Embed Size (px)
Citation preview

ANNALS O F CLINICAL AND LABORATOKY SCIEN CE, Vol. 19, No. 4 Copyright © 1989, Institute for Clinical Science, Inc.
The Role of Cholesterol Oxidation Products in the Pathogenesis of Atherosclerosis
ROBERT J. MORIN, M .D. and SHI-KAUNG PENG, M .D ., P h .D .
Harbor-UCLA Department o f Pathology Medical Center
Torrance, CA 90509
ABSTRACT
Cholesterol undergoes spontaneous autoxidation, leading to the production of potentially atherogenic oxidation derivatives. W hen 25-hydroxy- cho lestero l (25-OH) or cholestane-3p,5a,6pJ-trio l (triol) was in jec ted intravenously into rabbits, the aortic surfaces showed num erous balloonlike protrusions and crater-like defects indicative of endothelial damage. A lterations in m em brane function caused by these cholesterol oxides could be the mechanism for their cytotoxic effect. Carrier-m ediated hex- ose transport by cultured rabbit aortic smooth muscle cells, m easured using 2-deoxyglucose, was reversibly inhibited by triol within one hour. A m em brane-bound enzyme, 5'-nucleotidase, was inhibited after 24 to 48 hrs incubation with e ither 25-OH or triol. Endocytosis was also significantly inhibited by both 25-OH and triol. D epletion of m em brane cholesterol content by the cholesterol oxides could account for the m em brane functional alterations. Cholesterol biosynthesis is markedly inhibited by 25-OH. Triol has a lesser effect on cholesterol biosynthesis, bu t it is more poten t in blocking uptake of cholesterol by arterial cells in culture.
Cholesterol oxides may also influence cholesteryl ester accumulation by arterial smooth muscle cells. Incubation of cells with 25-OH resulted in a four-fold increase in cholesterol esterifying activity but no effect on cholesteryl ester hydrolytic activity.
The cholesterol oxides appear to be transported in the blood prim arily by very low density lipoproteins (VLDL) and low density lipoproteins (LDL). Oxidized LD L has cytotoxic effects and enhances m acrophage lipid accumulation. These be effects may be directly related to the cholesterol oxide content of these lipoproteins.
Introduction
Cholesterol now has a well established role in the pathogenesis of atherosclerosis. Cholesterol undergoes spontaneous autoxidation, leading to the production
225
0091-7370/89/0700-0225 $02.00 © Institute for Clinical Science, Inc.
of oxidized derivatives which may be more injurious to the arterial wall than ch o leste ro l itself. The ev idence and m ech an ism s fo r th is e ffec t w ill be reviewed.
The initial event in atherogenesis is believed to be an injury to arterial endo-

226 MORIN AND PENG
thelium , followed by an inflam m atory re s p o n s e in th e a r te r ia l w all. T he response includes increased endothelial tu rn o v e r and p e rm e a b ility , in tim a l edem a and infiltration of monocytes that ingest excessive intimal lipids and transform into foam cells. Endothelial denudation and replication may in fluence lesion progression, not only by allowing platelet adhesion and lipid penetration, bu t also by production of substances prom oting sm ooth m uscle p ro liferation . These substances stimulate migration of smooth muscle cells to the intim a and their modulation into a synthetic phase. Proliferating smooth muscle cells, which secrete collagen, elastin and proteoglycans to the surrounding matrix, and intimal lipid deposition eventually give rise to the typical atherosclerotic lesion.
W hile there are some suggestions that hypercholesterolemia may cause arterial injury and increased endothelial tu rn over, the mechanism is not well characterized . 14,55 In one earlier study, hyperc h o le s te ro le m ia w as e n d o g e n o u s ly induced in chickens on a cholesterol-free d iet by the subcutaneous implantation of d ie th y ls tilb es te ro l. T hese w ere com pared w ith chickens w ith com parable levels of hypercho lestero lem ia (about 400 mg per dl) induced by feeding a two percent USP grade cholesterol-containing diet. After six and one half months on these regimens, num erous large atherosclerotic lesions w ere observed in the thoracic aortas of the cho lestero l-fed group of chickens, w hereas the form er group with endogenous hypercholesterolem ia showed only m inor degrees of atherosclerosis.9 Cholesterol readily oxidizes in air at room tem peratu re . 2,13,24,58 In the preparation of cholesterol diets to induce experimental atherosclerosis, it is com m on practice to mix chow w ith a solution of cholesterol in organic solvent, then air dry it to rem ove the solvent, thereby exposing thin films of cholesterol to ideal conditions for autoxidation.
The m ore extensive atherosclerosis in the cholesterol-fed chicken may well have been due to cholesterol autoxidation products in the USP grade cholesterol.
Imai et al26 reported that purification of stock five-year-old USP grade cholesterol largely prevented its atherogenicity when fed to rabbits, whereas adm inistration o f th e c o n c e n tra te d im p u ritie s p resen t in USP cholestero l produced severe a rte ria l dam age, w ith sm ooth m uscle ce ll d e a th and focal in tim a l edem a. T hese im p u ritie s have been shown to be largely composed of cholestero l oxidation p ro d u c ts . 61 The m ajor components of cholesterol autoxidation w e re id e n t if ie d to b e c h o le s ta n e - 3(3,5a,6p-triol, 5a ,6a-and 5(3, 63-epoxy- cholesterol, 7-ketocholesterol, 7a-and 7(3-hydroxycholesterol and 25-hydroxy- cholesterol. Rabbits fed a cholesterol- con ta in ing d ie t for two m onths w ere found to have two to five times the normal plasma of the a and 3 epoxides and cholestanetriol and five to eight times th e norm al liver levels of th ese oxy- sterols . 62
In our earlier in vitro studies, a concentrate of cholesterol oxidation products was prepared by recrystallizing five- y e a r - o ld U S P c h o le s t e r o l fro m a m ethano l ex tract. Pathophysiological effects of these oxidation products were studied in rabbit aortic smooth muscle cell cu ltu res. The concen tra te of the cholesterol oxidation products showed rem arkable in v itro cytotoxic effects, whereas purified cholesterol at the same concentration produced no toxic effects. The concentrate of cholesterol oxidation products was fractionated further by thin layer chromatography (TLC). The effects of each TLC fraction were tested and the results showed that 25-hydroxycholes- terol and choIestane-3p,5a,6(3-triol were most toxic and probably responsible for the biological toxicity of the contam inants of cholesterol. 52,53 These two cho

CHOLESTEROL OXIDATION PRODUCTS 227
lestero l oxidation products w ere then tested for their angiotoxicity after intravenous administration. Injections of five mg p er kg of e ither com pound caused damaging effects in rabbits, including fibromuscular thickening and cell death in the aorta and segmental thickening in both major and m inor branches of the pulm onary artery .25 O f the identifiable com pounds of ch o le s te ro l ox idation p roducts used, the 25-hydroxycholes- terol and cholestane-3pJ,5a,63-triol were as po ten t as the concentrate of choleste ro l con tam inan ts in inducing these angiotoxic effects in rabbits. Both 5 a ,6 a- epoxycholesterol and 7-ketocholesteroI w ere less toxic. Intravenous injections of 25-hydroxycholesterol and cholestane- 3p,5a,6|3-triol in ethanol likewise caused aortic damage in rabbits. Purified chole s te ro l d id n o t p ro d u c e d am ag ing effects.
