Embed Size (px)
Citation preview

ORIGINAL ARTICLE
Taxol Directly Induces EndoplasmicReticulum-Associated Calcium Changes That PromoteApoptosis in Breast Cancer Cells
Zhi Pan, PhD and Lauren Gollahon, PhD
Department of Biological Sciences, Texas Tech University, Lubbock, Texas
n Abstract: Calcium, a key regulator of cell survival, is also important in regulating apoptosis. Although the chemothera-peutic agent Taxol employs apoptosis to induce cell death, the exact mechanism of how it induces apoptosis and the roleof calcium in this process remains unclear. The main intracellular calcium storehouse, the endoplasmic reticulum, was iden-tified as a new important gateway in apoptosis, possibly providing a target for Taxol. The goal of this study was to investi-gate whether calcium changes associated with the endoplasmic reticulum, were directly or indirectly generated by Taxol atclinically relevant doses, and related to Taxol-induced apoptosis in breast cancer cells. Time-lapsed imaging techniquesfollowed by an endoplasmic reticulum-targeted construct, cameleon D1ER, were used to monitor cytosol––endoplasmicreticulum calcium dynamics in MDA-MB-468 (Bcl-2 negative) and MCF 7 (Bcl-2 positive) breast carcinoma cells. Apoptosislevels were measured with Annexin V and Propidium Iodide (PI) using flow cytometry. In both cell lines, Taxol at 2.5 lM(�10)6 M) was observed to induce significant internal calcium changes, first a rapid endoplasmic reticulum calcium releaseand a transient cytosolic calcium increase upon Taxol addition. After several hours of Taxol treatment, the endoplasmicreticulum calcium store was gradually depleted, and a sustained cytosolic calcium elevation was observed before significantinduction of apoptosis. Inhibition of these calcium changes decreased Taxol-induced apoptosis levels. In contrast, 0.2 lMTaxol (�10)7 M) induced only a slight cellular calcium change, not enough to regulate apoptosis. Our findings demonstratethat endoplasmic reticulum calcium stores provide a direct target for Taxol action and are important for induction of apopto-sis, independent of Bcl-2 status. Furthermore, our results show for the first time, that the role of calcium in Taxol-inducedendoplasmic reticulum-mediated apoptosis is dependent on Taxol dosage. n
Key Words: apoptosis, breast cancer, calcium (Ca2+), endoplasmic reticulum, Taxol
The main mechanism for most chemotherapeutic
agent-induced cell death is apoptosis, which is a
highly regulated, multi-pathway process (1–3). Since
the exact mechanism of apoptosis is dependent upon
both cell and inducer types, it needs to be studied in
those specific cell-inducer models (2–4). Taxol, an
important chemotherapeutic agent, is widely used in
breast cancer treatment. However, how Taxol induces
apoptosis in breast cancer cells remains unclear.
Although calcium (Ca2+) disturbances in regulating
apoptosis have been targeted by various inducers in
different cell types (5), the role of calcium in Taxol-
induced apoptosis of breast cancer cells is not widely
considered and as a result, is under-explored. There
are three possible reasons for this: (a) since Taxol was
initially found to be a microtubule stabilizer, previous
studies primarily focused on the relationship between
the microtubule and apoptosis (5–10). (b) The techno-
logical difficulty to measure dynamic calcium changes
in living cells (11,12) also limits our ability to study
the Taxol-Ca2+ relationship. (c) Calcium regulation in
nonexcitable cell type such as breast epithelium is
much more poorly elucidated than in excitable cell
types such as muscle and nervous cells (13–18). Given
its potency, Taxol may induce apoptosis through other
mechanisms in addition to stabilizing microtubules.
Calcium may act as an important cellular regulator
for Taxol-induced apoptosis since it has a dual role in
the fate of the cell. While stable calcium homeostasis
is necessary for normal cell survival, impaired calcium
homeostasis is toxic to cells and may induce cell death
(14–16). Normally, the cell must maintain a low cyto-
solic calcium level ([Ca2+]C�0.1 lM) due to calcium
cytotoxicity. Most internal calcium is stored in the
endoplasmic reticulum (ER) pool ([Ca2+]ER�0.1–
1 mM) (15–17). The high calcium content within the
Address correspondence and reprint requests to: Lauren Gollahon, PhD,
Department of Biological Sciences, Texas Tech University, Box 43131. Lub-
bock, TX 79409-3131, USA, or e-mail: [email protected].
DOI: 10.1111/j.1524-4741.2010.00988.x
� 2010 Wiley Periodicals, Inc., 1075-122X/10The Breast Journal, Volume 17 Number 1, 2011 56–70

ER is maintained by the balance between passive
release through calcium channel inositol 1,4,5-tris-
phosphate receptor (IP3R) and active calcium pump-
ing by Sarco ⁄ Endoplasmic Reticulum Ca2+-ATPase
pumps, SERCA in epithelial cell type (13–18). In
regards to programmed cell death, the ER is important
in calcium-apoptosis regulation because its capability
to mobilize calcium determines the cell’s ability to
undergo apoptosis in response to specific inducers
(19–24). Therefore, the ER has been suggested as an
important upstream gateway in apoptosis. The higher
the basal ER calcium level, the more calcium may be
released from the ER through IP3R channel to the
cytosol, resulting in either sensitization of mitochon-
dria to apoptosis or activation of apoptosis-associated
enzymes (24,25).
Previous studies (26,27) showed that Taxol treat-
ment results in the production of intermediates such
as lipid ceramide through de novo pathways from the
lipid membranes and reactive oxygen species (ROS)
from the mitochondrial matrix after several hours. In
other studies, ceramide and ROS reportedly triggered
a massive calcium release from the ER, initiating a
downstream mitochondrial apoptotic pathway in a
strictly ER calcium-dependent manner (21,22,28).
Taken together these studies suggest that Taxol may
be engaged in the ER calcium pathway in an indirect
manner after long-term exposure. Another report also
showed that some ER calcium changes were observed
after long-term exposure Taxol treatment (29,30),
supporting the idea that Taxol may indirectly affect
the ER.
However, the Taxol-ER Ca2+-apoptosis relationship
in breast cancer cells was not tested or confirmed, and
calcium regulation in cancer derived from epithelial
cell origin is not understood. No studies address
whether clinically relevant doses (0.1–10 lM) of
Taxol can directly affect calcium homeostasis and thus
regulate apoptosis in breast cancer cells. The purpose
of this study was to elucidate the role of calcium in
regulating Taxol-induced apoptosis in breast cancer
cells by addressing four questions: (a) Whether Taxol
directly or indirectly affects calcium homeostasis, espe-
cially the ER calcium store. (b) Whether Taxol-
induced calcium changes are related to apoptosis. (c)
Whether Taxol application conditions such as dosage
and exposure time are critical for Taxol-induced cal-
cium changes and apoptosis. (d) Whether other factors
such as antiapoptotic protein Bcl-2 status affect the
Taxol-ER Ca2+ calcium-apoptosis relationship.
MATERIALS AND METHODS
Cell Culture and Reagents
For this study, the breast cancer cell lines MDA-
MB-468 and MCF 7 were analyzed. M468 cells are
negative for the endogenous antiapoptotic Bcl-2 pro-
tein (31). Thus, the effects of Bcl-2 on calcium homeo-
stasis are eliminated. In contrast, MCF 7 breast cancer
cells show strong Bcl-2 expression (32). In addition,
M468 (estrogen receptor negative) and MCF 7 (estro-
gen receptor positive) representative two major sub-
types of breast cancer based on their estrogen receptor
status and hormone dependence (33,34). Both cell
lines were obtained from the American Tissue Culture
Center (ATCC, Manassas, VA), and cultured in
Dulbecco’s Modified Eagle’s Medium (DMEM),
(Sigma, St. Louis, MO), supplemented with 10% calf
serum (Hyclone, Logan, UT), 2 mM l-glutamine
(Sigma) and 1% penicillin ⁄ streptomycin (Sigma) at
37�C in 5% CO2. After reaching 70% confluence in
T25 flasks, cells were harvested with 1X 0.05% Tryp-
sin- 0.53 mM EDTA (Invitrogen-Gibco, Grand Island,
NY) and counted with a hemocytometer (Hausser
Scientific, Horsham, PA). Cells were plated at 50K
into each well of 24-well plates (BD Falcon, Franklin
Lakes, NJ) for 36 hours before commencing treat-
ments.
Taxol Treatment
Since the actions of Taxol on the cells depend on
the dose administered and the greatest difference was
observed between 10)6 and 10)7 M (7,35,36), Taxol
doses tested in this study needed to be considered. In
this study, two specific clinically relevant Taxol doses,
2.5 lM (high dose) and 0.2 lM (low dose) were used.
These are the estimated plasma levels in patients after
being administered a common dosage 135 mg ⁄ m2
for 3 hours and 24 infusion schedules, respectively
(37–39). Although the in vivo conditions are much
more complicated, we tried to more closely simulate a
clinically relevant phenomenon in studying the Taxol-
calcium relationship in breast cancer cells by recapitu-
lating these Taxol concentrations. In this study, Taxol
treatment within 10 minutes was defined as ‘‘short-
term’’ to capture rapid calcium changes directly
induced by Taxol addition. Taxol treatment for
greater than 1 hour was defined as ‘‘long-term’’ since
it may involve indirect and stable cellular changes.
Taxol was dissolved in dimethyl sulfoxide (DMSO)
solution and then diluted to the desired concentration
Calcium Regulates Taxol-Induced ER-Associated Apoptosis • 57

with medium. DMSO concentrations were kept below
0.5% in all experiments. Both Taxol and DMSO were
purchased from Sigma. Bovine serum albumin (BSA)
was purchased from Fisher Biotech Chemical (Fair
Lawn, NJ). The cytosolic calcium chelator 1,2-bis (o-
aminophenoxy) ethane-N,N,N¢,N¢-tetraacetic acid
with acetoxymethyl esters (BAPTA-AM), the ER cal-
cium pump inhibitor Thapsigargin (TG), the calcium
calibrating agents Ionomycin and EDTA were all
obtained from Invitrogen (Carlsbad, CA). The ER cal-
cium channel inhibitor 2-APB was purchased from
EMD Biosciences (San Diego, CA).
