Embed Size (px)
Citation preview

THE ESSENTIALS OF METABOLIC NUTRITION:
WHAT TO DO WHEN YOU ARE JUST GETTING STARTED
Suzanne Hollander, MS, RD, LDN
Boston Children’s Hospital
Jessica Burfield, RD, CSP, LDN
Children’s Hospital of Philadelphia
1

DISCLOSURES
• Suzanne Hollander, MS, RD, LDN• I have served as a paid consultant to Vitaflo, Nutricia and
Horizon. I have served as study staff on research protocols sponsored by Biomarin, Homology, and TravereTherapeutics.
• Jessica Burfield, RD, CSP, LDN• I have served as a paid consultant to Abbott, Ultragenyx,
and Nutricia. I have served as study staff on a research protocol sponsored by Nutricia
2

OBJECTIVES
• Interactive session for RDs new to the field of metabolism
• Discuss the MNT for disorders “commonly” encountered by the Metabolic RD.
• Explore the application of MNT for these disorders through case presentations.
• Develop an understanding of MNT concepts through group discussion.
3

Outline
I’m new to metabolics!
Where do I turn for information?• GMDI Resources
• GMDI/SERN Nutrition Guidelines
• Additional resources: • Listserv
• Publications
• State NBS-sponsored materials
• Industry-sponsored materials
Overview of conditions:• PKU (SH)• MSUD (JB)• HCU, IVA (JB)• MMA/PA (SH)• GA1 (JB)• UCDs (SH)• FAODs (SH)• Galactosemia (JB)• GSD Resources (SH)

ADIME for the Metabolic Patient
Assessment
• Molecular diagnosis
• Biochemical findings
• Growth
• Feeding modality/ ability/ tolerance
• Educational needs
• Neurocognitive function
• Social & financial circumstances
Diagnosis
• INTAKE DOMAIN
• Intake of types of amino acids/fats/carbs inconsistent with needs…
• Inadequate or excessive [nutrient]intake…
• CLINICAL DOMAIN
• Impaired Nutrient Utilization…
• Altered nutrition related lab values…
• BEHAVIORAL/ENVIRONMENTAL
• Food and nutrition related knowledge deficit…
• Undesirable food choices…
• Self monitoring deficit…
• Limited access to nutrition related supplies...
Intervention
• Is there an evidence-based diet?
• Are there treatment guidelines?
• If no published evidence/guidelines, are there compelling published case studies?
• What have colleagues done?
• Is harm possible with or without an intervention?
• How will you monitor/ evaluate efficacy?
Monitoring/
Evaluation
• Growth
• Biochemical indices
• Neurocognitive outcomes
• Quality of life—how to measure?
• Patient/family ability to follow
• Functional improvements

GMDI Member Resources
• Clinical Practice Tools and Educational Resources
• Recent Publications
• Webinar Series
• Research Grant Program
• Newsletter
• Conference!
6
Gmdi.org/member-resources

GMDI/SERN Nutrition Management Guidelines
• PKU Guideline & Toolkit
• MSUD Guideline & Toolkit
• PROP Guideline & Toolkit
• VLCAD Guideline & Toolkit
• MCAD Guideline
7
https://gmdi.org/Members/Clinical-Practice-Tools/Nutrition-Guidelines https://southeastgenetics.org/ngp/guidelines.php
GuidelinesWhat and Why” Evidence-based, comprehensive approach to nutrition managementFollows the Nutrition Care Process
Toolkit“How”Step-by-step guidance for developing the nutrition plan Age-based case studies
Consumer Summary and FAQ (English and Spanish)

Other Resources
• Listserv: GNO-METAB Listserv• Register with GNO Metabolic Listserv Moderator: [email protected]
• Published literature, JIMD for example
• State NBS materials
• Industry-sponsored materials and webinars
• Met-Ed: http://www.met-ed.net/index.php
8

Phenylketonuria (PKU): Biochemical Pathway
Cause: Impaired synthesis or recycling of phenylalanine hydroxylase (PAH) or its cofactor, tetrahydrobiopterin (B4)
(SERN/GMDI Guidelines)
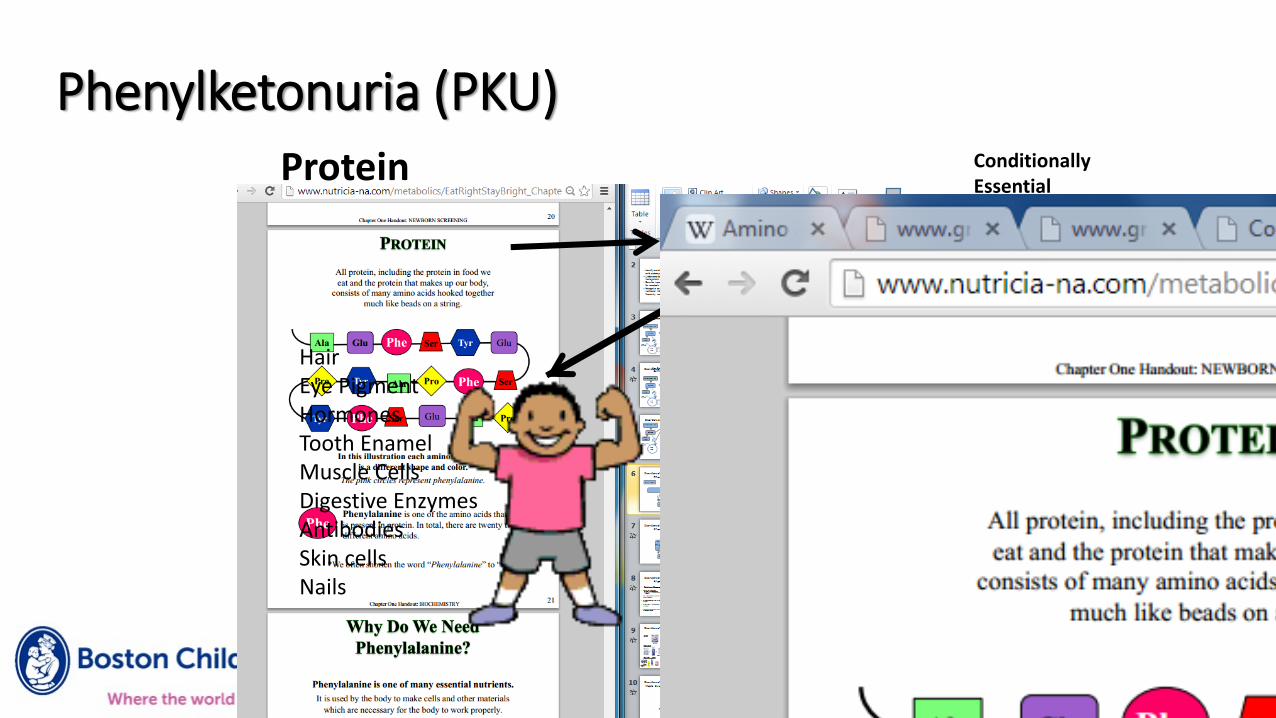
Phenylketonuria (PKU)Conditionally Essential
Protein
PAH
BH4
HairEye PigmentHormonesTooth EnamelMuscle CellsDigestive EnzymesAntibodiesSkin cellsNails
Neurotransmitters

PKU With and Without TreatmentWith Treatment
(Target PHE levels 120-360 umol/L)
Without Treatment(Especially with very high levels in early
childhood/infancy)
Normal Development Developmental Delay
Normal memory and executive function
Compromised memory and executive function, headaches
Normal behavior Agitated Behavior
Natural skin and hair color Light pigmentation
Clear skin (no rash) Eczema, skin rashes
Normal urine/sweat smell Musty smelling urine and sweat
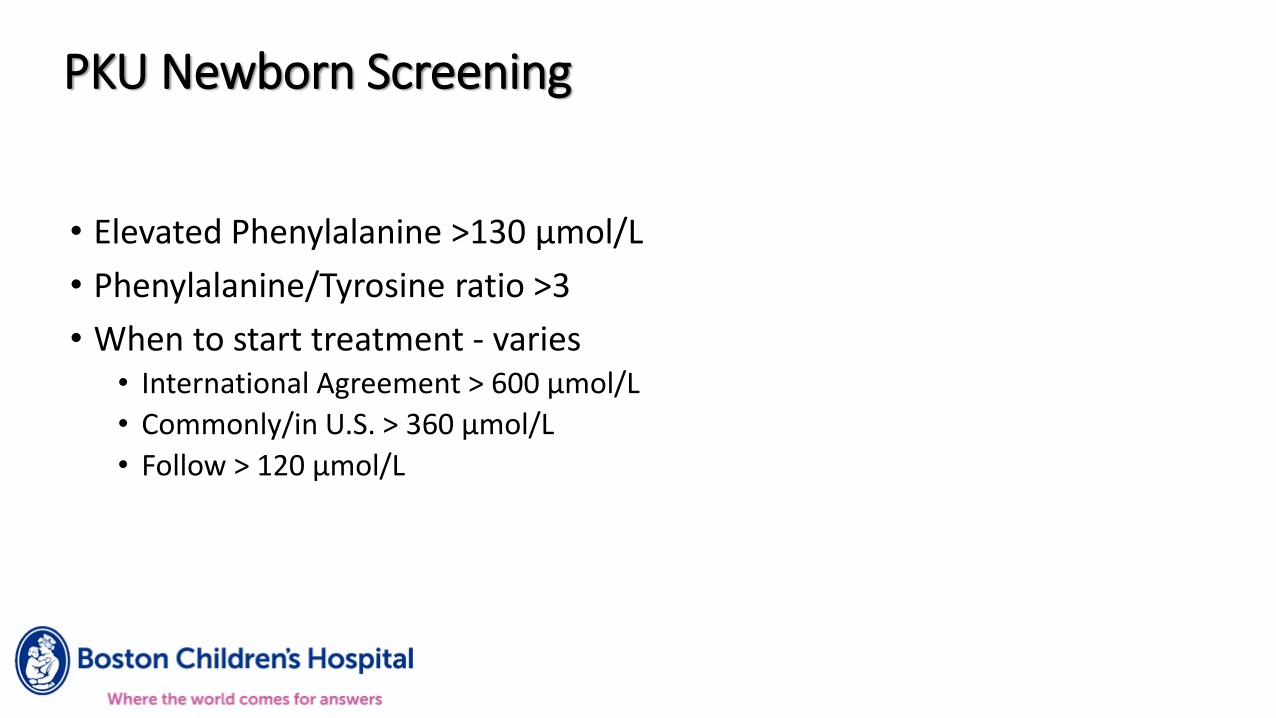
• Elevated Phenylalanine >130 µmol/L
• Phenylalanine/Tyrosine ratio >3
• When to start treatment - varies• International Agreement > 600 µmol/L
• Commonly/in U.S. > 360 µmol/L
• Follow > 120 µmol/L
PKU Newborn Screening
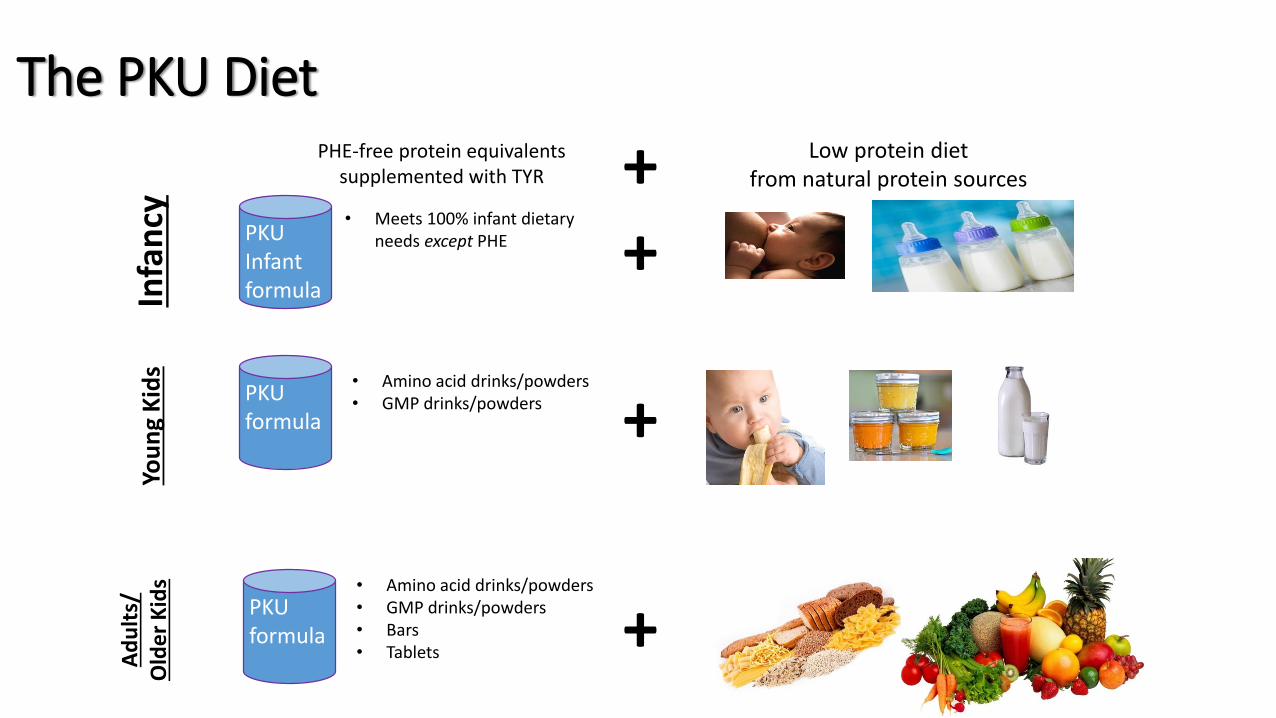
The PKU DietIn
fan
cy
+
+
Ad
ult
s/O
lde
r K
ids
+
PHE-free protein equivalentssupplemented with TYR + Low protein diet
from natural protein sources Yo
un
g K
ids
PKU Infant formula
PKU formula
• Amino acid drinks/powders• GMP drinks/powders
PKU formula
• Amino acid drinks/powders• GMP drinks/powders• Bars• Tablets
• Meets 100% infant dietary needs except PHE

