Embed Size (px)
Citation preview

The modern RNA world:Not all genes encode proteins.
Eddy labHHMI/Washington University, Saint Louis

GeneSweep; or, Ewan’s definition of a genehttp://www.ensembl.org/Genesweep
Footnote 1.
“We are restricting ourselves to protein coding genes to allow an effective assessment. RNA genes were considered too difficult to assess by 2003.”
Rule 2.
“A gene is a set of connected transcripts.... A transcript is a set of exons.... one transcript must encode a protein (see footnotes).”

Life with 6000 Genes
A. Goffeau, B.G. Barrell, H. Bussey, R.W. Davis, B. Dujon,H. Feldmann, F. Galibert, J.D. Hoheisel, C. Jacq, M. Johnston,
E.J. Louis, H.W. Mewes, Y. Murakami, P. Phillippsen,H. Tettelin, S.G. Oliver
Science 274:546-567, 1996
but besides the ~6000 protein-coding genes, there’s also:140 ribosomal RNA genes,275 transfer RNA genes,40 small nuclear RNA genes,~100 small nucleolar RNA genes,... ?
life with 6000 genes

Structure of the large ribosomal subunitHaloarcula marismortui
Ban et. al., Science 289:905-920, 2000

inside-out genes?
Human UHG (U22 host gene)no significant ORFs; not conserved with mouse; rapidly degraded
Eight intron-encoded snoRNAsconserved with mouse; stable
Tycowski, Shu, and SteitzNature 379:464-466, 1996

pRNA in 29 rotary packaging motor Simpson et al, Nature 408:745-750, 2000
“Structure of the bacteriophage 29 DNA packaging motor”

Cartilage-hair hypoplasia mapped to an RNARidanpaa et al. Cell 104:195-203, 2001
RMRP: Human RNase MRP, 267 nt

The human Prader-Willi critical regionCavaille et al., PNAS 97:14035-7, 2000

RNA genes can be hard to detect
UGAGGUAGUAGGUUGUAUAGU
C. elegans let-7; 21 ntPasquinelli et al. Nature 408:86-89, 2000
• often small• sometimes multicopy and redundant• often not polyadenylated (and remember EST libraries are poly-A selected)• immune to frameshift and nonsense mutation• no open reading frame or codon bias• often rapidly evolving in primary sequence

The Altuvia screen
“Over a period of about 30 years, only four bona fide regulatory RNAs have been discovered in E. coli. Here we report on the discovery of 14 novel small RNA-encoding genes....”
Argaman et al., Current Biology 11:941-50, 2001“Novel small RNA-encoding genes in the intergenic regions of E. coli”
sraA 120 ntsraB 149-168 ntrprA 105 ntsraC 234-249 ntsraD 70 ntgcvB 205 ntsraE 88 ntsraF 189 ntsraG 146-174 ntsraH 88-108 ntsraI 91-94 ntsraJ 172 ntsraK 245 ntsraL 140 nt
• start w/ “intergenic” regions
• computational identification of putative promoter and terminator, 50-400 nt apart
• select regions conserved with other bacteria by BLAST

The Gottesman screenWassarman et al., Genes Dev. 15:1637-51, 2001
“Identification of novel small RNAs using comparative genomics and microarrays”
rydB 60 ntryeE 86 ntryfA 320 ntryhA 45 nt (sraH)ryhB 90 nt (sraI)ryiA 210 ntryjA 92 ntrybB 80 ntryiB 270 nt (sraK, csrC)rybA 205 ntrygA 89 nt (sraE)rygB 83 ntryeA 275 ntryeB 100 ntryeC 107,143 ntryeD 102,137 ntrygC 107,139 nt
• intergenic regions >= 180 nt
• conserved w/ other bacteria by BLAST
• manual inspection of location & sequence
• expression detected on high-density oligo probe array
“... a multifaceted search strategy to predict sRNA genes was validated by our discovery of 17 novel sRNAs....”

Two computational analysis problems
1. Similarity search (e.g. BLAST): I give you a query; you find sequences in a database that look like the query.
For RNA, you want to take the secondary structure of the query into account.
2. Genefinding (e.g. GENSCAN): Based solely on a priori knowledge of what a “gene” looks like, find genes in a genome sequence.
For RNA – with no open reading frame and no codon bias – what do you look for?