In a subsequent study, three groups of New Zealand W hite male rabbits were given intravenously either 25 mg per kg of 25-hydroxycholesterol, cholestane- 3p ,5a ,6 (3 -trio l or vehicle only . 51 The luminal surfaces of aortas receiving 25- hydroxycholesterol w ere exam ined by s c a n n in g e le c tro n m ic ro sc o p y and showed num erous balloon-like p ro tru sions and crater-like defects as well as circulating formed elem ents adhering on the luminal surfaces. The lum inal surfaces of aortas of rabbits given choles- tane-3p,5a,6p-triol had similar bu t more frequen t lesions than the 25-hydroxyc h o le s te ro l g roup (figure 1). M icro throm bi w ere occasionally found. The aortas of the control group had significantly fewer lesions. Transmission electron microscopy studies showed intracy- t o p la s m ic v a c u o le s a n d d i f f u s e subendothelial edem a in the aortas of the two groups receiving the cholesterol oxidation products. The balloon-like protrusions and crater-like defects observed by sc a n n in g e le c tro n m ic ro sc o p y appeared to represent initial endothelial
cell injury induced by cholesterol oxidation products.
There are at least two possible mechanism s by which cholesterol oxidation products can cause cell injury. Inhibition of cholesterol biosynthesis by cholesterol oxidation products could result in several consequences. If phospholipid concentrations are not altered significantly by cholesterol oxidation products, the molar ratio of m em brane cholesterol to phospholipid would decline. D ecreased cellular cholesterol content could eventually suppress m em brane biogenesis and inhibit cell growth. Another mechanism could be that the cholesterol oxidation products, owing to their similar molecular structure, may be incorporated into cell m em branes or displace cholesterol from cell m em branes. Cholesterol in cell m em branes imparts the optimal rigidity e s s e n t ia l fo r m e m b ra n e fu n c t io n . D ecreased cellular cholesterol content or substitution of cholesterol oxidation p roducts for cholestero l in these cell m em branes would increase fluidity and alter a num ber of membrane-associated functions, resulting in cell death. Experim ents w ere designed by the p resen t au tho rs to te s t bo th of th ese m echanisms, i.e ., effects on cholesterol biosynthesis and on cholesterol up take, and effects on m em brane associated functions, including carrier m ediated hexose transport, ion transport and m em brane bound enzymes, large molecule uptake by endocytosis, and prostacyclin synthesis.
Effects on Cholesterol Biosynthesis
In cultured rabbit aortic smooth muscle cells, the activity of hydroxy methyl glutaryl coenzyme A (HMGCoA) reductase, the rate-limiting step in cholesterol biosynthesis, was reduced by 83 percent when cells were incubated with 3 ¡xg per ml of 25-hydroxycholesterol and by 63

228 MORIN AND PENG
FIGURE 1. A orta of ra b b it rece iv in g cholestane-3(3,5oi,6p5-triol show ing th e p re se n c e o f n u m b e ro u s b leb s a n d c ra te rs on th e lum inal surface. M any p la te le ts a re also a d h e r in g on th e su rface , x 4 ,700.
percen t when incubated w ith choles- tane-3(3,5a,6p-triol.53 In o ther studies, deoxyribonucleic acid (DNA) synthesis, as m easured by m ethyl-3H thym idine incorporation per mg of cellular protein, declined in parallel with the HMGCoA reductase activity, and cell division was blocked in the Gx phase of the cycle.30 This may be a mechanism for the cytotoxicity of the cholesterol oxidation products. Cholesterol biosynthesis as m easured by 14C-acetate incorporation, was proportionately decreased by the above two cholesterol oxidation products.47
Effects on Cholesterol Uptake
Labeled [4-14C]-cholesterol was added to culture media containing various con
centrations (0 to 50 (xg per ml) of cholesterol oxidation products.47 These media were transferred to culture flasks conta in ing m onolayers of aortic sm ooth muscle cells and then incubated for one hour at 37°C. Uptake of labeled cholesterol was m easured after the cu ltures w ere w ashed four tim es w ith H ank ’s solution. For rem oval of non-specific surface-bound sterol from the cells, the monolayers were incubated at 37°C for 45 m inutes in media containing pronase. The non-specific surface-bound cholesterol am ounted to 35 pe rcen t of total cho lestero l up take . The m ost p o ten t inhibitor of cholesterol uptake was cho- lestane-3p,5a,6(3-triol. W hen triol was p re se n t at 50 |xg p e r ml of m edium , uptake of cholesterol by the cu ltu red
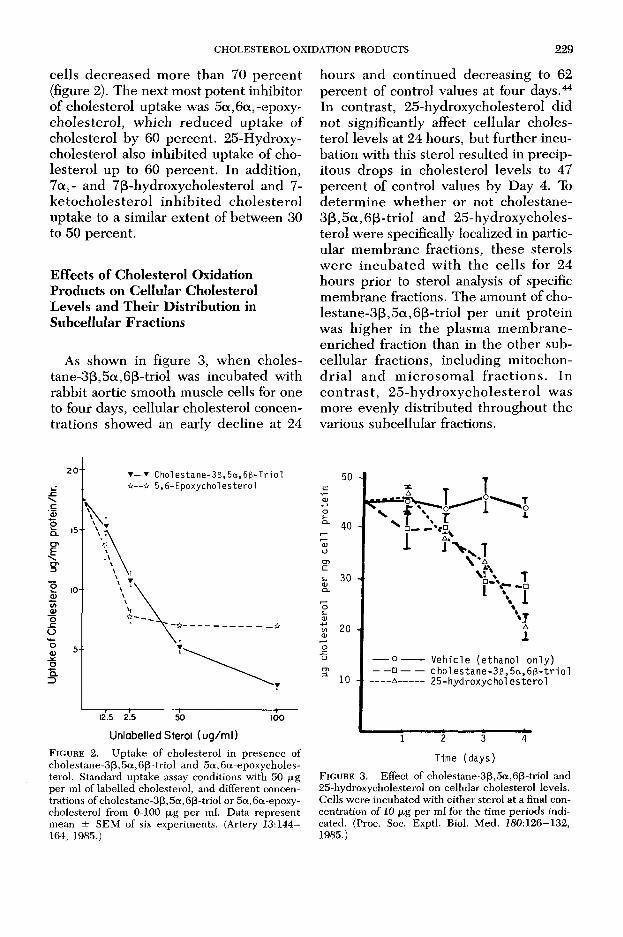
CHOLESTEROL OXIDATION PRODUCTS 229
cells decreased m ore than 70 pe rcen t (figure 2). The next most potent inhibitor of cholesterol uptake was 5 a ,6 a,-epoxy- cholesterol, which reduced uptake of cholesterol by 60 percent. 25-Hydroxy- cholesterol also inhibited uptake of cholesterol up to 60 percent. In addition, 7 a ,- and 7(3-hydroxycholesterol and 7- ke to ch o leste ro l in h ib ite d cho lestero l uptake to a similar extent of betw een 30 to 50 percent.
