Embed Size (px)
Citation preview

SYSTEMIC EFFECTS OF MINOR EXPRESSION IN ADIPOCYTES
by
ROSICKA GRACE WALTON
W. TIMOTHY GARVEY, COMMITTEE CHAIR
YUCHANG FU TIMOTHY R. NAGY PHILIP A. WOOD
A THESIS
Submitted to the graduate faculty of the University of Alabama at Birmingham, In partial fulfillment of the requirements for the degree of
Master of Science
BIRMINGHAM, ALABAMA
2008

SYSTEMIC EFFECTS OF MINOR EXPRESSION IN ADIPOCYTES
ROSICKA GRACE WALTON
NUTRITION SCIENCES
ABSTRACT
The NR4A family of orphan nuclear receptors function as early response
genes with numerous stimuli. The NR4A family consists of three known
members, Nur77, Nurr1, and Mitogen-inducible nuclear orphan receptor
(MINOR). Our lab has previously demonstrated that stable over-expression of
MINOR in 3T3-L1 adipocytes enhances insulin-stimulated glucose uptake. In
order to assess the in vivo affect of MINOR on adipocytes, we generated
transgenic mice with MINOR over-expression driven by the adipocyte- and
macrophage-specific AP2 promoter (AP2-MINOR mice). We initially
hypothesized that AP2-MINOR mice would display increased glucose tolerance
and insulin sensitivity. However, AP2-MINOR transgenic male and female mice
have increased fasting glucose and respond poorly to glucose challenge, with
males developing insulin resistance late in life. AP2-MINOR males are larger
than wild-type due to increases in both lean and fat mass. When males are
maintained on high fat chow, transgenic animals are larger, but do not differ from
wild-type when challenged with glucose or insulin. These results led us to
hypothesize that MINOR over-expression induces adipocyte hypersensitivity to β-
adrenergic agonism, leading to increased serum non-esterified fatty acids and
impairment of the pancreatic β-cell response to glucose. Indeed, insulin fails to
inhibit transgenic NEFA production below the level of wild-type in spite of
ii

significantly reduced serum catecholamines. Oddly, fasting serum total
cholesterol and LDL were increased and NEFA was decreased in AP2-MINOR
animals versus wild-type. Thus, it appears that MINOR over-expression effects
both glucose and lipid homeostasis. As a whole, these observations are
consistent with a MINOR-mediated down-regulation of Liver X receptor (LXR)
and its target, sterol regulatory element binding protein-1c (SREBP-1c).
iii

ACKNOWLEDGEMENTS
Firstly, I would like to express my gratitude to my committee members, W.
Timothy Garvey, M.D., Yuchang Fu, Ph.D., Timothy R. Nagy, Ph.D., and Philip A.
Wood, D.V.M. Dr. Garvey has been incredibly generous with his time and he has
provided steadfast support and mentorship. Dr. Fu oversaw preliminary work on
MINOR, enabling this project to exist. Doctors Garvey and Fu formulated our
original hypotheses, and their scientific rigor and vision are appreciated. I would
also like to offer special thanks Dr. Nagy, who has mentored me for 8 years, and
encouraged me to pursue graduate studies. As a member of my committee, Dr.
Nagy’s advice and expertise regarding body composition and statistics have
been invaluable. Finally, Dr. Wood has also mentored me for many years and
served as an irreplaceable committee member, providing unparalleled expertise
in murine lipidology.
I would also like to thank the members of the Garvey lab. In particular,
Liehong Luo, Nanlan Luo, and Xiaolin Zhu performed many of the background
experiments on MINOR. However, each member of the lab has contributed to the
project by helping with animal sacrifices, molecular biology, and numerous other
essential tasks. These lab members include Pedro del Corral, Helliner Hill,
Cristina Lara-Castro, Jiarong Liu, Kerry Lok, Doug Moellering, Sasha Smith, Ling
Tian, and Xuxia Wu. I would like to specifically thank Dr. B. Hong Chung for his
technical and physiological expertise in the area of apolipoproteins.
iv

I am also thankful to the following scientists who work in other labs: from
the Nagy lab, Maria Johnson, Meghana Gavand, and Amanda Watts for
performing DEXA scans; from the lab of Dr. Barbara Gower, Maryellen Williams
who performed insulin radio-immuno assays; Ray Johnson from the CMN/KC
Neurochemistry Core Lab, Vanderbilt University, performed catecholamine
assays; Kate Kosmac from William Britt’s lab helped with molecular biology and
thesis development; the members of Dr. Robert Kesterson’s lab and the
members of Dr. Wood’s lab were generous with advice and equipment. Many
others have had some hand in this project and I regret that I cannot name them
all.
Lastly, my family has been unfailing in their support of my academic
endeavors and I am forever grateful.
After all, it takes a village to raise a mouse.
v

TABLE OF CONTENTS
Page ABSTRACT ...........................................................................................................ii ACKNOWLEDGEMENTS.....................................................................................iv LIST OF TABLES ................................................................................................. v LIST OF FIGURES ...............................................................................................vi INTRODUCTION.................................................................................................. 1 METHODS............................................................................................................ 8 rtPCR............................................................................................................. 8 Western blotting............................................................................................. 8 Animals.......................................................................................................... 8 Serum Assays ............................................................................................. 10 RESULTS........................................................................................................... 12 Verification of transgene insertion and function ........................................... 12 Female AP2-MINOR transgenic mice.......................................................... 13 Male AP2-MINOR transgenic mice—standard chow ................................... 14 Male AP2-MINOR transgenic mice—high fat chow ..................................... 26 DISCUSSION ..................................................................................................... 29 REFERENCES................................................................................................... 34 APPENDIX: IACUC APPROVAL ........................................................................ 39
vi

LIST OF TABLES Table Page 1 Body composition in 8-week old female AP2-MINOR and wild-type mice maintained on standard chow............................................................................. 14 2 Body composition in 8-week old AP2-MINOR and wild-type mice................... 17 3 Body composition in 16-week old AP2-MINOR and wild-type mice................. 18 4 Body composition in 30-week old AP2-MINOR and wild-type mice................. 18 5 Fasting serum lipids in transgenic and wild-type mice maintained on standard chow ................................................................................................................... 23 6 Body composition in 16-week old, high-fat fed AP2-MINOR and wild-type Mice.................................................................................................................... 27 7 Summary of hypothesized protein expression or function changes in AP2-MINOR mice ....................................................................................................... 31
vii

LIST OF FIGURES Figure Page 1 Verification of transgene insertion and function............................................... 12 2 Fasting glucose is increased in female AP2-MINOR mice versus wild-type littermates........................................................................................................... 13 3 Body weight and food intake in AP2-MINOR and wild-type mice .................... 15 4 Glucose tolerance in 17-week old mice ........................................................... 19 5 At age 18 weeks, no differences were observed in insulin tolerance............... 20 6 Glucose and insulin parameters in 31 week-old mice...................................... 21 7 Insulin tolerance in 32-week old mice.............................................................. 22 8 Serum lipids in AP2-MINOR and wild-type mice.............................................. 24 9 Insulin’s ability to suppress lipolysis in AP2-MINOR and wild-type mice ......... 25 10 Body weight and food intake in mice fed high fat chow ................................. 26 11 Glucose and insulin tolerance tests in animals maintained on high fat chow 28 12 Fasting serum triglycerides............................................................................ 29
viii

1
INTRODUCTION
Worldwide, the number of people affected by diabetes has greatly
increased in the past 3 decades. The international Diabetes federation estimates
that diabetes care consumes 5-10% of total healthcare costs in many nations.
These costs are largely attributed to treatment of diabetes complications, which
may often be prevented through proper glycemic control. Many current anti-
hyperglycemic and insulin-sensitizing drugs have untoward side effects including
weight gain, resistance to weight loss, difficult dosing regimens, and
development of drug tolerance (Smyth & Heron, 2005). Insulin resistance is
strongly correlated to increased adipose tissue, particularly visceral adipose
tissue. Circulating and local levels of the relevant products of adipose tissue—
NEFA and adipocytokines—are often abnormal in patients with insulin resistance
and frank Type 2 diabetes (Farmer & Prusty, 2004). Thus, understanding the
regulation of adipocyte glucose and lipid metabolism provides one avenue for the
development of improved anti-diabetic drugs.
Mitogen-inducible nuclear orphan receptor (MINOR, also NOR-1, TEC,
CHN, NR4A3) is one of three known members of the NR4A subgroup of the
nuclear hormone receptor superfamily. Other NR4A members include Nur77
(TR3, NGFI-B, N10, DHR38, NAK-1, TIS1, NR4A1) and Nurr1 (HZF-3, RNR-3,
TINUR, NOT, NR4A2). NR4A family members are expressed in numerous
metabolically active tissues including brain, heart, skeletal muscle, kidney, liver,
certain leukocytes, and adipose. They are early response genes whose
expression is induced in a cell-type specific manner by numerous stimuli,