RNA structure: nested pairwise correlations

Context-free grammarspioneered in comp bio by David Searls
a CFG “derivation”Basic CFG “production rules”

HMM and SCFG algorithmsR Durbin, SR Eddy, GJ Mitchison, A Krogh
Biological Sequence Analysis:Probabilistic Models of Proteins and Nucleic AcidsCambridge Univ. Press, 1998
Goal
optimal alignmentP(sequence | model)
EM parameter estimation
memory complexity:time complexity (general):time complexity (as used):
HMM algorithm
ViterbiForward
Forward-Backward
O(MN)O(M2N)O(MN)
SCFG algorithm
CYKInside
Inside-Outside
O(MN2)O(M3N3)O(MN3)
• we can analyze target sequences with secondary structure models;• but the algorithms are computationally expensive.

SCFG-based RNA similarity search
COVE (Eddy and Durbin, 1994) structural profiles of RNA sequence families
tRNAscan-SE (Lowe and Eddy, 1997) fast prescreens + COVE model of tRNA – large scale tRNA detection
snoscan (Lowe and Eddy, 1998) C/D box small nucleolar RNA detection in yeast genome
snoRNAs detected in Archaea (Omer et al. 2000) C/D box snoRNA homologues detected in many Archaea collaboration with Pat Dennis’ lab at UBC Vancouver
FOLDALIGN (Jan Gorodkin) – automatic recognition and alignment of consensus secondary structure elements
also Y. Sakakibara, F. Lefebvre, B. Knudsen, I. Holmes, others...
profile HMMs from A. Krogh, D. Haussler “profile SCFGs”, or “covariance models”

SCFGs for RNA folding
• Minimum energy RNA folding by dynamic programming – Michael Zuker
• Partition function calculations (weighted summations over ensemble of all structures) – J. McCaskill
• SCFG analogue of the Zuker program; maximum likelihood folds by the CYK algorithm; summations by the Inside algorithm – E. Rivas and S.R. Eddy, 2000

Genefinding by comparative analysisJonathan Badger, Gary Olsen: CRITICA
The OTHER model:score with terms P(a,b | OTH)models divergence only
the CODING model:score with terms P(aaa,bbb | COD)models divergence, constrained byamino acid substitution matrix andcodon bias
Most comparative analysis relies just on differential rates of evolution.However, the pattern of mutation is also informative.

add: a comparative model of structural RNAs
The RNA model:terms: P(a-a’, b-b’ | RNA)models DNA divergence constrained by a secondary structure

Technical issues
- The structure is unknown; must do weighted sum over all possible structures. We extended an SCFG model of RNA folding (Rivas and Eddy, 2000) to a pair-SCFG, and we use an Inside algorithm to score it.
- model must deal with gapped alignments that are heterogeneous w.r.t. models – e.g. BLAST may align beyond the edge of the real RNA. We use pair-grammar formalisms for all three models, and include flanking models of conserved nonstructured alignment.
- though we want to sum over all structures, we also want to recover maximum likelihood start/end points of an RNA within a longer alignment. We use the generalized HMM parsing trick introduced by Stormo and Haussler (aka “semi Markov models” in Burge’s GENSCAN), and treat our RNA model as an i,j feature score in a generalized HMM.
- divergence times of the three models must be the same. We tie all model parameters to a choice of amino acid substitution matrix.
-

Three models – examples of their scores

A screen for novel ncRNAs in E. coliE. Rivas, R. Klein, T. Jones, S.R. Eddy, submitted
2367 E. coli intergenic sequences >50 nt in length
WUBLASTN vs. S. typhi, S. paratyphi, S. enteriditis, K. pneumoniaegave 23,674 WUBLASTN alignments w/ E<0.01, length >50 nt, >65% identity
QRNA classified: 556 candidate RNA loci 160 candidate small ORFs (not examined further)
281 candidate loci are explainable: cis-regulatory RNA structures (terminators, attenuators, etc.) and certain inverted repeat elements
leaves 275 candidate ncRNA gene loci
Northerns on 49 candidates: 11/49 are expressed as small stable RNAsin exponentially growing E. coli in rich media

Northern blots confirming E. coli RNAs

Summary of three E. coli screens
10/14 of the RNAs found by the Altuvia screen are in QRNA candidate list3 are just below 5 bit cutoff; one (sraI) completely missed
14/17 of the RNAs found by the Gottesman screen are in candidate list2 are just below cutoff; 1 was thrown out mistakenly (QRNAfound it, we thought it was just a terminator)
Conclusions: Sensitivity of QRNA is respectable; most E. coli ncRNAs conserve secondary structure
Only 4/11 of our confirmed ncRNAs are in the Altuvia or Gottesman genes
Conclusions: These screens have not saturated E. coli for new ncRNAs; A total of 34 new ncRNAs confirmed. We have >200 other candidates in testing; We have confirmed transcripts as short as 40 nt; The functions of these RNAs are unknown.

