Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1984 by The American Society of Biological Chemists, Inc.
Vol. 259, No. 11, Issue of June 10, pp. 7045-7055,1984 Printed in U.S.A.
The Physical and Catalytic Properties of Hydrogenase I1 of Clostridium pasteurianum A COMPARISON WITH HYDROGENASE I*
(Received for publication, January 10, 1984)
Michael W. W. Adams and Leonard E. Mortenson From Exxon Research and Engineering Company, Clinton Township, Annandale, New Jersey 08801
Hydrogenase I1 of Clostridium pasteurianum is a monomeric protein of Mr = 53,000 containing 8 iron and 8 acid-labile sulfide atoms/mol. It is distinct from hydrogenase I from the same organism (Mr = 60,000 12 Fe and 12 Sz-/mol). Metal analyses showed that neither hydrogenase contains nickel or any other met- als in significant amounts. The iron atoms of hydro- genase I1 resisted chelation by 2,Z”bipyridyl but all were susceptible when the enzyme was treated with ferricyanide. Core extrusion indicated the presence of two [4Fe-4S] clusters in hydrogenase I1 and EPR spec- troscopy showed two distinct paramagnetic species which could be interpreted as one [4Fe-4S]2+(z+3’+) and one [4Fe-4SI2+(’+r3+) per molecule. The absorption coef- ficient of Hz-reduced hydrogenase I1 at 420 nm was 23,000 M” cm“ with a A4z0/A276 ratio of 0.27. There were large differences between hydrogenase I and hy- drogenase I1 in the absorption spectra of the air-oxi- dized, H2-reduced, and dithionite-reduced forms of the enzymes. Hydrogenase I1 catalyzed Hz evolution with methyl viologen or ferredoxin as the electron carrier, and Hz oxidation with methylene blue or methyl viol- ogen as the electron acceptor. Apparent K, values were determined for all these reactions with both hy- drogenases. Hydrogenase I1 is a relatively inactive enzyme, except in the reduction of methylene blue by Hz. The pH dependencies of Hz oxidation were similar for both hydrogenases but were very different in Hz evolution. The activation energy values were much higher for Hz catalysis by hydrogenase I1 than for hydrogenase I. The two hydrogenases have the same sensitivity to inactivation by Oz but differ in their sensitivity to metal-chelating reagents and to CO. Hy- drogenase I is more readily inhibited by CO but hydro- genase I1 binds CO irreversibly. From the above data, a mechanism is proposed to account for the observed differences in the catalytic activities of hydrogenase I and hydrogenase 11.
The anaerobic Nz-fixing bacterium Clostridium pasteu- rianum produces large amounts of Hz as an end product of metabolism. These saccharolytic organisms metabolize car- bohydrates to organic acids, Hz, and COz and because they lack an electron transport chain, they obtain ATP by sub- strate level phosphorylation. The enzyme hydrogenase (EC class 1.12) which catalyzes the reversible activation of Hz has been purified from this organism and has been characterized
* The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
(Chen and Mortenson, 1974: Chen et al., 1976). It is an iron- sulfur protein of M , = 60,000 containing 12 iron atoms and 12 acid-labile sulfide atoms/mol. A second hydrogenase spe- cies was recently discovered in c. pmteurianurn Chen and Blanchard (1978), and we have now obtained this enzyme in a pure form.’ The latter hydrogenase, which we have termed hydrogenase 11, is also an iron sulfur protein but it has M, = 53,000 and contains 8 iron and 8 acid-labile sulfide atoms/ mol. The aim of the present work was to characterize hydro- genase I1 and compare its properties with those of hydrogen- ase I.
In the last few years, hydrogenases have been purified from a variety of microorganisms but a comparison of their prop- erties reveals only one common feature: all are iron-sulfur proteins. Other than this, they differ considerably in molec- ular composition, metal content, specific activity, and their sensitivity to inactivation by 0 2 (for review, see Adams et al., 1981). In addition, two recent developments in the field of iron-sulfur proteins in general and in the area of hydrogenases in particular have made a detailed characterization of the clostridial hydrogenases even more crucial. The first is the discovery of a new type of iron-sulfur center in proteins, the [3Fe-3S] cluster first identified in ferredoxins, from Azotobac- ter vinelandii (Emptage et al., 1980) and Desulfovibrio gigas (Huynh et al., 1980). A 3Fe center has now been reported in the hydrogenases isolated from Chromatium (Albracht et al., 1982b) and Desulfovibrio desulfuricans (Kruger et al., 1982). The other important finding is that many hydrogenases con- tain nickel atoms in addition to iron and inorganic sulfur as part of the active enzyme (see Kruger et al., 1982, and refer- ences therein). Spectroscopic studies have indicated that nickel functions as an additonal redox center (Cammack et al., 1982; LeGall et al., 1982). As yet, it is not known if nickel is obligatory for Hz catalysis by hydrogenase or the role that nickel plays in Catalysis.
We have previously shown that hydrogenase I1 of C. pas- teurianurn is extremely active in Hz oxidation with methylene blue as the electron acceptor but that it catalyzes Hz evolution from reduced methyl viologen at very low rates.’ On the other hand, hydrogenase I catalyzes both reactions at extremely high.rates (Chen and Mortenson, 1974) and is one of the most active hydrogenases so far isolated (see Adams et al., 1981). We therefore investigated the catalytic properties of hydro- genase I1 with the hope of gaining some insight into the mechanistic differences between the two clostridial hydrogen- ases and perhaps of H2 catalysis in general. In addition, we report some physical characteristics of hydrogenase I1 which are compared with those of hydrogenase I and of other well studied hydrogenases.
Adam, M. W. W., and Mortenson, L. E. (1984) Biochim. Biophys. Acta, in press.
7045

7046 Hydrogenase II of Clostridium pasteurianum
MATERIALS AND METHODS
Chemicals-All reagents and chemicals were of the highest purity commercially available. Hydrogenase I, hydrogenase 11, and ferre- doxin were purified from NZ-grown C. pasteurianum. W5 cells as described previously.’
Enzyme Assays-Hydrogenase activity was routinely determined spectrophotometrically by measuring the reduction of methylene blue as described by Adams and Mortenson.’ The assay mixture contained methylene blue (0.05 mM) in 50 mM Tris/HCI, pH 8.0, under HZ at 30 “C. One unit of hydrogenase activity catalyzes the reduction of 1 @mol of methylene blue/min under these conditions, which is equiv- alent to 1 pmol of Hz oxidized/min. When methyl viologen was used in place of methylene blue, the reaction was measured at 600 nm, and an absorption coefficient of 12,000 M” cm” was assumed for the semiquinone form. Results are also expressed as micromoles of Hz oxidized/min, equivalent to 2 pmol of methyl viologen reduced/min. For the determination of the K , values for Hz, degassed assay cuvettes were flushed with various H2/Ar mixtures and the pHz was quanti- tated by removing samples from the gas phase and analyzing by gas chromatography. A solubility coefficient of 0.01819 was used to de- termine the concentration of dissolved Hz (see Vignais et al., 1982). Hydrogenase activity was also measured by Hz production with methyl viologen (1 mM) as the electron carrier and sodium dithionite (20 mM) as the electron donor in 50 mM Tris/HCl, pH 8.0.’ Activity is expressed as micromoles of Hz evolved/min. For the assay of hydrogenase I, all reaction mixtures contained bovine serum albumin (1 mg/ml) in both assay systems.
Metal Analyses-Plasma emission spectroscopy (inductively cou- pled plasma/atomic emission spectroscopy) which gives a 32-element analysis was performed by the Analytical and Information Division of Exxon Research and Engineering Company. Extraneous iron in the hydrogenase preparation was measured by complexing with 2,2‘- bipyridyl under reducing conditions according to Walker and Mor- tenson (1973). The release of iron from hydrogenase I1 by potassium ferricyanide was measured by a modification of this method. The hydrogenase (2-5 PM) in 100 mM Tris/HCl, pH 8.0, was incubated for 10 min at 25 ”C under Ar in anaerobic serum-stoppered cuvettes (type C-030-3-0302, Perkin-Elmer) with potassium ferricyanide (1 mM). Sodium dithionite (100 mM in 0.5 M Tris/HCl, pH 8.0) and 2,2’-bipyridyl (25 mM) were then added to final concentrations of 3 and 5 mM, respectively. An absorption coefficient of 8400 M” cm” was used to determine the ferrous-2,2’-bipyridyl complex.
Extrusion of the iron-Sulfur Center-The core extrusion reaction in 80% HMPAz (v/v) was carried out as described by Gillum et al., (1977). C. pasteurianum ferredoxin was used as a reference and the extruded Fe4S, centers were quantitated using an absorption coeffi- cient at 458 nm of 17,200 M” cm” for [Fe4S,(SPh)4]2-. The extrusion of the iron-sulfur centers of ferredoxin and hydrogenase I1 in aqueous DMF/Triton X-100 mixtures was performed by the method of Kurtz (1982). All extrusion reactions were carried out in serum-stoppered cuvettes under Ar (see above), and all spectra were recorded on a Kontron Uvikon 810 spectrophotometer at 25 “C.
EPR Spectroscopy-EPR spectra were recorded on a Varian E-109 spectrometer (Varian Instruments, Palo Alto, CA). Samples were cooled by a flow of cold He gas in an ESR-900 Cryostat (Oxford Instruments Inc., Columbia, MD).
Other Methods-Where indicated, hydrogenase samples were de- salted by passing the protein through a column (1 X 20 cm) of Sephadex G-25 equilibrated with 50 mM Tris/HCl, pH 8.0. All ma- nipulations were carried out anaerobically with Hz as the gas phase. Protein was determined by the Lowry method using bovine serum albumin as the standard.