2
including: inflammation (LPS, oxidized lipids, cytokines), growth factors, HPA-
axis hormones, tyrosine-derived neurotransmitters, forskolin and cAMP analogs,
oxidized LDL, and many others. NR4A members modulate expression of
steroidogenic genes in both the HPA-axis and target tissues, with known mutual
transcriptional repression between NR4A members and the glucocorticoid
receptor (extensively reviewed by Maxwell & Muscat, 2006). They are also potent
stimulators of hepatic gluconeogenic enzyme transcription (Pei et al. 2006).
Finally, NR4A members are thought to have a high degree of transcriptional and
functional redundancy.
NR4A members appear to have atypical ligand binding domains. Two
activation functions are described for most nuclear receptors: the N-terminal
domain (AF1) and the ligand binding domain (LBD, AF2). The AF1 is highly
conserved across NR4A members and is likely to mediate the majority of their
protein-protein interactions. Indeed, MINOR AF1 can independently recruit
coactivators. NR4A members differ in their LBDs even though all members lack a
hydrophobic cleft typically found in the LBD of steroid hormone receptors.
However, a hydrophobic patch (distinct from the missing hydrophobic cleft) on
the LBD is believed to interact with cofactors in order to alter transcription of
target genes (Flaig et al. 2005). Nur77 and Nurr1 form functional heterodimers
with each other (Maira et al. 1999) as well as the retinoid X receptor (Perlmann &
Jansson, 1995), unlike MINOR, which does not appear to heterodimerize with
other nuclear receptors (Zetterstrom et al. 1996). NR4A members bind NGFI-B

3
response elements (NBRE) and Nur-responsive elements (an everted repeat of
the NBRE) as monomers or homodimers (Philips et al. 1997).
MINOR was first described in the mid-1990’s (Ohkura et al. 1994; Hedvat
& Irving, 1995). Two human MINOR transcripts have been described, with the
longer encoding a 626 amino acid residue (Ohkura et al. 1996). Both rats and
humans produce two isoforms of MINOR protein, the smaller of which has little
transcriptional activity (Ohkura et al. 1998). MINOR transcription can be induced
by the calcium/calmodulin-dependent protein kinase cascade (Inuzuka et al.
2002) and high levels of MINOR protein may repress MINOR transcription
(Hedvat & Irving, 1995). In adrenal fasciculata cells, MINOR transcription is
induced by ACTH and angiotensin II and is dependant on PKA and PKC
cascades (Fernadez et al. 2000). Platelet-derived growth factor (Nomiyama et
al. 2006) and oxidized LDL (Crespo et al. 2005) have also been shown to
increase MINOR transcription. Reported transactivators of MINOR include
prostaglandin A2 (Kagaya et al. 2005) and the homeotic protein Six3 (Laflamme
et al. 2003). Finally, inhibition of TNF-α in psoriatic skin decreases Nurr1
transcript and protein levels (O’kane et al. 2007). Since NR4A family members
are functionally related, it is reasonable to hypothesize that TNF-α increases
MINOR transcription.
No metabolic abnormalities have been reported in MINOR knockout mice.
Rather, one line of MINOR -/- exhibits abnormal hippocampal development,
increased predisposition to excitotoxic glutamate receptor kainic acid-induced
seizure (Ponnio & Conneely, 2004), inner ear defects, and aberrant circling

4
behavior (Ponnio et al. 2002). A second MINOR knockout was embryonic lethal
in homozygous mice due to incomplete gastrulation while heterozygous mice
appeared to be normal (DeYoung et al. 2003). Transgenic global over-expression
of MINOR produces dramatically reduced body weight (~50%), atrophy of the
spleen and thymus, and a live birth rate of less than 50% (Kagaya et al. 2005).
Adipocyte lipolysis is a major source of fuel during fasting. Lipolysis is
regulated by catecholamines, insulin, and natriuretic hormones in a species-
specific manner. Hormone sensitive lipase (HSL) is believed to mediate acute
changes in lipolytic activity, while adipocyte triglyceride lipase is believed to
mediate basal levels of lipolysis. Catecholamines generally stimulate lipolysis,
while insulin potently inhibits lipolysis. Human adipocytes express four
adrenoreceptors α2A, β1, β2, and β3, of which the first inhibits and the latter three
stimulate lipolysis. β1 and β2 are more active than β3 in humans. However, in
rodents, β3 is the primary mediator of adrenergic-induced lipolysis. β receptors
are Gs-coupled, leading to activation of PKA and finally to phosphorylation of
HSL, which hydrolyzes triglycerides into free fatty acids (NEFA) and glycerol
(Arner, 2005).
Elevated serum NEFA is a common correlate to obesity, insulin
resistance, and Type 2 diabetes. Increased basal lipolysis likely accounts for a
large portion of the elevated NEFA observed in obesity. Increased adipocyte size
and chronically high TNFα levels in the adipose tissue have been implicated in
increasing NEFA production (Arner, 2005). Larger adipocytes display increased:
FAS and LPL activity, GLUT4 and leptin expression, and overall lipolytic capacity

5
(Farnier et al. 2003). Furthermore, there is a strong positive correlation between
adipocyte size and circulating TNFα (Winkler et al. 2003). The precise
mechanisms of chronic-TNFα-induced lipolysis are currently unknown. However,
there is evidence that NEFA released by adipocytes elicits TNFα production by
local macrophages, creating a chronic inflammatory cycle (Suganami et al.
2005). Obesity-related perturbations in NEFA metabolism are both species- and
fat pad specific. However, it is clear that C57BL/6J mice maintained on high fat
diet exhibit elevated NEFA and plasma glucose (Ahren & Scheurink, 1998).
Pancreatic β-cells undergo progressive deterioration in type 2 diabetes.
Although certain free fatty acids are known to acutely increase β-cell response to
glucose, many experts believe that chronically elevated NEFA are harmful to β-
cells. Evidence suggests that NEFA can harm β-cells at physiological glucose
concentrations (Dubois et al. 2004). However, other experiments indicate that
hyperglycemia is necessary for any deleterious effects of NEFA (Roberston et al.
2004; Goh et al. 2007). In any event, it is clear that a chronically hyperlipidemic
and hyperglycemic environment reduces glucose-stimulated insulin secretion.
Proposed mechanisms of β-cell lipotoxicity include inhibition of insulin gene
transcription (Poitout et al. 2006) and increased ceramide synthesis and NO
production (Newsholme et al. 2007).
Particular interest should be paid to data from George Muscat’s laboratory
imputing MINOR as a target of β-adrenergic signaling. MINOR expression is
increased in C2C12 skeletal muscle cells and mouse skeletal muscle tissue after
incubation with isoprenaline or specific β2 or β3 agonists. Suppression of MINOR

6
in C2C12 cells by siRNA causes reductions in UCP2 and CAV3 transcription and
induction of UCP3 and myostatin transcription. In vivo observations confirm
induction of UCP2 in mouse skeletal muscle after treatment with β-adrenergic
agonist (Pearen et al. 2006). Muscat’s group made similar observations about
Nur77. Notably, suppression of Nur77 caused decreased lipid oxidation in C2C12
cells (Maxwell et al. 2005). Another group examined whether NR4A members
participate in brown adipose tissue thermogenesis. Nur77 was transiently
induced in brown adipose tissue following β-adrenergic stimulation or cold-
induced thermogenesis. They also reported a “superinduction” of MINOR in the
brown adipose tissue of cold-exposed Nur77 knockout mice (Kanzleiter et al.
2005).
MINOR is naturally expressed in adipocytes and our lab has demonstrated
that MINOR increases insulin-stimulated glucose uptake when overexpressed in
vitro (Fu et al. 2007). However, the physiological effects of MINOR
overexpression in adipocytes are unknown. Thus transgenic AP2-MINOR mice
that overexpress MINOR in adipocytes were produced. Surprisingly, preliminary
data indicated that these animals have an impaired rather than improved
response to glucose. This observation could result from elevated circulating free
fatty acids which are known to impair pancreatic β-cell response to glucose.
Since: a) elevations in free fatty acids are the result of adipocyte lipolysis, which
can be stimulated by β-adrenergic agonism and b) MINOR is a known target of β-
adrenergic signaling, I hypothesize that MINOR enhances lipolysis in adipocytes.