Apoptosis Measurements
Changes in apoptosis levels of cultured cells, under
different treatments, were measured by double label-
ing using an Annexin V-Fluorescein isothiocyanate
(FITC, green) and Propidium Iodide (PI, red) Apopto-
sis Kit (Biovision, Mountain View, CA). After trypsi-
nization, the suspended cells were stained with
Annexin V-FITC and PI according to the manufac-
turer’s instructions. The Annexin V-FITC green signal
(visualized at 526 nm) localized to the cell membrane,
signifying early apoptosis. PI signal (observed at
620 nm) bound to nuclear material with the accompa-
nying green fluorescence, indicated a later apoptotic
event. If PI alone was observed, this indicated a cell
necrotic event. An Epics XL-MCL flow cytometer
(Beckman Coulter, Miami, FL) was used to count the
populations of living, early apoptotic, late apoptotic
and necrotic cells based on their fluorescent proper-
ties. The Annexin V-FITC signal and the PI signal
were counted through flow cytometer channels FL1
and FL3, respectively. Between 5,000 and 10,000
cells were analyzed per run. A two-parameter dot plot
was constructed to show the log of FL1 on the X-axis
versus the log of FL3 on the Y-axis. The quadrant
region, based on FL1 ⁄ FL3 signal distribution, divided
the plot into four parts to calculate the percentages of
healthy, early apoptotic, late apoptotic and necrotic
cells.
ER Calcium Measurements
Since calcium fluctuates among different cellular
compartments to maintain its intracellular homeosta-
sis, it is difficult to detect the calcium dynamics and
determine the sources of calcium changes. In this
study, a time-lapsed imaging technique was used to
monitor the calcium dynamics to detect calcium in the
endoplasmic reticular (ER) lumen of cultured breast
cancer cells. A novel ER-targeted cameleon (kind gift
of Dr. Roger Tsien, University of California San
Diego) was used to specifically detect ER calcium lev-
els and provide direct results (40,41). The cameleon
D1ER plasmid was transfected into the cells using
Lipofectamine (Invitrogen) according to the manufac-
turer’s instructions. M468 and MCF 7 cells were
imaged 48 hours after transfection using an Olympus
IX 71-based deconvolving fluorescence microscope
with a digital cooled charge-coupled (CCD) camera
(C4742-95-12ER, Hamatatsu Photonics). The analysis
software used was SimplePCI, (Compix Imaging Cor-
poration, Sewickley, PA). Dual-emission ratio imaging
for D1ER was performed with a D436 ⁄ 20x excitation
filter, DCLP455 dichroic mirror, and two emission
filters D480 ⁄ 40 m and D535 ⁄ 30 m (Chroma Techno-
logy, Rockingham, VT) controlled by a lambda 10-2
filter wheel (Sutter Instruments, Novato, CA). For
each D1ER expressed cell, the FRET (535) ⁄ CFP (480)
emission ratio was fixed arbitrarily as 1 at the start of
the acquisition. Calibration, background and crosstalk
were corrected according to the protocol developed by
Palmer and Tsien (42). Images were collected every
15 seconds for 5 minutes at room temperature (RT).
Cytosolic Calcium Measurements
To detect cytosolic calcium levels, the calcium-spe-
cific fluorescent dye Fluo4-AM (Molecular Probes,
Eugene, OR), was loaded into M468 and MCF 7 cells
using a modified procedure adapted from the manu-
facturer (Molecular Probes). Briefly, cells were incu-
bated for 30 minutes at 37�C with 4 lM Fluo4-AM in
Hank’s buffered salt solution (HBSS) (Invitrogen,
Grand island, NY) with 1.3 mM calcium and 0.5%
BSA. The 0.02% nonionic detergent Pluronic F-127
(Molecular Probes) was added to assist in solubilizing
the nonpolar AM ester of Fluo4 in the loading solu-
tion. Cells were then washed two times with fresh
HBSS and incubated in HBSS at RT prior to detection.
The Fluo4-AM green fluorescence, ex 488 nm ⁄ em
526 nm, was proportional to cytosolic calcium levels.
The changes of Fluo4 intensity over time in cell popu-
lations, in response to different agents, were moni-
tored using flow cytometry (FL1 channel). After
measuring the basal intensity level of the unstimulated
Fluo4 cells, Taxol or DMSO was added into the tube
and acquisition continued for 5–10 minutes. In order
to evaluate the calcium release from the ER into the
cytosol in Taxol-treated cells, the ER calcium pump
inhibitor Thapsigargin (TG) was added into the media
58 • pan and gollahon

and the changes were recorded using a similar time-
lapsed fluorescence imaging technique with the same
microscope system described in ER calcium measure-
ments. Fluo4 imaging was accomplished by using an
S484 ⁄ 15x excitation filter, a BP505 dichroic mirror,
and an emission filter HQ525 ⁄ 50 m (Chroma Tech-
nology). The time scan was started in order to moni-
tor resting levels of Fluo4 for 30 seconds. TG was
added just prior to the next capture and monitoring
was continued every 15 seconds for 5 minutes. Off-
line data analysis was used to measure mean green
intensity for 10–15 cells at each time point to generate
the intensity plot over time. All imaging was done at
20X at RT.
Immunoblotting
Total protein was extracted from the M468 and
MCF 7 cells using M-PER mammalian cell protein
extraction reagent (Pierce, Rockford, IL). Protein con-
centration was determined with the BCA Assay
(Pierce). Protein samples were denatured by heating at
95�C for 5 minutes and then analyzed by SDS-PAGE
gel. A prestained molecular weight marker (Bio-Rad,
Hercules, CA) was used to monitor protein gel migra-
tion during the run and the subsequent transfer. The
membrane was incubated with anti-IP3R antibody
(Rabbit anti-human polyclonal IgG) from Santa Cruz
Biotechnology (Santa Cruz, CA) at a dilution of 1:200
to detect ER calcium channel IP3R levels. Anti-IP3R
antibody was raised against amino acids 2402-2701
mapped at the C terminus of human IP3R. The result-
ing band is around 115 kD in size. The signal was
visualized with horseradish peroxidase (HRP) second-
ary antibody (goat anti-rabbit IgG) at a dilution of
1:5000 and was developed using Supersignal Ultra
Chemiluminescence Kit (Pierce). b-Tubulin expression
was used as an internal control (1:1000 dilution) for
equal protein loading. The developed films were
scanned and images were processed using Photoshop
software.
Statistical Analyses
Data from this study were generated from three
independent experiments. Apoptosis levels measured
were represented as mean ± SD (standard deviation)
in the figures. In order to show the calcium changes
over time clearly, calcium curves were represented as
mean values in the figures, but peak levels were shown
as mean ± SD in the additional figures for comparison.
Statistical analysis was performed with a two-sided
independent Student’s t-test to compare two means,
and one-way analysis of variance (ANOVA) to com-
pare more than two means of one variable. If one-way
ANOVA demonstrated unequal means, the Tukey’s
honestly significant difference (HSD) test was used to
find which mean was different by conducting multiple
pair-wise comparisons. In order to determine whether
an interaction between two independent variables
existed, the two-way ANOVA test performed. For all
analyses, differences with p < 0.05 were considered
statistically significant and indicated with *, p < 0.01
was indicated with **.
RESULTS
Taxol Induces Dose and Time-Dependent Apoptosis
in Breast Cancer Cells
For total apoptosis in both cell lines, results using
two-way ANOVA indicated that low (0.2 lM) and
high (2.5 lM) Taxol doses showed parallel time-
dependent tendencies (Fig. 1a,d) with no significant
interaction between each other. For apoptotic effects
within each dose, one-way ANOVA analysis showed
that both doses induced a significant increase in apop-
tosis from 12 hours on (p < 0.01, Fig. 1a,d). For
M468 cells, differences in apoptosis levels between the
two doses was significant at 12 hours (p < 0.01,
Fig. 1a) and 24 hours (p < 0.05, Fig. 1a). However,
there was no significant dose difference observed in
MCF 7 cells.
Although the results for total apoptosis were com-
pelling, they did not allow delineation of cell death as
a result of early apoptosis or late apoptotic events.
Therefore the results were separated into early
(Fig. 1b,e) and late (Fig. 1c,f) stage apoptosis. Early
apoptosis was demonstrated to be the major form of
cell death induced by Taxol after a 12 hour exposure
(Fig. 1b,e, p < 0.01). However after 48 hours, most
cell death was attributed to late apoptosis and necrosis
(Fig. 1c,f). There is a dose-dependent difference in
early apoptosis similar to total apoptosis in M468
cells: significant at 12 hours (p < 0.01, Fig. 1b) and
24 hours (p < 0.05, Fig. 1b). In contrast, MCF 7 does
not demonstrate the same dose-dependence (Fig. 1e).