RESULTS
In a previous report’ detailing the purification of hydrogen- ase 11, we showed that the enzyme is a monomeric protein of M, = 53,000. Chemical analyses indicated the presence of approximately 8 iron and 8 acid-labile sulfide atoms/mol of enzyme. Hydrogenase I1 is therefore distinct from hydrogen- ase I of C. pasteurianum which has M, = 60,000 and contains 12 iron and 12 acid-labile sulfide atoms/mol (Chen and Mor- tenson, 1974).
* The abbreviations used are: HMPA, hexamethylphosphoramide; DMF, N,N’-dimethylformamide; Mops, 4-morpholinepropanesul- fonic acid; Pa, pascal.
Metal Analyses-To ascertain the presence of any other metals in hydrogenase 11, the enzyme was examined by plasma emission spectroscopy. This gave a 32-element analysis (two nonmetals). Results from three determinations with different hydrogenase I1 preparations gave values of 8.0 k 0.48 g atoms of iron/53,000 g of protein. Analyses for Ni, Mo, Cu, Zn, Mn, and Se were all less than 0.1 g atoms/mol of enzyme. Similar results were obtained with hydrogenase I: that is, iron was the only metal present in molar amounts equivalent to or greater than the moles of protein. Values of 11.4 +- 0.51 g atoms of Fe/mol of protein were found; with Ni, less than 0.1 g atoms/mol were found.
The iron atoms of hydrogenase I1 were resistant to chelation by 2,Z”bipyridyl under anaerobic conditions indicating that all the iron is part of the active enzyme. However, the addition of the protein-denaturating agent sodium dodecyl sulfate (l%, w/v) resulted in the immediate formation of the ferrous-2,2‘- bipyridyl compIex equivalent to the release of 7.8 f 0.3 g atoms of iron/mol of enzyme (from three determinations). All the hydrogenase activity (both Hz evolution and H, uptake) was lost immediately upon the addition of sodium dodecyl sulfate.
Effect of Potassium Ferricyanide-Potassium ferricyanide is a commonly used oxidant in biological redox systems, and its effect on the activity and integrity of hydrogenase I1 was investigated. When the enzyme was treated with a ZOO-fold excess of ferricyanide under anaerobic conditions, no hydro- genase activity could be detected after a 5-min incubation period in either the Hz uptake or the Hz evolution assay. Sodium dithionite and 2,2’-bipyridyl were then added as described under “Materials and Methods.” The immediate red coloration showed that iron had been released from the en- zyme by the ferricyanide treatment. Quantitation gave values of 7.92 k 0.25 g atoms of iron/mol of hydrogenase I1 indicating that ferricyanide treatment is nonselective; all the iron atoms were released. Control experiments in the absence of hydro- genase showed that potassium ferricyanide contains 0.77% free iron.
Actiue Site Extrusion-Kurtz (1982) reported a new me- dium for the extrusion of [2Fe-2S] clusters from ferredoxins which involved treating the protein with thiophenol in DMF (5-17%, v/v)-Triton X-100 (5%, v/v) mixtures. Advantages over the previous method (see Gillum et al., 1977) include avoidance of toxic HMPA, lower concentrations of organic solvents, and for spinach ferredoxin (2Fe) a decreased extru- sion time. To discern the nature of the active site of hydro- genase 11, the new extrusion procedure was first investigated with C. pasteurianum ferredoxin which contains two [4Fe-4S] centers (Que et aL, 1975). Fig. 1 shows the results of one experiment using a final solvent composition of 79:147 (buffer:DMF:Triton X-100, v/v). The solvent has little effect on the spectrum of the native protein(Fig. 1, a and b ) , but the addition of thiophenol slowly displaced the protein band near 400 nm to longer wavelengths until the time-invariant spec- trum shown in Fig. IC was obtained, typical of the [Fe4S4(SPh),I2- anion (Kurtz, 1982). This required an incu- bation period of 80-100 min but the time could be decreased if the DMF concentration was increased. In a solvent mixture of 64:30:6 (buffer:DMF:Triton X-100, v/v), complete extru- sion could be obtained in 20-25 min. Results from three such experiments gave values of 1.94 f 0.05 for the mole ratio [4Fe-4S]/ferredoxin. The DMF/Triton X-100 procedure therefore is applicable to simple proteins containing [4Fe-4Sl clusters.
Extrusion of the iron-sulfur center of hydrogenase I1 by the DMF/Triton X-100 method was not successful. In the solvent mixtures 5 4 0 % DMF (with 6% Triton X-100, V/V) or 5-896

Hydrogenase II of Clostridium pasteurianum 7047
for hydrogenase 11, the absorption coefficients at 275 and 420 nm were 86,900 and 23,000 M” cm”, respectively (values from three preparations). Hydrogenase I1 under Hz could be further reduced by sodium dithionite as indicated by the decrease in absorption in the 400-600-nm range (Fig. 4a). If the Hz gas phase was replaced by Ar (no dithionite present) there was no change in the absorption spectrum but the admission of air to the sample increased the absorption in this region (Fig. 4b), a characteristic of iron-sulfur proteins. For the air-oxidized enzyme, the absorption coefficient at 420 nm was 27,200 M” cm”, corresponding to 3400 M” cm”/ iron atom.
Chemical analysis showed that if hydrogenase I1 was treated with potassium ferricyanide, iron was released from the en- zyme. When hydrogenase I1 (0.12 mM) was incubated under Ar with potassium ferricyanide (10 mM), the greenish solution was visibly bleached within 2 min. The absorption spectrum
L I I I 400 500 600
X (nm) FIG. 1. Active site extrusion of ferredoxin in aqueous DMF/
Triton X-100. The experiment was carried out as described under “Materials and Methods.” a, ferredoxin (28 pM) in 100 mM Tris/HCl, pH 8.0; b, solution in Q diluted with DMF and Triton X-100 to give a final solvent composition of 79147 (buffer:DMFTriton X-100, v/ v); e, solution in b after the addition of thiophenol (20 mM) recorded after 100 min.
Triton X-100 (with 14% DMF, v/v) with thiophenol (20-100 mM), the protein precipitated. In DMF/Triton mixtures, the protein was soluble, but the addition of thiophenol caused rapid precipitation. This extrusion medium therefore appears not to be suitable for this particular protein. We therefore turned to the previously described HMPA method (Gillum et al., 1977) using the same preparations of hydrogenase 11. As shown in Fig. 2, the active centers of hydrogenase I1 were extruded in HMPA (80%, v/v). Upon addition of thiophenol, the spectrum was red-shifted (Fig. 2, a and b) and after 16 min a constant spectrum (Fig. 2c) was obtained. The latter unambiguously demonstrates the presence of [4Fe-4S] cen- ters, rather than [2Fe-2S] centers or mixtures of centers in hydrogenase 11. The peak at 458 nm is characteristic of [Fe4S4(SPh)4]2- and although the A458/A550 ratio is low (1.87 f 0.09 from three determinations) compared to the pure cluster (A458/A550 > 2.02), this ratio is similar to that obtained for the extrusion of hydrogenase I (Gillum et al., 1977). From the A458 values, the mole ratio of [4Fe-4S]/protein was 2.04 k 0.11 for hydrogenase 11. These results therefore indicate that hydrogenase I1 contains 2 [4Fe-4S] clusters/mol in agreement with the measured metal and acid-labile sulfide content.
Absorption Spectra of Hydrogenase IZ-Hydrogenase I1 as isolated in the presence of sodium dithionite (1 mM) was dark green in color and could be easily distinguished from other proteins during column chromatography. Dithionite was re- moved by anaerobic gel filtration under HZ and the UV-visible absorption spectrum of the hydrogenase under HZ is given in Fig. 3a. This shows increased absorption from 700 to 275 nm with a broad band around 420 nm, typical of many other hydrogenases (see Adams et al., 1981). Assuming M, = 53,000
m E 0 c s: 9
400 500 600 700
A (nm)
FIG. 2. Active site extrusion of hydrogenase I1 in aqueous HMPA. The experiment was carried out as described under “Mate- rials and Methods.” a, hydrogenase I1 (12.4 p ~ ) in HMPA (80%, v/ v); b, solution in Q recorded 1 min after the addition of thiophenol (50 mM); c, solution in b recorded after a further 15 min.
I I I I I 1
0.6
Q
c s s
0.4 -
0.2 - -
I I 1 I
300 400 500 600
A (nm) FIG. 3. Absorption spectra of hydrogenase 11. Q, hydrogenase
I1 (0.4 mg/ml) under H1 in 50 mM Tris/HCl, pH 8.0, after desalting under H2; b, hydrogenase I1 was treated with potassium ferricyanide as described in the text and after removal of the ferricyanide by gel filtration the spectrum of the isolated protein (0.35 mg/ml) was recorded.