7
This study sought to determine the effect of adipose tissue-specific over-
expression of MINOR on body composition parameters and physiological
responses to glucose and insulin. Experiments began with assessment of
MINOR transgene functionality and protein expression. I proceeded to determine:
body weight, body composition, and food intake in MINOR transgenic mice on
both low fat and high fat diets; differences in glucose tolerance and insulin
tolerance in transgenic versus wild-type mice on both diets; differences in insulin-
mediated suppression of adipocyte lipolysis in transgenic versus wild-type mice;
and serum lipid variations in transgenic versus wild-type mice.

8
METHODS
rtPCR RNA was extracted from WAT using TRIzol reagent (Invitrogen)
according to manufacturer’s protocol. Samples were treated with DNAse I (amp
grade, Invitrogen), and then used as template for the production of cDNA
(SuperScript III, Invitrogen). PCR was then performed on cDNA and amplification
product resolved on 1.5% agarose gel. Primers used were the same as those
used for genotyping mice. Primer sequences were: 5’-GGA TCC AAA CTC ATT
ACT AAC CGG TA-3’ and 5’-ATA TCC AAG CCT TAG CCT GCC TGT-3’.
Western blotting WAT protein was extracted with Sigma CelLytic
Mammalian Tissue Lysis Reagent and separated by SDS-polyacrylamide gel
electrophoresis. Protein was transferred onto nitrocellulose membranes and
incubated overnight at 4°C with 5% non-fat milk in TBS. Membranes were then
incubated with MINOR antibody (Perseus Proteomics, Tokyo, Japan) for 1 hour
followed by incubation with horseradish peroxidase secondary antibody for 1
hour. Membranes were washed with TBS (with 0.1% Tween 20) and protein was
detected by chemiluminescence (Enhance, NEN Life Science) and quantified
with appropriate software.
Animals In order to produce transgenic animals and wild-type littermate
controls, breeding pairs consisted of one heterozygous AP2-MINOR transgenic
animal and one pure C57/BL (Taconic Farms). All animals were maintained
under standard conditions (22±2°C, 12-hour light cycle) and given ad libitum

9
access to water and either standard rodent chow (Harlan Teklad 7913 Irradiated
Modified 6% Mouse/Rat Sterilizible Diet) or high fat chow (Research Diets D-
12492, 60% kcal from fat, 20% kcal from protein, 20% kcal from carbohydrate).
Body weight and food intake were measured weekly beginning at age 8 weeks
and ending at age 30 weeks.
Glucose tolerance test (GTT): 17 week old and 31 week old animals were
fasted overnight with free access to water. At 9:00 a.m., animals were weighed
and given an intraperitoneal injection of D-(+)-glucose (100g glucose/L; 10μl/g
body weight). Blood glucose was measured at baseline (approximately 1 minute
prior to injection), and at 30, 60, 90, and 180 minutes post-injection (HemoCue
glucose 201 glucometer, HemoCue USA).
Insulin tolerance test (ITT): 18 and 32 week old animals were fasted for 4
hours and then weighed. Animals were then given an intraperitoneal injection of
1.5U (male mice) or 1.0U (female mice) Humalog lispro insulin/kg body weight.
Blood glucose was measured at baseline (approximately 1 minute prior to
injection), and at 30, 60, 90, and 180 minutes post-injection (HemoCue glucose
201 glucometer, HemoCue USA). Animals were not given access to food or
water during GTT and ITT.
In order to assess insulin’s ability to suppress adipocyte lipolysis, male
animals (age 33-35 weeks, maintained on standard chow) were fasted for 4
hours, weighed, and injected with 1.5U Humalog lispro insulin/kg body weight.
Mice were sacrificed exactly 60 minutes after insulin injection, and whole trunk

10
blood was collected for analysis. Analytes included insulin, NEFA, epinephrine,
and norepinephrine (methods below).
Animals were euthanized by decapitation without anesthesia. Male
animals were sacrificed at 33-35 weeks of age. Tissues and whole trunk blood
were harvested and perigonadal fat pads were weighed. All tissues except trunk
blood were quickly placed into liquid nitrogen and stored at -80°C until needed.
Whole trunk blood was centrifuged for 30 minutes at 5000xG, 4°C and serum
was collected.
DEXA scans were performed in the lab of Dr. Timothy R. Nagy (University
of Alabama at Birmingham), according to previously published protocols (Nagy &
Clair, 2000). Animals were scanned at age 8 weeks (females); age 8, 16, and 30
weeks (males fed standard chow); or age 16 weeks (males fed high fat chow).
Mice were anesthetized with 2% isoflourane and body composition was analyzed
using dual-energy X-ray absorptiometry (GE Lunar Piximus (Madison, WI),
software version 1.45).
Serum assays Serum insulin was measured by double-antibody
radioimmunoassay (Sensitive rat insulin RIA, Linco, St. Charles, MO) in the lab of
Dr. Barbara Gower (University of Alabama at Birmingham).
Serum NEFA, total cholesterol, and TAG were assessed by in vitro
enzymatic colorimetry (HR Series NEFA-HR(2), Cholesterol-E, and L-Type TG H
kits from Wako Diagnostics).

11
Serum catecholamines were measured in the CMN/KC Neurochemistry
Core Lab, Vanderbilt University, Nashville, TN. First, serum catecholamines are
adsorbed onto solid Al2O3, and then desorbed from the Al203 using 200 μl 0.1 N
acetic acid. Biogenic amines are determined by a specific HPLC assay utilizing
an Antec Decade II (oxidation: 0.5) electrochemical detector. Twenty μl samples
were injected using a Water 717+ autosampler onto a Phenomenex Nucleosil
(5u, 100A) C18 HPLC column (150 x 4.60 mm).
Apolipoprotein assays were performed on four pooled serum samples
from 16 transgenic mice and four pooled samples from 16 wild-type mice. Pooled
samples were analyzed using the lipoprotein autoprofiler method (Chung et al.
1981). First, a 2.5 hour density gradient ultracentrifugation was used to separate
the major lipoprotein fractions in the serum. Next, effluent was continuously
removed from the bottom of the density gradient and analyzed using enzymatic
colorimetric cholesterol and phospholipid kits from Wako Diagnostics.
Measurement of effluent absorbance at the correct wavelength indicates HDL,
LDL, and VLDL concentrations.

12
RESULTS
Verification of transgene insertion and function Transgenic mice were
genotyped by PCR, with the 3’ primer annealing to a V5 coding region in order to
preclude amplification of wild-type DNA. Transcriptional activity of the MINOR
transgene was verified by reverse transcriptase PCR (fig. 1a). Increased MINOR
protein levels were observed in white adipose tissue (fig. 1b and 1c) (p<0.05,
paired t-test). Preliminary quantitative PCR results indicate numerous transgene
copies (data not shown).
a
b
c
0
2
4
6
8
10
AP2-MINOR Wild-type
MIN
OR
sig
nal (
Odu
*mm
2)
WT TG WT TG WT TG WT TG TG WT
WT TG WT TG WT TG WT TG TG WT
Figure 1. Verification of transgene insertion and function. a) reverse transcriptase PCR showing amplification of transgene mRNA, b) western blot using MINOR antibody against protein extracted from gonadal fat, and c) quantification of western blot shown in panel b (p<0.05, paired t-test). Error bars represent SEM. WT=wild-type, TG=AP2-MINOR transgenic
*

13
Female AP2-MINOR transgenic mice Transgenic female body weight did not
differ from wild-type (age 8-16 weeks, p=0.8, repeated measures ANOVA). Nor
were there any differences in body composition at age 8 weeks (table 1). Body
composition was not assessed at age 16 weeks because no differences in body
weight were observed. Fasting glucose at age 17 weeks was significantly higher
in transgenic mice (fig. 2) (134.1 versus 109.5 mg/dL, p<0.05, Wilcoxon rank
sum). When GTT was assessed by AUC analysis, AP2-MINOR females tended
to have lower scores than wild-type (p=0.052, WIlcoxon rank sum). However,
repeated measures ANOVA indicated no between-group difference in glucose
tolerance. Likewise, no differences in insulin tolerance were observed.
Blo
od g
luco
se (m
g/dL
)
100
110
120
130
140
150
160
170
AP2-MINOR Wild-type
Genotype
Figure 2. Fasting glucose is increased in female AP2-MINOR mice versus wild-type littermates (P<0.05, Wilcoxon). AP2-MINOR N=8, Wild-type N=8

