Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc
Vol. 269, NO. 12, Issue of March 25, pp. 9195-9204, 1994 Printed in U.S.A.
Identification of Regulatory Elements in the Cutinase Promoter from Fusarium solani f. sp, pisi (Nectria haematococca)*
(Received for publication, August 26, 1993, and in revised form, November 15, 1993)
Jorg T. KiimperSO, Ute Kiirnpera Linda M. Rogers, and Pappachan E. Kolattukudyll From Ohio State Biotechnology Center, Ohio State University, CoEumbus, Ohio 43210

9196 Promoter Analysis of the Cutinase Gene from Fusarium standard media as described in Sambrook et al. (14). Single spore iso- lates ofE solani f. sp.pisi T8 (2) were maintained in aliquots as glycerol stocks (15% glycerol) at -80 OC; spores from frozen stocks were plated on potato dextrose agar (Difco) supplemented with 0.5 g/liter ground lyophilized pea stems (PPDA-Medium) and incubated for 4-7 days at 24 "C. Spores collected from these plates were used for all other inocu- lations. E solani pisi cultures were grown in minimal medium (1) with 0.5 to 2% glucose, on a rotary shaker with 200 rpm. Transformants were grown at 24 "C on V8-agar (10% V8 juice, 0.2% CaC03, 2% agar, pH 6.3) supplemented with 100-300 mg/liter hygromycin B.
Standard buffers were prepared according to Sambrook et al. (14). Chemicals, unless otherwise indicated, were purchased from Sigma. [3ZP1Deoxynucleotide triphosphates and d-threo-[dichloroacet~l~l,2- 14ClchlorampheNcol were purchased from DuPont NEN. Enzymes, un- less otherwise indicated, were purchased from Life Technologies, Inc. Novozyme 234 was from Novo Industries.
RNA Manipulations-E. coli plasmid DNA was isolated by the "boil- ing" method (14). E. coli transformation was performed by the method of Hanahan and co-workers as described in Sambrook et al. (14). Plas- mid templates were sequenced using a Sequenase Kit from U. S. Bio- chemical Corp. Conditions for PCR were as described by Innis (15): 100 p~ dNTP, 50 pmol of each primer, 10 to 100 ng of template in 100-pl reactions, using the supplied Zbq buffer and 2 units of Taq DNA po- lymerase from either Promega or Life Technologies, Inc. Unless other- wise outlined, cycle conditions were the following: 5-min denaturation at 94 "C, followed by 30 cycles (1 min at 94 "C, 1 min at 60 "C, 2 min at 72 "C) and a final extension reaction (5 min at 72 "C).
Plasmid Constructions-Plasmid pBluescript KS.431 is a pBlue- script KS derivative containing the gene for chloramphenicol acetyl- transferase (cat ) under control of a promoter fragment of the cutinase gene from K sotanz pisi (16). Sequencing of the fragment carrying the cutinase promoter revealed three differences to the published sequence (17): a t position -386 an additional G and at position -376 an additional C were inserted a C at position -6 was changed to T. Due to the two additional nucleotides, the length of the promoter fragment in pBlue- script KS 431 changes from the previously indicated 431 nucleotides (16) to 433 nucleotides.
pBluescript KS COC Hyg is a pBluescript KS derivative carrying the gene for hygromycin phosphotransferase (hph) under control of a pro- moter from Cochliobolus heterostrophus facilitating the selection in K solanipisi (16). pBluescript KS.431 Exp is a pBluescript KS.431 deriva- tive with the insertion of the Cochliobotus-promoter/hph gene fusion of pBluescript KS COC Hyg (16).
In all plasmids described below, the digits in their names correlate with the promoter length respective to the ATG-start codon of the cu- tinase gene.
For construction of plasmids pCAT141, pCAT212, pCAT249, pCAT287, and pCAT310, plasmid pBluescript KS.431 was digested with XbaI (cuts 5' of the cutinase promoter), treated with exonuclease Ba131, blunted with T4 DNA polymerase, digested with Sat1 (cuts 3' of the cut gene), and cloned in EcoRV/Sa~I-digested pBluescript KS. Deletions were analyzed by sequencing with the universal M13 primer. The dif- ferent promoter/ca~ gene fusions were isolated as XbuPSaZI fragments and cloned into ~IISatI-digested pBluescript K$ Cos Hyg. Plasmids pCAT225 and pCAT360 were generated as described by Bajar et al. (16): pBluescript KS.431 was digested with Aut11 and PuuI, respectively, blunted with T4 DNA polymerase, and digested with SafI, and the fragments containing the promoter/ca~ gene fusion were cloned into S~uIISuZI-digested pBluescript KS Cos Hyg. Plasmids pCAT195, pCAT181, pCAT167, and pCATl53 were generated by PCR using pBlue- script KS.431 as template. Oligo 1 (CGG AAT TCC GGATGAGCATT), located in the cat gene, was used as first primer, and one of four differ- ent oligonucleotides co~sponding to the 5' ends of the promoter frag- ment as second primers; the latter oligonucleotides contain an addi- tional XbaI site (oligo 3 (-195): GCT CTA GAA ACC GAG GAA ATG; oligo 4 (-181k GCT CTA GAA TGG ATC GCG AGC CGA G oligo 5 (-153): GCT CTAGAG CTT GGA CGATGATA; oligo 6 (-169): GCT CTA GAC GAG GCT CGA TTT CAG AG). PCR reactions were performed under standard conditions. The PCR products were digested with EcoRI (cuts 3' of promoter) and XbaI and used to replace the EcoWXbaI fragment of pBluescript KS 431. The producb were sequenced using the universal primer 5' of the promoter and oligo 9 (AAC GGT GTA ACA AGG GTG AA) 3' of the EcoRI site. The different promoter/cat gene fusions were isolated as XbuPSaEI fragments and cloned into XbuIISatI- digested pBluescript KS Cos Hyg. pCAT433AInd, pCAT433Apal1, pCAT433Apall/2, and pCAT433Apa12 are plasmids containing different mutated regions in the cutinase promoter, generated by recombinant PCR techniques following the protocol of Higuchi (18). For all reactions,
oligo 1 and oligo 2 (TTCATATGGCAATGCGCAG) in the cat gene and in the Cochliobolus-promoter, respectively, were used as outside primers. As mutagenesis primers, the following oligonucleotide pairs were used: for pCAT433AInd: oligo 12: GAA GCG GAA GAG CTT GCA TCT AGA TAT CTA TGG TGG 'RT CCT GAA GCG and oligo 13: CGC TTC AGG AAA CCA CCA TAG ATA TCT AGA TGC AAG CTC TTC CGC TTC; for pCAT433ApallJ2: oligo 14: AGA AAC CGA GGA AAT GGC ATA GCC TCC GAG GCT CGA ""I' CAG and oligo 15: CTG AAA TCG AGC CTC GGA GGC TAT GCC ATT TCC TCG GTT TCT for pCAT433Apa12: oligo 16: AAT GGA TCG CGA GCC GAT ATG GCA 'RT CAG AGC TTG GAC G and oligo 17: CGT CCAAGC TCT GAAATG CCATAT CGG CTC GCG ATC CAT T; for pCAT433Apall: oligo 18: CTA GAAACC GAG GAAATA TGA GCC GAG CCG AGG CTC GAT T and oligo 19: AAT CGA GCC TCG GCT CGG CTC ATA T l T CCT CGG ITT CTA G. For the primary PCR, reaction conditions were 100 p~ dNTP, 50 pmol of each primer, 100 ng of pBluescript KS.431, 10% dimethyl sulfoxide, 5% glycerol, 1 x Pfu DNA polymerase buffer 1 (Stratagene), and 2.5 units of Pfu DNA polymerase (Stratagene) in a 100-pl volume. After an initial denaturing step (5 min at 94 "C), 30 cycles were performed (1 min at 94 "C, 1 min
at 74 "C). at 55 "C, 2 min at 74 "C), followed by a final extension reaction (5 min
Half of the primary PCR products were used as templates for the secondary PCR, using the same buffer conditions as before, with the following cycle conditions: denaturing 5 min at 94 "C, 5 cycles 1 min at 94 "C, 1.5 min at 50 "C, 2 min at 74 "C, and 25 cycles with an increased annealing temperature of 55 "C, followed by a final extension step 5 min at 74 "C. The products were digested with ClaYBamHI and used to replace the respective restriction fragment in pBluescript KS.431. The sequence of the PCR-generated fragments was confirmed using the universal primer and oligo 9 as primers. Fragments with the different promoterlcat gene fusions were excised either as XbaVSalI fragments (pCAT433ApaW2, pCAT433Apa12, pCAT433Apall) or as SmaYXhoI- fragments (pCAT433AInd) and cloned into, respectively, digested pBluescript KS COC Hyg. The entire promoter of pBluescript KS.431 Exp was removed by digestion with HindIII and XbaI, blunted with T4 DNA polymerase, and subsequently religated to form pCATAProm. Plasmids pCAT181Rep+ and pCAT181Rep- were created by cloning the XbaI-digested PCR product of oligo 29 (GCT CTA GAC TAG GTT GCC TGAAAC TAT C ) and oligo 30 (GCT CTA GAC TTG ATA CTG GAC CAT GCG C), spanning the region from -237 to -295 of the promoter, in both orientations into theXba1 site 5' of the promoter of pCAT181. The PCR reaction was performed using pBluescript KS.431 as template. The final construct was sequenced using oligo 24 (GGAATT CTT GAA CAG ATG AAA CTA) as primer. Plus and minus signs in the plasmid names indicate the orientation of the insert respective to its original orienta- tion. Plasmids pCAT287Ind+ and pCAT287Ind- were constructed by cloning the complementary oligonucleotides oligo 27 (CTA GAA CCA CCC CGT GCC CCT CCC GCA AGC TCT TT) and oligo 28 (CTA GAA AGA GCT TGC GGG GAG GGG CAC GGG GTG GTT) in both orienta- tions into the XbaI site 5' of the promoter fragment of pCAT287.