7048 Hydrogenase II of Clostridium pasteurianum
I I 1 I 1 400 500 600 400 500 800
Unm)
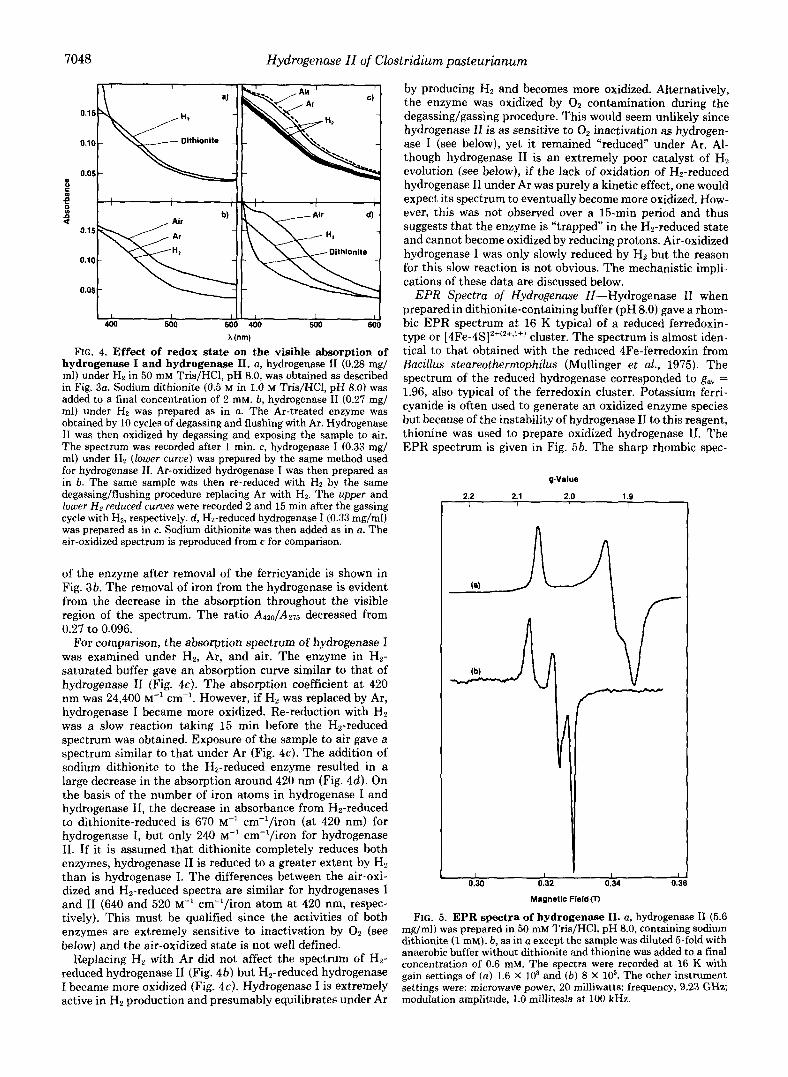
FIG. 4. Effect of redox state on the visible absorption of hydrogenase I and hydrogenase 11. a, hydrogenase I1 (0.28 mg/ ml) under Hz in 50 mM Tris/HCl, pH 8.0, was obtained as described in Fig. 3a. Sodium dithionite (0.5 M in 1.0 M TrislHC1, pH 8.0) was added to a final concentration of 2 mM. b, hydrogenase I1 (0.27 mg/ ml) under Hz was prepared as in a. The Ar-treated enzyme was obtained by 10 cycles of degassing and flushing with Ar. Hydrogenase I1 was then oxidized by degassing and exposing the sample to air. The spectrum was recorded after 1 min. c, hydrogenase I (0.33 mg/ ml) under Hz (lower curue) was prepared by the same method used for hydrogenase 11. Ar-oxidized hydrogenase I was then prepared as in 6. The same sample was then re-reduced with HZ by the same degassing/flushing procedure replacing Ar with Hz. The upper and lower H z reduced curves were recorded 2 and 15 min after the gassing cycle with HB, respectively. d, Hz-reduced hydrogenase I (0.33 mg/ml) was prepared as in c. Sodium dithionite was then added as in a. The air-oxidized spectrum is reproduced from c for comparison.
of the enzyme after removal of the ferricyanide is shown in Fig. 3b. The removal of iron from the hydrogenase is evident from the decrease in the absorption throughout the visible region of the spectrum. The ratio A4z0/A275 decreased from 0.27 to 0.096.
For comparison, the absorption spectrum of hydrogenase I was examined under Hz, Ar, and air. The enzyme in Hz- saturated buffer gave an absorption curve similar to that of hydrogenase I1 (Fig. 4c). The absorption coefficient a t 420 nm was 24,400 M“ cm”. However, if H, was replaced by Ar, hydrogenase I became more oxidized. Re-reduction with Hz was a slow reaction taking 15 min before the H2-reduced spectrum was obtained. Exposure of the sample to air gave a spectrum similar to that under Ar (Fig. 4c). The addition of sodium dithionite to the Hz-reduced enzyme resulted in a large decrease in the absorption around 420 nm (Fig. 4 4 . On the basis of the number of iron atoms in hydrogenase I and hydrogenase 11, the decrease in absorbance from Hz-reduced to dithionite-reduced is 670 M” cm”/iron (at 420 nm) for hydrogenase I, but only 240 M” cm“/iron for hydrogenase 11. If it is assumed that dithionite completely reduces both enzymes, hydrogenase I1 is reduced to a greater extent by Hz than is hydrogenase I. The differences between the air-oxi- dized and H2-reduced spectra are similar for hydrogenases I and I1 (640 and 520 M” cm”/iron atom at 420 nm, respec- tively). This must be qualified since the activities of both enzymes are extremely sensitive to inactivation by O2 (see below) and the air-oxidized state is not well defined.
Replacing Hz with Ar did not affect the spectrum of Hz- reduced hydrogenase I1 (Fig. 4b) but Hz-reduced hydrogenase I became more oxidized (Fig. 4c). Hydrogenase I is extremely active in H2 production and presumably equilibrates under Ar
by producing Hz and becomes more oxidized. Alternatively, the enzyme was oxidized by O2 contamination during the degassing/gassing procedure. This would seem unlikely since hydrogenase I1 is as sensitive to 0, inactivation as hydrogen- ase I (see below), yet it remained “reduced” under Ar. Al- though hydrogenase I1 is an extremely poor catalyst of Hz evolution (see below), if the lack of oxidation of Hz-reduced hydrogenase I1 under Ar was purely a kinetic effect, one would expect its spectrum to eventually become more oxidized. How- ever, this was not observed over a 15-min period and thus suggests that the enzyme is “trapped” in the Hz-reduced state and cannot become oxidized by reducing protons. Air-oxidized hydrogenase I was only slowly reduced by Hz but the reason for this slow reaction is not obvious. The mechanistic impli- cations of these data are discussed below.
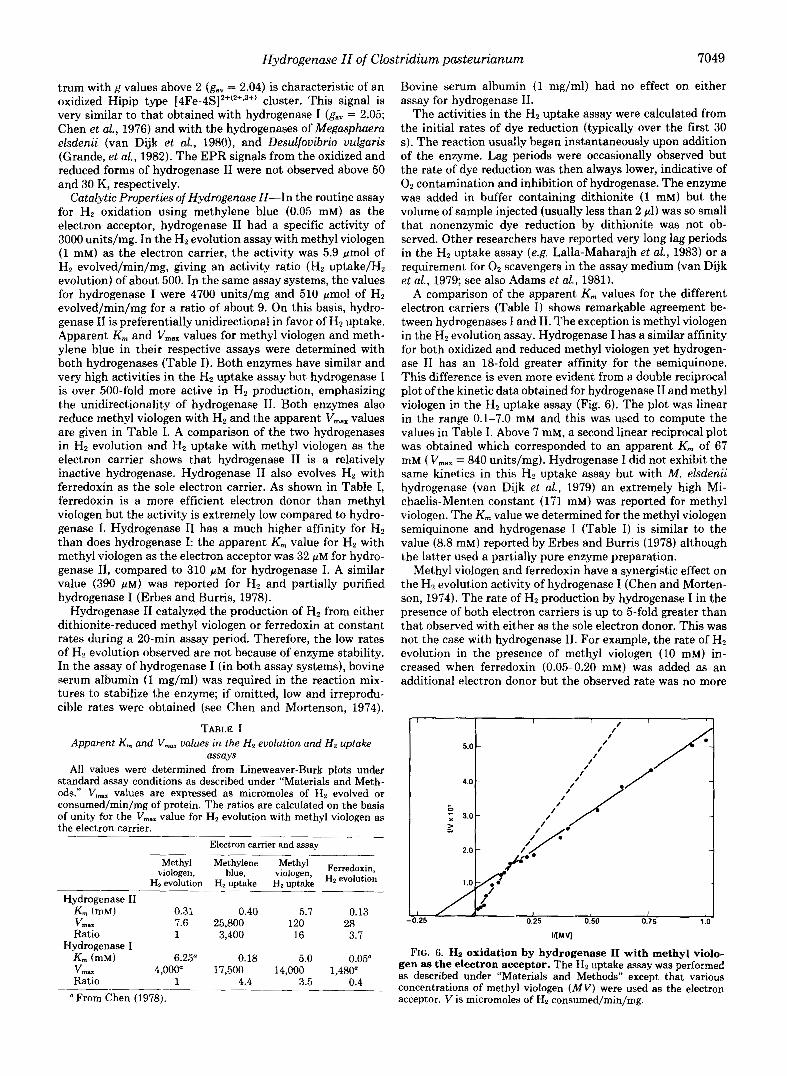
EPR Spectra of Hydrogenase II-Hydrogenase I1 when prepared in dithionite-containing buffer (pH 8.0) gave a rhom- bic EPR spectrum a t 16 K typical of a reduced ferredoxin- type or [4Fe-4S]2f(z+,1+) cluster. The spectrum is almost iden- tical to that obtained with the reduced 4Fe-ferredoxin from Bacillus steareothermophilus (Mullinger et al., 1975). The spectrum of the reduced hydrogenase corresponded to g,, = 1.96, also typical of the ferredoxin cluster. Potassium ferri- cyanide is often used to generate an oxidized enzyme species but because of the instability of hydrogenase I1 to this reagent, thionine was used to prepare oxidized hydrogenase 11. The EPR spectrum is given in Fig. 5b. The sharp rhombic spec-
gValue
2.2 2.1 2.0 1.9 I ( I I
I I I L 0.30 0.32 0.34 0.3
Magnetic F i e l d 0
FIG. 5. EPR spectra of hydrogenase 11. a, hydrogenase I1 (5.6 mg/ml) was prepared in 50 mM Tris/HCl, pH 8.0, cohtaining sodium dithionite (1 mM). b, as in a except the sample was diluted &fold with anaerobic buffer without dithionite and thionine was added to a final concentration of 0.6 mM. The spectra were recorded at 16 K with gain settings of ( a ) 1.6 X lo3 and ( b ) 8 X lo3. The other instrument settings were: microwave power, 20 milliwatts; frequency, 9.23 GHz; modulation amplitude, 1.0 millitesla at 100 kHz.