Effects of Cholesterol Oxidation Products on Cellular Cholesterol Levels and T heir D istribution in Subcellular Fractions
As shown in figure 3, when choles- tane-3]3,5a,6p-triol was incubated with rabbit aortic smooth muscle cells for one to four days, cellular cholesterol concentrations showed an early decline at 24
Unlabelled Sterol (ug/m l)
F ig u r e 2. Uptake of cholesterol in presence of cholestane-3ß,5a,6ß-triol and 5a,6a-epoxycholes- terol. Standard uptake assay conditions with 50 jjig per ml of labelled cholesterol, and different concentrations of cholestane-3ß,5a,6ß-triol or 5a,6a-epoxy- cholesterol from 0-100 jjig per ml. Data represent mean ± SEM of six experiments. (Artery J3:144— 164, 1985.)
hours and continued decreasing to 62 percent of control values at four days. 44 In contrast, 25-hydroxycholesterol did not significantly affect cellular cholesterol levels at 24 hours, but further incubation with this sterol resulted in precipitous drops in cholesterol levels to 47 percent of control values by Day 4. To determ ine w hether or not cholestane- 33 ,5a ,6 |3 -trio l and 25-hydroxycholesterol were specifically localized in particular m em brane fractions, these sterols w ere in cu b a ted w ith the cells for 24 hours prior to sterol analysis of specific m em brane fractions. The amount of cho- lestane-3p,5a,6(3-triol per unit protein was h igher in the plasm a m em brane- enriched fraction than in the o ther sub- cellular fractions, including m itochond r ia l and m icro som al fra c tio n s . In co n tra s t, 25 -h y d ro x y ch o leste ro l was more evenly distributed throughout the various subcellular fractions.
Time (days)
FIGURE 3. Effect of cholestane-3(3,5a,6(3-triol and 25-hydroxycholesterol on cellular cholesterol levels. Cells were incubated with either sterol at a final concentration of 10 |xg per mi for the time periods indicated. (Proc. Soc. Exptl. Biol. Med. 180:126—132, 1985.)

230 MORIN AND PENG
Effects on Carrier Mediated Hexose Transport
Carrier m ediated hexose transport is of critical importance in maintaining normal m em brane function. Because D-glu- cose moves b idirectionally , a glucose analogue, 2-deoxy-D-glucose, is utilized for D -glucose tran sp o rt studies. This compound is taken up by the same transp o rt m echan ism as D -g lucose; once inside the cell, it is phosphorylated and not fu rther m etabolized, and this p re vents reverse transport. The effects of various autoxidation derivatives on hexose transport in cu ltured rabbit aortic smooth muscle were studied. The most potent inhibitor of hexose transport was choiestane-3p ,5a,6 (i-triol (table 1). The inhibitory effect of the triol appeared to be dose dependen t, w ith a 58 percent decrease in hexose transport at the highest level (50 \xg p e r ml) te s te d .22 The inhibitory effect was not the result of cell toxicity, as the total protein content of the cultured cells did not vary and dye exclusion studies showed the cultured cells to be viable during the one hour incubation period . F u rth e rm o re , the
TABLE IEffect of Cholesterol Autoxidation Derivatives
on 2-Deoxy-D-glucose Transport
PercentChanges Transport ofCompared 2-Deoxy-D-glucose
Sterol to Control (nmoles/10 min/mg)
Control (vehicle only)* 225.29 + 45.78$25-Hydroxycholesterol f -15.51 190.34 + 47.3020a-Hydroxycholesterol - 3.86 216.58 + 29.075a,6a-Epoxy cholesterol - 1.87 221.08 + 21.80Cholest-3 6 » 5a-diol-6-one -27.25 § 163.89 + 25.477-Ketocholesterol - 2.67 219.26 ± 26.504-Cholesten-3-one + 1.24 228.09 ± 25.82Cholestane-38/5a,66-triol -30.56ff 156.44 + 38.00
*Vehicle was ethanol which was less than 0.4 percent of final volume of media,
fSterols were dispensed in basal culture medium supplemented with 10 percent fetal bovine serum at a final concentration of 25 yg per ml. All cells were incubated with 2 ml of medium containing a test sterol for one hour prior to transport assay.
^Values represent mean ± S.E.M. of at least two experiments.
§ p < 0.05 compared to control value.Ï p< 0.01 compared to control value.Reprinted from Exptl. Molec. Path. 41:249-257, 1984.
inhibitory effect of the triol on hexose tra n sp o rt was com ple te ly rev ers ib le . W hen duplicate cultures incubated with triol for 30 m inutes were transferred to control nu trien t m edia (triol-free), the rate of hexose transport retu rned to the control level by 1.5 hour.
Effects on Membrane-bound Enzymes
It has been suggested that slight p e rturbations in the m icroenvironm ent of intrinsic m em brane-bound enzymes can result in large changes in the activity of th e s e e n zy m es. P lasm a m e m b ra n e - bound enzym e activity was used as an index of the functional sta tus o f the plasm a m em branes of cu ltu red aortic smooth m uscle cells which w ere incubated for 24 to 48 hours with 10 |JLg per ml of e ith e r 25-hydroxycholesterol or cholestane-3[3,5a,63-triol. 43,44 The cells w ere h o m o g en ized , a fte r w hich th e plasma m em brane-enriched fraction was separated by ultracentrifugation using a discontinuous sucrose density gradient. C h o le s ta n e -3 p ,5 a ,6 (3 - tr io l an d 25- hydroxycholesterol both caused significant decreases in 5 '-nucleotidase activity at 24 and 48 hours (table II). Only cho- lestane-3p,5a,6(3-triol caused significant in h ib itio n o f N a+ , K + -adenosine t r i phosphate activity after incubation of 48 hours.