Plasmid pUC360 is a pUC19 derivative harboring a BgZIIIPuuI cuti- nase gene promoter fragment, spanning the region from +34 to -360. This plasmid was used as a source to generate different promoter frag- ments suitable as probes for gel retardation assays; different restriction fragments were blunted with T4 DNA polymerase and subcloned into the SmaI site of pUC18. The following plasmids carry the given pro- moter fragments (numbers in plasmid names indicate the first and last base pairs of the inserted fragment respective to the ATG-start codon):
-122: ~ ~ n d I I I I ~ t 1 ~ pUC -158/+34 TaqI; pUC -116/+34: EcoW HindIII. To form pUC -3101-230, t h e ~ u ~ A a t 1 1 fragment of pCAT310, containing a promoter fragment from nucleotides -310 to -230, was cloned into SmaI-digested pUCl8. Plasmid pBSM is a pBluescript KS
AAA TGG ATC GCG AGC CGA GGC TCG ATT TCA G) and oligo 21 derivative with the complemen~ry oligonucleotides oligo 20 (AAT TCG
(AAT TCT GAA ATC GAG CCT CGG CTC GCG ATC CAT TTC G) inserted in the EcoRI site of the polylinker.
Probes for Gel Re~ardat io~ Assays-Probes for gel retardation assays were labeled, unless otherwise mentioned, with four [ f f - 3 z P l ~ P s us- ing Klenow DNA polymerase after they were excised and purified by electropho~sis. For the generation of probes identical with the inserts of plasmids pUC -1161+34, pUC -158/+34, pUC -2251-122, pUC -2461 -161, pUC -3101-230, and pUC -3601-250, these plasmids were di- gested with EcoRI and BarnHI. Probe [-195/+341 is a PCR product of oligo 3 and the universal primer, with plasmid pUC360 as template. The PCR product was digested with EcoRI to yield a probe from -195 to +34. Probes [-225/-12OApall], [-225/120Apal~21, and ~-225/120Ap~21 were generated by PCR using oligo 23 (GGA AT7 CCT TTG ATG AAA
pUC -360/-250 BQ~~HIIAuuII; pUC -246,'-161: ~ ~ Y A u Q I I ; pUC -2251

Promoter Analysis of the Cutinase Gene from Fusarium 9197
ACT GCC) and oligo 24 (GGA ATT CTT GAA CAG ATG AAA CTA) as primers; both oligonucleotides have an internal EcoRI site allowing a labeling reaction with Klenow DNA polymerase after digestion of the PCR products with EcoRI. The PCR products contain the promoter re- gion from nucleotides -225 to -120. Using plasmids pCAT433ApalY2, pCAT433Apa12, and pCAT433Apall as templates allows the genera- tion of probes with the sequence modifications in the promoter frag- ment of the respective plasmid. Probe 1-1831-1511 was generated by digestion of plasmid pBSM with Xhol and Xbal. The probe contains the promoter sequence from nucleotides -183 to -151, bordered by the polylinker sequence of pBluescript KS. As nonspecific competitor for gel retardation assays, a 126-bp HinfI fragment of the coding region of the peZA gene from l? solani pisi (19) was used.
parat ti on of Nuclear Extracts-Mycelia were collected after growth until glucose depletion (1) from 1.2 liters of mineral medium containing 0.5% glucose, and protoplasts were prepared as described (16) except that STC buffer was replaced with ST buffer (1.2 M sorbitol, 10 mM Tris-HC1, pH 7.5) containing no CaCl,. The final protoplast pellet was resuspended in a minimum of 10 volumes of buffer A (25 rn HEPES, pH 7.9, 18% Rcoll, 0.5 mM CaC12, 1 r n ~ dithiothreitol (D'IT), I mM phenylmethylsulfonyl fluoride (PMSF), 10 mgJml leupeptin, 0.5% Triton X-100) by 3 to 4 strokes with a 15-mi Dounce homogenizer, pestle A. Nuclei, pelleted by centrifugation (16,000 x g at 4 "C for 10 min), were resuspended in 15 ml of buffer A and pelleted as before. Nuclei were checked microscopically by staining with mithramycin (20). Buffer B (25 m~ HEPES, pH 7.9, 20% glycerol, 0.1 mM EDTA, 420 m KCl, 1.5 mM MgC12, 1 mM DTT, 1 m~ PMSF, 10 mdml leupeptin) equivalent to half the packed volume of nuclei was added and the nuclei were gently resuspended with a glass ball a t the end o f a glass rod. Buffer B was then added to a final concentration of 330 m KCl. After 30 min on ice, nuclei were pelleted by centrifugation (16,000 x g at 4 "C for 15 min). Appropriate components were added to obtain the following composi- tion to the supernatant (buffer C): 20 mM HEPES, pH 7.9,l m DTT, 5 mM MgCl,, 20% glycerol, 1 m EDTA, 1 mM PMSF, 100 m KCl, 10 mdml leupeptin), and applied to a heparin-Sepharose column (bed vol- ume 5 ml) equilibrated with the same buffer. After washing, a linear gradient of 0.1 to 1 M KC1 in buffer C was applied. Fractions were pooled and concentrated 10-fold with a Centricon 30 filtration unit (Amicon), and the buffer was exchanged to storage buffer (100 mM KCl, 20 n m HEPES, pH 7.9, 20% glycerol, 1 rn EDTA, 1 nm PMSF, 1 m DTT). Aliquots were frozen in liquid nitrogen and stored at -80 "C.
Gel Retardation with Nuclear Extracts"Ge1 retardation assays were performed in 20-9 volumes with 25 rn HEPES, pH 7.9, 12% glycerol, 1 mrw EDTA, 1 mM PMSF, 1 rn EDTA, 80 m KCl, 0.3 mg/ml acetylated bovine serum albumin, 2 pg of dI-dC, and 1 to 4 PI of nuclear extracts; specifi~nonspecific competitors and 1.25 fmol of r2P-labeled probe were added last. After a 15-min incubation at 24 "C, samples were loaded on 4% polyacrylamide gels (32:l acry1ami~e:bisacrylamide in 0.25 x TEE) and run at 4 "C at 200 V for 2 to 4 h. Gels were dried on atm man No. 3MM paper and exposed to x-ray film for autoradio~aphy.
Preparation of Fungal DNA-About 2 g of mycelium grown in liquid culture was harvested by filtration and ground in liquid nitrogen using mortar and pestle, resuspended in 4 ml of lysis buffer (2% SDS, 62.5 m EDTA, 50 nm Tris, pH 8.2), and incubated for 1 h at 65 "C. After addition of 1.4 ml of 8 M potassium acetate, the mixture was incubated on ice for 30 min and centrifuged (9,000 x g, 10 min at 4 'e). To the supernatant, 1.4 ml of 8 M ammonium acetate and 4 ml of isopropyl alcohol were added, and, after 10 min at room temperature, the mixture was centrifuged as before. The pellet was dissolved in 600 pl of TE buffer, 100 p1 of RNase A (10 mg/ml) was added and incubated for 30 min at 37 "C, and 67 pl of 10 x proteinase K buffer (14) and 6 pl of proteinase K (20 mg/ml) were added and incubated for 30 min at 65 "C. After several extractions with phenoVch2orofon-n (l:l), DNA was pre- cipitated with ethanol. The pellet was dissolved in 200 pl of TE buffer.
