Embed Size (px)
Citation preview

Syndrome of the month
Journal of Medical Genetics 1989, 26, 119-126
Stickler's syndromeI K TEMPLEMothercare Department of Genetics, Institute of Child Health, London WCIN IEH.
In 1965, Stickler et a1l documented the associationof severe myopia and degenerative joint changes ina five generation family. They termed this auto-somal dominant condition hereditary progressivearthro-ophthalmopathy, but it is now morecommonly called Stickler's syndrome. Two yearslater Stickler and Pugh2 noted deafness in theoriginal proband and his mother and also remarkedon a characteristic flat facial appearance.
In 1972, Opitz et a13 pointed out the associationwith the Pierre-Robin sequence and Herrmann et aP4made an important contribution when theydescribed 64 cases of Stickler's syndrome, stressingthe variable manifestations of the condition.
Controversy remains as to whether the syndromedescribed by Stickler is a distinct entity or should beincorporated into part of a larger connective tissuedisorder which includes Marshall's,5 Wagner's,6 andWeissenbacher-Zweymuller syndromes.7 8
Clinical features
The variable manifestations of Stickler's syndromecan lead to diagnostic difficulties. The clinical find-ings can be divided into three groups-signs relatingto the eyes, joints, and facial appearance.,. In anysubject, signs from one group can predominate andlead to presentation to a number of specialities. Infamilies, the pattern of findings cannot be accuratelypredicted nor the severity assumed from previouslyaffected relatives.
OCULAR MANIFESTATIONSMyopia is generally severe (>-8 diopters),probably congenital, and progression is minimal.9Myopic degeneration of the retina can occur withlattice degeneration and myopic crescents visible onfundoscopy.
Chorioretinal degeneration is characterised byareas of abnormal retinal pigmentation, choroidal
Received for publication 27 June 1988.Revised version accepted for publication 5 July 1988.
atrophy, retinoschisis, and retinal holes. 10 These canoccur independently of myopia. Degeneration mayprogress to retinal detachment and when extensivethis leads to blindness. Detachment occursspontaneously and can be bilateral. It is more likelyin patients with a family history of detachment andin those under 30 years of age.When vitreal degeneration occurs, the vitreous
appears optically empty on slit lamp examinationwith a few floating strands.'1
There can be nuclear sclerotic cataracts, whichtend to occur in a younger than expected age group,cortical cataracts,4 or cataracts secondary to retinalsurgery.
JOINT CHANGESAlthough arthropathy was stressed by Stickler et al,'symptoms are very variable, age dependent, andoften so mild that only x ray changes are present.
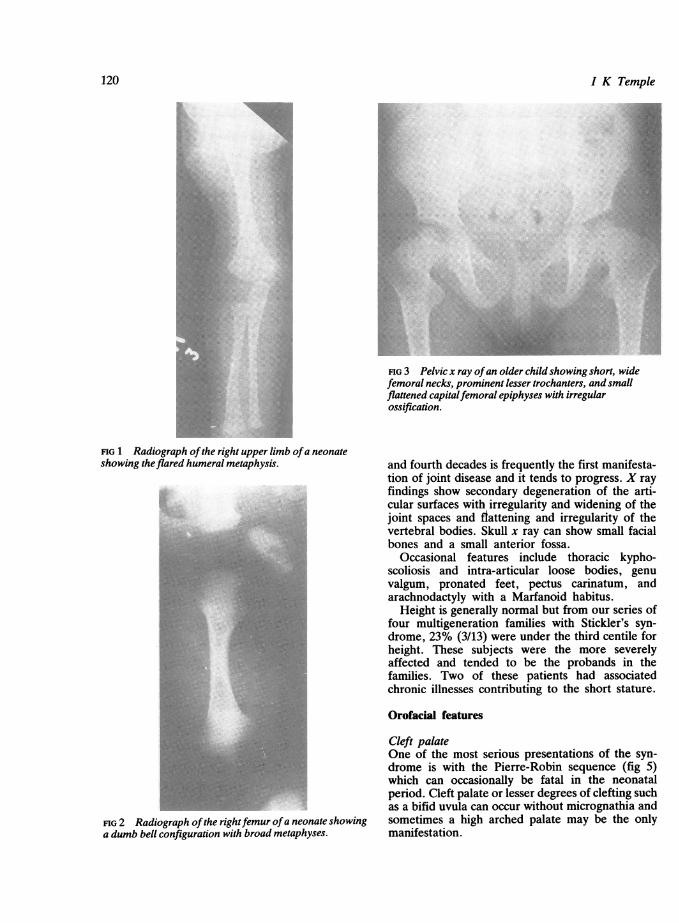
Birth to infancyClinical findings include prominent joints andhyperextensibility. Talipes equinovarus can occur.X ray findings include enlargement of epiphyses andmetaphyses (fig 1). If severe the long bones appear'dumb bell' shaped (fig 2). These findingscharacteristically improve with age and there isusually a period of several years when the longbones appear normal.