14
Table 1. Body composition in 8-week old female AP2-MINOR and wild-type mice maintained on standard chow
N=11/group Body weight (g) (95%CI)
Lean (g) (95%CI) Fat (g) (95%CI) % Fat
Mean, median
AP2-MINOR 18.9 (18.2-19.5)
15.3 (14.7-15.8)
1.77 (1.65-1.89) 10.4, 10.2
Wild-type 19.0 (18.4-19.7)
15.5 (14.9-16.0)
1.81 (1.69-1.93) 10.5, 10.9
P 0.68 0.60 0.67 0.83 (Wilcoxon)
Male AP2-MINOR transgenic mice—standard chow AP2-MINOR males
maintained on standard chow were significantly larger than their wild-type
littermates (fig. 3a) from age 8 to 16 weeks (p<0.05), with this pattern persisting
from age 20 to 30 weeks (fig. 3b) (p<0.01). Food intake tended to be slightly
higher in transgenic males from 9 to 16 weeks of age (fig. 3c) (p=0.1) and from
20 to 29 weeks of age (fig. 3d) (p=0.09). However, average weekly food intake
per gram body weight tended to be lower in transgenic animals from 9 to 16
weeks of age (p=0.07, Wilcoxon rank sum) (fig. 3e) and from 20 to 29 weeks of
age (p<0.01, Wilcoxon rank sum) (fig. 3f).

15
a
20
25
30
35
40
45
50
8 9 10 11 12 13 14 15 16
Age (weeks)
Bod
y w
eigh
t (g)
AP2-MINOR
Wild-type
b
20
25
30
35
40
45
50
20 21 22 23 24 25 26 27 28 29 30
Age (weeks)
Bod
y w
eigh
t (g)
Figure 3, panels a and b. Body weight and food intake in AP2-MINOR and wild-type mice. a) transgenic mice are larger than wild type mice from ages 8 to 16 weeks (p<0.05), b) transgenic mice are larger than wild-type from 20 to 30 weeks of age (<0.01). All body weight and food intake analyses are repeated measures ANOVA. Error bars represent SD. AP2-MINOR N=21, Wild-type N=19

16
c
20.0
25.0
30.0
35.0
40.0
9 10 11 12 13 14 15 16
Age (weeks)
Food
inta
ke (g
)
AP2-MINOR
Wild-type
d
20.0
25.0
30.0
35.0
40.0
20 21 22 23 24 25 26 27 28 29
Age (weeks)
Food
inta
ke (g
)
e
Ave
rage
wee
kly
food
inta
ke (g
)/bod
y w
eigh
t (g)
0.9
1
1.1
1.2
1.3
1.4
AP2-MINOR Wild-type
Genotype f
Ave
rage
wee
kly
food
inta
ke (g
)/bod
y w
eigh
t (g)
0.75
0.8
0.85
0.9
0.95
1
AP2-MINOR Wild-type
Genotype
Figure 3, panels c, d, e and f. Transgenic mice tend to consume slightly more food from 9 to 16 weeks of age (p=0.1) (c), and (d) from 20 to 29 weeks of age (p=0.09). All body weight and food intake analyses are repeated measures ANOVA. Error bars represent SD. Average weekly food intake (grams)/body weight (grams) is decreased in AP2-MINOR mice from ages 9 to 16 weeks (p=0.07, Wilcoxon) (e), and from ages 20 to 29 weeks (f) (p<0.01, Wilcoxon). AP2-MINOR N=21, Wild-type N=19

17
Body composition data indicate that male AP2-MINOR mice fed standard
chow are larger than wild-type at 8 weeks of age, with trends toward greater total
mass (p=0.16) and lean mass (p=0.12). However, no differences in fat mass or
percent fat were observed at 8 weeks of age (table 2). At 16 weeks of age,
transgenic mice displayed more pronounced trends toward greater total mass
(p=0.08) and lean mass (p=0.054). At 16 weeks, transgenic mice also displayed
a trend toward greater fat mass (p=0.18), with no difference in percent fat (table
3). At 30 weeks of age, AP2-MINOR mice again displayed a trend toward greater
total mass, with lean mass becoming significantly greater than wild-type (p<0.05).
Transgenic animals continued to tend toward greater fat mass (p=0.1), with no
differences observed in percent fat (table 4). When animals were sacrificed
(between 33 and 35 weeks of age), AP2-MINOR mice tended to have larger
gonadal fat pads, but this difference was not statistically significant (AP2-MINOR
mean fat pad weight=0.92 g, wild-type mean fat pad weight=0.82 g, p=0.14).
Table 2. Body composition in 8-week old AP2-MINOR and wild-type mice Body weight (g)
(95% CI) Lean mass (g)
(95% CI) Fat mass (g)
(95% CI) % Fat (g)
Mean, median AP2-MINOR
(n=20) 24.9
(24.0-25.8) 19.6
(19.0-20.1) 3.1
(2.7-3.4) 13.4, 13.0
Wild-type (n=19)
24.0 (23.1-24.9)
18.9 (18.3-19.5)
2.9 (2.5-3.3)
13.1, 12.8
P 0.16 0.12 0.51 0.52
(Wilcoxon rank sum)

18
Table 3. Body composition in 16-week old AP2-MINOR and wild-type mice Body weight (g)
(95% CI) Lean mass (g)
(95% CI) Fat mass (g)
(95% CI) % Fat (g)
mean, median AP2-MINOR
(n=19) 34.2
(32.5-35.9) 23.1
(22.4-23.9) 8.2
(7.1-9.3) 25.7, 25.7
Wild-type (n=19)
32.0 (30.3-33.8)
22.1 (21.4-22.8)
7.2 (6.1-8.2) 23.9, 25.1
P 0.08 0.054 0.18 0.34
(Wilcoxon rank sum)
Table 4. Body composition in 30-week old AP2-MINOR and wild-type mice Body weight (g)
(95% CI) Lean mass (g)
(95% CI) Fat mass (g)
(95% CI) % Fat
Mean, median Transgenic
(n=16) 42.8
(40.4-45.2) 25.6
(23.7-25.4) 14.3
(12.7-15.9) 36.5, 37.6
Wild-type (n=17)
39.6 (37.3-41.9)
23.2 (22.5-24.0)
12.5 (11.0-14.0) 34.4, 36.3
P 0.06 0.02 0.10 0.26
(Wilcoxon rank sum)
AP2-MINOR transgenic mice maintained on standard chow had elevated
fasting glucose compared to wild-type littermates at age 17 weeks (p=0.01) (fig.
4a). Additionally, glucose tolerance was significantly impaired in transgenic
animals at age 17 weeks. Two outliers were removed from the following analyses
due to glucose tolerance test AUC blood glucose that was greater than two
standard deviations from the mean. Both outlier animals were transgenic, with
AUC glucoses equaling 200 and 1530. Statistical significance was reduced
following the removal of these outliers. After removing outliers, AUC blood
glucose was still significantly greater in transgenic animals compared to wild-type
(p<0.05) (fig. 4b). Accordingly, repeated measures ANOVA analysis indicated
significantly different glucose responses between groups (p=0.01) (fig. 4c).