Effects on Horseradish Peroxidase Uptake by Endocytosis
Rabbit vascular endothelial cells were in cu b a ted w ith cho lestan e-3 |3 ,5 a ,6 3 - trio l, 2 5 -h y d ro x y ch o leste ro l, 5 a , 6 a - epoxycholesterol, or 7-ketocholesterol for 15 m inutes to 48 hours .46,57 Subsequently, the cells w ere incubated with ho rserad ish peroxidase (HRP) before finally lysing with 0.05 percen t Triton X-100. The cell lysates were assayed for HRP by adding a substrate m ixture of H 20 2 and O-dianisidine and measuring

CHOLESTEROL OXIDATION PRODUCTS 231
E f f e c t o f C h o l e s t a n e - 3 0 , S a , 6 6 - t r i o l a n d 2 5 - H y d r o x y c h o l e s t e r o l o n 5 ' - N u c l e o t i d a s e
A c t i v i t y i n C u l t u r e d A o r t i c S m o o t h M u s c l e C e l l s
TABLE II
5'NucleotidaseActivity Percent Change
Time (nM/min/ Compared toSterol (Hours) mg x 10~2) the Control
Control(vehicle only)* — 30.76±6.75* --
25-Hydroxy- 24 18.11±5.4lf -41.12cholesterol 48 22.60±3.47f -26.52
Cholestane- 24 18.82t5.89f -38.8138,5a,6g-triol 48 15.83±3.09f -48.53
* Control values represent the average values obtainedfrom zero time , 24 and 48 hour incubation with
f Values represent the mean ± S.E.M. of at least sixexperiments.
f p<0.01 compared to control value.Reprinted from Proc. Soc. Exp. Biol. Med. 180:126-132,1985.
the am ount of oxidized product. Pure cholesterol at 40 jxg per ml had no effect on the rate of endocytosis, whereas incubation with triol at the same concentration decreased the rate of endocytosis in 15 m inutes. After one hour of incubation, the effect of trio l leveled off at about 35 percent of the control rate. 7- Ketocholesterol had a slower and less dram atic effect, leveling off at 55 percent of the control rate after 24 hours. 25- H ydroxycholesterol and 5a ,6a-epoxy- cholestero l had very little im m ediate effect on endocytosis.
M echanisms of Cytotoxic Effects of Cholesterol Oxidation Products
The po ten t inhibition of cholesterol b iosyn thesis by cho lestero l oxidation products could result in several consequences. The decrease in cellular cholesterol levels as shown in our study44 and others29 38 would deplete the cholesterol available for m em brane synthesis. The molar ratio of m em brane cholesterol to p h o sp h o lip id w ould d ec lin e , and m em brane associated functions could be disturbed. Since the inhibitory effects on many m em brane-associated functions, including carrier-m ediated hexose trans
port and endocytosis of horseradish per- o x id a s e i n d u c e d b y c h o le s t a n e - 33,5a,6(3-triol, occurred as early as 15 m inutes, it is unlikely that this im m ediate effect was due to loss of m em brane cho lestero l secondary to inh ib ition of ch o leste ro l b iosyn thesis. In fact, th e inh ib ito ry effect of th e triol on m em b ran e function occurred even in th e presence of an exogenous source of cholesterol (fetal bovine serum).
C h o le s ta n e -3 3 ,5 a ,6 3 -tr io l has also been shown to affect o ther m em brane functions. G ordon e t al38 have shown that several cholesterol oxidation products, including triol, inhibit leukocyte chemotaxis in vitro. Streuli et al58 have dem onstra ted that the trio l inh ib ited lymphocyte-E rosette formation. In both of these studies, the inhibitory effects were observed after a short exposure to the sterols. C holestane-3p ,5a,6p-trio l was found to increase in th e p lasm a m em brane fraction five-fold more than in other cell fractions when the arterial smooth muscle cells were incubated with the trio l . 44 It would appear, therefore, that triol may displace cholesterol from cell m em branes and th e re b y d is ru p t their hydrophobic interior. In contrast to th e trio l, 25-hydroxycholestero l, th e most potent inhibitor of cholesterol biosynthesis, had a delayed effect on m em b rane fu n c tio n s , 44 and in h ib ite d cell growth only after prolonged incubation of m ore than 24 hours . 1 These effects could be reversed by adding mevalonate or cholesterol to the culture m edium , 10 substantiating that the delayed effects of 25-OH on m em brane function may be due to m em brane cholesterol depletion secondary to reduced synthesis.
Prostaglandin M etabolism and Cholesterol Oxidation Products
Recently, the balance or imbalance of prostacyclin (PG I2) and th rom boxane

232 MORIN AND PENG
production has been implicated in ath- erogenesis. Many lipid peroxides, particularly fatty acid peroxides, were shown to be potent inhibitors of PG I2 synthesis39'56 and may play an im portant role in the developm ent of a therosclerosis . 17 Martinez-Sales e t al35 dem onstrated that the aortas of rats fed a diet enriched with two p e rc e n t ch o leste ro l au toxidation products for 24 hours prior to sacrifice showed a slight increase in both lipid peroxide and prostacyclin production. They believed the results suggested that the increase in the prostacyclin production might be a protective compensatory m echanism in response to vessel wall injury.
Our study using cultured rabbit aortic smooth muscle cells preincubated with cho lestero l oxidation p roducts for 24 hours show ed th a t th e effect v a ried depending on the type of cholesterol oxidation products . 54 Although cholestane- 3p ,5a ,6p -trio l had an inhibitory effect on PGI2 synthesis (11.1 ± 1.7 pg per fxg p ro te in vs. 16.2 ± 1.9 control), 25- h y d ro x y c h o le s te ro l a p p e a re d to be somewhat stimulatory (35.5 ± 4.8). The explanation could be the p rom p t and direct m em brane effect by cholestane- 33,5a,6|3-triol, resulting in an im m ediate inhibition of PGI2 production versus the somewhat delayed m em brane effect of 25-hydroxycholesterol.
Cholesteryl Ester Metabolism and Cholesterol Oxidation Products
A nother possible m ode of action of cholesterol oxidation products in athero- genesis might be at the stage of cholesteryl ester accum ulation and foam cell formation.
L ipopro te ins from plasm a or o th e r sources en ter cell lysosomes where cholesteryl esters are hydrolyzed and the free cholesterol subsequently resynthesized into lipid droplets of cholesteryl
esters by the enzyme acyl CoA cholestero l acyltransferase (ACAT). C holesteryl esters in lipid droplets are continuously be ing ac ted on by hydro ly tic enzym es . 8 F ree cholesterol derived from this hydrolysis has the ability to migrate through the cell m em brane and is available for transport out of the cell by high density lipoproteins (HDL).