Options for PKU Dietary Management
• Count dietary intake of phe (mg) in all foods
• Count total protein (grams) in all foods
• Simplified Diet Approach—only count phe (mg) or protein (grams) in foods with >75 mg phe per 100g food weight (approach varies)
Approach depends on:
• Family preference and experience
• Type/severity?
• Metabolic control ?14
Hansen et al. JIMD, 2020.

Goals of PKU Diet Treatment • PHE levels between 120-360 µmol/L or 2-6 mg/dL
• Promote normal growth and development
• Prevent catabolism
• Provide adequate macro and micronutrients• Caloric balance
• Adequate phe and tyr
• Total protein
• DHA/ALA?
• B12, iron, calcium

PKU Patient Concerns
• Diet for Life• “New” ACMG/GMDI guidelines
• Diet fatigue
• Formula fatigue
• Neurological involvement• Executive functioning
• Depression, anxiety
• Life changes, age related complications

PKU Adjunctive/Alternative Therapies
• Large Neutral Amino Acids
• “Compete” with phenylalanine to cross blood brain barrier
• Over the age of 12; not recommended for women of child bearing age
• Large volume of pills/powder
• More relaxed diet but don’t lower blood levels
• Cofactor Therapy (BH4)
• One dose daily
• Not all responsive; not all responsive enough
• Pegvaliase – PEGeylated recombinant phenylalanine ammonia lyase(enzyme substitute)
• Future? Gene therapy

PKU Diet Initiation
• Eliminate phe from diet for 0-48 (72?) hours
• Considerations for breastfeeding?
• Think about phe level turnaround time
Phenylalanine Levels Initiate Phenylalanine After
360-600 µmol/L (6-10 mg/dL) 24 hours
600-1200 µmol/L (10-20 mg/dL) 48 hours
1200-2400 µmol/L (20-40 mg/dL) 72 hours
>2400 µmol/L (>40 mg/dL) 96 hours
Bernstein et al
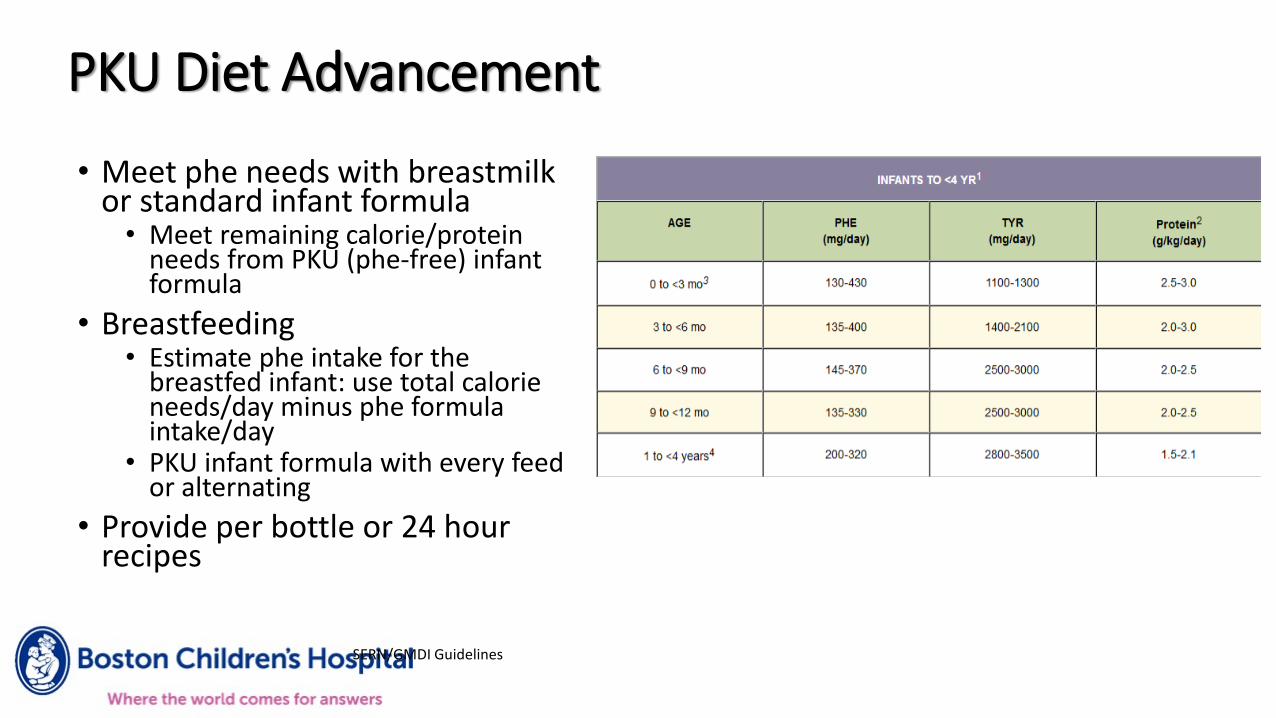
PKU Diet Advancement
SERN/GMDI Guidelines
• Meet phe needs with breastmilk or standard infant formula
• Meet remaining calorie/protein needs from PKU (phe-free) infant formula
• Breastfeeding• Estimate phe intake for the
breastfed infant: use total calorie needs/day minus phe formula intake/day
• PKU infant formula with every feed or alternating
• Provide per bottle or 24 hour recipes

PKU Diet Progression: Using Simplified Diet in Infancy
20
5-7 months4 months 7-9 months 9-12 months
• 3 structured meals per day
• Increase food protein
• Continue same PKU infant formula,
decrease breastmilk/standard
formula (24-30 oz total liquid
volume)
• May use semi-solid medical food
• 1-2 structured meals per day
• 0 g protein foods
• Introduce a cup!
• Continue same breastmilk/formula+
PKU infant formula (~28-36 oz total
liquid volume)
• May use semi-solid medical food
• 3 meals, 1 snack
• Add and vary herbs, spices,
textures
• 80-100% intact protein from
foods
• Continue PKU infant formula,
wean breastmilk/standard
formula (14-20 oz total liquid
volume)
• May use semi-solid medical food
• Teach Simplified Diet
• Answer questions

Maternal PKU
SERN/GMDI Guidelines
• High phe is teratogenic and can cause harm to the fetus• Mental retardation• Microcephaly• Congenital heart disease• IUGR
• Goals for pregnancy are PHE 120-360 umol/L throughout pregnancy (including early pregnancy)
• Challenges• Some women have liberalized diets and are returning to diet for pregnancy• Caloric adequacy on protein-restricted diet• Medical Food tolerance with nausea symptoms• Special nutritional needs (i.e. multiple pregnancies, gestational diabetes)

Maternal PKU Case Study:35 year old women planning pregnancy• 35 year old women off diet x15 years
• Estimated 70-80g protein intake/day
• Returns to clinic with interest in pregnancy• PHE levels >1600 umol/L
• *Encouraged/advised to WAIT to conceive* (risks outlined by metabolic provider)
• 4 week Kuvan trial unresponsive
• Started on medical food + diet
22
Maternal PKU Case Study:35 year old woman restarting diet
Starting prescription:
• GMP Medical Food: 60 grams per day
• Low protein diet: 10 grams per day (5 grams counted using “Simplified Diet”)
• Weekly phe levels w/ 3-day diet analysis 23

Maternal PKU Case Study:35 year old woman restarting diet
Troubleshooting challenges:
• Too tired to cook—take out options discussed
• Portions of “0g” foods on Simplified Diet—took a few things off the “free” list
• Medical Food!
24
PHE (umol/L)
TYR (umol/L)
Pre-pregnancy 1700 45 Return to clinicPre-pregnancy 1750 34 BH4Pre-pregnancy 1630 39 BH4Pre-pregnancy 673 42 10g pro, 60 g MFPre-pregnancy 535 55 8g pro, 70g MFPregnant wk 5 435 53 6g pro, 80g MFPregnant wk 12 225 87 6g pro, 80g MFPregnant wk 20 341 76 8 g pro, 80g MFPregnant wk 28 278 64 15g pro, 80g MFPregnant wk 36 236 45 25g pro, 60g MF