ChildhoodClinical findings include pain and stiffness in anyjoint on overuse and symptoms may mimic juvenilearthritis when severe. There is hypermobility ofjoints. X ray findings include mild spondylo-epiphyseal dysplasia with widening of the ends oflong bones. The femoral epiphysis is commonly flatand irregular and associated with a broad femoralneck (fig 3). In the spine the changes can be severewith irregularity of the vertebral end plates (fig 4).
AdulthoodOsteoarthritis of large joints developing in the third
119
I K Temple
::o
;...::E_
.-..:-. _.a_.%%.....2 _
:. ..5
::. ,...'.s,',..S. ,...
:42.. ' r.'' '.
::
,.:.:
:" i
FIG 3 Pelvic x ray ofan older child showing short, widefemoral necks, prominent lesser trochanters, and smallflattened capitalfemoral epiphyses with irregularossification.
FIG 1 Radiograph ofthe right upper limb ofa neonateshowing theflared humeral metaphysis.
FIG 2 Radiograph ofthe rightfemur ofa neonate showinga dumb bell configuration with broad metaphyses.
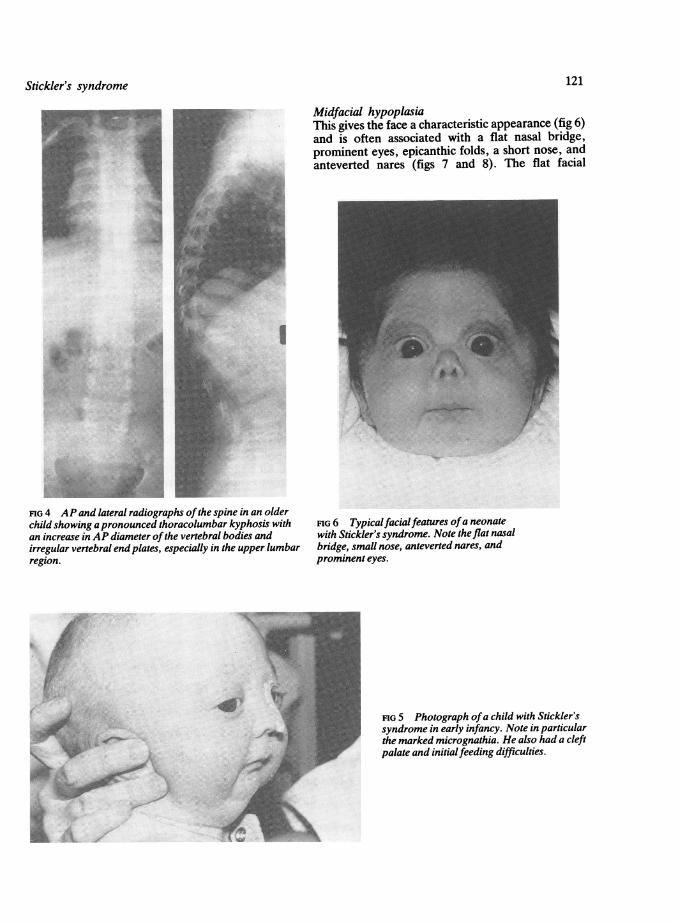
and fourth decades is frequently the first manifesta-tion of joint disease and it tends to progress. X rayfindings show secondary degeneration of the arti-cular surfaces with irregularity and widening of thejoint spaces and fiattening and irregularity of thevertebral bodies. Skull x ray can show small facialbones and a small anterior fossa.