Fungal Dansforrnation-Transformation of F: solani pisi was done using protoplasts as described 117). Transformants were identified by their ability to grow on V8 medium plates with 300 mglliter hygromycin and further analysis by Southern hybridization of genomic DNA.
CAT Assays of I;: solani pisi Ransforman~s-Transformants were grown in 300 ml of minimal medium with 1.5% glucose for 36 h at 24 "C at 200 rpm on a rotary shaker. Protoptasts were prepared as described before (171; instead of STC, ST buffer lacking CaCI, was used. Proto- plasts (5 x lo*) were incubated in 5 mi of minimal medium with 1.2 M sorbitol and 10 IIUI Tris/HCl, pH 7.5, with gentle shaking at 24 "C. To test for repression, 2% glucose was added to the medium; after incuba- tion for 2 h, cutin hydrolysate (1) was added to a final concentration of 80 pglml. For induction, the cutin hydrolysate was added after a pre- incubation of 2 h without the addition of glucose. After a further 12-h
incubation, protoplasts were pelleted by centrifugation (600 x g, 7 min, 4 "C), resuspended in 500 pl of TridHC1, pH 7.8, and frozen in liquid nitrogen. After thawing on ice, the lysate was centrifuged (10 min, 16,000 x g, 4 "C), and the supernatant was incubated for 10 min at 65 "C and centrifuged as before. Protein concentration of the superna- tant was determined using the Bio-Rad protein assay (21) using bovine serum albumin as standard. CAT assays were done with d-threo-Idi- ch~or~ce~yl-l,2-14Clchloramphenico~ as substrate, and the products were analyzed by thin layer chromatography (22) and a PhosphorIm- ager {Molecular Dynamics). One relative unit of CAT activity equals a conversion of 1% chloramphenicol by 50 pg of protein.
~ e ~ h y l a t ~ o n Interference Assay-For methylation interference as- says, the gel retardation reaction was scaled up 5- to 10-fold. All fol- lowing steps were according to the "Basic Protocol for Methylation In- terference Assays" described in Ausubel et al. 123). For detection of CTFl and CTF2, probes [-225/-1221 and 1-3101-2301 were used, re- spectively, labeled either with Ia-"2PldATP at the EcoRI site or with [a-32P]dGTP and [a-32PldCTP at the BamHI site, allowing the analysis of methylation interference with both strands.
W-Cross-linking Experiments-A double-stranded DNA fragment was generated by extending oligo 33 (CAA GCT CTG A) with Klenow DNA polymerase and oligo 32 (GGA AAT GGA ATC GCG AGC CGA GGC ACG ATT TCA GAG CTT G ) as template using 5'-bromodeoxyuri- dine instead of WP, dATP, [a-32PldGTP and [a-32PldCTF'. W cross- linking experiments were done according to Ausubel et al. (23). In brief, gel retardation reactions were scaled up 5-fold, W-treated for 30 min at 4 "C under an inverted transilluminator (312 nm), and 10 mM CaCl,, 23 units of DNase I, and 2.5 units of micrococcal nuclease were added. After incubation for 30 min at 37 "C, the samples were denatured by boiling and electrophoresed through a 10% SDS-polyacrylamide gel. The gel was dried on Whatman No. 3MM paper and exposed for up to 5 days.
Determination of the Dunscription Initiation Sites-Nuclease S1 analysis and primer extension experiments were performed according to Sambrook (14). In both experiments, oligo 38 (GTG AGA GCG AAG AAT TTC ATG GTG AAG ATG) from +20 t o -10 of the cutinase gene was used as primer. For nuclease S1 analysis, pUC360 was used as template to generate the single-stranded probe complementary to the cutinase mRNA. After hybridization of 50 pg of total RNA from cutin monomer- induced l? solani pisi or from Saccharomyces cereuisiae with the respec- tive radioactive labeled probes in the hybridization buffer used for nuclease S1 analysis a t 30 "C overnight, the samples were treated with nuclease S1 and avian myeloblastosis virus reverse transcriptase, re- spectively, phenol-extracted, precipitated, and analyzed on a 5% dena- turing polyacrylamide gel.
RESULTS Identi~cation of Promoter Elements I n v o ~ ~ e d in the Regula-
tion of 7kmscription of the Cutinase Gene-In order to study the elements involved in regulation of the cutinase gene, dif- ferent deletions of the cutinase promoter were fused to the gene for chloramphenicol acetyltransferase (cat) as a marker gene, allowing the quantitative determination of the promoter strength in vivo. The plasmid also contained the gene for hy- gromycin phosphotransferase (hph) linked to a constitutive promoter from C. heterostrophus. R solani pisi transformants were selected by their ability to grow on medium containing 300 mgfliter hygromycin and analyzed by Southern analysis of genomic DNA. In a11 cases, the vector was found to be inte- grated into the FI solani pisi genome as tandem repeats; for further analysis, transformants with a comparable copy num- ber of the vector were chosen (data not shown). For reasons that are not understood, we could not find any CAT activity in protein extracts prepared from intact mycelia of transformants. However, protoplasts from stable transformants could be readily analyzed for regulated CAT expression. Protoplasts were incubated with cutin hydrolysate to measure induction and with cutin hydrolysate and glucose to detect repression. For each construct, usually six independent t r a n s f o ~ ~ t s were used. Protein extracts were prepared by lysis of the pro- toplasts and assayed for CAT activity. Fig. VI shows the differ- ent constructs used for this analysis. Plasmid pCAT433 con- tains a 433-bp promoter fragment that was previously shown to

9198 Promoter Analysis of the Cutinase Gene from Fusarium
B
8" t i i i f i i FIG. 1. Deletion analysis of the cutinase promoter in uiuo. Anal-
ysis of promoter region from -433 to -141 is shown in A and from -209 to -141 in B. R solani pisi was transformed with different promoter-cat gene fusions; the number designations on plasmids refer to the pro- moter length respective to the start codon of the cutinase gene. Proto- plasts from stable transformants were incubated either without any addition (noninduced; filled bars), with the addition of cutin hydroly- sate (induced; open bars) or with the addition of cutin hydrolysate and glucose (repressed; stippled bars). The mean values (and deviations) from CAT assays performed with extracts usually from six independent transformants are shown; the deviations found are normal for fungal transformants.
confer induction and repression (16). In a first set of experi- ments, we used different 5' deletions of the promoter fragment of plasmid pCAT433 generated by digestion with exonuclease Ba131. This included plasmids pCAT310, pCAT287, pCAT249, pCAT209, and pCAT141. The data from the CAT assays of these transformants, together with those of plasmids pCAT433, pCAT360, and pCAT225 (16) are summarized in Fig. lA.
Promoter fragments 209 bp and longer conferred inducibility by cutin hydrolysate and glucose repression, although quanti- tative expression vaned between the different fragments. CAT activity after induction was found to be in the same range for transformants harboring pCAT433 and pCAT360. The activity, however, decreased drastically from 120-130 units to values less than 1 unit when transformants with a promoter shorter than 360 bp were assayed; it increased again to values up to 111 units for promoter fragments between 249 bp and 209 bp. Re- pression of CAT activity by glucose was found for all the con- structs. Transformants with a 141-bp promoter fragment showed neither inducibility nor repression, but basal activity was still detectable. Interestingly, the promoter length also influenced constitutive CAT activity; while transformants har- boring pCAT433, pCAT360, pCAT310, and pCAT287 showed
hardly any detectable constitutive CAT expression, transfor- mants harboring constructs with shorter promoter fragments, including the noninducible pCAT141, had a significant basal CAT activity of up to 5.3 units of conversion. Transformants with a promoterless cut gene had, as expected, no measurable CAT activity. These experimental results showed that inducible CAT expression was conferred by a 209-bp promoter fragment, while a 141-bp fragment did not allow regulated gene expres- sion; the elements responsible for induction by cutin hydroly- sate therefore have to be located between -209 and -141.
The results also indicate the existence of two elements influ- encing quantitative expression. A negative-acting element be- tween positions -287 and -249 keeps basal transcription low but also reduces inducibility. An activator between positions -360 and -310 functions as an antagonist to the negative ele- ment and thus restores inducibility, without affecting the level of constitutive gene expression. In a previous study (16), trans- formants harboring a 225-bp promoter fragment linked to the CAT gene did not show significant inducibility, possibly be- cause the transformants harboring different deletion con- structs used in that study gave only very low CAT activity in the range of 2% conversion.