Hydrogenase 11 of Clostridium pasteurianum 7049
trum with g values above 2 (gav = 2.04) is characteristic of an oxidized Hipip type [4Fe-4S]2+(z+,3+) cluster. This signal is very similar to that obtained with hydrogenase I (gay = 2.05; Chen et al., 1976) and with the hydrogenases of Megasphuera elsdenii (van Dijk et al., 1980), and Desulfouibrio vulgaris (Grande, et al., 1982). The EPR signals from the oxidized and reduced forms of hydrogenase I1 were not observed above 50 and 30 K, respectively.
Catalytic Properties of Hydrogenase II-In the routine assay for Hz oxidation using methylene blue (0.05 mM) as the electron acceptor, hydrogenase I1 had a specific activity of 3000 units/mg. In the Hz evolution assay with methyl viologen (1 mM) as the electron carrier, the activity was 5.9 pmol of Hz evolved/min/mg, giving an activity ratio (Hz uptake/Hz evolution) of about 500. In the same assay systems, the values for hydrogenase I were 4700 units/mg and 510 pmol of Hz evolved/min/mg for a ratio of about 9. On this basis, hydro- genase I1 is preferentially unidirectional in favor of Hz uptake. Apparent K, and Vma, values for methyl viologen and meth- ylene blue in their respective assays were determined with both hydrogenases (Table I). Both enzymes have similar and very high activities in the Hz uptake assay but hydrogenase I is over 500-fold more active in Hz production, emphasizing the unidirectionality of hydrogenase 11. Both enzymes also reduce methyl viologen with Hz and the apparent V,,, values are given in Table 1. A comparison of the two hydrogenases in Hz evolution and Hz uptake with methyl viologen as the electron carrier shows that hydrogenase 11 is a relatively inactive hydrogenase. Hydrogenase I1 also evolves HP with ferredoxin as the sole electron carrier. As shown in Table I, ferredoxin is a more efficient electron donor than methyl viologen but the activity is extremely low compared to hydro- genase 1. Hydrogenase I1 has a much higher affinity for Hz than does hydrogenase I: the apparent K, value for HP with methyl viologen as the electron acceptor was 32 p~ for hydro- genase 11, compared to 310 PM for hydrogenase I. A similar value (390 p ~ ) was reported for Hz and partially purified hydrogenase I (Erbes and Burris, 1978).
Hydrogenase I1 catalyzed the production of Hz from either dithionite-reduced methyl viologen or ferredoxin a t constant rates during a 20-min assay period. Therefore, the low rates of Hz evolution observed are not because of enzyme stability. In the assay of hydrogenase I (in both assay systems), bovine serum albumin (1 mg/ml) was required in the reaction mix- tures to stabilize the enzyme; if omitted, low and irreprodu- cible rates were obtained (see Chen and Mortenson, 1974).
TABLE I Apparent K,,, and V,, values in the Hz evolution and Hz uptake
assays All values were determined from Lineweaver-Burk plots under
standard assay conditions as described under "Materials and Meth- ods." VmaX values are expressed as micromoles of Hz evolved or consumed/min/mg of protein. The ratios are calculated on the basis of unity for the VmaX value for H, evolution with methyl viologen as the electron carrier.
Electron carrier and assay
viologen, blue, viologen, Methyl Methylene Methyl
H2 evolution Hz uptake H2 uptake ~
Hydrogenase I1 K, (mM) 0.31 0.40 5.7 0.13 V k X 7.6 25,800 120 Ratio 1 3,400 16 3.7
K m (mM) 6.25" 0.18 5.0 0.05"
Ratio 1 4.4 3.5 0.4
"From Chen (1978).
28
Hydrogenase I
Vmax 4,000" 17,500 14,000 1,480" -
Bovine serum albumin (1 mg/ml) had no effect on either assay for hydrogenase 11.
The activities in the Hz uptake assay were calculated from the initial rates of dye reduction (typically over the first 30 s). The reaction usually began instantaneously upon addition of the enzyme. Lag periods were occasionally observed but the rate of dye reduction was then always lower, indicative of Oz contamination and inhibition of hydrogenase. The enzyme was added in buffer containing dithionite (1 mM) but the volume of sample injected (usually less than 2 pl) was so small that nonenzymic dye reduction by dithionite was not ob- served. Other researchers have reported very long lag periods in the Hz uptake assay (e.g. Lalla-Maharajh et al., 1983) or a requirement for 0, scavengers in the assay medium (van Dijk et al., 1979; see also Adams et al., 1981).
A comparison of the apparent K,,, values for the different electron carriers (Table I) shows remarkable agreement be- tween hydrogenases I and 11. The exception is methyl viologen in the Hz evolution assay. Hydrogenase I has a similar affinity for both oxidized and reduced methyl viologen yet hydrogen- ase I1 has an 18-fold greater affinity for the semiquinone. This difference is even more evident from a double reciprocal plot of the kinetic data obtained for hydrogenase I1 and methyl viologen in the Hz uptake assay (Fig. 6). The plot was linear in the range 0.1-7.0 mM and this was used to compute the values in Table I. Above 7 mM, a second linear reciprocal plot was obtained which corresponded to an apparent K, of 67 mM ( Vmax = 840 units/mg). Hydrogenase I did not exhibit the same kinetics in this H B uptake assay but with M . elsdenii hydrogenase (van Dijk et al., 1979) an extremely high Mi- chaelis-Menten constant (171 mM) was reported for methyl viologen. The K, value we determined for the methyl viologen semiquinone and hydrogenase I (Table I) is similar to the value (8.8 mM) reported by Erbes and Burris (1978) although the latter used a partially pure enzyme preparation.
Methyl viologen and ferredoxin have a synergistic effect on the HP evolution activity of hydrogenase I (Chen and Morten- son, 1974). The rate of HZ production by hydrogenase I in the presence of both electron carriers is up to 5-fold greater than that observed with either as the sole electron donor. This was not the case with hydrogenase 11. For example, the rate of H2 evolution in the presence of methyl viologen (10 mM) in- creased when ferredoxin (0.05-0.20 mM) was added as an additional electron donor but the observed rate was no more
Il A .I / I ! I 1
-0.25 0.25 0.50 0.75 1.0
IIIMV]
FIG. 6. Hz oxidation by hydrogenase I1 with methyl violo- gen as the electron acceptor. The HZ uptake assay was performed as described under "Materials and Methods" except that various concentrations of methyl viologen ( M V ) were used as the electron acceptor. V is micromoles of HZ consumed/min/mg.

7050 Hydrogenase I1 of Clostridium pasteurianum
than the sum of the rates obtained with each as the sole electron donor.
Effect of p H on Catalytic Actiuity-The rate of Hz oxidation catalyzed by hydrogenase I1 with methylene blue as the elec- tron acceptor showed pH optima at 7.0 and 10.5 (Fig. 7). In contrast, the rate of Hz uptake with methyl viologen as the electron acceptor increased slowly with increasing pH below 9 but sharply increased to a relatively high optimum value of 11.4. For comparison, the effect of pH on hydrogenase I- catalyzed Hz uptake with methyl viologen as the electron carrier was also determined (Fig. 7). This gave a similarly shaped curve to hydrogenase I1 but was slightly acid-shifted with an optimum at pH 9.8. This may indicate some mechan- istic similarities between the two enzymes although the rates of Hz oxidation obtained with hydrogenase I are about 10 times higher. The absorption coefficient of the methyl violo- gen semiquinone decreases below pH 7.0 (Erbes and Burris, 1978). The activities in Fig. 7 are uncorrected for this effect and are therefore slight overestimates of the rate of H2 oxi- dation. However, the Hz uptake assay of hydrogenase I1 at pH 8.0 measures minimal activity with both methyl viologen and methylene blue as the electron carriers (Fig. 7).
The oxidized forms of methyl viologen and methylene blue under Hz do not affect the pH of the media; therefore, the pH of the buffer used in the Hz uptake assay is the true pH of the assay medium (prior to H, catalysis). However, this is not the case with the Hz evolution assay using dithionite-reduced methyl viologen as the electron donor to hydrogenase. The reduction of methyl viologen by dithionite can be represented by the following set of equations where MVz+ and MV+ represent the oxidized and reduced forms of methyl viologen, respectively:
” 2 0 : - G= so; (1)
MV2+ + SO; + H20 G= MV’ + HS05 + H’ (2)
HS05 + SO? + H’ (3)
Dithionite oxidation proceeds via the radical SO; (Equation 1) with a subsequent one-electron transfer to yield bisulfite and the reduced dye (Equation 2). The ionization of bisulfite (Equation 3) has a pK., of 6.9 (see Mayhew, 1978). The reduction of methyl viologen by sodium dithionite will there- fore increase the acidity of the media via Equations 2 and 3.
pH
FIG. 7. Effect of pH on Ha oxidation activity. The HZ uptake assay was performed as described under “Materials and Methods” using methylene blue (0.05 mM, 0) or methyl viologen (10 mM; 0 and 0) as the electron acceptor. The activities were determined with hydrogenase I1 (0, U) and hydrogenase I (0). The buffers used were: citrate, pH 5.0-6.0; Mops, pH 6.2-7.0; Tris/HCl, pH 7.5-8.4; glycine/
phate/NaOH, pH 11.5-12.0. All buffers were at a final concentration NaOH, pH 8.8-10.2; e-aminocaproic acid, pH 10.3-11.4; and phos-
Of 50 mM.