As with the reports from previous studies (43,44), our
results showed that MCF 7 cells are more resistant to
Taxol treatment. After a 12 hours exposure, the per-
centage of early apoptotic cells increased only 2 fold
and 2.5 fold for low and high dose Taxol respectively,
Calcium Regulates Taxol-Induced ER-Associated Apoptosis • 59
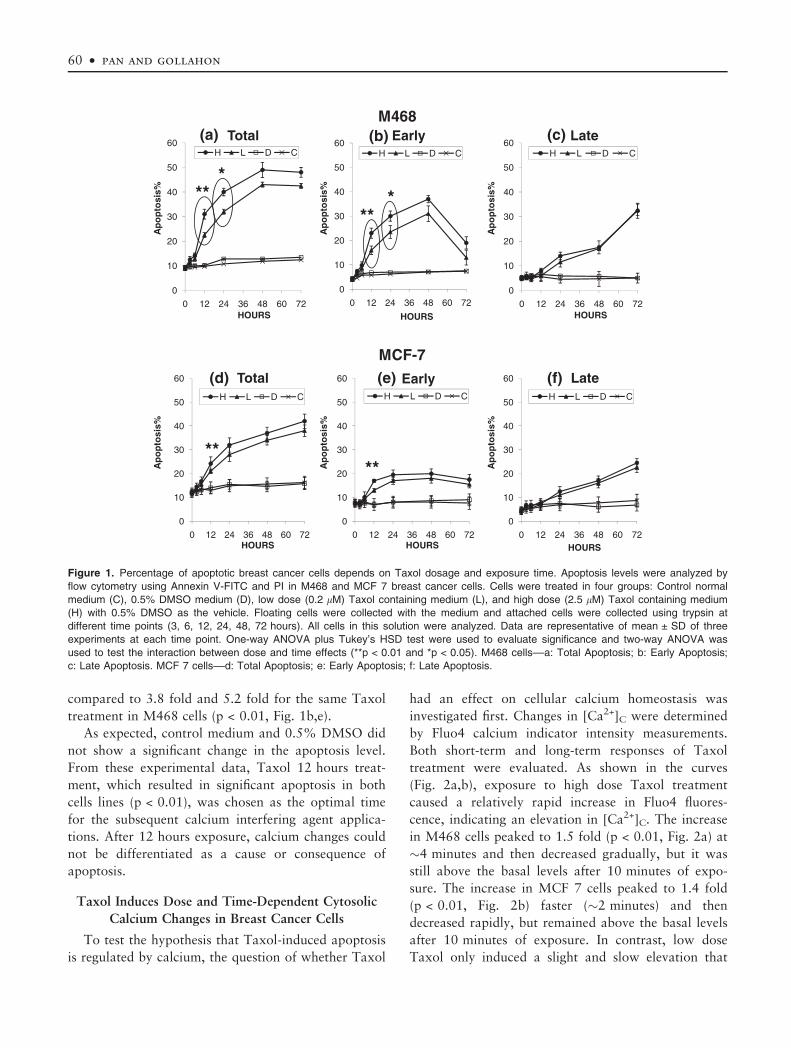
compared to 3.8 fold and 5.2 fold for the same Taxol
treatment in M468 cells (p < 0.01, Fig. 1b,e).
As expected, control medium and 0.5% DMSO did
not show a significant change in the apoptosis level.
From these experimental data, Taxol 12 hours treat-
ment, which resulted in significant apoptosis in both
cells lines (p < 0.01), was chosen as the optimal time
for the subsequent calcium interfering agent applica-
tions. After 12 hours exposure, calcium changes could
not be differentiated as a cause or consequence of
apoptosis.
Taxol Induces Dose and Time-Dependent Cytosolic
Calcium Changes in Breast Cancer Cells
To test the hypothesis that Taxol-induced apoptosis
is regulated by calcium, the question of whether Taxol
had an effect on cellular calcium homeostasis was
investigated first. Changes in [Ca2+]C were determined
by Fluo4 calcium indicator intensity measurements.
Both short-term and long-term responses of Taxol
treatment were evaluated. As shown in the curves
(Fig. 2a,b), exposure to high dose Taxol treatment
caused a relatively rapid increase in Fluo4 fluores-
cence, indicating an elevation in [Ca2+]C. The increase
in M468 cells peaked to 1.5 fold (p < 0.01, Fig. 2a) at
�4 minutes and then decreased gradually, but it was
still above the basal levels after 10 minutes of expo-
sure. The increase in MCF 7 cells peaked to 1.4 fold
(p < 0.01, Fig. 2b) faster (�2 minutes) and then
decreased rapidly, but remained above the basal levels
after 10 minutes of exposure. In contrast, low dose
Taxol only induced a slight and slow elevation that
0
10
20
30
40
50
60
0 12 24 36 48 60 72HOURS
H L D C
0
10
20
30
40
50
60
0 12 24 36 48 60 72
HOURS
H L D C
0
10
20
30
40
50
60
0 12 24 36 48 60 72HOURS
H L D C
***
***
LateEarlyTotal(a) (b) (c)M468
Ap
op
tosi
s%
Ap
op
tosi
s%
Ap
op
tosi
s%
0
10
20
30
40
50
60
0 12 24 36 48 60 72HOURS
H L D C
0
10
20
30
40
50
60
0 12 24 36 48 60 72HOURS
H L D C
0
10
20
30
40
50
60
0 12 24 36 48 60 72HOURS
H L D C
LateEarlyTotal (f)(e)(d)
MCF-7
Ap
op
tosi
s%
Ap
op
tosi
s%
Ap
op
tosi
s%
****
Figure 1. Percentage of apoptotic breast cancer cells depends on Taxol dosage and exposure time. Apoptosis levels were analyzed by
flow cytometry using Annexin V-FITC and PI in M468 and MCF 7 breast cancer cells. Cells were treated in four groups: Control normal
medium (C), 0.5% DMSO medium (D), low dose (0.2 lM) Taxol containing medium (L), and high dose (2.5 lM) Taxol containing medium
(H) with 0.5% DMSO as the vehicle. Floating cells were collected with the medium and attached cells were collected using trypsin at
different time points (3, 6, 12, 24, 48, 72 hours). All cells in this solution were analyzed. Data are representative of mean ± SD of three
experiments at each time point. One-way ANOVA plus Tukey’s HSD test were used to evaluate significance and two-way ANOVA was
used to test the interaction between dose and time effects (**p < 0.01 and *p < 0.05). M468 cells––a: Total Apoptosis; b: Early Apoptosis;
c: Late Apoptosis. MCF 7 cells––d: Total Apoptosis; e: Early Apoptosis; f: Late Apoptosis.
60 • pan and gollahon
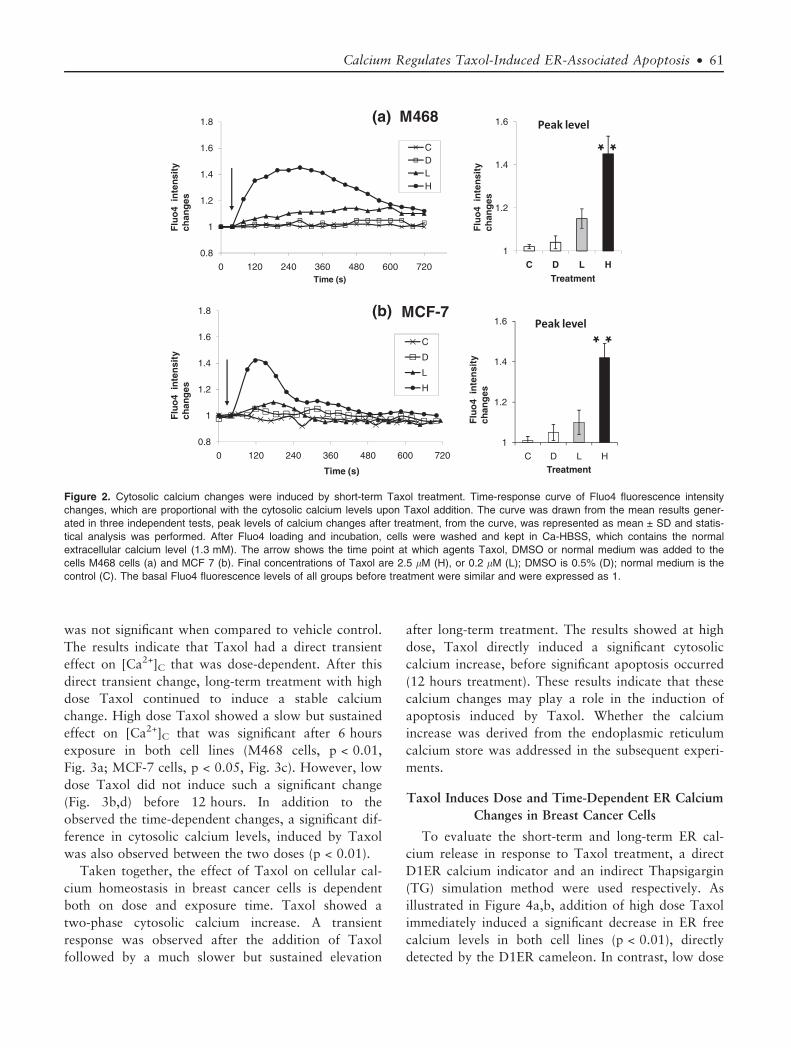
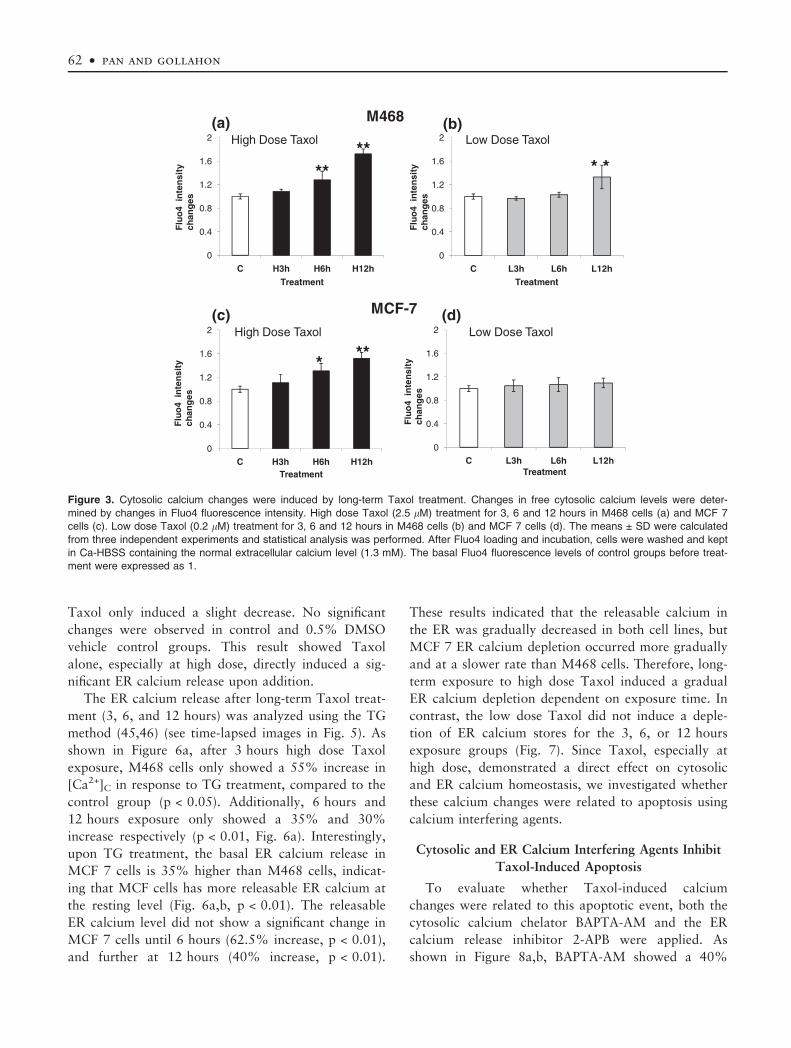
was not significant when compared to vehicle control.