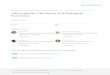
Identification of a Positive Promoter Element Essential for High Levels of Induction-The sequence from -360 to -310 of the cutinase promoter, found to contain the positive-acting pro- moter element, includes a G-rich region (Fig. 2A) . Bajar et al. (16) describe the similarity of this region (position -316 to -325) to an Spl transcription factor-binding element, known to function as an enhancer in mammalian systems. More striking is the similarity to promoter sequences in Aspergillus, found to act as a positive element in the A. niger gpdA gene in vivo (24) (Fig. 2 B ) . In order to determine if the G-rich sequence is the positive-acting promoter element in Fusarium, it was dis- rupted by PCR-directed mutagenesis (Fig. 2.4). A 433-bp pro- moter fragment carrying this mutation (pCAT433AInd) gave very little induction, the same as promoter fragments (pCAT287 and pCAT310) lacking this region (Fig. 2C). Thus, the G-rich element is indeed essential for the high level of cutinase gene expression in li: solani pisi.
The homology of this element to the Spl binding site might indicate that it also acts as an enhancer. If this is the case, it should function independently of its position and orientation. To test this hypothesis, we integrated a 31-bp fragment (position -305 to -335) with the entire G-rich region in both orientations 5' of the promoter fragment of plasmid pCAT287, leading to plasmids pCAT287Ind+ and pCAT287Ind-. Transformants har- boring plasmid pCAT287 show almost no inducibility; if the G- rich element acts as an enhancer, an increased CAT expression should easily be detectable. As shown in Fig. 2C, the G-rich element did not increase CAT inducibility. CAT activities of pCAT287Ind+ and pCAT287Ind- transformants were in the same range as for pCAT287 (Fig. 2C, inset). Thus, unlike Spl , the G-rich element does not act as a true enhancer.
Further evidence for the role of the G-rich sequence as a functional promoter element of the cutinase gene came from the analysis of its capability to bind nuclear proteins in vitro. To perform mobility shift assays, nuclear protein extracts were prepared from nuclei isolated from protoplasts; proteins with affinity to DNA were then enriched on a heparin-Sepharose column. Using a 31-bp DNA fragment (position -305 to -335) harboring the entire G-rich sequence as a probe, several re- tarded complexes were formed (Fig. 3). The specificity of the retarded complexes was determined by competition with a 500- fold molar excess of an unlabeled DNA fragment also harboring the G-rich sequence (fragment [-360/-2471). The two com- plexes with the slowest migration were reduced drastically. In contrast, the addition of a 500-fold molar excess from a non-
Promoter Analysis of the Cutinase Gene from Fusarium
A
. . C G G C A C G G G T T G C A A C T A G A A K G ~ ~ + ~ T C C T ~ . I
F. solmi pisi CUIA
A . niner ~pd.4
A . nidulam fpdA
A. nidulamp&4
GGAGGGGCACGGGG'E -3nx ............ GGAGGGGCACTGGACT -593
TGAGGTGCAGTGGAX -750/700
TGAGGTGTAATGCA'E -499
...........
.........
K A T 4 3 1 pCAT433Alnd pCAT2R7Ind+ pCARR7lnd. pCAT287
FIG. 2. Functional analysis of a G-rich promoter element from cutinase gene. A, sequence of the cutinase promoter from -300 to -360. The nucleotides given in hold were mutated in plasmid pCAT433hInd. The arrow indicates the fragment cloned 5' of the pro- moter fragment in plasmid pCAT287 to form plasmids pCAT287Ind+ (wild type orientation) and pCAT287Ind- (reverse orientation), B, se- quence comparison between the G-rich sequence in the cutinase pro- moter and different promoter elements of A. niger and A. nidulans. Identity with cut A sequence is indicated by asterisks placed on top of each base. The numbers indicate the position of the 3' nucleotide of the given sequence respective to the ATG codon. C, CAT activities of trans- formants with pCAT433. compared to pCAT433AInd with a mutated
formants of plasmids pCAT287Ind+, pCAT287Ind- (see A above). and G-rich element (region in hold in part A ) and CAT activities of trans-
pCAT287. CAT assays were performed as described in Fig. 1. In pCAT287 derivatives, CAT activity was barely detectable under any conditions. as shown in the insrt with rescaled y axis.
specific competitor (a 126-bp fragment of the coding region of the pectate lyase gene (pelA) from F: solani pisi (19)) had no influence on complex formation. Thus, the upper two bands were the result of specific DNA-protein interaction, indicating the capability of the G-rich sequence to bind trans-acting pro- tein factors.
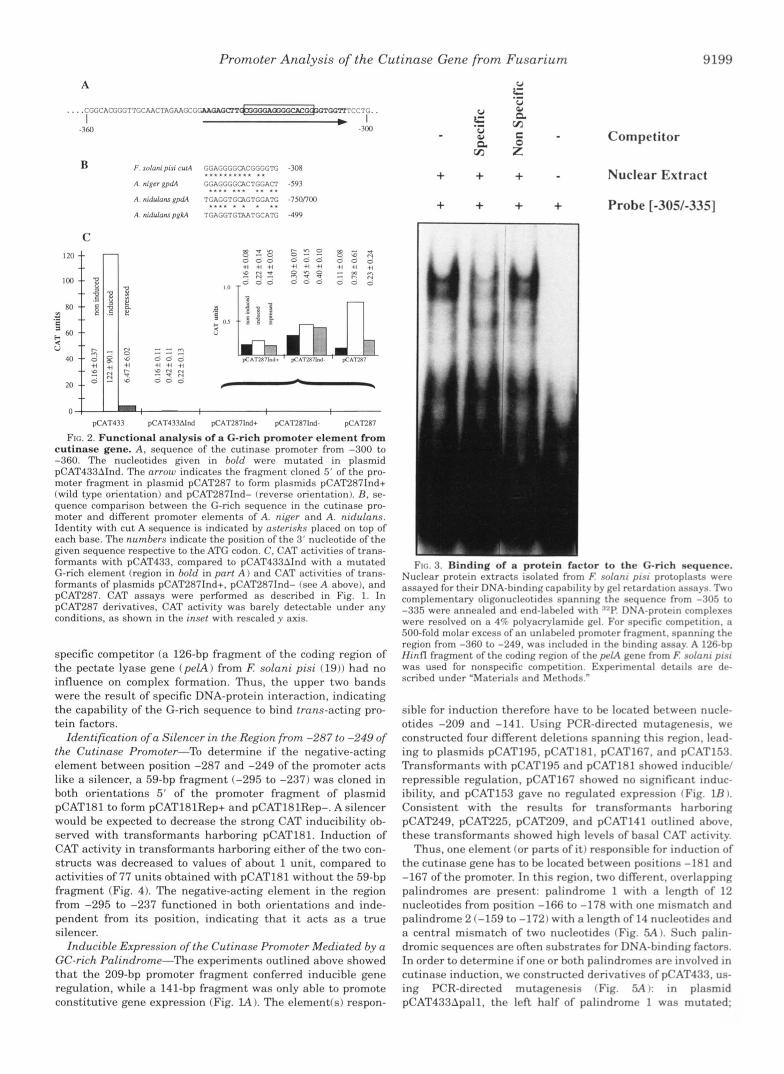
Identification of a Silencer in the Region from -287 to -249 of the Cutinase Promoter-To determine if the negative-acting element between position -287 and -249 of the promoter acts like a silencer, a 59-bp fragment (-295 to -237) was cloned in both orientations 5' of the promoter fragment of plasmid pCAT181 to form pCAT181Rep+ and pCAT181Rep-. A silencer would be expected to decrease the strong CAT inducibility ob- served with transformants harboring pCAT181. Induction of CAT activity in transformants harboring either of the two con- structs was decreased to values of about 1 unit, compared to activities of 77 units obtained with pCAT181 without the 59-bp fragment (Fig. 4). The negative-acting element in the region from -295 to -237 functioned in both orientations and inde- pendent from its position, indicating that it acts as a t rue silencer.