Maternal PKU with Complications
• Nausea/vomiting• Find tolerable medical food• Smaller, more frequent meals and medical food volumes• Refer to OB for anti-emetics if vomiting persists and/or affects blood phe control
• Gestational Diabetes• Manage expectations--insulin may be required to manage both PKU and GDM together
successfully • Work with OB, endo, GDM dietitian to explain limited ability to adjust diet• Consider calorie and weight gain needs• Reduce added sugar where possible (switch medical food?)• Increase healthy fats• Promote physical activity
25

Resources For Patients & Families• PKU News
• National PKU Alliance
• Formula and food companies
• How Much Phe
• Diet tracking apps and online
• Low Protein Cookery for PKU
• Cook for Love
• Family support groups, Facebook groups
• GMDI/SERN Consumer page

PKU References Acosta, P. B. & Matalon, K. M. (2010). Nutrition Management of Patients with Inherited Disorders of Aromatic Amino Acid Metabolism. In P. B. Acosta, Nutrition Management of patients with Inherited Metabolic Disorders (pp. 119-127). Sudbury, MA: Jones and Bartlett Publishers.
GMDI/SERN Management Guidelines Portal. PKU Nutrition Management Guidelines. https://southeastgenetics.org/ngp/guidelines.php/90/PKU%20Nutrition%20Guidelines/Version%201.12. March 2015.
Ross Products Division (2001) DISORDERS OF AMINO ACID METABOLISM Aromatic Amino Acids.PROTOCOL 1 Phenylketonuria (PKU) Nutrition Support of Infants, Children, and Adults With PHENEX™-1 and PHENEX™-2 Amino Acid-Modified Medical Foods. In The Ross Metabolic Formula System Nutrition Support Protocols, 4th Edition (pp.1-32). Columbus, OH.
Singh RH, Rohr F, Frazier D, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med. 2014;16(2):121-131.
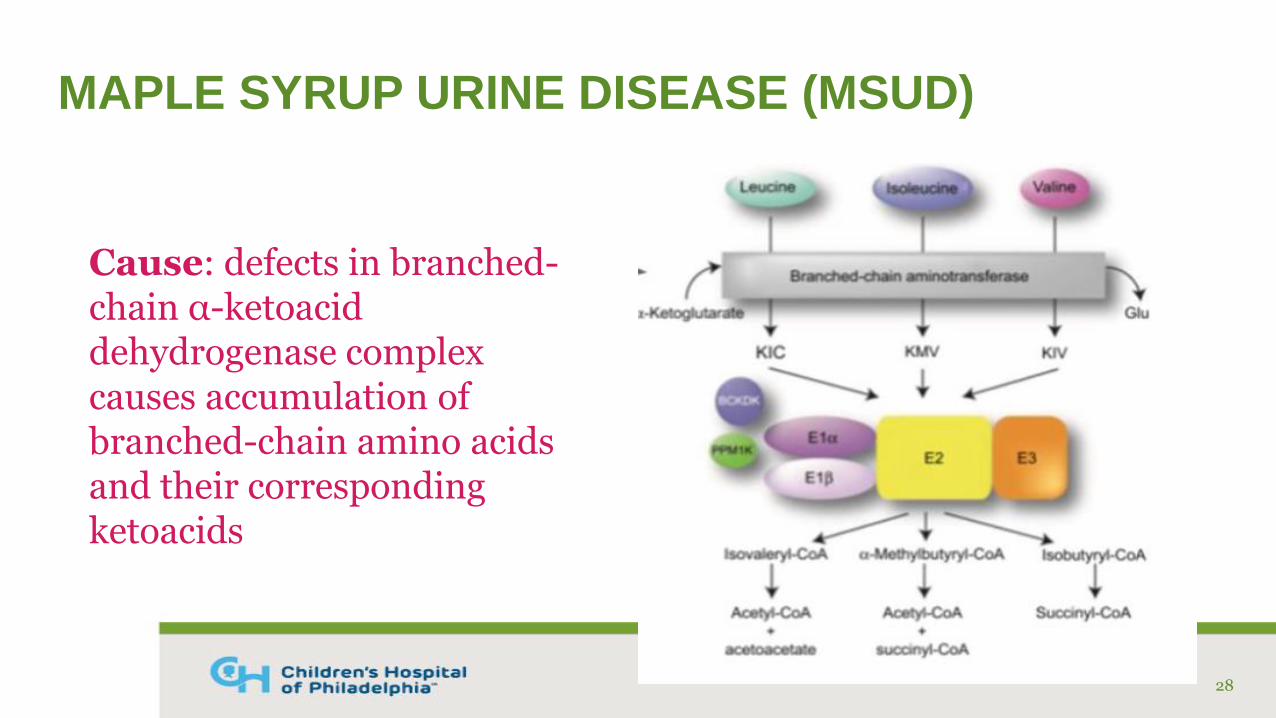
MAPLE SYRUP URINE DISEASE (MSUD)
28
Cause: defects in branched-chain α-ketoacid dehydrogenase complex causes accumulation of branched-chain amino acids and their corresponding ketoacids

MSUDNEWBORN SCREEN
29
• Newborns may be symptomatic before you receive the positive screen
• Symptomatic: elevated BCAA and Leu:Phe ratio >4.5-5• Progressive encephalopathy, poor feeding, somnolence, cerebral edema, coma
• Asymptomatic: normal or slightly elevated BCAA• Mild/intermittent form: psychomotor delay, neurological disease,
ketoacidosis
• Maple syrup smell is first evident in cerumen rather than urine

MSUD MEDICAL NUTRITION THERAPY
• Goals of MNT• Reduce toxic metabolites by restricting BCAAs to amount necessary
to maintain appropriate plasma concentrations • Leucine
• < 5 years old: 75-200 umol/L (1 - 2.6 mg%)
• > 5 years old: 75-300 umol/L (1 – 4 mg%)
• Isoleucine & Valine: 60% of leucine level
• Encourage anabolism and prevent catabolism
• Allow for normal growth and development
• Determine if thiamin responsive
30

NUTRITION ASSESSMENTMSUD
• First, tell the team to stop all sources of BCAAs and start dextrose containing IV fluids
• Then, proceed with your nutrition assessment• Relevant metabolic studies
• Calories, electrolytes provided by current fluids, intralipids
• Feeding history
• Anthropometrics: weight, length, HC, weight for length, etc.
• Nutrition focused physical findings: tone, vomiting, diarrhea, odor
• Other considerations: coma, dialysis
31

MSUD CASE STUDY NEWBORN MANAGEMENT
• 4-day-old full-term female
• Labs in the ED• Blood glucose 36, bicarb 16, +large ketones in urine
• Team obtained central access• D20 ¼ NS with 10 mEq/L KCl• 2 g/kg intralipids, amino acids held
• Confirmatory PAA drawn in ED• Leucine: 20.6 mg%, Isoleucine: 6.3 mg%, Valine: 10.6
mg%, Alloisoleucine: 1.8 mg%
32

MSUD CASE STUDY NEWBORN MANAGEMENT
• Enteral feeds PO/NG vs. TPN• Begin with medical food free of BCAAs; hold breast milk and/or
standard infant formula
• Meet nutrition goals per GMDI guidelines• 118-130 kcal/kg, 2.5-3 g/kg protein (BCAA-free), fluids per TFL
• Use calorie modulars to meet needs, if needed
• May need to use combination of PN and enteral nutrition • MSUD PN Integrity Pharmacy in Atlanta, GA; turnaround time 24-48 hours
• Standard PN with limited/no protein
• Introduce breast milk or standard infant formula once leucine is <6 mg%
33

NUTRITION INTERVENTIONPN SPECIAL CONSIDERATIONS
• Energy goals• Infant: 118-130 kcal/kg, 2.5-3 g/kg/day (BCAA-free amino acid solution)
• Children: 80-100 kcal/kg/day
• Adults: 40-45 kcal/kg/day
• Serum bicarbonate• Closely monitor CO2 levels
• Correct metabolic acidosis by adding Na Acetate or K Acetate in TPN
• Serum sodium• Maintain normal serum Na+ levels as hyponatremia enhances brain
edema in MSUD
• Typically 4 mEq/kg/d Na total (from NaCl, Na Acetate, & Na PO4)
34
* Adapted from New England Consortium of Metabolic Physicians
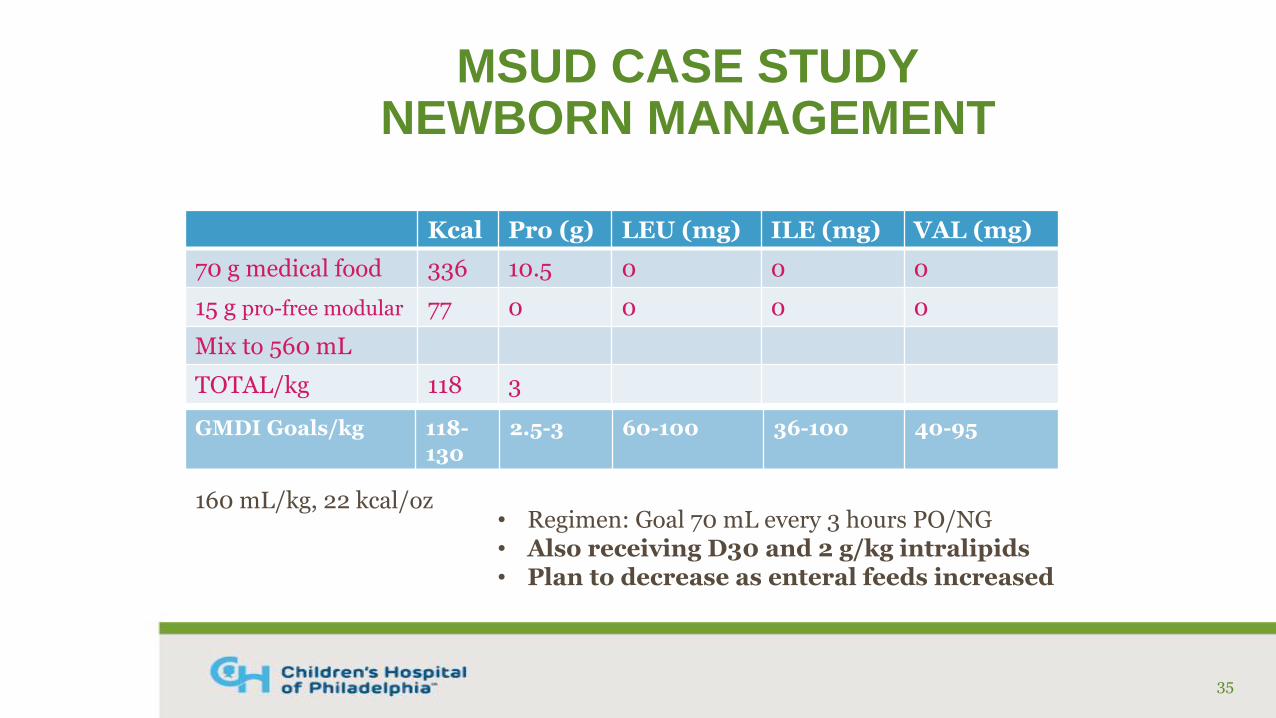
MSUD CASE STUDYNEWBORN MANAGEMENT
Kcal Pro (g) LEU (mg) ILE (mg) VAL (mg)
70 g medical food 336 10.5 0 0 0
15 g pro-free modular 77 0 0 0 0
Mix to 560 mL
TOTAL/kg 118 3
35
160 mL/kg, 22 kcal/oz
GMDI Goals/kg 118-130
2.5-3 60-100 36-100 40-95
• Regimen: Goal 70 mL every 3 hours PO/NG• Also receiving D30 and 2 g/kg intralipids• Plan to decrease as enteral feeds increased