The results indicate that Taxol had a direct transient
effect on [Ca2+]C that was dose-dependent. After this
direct transient change, long-term treatment with high
dose Taxol continued to induce a stable calcium
change. High dose Taxol showed a slow but sustained
effect on [Ca2+]C that was significant after 6 hours
exposure in both cell lines (M468 cells, p < 0.01,
Fig. 3a; MCF-7 cells, p < 0.05, Fig. 3c). However, low
dose Taxol did not induce such a significant change
(Fig. 3b,d) before 12 hours. In addition to the
observed the time-dependent changes, a significant dif-
ference in cytosolic calcium levels, induced by Taxol
was also observed between the two doses (p < 0.01).
Taken together, the effect of Taxol on cellular cal-
cium homeostasis in breast cancer cells is dependent
both on dose and exposure time. Taxol showed a
two-phase cytosolic calcium increase. A transient
response was observed after the addition of Taxol
followed by a much slower but sustained elevation
after long-term treatment. The results showed at high
dose, Taxol directly induced a significant cytosolic
calcium increase, before significant apoptosis occurred
(12 hours treatment). These results indicate that these
calcium changes may play a role in the induction of
apoptosis induced by Taxol. Whether the calcium
increase was derived from the endoplasmic reticulum
calcium store was addressed in the subsequent experi-
ments.
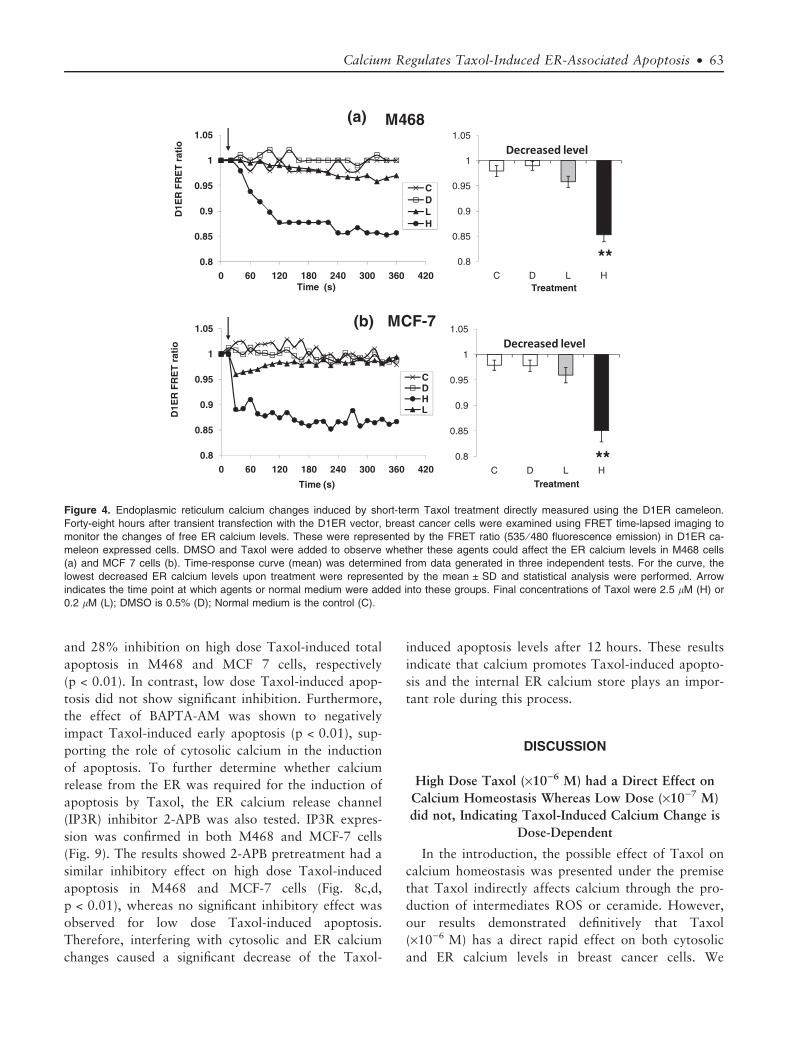
Taxol Induces Dose and Time-Dependent ER Calcium
Changes in Breast Cancer Cells
To evaluate the short-term and long-term ER cal-
cium release in response to Taxol treatment, a direct
D1ER calcium indicator and an indirect Thapsigargin
(TG) simulation method were used respectively. As
illustrated in Figure 4a,b, addition of high dose Taxol
immediately induced a significant decrease in ER free
calcium levels in both cell lines (p < 0.01), directly
detected by the D1ER cameleon. In contrast, low dose
0.8
1
1.2
1.4
1.6
1.8
0 120 240 360 480 600 720Time (s)
CDLH
1
1.2
1.4
1.6
C D L H
* *
(a)
Flu
o4
inte
nsi
ty
chan
ges
Flu
o4
inte
nsi
ty
chan
ges
Treatment
M468
0.8
1
1.2
1.4
1.6
1.8
0 120 240 360 480 600 720
C
D
L
H
Flu
o4
inte
nsi
ty
chan
ges
Time (s)
1
1.2
1.4
1.6
C D L HTreatment
* *
Flu
o4
inte
nsi
ty
chan
ges
MCF-7(b)
Figure 2. Cytosolic calcium changes were induced by short-term Taxol treatment. Time-response curve of Fluo4 fluorescence intensity
changes, which are proportional with the cytosolic calcium levels upon Taxol addition. The curve was drawn from the mean results gener-
ated in three independent tests, peak levels of calcium changes after treatment, from the curve, was represented as mean ± SD and statis-
tical analysis was performed. After Fluo4 loading and incubation, cells were washed and kept in Ca-HBSS, which contains the normal
extracellular calcium level (1.3 mM). The arrow shows the time point at which agents Taxol, DMSO or normal medium was added to the
cells M468 cells (a) and MCF 7 (b). Final concentrations of Taxol are 2.5 lM (H), or 0.2 lM (L); DMSO is 0.5% (D); normal medium is the
control (C). The basal Fluo4 fluorescence levels of all groups before treatment were similar and were expressed as 1.
Calcium Regulates Taxol-Induced ER-Associated Apoptosis • 61
Taxol only induced a slight decrease. No significant
changes were observed in control and 0.5% DMSO
vehicle control groups. This result showed Taxol
alone, especially at high dose, directly induced a sig-
nificant ER calcium release upon addition.
The ER calcium release after long-term Taxol treat-
ment (3, 6, and 12 hours) was analyzed using the TG
method (45,46) (see time-lapsed images in Fig. 5). As
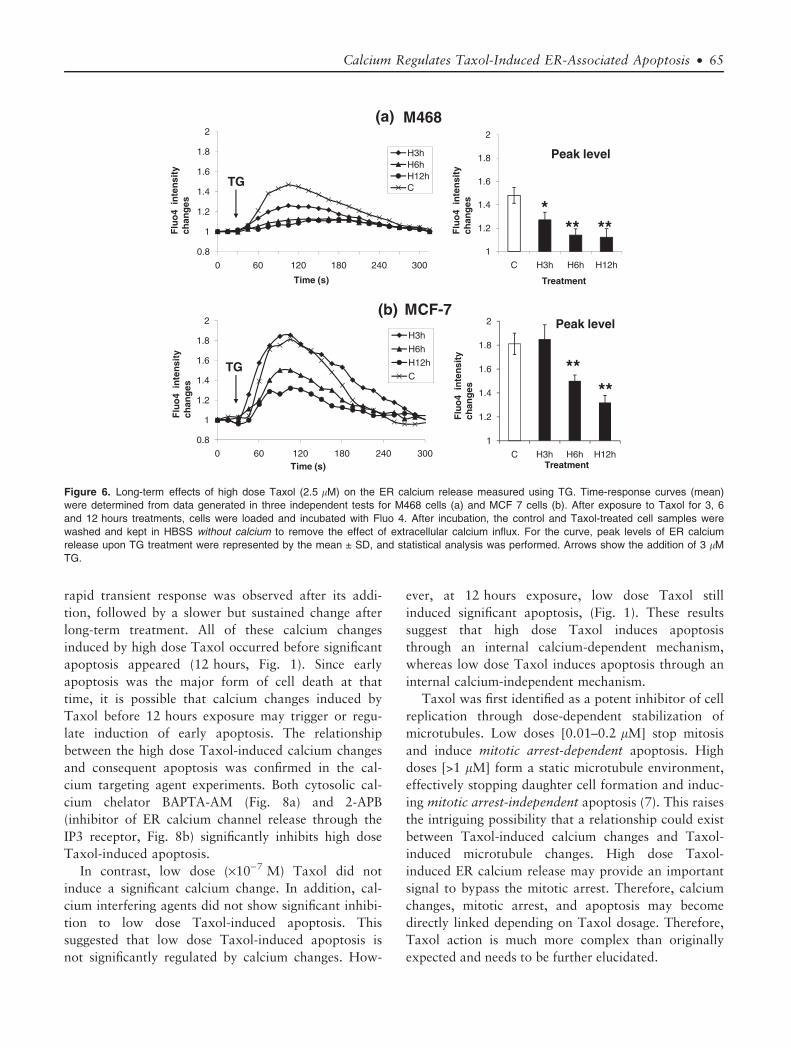
shown in Figure 6a, after 3 hours high dose Taxol
exposure, M468 cells only showed a 55% increase in
[Ca2+]C in response to TG treatment, compared to the
control group (p < 0.05). Additionally, 6 hours and
12 hours exposure only showed a 35% and 30%
increase respectively (p < 0.01, Fig. 6a). Interestingly,
upon TG treatment, the basal ER calcium release in
MCF 7 cells is 35% higher than M468 cells, indicat-
ing that MCF cells has more releasable ER calcium at
the resting level (Fig. 6a,b, p < 0.01). The releasable
ER calcium level did not show a significant change in
MCF 7 cells until 6 hours (62.5% increase, p < 0.01),
and further at 12 hours (40% increase, p < 0.01).