To investigate the effectiveness of the buffers to be used in resisting this pH change, the pH values of the reaction mix- tures were determined by adding methyl viologen (to 1 or 10 mM final concentrations) to sodium dithionite (20 mM) in the buffer. The buffers used were as in Fig. 7, all at final concen- trations of 50 mM. With an initial pH of 7 or less, the pH of the medium was unaffected by the reduction of methyl viol- ogen (1 mM). Above pH 7.0, 1 mM methyl viologen caused a small decrease in pH but with 10 mM methyl viologen large pH changes were observed. For example, at initial pH values of 7.03, 8.08, 9.00, 10.40, and 11.40, the pH values after the addition of 1 mM (or 10 mM) methyl viologen were 7.02 (6.72), 7.93 (7.37), 8.20 (6.76), 9.97 (9.35), and 11.11 (10.60), respec- tively. Increasing the buffer concentration to 100 mM negated the pH change with 1 mM methyl viologen and this was used to determine the effect of pH on hydrogenase activity. How- ever, large pH changes were still observed using 10 mM methyl viologen at pH 8 and above, even with the increased buffering capacity of the media. It should therefore be noted that the reduction of methyl viologen by dithionite can have a drastic effect on the pH of the assay medium. The change in pH predicted by the above equations is observed and, as expected, ApH is dependent on the concentration of reduced methyl viologen.
The effect of pH on hydrogenase-catalyzed Hz evolution is shown in Fig. 8. The activity of hydrogenase I increased steadily with decreasing pH with an optimum at pH 6.3 but a very different curve was obtained with hydrogenase 11. The latter showed two pH optima at 9.1 and 5.8. This suggests a different mechanism of Hz evolution for the two enzymes although from Fig. 7 they have a similar pH dependency in HZ oxidation. The midpoint potential (EM) of methyl viologen is independent of pH but the E M of sodium dithionite varies with pH (Mayhew, 1978). Below pH 7.0, the latter limits the extent of methyl viologen reduction, i.e. the substrate concen- tration decreases. Increasing the methyl viologen concentra- tion to 10 mM resulted in an increase in the activity of hydrogenase I1 below pH 8.0 (Fig. 8). At pH 9.0 and 10.3 (measured pH values before enzyme addition), the activity was less than 20% above that measured with 1 mM methyl viologen at the same pH, which suggests that the increased activity below pH 7.0 reflects the increased concentration of reduced methyl viologen rather than a change in the K, value.
FIG. 8. Effect of pH on Ha evolution activity. The Hz evolution PH
assay was performed as described under “Materials and Methods” using methyl viologen as the electron carrier. The final concentrations of methyl viologen were 1.0 (0, A) and 10 mM (0). The activities were determined with hydrogenase I1 (0, 0) and hydrogenase I (A). The buffers used were as in Fig. 7 except the final concentrations were 100 mM.

Hydrogenase II of Clostridium pasteurianum 7051
The K , value for the methyl viologen semiquinone with hydrogenase I was not significantly changed by pH (Erbes and Burris, 1978). As was the case with the Hz uptake assay, the routine Hz evolution assay at pH 8.0 measures the minimal activity of hydrogenase I1 (except at the extremes of pH). It is remarkable that the enzyme catalyzes Hz evolution at a similar rate at both pH 8.0 and 10.7 in spite of the almost three orders of magnitude difference in proton concentration.
Effect of OZ-Hydrogenase I is extremely sensitive to inac- tivation by Oz. The time required for a 50% loss in HZ uptake activity after exposing the enzyme to air is 2-5 min (Erbes and Burris, 1978; Chen, 1978). Hydrogenase I1 was similarly sensitive to O2 inactivation. For the pure enzyme, a 50% loss of activity occurred in about 2 min after air was admitted to the sample (Fig. 9). Chen (1978) reported a half-time value for activity loss of 30 min for a partially purified preparation of hydrogenase 11. Hydrogenase I1 was not protected from Oz inactivation to any extent by methyl viologen (10 mM) or ferredoxin (0.5 mM). Ferredoxin has been reported to stabilize hydrogenase I against O2 (Khan et al., 1981) although the enzyme preparation used was only about 3% pure. Routinely, hydrogenase I1 was stored as pellets in liquid NS and there was no loss of activity over a 4-month period. In buffer (pH 7-8) containing dithionite (1 mM) at 4 “C, hydrogenase I1 lost no significant amount of activity after a 1-week incubation period.
Effect of Temperature-To determine the effect of temper- ature on the rate of Hz catalysis by hydrogenases I and 11, methyl viologen was used as the electron carrier in both the Hz uptake and Hz evolution assays. The Arrhenius plot for hydrogenase I-catalyzed Hz uptake is given in Fig. 10. With both enzymes in both assay systems, the plots were linear in the range 15-50 “C. Between 50 and 70 “C, the rates of reac- tion decreased (Fig. 10) and the Arrhenius plots were used to calculate the activation energy ( E A ) values and the tempera- ture optimum for that reaction. The values are given in Table I1 for both hydrogenases. The E A values were obtained using
Time (min)
FIG. 9. Inactivation of hydrogenase I1 by 0 2 . Hydrogenase I1 (1 ml) in 50 mM Tris/HCl, pH 8, in a serum-capped vial (8 ml) under Ar was incubated at 25 “C in a shaking water bath at 150 rpm. At zero time, the serum stopper was removed to expose the sample to air. At intervals, samples were removed and injected into cuvettes to determine the residual Hz uptake activity as described under “Mate- rials and Methods.” The hydrogenase I1 samples were: 0, specific activity of 2950 units/mg, 0.015 mg/ml; e, specific activity of 390 units/mg, 0.028 mg/ml.
1 x 1 OVK
FIG. 10. Arrhenius plot of hydrogenase I-catalyzed Ha up- take. The HP uptake activity of hydrogenase I was determined at various temperatures under the assay conditions described under “Materials and Methods” except that the electron carrier was methyl viologen (10 mM) and 50 rnM Mops, pH 7.2, was used as the buffer. Hydrogenase I (0.11 fig) was added to initiate the reaction after the reaction mixture had been equilibrated at the desired temperature for 15 min.
TABLE I1 Activation energy values and temperature optima for hydrogenase I
and hydrogenase II H2 evolution and Hz oxidation were both measured using methyl
viologen (1-10 mM) as the electron carrier. Results are calculated from the Arrhenius plots. See the text and Fig. 10 for details.
Hz evolution Hz uptake
EA 1, EA T- k J / m o l “C kJ lmo l “C
Hydrogenase I 35.9-38.4 48 24.0-27.2 50 Hydrogenase I1 48.3-50.0 60 49.9-53.3 65
concentrations of methyl viologen between 1.0 and 10.0 mM in both assay systems. The small variation in the values suggests that the activation energy is not dependent on the electron carrier concentration. All assays were carried out in Mops buffer since this is resistant to significant changes in pH with temperature. Between 25 and 45 “C the measured ApH was 0.10. Over the same temperature change, the pH of 50 mM Tris/HCl buffer decreased form 8.0 to 7.4; thus, any EA values determined with this buffer, e.g. Colbeau and Vig- nais (1981), should take the ApH into account.
Hydrogenase I1 catalyzed Hz production and Hz oxidation at about 1% of the rates observed with hydrogenase I with the methyl viologen concentrations used (cf. Table I) and in both assay systems the E A values for hydrogenase I1 were almost twice those obtained for hydrogenase I. The lower activity of hydrogenase I1 may therefore be due in part to the much higher energy barrier to be overcome in carrying out these reactions. This is substantiated to a large extent by a comparison with other hydrogenases. The specific activities in the Hz evolution assay (using reduced methyl viologen) for Thiocapsa hydrogenase (Gogotov et al., 1978), hydrogenase 11,
7052 Hydrogenase 11 of Clostridium pasteurianum
Rhodospirillum hydrogenase (Adams and Hall, 1979a), Esch- erichia coli hydrogenase (Adams and Hall, 1979b), and hydro- genase I, are 5, 6, 26, 100, and 500 pmol of Hz evolved/min/ mg. The reported EA values are 66,50,57,37, and 36 kJ/mol, respectively. A comparison with other hydrogenases is diffi- cult because of the different assay conditions and electron carriers that were used but the EA values reported all lie in the range 26-70 kJ/mol (see Adams et al., 1981).
The To,, values given in Table I1 are from the initial rates of HP catalysis. Hydrogenase I1 was slightly more thermosta- ble than hydrogenase I but both are much more sensitive to high temperatures than, for example, the hydrogenases of the photosynthetic bacteria (Gogotov et al., 1978; Adams and Hall, 1979a). Thiocapsa hydrogenase has a reported temper- ature optimum of 70 "C for Hz evolution over 10 min, showing that the decreased rates of Hz production observed with hydrogenases I and I1 were not due to the instability of the assay system. The absorbance of the methyl viologen semi- quinone does not significantly change with temperature (May- hew, 1978); thus, the H2 uptake assay is also satisfactory at elevated temperatures.