36
Source: GMDI MSUD Guidelines
GOAL NUTRIENT INTAKE IN NEWBORN PERIOD
Alanine – 100 mg/kg if low or LEU persistently high
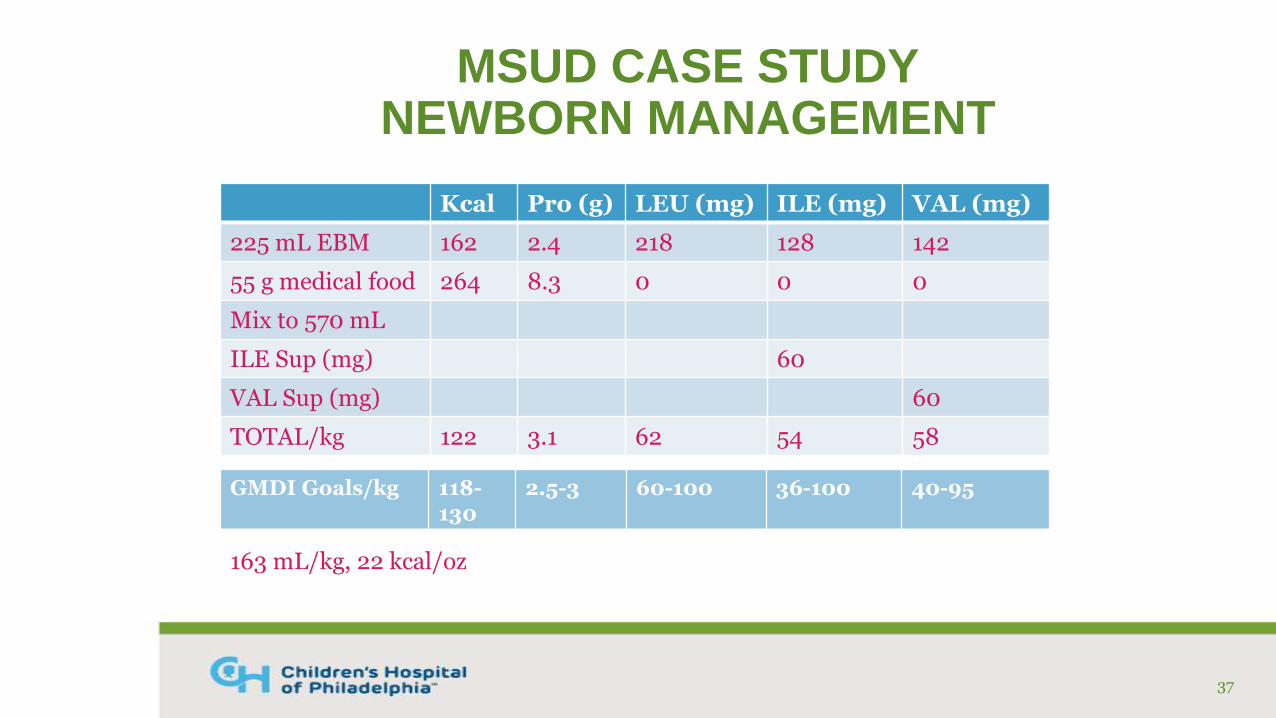
MSUD CASE STUDYNEWBORN MANAGEMENT
Kcal Pro (g) LEU (mg) ILE (mg) VAL (mg)
225 mL EBM 162 2.4 218 128 142
55 g medical food 264 8.3 0 0 0
Mix to 570 mL
ILE Sup (mg) 60
VAL Sup (mg) 60
TOTAL/kg 122 3.1 62 54 58
37
163 mL/kg, 22 kcal/oz
GMDI Goals/kg 118-130
2.5-3 60-100 36-100 40-95
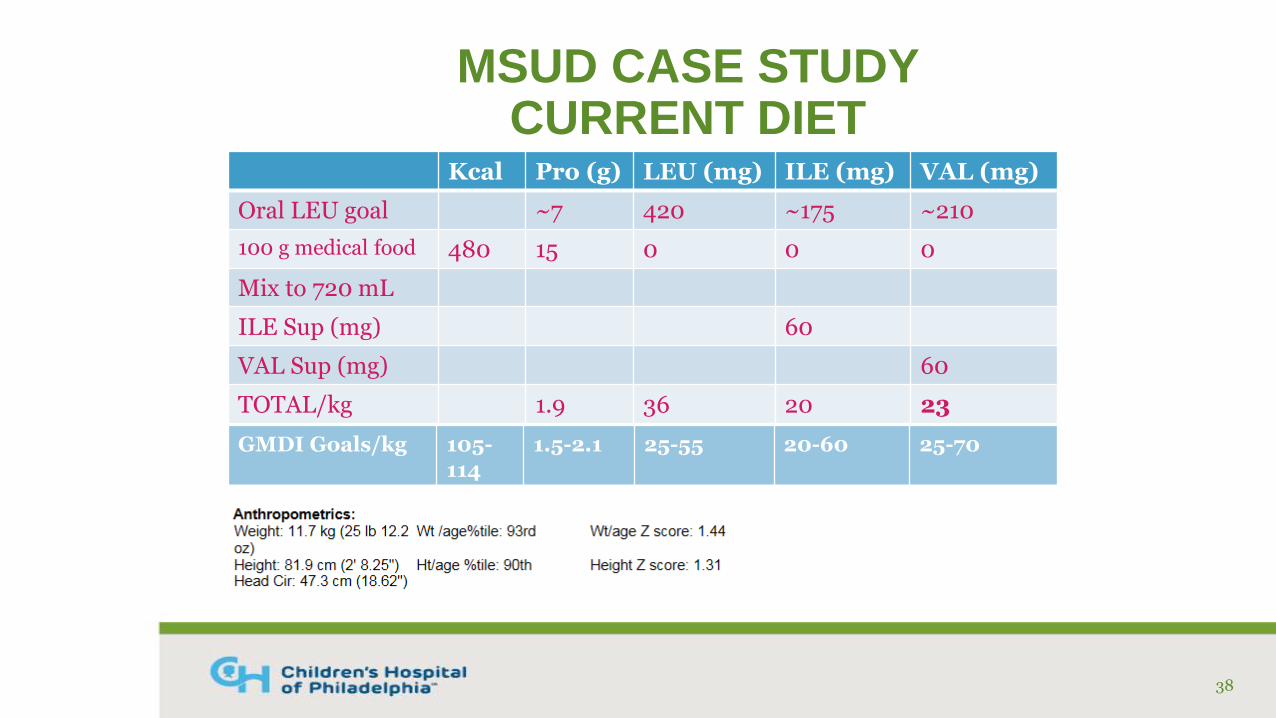
MSUD CASE STUDY CURRENT DIET
Kcal Pro (g) LEU (mg) ILE (mg) VAL (mg)
Oral LEU goal ~7 420 ~175 ~210
100 g medical food 480 15 0 0 0
Mix to 720 mL
ILE Sup (mg) 60
VAL Sup (mg) 60
TOTAL/kg 1.9 36 20 23
38
GMDI Goals/kg 105-114
1.5-2.1 25-55 20-60 25-70

MSUD ONGOING MANAGEMENTSICK DAY DIET
• Stop all sources of BCAAs
• Provide 120% of calorie and fluid needs• Be sure to account for any IV fluids
• Meet 100% of protein needs via metabolic formula
• Double amino acid supplements
• Consider alanine supplement (100 mg/kg) if not already taking
• May require PN depending on illness type and severity
39

MSUD ONGOING MANAGEMENTLIVER TRANSPLANT
• “Cure”
• Relieves the burden of dietary treatment
• Patients typically have BCAA levels within treatment range for MSUD, but not “normal”
• Other considerations
40

REFERENCES
Berry GT, Heindenreich R, Kaplan P, et al. Branched-chain amino acid-free parenteral nutrition in the treatment of acute metabolic decompensation in patients with maple syrup urine disease. N Engl J Med. 1991; 324(3):175–179.
Frazier DM, Allgeier C, Homer C, et al. Nutrition management guidelines for maple syrup urine disease: An evidence- and consensus-based approach. Mol Genet Metab. 2014; 112(3):210-217.
Genetic Metabolic Dietetians International. MSUD Nutrition Management Guidelines. https://southeastgenetics.org/ngp/guidelines.hp/103/MSUD%20 Nutrition%20Guidelines/Version%201.53. September 2017. Accessed February 27, 2018.
Mazariegos GV, Morton DH, Sindhi R, et al. Liver transplantation for classical maple syrup urine disease: Long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. J Pediatri. 2012; 160(1):116-121.
41
Methylmalonic and propionic acidemia
GMDI/SERN Guidelines

Propionic Acidemia Methylmalonic Acidemia
Signs/symptomsat initial presentation
Poor feeding, vomiting, lethargy, tachypnea, abnormal tone, acidosis
Progressing to respiratory distress, seizures, coma and death
Biochemical indices
• Elevated C3 (propionylcarnitine)• Total and Free carnitine – low• Plasma amino acids – elevated glycine, alanine; Methyl/propionic precursors valine, methionine,
isoleucine, threonine• Ammonia• Urine organic acids – 3-OH-propionic acid; methylcitric acid; methylmalonic acid (MMA only)
Acute Treatment (diagnosis or illness)
• Carglumic acid or dialysis to lower ammonia• Caloric adequacy to promote anabolism/limit catabolism
• IV dextrose/lipids• Oral or enteral feeds
• Insulin to promote anabolism prn• Protein restriction (short-term) prn to limit total protein load
Chronic Dietary Treatment
• Protein restriction + MMA/PROP medical food (or may tolerate RDA protein)• Emergency management protocol• Considerations for comorbidities—pancreatitis, renal involvement, tone and functional impairments• Gtube for feeding adequacy and/or feeding access in acute setting
Complications Pancreatitis, cardiomyopathy, cognitive impairmentLiver transplant?
Renal tubular acidosis, cognitive impairmentLiver/kidney transplant?