In cultured mouse L fibroblast cells, incubation with 25-hydroxycholesterol or ch o lestan e-3 3 ,5 a ,6 p -trio l resu lted in intracellular lipid accum ulation . 22 The cho lestery l e s te r co n te n t of c u ltu red hepatocytes from rabbits fed oxidized c h o le s te ro l d e r iv a tiv e s was g rea tly increased as com pared to hepatocytes from control or pure cholesterol-fed rabbits . 33 The effects have been examined by us of cholesterol oxidation products on enzymes that participate in accumulation and removal of cellular cholesteryl e s te rs . 41 R abbit aortic sm ooth m uscle cells in culture were pre-incubated for 24 hours in lipoprotein deficient media and then incubated for 15 m inutes, one or five hours with 5 fxg per ml media of e ith e r 25-hydroxycholesterol, choles- tane-3fS,5a,63-triol cholesterol, or e th anol control. After one hour of incubation, ACAT activity showed a four-fold increase in the 25-hydroxycholesterol group (7.63 p e rc e n t ± 2.24 p e rc e n t esterified p e r mg p ro te in p e r hour as compared to the control 1.96 percent ± 1.27 percent). Cholestane-3(3,5a,6(3-triol did not affect ACAT activity after any of the incubation time intervals. The activities of acid cholesteryl este r hydrolase and neutral cholesteryl ester hydrolase were not significantly affected by either 25-hydroxycholestero l or cho lestane- 3(3 ,5a,6(3-trio l a t any tim e in te rv a l. Enhancem ent of ACAT by 25-hydroxycholesterol without change in cholesteryl ester hydrolase may predispose to cholesteryl este r accum ulation by arterial smooth muscle cells.

CHOLESTEROL OXIDATION PRODUCTS 233
Absorption and T ransport of Cholesterol Oxidation Products
Since cholesterol oxidation products are p resent in our diets, it is im portant to determ ine to what extent these compounds can be absorbed and w hether or not they can be transported by lipoproteins which can bind and internalize into arterial cells. The absorption and transport of one of the most toxic cholesterol oxidation products, 25-hydroxy cholesterol, was studied previously in squirel m o n k e y s . 50 U sing th e d u a l- iso to p e plasma ratio technique, the absorption of25-hydroxycholesterol was demonstrated to be ap p ro x im ate ly 30 p e rc e n t. Its transport in plasma lipoproteins has also been studied. W hen squirrel monkeys w ere fed radioactive sterols (both purified cholesterol and 25-hydroxycholesterol), the distribution of labeled cholestero l in very low density lipoproteins (V L D L ), low d e n s ity l ip o p ro te in s (LDL), and high density lipopro teins (H D L) was alm ost identical to that of u n lab e led cho lestero l. O n the o th e r hand, the majority of 25-hydroxycholes- terol radioactivity was located in LD L and VLDL (55.7 percent ± 2 .9 and 34.1 p ercen t ± 2.9, respectively) and only 10.2 percent ± 0 .5 was present in HDL. These results indicated that the majority of 25-hydroxycholesterol is transported to the pe riphera l tissues, such as the arterial wall, by VLDL and LDL. The high density lipoproteins carry only a relatively m inor amount of 25-hydroxycholesterol.
O u r re c e n t stud ies have shown to what extent cholesterol oxidation products can be absorbed and transported w hen p resen t together w ith a norm al d ietary cholesterol in take . 45,48 Radiolabeled pure [4-14C]-cholesterol was kept at 60°C under air to autoxidize for five w eeks, a fte r w hich approxim ately 1 2 p ercen t cholesterol oxidation products
were formed. The mixture, suspended in gelatin, was given to rabbits by gastric gavage. Rabbits were sacrificed at four, 24, and 48 hours after treatm ent. Cholestero l and its autoxidation products w ere separated by thin layer chromatography into five fractions. Radioactivities of each fraction of cholesterol oxidation products and cholesterol in the original mixture before administration and in the rabb it sera after adm inistration w ere sim ilar, sug g estin g th a t th e ra te s of absorption of cholesterol oxidation products are not significantly different from that of cholesterol.
L ipop ro te in s w ere frac tiona ted by u ltracentrifugation into VLDL, LD L, and HDL. Radioactivities of each lipoprotein sterol fraction separated by thin layer chromatography showed that fractions contain ing cho Iestane-33 ,5a ,63 - triol, 7a-and 7|3-hydroxycholesterol, and 7-ketocholesterol were selectively transported in VLDL, w hereas m ost of the25-hydroxycholesterol was p resen t in LDL. In addition, H D L contained only m inute amounts of cholesterol oxidation products. Oxygenated sterols, including epoxycholesterol, 7-ketocholesterol, and26-hydroxycholesterol, have previously been found in hum an p lasm a . 6 These oxygenated sterols w ere also predom inantly located in VLDL and LDL. The concentrations of a - and p-epoxycholes- terol in VLDL were three times greater than in H D L, and the concentration of 7 -ketocho leste ro l in b o th V L D L and LD L was also three times greater than in H D L . C hron ically e lev a ted levels of plasma VLDL and LD L have been assoc iated w ith an increased incidence of atherosclerosis in num erous epidem iological studies . 31 The selective transport of ch o leste ro l oxidation p roducts by VLDL and LD L may account in part for the atherogenicity of these lipoproteins. This hypothesis is strengthened by the demonstration that under certain condi

234 MORIN AND PENG
tions LD L and VLDL are injurious or toxic to cu ltured endothelial cells and arterial smooth muscle cells . 18,21
It has also been shown that toxic LD L and VLDL can be formed by free radical-induced oxidation of a lipid component of the lipoprotein . 20,40 The mechanism by which the oxidized lipid fraction of LD L and VLDL might damage cultu red cells and the identity of the toxic agent are not yet established. It is possible that these toxic components are chole s te ro l o x id a tio n p ro d u c ts . W h e n K rieger e t al32 rep laced the free and esterified cholestero l of plasm a LD L with 25-hydroxycholesterol oleate, the resulting particle bound to LD L recepto rs on h u m an f ib ro b la s ts an d was hydrolyzed in lysosom es in a m anner similar to that of native LDL. This oxy- sterol-contain ing L D L suppressed 3- hydroxy-3-m ethylglutaryl coenzym e A (HMG CoA) reductase. W hen fibroblasts were incubated with the oxysterol-con- taining LD L without added cholesterol, the cells developed structural abnormalities, their growth was inhibited, and the cells died. The toxic effects of the oxy- sterol-containing LD L were prevented when the growth m edium was supplem ented with cholesterol or mevalonate. In another study, treatm ent of LD L with cholesterol oxidase, resulting in oxidatio n of ch o le s te ro l to c h o le s ten o n e , resulted in the LD L becoming toxic to cultured bovine aortic endothelial cells . 4
The earliest cellular interaction that occurs in hypercholesterolemia has been shown to be adhesion of monocytes to dysfunctional endothelial cells . 55 Monocytes have very few binding sites for native LD L but ingest large amounts of oxidized or charge-m odified L D L via h igh-affin ity scavenger re c e p to rs 15,19 which are no t dow n-regulated in the presence of excess cholesterol . 7
It therefore appears that the presence of lipopro teins containing cholesterol oxidation products form dietary sources
or th rough oxidative m odification by arterial cells, not only could cause arterial cell injury bu t could also enhance lipid accumulation and foam cell formation.