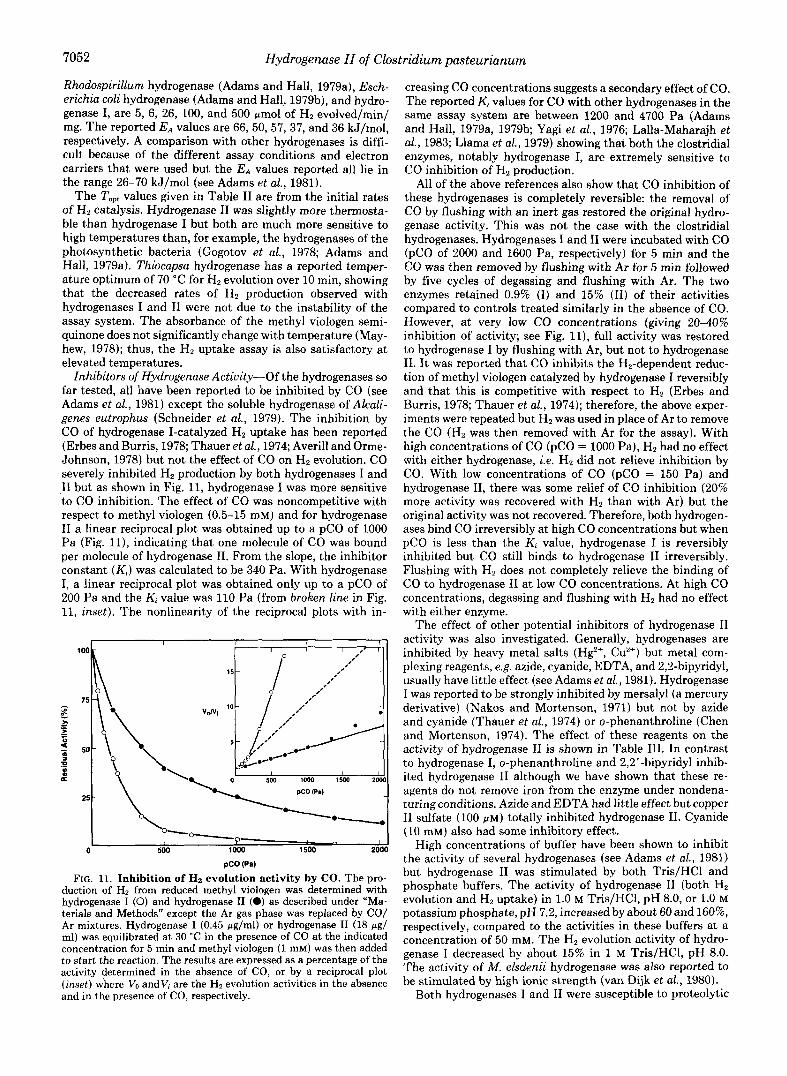
Inhibitors of Hydrogenase Actiuity-Of the hydrogenases so far tested, all have been reported to be inhibited by CO (see Adams et al., 1981) except the soluble hydrogenase of Alcali- genes eutrophus (Schneider et al., 1979). The inhibition by CO of hydrogenase I-catalyzed Hz uptake has been reported (Erbes and Burris, 1978; Thauer et al., 1974; Averill and Orme- Johnson, 1978) but not the effect of CO on Hz evolution. CO severely inhibited Hz production by both hydrogenases I and I1 but as shown in Fig. 11, hydrogenase I was more sensitive to CO inhibition. The effect of CO was noncompetitive with respect to methyl viologen (0.5-15 mM) and for hydrogenase I1 a linear reciprocal plot was obtained up to a pC0 of 1000 Pa (Fig. 11), indicating that one molecule of CO was bound per molecule of hydrogenase 11. From the slope, the inhibitor constant ( K , ) was calculated to be 340 Pa. With hydrogenase I, a linear reciprocal plot was obtained only up to a pC0 of 200 Pa and the Ki value was 110 Pa (from broken line in Fig. 11, inset). The nonlinearity of the reciprocal plots with in-
,7 I
0 500 lo00 1500 2000
PCO (pa)
FIG. 11. Inhibition of Hz evolution activity by CO. The pro- duction of H, from reduced methyl viologen was determined with hydrogenase I (0) and hydrogenase I1 (0) as described under "Ma- terials and Methods" except the Ar gas phase was replaced by CO/ Ar mixtures. Hydrogenase I (0.45 pglml) or hydrogenase I1 (18 pg/ ml) was equilibrated at 30 "C in the presence of CO at the indicated concentration for 5 min and methyl viologen (1 mM) was then added to start the reaction. The results are expressed as a percentage of the activity determined in the absence of CO, or by a reciprocal plot (inset) where VO and V; are the Ha evolution activities in the absence and in the presence of CO, respectively.
creasing CO concentrations suggests a secondary effect of CO. The reported Ki values for CO with other hydrogenases in the same assay system are between 1200 and 4700 Pa (Adams and Hall, 1979a, 197913; Yagi et al., 1976; Lalla-Maharajh et ai., 1983; Llama et al., 1979) showing that both the clostridial enzymes, notably hydrogenase I, are extremely sensitive to CO inhibition of Hz production.
All of the above references also show that CO inhibition of these hydrogenases is completely reversible: the removal of CO by flushing with an inert gas restored the original hydro- genase activity. This was not the case with the clostridial hydrogenases. Hydrogenases I and I1 were incubated with CO ($0 of 2000 and 1600 Pa, respectively) for 5 min and the CO was then removed by flushing with Ar for 5 min followed by five cycles of degassing and flushing with Ar. The two enzymes retained 0.9% (I) and 15% (11) of their activities compared to controls treated similarly in the absence of CO. However, at very low CO concentrations (giving 20-40% inhibition of activity; see Fig. 11), full activity was restored to hydrogenase I by flushing with Ar, but not to hydrogenase 11. I t was reported that CO inhibits the Hz-dependent reduc- tion of methyl viologen catalyzed by hydrogenase I reversibly and that this is competitive with respect to Hz (Erbes and Burris, 1978; Thauer et al., 1974); therefore, the above exper- iments were repeated but Hz was used in place of Ar to remove the CO (Ha was then removed with Ar for the assay). With high concentrations of CO (pC0 = 1000 Pa), Ha had no effect with either hydrogenase, i.e. Hz did not relieve inhibition by CO. With low concentrations of CO (pC0 = 150 Pa) and hydrogenase 11, there was some relief of CO inhibition (20% more activity was recovered with Hz than with Ar) but the original activity was not recovered. Therefore, both hydrogen- ases bind CO irreversibly at high CO concentrations but when pC0 is less than the Kt value, hydrogenase I is reversibly inhibited but CO still binds to hydrogenase I1 irreversibly. Flushing with Hz does not completely relieve the binding of CO to hydrogenase I1 a t low CO concentrations. At high CO concentrations, degassing and flushing with H2 had no effect with either enzyme.
The effect of other potential inhibitors of hydrogenase I1 activity was also investigated. Generally, hydrogenases are inhibited by heavy metal salts (H$+, Cu2+) but metal com- plexing reagents, e.g. azide, cyanide, EDTA, and 2,2-bipyridyl, usually have little effect (see Adams et al., 1981). Hydrogenase I was reported to be strongly inhibited by mersalyl (a mercury derivative) (Nakos and Mortenson, 1971) but not by azide and cyanide (Thauer et al., 1974) or o-phenanthroline (Chen and Mortenson, 1974). The effect of these reagents on the activity of hydrogenase 11 is shown in Table III. In contrast to hydrogenase I, o-phenanthroline and 2,2'-bipyridyl inhib- ited hydrogenase I1 although we have shown that these re- agents do not remove iron from the enzyme under nondena- turing conditions. Azide and EDTA had little effect but copper I1 sulfate (100 p ~ ) totally inhibited hydrogenase 11. Cyanide (10 mM) also had some inhibitory effect.
High concentrations of buffer have been shown to inhibit the activity of several hydrogenases (see Adams et al., 1981) but hydrogenase I1 was stimulated by both Tris/HCl and phosphate buffers. The activity of hydrogenase I1 (both HZ evolution and Hz uptake) in 1.0 M Tris/HCl, pH 8.0, or 1.0 M potassium phosphate, pH 7.2, increased by about 60 and 160%, respectively, compared to the activities in these buffers at a concentration of 50 mM. The H, evolution activity of hydro- genase I decreased by about 15% in 1 M Tris/HCl, pH 8.0. The activity of M. elsdenii hydrogenase was also reported to be stimulated by high ionic strength (van Dijk et al., 1980).
Both hydrogenases I and I1 were susceptible to proteolytic

Hydrogenase II of Clostridium pasteurianurn 7053
TABLE 111 Inhibition of hydrogenase I I
Hydrogenase I1 was assayed by Hz uptake with methylene blue as the electron acceptor as described under “Materials and Methods” except the reaction mixture also contained the inhibitor (1.0 or 10 mM). All reactions were performed in 50 mM potassium phosphate buffer, pH 7.2, except for copper 11 sulfate where 50 mM Tris/HCl, pH 8.0, was used. Results are expressed as a percentage of the activity observed in the absence of inhibitor.
Residual activity
1.0 mM 10.0 mM
%
Inhibitor -
o-Phenanthroline 36 6 EDTA 108 94 2,2‘-Bipyridyl 66 38 Phenylmethanesulfonyl fluoride 61 15 Copper I1 sulfate 0 0 Potassium cyanide 108 I7 Sodium azide 107 98
digestion. When hydrogenase I1 (0.5 mg/ml) was incubated a t 25 “C with trypsin, chymotrypsin, or subtilisin (all 0.1 mg/ ml), over 90% of the Hz uptake activity was lost within 20 min. Hydrogenase I was less sensitive since under the same conditions less than 10% of the activity was lost with chy- motrypsin and subtilisin and about 20% was lost with trypsin. E. coli hydrogenase has been reported to be unaffected by chymotrypsin or trypsin (Adams and Hall, 1979b).