human/mouse ncRNA detection
the cartilage-hair hypoplasia region:
QRNA is a general genefinder for structural ncRNA genes.
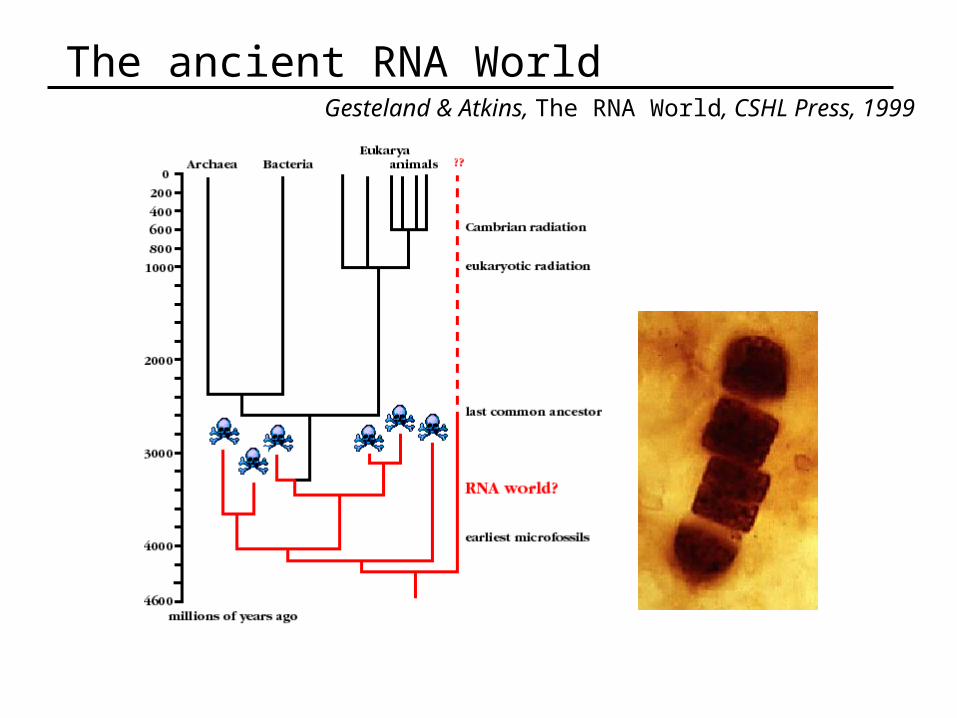
The ancient RNA WorldGesteland & Atkins, The RNA World, CSHL Press, 1999

RNA is very good at recognizing RNA
RA Lease & M Belfort, PNAS 97:9919-24, 2000“A trans-acting RNA as a control switch in Escherichia coli...”

A closing idea: The modern RNA world
Hypothesis:When a cell needs to make a molecule X that specifically recognizes a target RNA molecule, and the function of X is either:
- catalytically unsophisticated (e.g. steric repression of translation); or- something that can be abstracted onto a single protein (e.g.
many guide snoRNAs, one catalytic methylase)
then RNA may be the material of choice. Small, highly specific complementary RNAs can be generated by simply duplicating part of the antisense strand of the target RNA. Specific RNA-binding proteins are big, expensive, and more difficult to evolve.

Summary
• Noncoding RNAs are genes too.
• Methods to find homologous RNAs by structural similarity have been greatly improved, using stochastic context free grammar algorithms.
• Methods to find novel RNAs by de novo genefinding are our current aim. Two different screens detect new structural RNAs:
- a simple GC screen in AT-rich hyperthermophile genomes;
- QRNA, an RNA genefinder using comparative sequence analysis.
[SR Eddy, Curr Opin Genet Dev 9:965, 1999]
[R Durbin et al., Biological Sequence Analysis, Cambridge U. Press 1998]
[RJ Klein, Z Misulovin, SR Eddy, in preparation]
[E Rivas, RJ Klein, TA Jones, SR Eddy, submitted]

Acknowledgementshttp://www.genetics.wustl.edu/eddy/
The QRNA comparative analysis screen:
Elena RivasTom JonesRobbie Klein





