Experim ental Atherosclerosis Induced by Cholesterol Oxidation Products
A few investiga to rs have p roduced experim ental atherosclerosis by feeding p u re c h o le s te ro l ox idation p ro d u c ts alone or in combination with pure cholesterol. Cook et al in 1968 were the first to use cholesterol oxidation products to induce experim ental atherosclerosis . 11 They had tested 103 compounds or organ cultures of chicken hearts5 and rabbit aorta . 34 C holestane-3p,5a,6 |3-triol was found to be the m ost toxic. This compound, therefore, was chosen to be fed to the New Zealand white male rabbits in doses of 0 .0 1 p ercen t of a standard commercial diet, corresponding to about 30 mg per kg of body w eight p e r day. The duration of feeding ranged from 27 to 350 days. Lesions w ere observed in the aorta, starting w ith deposition of su d a n o p h ilic m a te ria l, p ro g re ss in g through various pathological changes in the intima and media, and term inating w ith the ao rta show ing considerab le fibrosis and calcification.
Cook et al11 po in ted out that lesions induced in the rabbit aorta by feeding a high cholesterol diet bear only a partial resem blance to hum an atherosclerosis. In the hum an type, not only are lipid deposits p resen t, b u t th e re are o ther changes such as disruption of elastic tissue, alteration in ground substance, tissue necrosis, and calcification. These a u th o r s c la im e d th a t th e c h a n g e s induced by prolonged feeding of relative ly sm all am oun ts of ch o lestan e- 3p,5a,6(3-triol produced lesions in rabbits with a closer resem blance to those of hum an atherosclerosis. M ore recently, cholestane-3(3,5a,6 fJ-triol adm inistered

CHOLESTEROL OXIDATION PRODUCTS 235
orally to rats produced toxic damage to aortic endo thelia l and sm ooth m uscle cells. 36 The endothelium showed extensive areas of cellular degeneration and desquam ation, w ith p late le t and fibrin ad h eren ce . The sm ooth m uscle cells show ed nuclear dam age, cytoplasm ic vacuo lar d e g e n e ra tio n , and c e llu la r necrosis in w ide areas of th e m edia. Jacobson et al28 have recently studied W h ite C a rn e a u p ig eo n s gavage-fed either 0.05 percent pure cholesterol or 0.05 percent pure cholesterol with trace levels of cholestane-3(3,5a,6p-triol for three months. These are amounts similar to th e es tim a ted U .S . d ie ta ry in take levels. Aortic lipids, aortic calcium and coronary a rte ry h istopatho logy w ere assessed. Aortic lipids showed no signific a n t d if fe re n c e s b e tw e e n th e tw o groups, bu t aortic calcium accumulation in the cholesterol with triol group was 1.16 ± 0.35 mg per g, as compared to 0 .82 ± 0 .27 in th e c h o le s te ro l- fe d groups, an increase of 42 percent (P < 0.01). Coronary artery atherosclerosis, as m easured by percent mean luminal stenosis, was 5.23 + 5.4 percent in the cholesterol with triol group as compared to 2.80 + 1.4 percent in the cholesterol-fed group, a significant difference of 87 percent (P < 0.01). These results indicate that low level d ietary intake of choles- tan e -tr io l is a th e ro g en ic to a g rea te r degree than pure cholesterol alone. The data suggest that cholestane-triol is an atherogenic risk factor in doses approximating U.S. dietary exposure to cholesterol and its auto-oxides. Furtherm ore, the resu lts call for re in te rp re ta tio n of data from studies using animal models fed U.S. P. cholesterol and of hum an epidemiologic data not accounting for cholesterol oxide intake.
Two re c e n t re p o r ts in d ic a te th a t Indian im m igrants to London and the W est Indies have higher rates of atherosclerosis than would be expected from their lack of the common risk factors. 3,36
The d ie t of this population is high in G hee, a b u tte r p roduct contain ing 12 p e rc e n t o f its s te ro ls as ch o le s te ro l ox ides . 27 S ince cho lestero l oxidation products are found in a num ber of other hum an foodstuffs, particu la rly d ried eggs, dairy products, or deep-fat frying m edia , 12,42,49 further experimental studies are im perative and should provide knowledge highly relevant to the p re vention of hum an atherosclerotic cardiovascular disease.
Acknowledgments
These studies were supported by NIH Grant HL 32527, grant awards from the Paul Glenn Foundation, the Candle Foundation, and Research Grant Award 895GI-1 from the American Heart Association, Greater Los Angeles Affiliate.
References
1. Ba r a n o w sk e .A ., A d a m s , C. W. M ., B a yliss- H ig h . O . B ., a n d Bo w y er , D . B .: C o n n e c tiv e tissu e re sp o n ses to oxysterols. A theroscleros is 41:255-266, 1982.
2. B e c k a r t h , A. L . J . : The oxidation of crystalline cholesterol. Proc. Chem. Soc. 194-195, 1958.
3. B e c k l e s , G. L. A, K ir k w o o d , B. R., and C arso n , D . C .: High total and cardivoascular disease mortality in adults of Indian descent in Trinidad, unexplained by major risk factors. Lancet i:1298-1301, 1986.
4. B e r n h e i m e r , A. W., R o b i n s o n , W. G., L i n d e r , B . , M u l l i n s , D ., Y i p , Y . K., C o o p e r , N. S., S E I D M A N , I., and U W A J I M A , T.: Toxicity of enzy- mically-oxidized low-density lipoprotein. Bio- chim. Biophys. Res. C o m m u n . 148:260-266,1987.
5. B isw a s , S ., M a c D o u g a l l , J. D . B., and C o o k , R. P.: The effect of various steroids, mainly of the C27 Series, on the growth of chick heart explants. Brit. J. Exptl. Path. 45:13-20, 1964.
6. B r o o k s , C . J. W ., M c K e n n a , R. M ., C o l e , W. J., M a cLa c h l a n , J., and La w r ie , T. D. V.: “Profile” analysis of oxygenated sterols in plasma and serum. Biochem. Soc. Trans. 11:700-701,1983.
7. B r o w n , M. L. and G o l d s t e i n , J. L.: Lipoprotein metabolism in the macrophage; implications for cholesterol deposition in atherosclerosis. Ann. Rev. Biochim. 52:223- 261, 1983.
8. B r o w n , M. S., H o, Y. K ., and G o l d s t e i n , J. L .: The cholesterol ester cycle in macrophage foam cells. J. Biol Chem.255:9344-9352, 1980.