DISCUSSION
The separation during purification of more than one hydro- genase activity has been found with many organisms, e.g. M. elsdenii (van Dijk et al., 1979), Chromatium D (Llama et al., 1981), Methanobacterium thermoautotrophicum (Jacobson et al., 1982), Anabaena cylindrica (Tel-Or et al., 1978), and D. desulfuricam (Kruger et al., 1982) but it is not clear if the different activities represent different enzymes or complexes of the same enzyme (see also Adams et al., 1981). I t is only from the aerobic hydrogen bacterium Alcaligenes eutrophus (Schneider et at., 1979; Schink and Schlegel, 1979) and from the methanogenic bacterium Methanobacterium formicicum (Jin et al., 1983) that two hydrogenases have been purified and characterized and have been shown unambiguously to be two distinct enzymes. Our data with C. pasteurianum shows that this organism also contains two very distinct hydrogenase species that differ in molecular structure, metal content, catalytic activity, and sensitivity to inhibitors.
Neither hydrogenase I or hydrogenase I1 of C. pasteurianum contained metal atoms other than iron in significant amounts. Nickel has recently been implicated to be part of the active center of several hydrogenases. Isotopic substitution analyses and/or EPR studies have shown that nickel is part of the hydrogenases from D. gigas (LeGall et al., 1982; Cammack et aL, 1982), M . thermoautotrophicum (Albracht et al., 1982a) Vibrio succinogenes (Unden et al., 1982), A. eutrophus (Fried- rich et al., 1982), and from two strains of D. desulfuricans (Kmger et al., 1982; Lalla-Maharajh et al., 1983). Nickel is also required for the biosynthesis of hydrogenase in some other organisms (Takakuwa and Wall, 1981; Partridge and Yates, 1982; Albracht et al., 1982b). The two hydrogenases of C. pasteurianum are so far the only reported hydrogenases that do not contain nickel, showing that nickel is not obliga- tory for Hz catalysis by hydrogenase. All the above enzymes function in uiuo to primarily consume Hz, whereas hydrogen- ase I of C. pasteurianum has the well defined physiological role of HZ production. The latter enzyme is also much more
active in Hz evolution in vitro than the other hydrogenases. One could speculate that nickel serves to modulate hydrogen- ase activity in favor of Hz consumption and the data on the nickel content of hydrogenases from other HZ producing or- ganisms, e.g. M . elsdenii, E. coli, are eagerly awaited. The physiological role of hydrogenase 11 is at present unknown.
Our analyses showed that hydrogenase I1 of c. pasteu- rianum contains 8 iron and 8 acid-labile sulfide g atoms/ 53,000 g of protein. Accepting the inherent error in these analyses, we often found values of slightly above eight in both cases although no iron could be removed from the enzyme by chelators under nondenaturing conditions. Analyses of iron- sulfur proteins for iron and acid-labile sulfide typically give values slightly less than the actual value and one “rounds up’’ to the nearest integer. However, extrusion studies with hydro- genase I1 indicated the presence of two [4Fe-4S] clusters in agreement with 8 iron atoms/molecule. We therefore feel that rather than hydrogenase I1 containing an additional iron center, either the molecular weight is slightly underestimated or the enzyme does not give a true value in the determination of protein.
The new medium for the extrusion of 2Fe clusters reported by Kurtz (1982) was modified to extrude [4Fe-4S] centers from C. pasteurianum ferredoxin but this method was not applicable to hydrogenase 11. Analysis by the HMPA method unequivocably showed the presence of [4Fe-4S] centers in hydrogenase 11. Several iron-sulfur proteins have recently been reported to contain 3Fe-clusters (see Beinert et al., 1983) but these extrude as [2Fe-2S] clusters (Kurtz et al., 1979); therefore hydrogenase I1 does not contain 2Fe or 3Fe centers. The EPR spectra of hydrogenase I1 can be nicely interpreted assuming the presence of two [4Fe-4S] clusters. The reduced enzyme gives a spectrum (gav < 2) typical of a reduced ferre- doxin [4Fe-4S]2+(’+r1+) cluster which disappears upon oxida- tion to give a sharp rhombic signal with g., > 2, typical of a Hipip or [4Fe-4S]2+(z+,3+) cluster. The lack of complexity in both spectra is indicative of each signal arising from a single noninteracting cluster (Fig. 5). The reduced enzyme is there- fore equivalent to (C+, ,’+) and the oxidized form is (C2+, C3’) (see Adams et al., 1981). The EPR spectra of hydrogenase I1 are very similar to those obtained from hydrogenase I (Chen et al., 1976) except that the latter has two spin-coupled reduced ferredoxin (C+) clusters which give rise to a complex EPR signal. The EPR signals with g,, > 2 from the oxidized forms of the two clostridial hydrogenases are remarkably similar. However, an oxidized 3Fe cluster also exhibits an EPR signal with g > 2 (see Beinert et al., 1983 and references therein). This signal is isotropic and is therefore distinct from the strong rhombic signals observed with the clostridial hy- drogenases. The absence of 3Fe cluster in these hydrogenases is also supported by the metal analyses and the extrusion data. The 4Fe cluster giving rise to the rhombic EPR signal has been proposed as the site of HZ catalysis (Chen et al., 1976; Chen, 1978). A mechanism of catalysis involving the “superreduced state of this cluster, i.e. (1+) state, has re- cently been proposed by Veeger and eo-workers (Grande et al., 1982).
When hydrogenase I1 of C. pasteurianum was treated with ferricyanide, all of the iron atoms were lost from the enzyme. Treatment of C. pasteurianum ferredoxin with ferricyanide causes the conversion of 4Fe clusters to 3Fe clusters (Thom- son et al., 1981) but hydrogenase I1 was particularly suscep- tible to this oxidant. Preliminary experiments have shown that, after exposure to ferricyanide for less than 1 min hydro- genase I1 exhibited an isotropic EPR signal at g > 2.3 The
M. W. W. Adams, unpublished data.

7054 Hydrogenase II of Clostridium pasteurianum
destruction of the enzyme by ferricyanide may therefore in- volve the intermediate formation of 3Fe centers. The effect of ferricyanide on hydrogenase I has not been investigated but the enzyme can be reversibly oxidized and reduced with ferricyanide and Hz or dithionite (Chen et al., 1976).
The catalytic properties of hydrogenases I and I1 are very different but the term “uptake” hydrogenase is not fully descriptive of hydrogenase 11. With methyl viologen as the electron carrier, hydrogenase I1 is a relatively inactive hydro- genase in catalyzing both Hz evolution and Hz uptake. Its activity is comparable to the very low values reported for the hydrogenases of the photosynthetic bacteria (Gogotov et al,, 1978; Adams and Hall, 1979a; Llama et al., 1981). It is only in the Hz uptake assay with methylene blue as the electron acceptor that hydrogenase I1 is extremely active, comparable to hydrogenase I. The apparent K , values for the electron carriers tested are remarkably similar for the two enzymes, except for the methyl viologen semiquinone where there is a 30-fold difference. The ease with which hydrogenase I1 satu- rates with reduced methyl viologen may help explain the low activity. The hydrogenase from M. ekdenii is extremely active in Hz production and is not easily saturated with methyl viologen (van Dijk et al,, 1979). The pH dependence of HZ evolution catalyzed by hydrogenase I1 is very unusual in that it is biphasic and optimal at pH 9.1. For all other hydrogenases so far examined, a typical bell-shaped curve is obtained with an optimum pH value of 7 or less (see Adams et al., 1981 and references therein).
The differences in pH dependency between the two clostri- dial hydrogenases presumably reflect differences in the mech- anism of Hz catalysis. This is further supported by the large discrepancies in the activities of the two enzymes, in their respective activation energies in both assay systems and in their sensitivities to inactivation by CO. Hydrogenase I was more sensitive to CO than hydrogenase 11, yet the latter appears to bind CO irreversibly, even at low CO concentra- tions. There was partial relief of the CO inhibition of hydro- genase I1 by flushing with Hz, but not with Ar. All these data can be considered in terms of the mechanism of HZ catalysis. A mechanism for Hz activation is given by Equations 4-8 which is a simplified version of that proposed by Adams et al. (1981). E, E-, and EH- are different redox states of hydro- genase and Cox and C!& are the oxidized and reduced forms of a one-electron carrier:
E + C d + E - + C,, kl
k2 (4)
k3
k, E - + H+ == EH (5)
ks 4
EH + C d e EH- + Cox (6)
k7
ka EH- + H+ + EHz (7)
ke k,o
EH, + E + HZ (8)
It is proposed that hydrogenase I1 is a poor catalyst of HZ production because Hz binds so strongly to the enzyme that the limiting step in Hz evolution (Equations 4 to 8) is the removal of H, from the catalytic site, i.e. klo > kg. A strong affinity between hydrogenase I1 and Hz is suggested by the 10-fold difference in the K , values for Hz and hydrogenases I and I1 (310 and 32 PM, respectively), and by the partial relief by Hz of the strong and irreversible binding of CO to hydro- genase 11. This is further supported by the data on the
absorption spectra of the Hz-reduced forms of hydrogenases I and I1 (Fig. 4). The spectrum of hydrogenase I became more oxidized when Hz was replaced by Ar, presumably by Hz production (equivalent to Equations 7 and 8). The spectrum of hydrogenase I1 did not change under these conditions, i.e. klo > kg. In the reverse reaction, HZ oxidation by hydrogenase I1 (Equations 8 to 4) is limited by the rate of electron transfer to the electron acceptor (Equations 4 and 6) which could be thermodynamically controlled. Both hydrogenases showed similar K , values for the electron carriers in the Hz uptake assay. A low potential electron acceptor, e.g. methyl viologen (EM = -440 mV), is therefore reduced much less efficiently than an electron carrier with a higher midpoint potential, e.g. methylene blue (EM = +11 mV). Hydrogenase I1 is obviously not limited by the rate of Hz catalysis, i.e. the cleavage or formation of Hz (Equation 7), because of the high rates of Hz oxidation observed with methylene blue as the electron accep- tor. Hydrogenase I reduced methylene blue and methyl viol- ogen with H2 at high rates (methyl viologen was slightly less active; Table I); therefore, hydrogenase 1 may well be limited by the rate of covalent bond formation or breakage (Equation 7). That is, the removal of HZ from the catalytic site (Equation 8) does not limit Hz production by hydrogenase I and the redox potential of the electron acceptor (which influences Equations 4 and 6) does not dramatically affect the observed rates of HI? oxidation. Hydrogenase I is thus an extremely active enzyme limited by Equation 7. Hydrogenase I1 is also an extremely active catalyst (uiz. Equation 7) but is limited by Equation 8. High rates of Hz catalysis by hydrogenase I1 are therefore only observed in Hz oxidation using an electron acceptor with a much more positive potential than the Hz electrode (EM = -420 mV). Experiments to further probe the mechanistic differences between the two enzymes are cur- rently in progress.