19
Insulin tolerance did not differ between groups at age 18 weeks when assessed
by either AUC or repeated measures ANOVA (fig. 5).
a
100110120130140150160170180190200
AP2-MINOR Wild-typeFast
ing
bloo
d gl
ucos
e (m
g/dL
)
b
400
600
800
1000
1200
1400
AP2-MINOR Wild-type
Glu
cose
tole
ranc
e te
stA
UC
*
*
c
100
150
200
250
300
350
400
450
0 30 60 90 180
Time (minutes)
Bloo
d gl
ucos
e (m
g/dL
)
AP2-MINOR
Wild-type
Figure 4. Glucose tolerance in 17-week old mice. a) transgenic mice display elevated fasting glucose at age 17 weeks (p=0.01), b) glucose tolerance test AUC is significantly higher in transgenic mice (p<0.05), c) AP2-MINOR mice display reduced glucose tolerance (p=0.01, repeated measures ANOVA). Error bars represent SD. AP2-MINOR N=15, Wild-type N=16

20
75100125150175200225250275
0 30 60 90 180
Time (minutes)
Blo
od g
luco
se (m
g/dL
)
AP2-MINOR
Wild-type
Figure 5. At age 18 weeks, no differences were observed in insulin tolerance (repeated measures ANOVA). Error bars represent SD. AP2-MINOR N=15, Wild-type N=16
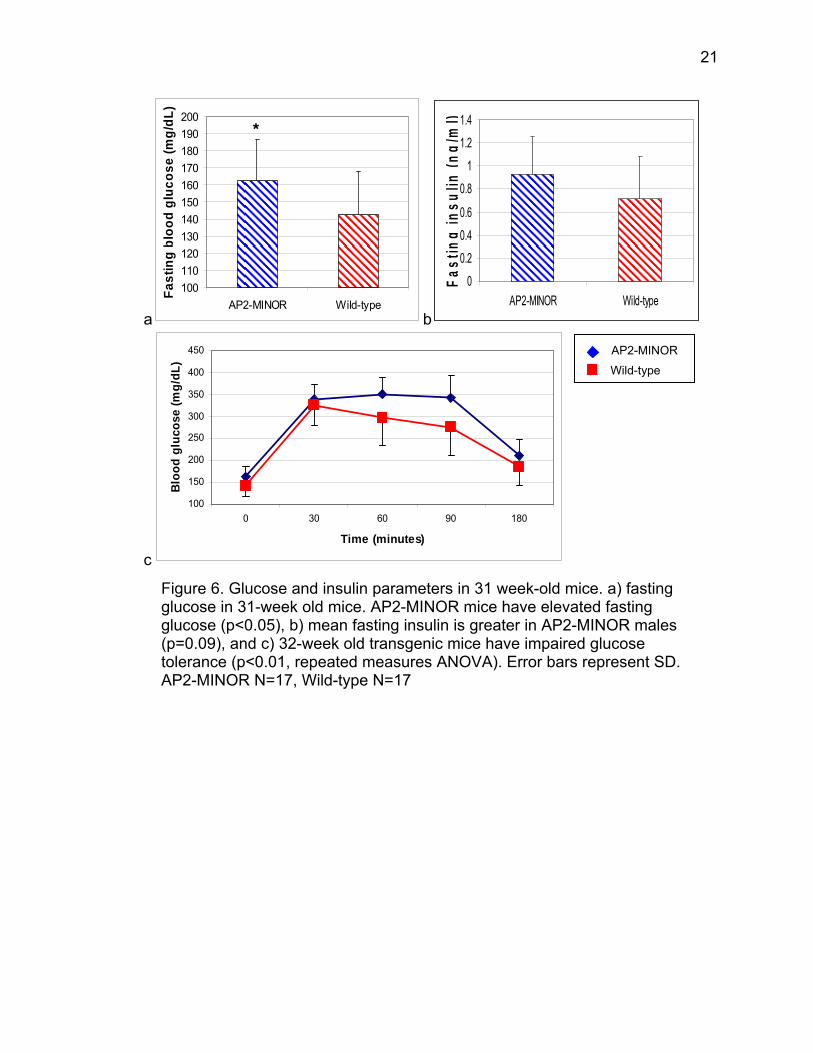
At 31 weeks of age, transgenic animals continued to display increased
fasting glucose compared to wild-type littermates (p<0.05) (fig. 6a). At 33-35
weeks of age, fasting insulin was also slightly elevated in AP2-MINOR animals
compared to wild-type (mean values 0.93 versus 0.72 ng/ml, p=0.09) (fig. 6b).
Furthermore, transgenic animals were glucose intolerant compared to wild-type
(p<0.01), with a significant interaction between time and genotype (p<0.01) (fig.
6c). However, glucose tolerance test AUC was not significantly different at age
31 weeks (mean AUC value for transgenic animals=694.7, mean AUC value for
wild-type animals=604.2). Transgenic mice appeared to be slightly insulin
resistant at age 32 weeks; insulin tolerance test AUC absolute values were
significantly smaller in AP2-MINOR mice (mean AUC value for transgenic
animals=-237.5, mean AUC value for wild-type animals=-388.33, p<0.05) (fig.
7a). However, repeated measures ANOVA did not indicate a significant
difference in insulin tolerance (p=0.2) (fig. 7b).
21
a
100110120130140150160170180190200
AP2-MINOR Wild-type
Fast
ing
bloo
d gl
ucos
e (m
g/dL
)
b
0
0.2
0.4
0.6
0.8
1
1.2
1.4
AP2-MINOR Wild-type
Fast
ing
insu
lin (n
g/m
l)
*
c
100
150
200
250
300
350
400
450
0 30 60 90 180
Time (minutes)
Bloo
d gl
ucos
e (m
g/dL
)
AP2-MINOR
Wild-type
Figure 6. Glucose and insulin parameters in 31 week-old mice. a) fasting glucose in 31-week old mice. AP2-MINOR mice have elevated fasting glucose (p<0.05), b) mean fasting insulin is greater in AP2-MINOR males (p=0.09), and c) 32-week old transgenic mice have impaired glucose tolerance (p<0.01, repeated measures ANOVA). Error bars represent SD. AP2-MINOR N=17, Wild-type N=17

22
a-500
-400
-300
-200
-100
Blo
od g
luco
se A
UC
(mg/
dL)
AP2-MINOR Wild-type
*
b
75100125150175200225250275300
0 30 60 90 180
Time (minutes)
Blo
od g
luco
se (m
g/dL
)
AP2-MINOR
Wild-type
Figure 7. Insulin tolerance in 32-week old mice. a) AP2-MINOR mice display decreased absolute value AUC blood glucose following insulin challenge (p<0.05), b) blood glucose curves do not differ between groups following insulin delivery (p=0.2, repeated measures ANOVA). Error bars represent SD. AP2-MINOR N=17, Wild-type N=17
Fasting serum lipid values are summarized in table 5. We were surprised
to observe significantly decreased non-esterified fatty acids (NEFA) in transgenic
mice versus wild-type (p<0.05) (figure 8a). AP2-MINOR animals also tended to
have lower mean triglycerides (p=0.16). Paradoxically, total cholesterol was
significantly higher in transgenic mice (p=0.001) (fig. 8b), with increased LDL
accounting almost completely for the difference in total cholesterol (AP2-MINOR

23
mean LDL=35.05 mg/dL, wild-type mean LDL=22.0 mg/dL, p=0.0002 two-way
ANOVA, Brown-Forsythe test p=0.42) (fig. 8c). Mean VLDL was greater in AP2-
MINOR mice (p=0.16), and HDL was not different between groups.
Table 5. Fasting serum lipids in transgenic and wild-type mice maintained on standard chow
AP2-MINOR Wild-type P Triglycerides (N=15/group) 105.9 118.4 0.16
NEFA (N=15/group) 1.15 1.32 0.02
Total cholesterol (N=15/group) 117.9 96.1 0.001
HDL (N=4 pooled
samples/group) 73.7 69.6 0.59
LDL (N=4 pooled
samples/group) 35.05 22.0 0.0002
VLDL (N=4 pooled
samples/group) 6.475 5.4 0.16

24
a
00.20.40.60.8
11.21.41.61.8
AP2-MINOR Wild-type
Non
-est
erifi
ed fa
tty a
cid
(mm
ol/L
)
b
0
30
60
90
120
150
AP2-MINOR Wild-type
Tota
l ser
um c
hole
ster
ol
(mg/
dL)
* **
c
05
10152025303540
AP2-MINOR Wild-type
LDL
(mg/
dL)
***
Figure 8. Serum lipids in AP2-MINOR and wild-type mice. a) transgenic mice have significantly lower fasting serum NEFA (p<0.05), b) total cholesterol is significantly increased in transgenic animals (p=0.001), and c) increased total cholesterol is explained by highly increased low density lipoprotein (p=0.0002, 4 pooled samples/group). Error bars represent SD. AP2-MINOR N=15, Wild-type N=15
Insulin’s ability to suppress adipocyte lipolysis appears somewhat
impaired in AP2-MINOR transgenic mice. When injected with insulin and
sacrificed 1 hour later, transgenic animals had slightly higher serum NEFA than
wild-type (mean value 1.2 mmol/L versus 1.1 mmol/L, p=0.17) (fig. 9b). This
occurred in spite of significantly decreased serum catecholamines in transgenic
animals; mean serum norepinephrine was 35.7 pg/μl in transgenic mice versus
43.9 pg/μl in wild-type mice (p<0.05) (fig. 9c). Mean serum epinephrine was 5.1