9. C h a id o f f , I. L ., L indsay , S., L o r e n z , F. W ., and E n t e n m a n , C .: Production of atherosclerosis in the aorta of the bird by administration of diethylstilbesterol. J. Exptl. Med. 88:3 7 3 -3 8 8 , 1948.

236 MORIN AND PENC
10. C h e n , H. W . , K a n d u t s c h , A. A., and W e y m o u t h , C . : Inhibtion of cell growth by oxygenated derivates of cholesterol. Nature 251:419- 421, 1974.
11. C o o k , R. P., M a c D o u g a l , J . D . B ., and B i s w a s , S . : Experiemtal atherosclerosis in rabbits after feeding cholestant-triol. Brit. L. Exptl. Pathol. 49:265-271, 1968.
12. F in o c c h ia r o , E . T ., L e e , D ., and Richard, T.: Identification and quantitation of cholesterol oxides in grated cheese and bleached butteroil. J. Amer. Oil Chem. Sox. 61:877, 1984.
13. F l O R I T Y , J. A. and S i m s , R. J . : Autoxidation products of cholesterol. J. Am. Oil Chem. Soc. 44:221—224, 1967.
14. F l o r e n t in , R. A ., N a m , S. C., L e e , K. T ., and THOMAS, W. A .: Increased 3H-thymidine incorporation into endothelil cells of swine fed cholesterol for 3 days. Expt. Mol. Path. 10:250- 255, 1969.
15. G o l d s t e i n , J. L., Ho, Y. K., B a s u , S. K., and B r o w n , M . S.: Binding site on marcrophages that mediated uptake and degradation of acety- lated low density lipoprotein, producing massive c h o le s te ro l d ep o stio n . P roc. N atl. Acad. Sci. 76:333- 337, 1979.
16. G o r d o n , L I., B a s s , J., and Y a c h n i n , S.: Inhibition of polymorphonuclear leukocyte (PMN) chemotaxis by oxygenated sterol compounds (OSC). Clin. Res. 27:688A, 1979.
17. G r y g l e w s k e , R. J. and S Z C Z E K L I K , A.: Inhibition of prostacylin formation by lipid peroxides in the arterial wall: hypothetical step in developm en t o f a therosclerosis. M ateria. M edica. Polona. Fasc.4('37/):328-341,1978.
18. H e n r i k s e n , T ., E v e n s e n , S. S., and C a r - L A N D E R , B.: Injury to human endothelial cells in culture induced by low density lipoproteins. Scand. J. Clin. Lab. Invest. 39:361—368, 1979.
19. H e n r i k s e n , T., M a h o n e y , E. M . , and S t e i n b e r g , D . : Enhanced macrophage degradation of biologically modified low density lipoprotein. Arteriosclerosis 3:149-159, 1983.
20. H e s s l e r , J. R ., M o r e l , D . W ., L e w is , L .J ., and C h is o l m , G. M .: Lipoprotein oxidation and lipoprotein-induced cytotoxicity. Arteriosclerosis 3 :2 1 5 -2 2 2 , 1983.
21. H e s s l e r , J . R . , R o b e r t s o n , A. L ., J r . , and C H I S O M , G . M . : LDL-induced cytotoxicity and its inhibition by H DL in human vascular smooth muscle and endothelial cells in culture. Atherosclerosis 32:213-229, 1979.
22. H i g h l e y , N. A. and T a y l o r , S. L .: T h e steatotic and cytotoxic effects of cholesterol oxides in cultured L cells. Food. Chem. Toxicol. 22:983-992,1984.
23. H i l l , J. C ., P e n g , S. K., M o r i n , R. J., and T a y l o r , C. B.: Effects of cholesterol autoxidation derivatives on hexose transport in cultured aortic sm ooth m uscle cells. Exptl. Molec. Pathol. 4J:249-257, 1984.
24. H o r v a t h , C: Quantitative determination of cholesterol in autoxidation mixtures by thin layer chrom atography. J. Chromatogr. 22:52-59 , 1966.
25. I m a i, H., W e r t h e s s e n , N. T., Subram anyam , V., L e q u o s n e , P. W ., Solow ay , A.H., and K an- isaw a , M.: Angiotoxicity of oxygenated sterols and possible percursors. Science 207:651—653,1980.
26. I m a i, H ., W e r t h e s s e n , N. T., Taylor , C. B., and L e e , K. T .: Angiotoxicity and atherosclerosis due to contaminants of U.S.P. grade cholestero l. Arch. Path. Lab. Med. 100:565-572 , 1976.
27. Ja c o b so n , M . S.: Cholesterol oxides in indian ghee: Possible cause of unexplained high risk of atherosclerosis in Indian immigrant populations. Lancet ii:656—658, 1987
28. J a c o b s o n , M. S ., P r ic e , M. G ., S h a m o o , A. E ., and H e a l d , F. P .: A therogenesis in W hite Carneau pigions, Effects of low-level cho- lestane-triol feeding. Atherosclerosis 57:209- 217, 1985.
29. KANDUTCH, A. A. and CHEN, H. W.: Consequences of blocked sterol synthesis in cultured cells. J. Biol. Chem. 252:409-415, 1977.
30. K a n d u t c h , A. A., C h e n , H . W., and H e i n - IGER, H . J .: Biological activity of some oxygenated sterols. Science 201:498—501, 1978.
31. Ka n n e l , W. B., C a st e l l i, W. P., and G o r d o n , T .: Cholsterol in the prediction of atherosclerotic disease. Ann. Int. Med. 90:85-91, 1979.
32. K r ie g e r , M ., G o l d s t e in , J. L ., and B r o w n , M. S.: Receptor-mediated uptake of low density lipoproteins reconstituted with 25-hydroxycho- lesteryl oleate suppresses 3-hydroxy-3-methyl- glutaryl-coenzym e a reductase and inhibits growth of human fibroblasts. Proc. Natl. Acad. Sci. 75:5052-5056, 1978.
33. Ko s y k h , V. A., L a n k in , V., P o d r e z , E. A., D m itriy , K. N. Vo l g u s h e v , S. A. Vic t o r o v , A. V., R e p in , V. S. and S m ir n o v , V. N.: Very low density lipoproteins secretion by cultured hepatocytes of rabbits receiving old commerical and purified cholesterol. In press, Lipids, 1989.
34. M a cD o u g a l l , J. D . B ., B isw a s , S., and C o o k , R. P.: The effects of certain C27 steroids on organ cultures of rabbit aortas. Brit. J. Exptl. Path. 46:549-553, 1965.
35 . M a r t i n e z - S a l e s , V., F o r n a s , E ., a n d CAMANAS, A.: Prostacyclin production and lipid peroxidation in the aorta of rats fed with cholestero l oxidation products. A rtery 12:213-219,1983.
36. M a tth ia s , D., B e c k e r , C.-H ., G o d ic k e , W., Sc h m id t , R., and Po n s o l d , K.: Action of cho- lestane-3,5,6-triol on rats with particular reference to the aorta. Atherosclerosis 63:115-124,1987.