Nutrition Management of PROPGMDI/SERN Nutrition Guidelines
• PROTEIN: Reduce intake of propiogenic amino acids while maintaining normal blood concentrations of ILE, VAL, MET and THR
o 60-100% age-appropriate protein intake from intact source (Recommendation 1.1)
o Meet remaining protein need (100-120%) with medical food (Recommendation 1.2)
• CALORIES:
o Provide 80-120% of total energy requirements for age to spare protein catabolism and promote normal growth (Recommendation 1.5)

Nutrition Management of PROP
• Enteral tubes may be required for intake adequacy• ~65% PROP patients have G-Tubes
• Oral aversion often reported
• Emergency management requires caloric adequacy
• Management of constipation• Reported frequently
• Enhanced gut motility may improve metabolic stability (Prasad C, Nurko S, Borovoy J,
Korson MS.,J Pediatr. 2004 Apr;144(4):532-5.)
• Nutritional strategies may support medical management

Acute Decompensation/Illness Management• Recognize potential stress/risks
• Monitor growth—inadequate weight gain?• Labs—acidosis, PAAs• GI symptoms and intake adequacy• Illness• Teething, immunizations?
• Caloric adequacy is critical to break or prevent catabolic cascade, but most challenging during periods of illness
• Enteral tubes• IV fluids—D10+ %, lipids?• Limit holding of feeds and reduction in protein to as short as possible• Increase supplements/medications if needed
• Hospital versus home management plans

Troubleshooting Challenges• Low ILE, VAL
• Increase intact protein• But may need to ALSO decrease medical food due to high relative leucine content
• Frequent decompensation and illness management • Caution with excessive use of a reduced protein regimen
• Nausea, vomiting, constipation, diarrhea• Co-manage with GI providers• Blenderized tube feeds? • Medication management to promote intake adequacy
• Kidney disease and pancreatitis• Adjust medical food products as needed
• Transplant• Liberate dietary protein?

MMA/PA References GMDI/SERN Management Guidelines Portal. PROP Nutrition Management Guidelines. https://southeastgenetics.org/ngp/guidelines.php/104/PROP%20Nutrition%20Guidelines/Version%201.2. September 2017.
Manoli, I., Myles, J.G., Sloan, J.L., Shchelochkov, O.A., & Venditti, C.P. (Apr 2016). A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonicacidemias. Genet. Med., 18, 386-95. doi:10.1038/gim.2015.102
Ross Metabolic System (2001). Nutrition Support Protocols Ross Metabolic Formula System Abbott Labs 4th edition. Abbott Labs.

HOMOCYSTINURIA
• Genetic defect in cystathionine beta-synthase (CBS)• CBS + vitamin B6 convert homocysteine cystathionine cysteine
• Accumulation of homocysteine causes symptoms• Wide spectrum of severity
• Optic lens dislocation, myopia, osteoporosis and a ‘marfanoid’ habitus, bone deformities, learning difficulties, psychiatric disturbances, seizures, predisposition to thromboembolism
• Individuals who are vitamin B6 responsive have milder phenotype and later onset of symptoms
• 10-500 mg/day vitamin B6 for 6 weeks with at least 2 measures of tHcy on treatment
• Some patients may be partially responsive
49

HOMOCYSTINURIA TREATMENT• Methionine-restricted diet + methionine-free medical food
• Level of protein restriction varies; start @ 90-120 mg Met/day• Adjust protein (methionine) allowance to achieve goal tHcy
• Vitamin B12 and folate supplements
• Betaine• 50-100 mg/kg/day• Lower tHcy levels by donating methyl group to convert Hcy Met• Must be used in coordination with low protein diet to reduce tHcy
• Treatment Goals• tHcy <50 μmol/L• Methionine <1000 μmol/L (Betaine)• Normal vitamin B12, folate, vitamin D, cystine
50

HOMOCYSTINURIA MANAGEMENT CHALLENGES
• Current treatment options are limited and inefficient
• Treatment compliance is suboptimal• Effective treatment requires very low protein diet and medical food• Late-diagnoses are not uncommon
• Very difficult to initiate low protein diet and medical food• Medical food does not taste good• Quality of life, social stigmatization, exclusion• Possible intellectual delay may impact ability to adhere to recommendations
• Patients “feel fine.” Often no outward symptoms associated with non-compliance
• Outcomes are poor without adequate treatment
• Novel therapy: OT-58 enzyme replacement therapy (phase 1/2)
51

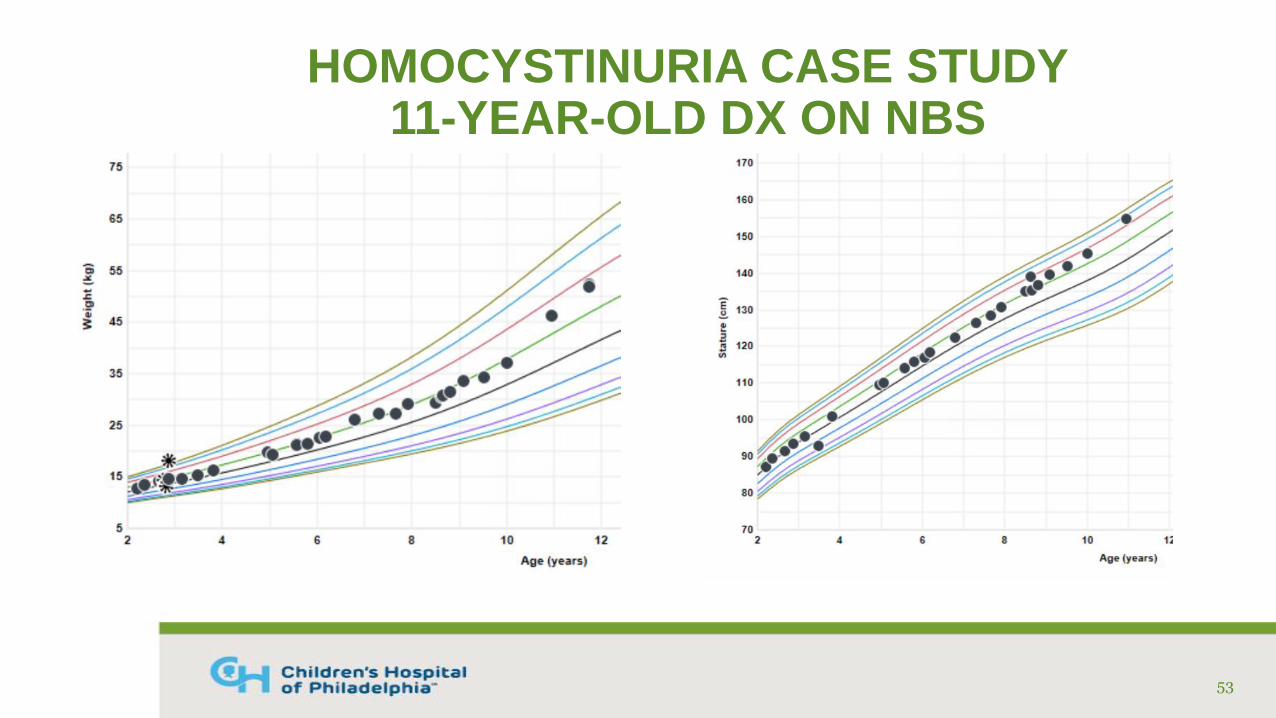
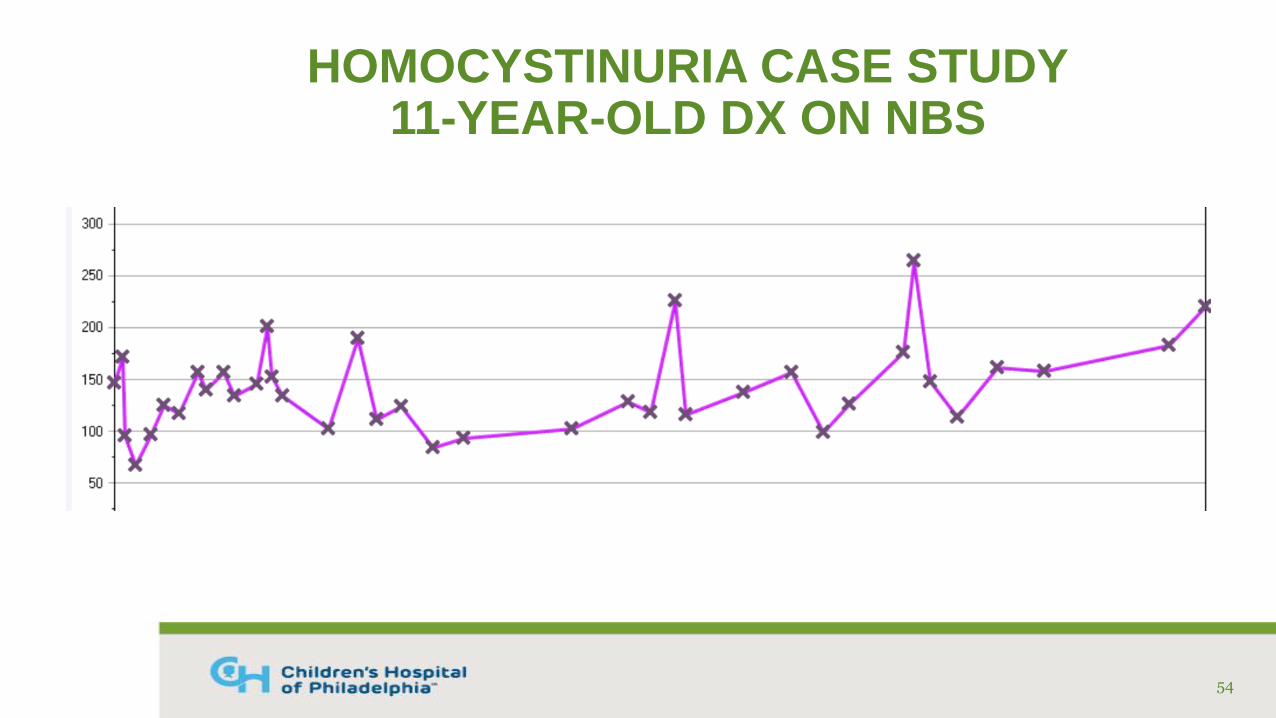
HOMOCYSTINURIA CASE STUDY11-YEAR-OLD DX ON NBS
• Diagnosed via NBS on DOL #11
• Confirmatory labs (umol/mL): Free Hcy: 32, Total Hcy: 147, Methionine: 1280
• Genetic testing: compound het. for 2 known mutations in CBS gene
• Treated with cystadane, pyridoxine, B12, folic acid, and low protein diet with medical food
• Fairly good compliance with medications and supplements
52
HOMOCYSTINURIA CASE STUDY11-YEAR-OLD DX ON NBS
53
HOMOCYSTINURIA CASE STUDY11-YEAR-OLD DX ON NBS
54

HOMOCYSTINURIA CASE STUDY11-YEAR-OLD DX ON NBS
• What interventions have we tried to increase dietary compliance?
• Education/reinforcement of diet at every clinic visit
• Suggestions on low protein foods available in local supermarkets
• Recipes, meal plans and ideas
• Formula samples, mixing ideas
• Networking, group education sessions
Is it effective?
55