25
pg/μl in transgenic mice versus 10.3 pg/μl in wild-type mice (p<0.001) (fig. 9d).
Post-injection insulin levels did not differ between groups (AP2-MINOR mean
insulin=3.9 ng/ml, wild-type mean insulin=3.8 ng/ml, p=0.9) (fig. 9a). There was
no correlation between serum insulin and NEFA for either group alone or for both
groups together. However, there was a slight negative correlation between serum
eprinephrine and NEFA, ostensibly due to deranged epinephrine values in
transgenic mice (R squared=0.19, p=0.07).
a
0
1
2
3
4
5
6
7
AP2-MINOR Wild-type
Post
-inje
ctio
n in
sulin
(n
g/m
l)
b
00.20.40.60.8
11.21.41.61.8
AP2-MINOR Wild-type
NEF
A (m
mol
/L)
c
0
10
20
30
40
50
60
AP2-MINOR Wild-typeSeru
m n
orep
inep
hrin
e (p
g/ul
)
d
0
2
4
6
8
10
12
14
16
AP2-MINOR Wild-typeSeru
m e
pine
phrin
e (p
g/ul
)
* ***
Figure 9. Insulin’s ability to suppress lipolysis in AP2-MINOR and wild-type mice. a) insulin levels attained following injection did not differ between groups (p=0.9), b) NEFA levels were slightly higher in transgenic animals (p=0.17). c) serum norepinephrine is significantly decreased in transgenic mice compared to wild-type (p<0.05), d) serum epinephrine is highly significantly decreased in AP2-MINOR mice compared to wild-type (p<0.001). Error bars represent SD. AP2-MINOR N=9, Wild-type N=9

26
Male AP2-MINOR transgenic mice—high fat diet Male AP2-MINOR mice
maintained on high fat chow were also larger than their wild-type littermates from
age 9 to 16 weeks (p<0.05) (fig 10a). From age 20 to 30 weeks, transgenic
animals had higher mean body weights that did not reach statistical significance
(p=0.15) (fig 10b). Food intake did not differ between groups (p=0.3) (fig. 10c).
a
2025303540455055
1 2 3 4 5 6 7 8
Age (weeks)
Bod
y w
eigh
t (g)
AP2-MINOR
Wild-type
b
20
25
30
35
40
45
50
55
20 21 22 23 24 25 26 27 28 29 30
Age (weeks)
Bod
y w
eigh
t (g)
Figure 10, panels and b. Body weight and food intake in mice fed high fat chow. a) AP2-MINOR body weight was significantly higher from age 9 to 16 weeks (p<0.05), b) body weight in AP2-MINOR mice was not significantly greater from age 20 to 30 weeks (p=0.15). Body weight and food intake analyses are repeated measures ANOVA. Error bars represent SD. AP2-MINOR N=13, Wild-type N=14

27
c
15171921232527
10 11 12 13 14 15
Week
Food
inta
ke (g
)
AP2-MINOR
Wild-type
Figure 10, panel c. Food intake did not differ between transgenic and wild-type mice (p=0.3) maintained on high fat chow (p=0.3, repeated measures ANOVA). Error bars represent SD. AP2-MINOR N=13, Wild-type N=14
Table 6 summarizes body composition measures in mice maintained on
high fat chow. At age 16 weeks, no significant differences were observed in body
weight (on the day of DEXA scan), lean mass, fat mass, or percent fat. Gonadal
fat pad weight at age 33-35 weeks of age also did not differ between groups
(p=0.15), although transgenic mice had lower mean fat pad weight than wild-type
littermates (0.52 g versus 0.57 g).
Table 6. Body composition in 16-week old, high-fat fed AP2-MINOR and wild-type mice
Body weight (g) (95% CI)
Lean mass (g) (95% CI)
Fat mass (g) (95% CI)
% Fat mean,
median AP2-MINOR
N=8 45.0
(43.0-46.9) 22.2
(21.0-23.4) 18.8
(17.8-19.8) 45.0, 45.0
Wild-type N=12
43.7 (42.1-45.3)
21.5 (20.5-22.5)
18.2 (17.3-19.0) 45.0, 46.0
P 0.29 0.38 0.32 1.0 (Wilcoxon)

28
Transgenic mice maintained on high fat chow exhibited no difference in
fasting glucose, fasting insulin, glucose tolerance, or insulin tolerance. Results of
glucose and insulin tolerance tests are shown in figure 11.
a
100150200250300350400450
0 30 60 90 180
Time (minutes)
Blo
od g
luco
se (m
g/dL
)
AP2-MINOR
Wild-type
b
75100125150175200225250275300
0 30 60 90 180
Time (minutes)
Blo
od g
luco
se (m
g/dL
)
Figure 11. Glucose (a) and insulin (b) tolerance tests in animals
maintained on high fat chow. Results are not significant when assessed by either AUC student’s t-test or repeated measures ANOVA. Error bars represent SD. AP2-MINOR N=10, Wild-type N=11
Additionally, fasting serum lipids did not differ significantly between AP2-
MINOR and wild-type animals raised on high fat chow. Thus, NEFA, total
cholesterol, HDL, LDL, and VLDL were virtually identical between groups.
However, fasting triglycerides displayed a significant difference in variance

29
(p<0.05, Brown-Forsythe), with transgenic animals having less variance about
the mean (fig. 12).
Tr
igly
cerid
es (m
g/dL
)
60
70
80
90
100
110
120
130
AP2-MINOR Wild-type
Genotype
Figure 12. Fasting serum triglycerides. Means do not differ significantly (p=0.16, Welch ANOVA), but variance is significantly different (p<0.05, Brown-Forsythe). AP2-MINOR N=11, Wild-type N=14
DISCUSSION AP2-MINOR transgenic mice exhibit abnormal glucose and lipid
homeostasis. At age 16 weeks, female mice maintained on standard chow have
elevated fasting glucose and impaired glucose tolerance. Male transgenic mice
maintained on standard chow were larger than wild-type, with no significant
differences in body composition at 8 or 16 weeks of age. For the duration of the
study, transgenic mice ate less per gram body weight in spite of significantly
increased lean mass at age 30 weeks. This finding is particularly interesting
when considering the fact that transgenic mice have slightly higher day-time body
temperatures (data not shown). So, it would be plausible to hypothesize that
AP2-MINOR mice are less active than wild-type. AP2-MINOR males fed standard
chow exhibited impaired glucose tolerance at ages 17 and 31 weeks, and
impaired insulin tolerance at age 32 weeks. Fasting serum triglyceride was
slightly decreased in transgenic animals while NEFA, total cholesterol, and LDL
were significantly elevated.

30
The adipocytes of AP2-MINOR transgenic mice appear to be either
exquisitely sensitive to serum catecholamines or resistant to the effects of insulin,
or both; insulin failed to decrease transgenic serum NEFA below the level of wild-
type animals in spite of significantly lower catecholamines in transgenics. It is
unclear whether decreased catecholamines following insulin injection indicate a
chronic depression of the sympathetic nervous system or a decreased response
to the acute stress associated with human handling. It also unclear whether this
change is mild or severe; reports on sympathetic function in adipocytes employ
synthetic β-adrenergic agonists and antagonists rather than epinephrine and
norepinephrine, preventing meaningful comparisons with our experiment. In any
event, alterations in either acute or chronic stress responses could have lasting
physiological consequences. Furthermore, perturbations in adrenergic tone could
account for elevated fasting glucose and poor glucose tolerance in transgenic
mice since β-adrenoreceptor agonism potentiates pancreatic production of insulin
and both α- and β-adrenoreceptors modulate glucagon secretion (Nonogaki
2000).
It would also be reasonable to hypothesize that the physiological
characteristics observed in AP2-MINOR mice are due to down-regulation of liver
X receptor (LXR) and its direct target, sterol response element binding protein-1c
(SREBP-1c), important transcriptional regulators of lipid, sterol, and glucose
metabolic enzymes in adipocytes. It is interesting to note that adenoviral-driven
over-expression of NR4A family member Nur77 inhibits SREBP-1c expression
and activity in mouse hepatocytes, inducing a 98% increase in serum LDL and a
67% increase in serum triglycerides (Pols et al. 2008). Pols et al. also observed
decreased expression of LXR target genes, ABCg5 and ABCg8, leading them to
hypothesize that down-regulation of SREBP-1c is effected through inhibition of
LXR. In vivo over-expression of SREBP-1c driven by the AP2 promoter induces a
severe lipodystrophic phenotype in mice, with enlarged fatty liver, atrophied white
adipose tissue depots, and “WAT” phenotype in intrascapular brown fat pads.
These changes are mediated by a host of protein expression abnormalities,
including: down-regulation of leptin, fatty acid synthase (WAT), PPAR-γ, C/EPB-

