Physiol
1989;257:E301–E308.202. Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose dis-
posal and hepatic glucose output. Endocrinology 1992;130:43–52.203. Van der Poll T, Romijnm A, Endert E, Borm JJJ, Buller HR, Sauerwein HP. Tumor necrosis factor mim-
ics the metabolic response to acute infection in healthy humans. Am J Physiol 1991;261: E457–E465.204. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of
tumor necrosis factor-a in human obesity and insulin resistance. J Clin Invest 1995;95:2409–2415.205. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-a: direct role
in obesity-linked insulin resistance. Science 1993;259: 87–91.206. Stephens JM, Pekala PH. Transcriptional repression of the GLUT4 and C/EBP genes in 3T3-L1 adipocytes
by tumor necrosis factor-a . J Biol Chem 1991;266:21839–21845.207. Pekala P, Kawakami W, Angus CW, Lane A, Cerami A. Selective inhibition of synthesis for de novo fat-
ty acid biosynthesis by an endotoxin-induced mediator from exudate cells. Proc Natl Acad Sci USA 1983;80:2743–2747.
208. Spiegelman BM, Hotamisligil GS. Through thick and thin: wasting, obesity, and TNFa. Cell 1993;73:625–627.
209. Saghizadeh M, Ong JM, Garvey WT, Henry RR, Kern PA. The expression of TNF-a by human muscle.Relationship to insulin resistance. J Clin Invest 1996;97:1111–1116.
210. Hofmann C, Lorenz K, Braithwaite SS, Colca JR, Palazuk BJ, Hotamisligil GS, Spiegelman BM. Alteredgene expression for tumor necrosis factor-a and its receptors during drug dietary modulation of insulinresistance. Endocrinology 1994;134:264–270.
211. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF Spiegelman BM IRS-mediated inhibition ofinsulin receptor tyrosine kinase activity in TNF-a and obesity-induced insulin resistance. Science1996;271:665–668.
246 • ARGILÉS AND LÓPEZ-SORIANO

212. Balazs C, Kiss E. Immunological aspects of the effect of pentoxifylline (Trental). Acta Microbiol ImmunolHung 1994;41:121–126.
213. Goldberg RM, Loprinzi CL, Mailliard JA, O’Fallon JR, Krook JE, Ghosh C, Hestorff RD, Chong SF,Reuter NF, Shanahan TG. Pentoxifylline for treatment of cancer anorexia and cachexia: A randomized,double-blind, placebo-controlled trial. J Clin Oncol 1995;13:2856–2859.
214. Semmler J, Wachtel, H, Endres S. The specific type IV phosphodiesterase inhibitor rolipram suppressestumor necrosis factor-a production by human mononuclear cells. Int J Immunopharmacol 1993;15:409–413.
215. Badger AM, Olivera DL, Esser KM. Beneficial effects of the phosphodiesterase inhibitors BRL 61063,pentoxifylline, and rollipram in a murine model of endotoxic shock. Circ Shock 1994;44:188–195.
216. Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosisfactor-a production in stimulated human monocytes. J Exp Med 1991;173:699–703.
217. Sampaio EP, Kaplan G, Miranda A, Nery JAC, Miguel CP, Viana SM, Sarno EM. J Infect Dis1993;168:408–414.
218. Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G. Thalidomide exerts its inhib-itory action on tumor necrosis factor-a by enhancing mRNA degradation. J Exp Med 1993;177:1675–1680.
219. Tramontana JM, Utaipat U, Molloy A, Akarasewi P, Borroughs M, Makonkawkeyoon S, Johnson B,Klausner JD, Rom W, Kaplan G. Thalidomide treatment reduces tumor necrosis factor-α production andenhances weight gain in patients with pulmonary tuberculosis. Molec Med 1995;1:384–397.
220. Reinhart K, Wiegand-Lohnert C, Grimminger F, Kaul M, Whington S, Treacher D, Eckart J, Willatts S,Bouza C, Krausch D, Stockenhuber F, Eiselstein J, Daum L, Kempeni J. Assessment of the safety and ef-ficacy of the monoclonal anti-tumor necrosis factor antibody fragment, MAK 195F, in patients with sep-sis and septic shock: a multicenter, randomized, placebo-controlled, dose-ranging study. Crit Care Med1996;24:733–742.
221. Yasumoto K, Mukaida N, Harada A, Kuno K, Akiyama M, Nakashima E, Fujioka N, Mai M, Kasahara T,Fujimoto-Ouchi K. Molecular analysis of the cytokine network involved in cachexia in colon 26 adeno-carcinoma-bearing mice. Cancer Res 1995;55:921–927.
222. Emilie D, Wijdenes J, Gisselbrecht C, Jarrousse B, Billaud E, Blay JY, Gabarre J, Gaillard JP, Brochier J,Raphael M. Administration of an interleukin-6 monoclonal antibody to patients with acquired immunod-eficiency syndrome and lymphoma: effect on lymphoma growth and on B clinical symptoms. Blood1994;84:2472–2479.
223. Mori K, Fujimoto-Ouchi K, Ishikawa T, Sekiguchi F, Ishitsuka H, Tanaka Y. Murine interleukin-12 pre-vents the development of cancer cachexia in a murine model. Int J Cancer 1996;67:849–855.
224. Tanaka Y, Eda H, Tanaka T, Udagawa T, Ishikawa T, Horii I, Ishitsuka H. Experimental cancer cachexiainduced by transplantable colon 26 adenocarcinoma in mice. Cancer Res 1990;50:2290–2295.
225. Ouchi KF, Tamura S, Mori K, Tanaka Y, Ishitsuka H. Establishment and characterization of cachexia-inducing and non-inducing clones of murine colon. Int J Cancer 1995;61:522–528.
226. Bielefeldt-Ohmann H, Marzo AL, Himbeck RP, Jarnicki AG, Robinson BW, Fitzpatrick DR. Interleukin-6 involvement in mesothelioma pathobiology: inhibitin by interferon-α immunotherapy. Cancer ImmunolImmunother 1995;40:241–250.
227. Sakurai Y, Zhang XJ, Wolfe RR. Insulin-like growth factor-I and insulin reduce leucine flux and oxida-tion in conscious tumor necrosis factor-infused dogs. Surgery 1995;117:305–313.
228. Ng EH, Rock CS, Lazarus DD, Stiaino-Coico L, Moldawer LL, Lowry SF. Insulin-like growth factor Ipreserves host lean tissue mass in cancer cachexia. Am J Physiol 1992;262:R426–R431.
229. Quinn LS, Haugk KL, Grabstein KH. Interleukin-15: a novel anabolic cytokine for skeletal muscle. En-docrinology 1995;136:3669–3672.
230. Carbó N, López-Soriano J, Costelli P, Busquets S, Alvarez B, Baccino FM, Quinn LS, López-Soriano FJ,Argilés JM. Interleukin-15 mediates reciprocal regulation of adipose and muscle mass. A potential role inbody weight control. To appear.
231. Hill AG, Jacobson L, Gonzalez J, Rounds J, Majzoub JA, Wilmore DW. Chronic central nervous systemexposure to interleukin-1 causes catabolism in the rat. Am J Physiol 1996;271:R1142–R1148.
232. Ling PR, Schwartz JH, Bistrian BR. Mechanisms of host wasting induced by administration of cytokinesin rats. Am J Physiol 1997;272:E333–E339.
CYTOKINES AND CANCER CACHEXIA • 247