37. M c K e ig u e , P. M ., A d e l s t e in , A. M ., and Sh ipley , M . J . : D iet and risk factors for coronary heart disease in Asians in Northwest London. Lancet ii: 1086-1090, 1985.
38. M il l s , J. T., M h e l e r , M . F., and A damany , A. M .: Effect of 25-hydroxycholesterol on disposition and turnover of membrane components in cultured aortic smooth muscle cells. Fed. Proc. 39:3009,1980.
39. M o n c a d a , S., G r y g lew sk i, R. J., B u n t in g , S.,

CHOLESTEROL OXIDATION PRODUCTS 237
et al.: A lipid peroxide inhibits the enzyme in blood vessel microsomes that generated from prostaglandin endoperoxides the substance which prevents platelet aggregation. Protaglan- dins 12:715-737, 1976.
40. M o r e l , D. W., H e ssle r , J. R., and C h is o l m , G. M .: Low density lipoprotein cytotoxicty induced by free radical peroxidation of lipid. J. Lipid Res. 24:1070-1076, 1983.
41. M o r in , R. J., R iley , N. E., G a o , D. W ., and P e n g , S. K .: Effects of cholesterol oxides on enzymes of cholesteryl ester metabolism in rabbit aortic smooth muscle cells. FASEB J. 2:1190,1988.
42. N aber , E. C. and B ig g e r t , M. D.: Cholesterol oxidation products in fresh and heat-treated egg yolk lipid. Fed. Proc. 33:581, 1982.
43. P e n g , S. K., H il l , J. C ., M o r in , R. J., and SAFARIC, J.: Electron microscopic cytochemistry of membrane bound enzymes of cultured aortic cells treated with cholesterol oxidation derivatives. Proc. E lectron Microsc. Soc. of Am. 42:714-715, 1984.
44. P e n g , S. K., H il l , J. C ., M o r in , R. J., and Taylor , C. B .: Influence of cholesterol oxidation derivatives on membrane bound enzymes in cultured aortic smooth muscle cells. Proc. Soc. Exptl. Bio. Med. i80:126-132, 1985.
45. P e n g , S. K., M o r in , R. J., P h il l ip s , G. A., and Xia, G. Z.: Absorption and transport of cholesterol autoxidation derivatives in rabbits. Fed. Proc. 43(3):230, 1986.
46. P e n g , S. K., M o r in , R. J., Se n t o v ic h , S., and T aylor, C. B.: The effect of cholesterol oxidation products on membrane functions. J. Am. Oil Chem. Soc. 62:634- 635, 1985.
47. P e n g , S. K., M o r in , R. J., T h a m , P., and T aylo r , C. B .: The effects of oxygenated derivatives of cholesterol on cholesterol uptake by aortic smooth muscle cells. Artery 73:144-164, 1985.
48. P e n g , S. K., P h il l ip s , G. A., Xla, G. Z., and MORIN, R. J.: Transport of cholesterol autoxidation products by rabbit lipoproteins. Atherosclerosis 64:1 -6 , 1987.
49. P e n g , S. K. and T aylor, C. B .: Atherogenic effect of oxidized cholesterol. Dietary Fat and Health. Perkin, E. and Visek, V., eds. Am. Oil Chem. Soc. 1983, pp. 919- 933.
50. P e n g , S. K., T ay lo r , C. B., H a u n g , W. Y., H il l , J. C., and M ik k e l so n , B.: Distribution of 25-hydroxycholesterol in the lipoprotein and its role in atherogenesis. A study in squirrel monkeys. Atherosclerosis 41:395—402, 1982.
51. P e n g , S. K., T aylor, C. B., H il l , J. C., and M o r in , R. J . : Cholesterol oxidation derivates and arterial endothelial damage. Atherosclerosis 54:121-133, 1985.
52. P e n g , S. K., T aylor , C . B., T h a m , P., W e r t h - e s s e n , N. T ., and M ik k e l s o n , B.: Effect of auto-oxidation products from cholesterol on aortic smoth muscle cells an in vitro study. Arch. Path. Lab. Med. i 02:57-61, 1978.
53. P e n g , S. K., T h a m , P ., T ay lo r , C. B., and M ik k e l s o n , B.: Cytoxicity of cholesterol oxidation derivatives on cultured aortic smooth muscle cells and their effect on cholesterol biosynthesis. Am. J. Clin. Nutr. 32:1033-1042, 1979.
54. P e n g , S. K., W u, A. Y., C o l a c ic c o , L. A., M ik k e l s o n , B., and Taylor , C . B.: Inhibition of prostaglandin synthesis by cholesterol oxidation products in cultured aortic smooth muscle cells. Fed. Proc. 42:809, 1983.
55. ROSS, R .: The pathogenesis of atherosclerosis— An update. New Engl. J. Med. 314:488-500, 1986.
56. Sa l m o n , J. A., Sm it h , D. R ., F l o w e r , R. J., M o n c a d a , S ., and Va n e , J. R .: Futher studies on the enzymatic coversion o f prostaglandin endoperoxide into prostacylclin by porcine aorta microsomes. Biochim. Biophys. Acta 523:250— 262, 1978.
57. Se n t o v ic h , S ., M o r in , R. J., and P e n g , S. K.: Effect of cholesterol oxidation derivates on endocytosis of rabbit vascular endothelium. Fed. Proc. 44:1263, 1985.
58. Sm it h , L. L ., M a t t h e w s , W. S ., P r ic e , J. C., Ba c h m a n , R. C., and Re y n o l d s , B.: Thin layer chrom atographic examination of cholesterol autoxidation. J. Chromatogr. 27:187-205, 1976.
59. St r e u l i , R. A., C h u n g , J ., Sc a n u , A. M., and Ya c h n in , S.: Inhibition of human E-rosette formation by oxygenated sterols. J. Im munol. 123:2897-2902, 1979.
60. T aylor, C. B., P e n g , S. K., H il l , J. C., an d MIKKELSON, B.: S cann ing e lec tro n m icroscopic stu d y on a r te r ia l in ju ry b y ox idation p ro d u c ts o f ch o les te ro l in rab b its . F ed . P roc . 39:771, 1980.
61. Ta y lo r , C. B ., P e n g , S. K., W e r t h e s s e n , N. T ., T h a m , P., and L e e , K. T.: Spontaneously occurring angiotoxic derivatives of cholesterol. Am. J. Clin. Nutr. 32:40-57, 1979.
62. Wata b e , T., I so b e , M., and D a n a i, M.: Cholesterol diet increases plasma and liver concentrations of cholesterol epoxides and cholestanetriol.J. Pharm. Dyn.3:553-556, 1980.





