31
α, adipocyte FABP (AP2), Insulin receptor, IRS-1, IRS-2, GLUT-4, and UCP-1
(BAT), and concomitant up-regulation of LDL receptor, acetyl Co-A carboxylase,
HMG-CoA reductase, TNF- α, stearoyl Co-A desaturase (liver, BAT) and fatty
acid synthase (liver, BAT) (Shimomura et al. 1998). It is possible that
overexpression of MINOR in adipocytes is driving decreased expression or
activity of SREBP-1c, causing protein expression changes that are more subtle,
yet roughly inverse to those seen in AP2-SREBP-1c mice (summarized in table
7).
Table 7. Summary of hypothesized protein expression or function changes in AP2-MINOR mice Direction Protein Increased Leptin FAS (WAT) PPAR-γ C/EBP-α AP2 (FABP) UCP-1 (BAT) Insulin receptor IRS-1 IRS-2 GLUT-4 Decreased LDL receptor TNF-α ACC SCD HMGCoA reductase FAS (liver, BAT)
Thus, MINOR-mediated SREBP-1c down-regulation could cause
increased leptin expression leading to an overall decrease in sympathetic
nervous system tone and a blunted pancreatic response to glucose. Contrary to
observations in rats and humans, chronically elevated leptin in mice is known to
decrease sympathetic output through direct actions of leptin on the hypothalamus
(Malendowicz et al. 2007). Furthermore, pancreas-specific knockout of leptin

32
receptor in mice results in improved glucose tolerance, an effect which is
dampened by high-fat feeding (Morioka et al. 2007).
MINOR-induced SREBP-1c could also drive decreased TNF-α production
resulting in decreased sympathetic tone. Indeed, TNF-α administration to mice
(at doses too small to affect blood pressure, food intake, or prolactin release)
causes increased ACTH production within minutes (Dunn, 2000).
Furthermore, decreased SREBP-1c could account for increased serum
LDL and decreased NEFA in AP2-MINOR mice. SREBP-1c is a known
transcriptional activator of LDL receptor (Pols et al. 2008). Mice lacking LDL
receptor are well characterized, with increased serum LDL and IDL (Breslow,
1996). However, LDL levels observed in AP2-MINOR mice are not sufficiently
elevated to promote atherosclerotic plaque formation; in LDL-receptor knockout
and ApoE knockout mice, non-HDL cholesterol of approximately 300 mg/dL is
required for atherogenesis (Hsueh et al. 2007). Increased expression of FABP in
AP2-MINOR mice could account for decreased serum NEFA. Mice lacking FABP
display slightly increased NEFA and decreased triglycerides when reared on
standard chow. Additionally, transgenic AP2-endothelial-FABP mice display
increased serum glucose and decreased insulin sensitivity (Furuhashi &
Hotamisligil, 2008).
Since MINOR overexpression in cultured 3T3-L1 cells led to enhanced
insulin-stimulated glucose uptake (Fu et al. 2007), we were surprised to find that
AP2-MINOR mice responded poorly when challenged with glucose and insulin.
However, the apparent contradiction between our in vitro and in vivo findings
could be explained by SREBP-1c modulation of a wide array of proteins required
for lipid metabolism, insulin-signaling, and glucose metabolism in adipocytes. In
vitro, the effects of the lipid modulators, LDL-receptor and FABP, would be
assumed to be minimal, while the effects of insulin- and glucose-related
pathways would have more obvious effects. Thus decreased SREBP-1c function
in vitro could allow increased expression of insulin receptor, IRS-1, IRS-2, GLUT-
4, PPAR-γ, and C/EBP-α, producing increased insulin sensitivity.

33
The AP2 promoter is known to be functional in macrophages as well as
adipocytes. Expression of the AP2 gene product, adipocyte fatty acid binding
protein (FABP), is approximately 10,000X higher in adipocytes than THP-1
monocytes (Furuhashi & Hotamisligil, 2008). Yet, our lab has demonstrated that
oxidized lipid-mediated induction of foam cell formation is accompanied by
extreme up-regulation of FABP (Fu et al. 2006). Thus, AP2-driven transgene
expression in macrophages could be a significant contributor to the phenotype of
the AP2-MINOR mice.
Finally, it is clear that NR4A family members are not required for
adipogenesis; stable expression of a dominant negative Nur77 suppresses the
function of all NR4A family members without affecting lipid accumulation or
mature adipocyte gene production (Au et al. 2008). Thus it appears that MINOR
and other NR4A family members function as important modulators of other
nuclear receptors in differentiating and mature adipocytes. Over-expression of
MINOR in mouse adipocytes causes decreased glucose tolerance and mild
dislipidemia, and these effects are likely due to complex interactions between
MINOR and other nuclear receptors.

34
REFERENCES Ahren B, Scheurink AJ. Marked hyperleptinemia after high-fat diet associated with severe glucose intolerance in mice. Eur J Endocrinol. 1998 Oct;139(4):461-7. Arner P. Human fat cell lipolysis: biochemistry, regulation and clinical role. Best Pract Res Clin Endocrinol Metab. 2005 Dec;19(4):471-82. Au WS, Payne VA, O'Rahilly S, Rochford JJ. The NR4A family of orphan nuclear receptors are not required for adipogenesis. Int J Obes (Lond). 2008 Feb;32(2):388-92. Breslow, Jan L. Mouse models of atherosclerosisScience. Washington: May 3, 1996. Vol. 272, Iss. 5262; pg. 685 Chung BH, Segrest JP, Cone JT, Pfau J, Geer JC, Duncan LA. High resolution plasma lipoprotein cholesterol profiles by a rapid, high volume semi-automated method. J Lipid Res. 1981 Aug;22(6):1003-14. Crespo J, Martinez-Gonzalez J, Rius J, Badimon L. Simvastatin inhibits NOR-1 expression induced by hyperlipemia by interfering with CREB activation. Cardiovasc Res. 2005 Aug 1;67(2):333-41.
DeYoung RA, Baker JC, Cado D, Winoto A. The orphan steroid receptor Nur77 family member Nor-1 is essential for early mouse embryogenesis. J Biol Chem. 2003 Nov 21;278(47):47104-9.
Dubois M, Kerr-Conte J, Gmyr V, Bouckenooghe T, Muharram G, D'Herbomez M, Martin-Ponthieu A, Vantyghem MC, Vandewalle B, Pattou F. Non-esterified fatty acids are deleterious for human pancreatic islet function at physiological glucose concentration. Diabetologia. 2004 Mar;47(3):463-9.
Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci. 2000;917:608-17. Farmer SR, Prusty D. Role of the Adipocyte in Type 2 Diabetes. International Textbook of Diabetes Mellitus. DeFronzo et al., editors. John Wiley & Sons. 2004. pp. 641-654.

35
Farnier C, Krief S, Blache M, Diot-Dupuy F, Mory G, Ferre P, Bazin R. Adipocyte functions are modulated by cell size change: potential involvement of an integrin/ERK signalling pathway. Int J Obes Relat Metab Disord. 2003 Oct;27(10):1178-86. Fernandez PM, Brunel F, Jimenez MA, Saez JM, Cereghini S, Zakin MM. Nuclear receptors Nor1 and NGFI-B/Nur77 play similar, albeit distinct, roles in the hypothalamo-pituitary-adrenal axis. Endocrinology. 2000 Jul;141(7):2392-400. Flaig R, Greschik H, Peluso-Iltis C, Moras D. Structural basis for the cell-specific activities of the NGFI-B and the Nurr1 ligand-binding domain. J Biol Chem. 2005 May 13;280(19):19250-8. Fu Y, Luo L, Luo N, Garvey WT. Lipid metabolism mediated by adipocyte lipid binding protein (ALBP/aP2) gene expression in human THP-1 macrophages. Atherosclerosis. 2006 Sep;188(1):102-11. Fu Y, Luo L, Luo N, Zhu X, Garvey WT. NR4A orphan nuclear receptors modulate insulin action and the glucose transport system: Potential role in insulin resistance. J Biol Chem. 2007 Sep 4; Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008 Jun;7(6):489-503. Goh TT, Mason TM, Gupta N, So A, Lam TK, Lam L, Lewis GF, Mari A, Giacca A. Lipid-induced beta-cell dysfunction in vivo in models of progressive beta-cell failure. Am J Physiol Endocrinol Metab. 2007 Feb;292(2):E549-60. Hedvat CV, Irving SG. The isolation and characterization of MINOR, a novel mitogen-inducible nuclear orphan receptor. Mol Endocrinol. 1995 Dec;9(12):1692-700. Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, Fisher EA, Dansky H, McClain DA, McIndoe R, Wassef MK, Rabadán-Diehl C, Goldberg IJ. Recipes for creating animal models of diabetic cardiovascular disease. Circ Res. 2007 May 25;100(10):1415-27.
Inuzuka H, Tokumitsu H, Ohkura N, Kobayashi R. Transcriptional regulation of nuclear orphan receptor, NOR-1, by Ca(2+)/calmodulin-dependent protein kinase cascade. FEBS Lett. 2002 Jul 3;522(1-3):88-92.
Kagaya S, Ohkura N, Tsukada T, Miyagawa M, Sugita Y, Tsujimoto G, Matsumoto K, Saito H, Hashida R. Prostaglandin A2 acts as a transactivator for