Inducible Expression of the Cutinase Promoter Mediated by a GC-rich Palindrome-The experiments outlined above showed tha t t he 209-bp promoter fragment conferred inducible gene regulation, while a 141-bp fragment was only able to promote constitutive gene expression (Fig. lA). The elementfs) respon-
5 V
w V
v1 a
+ + + + + + +
9 199
Competitor
Nuclear Extract
Probe [-30Y-3351
FIC, :1 l%incling o f a p r o t r i n f;tctor to the G-rich Requence. Nuclrnr p r ~ ~ i r , ~ n <.utr:lct-: I . o l ; ~ t t d frllrn E solanr pist protoplasts wrrr assayed for thmr I)NA-tnnd~np c:lpahl~ty by gel retardation assays. Two complementap ol~~onuclrot~drs spanning the sequence from -905 to -335 were annealrd and end-labelrd with Y? DNA-protrin complrxes were resolved on a 4? polyacrylamide gel. For sprc~fic comprtitwn. a 500-fold molar excess of an unlabeled promoter frapent. spnnning the region from -360 to -249. was included in the bindlng assay. A 126-hp HInfI fragment of the coding region of the prlA g e m from I: solanr P I S I
scribed under "Materials and Mrthoda." was used for nonspecific competition. Exprrimrntnl details arr dr-
sible for induction therefore have to be located between nucle- otides -209 and -141. Using PCR-directed mutagenesis. we constructed four different deletions spanning this region, lead- ing to plasmids pCAT195, pCAT181, pCATl67, and pCAT15.7. Transformants with pCAT19.5 and pCATl8l showed inducible/ repressible regulation. pCAT167 showed no significant induc- ibility, and pCAT153 gave no regulated expression fFig. 1B 1.
Consistent with the results for transformants harboring pCAT249, pCAT225, pCAT209, and pCAT141 outlined above, these transformants showed high levels of basal CAT activity
Thus, one element for parts of it) responsible for induction of the cutinase gene has to be located between positions -181 and -167 of the promoter. In this region, two different, overlapping palindromes are present: palindrome 1 with a length of 12 nucleotides from position -166 to -178 with one mismatch and palindrome 2 (-159 to -172) with a length of 14 nucleotides and a central mismatch of two nucleotides (Fig. FiA ). Such palin- dromic sequences are often substrates for DNA-binding factors. In order to determine ifone or both palindromes are involved in cutinase induction, we constructed derivatives of pCAT4.73, us- ing PCR-directed mutagenesis (Fig. SA): in plasmid pCAT433Apal1, the left half of palindrome 1 was mutated;
9200 Promoter Analysis of the Cutinase Gene from Fusarium
I pCATIhl pCATIRIRcp+ pCATIRIRcp
FIG. 4. Effect of a n e g a t i v e promoter e l e m e n t f r o m E nolani pini c u t i n a s e gene on CAT ac t iv i ty o f an inducih le CAT gene construct i n F. nolani pini t r ans fo rman t s . A 59-hp promoter frag- ment 1-237 to -2951 was cloned in hoth orirntations 5' o f the 181-hp promoter fragment of plasmid pCAT181. Transformants with the rr- sulting plasmids pCATIRlRrp+ (same orientation as in the wild type promoter) and pCATlRI- (inverse orientation) and control plasmid pCAT181 were assayed for CAT activity as dcscrihrd under "Materials and Methods."
A
R
.! * I
I- V
IM
888 0 06
0 00
'I +I +I 00-
pCAT433 pCATJ.\.L pCAT433- pCAT433- Apoll Apolli2 ApalZ
FIG. 5. I d e n t i t y o f a GC-r i ch pa l ind rome that med ia t e s i nduc - ible expres s ion o f the cu t inase p romote r . A, sequence of the cuti- nase promotcv from position -137 to -185. respective to the ATC s ta r t codon. The 5' ends of the insets used to construct the plasmids utilized to test the functional role of this region are indicated. The two different, overlapping palindromic sequences are highltghfrd by different boxes; nucleotides matching the dyad symmetries o f the palindromes are given in hold. Plasmids pCAT433Apall. pCAT433Apa11/2, and pCAT433Apa12 carry the 433-hp promoter fragment with the mutations indicated. B . constitutive, induced, and glucose-repressed CAT activity of transfor- mants with plasmids pCAT43.1, pCAT433Apall. pCAT433Apal1/2. and pCAT433Apn12. CAT assays were done as described under "Materials and Methods." Constitutive expression was hardly detectable; oprn hnrs show CAT activity induced by cutin hydrolysate. and sfipplrd hnrs show glucose-repressed activities.
plasmid pCAT433Apal1/2 carried a mutation in the overlap- ping part of both palindromes, and in plasmid pCAT433Apa12, the right half of palindrome 2 was mutated. Fig. 5B shows the
- + - + - +
4
4
V I , . 1; ( k l shift expc.rirnc*nt\ shc)wing specific hinding of a n u c l e a r p r o t e i n to p;llindromc. 2 in cu t inase p romoter . Thrr r diffrrrnt pron1otc.r fraj:ml.n1i spanninL: th<. r c * ~ ~ o n from nurlrotidrs -22 . ) c ' r to -120 with the same mut:ltions as In plasmids p('AT433Apall. p(XTT433Apa1112. and p('AT4XUpaI2 wcrr rnd-lahvled with '.'I' and used as prohes in gel shift assays undrr thr samr rnndrtinns as in Fig. 3. Promotrr fragment [-225/-1381 with thr wild type srqurnrr was used as a control. Exprrimrntal drtails arr drscrihrd undrr "Slatcrials and Methods."
results of CAT assays performed with transformants harhoring these constructs. The mutation of the left half of palindrome 1 increased inducibility, while mutation of the common part of both palindromes reduced inducihilitv drasticallv; with this extremely low level of expression, repression could not be de- tected. Mutation of the right part of palindrome 2 ahnlished any detectahle CAT activity. Thus, palindrome 2 is responsible for regulation of cutinase gene induction hy cutin monomers.
Specific Binding of Nuclear Protrin/s/ in Vitro to the GC-rich Palindrome Involwd in Cutinase Gene Induction-In order to analyze hinding of a trans-acting protein factor to palindrome 2 in vitro, we performed mobility shift assays, using nuclear ex- tracts prepared from isolated nuclei (Fig. 6). Prohe [-225/-1201 which contains the GC-rich palindrome 2 was specifically bound by nuclear proteins. To demonstrate that palindrome 2 was indeed the binding site of these nuclear proteins, deriva- tives of probe 1-225/-1181 containing the same mutations as the promoter-cat fusions analyzed in vivo indicated a h v r were used as probes in gel retardation assays. Compared to probe 1-225/-118], binding of nuclear proteins to prnhc 1-22.V -120Apa11/2] was diminished, to prohe [-225/-120.lpa121 hind- ing was drastically decreased, and to probe [-225/-120~palll binding was even stronger than to the unmodified probe 1-2251 -1181 (Fig. 6). These data show that a nuclear factor specifically interacts with palindrome 2 that was found to mediate regu- lated expression of the cutinase gene in v i m The factor was named CTFl for Cutinase Transcription Factor 1. The different mutations of probe 1-225/-1201 did not onlv affect the intensity of the retarded complexes but, in addition, resulted in the for-

Promoter Analysis of the Cutinase Gene from Fusarium
A 0, Y
FIG. 7. Identification of the cutinase promoter region that binds CTF1. A, methylation interference assay. In the promoter fragment 1-225/-1181. either the lower strand or the upper strand was end-labeled with 92P, methylated, and subjected to gel shift assays. After piper- idine cleavage, the free and retarded frag- ments were analyzed on 8% denaturing polyacrylamide gels. The sequences of the upper and lower strand are given; the methylated G-residues interfering with binding are indicated by asterisks. In R . the sequence of the cutinase promoter from nucleotide -148 to -184 is given; pal- indrome 2. the CTFl binding site, is high- lighted by a box. The G-residues identified by the methylation interference assay are indicated by astrrisks.