Hydrogenases I and I1 differ considerably from the point of view of Hz catalysis as described above but structurally the similarities are striking. Hydrogenase I1 is of slightly lower molecular weight and lacks what appears to be one
previously shown that the cellular concentration of hydrogen- ase I1 seems to increase with cell growth, whereas the content of hydrogenase I remains unchanged.’ A tempting speculation is therefore that hydrogenase I1 is a primary breakdown product of hydrogenase I which accumulates during cell growth. The loss of a single [4Fe-4S] center and a small stabilizing peptide from hydrogenase I might confer the de- scribed properties on hydrogenase 11. We are currently per- forming peptide analysis and immunological studies to inves- tigate this possibility.
[4Fe_4S]Z+(z+J+) cluster compared to hydrogenase I. We have
Acknowledgments-The excellent technical assistance of Judy Ki- mak-Savas is gratefully acknowledged. We thank Dr. J-S. Chen for helpful discussions.
REFERENCES Adams, M. W. W., and Hall, D. 0. (1979a) Arch. Biochern. Biophys.
Adams, M. W. W., and Hall, D. 0. (1979b) Biochern. J. 183, 11-22 Adams, M. W. W., Mortenson, L. E., and Chen, J-S. (1981) Biochem.
Albracht, S. P. J., Graf, E-G., and Thauer, R. K. (1982a) FEES Lett.
Albracht, S. P. J., Albrecht-Ellmer, K. J., Schmedding, D. J. M., and
Averill, B. A., and Orme-Johnson, W. H. (1978) J . Am. Chem. SOC.
Beinert, H . , Emptage, M. H., Dryer, J-L., Scott, R. A., Hahn, J. E. Hodgson, K. O., and Thomson, A. J. (1983) Proc. Natl. Acad. Sci.
195, 288-299
Bwphys. Acta 594,105-176
140,311-313
Slater, E. C. (1982b) Biochim. Biophys. Acta 681,330-334
100,5234-5235
U. S. A. 80,393-396

Hydrogenase II of Cl05
Cammack, R., Patil, D., Aquirre, R., and Hatchikian, E. C. (1982)
Chen, J-S. (1978) in Hydrogenases: Their Catalytic Activity, Structure and Function (Schlegel, H. G., and Schneider, K., eds) pp. 57-82, E. Goltze K. G., Gottingen
Chen, J-S., and Blanchard, D. K. (1978) Biochem. Biophys. Res. Commun. 8 4 , 1144-1150
Chen, J-S., and Mortenson, L. E. (1974) Biochim. Biophys. Acta 371 ,
Chen, J-S., Mortenson, L. E., and Palmer, G. (1976) in Iron and Copper Proteins (Yasunobu, K. T., Mower, H. F., and Hayaishi, O., eds) pp. 68-83, Plenum Press, New York
Colbeau, A., and Vignais, P. M. (1981) Biochim. Biophys. Acta 6 6 2 ,
Emptage, M. H., Kent, T. A., Huynh, B. H., Rawlings, J., Orme- Johnson, W. H., and Munck, E. (1980) J. Biol. Chem. 2 5 5 , 1793- 1796
Erbes, D. L., and Burris, R. H. (1978) Biochim. Biophys. Acta 525 , 45-54
Friedrich, C. G., Schneider, K., and Friedrich, B. (1982) J. Bacterwl. 152,42-48
Gillum, W. O., Mortenson, L. E., Chen, J-S., and Holm, R. H. (1977) J. Am. Chem. Soc. 99,584-595
Gogotov, I. N., Zorin, N. A., Serebriakova, L. T., and Kondratieva, E. N. (1978) Biochim. Biophys. Acta 523, 335-343
Grande, H. J., van Dijk, C., Dunham, W. R., and Veeger, C. (1982) in The Biological Chemistry of Iron (Dunford, H. B., ed) pp. 193- 206, D. bide1 Publishing Co., Boston
Huynh, B. H., Moura, J. J. G., Moura, I., Kent, T. A., LeGall, J., Xavier, A. V., and Munck, E. (1980) J. Bwl. Chem. 2 5 5 , 3242- 3244
Jacobson, F. S., Daniels, L., Fox, J. A., Walsh, C. T., and Orme- Johnson, W. H. (1982) J. Biol. Chem. 2 5 7 , 3385-3388
Jin, S-L. C., Blanchard, D. K., and Chen, J-S (1983) Biochim. Biophys. Acta 748,8-20
Khan, S. M., Klibanov, A. M., Kaplan, N. O., and Kamen, M. D. (1981) Biochim. Biophys. Acta 659,457-465
Kriiger, H-J., Huynh, B. H., Ljungdahl, P. O., Xavier, A. V., Der-
J. J. G., and LeGall, J. (1982) J. Bwl. Chem. 2 6 7 , 14620-14623 Vartanian, D. V., Moura, I., Peck, H. D., Jr., Teixeira, M., Moura,
Kurtz, D. M. (1982) Biochem. Biophys. Res: Commun. 104 , 437-442 Kurtz, D. M., Holm, R. H., Ruzicka, F. J., Beinert, H., Coles, C. J.,
FEBS Lett. 142,289-292
283-298
271-284
and Singer, T. P. (1979) J. Biol. Chem. 254,4967-4969
;tridium pasteurianum 7055
Lalla-Maharajh, W. V., Hall, D. O., Cammack, R., Rao, K. K., and LeGall, J. (1983) Bwchem. J. 209,445-454
LeGall, J., Ljungdahl, P. O., Moura, I., Peck, H. D., Jr., Xavier, A. V., Moura, J. J. G., Teixera, M., Huynh, B. H., and DerVartanian, D. V. (1982) Biochem. Biophys. Res. Commun. 106,610-616
Llama, M. J., Serra, J. L., Rao, K. K., and Hall, D. 0. (1979) FEBS
Llama, M. J., Serra, J. L., Rao, K. K., and Hall, D. 0. (1981) Eur. J.
Mayhew, S. G. (1978) Eur. J. Biochem. 85,535-547 Mullinger, R. N., Cammack, R., Rao, K. K., Hall, D. O., Dickson, D.
P. E., Johnson, C. E., Rush, J. D., and Simpopoulos, A. (1975) Biochem. J. 151, 75-83
Nakos, G., and Mortenson, L. E. (1971) Biochemistry 10, 2442-2449 Partridge, C. D. P., and Yates, M. G. (1982) Biochem. J. 2 0 4 , 339-
Que, L., Jr., Holm, R. H., and Mortenson, L. E. (1975) J. Am. Chem.
Schink, B., and Schlegel, H. G. (1979) Biochim. Biophys. Acta 567 ,
Schneider, K., Cammack, R., Schlegel, H. G., and Hall, D. 0. (1979)
Takakuwa, S., and Wall, J. D. (1981) FEMS Microbwl. Lett. 12,359-
Tel-Or, E., Luijk, L. W., and Packer, L. (1978) Arch. Biochem.
Thauer, R. K., Kaufer, B., Zahringer, M., and Jungermann, K. (1974) Eur. J. Bwchem. 42,441-452
Thomson, A. J., Robinson, A. E., Johnson, M. K., Cammack, R., Rao,
432 K. K., and Hall, D. 0. (1981) Biochim. Biophys. Acta 637 , 423-
Lett. 98,342-346
Biochem. 114,89-96
344
SOC. 97,463-464
315-324
Biochim. Biophys. Acta 578,445-461
363
Bkphys. 185,185-194
Unden, G., Bijcher, R., Knecht, J., and Kroger, A. (1982) FEBS Lett. 145,230-234
van Dijk, C., Mayhew, S. G., Grande, H. J., and Veeger, C. (1979)
van Dijk, C., Grande, H. J., Mayhew, S. G., and Veeger, C. (1980)
Vignais, P. M., Henry, M. F., Berlier, Y., and Lespinat, P. A. (1982)
Walker, G. A., and Mortenson, L. E. (1973) Biochem. Biophys. Res.
Yagi, T., Kimura, K., Daidoji, H., Sakai, F., Tamura, S., and Inokuchi,
Eur. J . B k h e m . 102,317-330
Eur. J . Biochem. 107,251-261
Biochim. Biophys. Acta 681 , 519-529
Commun. 53,904-909
M. (1976) J. Biochem. (Tokyo) 79,661-671







![How oxygen attacks [FeFe] hydrogenases from photosynthetic ... · How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms Sven T. Strippa, Gabrielle Goldetb, Caterina](https://img.pdfslide.us/doc/110x75/603ba0c1ea10106e07149762/how-oxygen-attacks-fefe-hydrogenases-from-photosynthetic-how-oxygen-attacks.jpg)





