233. Roe SY, Cooper AL, Morris ID, Hopkins SJ, Rothwell NJ. Involvement of cytokines in cachexia inducedby T-cell leukaemia in the rat. Eur Cytokine Netw 1997;8:45–49.
234. Sheen-Chen SM, Chen WJ, Eng HL, Chou FF. Serum concentration of tumor necrosis factor in patientswith breast cancer. Breast Cancer Res Treat 1997; 43:211–215.
Josep M. Argilés obtained his Doctorate from the University of Barcelona in 1981. He then became Lecturerin Biochemistry and carried out postdoctoral work on acetone metabolism in mammals. In 1987 he undertookfurther postdoctoral training in the Laboratory of Molecular Biology at the National Institute of Health, Bethes-da, Maryland. In 1988 he became interested in the role of cytokines in the modulation of the host response andwent to the Metabolic Research Laboratory, Radcliffe Infirmary, Oxford, U.K., to work on the metabolic effectsof tumor necrosis factor and interleukin-1. At present he is Associate Professor of Biochemistry and MolecularBiology at the Faculty of Biology, University of Barcelona, being the leader of a research group interested intumor-host interrelationships.
Francisco J. López-Soriano obtained his doctorate from the University of Barcelona in 1987 investigating therole of amino acid metabolism in brown adipose tissue thermogenesis. Later, he undertook postdoctoral re-search in the Metabolic Research Laboratory, Radcliffe Infirmary, Oxford, U.K., working on different aspectsof cytokines during the perinatal period. In 1988 he was promoted to Associate Professor of Biochemistry andMolecular Biology at the Faculty of Biology, University of Barcelona, where he is at present investigating therole of cytokines in muscle wasting and its relation with cachexia.
248 • ARGILÉS AND LÓPEZ-SORIANO








![Nitrogen Excretion in Cancer Cachexia and Its …...[CANCER RESEARCH 49. 3800-3804. July 15. 1989] Nitrogen Excretion in Cancer Cachexia and Its Modification by a High Fat Diet in](https://img.pdfslide.us/doc/110x75/5eaa4ca79f50116bbd270637/nitrogen-excretion-in-cancer-cachexia-and-its-cancer-research-49-3800-3804.jpg)




