These results indicated that the releasable calcium in
the ER was gradually decreased in both cell lines, but
MCF 7 ER calcium depletion occurred more gradually
and at a slower rate than M468 cells. Therefore, long-
term exposure to high dose Taxol induced a gradual
ER calcium depletion dependent on exposure time. In
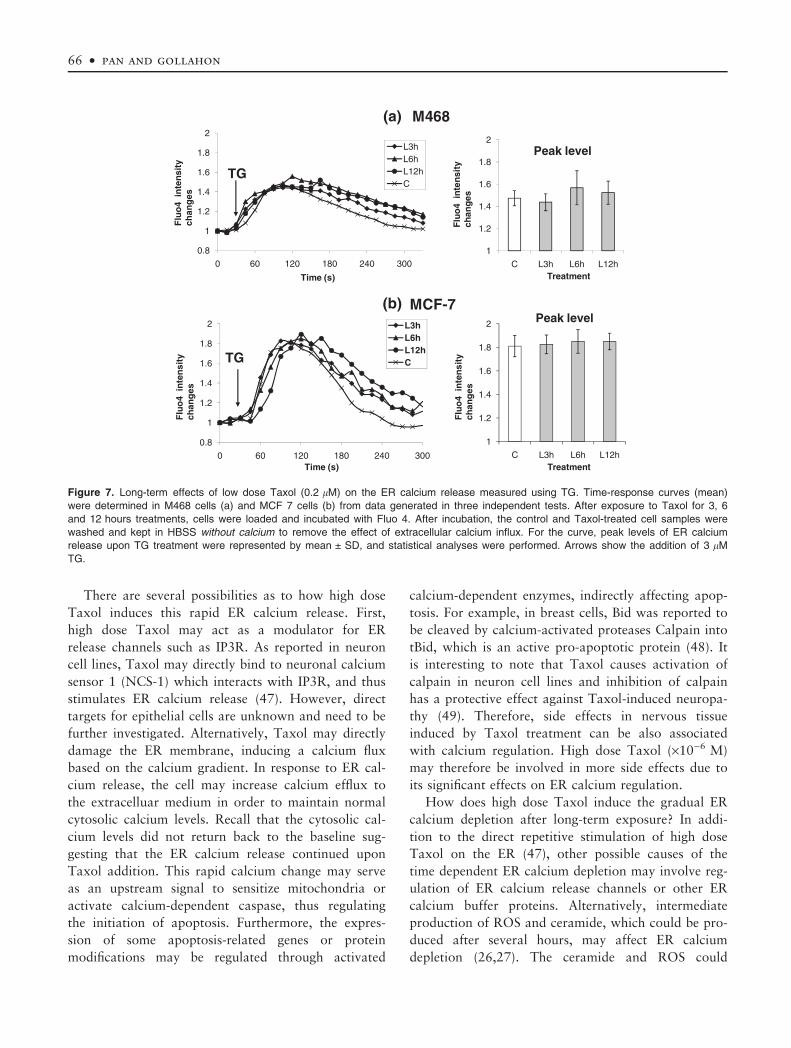
contrast, the low dose Taxol did not induce a deple-
tion of ER calcium stores for the 3, 6, or 12 hours
exposure groups (Fig. 7). Since Taxol, especially at
high dose, demonstrated a direct effect on cytosolic
and ER calcium homeostasis, we investigated whether
these calcium changes were related to apoptosis using
calcium interfering agents.
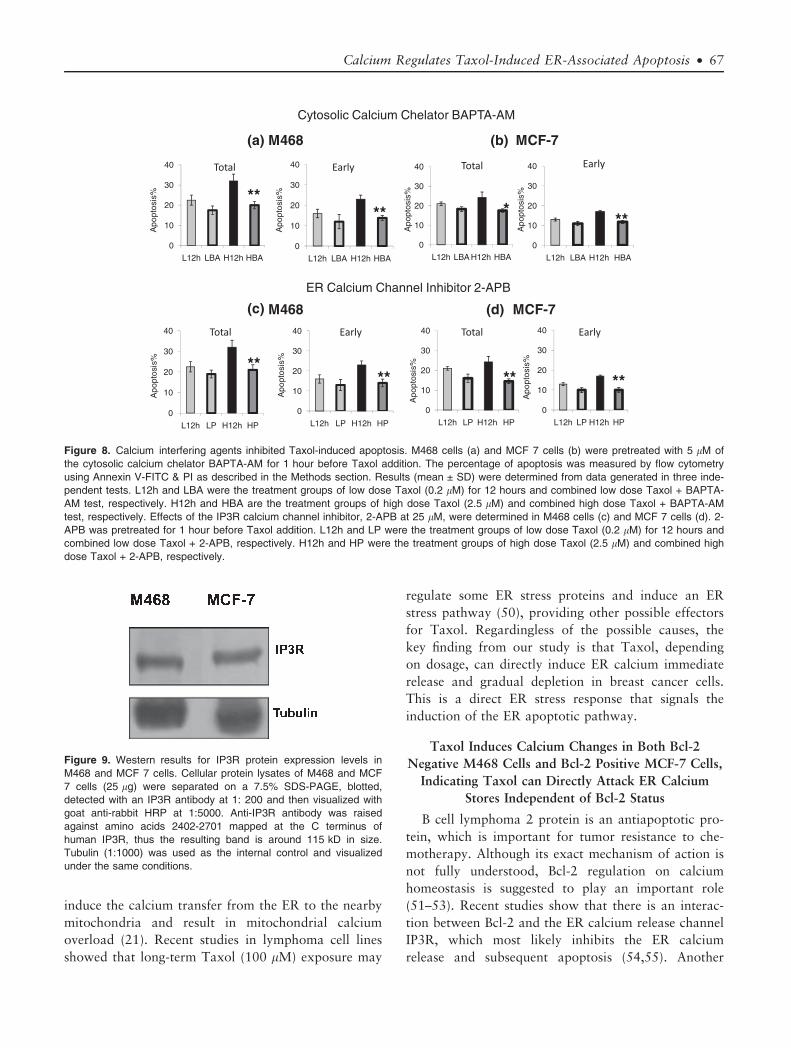
Cytosolic and ER Calcium Interfering Agents Inhibit
Taxol-Induced Apoptosis
To evaluate whether Taxol-induced calcium
changes were related to this apoptotic event, both the
cytosolic calcium chelator BAPTA-AM and the ER
calcium release inhibitor 2-APB were applied. As
shown in Figure 8a,b, BAPTA-AM showed a 40%
Treatment
0
0.4
0.8
1.2
1.6
2
C L3h L6h L12h0
0.4
0.8
1.2
1.6
2
C H3h H6h H12h
loxaTesoDwoLloxaTesoDhgiH
M468
****
* *
TreatmentTreatment
Flu
o4
inte
nsi
ty
chan
ges
Flu
o4
inte
nsi
ty
chan
ges
(a) (b)
0
0.4
0.8
1.2
1.6
2
C H3h H6h H12h
***
High Dose Taxol
0
0.4
0.8
1.2
1.6
2
C L3h L6h L12h
Low Dose Taxol
MCF-7
Flu
o4
inte
nsi
ty
chan
ges
Flu
o4
inte
nsi
ty
chan
ges
(d)(c)
Treatment
Figure 3. Cytosolic calcium changes were induced by long-term Taxol treatment. Changes in free cytosolic calcium levels were deter-
mined by changes in Fluo4 fluorescence intensity. High dose Taxol (2.5 lM) treatment for 3, 6 and 12 hours in M468 cells (a) and MCF 7
cells (c). Low dose Taxol (0.2 lM) treatment for 3, 6 and 12 hours in M468 cells (b) and MCF 7 cells (d). The means ± SD were calculated
from three independent experiments and statistical analysis was performed. After Fluo4 loading and incubation, cells were washed and kept
in Ca-HBSS containing the normal extracellular calcium level (1.3 mM). The basal Fluo4 fluorescence levels of control groups before treat-
ment were expressed as 1.
62 • pan and gollahon
and 28% inhibition on high dose Taxol-induced total
apoptosis in M468 and MCF 7 cells, respectively
(p < 0.01). In contrast, low dose Taxol-induced apop-
tosis did not show significant inhibition. Furthermore,
the effect of BAPTA-AM was shown to negatively
impact Taxol-induced early apoptosis (p < 0.01), sup-
porting the role of cytosolic calcium in the induction
of apoptosis. To further determine whether calcium
release from the ER was required for the induction of
apoptosis by Taxol, the ER calcium release channel
(IP3R) inhibitor 2-APB was also tested. IP3R expres-
sion was confirmed in both M468 and MCF-7 cells
(Fig. 9). The results showed 2-APB pretreatment had a
similar inhibitory effect on high dose Taxol-induced
apoptosis in M468 and MCF-7 cells (Fig. 8c,d,
p < 0.01), whereas no significant inhibitory effect was
observed for low dose Taxol-induced apoptosis.
Therefore, interfering with cytosolic and ER calcium
changes caused a significant decrease of the Taxol-
induced apoptosis levels after 12 hours. These results
indicate that calcium promotes Taxol-induced apopto-
sis and the internal ER calcium store plays an impor-
tant role during this process.
DISCUSSION
High Dose Taxol (·10)6 M) had a Direct Effect on
Calcium Homeostasis Whereas Low Dose (·10)7 M)
did not, Indicating Taxol-Induced Calcium Change is
Dose-Dependent
In the introduction, the possible effect of Taxol on
calcium homeostasis was presented under the premise
that Taxol indirectly affects calcium through the pro-
duction of intermediates ROS or ceramide. However,
our results demonstrated definitively that Taxol
(·10)6 M) has a direct rapid effect on both cytosolic
and ER calcium levels in breast cancer cells. We
0.8
0.85
0.9
0.95
1
1.05
0 60 120 180 240 300 360 420
D1E
R F
RE
T r
atio
Time (s)
CDLH
0.8
0.85
0.9
0.95
1
1.05
C D L H
M468
**
(a)
Treatment
0.8
0.85
0.9
0.95
1
1.05
0 60 120 180 240 300 360 420
D1E
R F
RE
T r
atio
Time (s)
CDHL
0.8
0.85
0.9
0.95
1
1.05
C D L H**
(b)
Treatment
MCF-7
Figure 4. Endoplasmic reticulum calcium changes induced by short-term Taxol treatment directly measured using the D1ER cameleon.