B
Upper Strand Lower Strand
Lower Strand
mation of complexes which differed in migration behavior. With the wild type sequence or [-225/-120Apall] as a probe, a double band was detected (Fig. 6). In contrast, binding to [-225/ -120Apa11/2] or [-225/-120Apa12] resulted in the formation of one band only, which migrated between the two bands obtained by using fragments with intact palindrome 2. To determine more precisely the interaction of CTFl with its
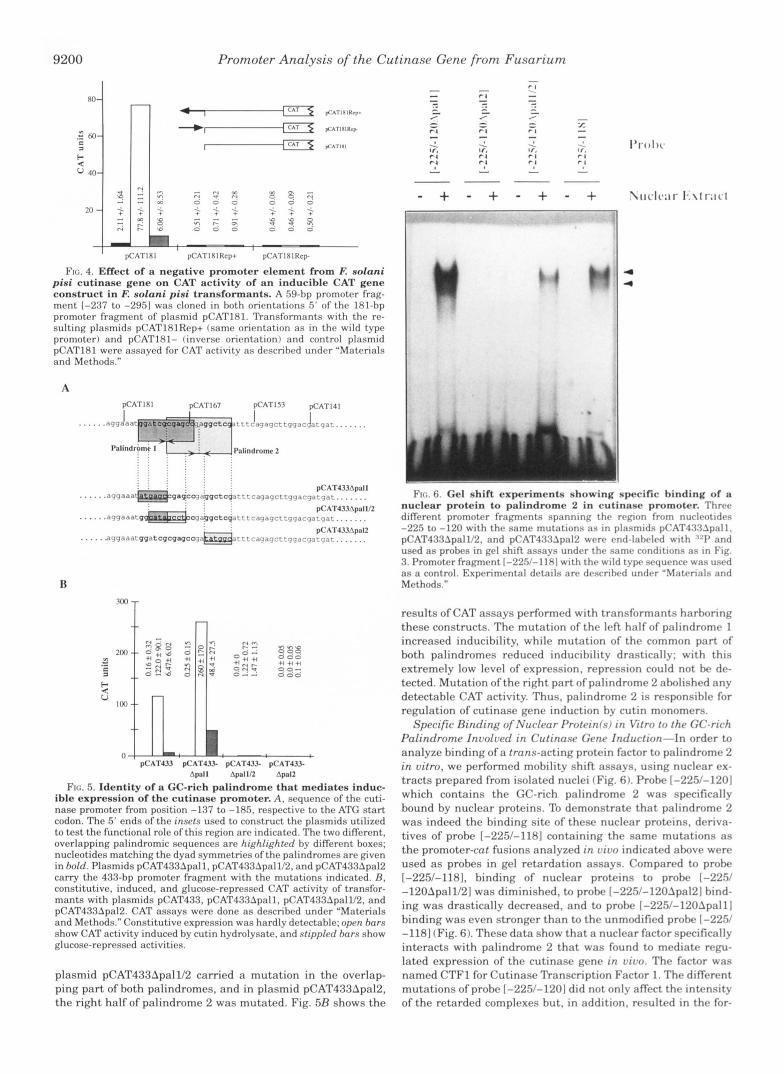
binding site, methylation interference assays were performed (Fig. 7). As expected, binding of CTFl was eliminated by methylation of guanine residues present in palindrome 2. Resi- dues involved in binding were symmetrically distributed in both half-sites of the CTFl binding site (positions -171, -169, -164, and -163 of the upper and -168, -167, -162, and -160 of the lower strand). Methylation of the outermost guanine resi- dues of the palindrome did not affect binding (positions -159 of the upper and -172 of the lower strand). In addition, methyla- tion of the G-residue at position -166 of the upper strand in the central mismatch between the palindromic half-sites interfered with binding of CTF1.
To further characterize CTF1, UV-cross-linking experiments were performed. A 5-bromodeoxyuridine-containing probe with a modified CTFl binding site was used for selective labeling of polypeptides by photochemical cross-linking. The wild type CFTl binding site contains only 2 thymidine residues on one strand. To increase the probability of cross-linking, we used a modified binding site with a n A - > T transversion, which al- lowed the incorporation of an additional 5-bromodeoxyuridine per molecule. Gel retardation experiments confirmed that this probe was bound with a similar afinity as the wild type bind- ing site (data not shown). DNA-protein complexes obtained with the internally :'2P-labeled modified probe were subjected to UV-irradiation and, after DNase treatment, were resolved by SDS-polyacrylamide gel electrophoresis (Fig. 8). After photo- chemical cross-linking, a major band of approximately 49 kDa,
920 1
1,'ppc.r Strand
5 ' G G A
T G A A
C G C
G
5 I GGAAATGGATC+-G~=~ATTTCAGAGCT * * * *
3'CCl"l'TACCTAGC@CTC~C~CCGA~TMGTCTCGA * * * *
three minor bands of approximately 63, 73, and 91 kDa, and a smear at 33 kDa were selectively labeled hy the attached oli- gomers. The relative intensities of the signal at 49 kDa and of the signals of higher molecular mass vaned between different experiments, but all signals resulted from sequence-specific binding to the CTFl binding site as shown hy competition ex- periments with specific and nonspecific competitors (Fig. 8). The signal at 33 kDa probably represents degradation products of the 49-kDa band, as we never detected a concrete hand in the variable labeling observed in this region.
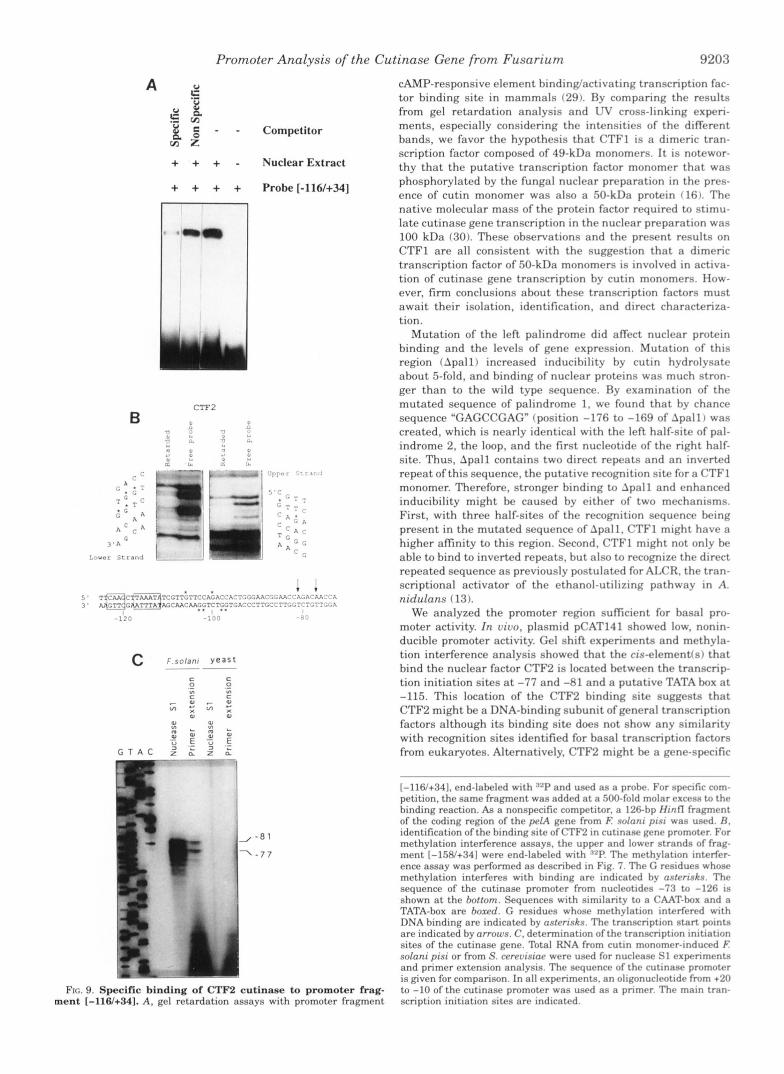
Specific Binding of a Protein (CTF2) to a Sitr Near tha 7 h n - scription Start Site ofthe Cutinasa Gena-A second DNA-hind- ing protein, CTF2, was identified by gel shift assays. which showed specific binding to promoter fragment 1-116/+34] (Fig. 9A ). The binding site of CTF2 was determined by methylation interference experiments (Fig. 9R ). The guanine residues that are in close contact to this DNA-binding protein are located a t positions -97, -98, -102, and -103 of the lower strand and positions -100 and -106 of the upper strand.
To elucidate the relative position of this DNA-binding factor with respect to the transcription initiation site of the cutinase gene, nuclease S1 experiments and primer extension analysis were done to identify the transcription initiation site. Both types of experiments showed that nucleotides -77 and -81 upstream from the ATG-codon are used as the major transcrip- tion initiation sites (Fig. 9C). The existence of several start sites is commonly found in fungal genes (25,. The length of the 5'-nontranslated region, which is less than 100 nucleotides in the majority of fungal genes, fits well to puhlished data (25). Approximately 40 nucleotides upstream from the transcription initiation sites, a TATA-like sequence can be ohserved f Fig. 9B 1. The significance of this box is not clear as fungal promoters without any homology to TATA boxes have been identified (2%. There is no sequence in the cutinase promoter fitting the con-

9202 Promoter Analysis of the Cutinase Gene from Fusarium
kDa
200
97
68
43
29
18
+ + + + + + + +
Competitor
Nuclear extracts
uv
FIG. 8. Identification o f po1ypeptidc.s hinding to the CTFl rec- ognition scquencc. hy UV cross-linking. An internally "P-labeled, double-strantld olignnuclcotitlr with a modified CTFl binding site which contained 5-hromodeoxyuridine was used in binding reactions. After UV-irradiation and IINase treatment. the samples were subjected to SDS-polyacrylamide gel electrophoresis with 10Q polyacrylamide gel. As competitors (500-fold molar excess). promoter fragment 1-2251 -1221 was used for specific and a 126-bp H i n n fragment of the coding region o f the pelA gene from E solnni pisi was used for nonspecific competition. The position of the molecular mass markers (high molecu- lar mass markers, Life Technologies, Inc.) are indicated.
sensus for a CAAT box. At -240 and -125 (approximately 160 and 45 bp upstream from the transcription start sites, respec- tively) "CAAG" sequences can be found. Although one of these sequences might be used as CAAT box, fungal promoters with no homology to a CAAT sequence have been reported (25). CTF2 hinds between the transcription initiation sites and the putative TATA box. I ts recognition sequence does not match any known binding site of other transcription factors.