36
NOR1 (NR4A3) within the nuclear receptor superfamily. Biol Pharm Bull. 2005 Sep;28(9):1603-7.
Kanzleiter T, Schneider T, Walter I, Bolze F, Eickhorst C, Heldmaier G, Klaus S, Klingenspor M. Evidence for Nr4a1 as a cold-induced effector of brown fat thermogenesis. Physiol Genomics. 2005 Dec 14;24(1):37-44.
Laflamme C, Filion C, Bridge JA, Ladanyi M, Goldring MB, Labelle Y. The homeotic protein Six3 is a coactivator of the nuclear receptor NOR-1 and a corepressor of the fusion protein EWS/NOR-1 in human extraskeletal myxoid chondrosarcomas. Cancer Res. 2003 Jan 15;63(2):449-54
Maira M, Martens C, Philips A, Drouin J. Heterodimerization between members of the Nur subfamily of orphan nuclear receptors as a novel mechanism for gene activation. Mol Cell Biol. 1999 Nov;19(11):7549-57. Malendowicz LK, Rucinski M, Belloni AS, Ziolkowska A, Nussdorfer GG. Leptin and the regulation of the hypothalamic-pituitary-adrenal axis. Int Rev Cytol. 2007;263:63-102.
Maxwell MA, Cleasby ME, Harding A, Stark A, Cooney GJ, Muscat GE. Nur77 regulates lipolysis in skeletal muscle cells. Evidence for cross-talk between the beta-adrenergic and an orphan nuclear hormone receptor pathway. J Biol Chem. 2005 Apr 1;280(13):12573-84.
Maxwell MA, Muscat GE. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal. 2006;4:e002. Morioka T, Asilmaz E, Hu J, Dishinger JF, Kurpad AJ, Elias CF, Li H, Elmquist JK, Kennedy RT, Kulkarni RN. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J Clin Invest. 2007 Oct;117(10):2860-8. Nagy TR, Clair AL. Precision and accuracy of dual-energy X-ray absorptiometry for determining in vivo body composition of mice. Obes Res. 2000 Aug;8(5):392-8
Newsholme P, Keane D, Welters HJ, Morgan NG. Life and death decisions of the pancreatic beta-cell: the role of fatty acids. Clin Sci (Lond). 2007 Jan;112(1):27-42.
Nomiyama T, Nakamachi T, Gizard F, Heywood EB, Jones KL, Ohkura N, Kawamori R, Conneely OM, Bruemmer D. The NR4A orphan nuclear receptor NOR1 is induced by platelet-derived growth factor and mediates vascular smooth muscle cell proliferation. J Biol Chem. 2006 Nov 3;281(44):33467-76.

37
Nonogaki K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia. 2000 May;43(5):533-49. Ohkura N, Hijikuro M, Yamamoto A, Miki K. Molecular cloning of a novel thyroid/steroid receptor superfamily gene from cultured rat neuronal cells. Biochem Biophys Res Commun. 1994 Dec 30;205(3):1959-65. Ohkura N, Ito M, Tsukada T, Sasaki K, Yamaguchi K, Miki K. Structure, mapping and expression of a human NOR-1 gene, the third member of the Nur77/NGFI-B family. Biochim Biophys Acta. 1996 Sep 11;1308(3):205-14
Ohkura N, Ito M, Tsukada T, Sasaki K, Yamaguchi K, Miki K. Alternative splicing generates isoforms of human neuron-derived orphan receptor-1 (NOR-1) mRNA. Gene. 1998 Apr 28;211(1):79-85.
O'kane M, Markham T, McEvoy AN, Fearon U, Veale DJ, Fitzgerald O, Kirby B, Murphy EP. Increased Expression of the Orphan Nuclear Receptor NURR1 in Psoriasis and Modulation following TNF-alpha Inhibition. J Invest Dermatol. 2007 Aug 2; [Epub ahead of print]
Pearen MA, Ryall JG, Maxwell MA, Ohkura N, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, is a target of beta-adrenergic signaling in skeletal muscle. Endocrinology. 2006 Nov;147(11):5217-27.
Pei L, Waki H, Vaitheesvaran B, Wilpitz DC, Kurland IJ, Tontonoz P. NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat Med. 2006 Sep;12(9):1048-55. Perlmann T, Jansson L. A novel pathway for vitamin A signaling mediated by RXR heterodimerization with NGFI-B and NURR1. Genes Dev. 1995 Apr 1;9(7):769-82. Philips A, Maira M, Mullick A, Chamberland M, Lesage S, Hugo P, Drouin J. Antagonism between Nur77 and glucocorticoid receptor for control of transcription. Mol Cell Biol. 1997 Oct;17(10):5952-9. Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS. Regulation of the insulin gene by glucose and fatty acids. J Nutr. 2006 Apr;136(4):873-6. Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS. Regulation of the insulin gene by glucose and fatty acids. J Nutr. 2006 Apr;136(4):873-6. Pols TW, Ottenhoff R, Vos M, Levels JH, Quax PH, Meijers JC, Pannekoek H, Groen AK, de Vries CJ. Nur77 modulates hepatic lipid metabolism through suppression of SREBP1c activity. Biochem Biophys Res Commun. 2008 Feb 22;366(4):910-6. Epub 2007 Dec 18.

38
Ponnio T, Burton Q, Pereira FA, Wu DK, Conneely OM. The nuclear receptor Nor-1 is essential for proliferation of the semicircular canals of the mouse inner ear. Mol Cell Biol. 2002 Feb;22(3):935-45. Ponnio T, Conneely OM. nor-1 regulates hippocampal axon guidance, pyramidal cell survival, and seizure susceptibility. Mol Cell Biol. 2004 Oct;24(20):9070-8. Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004 Oct 8;279(41):42351-4. Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL, Brown MS. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. 1998 Oct 15;12(20):3182-94. Smyth S, Heron A. Nat Med. Diabetes and Obesity: the twin epidemics. 2006 Jan;12(1):75-80. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol. 2005 Oct;25(10):2062-8. Winkler G, Kiss S, Keszthelyi L, Sapi Z, Ory I, Salamon F, Kovacs M, Vargha P, Szekeres O, Speer G, Karadi I, Sikter M, Kaszas E, Dworak O, Gero G, Cseh K. Expression of tumor necrosis factor (TNF)-alpha protein in the subcutaneous and visceral adipose tissue in correlation with adipocyte cell volume, serum TNF-alpha, soluble serum TNF-receptor-2 concentrations and C-peptide level. Eur J Endocrinol. 2003 Aug;149(2):129-35. Zetterstrom RH, Solomin L, Mitsiadis T, Olson L, Perlmann T. Retinoid X receptor heterodimerization and developmental expression distinguish the orphan nuclear receptors NGFI-B, Nurr1, and Nor1. Mol Endocrinol. 1996 Dec;10(12):1656-66.

39
APPENDIX: IACUC APPROVAL

![[2 Timothy] Leader’s Guide - Cru at UNCcruatunc.com/hp_wordpress/wp-content/uploads/2013/08/2-Timothy... · [2 Timothy] Leader’s Guide ... Timothy Introduction Welcome to](https://img.pdfslide.us/doc/110x75/5a7906cc7f8b9a9d218d1646/2-timothy-leaders-guide-cru-at-2-timothy-leaders-guide-timothy-introduction.jpg)



