Forty-eight hours after transient transfection with the D1ER vector, breast cancer cells were examined using FRET time-lapsed imaging to
monitor the changes of free ER calcium levels. These were represented by the FRET ratio (535 ⁄ 480 fluorescence emission) in D1ER ca-
meleon expressed cells. DMSO and Taxol were added to observe whether these agents could affect the ER calcium levels in M468 cells
(a) and MCF 7 cells (b). Time-response curve (mean) was determined from data generated in three independent tests. For the curve, the
lowest decreased ER calcium levels upon treatment were represented by the mean ± SD and statistical analysis were performed. Arrow
indicates the time point at which agents or normal medium were added into these groups. Final concentrations of Taxol were 2.5 lM (H) or
0.2 lM (L); DMSO is 0.5% (D); Normal medium is the control (C).
Calcium Regulates Taxol-Induced ER-Associated Apoptosis • 63

showed that addition of Taxol alone at a dose compa-
rable to clinical plasma levels (2.5 lM) induced a
rapid ER calcium release (Fig. 4) as well as a transient
increase in cytosolic calcium levels (Fig. 2) in both
M468 and MCF-7 breast cancer cell lines. After this
transient, rapid increase, a gradual ER calcium deple-
tion (Fig. 6) and sustained cytosolic calcium increase
(Fig. 3) occurred after long Taxol exposure. For both
short-term and long-term Taxol exposure, ER calcium
release occurred before the cytosolic calcium increases,
suggesting the ER provides a direct target for Taxol.
In contrast, the effects of Taxol at 0.2 lM
(·10)7 M) on calcium were much slower and less dra-
matic. Cytosolic calcium levels only slightly increased
in response to Taxol addition (Fig. 2). Nor was a sta-
ble cytosolic calcium increase observed until 12 hours
exposure to Taxol in M468 cells (Fig. 3b). By this
time, significant apoptosis had already occurred and
thus it became difficult to exclude whether calcium
release was a cause or an effect of apoptosis. In MCF
7 cells, low dose Taxol did not induce a significant
cytosolic calcium increase after long-term treatment.
The ER calcium responses were consistent with cyto-
solic calcium responses. Only a slow and slight
calcium release from the ER calcium store in M468
cells, and a transient calcium release in MCF 7 cells
were observed (Fig. 4a,b). Neither was significant. The
ER depletion did not occur during long-term exposure
of low dose Taxol treatment for both cell lines
(Fig. 7). Therefore, there is an obvious difference
between the dose effects of 10)6 M and 10)7 M Taxol
on calcium homeostasis with high dose directly and
rapidly changing calcium levels by triggering calcium
release from the ER, and low dose demonstrating a
nonsignificant change.
High Dose Taxol (·10)6 M) -Induced Calcium
Changes Promote Apoptosis, Whereas Low Dose
Taxol (·10)7 M) did not, Indicating an Induction of
Apoptosis Through Different Mechanisms
High dose (·10)6 M) Taxol induced significant ER
and cytosolic calcium changes in a biphasic manner. A
Figure 5. Time-lapsed images of Thapsigargin (TG)-induced Fluo4 changes in untreated M468 and MCF 7 cells. In order to evaluate the
calcium release from the ER into the cytosol in Taxol-treated cells, the ER calcium pump SERCA inhibitor Thapsigargin (TG) was added
into the media at the indicated ‘‘0 seconds’’ time point and the changes were recorded using a time-lapsed fluorescence imaging technique.
The Fluo4-AM fluorescence intensity changes were proportional to cytosolic calcium levels. Images were collected every 15 seconds for
5 minutes at room temperature (RT) at 20X. The ineffective SERCA pump failed to compensate for the ER passive release and resulted in
an increase in cytosolic calcium, visualized as an increase in signal intensity. Therefore, in the absence of extracellular calcium, upon TG
addition, all the observed cytosolic calcium increase originated from the ER store. This provided an indirect method to evaluate the ER cal-
cium release.
64 • pan and gollahon
rapid transient response was observed after its addi-
tion, followed by a slower but sustained change after
long-term treatment. All of these calcium changes
induced by high dose Taxol occurred before significant
apoptosis appeared (12 hours, Fig. 1). Since early
apoptosis was the major form of cell death at that
time, it is possible that calcium changes induced by
Taxol before 12 hours exposure may trigger or regu-
late induction of early apoptosis. The relationship
between the high dose Taxol-induced calcium changes
and consequent apoptosis was confirmed in the cal-
cium targeting agent experiments. Both cytosolic cal-
cium chelator BAPTA-AM (Fig. 8a) and 2-APB
(inhibitor of ER calcium channel release through the
IP3 receptor, Fig. 8b) significantly inhibits high dose
Taxol-induced apoptosis.
In contrast, low dose (·10)7 M) Taxol did not
induce a significant calcium change. In addition, cal-
cium interfering agents did not show significant inhibi-
tion to low dose Taxol-induced apoptosis. This
suggested that low dose Taxol-induced apoptosis is
not significantly regulated by calcium changes. How-
ever, at 12 hours exposure, low dose Taxol still
induced significant apoptosis, (Fig. 1). These results
suggest that high dose Taxol induces apoptosis
through an internal calcium-dependent mechanism,
whereas low dose Taxol induces apoptosis through an
internal calcium-independent mechanism.
Taxol was first identified as a potent inhibitor of cell
replication through dose-dependent stabilization of
microtubules. Low doses [0.01–0.2 lM] stop mitosis
and induce mitotic arrest-dependent apoptosis. High
doses [>1 lM] form a static microtubule environment,
effectively stopping daughter cell formation and induc-
ing mitotic arrest-independent apoptosis (7). This raises
the intriguing possibility that a relationship could exist
between Taxol-induced calcium changes and Taxol-
induced microtubule changes. High dose Taxol-
induced ER calcium release may provide an important
signal to bypass the mitotic arrest. Therefore, calcium
changes, mitotic arrest, and apoptosis may become
directly linked depending on Taxol dosage. Therefore,
Taxol action is much more complex than originally
expected and needs to be further elucidated.
0.8
1
1.2
1.4
1.6
1.8
2
0 60 120 180 240 300
Time (s)
H3hH6hH12hC
1
1.2
1.4
1.6
1.8
2
C H3h H6h H12h
TG
*** **
(a) M468
Peak level
Treatment
Flu
o4
inte
nsi
ty
chan
ges
Flu
o4
inte
nsi
ty
chan
ges
0.8
1
1.2
1.4
1.6
1.8
2
0 60 120 180 240 300
H3h
H6h
H12h
C
Time (s)
1
1.2
1.4
1.6
1.8
2
C H3h H6h H12h
****
Peak level
TG
Treatment
Flu
o4
inte
nsi
ty
chan
ges
MCF-7(b)
Flu
o4
inte
nsi
ty
chan
ges
Figure 6. Long-term effects of high dose Taxol (2.5 lM) on the ER calcium release measured using TG. Time-response curves (mean)
were determined from data generated in three independent tests for M468 cells (a) and MCF 7 cells (b). After exposure to Taxol for 3, 6
and 12 hours treatments, cells were loaded and incubated with Fluo 4. After incubation, the control and Taxol-treated cell samples were
washed and kept in HBSS without calcium to remove the effect of extracellular calcium influx. For the curve, peak levels of ER calcium
release upon TG treatment were represented by the mean ± SD, and statistical analysis was performed. Arrows show the addition of 3 lM
TG.
Calcium Regulates Taxol-Induced ER-Associated Apoptosis • 65
There are several possibilities as to how high dose
Taxol induces this rapid ER calcium release. First,
high dose Taxol may act as a modulator for ER
release channels such as IP3R. As reported in neuron
cell lines, Taxol may directly bind to neuronal calcium
sensor 1 (NCS-1) which interacts with IP3R, and thus
stimulates ER calcium release (47). However, direct
targets for epithelial cells are unknown and need to be
further investigated. Alternatively, Taxol may directly
damage the ER membrane, inducing a calcium flux
based on the calcium gradient. In response to ER cal-
cium release, the cell may increase calcium efflux to
the extracelluar medium in order to maintain normal
cytosolic calcium levels. Recall that the cytosolic cal-
cium levels did not return back to the baseline sug-
gesting that the ER calcium release continued upon
Taxol addition. This rapid calcium change may serve
as an upstream signal to sensitize mitochondria or
activate calcium-dependent caspase, thus regulating
the initiation of apoptosis. Furthermore, the expres-
sion of some apoptosis-related genes or protein
modifications may be regulated through activated
calcium-dependent enzymes, indirectly affecting apop-
tosis. For example, in breast cells, Bid was reported to
be cleaved by calcium-activated proteases Calpain into
tBid, which is an active pro-apoptotic protein (48). It
is interesting to note that Taxol causes activation of
calpain in neuron cell lines and inhibition of calpain
has a protective effect against Taxol-induced neuropa-
thy (49). Therefore, side effects in nervous tissue
induced by Taxol treatment can be also associated
with calcium regulation. High dose Taxol (·10)6 M)
may therefore be involved in more side effects due to
its significant effects on ER calcium regulation.