DISCUSSION Analysis of a series of cutinase promoter-cat gene transcrip-
tional fusions in transgenic E solani pisi revealed four different promoter regions involved in cutinase gene regulation. The positive-acting element that lies between -310 and -360 con- tained an element that showed similarity to the Spl binding site, an enhancer in several viral and mammalian promoters (26), and striking homology to the G-rich elements from pro- moters of the A . nidulans pgkA gene and from the A. nidulans and A. niger gpdA genes, encoding phosphoglycerate kinase and glyceraldehyde-3-phosphate dehydrogenase, respectively (24). Mutations in this sequence (pCAT433-AInd) caused a 300- fold decrease in promoter activity. Consistent with the data from promoter deletions, this mutation modified only the quan- titative level of promoter activity while inducibility and catabo- lite repression remained unaffected. Contrary to the mamma- lian G-rich enhancer, the Spl binding site, the G-rich activator from E solani pisi did not function as a true enhancer. The functional importance of the G-rich site was further supported by in vitro analysis showing sequence-specific protein-DNA in- teraction. The virtual abolition of promoter activity by muta-
tion in the G-rich element was somewhat surprising as deletion of the pgk box from the A. nidulans gpa!A promoter only re- duced promoter activity to 70% (5 ) . The pgk box in the gpdA promoter as well as the two similar boxes in the other Aspvr- gillus promoters are located at a much longer distance from the transcriptional start site than the G-rich box in thr cutinase gene (500 to 700 bp compared with 230 hp), and, thus, posi- tional effects might influence promoter activity exerted by the similar boxes. In addition, these differences might he due to the fact that the gpdA gene is a constitutively rxprrssed gene. whereas the cutinase gene is induced.
Secondly, a negative-acting element was identified hetween -287 and -249 of the cutinase gene. Deletion of this region restored high levels of inducibility and in addition caused an increase in basal promoter activity. Unlike the C-rich site, this negative element is a true silencer, a s its activity was found to be independent of distance and direction relativr to thr tran- scription start site. The present results suggest that the acti- vating G-rich element and the repressor prohahly act together to quantitatively regulate cutinase gene transcription: the re- pressor's task is to keep basal transcription low but it also reduces the extent of inducihility. Therefore, the G-rich element acts as its antagonist to restore the level of inducibility.
In vivo and in vitro analyses of cutinase promoter deletions revealed that a cutinase transcription factor (CTFl J hinding at a specific site in the promoter mediates induction of the cuti- nase gene. The promoter element responsible for gene induc- tion by cutin hydrolysate contains two overlapping palin- dromes, sequence elements which are often the target of specific DNA-binding proteins. Consistently, mutation of the right palindrome (palindrome 2) severely diminished or com- pletely abolished inducibility as well as sequence-specific hind- ing of nuclear protein(s). In addition, methylation interfrrence experiments identified the right palindrome as the targr t of DNA-protein interaction. Our results do not reveal any func- tion for the left palindrome in cutinase gene remlation.
The formation of two bands in the gel retardation analysis with intact palindrome 2 might he due to binding of diffrrrnt transcription factors to palindrome 2. one of them hring ca- pable of binding to the intact palindrome only, whereas the other might also recognize one single half-site. Alternatively. CTFl might bind its symmetric binding site as a dimrr. If this was the case, probes with both half sites of the palindrome intact (wild type and [-225/-1201pnlll~ would allow complex formation with the CTFl dimer (upper band) or a CTFl mon- omer (lower band). After mutation of parts of the palindrome ([-225/-120Apa11/2] and [-225/-120Apa121) only one subunit of CTFl can bind to the DNA. As the two complexes formed with the intact palindrome would have very similar migration prop- erties, monomer-dimer formation can only cause this pattern if. in addition, binding of one or both subunits causes different bending ofthe DNA(see, for example, Laned 01. ( 2 7 ) ) . I h d i n g of the DNA may also explain the different migration pattern of the bands formed with [-225/-120Apa11/21 and 1-2251 -120Apa121.
Additional information about CTFl came from W cross- linking experiments. The major band at 49 kDa could mean tha t CTFl i s a protein of 49-kDa binding as a dimer to palin- drome 2. The signal at 91 kDa could result from W cross- linking of both subunits in a dimer to the same DNA strand. As the probability of this would he much lower than photochemical cross-linking of only one subunit, the resulting hand is expected to be much weaker as actually found. The hands at 73 kDa and 63 kDa might correspond to degradation products of one or two subunits of the dimer. Alternatively, palindrome 2 might he recognized by several proteins. A similar situation is found. for example, for the CG-1 box in Nicotiana fahacum (28) or the
Promoter Analysis of the Cutinase Gene from Fusarium 9203
CAMP-responsive element binding/activating transcription fac-
+ + + - Nuclear Extract
+ + + + Probe [-116/+34]
FIG. 9. Specific binding of CTF2 cutinase to promoter ment [-116/+341. A. gc.1 rctnrdation assays with promoter frag
tor binding site in mammals (29). By comparing the results from gel retardation analysis and W cross-linking experi- ments, especially considering the intensities of the different bands, we favor the hypothesis that CTFl is a dimeric tran- scription factor composed of 49-kDa monomers. It is notewor- thy that the putative transcription factor monomer that was phosphorylated by the fungal nuclear preparation in the pres- ence of cutin monomer was also a 50-kDa protein f 161. The native molecular mass of the protein factor required to stimu- late cutinase gene transcription in the nuclear preparation was 100 kDa (30). These observations and the present results on CTFl are all consistent with the suggestion that a dimeric transcription factor of 50-kDa monomers is involved in activa- tion of cutinase gene transcription by cutin monomers. How- ever, firm conclusions about these transcription factors must await their isolation, identification, and direct characteriza- tion.
Mutation of the left palindrome did affect nuclear protein binding and the levels of gene expression. Mutation of this region (Apall) increased inducibility by cutin hydrolysate about 5-fold. and binding of nuclear proteins was much stron- ger than to the wild type sequence. Ry examination of the mutated sequence of palindrome 1, we found that by chance sequence "GAGCCGAG" (position -176 to -169 of Apall) was created, which is nearly identical with the left half-site of pal- indrome 2, the loop, and the first nucleotide of the right half- site. Thus, Apall contains two direct repeats and an inverted repeat of this sequence, the putative recognition site for a CTFl monomer. Therefore, stronger binding to Apall and enhanced inducibility might be caused by either of two mechanisms. First, with three half-sites of the recognition sequence being present in the mutated sequence of Apall, CTFl might have a higher affinity to this region. Second, CTFl might not only be able to bind to inverted repeats, but also to recognize the direct repeated sequence as previously postulated for ALCR. the tran- scriptional activator of the ethanol-utilizing pathway in A. nidulans (13).