HOMOCYSTINURIA CASE STUDY11-YEAR-OLD DX ON NBS
• Admitted to the hospital last week with recurrent pancreatitis• Prescribed diet: 15 grams protein from food and 45 grams protein
equivalents from medical food• Normal growth, weight 51.9 kg
• Unclear if protein goal is appropriate due to history of non-compliance
• Only taking 1 serving/15-gram PE from medical food
• Patient and mother received education on low protein diet and importance of medical food
• An hour after meeting with patient, RN notified RD that patient was trying to order high protein foods from the cafeteria
56

BARRIERS TO CARE
• WHY?
• Failure of patient/caregiver to fully understand the IEM• “My child feels fine” or “Nothing has happened, what’s the
point”
• “I can’t force my 11-year-old to eat these foods”
• Financial constraints or lack of insurance coverage
• Inadequate communication due to language barriers, low literacy, or lack of access to required technology (telephone, internet, etc.)
57

HOMOCYSTINURIARDN RESOURCES
• Diagnosis and treatment guidelines• Morris AA, Kožich V, Santra S, et al. Guidelines for the diagnosis
and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017;40(1):49-74.
• MU textbook• Thomas JA. Homocystinuria: Diagnosis and management. In:
Bernstein LE, Rohr F, Helm JR, eds. Nutrition Management of Inherited Metabolic Diseases. Springer; 2015:149-158.
58

REFERENCES
Morris AA, Kožich V, Santra S, et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017;40(1):49-74.
Bublil EM, Majtan T. Classical homocystinuria: From cystathionine beta-synthase deficiency to novel enzyme therapies. Biochimie. 2020; 173:48-56.
Thomas JA. Homocystinuria: Diagnosis and management. In: Bernstein LE, Rohr F, Helm JR, eds. Nutrition Management of Inherited Metabolic Diseases. Springer; 2015:149-158.
59
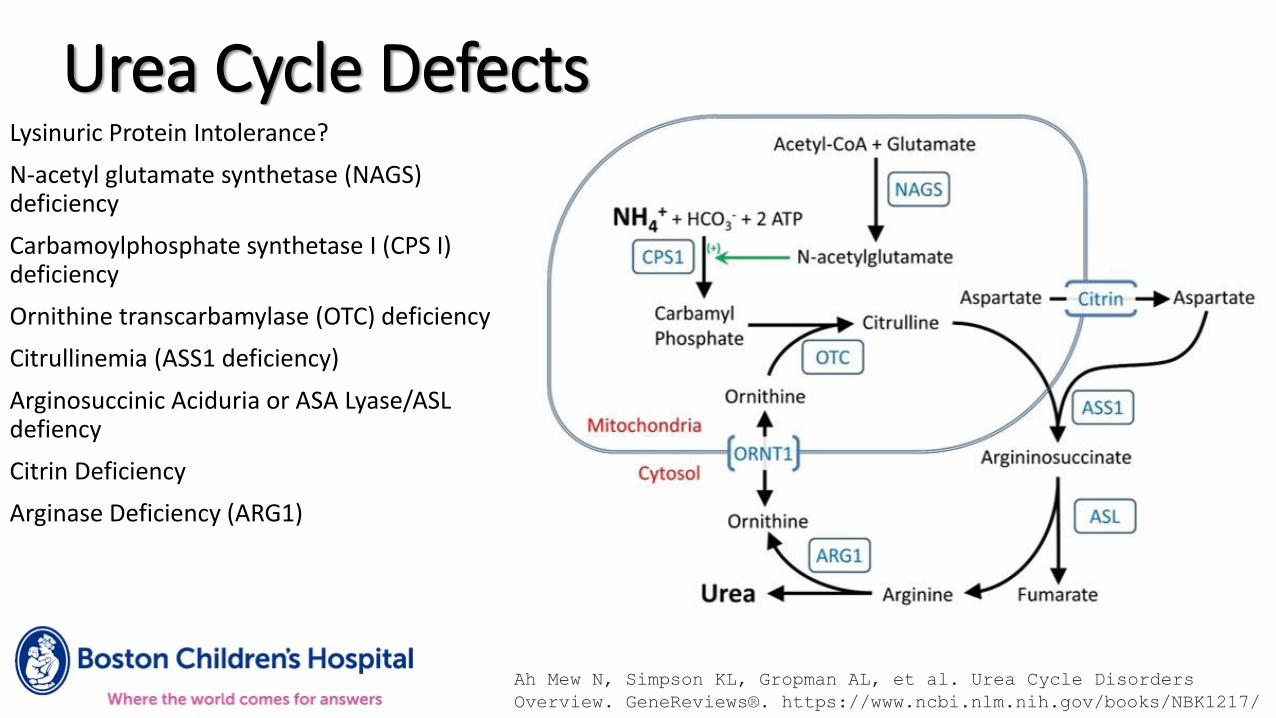
Urea Cycle DefectsLysinuric Protein Intolerance?
N-acetyl glutamate synthetase (NAGS) deficiency
Carbamoylphosphate synthetase I (CPS I) deficiency
Ornithine transcarbamylase (OTC) deficiency
Citrullinemia (ASS1 deficiency)
Arginosuccinic Aciduria or ASA Lyase/ASL defiency
Citrin Deficiency
Arginase Deficiency (ARG1)
Ah Mew N, Simpson KL, Gropman AL, et al. Urea Cycle Disorders
Overview. GeneReviews®. https://www.ncbi.nlm.nih.gov/books/NBK1217/

Urea Cycle DefectsInitial Presentation• Often undetected by NBS• Severe hyperammonemia in infancy with CPS1, OTC, ASS, ASL, NAGS
o Failure to feed, loss of thermoregulation, somnolenceo Progressing to lethargy, hyper/hypoventilation, seizures, coma
• In less severe defects, initial presentation may appear later in childhood or later in life
Early Onset Late Onset
Poor feedingVomitingTachypneaIrritabilityLethargyComa, death
Failure to thriveVomitingLiver dysfunctionLethargy IrritabilityBehavioral changesSeizures Protein avoidancePsychiatric symptoms

Urea Cycle Defects:Chronic treatment and management
• Meds use alternative pathways to excrete nitrogeno Sodium benzoate – binds glycineo Sodium phenylacetate – binds glutamineo Glycerol phenylacetate – binds glutamineo Ammunol – IV form phenylbuterate/ phenylacetate
• ILLNESS/EMERGENCY MANAGEMENT plans!• Social support and intervention services• Liver transplant in some cases

Urea Cycle Defects:Chronic treatment and management
• Diet o Protein limitation--not specific to any 1 amino acid and tolerance is variable by individual, disorder,
phenotypeo If less than RDA for protein tolerated, use essential amino acid formula because it is less ureagenico Protein-free formula prn for caloric/micronutrient adequacy
Adapted from R.H. Singh Nutritional Management of Urea Cycle Disorders
Age Intact Protein (g/kg/d)
Essential Amino Acid (g/kg/d)
Total Protein(g/kg/d)
0-1 yr 0.6-1.1 0.6-1.1 1.2-2.2
1-7 yr 0.4-0.5 0.6-0.7 1.0-1.2
7-19 yr 0.3-0.7 0.4-0.7 0.7-1.4
> 19 yr 0.2-0.5 0.3-0.5 0.5-1.0

Urea Cycle Defects:Treatment at Diagnosis or Following Acute Hyperammonemia
• Ammonia scavengerso May lead to low BCAAs and further catabolism
• Dialysis o Requires aggressive nutrition intervention to prevent worsening catabolic cascade
• Calories to halt catabolism and promote anabolismo IV fluids—dextrose, lipids (+ insulin?)o TPN as needed (usually w/ lower amino acid component)o Enteral feeds as early as possible
• Resuming enteral feedso High calories as much as toleratedo Reintroduce protein progressively but swiftly
Withholding >24 hrs may result in worsened catabolism Intact protein & essential amino acids
o Eventual diet goal for severe defect= 50:50 to 70:30 intact: essential amino acid ratio

Urea Cycle Defects
Monitoring• Neurocognitive outcomes• Labs—plasma amino acids, NH3, other nutritional markers• Growth
o Caloric adequacy critical, so excessive weight gain can be problematic after early infancyo Caution with weight loss even in older patients
Particular Challenges• Feeding adequacy without overfeeding• Family anxiety• Neurocognitive deficits, in patient and caregiver in some cases• Adherence to diet/formula• Transition to adulthood and adult medical care• Establishing healthy eating pattern and healthy relationship to food

UCD References Singh RH, Rhead WJ, Smith W, Lee B, Sniderman King L, Summar M. Nutritional management of urea cycle
disorders. Crit Care Clin. 2005 Oct;21(4 Suppl):S27-35. doi: 10.1016/j.ccc.2005.08.003. PMID: 16227113.
Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. Published 2012 May 29. doi:10.1186/1750-1172-7-32
Berry GT, Steiner RD. Long-term management of patients with urea cycle disorders. J Pediatr. 2001 Jan;138(1 Suppl):S56-60; discussion S60-1. doi: 10.1067/mpd.2001.111837. PMID: 11148550.