How does high dose Taxol induce the gradual ER
calcium depletion after long-term exposure? In addi-
tion to the direct repetitive stimulation of high dose
Taxol on the ER (47), other possible causes of the
time dependent ER calcium depletion may involve reg-
ulation of ER calcium release channels or other ER
calcium buffer proteins. Alternatively, intermediate
production of ROS and ceramide, which could be pro-
duced after several hours, may affect ER calcium
depletion (26,27). The ceramide and ROS could
0.8
1
1.2
1.4
1.6
1.8
2
0 60 120 180 240 300
L3hL6hL12hCTG
Time (s)
1
1.2
1.4
1.6
1.8
2
C L3h L6h L12h
Peak level(b)
TreatmentF
luo
4 in
ten
sity
ch
ang
es
Flu
o4
inte
nsi
ty
chan
ges
0.8
1
1.2
1.4
1.6
1.8
2
0 60 120 180 240 300
Time (s)
L3hL6hL12hC
1
1.2
1.4
1.6
1.8
2
C L3h L6h L12h
TG
M468(a)
Peak level
Treatment
Flu
o4
inte
nsi
ty
chan
ges
Flu
o4
inte
nsi
ty
chan
ges
MCF-7
Figure 7. Long-term effects of low dose Taxol (0.2 lM) on the ER calcium release measured using TG. Time-response curves (mean)
were determined in M468 cells (a) and MCF 7 cells (b) from data generated in three independent tests. After exposure to Taxol for 3, 6
and 12 hours treatments, cells were loaded and incubated with Fluo 4. After incubation, the control and Taxol-treated cell samples were
washed and kept in HBSS without calcium to remove the effect of extracellular calcium influx. For the curve, peak levels of ER calcium
release upon TG treatment were represented by mean ± SD, and statistical analyses were performed. Arrows show the addition of 3 lM
TG.
66 • pan and gollahon
induce the calcium transfer from the ER to the nearby
mitochondria and result in mitochondrial calcium
overload (21). Recent studies in lymphoma cell lines
showed that long-term Taxol (100 lM) exposure may
regulate some ER stress proteins and induce an ER
stress pathway (50), providing other possible effectors
for Taxol. Regardingless of the possible causes, the
key finding from our study is that Taxol, depending
on dosage, can directly induce ER calcium immediate
release and gradual depletion in breast cancer cells.
This is a direct ER stress response that signals the
induction of the ER apoptotic pathway.
Taxol Induces Calcium Changes in Both Bcl-2
Negative M468 Cells and Bcl-2 Positive MCF-7 Cells,
Indicating Taxol can Directly Attack ER Calcium
Stores Independent of Bcl-2 Status
B cell lymphoma 2 protein is an antiapoptotic pro-
tein, which is important for tumor resistance to che-
motherapy. Although its exact mechanism of action is
not fully understood, Bcl-2 regulation on calcium
homeostasis is suggested to play an important role
(51–53). Recent studies show that there is an interac-
tion between Bcl-2 and the ER calcium release channel
IP3R, which most likely inhibits the ER calcium
release and subsequent apoptosis (54,55). Another
(b)
0
10
20
30
40
L12h LBA H12h HBA
*
0
10
20
30
40
L12h LBA H12h HBA
**
Apo
ptos
is%
Apo
ptos
is%
0
10
20
30
40
L12h LBA H12h HBA0
10
20
30
40
L12h LBA H12h HBA
Apo
ptos
is%
Apo
ptos
is%
****
0
10
20
30
40
L12h LP H12h HP
**
(d)
0
10
20
30
40
L12h LP H12h HP
.. **
Apo
ptos
is%
Apo
ptos
is%
(c) MCF-7
0
10
20
30
40
L12h LP H12h HP0
10
20
30
40
L12h LP H12h HP
Apo
ptos
is%
Apo
ptos
is%
****
ER Calcium Channel Inhibitor 2-APB
M468(a)
Cytosolic Calcium Chelator BAPTA-AM
MCF-7
M468
Figure 8. Calcium interfering agents inhibited Taxol-induced apoptosis. M468 cells (a) and MCF 7 cells (b) were pretreated with 5 lM of
the cytosolic calcium chelator BAPTA-AM for 1 hour before Taxol addition. The percentage of apoptosis was measured by flow cytometry
using Annexin V-FITC & PI as described in the Methods section. Results (mean ± SD) were determined from data generated in three inde-
pendent tests. L12h and LBA were the treatment groups of low dose Taxol (0.2 lM) for 12 hours and combined low dose Taxol + BAPTA-
AM test, respectively. H12h and HBA are the treatment groups of high dose Taxol (2.5 lM) and combined high dose Taxol + BAPTA-AM
test, respectively. Effects of the IP3R calcium channel inhibitor, 2-APB at 25 lM, were determined in M468 cells (c) and MCF 7 cells (d). 2-
APB was pretreated for 1 hour before Taxol addition. L12h and LP were the treatment groups of low dose Taxol (0.2 lM) for 12 hours and
combined low dose Taxol + 2-APB, respectively. H12h and HP were the treatment groups of high dose Taxol (2.5 lM) and combined high
dose Taxol + 2-APB, respectively.
Figure 9. Western results for IP3R protein expression levels in
M468 and MCF 7 cells. Cellular protein lysates of M468 and MCF
7 cells (25 lg) were separated on a 7.5% SDS-PAGE, blotted,
detected with an IP3R antibody at 1: 200 and then visualized with
goat anti-rabbit HRP at 1:5000. Anti-IP3R antibody was raised
against amino acids 2402-2701 mapped at the C terminus of
human IP3R, thus the resulting band is around 115 kD in size.
Tubulin (1:1000) was used as the internal control and visualized
under the same conditions.
Calcium Regulates Taxol-Induced ER-Associated Apoptosis • 67

recent study indicated that Taxol can directly interact
and attack Bcl-2 protein as it polymerizes tubulins
(56). Therefore, it is difficult to clarify whether the
calcium responses are directly induced by Taxol or by
a Taxol-Bcl-2 interaction in Bcl-2 positive cell lines.
To address this question, both the Bcl-2 negative cell
line MDA-MB-468 and the Bcl-2 positive cell line
MCF-7 were investigated in our study. We observed
similar dose-dependent Taxol-induced calcium changes.
High dose Taxol treatment induced ER calcium release
and then cytosolic calcium increase, which promoted
apoptosis, whereas low dose Taxol did not. Therefore,
this result is not limited to one cell line or breast
cancer type, but more likely indicates a general
phenomenon.
Although the patterns are similar, there are some
differences between Bcl-2 negative and positive cell
lines. First, our results showed that MCF-7 is more
resistant to Taxol treatment, which indicates Bcl-2
expression had an inhibitory effect on apoptosis.
Additionally, the releasable ER calcium levels were
much higher in MCF 7 cells at rest, which supports
Bcl-2 protein inhibits ER calcium release and conse-
quently increases the basal level of releasable ER cal-
cium. Upon Taxol treatment, the transient cytosolic
increase in MCF 7 cells is faster, consistent with the
faster ER calcium release observed. However, the
amplitude of the calcium changes is similar to M468
cells, indicating that Taxol make attack multiple tar-
gets associated with the ER, and its direct inhibition
on Bcl-2 action may accelerate its attack on the ER
calcium store. Although the transient phase is faster,
the ER calcium depletion and sustained cytosolic cal-
cium increase occurring in MCF 7 cells is slower and
smaller. Since no significant difference in expression
levels of IP3R was observed between M468 and
MCF-7 cells by Western blot (Fig. 9), this indicates
that the slower ER calcium depletion observed in
MCF-7 cells is most likely related to its higher releas-
able ER calcium levels, making the ER calcium store
more difficult to deplete. Thus, Taxol can directly
attack ER calcium independent of Bcl-2 status, the
Bcl-2 regulation of basal ER calcium release, and the
interaction of Taxol on Bcl-2 together, forming a
complex picture. The direct assault of Taxol of Bcl-2
may speed up the ER calcium release, but it does not
increase the amplitude. The final outcome is depen-
dent upon the competition between Bcl-2 and Taxol
for the ER calcium release, the mechanism of which
provides a new field need to be further investigated.
In summary, several interesting new observations
were revealed regarding the role of calcium in Taxol-
induced apoptosis in breast cancer cells. Within the
scope of this study, two major subtypes of breast can-
cer cell lines, hormone-dependent, Bcl-2 positive
MCF-7 and hormone-indpendent, Bcl-2 negative
MDA-MB-468, were investigated. The major finding
is that the addition of Taxol (10)6 M) can directly
induce a significant ER calcium release and a subse-
quent transient cytosolic calcium increase. Further-
more, ER calcium release induced by Taxol appeared
before the cytosolic calcium increase––after either
immediate addition or long-term exposure of Taxol
treatment, suggesting that the ER calcium store
provides Taxol a direct upstream target. These Taxol-
induced calcium changes significantly promoted
apoptosis, confirming calcium (especially ER calcium)
plays an important role in Taxol-induced apoptosis in
breast cancer cells. Interestingly, the same results were
not observed for low dose Taxol (10)7 M), although
low dose Taxol can also induce significant apoptosis.
Therefore, Taxol dosage is a key element in determin-
ing its involvement in the ER apoptotic pathway,
independent of Bcl-2 status. Bcl-2 acts more as a mod-
ulator on the ER calcium release and is not the only
target on the ER that Taxol can attack directly. These
findings provide new insights into the mechanism of
Taxol action and may aid in the development of more
effective breast cancer therapies. In addition, our
results indicate that calcium regulation of breast can-
cer cells is important for both tumor resistance and
side effects to chemotherapy. This information may
aid clinicians in devising more effective strategies for
the management of cancer treatment.
Acknowledgments
We thank Prof. R.Y. Tsien, University of California
San Diego for providing the D1ER cameleon plasmid.
We also thank the Imaging Center of Texas Tech
University for providing the necessary microscope
system and training for this research. Financial
support was provided by the Department of Biological
Sciences, Texas Tech University. This work was also
supported by research grants from the Texas Tech
University Association of Biologists.
REFERENCES
1. Igney FH, Krammer PH. Death and anti-death: tumour
resistance to apoptosis. Nat Rev Cancer 2002;2:277–88.
68 • pan and gollahon

2. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link
between cancer genetics and chemotherapy. Cell 2002;108:153–64.
3. Brown JM, Wouters BG. Apoptosis: mediator or mode of cellkilling by anticancer agents? Drug Resist Updat 2001;4:135–6.
4. Brown M, Wilson G. Apoptosis genes and resistance to can-
cer therapy. Canc Biol Ther 2003;2:477–90.5. Milross CG, Mason KA, Hunter NR, Chung WK, Peters LJ,
Milas L. Relationship of mitotic arrest and apoptosis to antitumor
effect of paclitaxel. J Natl Cancer Inst 1996;88:1308–14.