We analyzed the promoter region sufficient for basal pro- moter activity. In oivo, plasmid pCAT141 showed low. nonin- ducible promoter activity. Gel shift experiments and methyla- tion interference analysis showed that the cis-elementfs) that bind the nuclear factor CTFB is located between the transcrip- tion initiation sites at -77 and -81 and a putative TATA box a t -115. This location of the CTFB binding site suggests that CTF2 might be a DNA-binding subunit of general transcription factors although its binding site does not show any similarity with recognition sites identified for basal transcription factors from eukaryotes. Alternatively, CTFB might be a gene-specific
1-116/+341. end-labeled with RZP and used as prohr. For sprcific com- petition, the same fragment was added at a 500-fold molar rxcrss to t h r binding reaction. A$ a nonspecific competitor. a 126-hp IIintl f r a p r n t of the coding region of thr prlA grnr from E solnni pint was used. fl. identification of the binding site of CTF2 in cutinasr gene promotrr. For methylation interference assays. the upper and lowrr strands of frag- ment [-158/+341 were end-labeled with "P. Thr mrthyla t~on interfrr- ence assay was performed as drscrihed in Fig. 7 . Thr <; rrsidurcr whonr methylation interfrres with hinding are indicatrd hy nsfrrrskn. Thr sequence of the cutinase promotrr from nuclrotides -73 to -126 IS
shown at the hottom. Sequences with similarity to a CAAT-hox and a TATA-box are hoxrd. G rrsidues whose methylation intrrfrred with DNA binding are indicated by osfrrisk.s. The transcription start pmnts are indicated by arrows. C . determination of the transcription Initlation sites of the cutinase gene. Total RNA from cutin monomrr-inducrd E solan; pis; or from S . crrruismp were used for nuclrasr SI exprrimrntq and primer extension analysis. The sequrncr of t h r cutlnasc promotrr
frag- to -10 of the cutinase pmmoter was usrd as a pnmrr. Thr main tran- is given for comparison. In all experimrnts. an oligonuclrotlde from +20
v e n t scription initiation sites are indicated.

9204 Promoter Analysis of the Cutinase Gene from Fusarium
activating or repressing DNA-binding protein involved in cuti- eds) pp. 217-226, Foundation for Biotechnical and Industrial Fermentation
Preliminary data suggest that CTF2 binds to its recognition H., and van den Hondel, C. A. M. J. J. (1990) Gene (Amst.) 93, 101-109 lIaSe gene possibly mediating glucose repression. 5. Punt, P. J., Dingemanse, M. A., Kuyvenhoven, A,, Soede, R. D. M., Pouwels, P.
sequence in glucose nondepleted, depleted, and cutin hydroly- 6. Burger, G., Tilburn, J., and Scazzocchio, C. (1991) Mol. Cell. Biol. 11,795402
sate-induced supporting the that CTF2 is a 8. Felenbok, B., Sequeval, D., Mathieu, M., Sibley, S., Gwynne, D. I., and Davies,
tivity Of CTF1 appears to be to repression; 10. Baum, J. A., Geever, R., and Giles, N. H. (1987) Mol. Cell. B i d . 7,1256-1266 9. Yuan, G. F., Fu, Y. H., and Marzluf, G. A. (1991) Mol. Cell. Biol. 11,5735-5745
Research, Helsinki, Finland
7. Dowzer, C. E. A,, and Kelly, J. M. (1991) Mol. Cell. Biol. 11, 5701-5709
basal transcription factor. On the other hand, the binding ac- R. W. (1988) Gene (Amst.) 73, 385396
only nuclear extracts from glucose-depleted cultures showed 11. Fu, Y.-H., and Marzluf, G. A. (1990) Proc. Natl. Acad. s~i. U. s. A. 87,5331- binding to the palindrome region.' Expression of CTFl might 5335 be regulated by catabolite repression. Alternatively, CTFl may be constitutively expressed but binds its recognition sequence 13. Kulmburg, P., Sequeval, D., Lenouvel, F., Mathieu, M., and Felenbok, B. (1992)
level. Examples for activation of transcription factors include Laboratow Manual. Cold Surine Harbor Laboratorv Press. Cold Surine
12. Kulmburg, P., Sequeval, D., Lenouvel, F., Mathieu, M., and Felenbok, B. ( 1992) Mol. Cell. Biol. 12, 1932-1939
J. Biol. Chem. 267,21146-21153 Only after an activation which is by the 14. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: A
phosphorylation or reduction of the transcription factor itself regulating its DNA binding activity (31, 32) or allowing its translocation in the nucleus (33), or phosphorylation and thereby inactivation of a dominant inhibitor (34). Protein ki- nase A might be involved in the activation of CTFl binding, as the CAMP level in glucose-depleted cells from R solani pisi was found to be about 2-3-fold higher than that in nondepleted culture^.^ CTFl binds to its target sequence in depleted cells, whether induced by cutin hydrolysate or not. Thus, gene induc- tion of cutinase genes is probably regulated by an additional post-translational mechanism, possibly phosphorylation of CTF1. Involvement of phosphorylated transcription factor was suggested by the previous observation that treatment of DNA- binding fungal protein preparation with immobilized phospha- tase prevented its binding to cutinase promoter (16). We have evidence that Ca'+/calmodulin and a tyrosine protein kinase are involved in regulation of cutinase tran~cription.~ Isolation and characterization of CTFl and CTF2 as well as of the si- lencer- and activator-binding proteins are necessary to further elucidate the mechanisms of cutinase gene regulation.
REFERENCES 1. Lin, T. S., and Kolattukudy, P. E. (1978) J. Bacterid. 133, 942-951 2. Woloshuk, C. P., and Kolattukudy, P. E. (1986) Proc. Natl. Acad. Sci. U. S. A.
83, 1704-1708 3. Koller, W., Allan, C. R., and Kolattukudy, P. E. (1982) Physiol. Plant Pathol. 20,
47-60 4. Podila, G. K., Dickman, M. B., Rogers, L. M., and Kolattukudy, P. E. (1989) in
Molecular Biology of Filamentous Fungi (Nevalainen, H., and Penttila, M.,
J. Kzimper, U. Klimper, and P. E. Kolattukudy, unpublished results. U. Klimper and P. E. Kolattukudy, unpublished results.
Harbor, N% 15. Innis, M. A,, and Gelfand, D. H. (1990) in PCR Protocols: A Guide to Methods
and Applications (Innis, M. A., Gelfand, D. H., Sninsky, J. J., and White, T.
16. Bajar, A., Podila, G. K., and Kolattukudy, P. E. (1991) Proc. Natl. Acad. Sei. J., eds) pp. 3-20, Academic Press, Inc., San Diego
17. Soliday, C. L., Dickman, M. B., and Kolattukudy, P. E. (1989) J. Bacteriol. 171, U. S. A. 88,8208-8212
18. Higuchi, R. (1990) in PCR Protocols: A Guide to Methods and Applications 1942-1951
(Innis, M. A., Gelfand, D. H., Sninsky, J. J., and White, T. J. eds) pp. 177-183, Academic Press, Inc., San Diego
19. GonzBlez-Candelas, L., and Kolattukudy, P. E. (1992) J. Bacterid. 174,6343- 6349
20. Slater, M. L. (1978) Methods Cell Biol. 20, 135-140 21. Bradford, M. M. (1976)Anal. Biochem. 72,248-254 22. Gorman, C. M., Moffat, L. F., and Howard, B. H. (1982) Mol. Cell. Biol. 2,
1044-1051 23. Ausubel, G. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith,
J. A,, and Struhl, K. (1989) Current Protocols in Molecular Biology, Greene
24. Punt, P. J., and van den Hondel, C. (1991) in Molecular Biology ofFilamentous Publishing Associates and Wiley-Interscience, New York
Fungi (Stahl, U., and Tudzynski, P., eds) pp. 177-187, VCH Verlagsgesell-
. - ~I
~~
schaft, m.b.H., Weinheim
Eukaryotic Microbes (Kinghorn, J. D., ed) pp. 93-139, IRL Press, Oxford
20-23
~~
25. Gurr, S. J., Unkles, S. E., and Kinghorn, J. R. (1987) in Gene Structure in
26. Kadonaga, J. T., Jones, K. A., and Tjian, R. (1986) %ends Biochem. Sci. 11,
27. Lane, D., Prentki, P., and Chandler, M. (1992) Microbiol. Rev. 56,509-528 28. Staieer, D., Becker, E , Schell, J., Koncz, C., and Palme, K. (1991) Eur J.
29. Hai, T., Liu, F., Allegretto, E. A,, Karin, M., and Green, M. R. (1988) Genes & B&hem. 199,519-527
30. Kolattukudy, P. E. (1992) in Molecular Signals in Plant-Microbe Communica-
31. Abate, C., Patel, L., Rauscher, G. J., 111, and Curran, T. (1990) Science 249,
32. Englander, E. W., Widen, S. G., and Wilson, S. H. (1991) Nucleic Acids Res. 19,
33. Metz, R., and Ziff, E. (1991) Genes & Deu. 6, 1754-1766 34. Auwerx, J., and Sassone-Corsi, P. (1991) Cell 64,983-993 35. Focus (1986) 8,2
Deu. 2, 1216-1226
tions (Verma, D. P. S., ed) pp. 65-83, CRC Press, Boca Raton, FL
1157-1161
3369-3375