GLUTARIC ACIDEMIA TYPE I
• Genetic defect in glutaryl-CoA dehydrogenase• Lysine, hydroxylysine, and tryptophan metabolism
• Accumulation of 3-hydroxyglutaric and glutaric acid
• Diagnosed via newborn screen or based on symptoms• Typically presents in first 6 years of life
• Brain atrophy, macrocephaly, acute dystonia, stroke
• Often triggered by intercurrent infection
• Especially vulnerable between 6-18 months of age
67
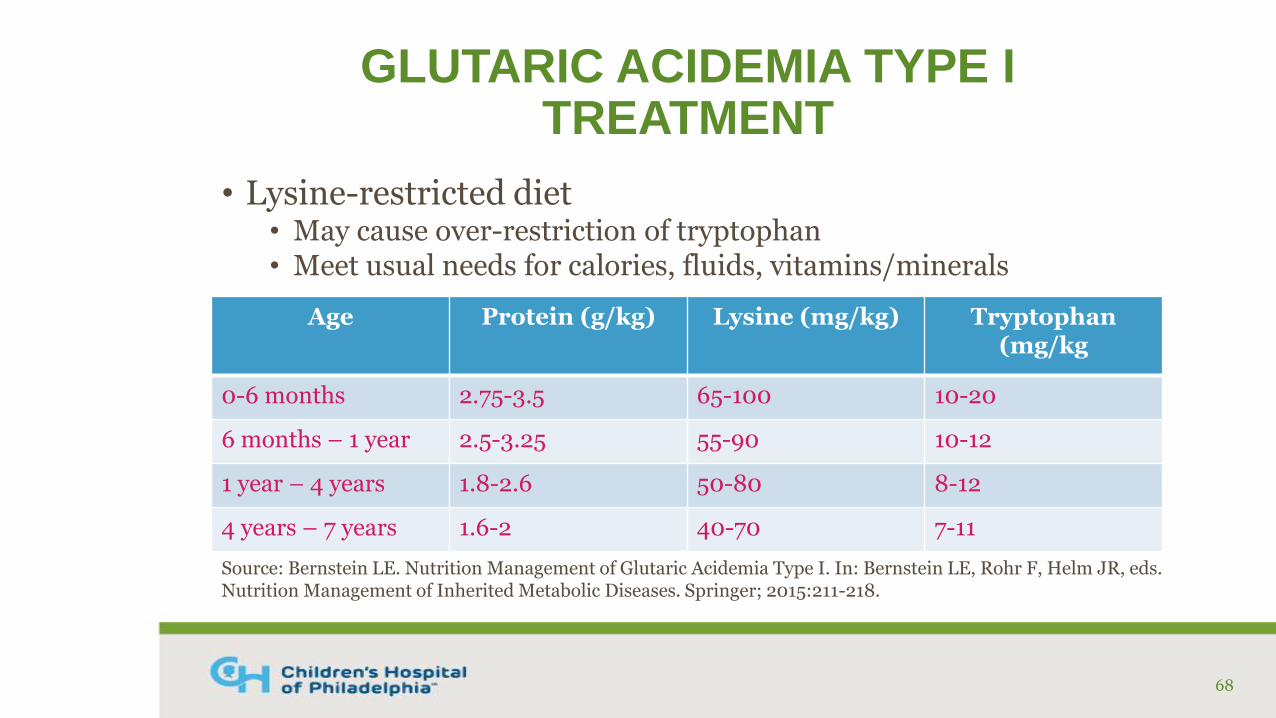
GLUTARIC ACIDEMIA TYPE ITREATMENT
• Lysine-restricted diet• May cause over-restriction of tryptophan• Meet usual needs for calories, fluids, vitamins/minerals
68
Age Protein (g/kg) Lysine (mg/kg) Tryptophan(mg/kg
0-6 months 2.75-3.5 65-100 10-20
6 months – 1 year 2.5-3.25 55-90 10-12
1 year – 4 years 1.8-2.6 50-80 8-12
4 years – 7 years 1.6-2 40-70 7-11
Source: Bernstein LE. Nutrition Management of Glutaric Acidemia Type I. In: Bernstein LE, Rohr F, Helm JR, eds. Nutrition Management of Inherited Metabolic Diseases. Springer; 2015:211-218.

GLUTARIC ACIDEMIA TYPE ITREATMENT
• Lysine to Arginine ratio 1:1.5 – 1:2• Arginine competes with lysine for uptake in the brain
• Carnitine (75-100 mg/kg/day)• Adjust dose to maintain normal free carnitine levels; may cause
diarrhea and fishy odor
• Riboflavin (100 mg/day), Pantothenic acid (400-600 µg/kg)
• Early initiation of treatment reduces risk of neurological complications
69

GLUTARIC ACIDEMIA TYPE ISAMPLE DIET PLAN
• 5 day old, 3.2 kg male
70
• Add water to make 17 fluid ounces (22 cal/oz formula)• Provides: 160 mL/kg, 110 kcal/kg, 2 g/kg total protein, 78 mg/kg lysine,
25 mg/kg tryptophan• Arginine:lysine ratio 1.6

GLUTARIC ACIDEMIA TYPE IILLNESS MANAGEMENT
• Aggressive illness management is essential • Goal is to prevent encephalopathic crisis and subsequent injury to basal
ganglia and neurological sequelae
• Any illness (especially if febrile) requires consultation with Metabolism team and possible ED visit/admission
• Consider 23-hour admission for routine vaccinations
• D10 IV fluids, limit/stop complete protein, extra carnitine, treat the source of illness/infection
• If tolerating PO, encourage intake of extra medical food and protein-free foods and drinks
• Use protein-free calorie modular to meet increased needs to curb catabolism
71

REFERENCES
Bernstein LE. Nutrition Management of Glutaric Acidemia Type I. In: Bernstein LE, Rohr F, Helm JR, eds. Nutrition Management of Inherited Metabolic Diseases. Springer; 2015:211-218.
Coughlin CR. Glutaric Acidemia Type I: Diagnosis and Management. In: Bernstein LE, Rohr F, Helm JR, eds. Nutrition Management of Inherited Metabolic Diseases. Springer; 2015:203-210.
Strauss KA, Puffenberger EG, Robinson DL, Morton DH. Type I glutaricaciduria, part 1: natural history of 77 patients. Am J Med Genet C Semin Med Genet. 2003 Aug 15;121C(1):38-52.
Strauss KA, Williams KB, Carson VJ, et al. Glutaric acidemia type 1: Treatment and outcome of 168 patients over three decades. Mol Genet Metab. 2020 Nov;131(3):325-340
72

ISOVALERIC ACIDEMIA
• Genetic defect in isovaleryl-CoA dehydrogenase• Leucine metabolism
• Accumulation of organic acids causes “sweaty feet” odor
• Highly variable clinical presentation• Acute neonatal form: acute metabolic acidosis, coma, death
• Chronic intermittent form: developmental delay, acidosis during periods of illness/catabolism
• Mild/asymptomatic form
• Neurocognitive outcomes improved with early diagnosis and treatment
73

ISOVALERIC ACIDEMIATREATMENT
• Leucine-restricted (low protein) diet
• Leucine-free medical food (?)
• Carnitine (50-100 mg/kg)
• Glycine (150-250 mg/kg)
• Sick Days: D10 IV fluids, ammonia scavengers, dialysis
74
Source: Abbott Nutrition Support Protocols, 4th ed, page 113

ISOVALERIC ACIDEMIARDN RESOURCES
• Nutrition Management Protocol• Abbott Nutrition Support Protocols, 4th edition (protocol 6)
• MU Textbook• Thomas JA. Organic Acidemias. In: Bernstein LE, Rohr F, Helm JR,
eds. Nutrition Management of Inherited Metabolic Diseases. Springer; 2015:187-201.
75

REFERENCES
Grünert SC, Wendel U, Lindner M. et al. Clinical and neurocognitive outcome in symptomatic isovalericacidemia. Orphanet J Rare Dis. 2012; 7:9. doi: 10.1186/1750-1172-7-9.
Thomas JA. Organic Acidemias. In: Bernstein LE, Rohr F, Helm JR, eds. Nutrition Management of Inherited Metabolic Diseases. Springer; 2015:187-201.
76

Fatty Acid Oxidation Disorders
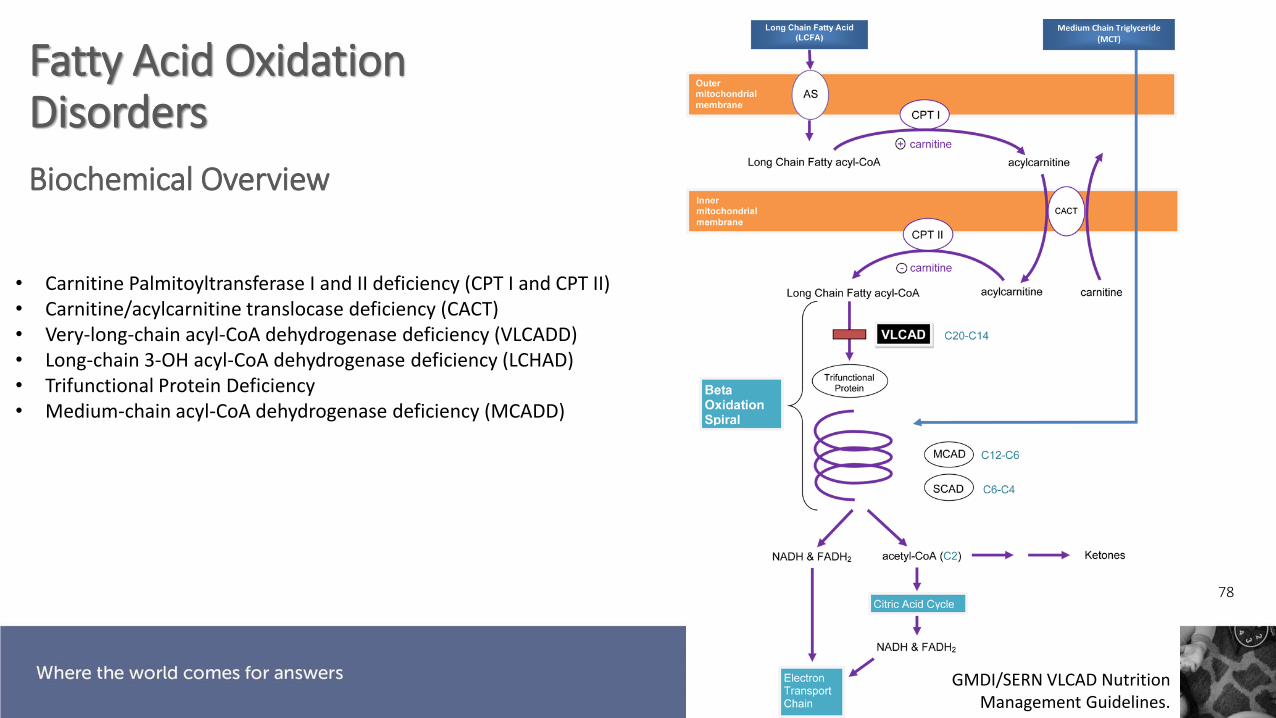
78
Fatty Acid Oxidation Disorders
Biochemical Overview
GMDI/SERN VLCAD Nutrition Management Guidelines.
• Carnitine Palmitoyltransferase I and II deficiency (CPT I and CPT II)• Carnitine/acylcarnitine translocase deficiency (CACT)• Very-long-chain acyl-CoA dehydrogenase deficiency (VLCADD)• Long-chain 3-OH acyl-CoA dehydrogenase deficiency (LCHAD)• Trifunctional Protein Deficiency• Medium-chain acyl-CoA dehydrogenase deficiency (MCADD)

Fatty Acid Beta Oxidation
MCAD No defect
EnergyProduced
Toxicity/Unusable substrate
LCHAD/VLCAD
C20
C18
C16
C14
C12
C10
C8
C6
C4
C2
Slide adapted from Derek Wong, MD
MCT

Signs/Symptoms of FAODs
LC-FAOD (early onset) LC-FAOD (late onset) MCAD
• Hypoketotic hypoglycemia• Encephalopathy• Liver dysfunction• Cardiomyopathy• Cardiac dysrhythmias• Sudden death
LCHAD/TFP deficiency – maternal HELLP (hemolysis, elevated liver enzymes, low platelets)
• Exercise or illness induced rhabdomyolysis
• Chronic myopathy• May remain asymptomatic
• Typically asymptomatic when well• Crisis symptoms:• hypoglycemia, Reye-like syndrome,
arrhythmia, seizures, coma, death