6. Shtil AA, Mandlekar S, Yu R, et al. Differential regulation ofmitogen-activated protein kinases by microtubule-binding agents in
human breast cancer cells. Oncogene 1999;18:377–84.
7. Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell
death: where the cell cycle and apoptosis come together. Cancer2000;88:2619–28.
8. Mollinedo F, Gajate C. Microtubules, microtubule-interfering
agents and apoptosis. Apoptosis 2003;8:413–50.
9. Masuda A, Maeno K, Nakagawa T, Saito H, Takahashi T.Association between mitotic spindle checkpoint impairment and sus-
ceptibility to the induction of apoptosis by anti-microtubule agents
in human lung cancers. Am J Pathol 2003;163:1109–16.10. Dziadyk JM, Sui M, Zhu X, Fan W. Paclitaxel-induced
apoptosis may occur without a prior G2 ⁄ M-phase arrest. AnticancerRes 2004;24:27–36.
11. Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Looking for-ward to seeing calcium. Nat Rev Mol Cell Biol 2003;4:579–86.
12. McConkey DJ, Nutt L. Measurement of changes in intracel-
lular calcium during apoptosis. Methods Mol Biol 2004;282:117–30.
13. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of celldeath: the calcium-apoptosis link. Nat Rev Mol Cell Biol2003;4:552–65.
14. Rizzuto R, Pinton P, Ferrari D, et al. Calcium and apoptosis:facts and hypotheses. Oncogene 2003;22:8619–27.
15. Cerella C, D’Alessio M, De Nicola M, Magrini A, Berga-
maschi A, Ghibelli L. Cytosolic and endoplasmic reticulum
Ca2+concentrations determine the extent and the morphological typeof apoptosis, respectively. Ann N Y Acad Sci 2003;1010:74–7.
16. Hajnoczky G, Davies E, Madesh M. Calcium signaling and
apoptosis. Biochem Biophys Res Commun 2003;304:445–54.
17. Berridge MJ, Bootman MD, Roderick HL. Calcium signal-ing: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol2003;4:517–29.
18. Monteith GR, McAndrew D, Faddy HM, Roberts-ThomsonSJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer2007;7:519–30.
19. Berridge MJ. The endoplasmic reticulum: a multifunctional
signaling organelle. Cell Calcium 2002;32:235–49.20. Szabadkai G, Rizzuto R. Participation of endoplasmic reticu-
lum and mitochondrial calcium handling in apoptosis: more than
just neighborhood? FEBS Lett 2004;567:111–5.
21. Demaurex N, Distelhorst C. Cell biology. Apoptosis-thecalcium connection. Science 2003;300:65–7.
22. Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore
GC. Regulation of apoptosis by endoplasmic reticulum pathways.
Oncogene 2003;22:8608–18.23. Hitomi J, Katayama T, Taniguchi M, Honda A, Imaizumi
K, Tohyama M. Apoptosis induced by endoplasmic reticulum stress
depends on activation of caspase-3 via caspase-12. Neurosci Lett2004;357:127–30.
24. Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic
reticulum stress to the cell death program. Cell Death Differ2004;11:372–80.
25. Lamkanfi M, Kalai M, Vandenabeele P. Caspase-12: an
overview. Cell Death Differ 2004;11:365–8.
26. Varbiro G, Veres B, Gallyas F Jr, Sumegi B. Direct effect of
Taxol on free radical formation and mitochondrial permeability
transition. Free Radic Biol Med 2001;31:548–58.27. Charles AG, Han TY, Liu YY, Hansen N, Giuliano AE,
Cabot MC. Taxol-induced ceramide generation and apoptosis in
human breast cancer cells. Cancer Chemother Pharmacol2001;47:444–50.
28. Colina C, Flores A, Rojas H, et al. Ceramide increase cyto-
plasmic Ca2+concentration in Jurkat T cells by liberation of calcium
from intracellular stores and activation of a store-operated calciumchannel. Arch Biochem Biophys 2005;436:333–45.
29. Padar S, Thomas DW, Liversey JC, Rahimian R. Differential
regulation of calcium homeostasis in adenocarcinoma cell line A549
and its Taxol-resistant subclone. Br J Pharmacol 2004;142:305–16.30. Mironov SL, Ivannikov MV, Johansson M. [Ca2+]i signaling
between mitochondria and endoplasmic reticulum in neurons is reg-
ulated by microtubules. From mitochondrial permeability transition
pore to Ca2+-induced Ca2+ release. J Biol Chem 2005;280:715–21.31. Wang NS, Unkila MT, Reineks EZ, Distelhorst CW. Tran-
sient expression of wild-type or mitochondrially targeted Bcl-2
induces apoptosis, whereas transient expression of endoplasmicreticulum-targeted Bcl-2 is protective against Bax-induced cell death.
J Biol Chem 2001;276:44117–28.
32. Yde CW, Issinger OG. Enhancing cisplatin sensitivity in
MCF-7 human breast cancer cells by down-regulation of Bcl-2 andcyclinD1. Int J Oncol 2006;29:1397–404.
33. Breast Cancer Facts and Figures 2007-2008. American Can-
cer Society
34. Cailleau R, McGrath CM. MDA-MB-468 & MCF-7 BreastCarcinoma Cell Line Product Description. Manassas, VA: American
Tissue Culture Center, 2008.
35. Blajeski AL, Kottke TJ, Kaufmann SH. A multistep modelfor paclitaxel-induced apoptosis in human breast cancer cell lines.
Exp Cell Res 2001;270:277–88.
36. Fan W. Possible mechanisms of paclitaxel-induced apoptosis.
Biochem Pharmacol 1999;57:1215–21.37. Rowinsky EK. Clinical Pharmacology of Taxol. J Natl Can-
cer Inst 1993;15:25–38.
38. McGuire WP, Rowinsky EK. Pharmacology and Meta-bolism, Paclitaxel in Cancer Treatment. New York, NY: MarcelDekker, Inc, 1995:91–120.
39. Taxol (Paclitaxel) Injection Product Information. Princeton,
NJ: Bristol-Myers Squibb Company, 2007.40. Palmer AE, Giacomello M, Kortemme T, et al. Ca2+ indica-
tors based on computationally redesigned calmodulin-peptide pairs.
Chem Biol 2006;13:521–30.
41. Demaurex N, Frieden M. Measurements of the free luminalER Ca2+ concentration with targeted ‘‘cameleon’’ fluorescent pro-
teins. Cell Calcium 2003;34:109–19.
42. Palmer AE, Tsien RY. Measuring calcium signaling using
genetically targetable fluorescent indicators. Nat Protoc2006;1:1057–65.
43. Liu G, Hu X, Chakrabarty S. Calcium sensing receptor
down-regulates malignant cell behavior and promotes chemo-
sensitivity in human breast cancer cells. Cell Calcium 2009;45:216–25.
44. McCloskey DE, Kaufmann SH, Prestigiacomo LJ, Davidson
NE. Paclitaxel induces programmed cell death in MDA-MB-468human breast cancer cells. Clin Cancer Res 1996;2:847–54.
45. Ichimiya M, Chang SH, Liu H, Berezesky IK, Trump BF,
Amstad PA. Effect of Bcl-2 on oxidant-induced cell death and intra-
cellular Ca2+ mobilization. Am J Physiol 1998;275:C832–9.46. Kim BC, Kim HT, Mamura M, Ambudkar IS, Choi KS, Kim
SJ. Tumor necrosis factor induces apoptosis in hepatoma cells by
Calcium Regulates Taxol-Induced ER-Associated Apoptosis • 69

increasing Ca2+ release from the endoplasmic reticulum and sup-
pressing Bcl-2 expression. J Biol Chem 2002;277:31381–9.
47. Boehmerle W, Splittgerber U, Lazarus MB, et al. Paclitaxelinduces calcium oscillations via an inositol 1, 4, 5-trisphosphate
receptor and neuronal calcium sensor 1-dependent mechanism. ProcNatl Acad Sci 2006;103:18356–61.
48. Mandic A, Viktorsson K, Strandberg L, et al. Calpain-medi-
ated Bid cleavage and calpain-independent Bak modulation: two
separate pathways in cisplatin-induced apoptosis. Mol Cell Biol2002;22:3003–13.
49. Wang MS, Davis AA, Culver DG, Wang Q, Powers JC,
Glass JD. Calpain inhibition protects against Taxol-induced sensory
neuropathy. Brain 2004;127:671–9.
50. Liao PC, Tan SK, Lieu CH, Jung HK. Involvement of endo-plasmic reticulum in paclitaxel-induced apoptosis. J Cell Biochem2008;104:1509–23.
51. Distelhorst CW, Shore GC. Bcl-2 and calcium: controversy
beneath the surface. Oncogene 2004;23:2875–80.
52. Brichese L, Barboule N, Heliez C, Valette A. Bcl-2
phosphorylation and proteasome-dependent degradation induced by
paclitaxel treatment: consequences on sensitivity of isolated mito-chondria to Bid. Exp Cell Res 2002;278:101–11.
53. Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ.
Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apop-tosis. EMBO J 2004;23:1207–16.
54. Rong YP, Bultynck G, Aromolaran AS, et al. The BH4
domain of Bcl-2 inhibits ER calcium release and apoptosis by bind-
ing the regulatory and coupling domain of the IP3 receptor. ProcNatl Acad Sci 2009;106:14397–402.
55. Rodi DJ, Janes RW, Sanganee HJ, Holton RA, Wallace BA,
Makowski L. Screening of a library of phage-displayed peptides
identifies human bcl-2 as a taxol-binding protein. J Mol Biol1999;285:197–203.
56. Ferlini C, Cicchillitti L, Raspaglio G, et al. Paclitaxel
directly binds to Bcl-2 and functionally mimics activity of Nur77.
Cancer Res 2009;69:6906–14.
70 • pan and gollahon








![Endoplasmic reticulum[1]](https://img.pdfslide.us/doc/110x75/58ed5fc71a28aba1678b4611/endoplasmic-reticulum1.jpg)




