General Treatment of LC-FAODs and MCADD
• Reduce reliance on fat as energy source prevent fasting• 0-4 months = < 4 hours• 5-12 months = +1 hour per month• 12+ months = 10-12 hours maximum
• Provide sufficient energy to prevent catabolism
• Adequate EFAs and total fat diet for age• For LC-FAODs, reduce LCF and supplement with MCT to meet total fat goals
• Hydration• normal requirements plus sports, work• Increased for heat/outside weather
• Prompt treatment of illness, infections & emergency plan• Reduce fasting time with frequent glucose/carbohydrate feeds as able• Emergency room for IV fluids with dextrose prn
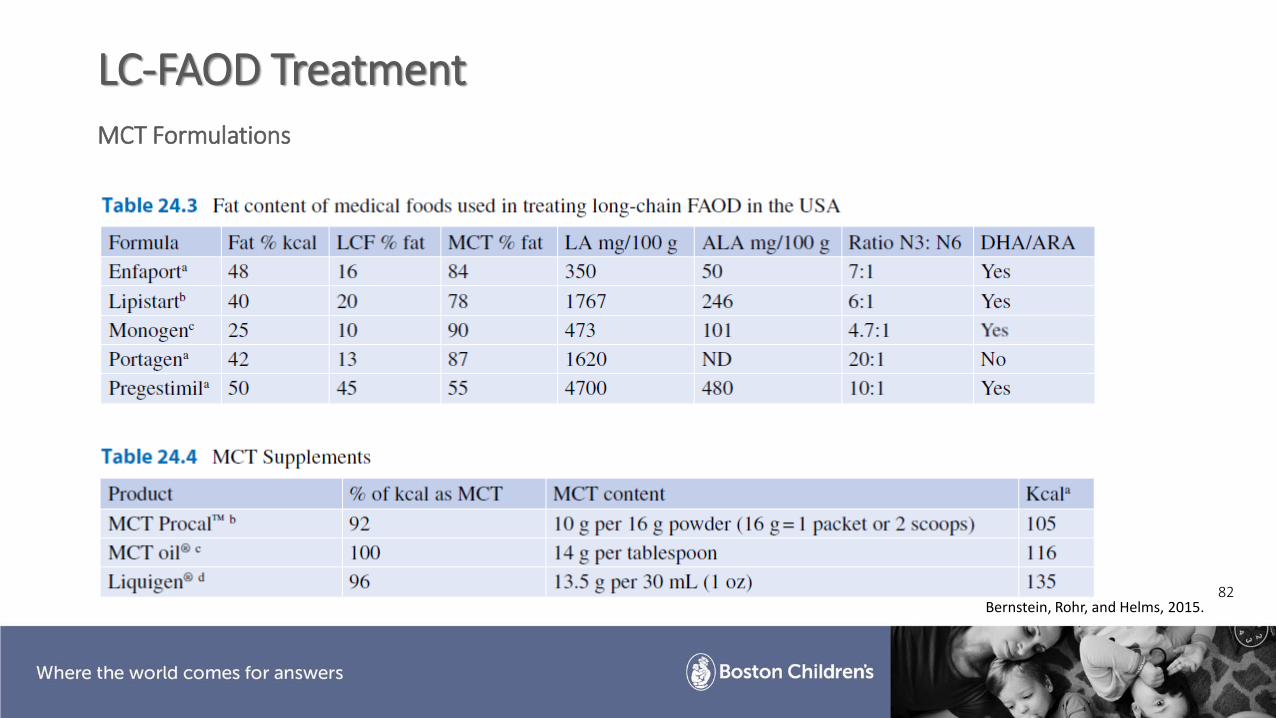
82
LC-FAOD TreatmentMCT Formulations
Bernstein, Rohr, and Helms, 2015.

83
Risks Associated with LC-FAOD Diet Interventions
• EFA deficiency due to low LCFA intake
oCheck essential fatty acid profiles, use total values versus ratios
oConsider supplementation to meet RDAs or use food sources carefully
• Fat Soluble Vitamin deficiencies
oMonitor blood levels in regular intervals
o Supplement (w/ H2O soluble forms?) as needed
• Poor growth/excessive weight gain
• Nausea, GI distress due to rapid advancement of MCT or a large total volume of MCT
• Over-medicalization

84
VLCAD Diet in Infancy
• Breastmilk or standard infant formula may be used for LCF source if prescription allows
• May pair with lowest LCF, highest MCT formula available to allow for greatest % breastmilk possible
• Potential risks of breastfeeding
• Intake adequacy difficult to assess especially in neonatal period
• Increased LCF intake
• Potential benefits of breastfeeding
• Overall health benefits
• Feeding frequency may be higher
• Feeding during illness may be more easily achieved?
Breastmilk
Low LCF, high MCT Medical Food
Mild Yes
Moderate Yes Consider
Severe Yes
Symptomaticreturned to asymptomatic state
Consider
Recommendations for Asymptomatic
VLCAD Infant1
1Adapted from GMDI/SERN Nutrition Management
Guidelines Recommendation 1.6
*
*

85
VLCAD Diet in Childhood:How do we administer MCT?
MCT oil Emulsified MCT
Powdered MCT
• Meet remaining 35% total calorie from fat
• Reasonable quantity to consume
• Timed to activity
• A change in activity or symptoms re-time an existing dose versus increase/add dose
• Not necessary at bedtime (use a carb + protein snack instead)
• For example:
• 25% LCF, 10% MCT = ~ a single dose timed with activity/exercise
• 20% LCF, 15% MCT = ~ 2 daily doses timed before school and before afternoon playtime
• 10% LCF, 25% MCT = ~3 daily doses timed before school, with lunch, and mid-afternoon
MCT Formulations
When to adjust MCT?

FAOD References GMDI/SERN Management Guidelines Portal. VLCAD Nutrition Management Guidelines. https://southeastgenetics.org/ngp/guidelines.php/106/VLCAD%20Nutrition%20Guidelines/Version%201.0. February 2019.
Gillingham MB. (2009). Nutrition Management of Patients with Inherited of Mitochondrial Fatty Acid Oxidation. In P. Acosta (Ed.), Nutrition Management of Patients with Inherited Metabolic Disorders (pp 369-403). Sudbury, Massachusetts: Jones & Bartlett Learning.
Behrend, A.M., Harding, C.O., Shoemaker, J.D., Matern, D., Sahn, D.J., Elliot, D.L., & Gillingham, M.B. (Jan 2012). Substrate oxidation and cardiac performance during exercise in disorders of long chain fatty acid oxidation. Mol. Genet. Metab., 105, 110-5. doi:10.1016/j.ymgme.2011.09.030

GALACTOSEMIA
• Clinical variant form: neonatal complications• Vomiting, diarrhea, jaundice, liver dysfunction (decreased INR), bilateral
cataracts, sepsis, death from hepatic and renal failure• Progressive after initiation of galactose containing feeds (typically present on
DOL 3-4)• Requires treatment with galactose restricted diet• S135L in African Americans have neonatal symptoms but no long-term
complications
• Biochemical variant (Duarte): asymptomatic• Residual enzyme activity; typically, only detected due to newborn screen• Moderate elevations in total galactose and decreased GALT activity (15-35%)• Do not need to be treated with galactose restricted diet or galactose challenge• May continue to receive breast milk or standard infant formula
• Epimerase and Galactokinase deficiency
87

CLASSICAL GALACTOSEMIADIETARY INTERVENTION
• Life-long lactose free, low galactose diet
• Soy formula (powder or ready to feed) in the first year• Elemental formula may be used for those with persistently elevated
metabolites
• Long-term benefit unclear; increased cost
• Transition to a toddler soy formula or a milk alternative (soy, rice, almond, coconut, hemp, etc.) depending on growth and dietary intake
88

CLASSICAL GALACTOSEMIADIETARY MANAGEMENT
• Food ingredient labels need to be read to check for the presence of restricted ingredients
• Ingredients change frequently
• Lactose may be used as a base component, filler, or diluent in medications/dietary supplements
89

CLASSICAL GALACTOSEMIADIETARY MANAGEMENT
Restricted ingredients
Breast milk, milk based infant formulas
Milk-based foods and beverages: low lactose milk, cream, ice cream, sour cream, yogurt, buttermilk, milk chocolate
Milk-based ingredients: buttermilk solids, casein, dry milk protein, dry milk solids, hydrolyzed whey protein, hydrolyzed casein protein, lactose, lactalbumin, lactoglobulin, lactostearin, whey, whey solids, curds, milk derivatives
Cheese and cheese based products, except as noted on following slide
Butter, some margarines
Processed meats using lactose
90

CLASSICAL GALACTOSEMIADIETARY MANAGEMENT
Allowed foods & ingredients
Soy-based infant formulas, amino acid-based elemental formulas
All fruits, vegetables and their juices, pickled fruit and vegetables
All legumes
Non-fermented soy-based products (soy milk, tofu, textured soy protein, hydrolyzed vegetable protein, soy protein concentrate, meat analogs
Aged cheeses: Jarlsberg, Emmentaler, Swiss, Gruyere, Tilsiter, mature Parmesan, mature Cheddar cheese
Sodium & calcium caseinate
All cocoa except milk chocolate
Natural & artificial flavorings, all gums, carrageenan
91
Allowed in moderation: soy sauce, fermented soy products, meat by-products, organ meats

CLASSICAL GALACTOSEMIABONE HEALTH
• Calcium and vitamin D intake may be limited due to dietary restrictions
• Calcium: fortified dry cereal, enriched dairy free milk alternatives, sardines, tofu, fortified orange juice, salmon, leafy greens
Calcium citrate or carbonate are preferred forms
• Vitamin D: sun, cod liver oil, fatty fish, canned tuna, enriched dairy free milk alternatives, fortified orange juice
• May require supplements if unable to meet needs via food sources
• Consider monitoring 25(OH) vitamin D levels annually
92

RESOURCES FOR PATIENTS
• Galactosemia Foundation @ www.Galactosemia.org
• Resources available through University of Colorado IMD Nutrition program (The Diet, School Age Children, New Parents, Making Healthy Choices cookbook & activity book)
• New England Consortium of Metabolic Programs
93

REFERENCESFiciciglu C, Hussa C, Gallagher PR, Thomas N, Yager C. Monitoring of biochemical status in children with Duarte Galactosemia: Utiilty of Galactose, Galactitol, Galactonate, and Galactose-1-Phosphate. Clin Chem. 2010; 56(7):1177-1182.
Ficicioglu C, Hussa C, Yager C, Segal S. Effect of galactose free formula on galactose-1-phosphate in two infants with classical galactosemia. Eur J Pediatr. 2008; 167(5):595-596.
Van Calcar SC, Bernstein LE, Rohr FJ, Scaman CH, Yannicelli S, Berry GT. A re-evaluation of life-long severe galactose restriction for the nutrition management of classic galactosemia. Mol Genet Metab. 2014; 112(3):191-197.
Welling L, Bernstein LE, Berry GT, et al. International clinical guideline for the management of classical galactosemia; diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017; 40(2):171-176.
94

GSD Resources
• “Resources for GSD”, Vitaflo VIA: https://www.vitaflo-via.com/disorder-resources/glycogen-storage-disease
• ACMG Practice Guidelines
• NORD: https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/

Discussion Topics and Questions
• Adolescent compliance and diet palatability
• Implementing diet after newborn screen—when is ideal to do it? When does the RD get involved?
• New to NBS disorders (i.e. Pompe) or other “non-diet” IEMs
• “What alternative food should be given when medical food is not available to the patient?”





