Occasional features include thoracic kypho-scoliosis and intra-articular loose bodies, genuvalgum, pronated feet, pectus carinatum, andarachnodactyly with a Marfanoid habitus.Height is generally normal but from our series of
four multigeneration families with Stickler's syn-drome, 23% (3/13) were under the third centile forheight. These subjects were the more severelyaffected and tended to be the probands in thefamilies. Two of these patients had associatedchronic illnesses contributing to the short stature.
Orofacial features
Cleft palateOne of the most serious presentations of the syn-drome is with the Pierre-Robin sequence (fig 5)which can occasionally be fatal in the neonatalperiod. Cleft palate or lesser degrees of clefting suchas a bifid uvula can occur without micrognathia andsometimes a high arched palate may be the onlymanifestation.
120
*. .:
:f.
121
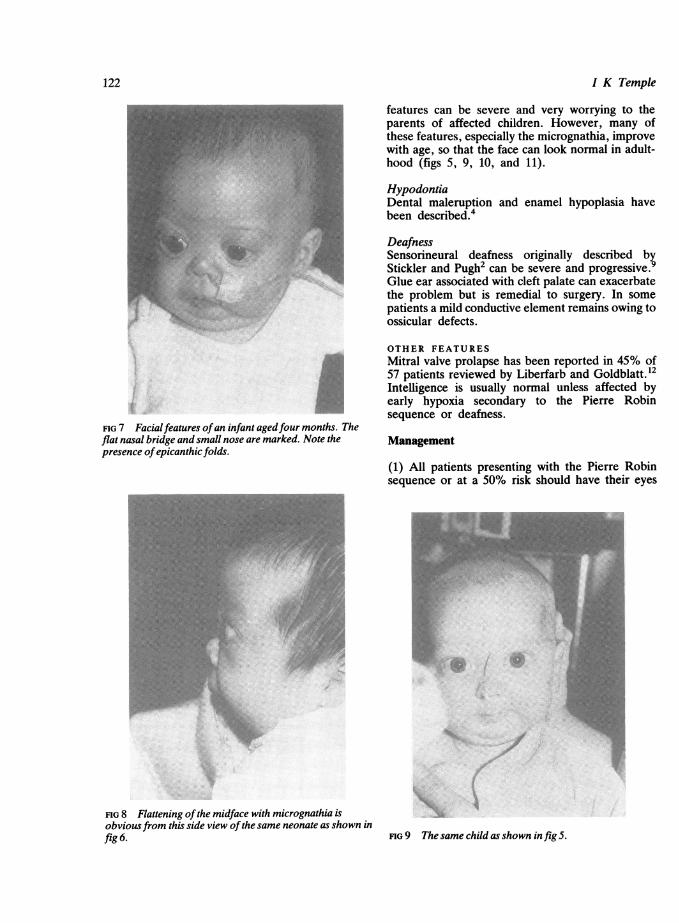
Midfacial hypoplasiaThis gives the face a characteristic appearance (fig 6)and is often associated with a flat nasal bridge,prominent eyes, epicanthic folds, a short nose, andanteverted nares (figs 7 and 8). The flat facial
I~~~~~..SP
FIG 4 AP and lateral radiographs ofthe spine in an olderchild showing apronounced thoracolumbar kyphosis withan increase in AP diameter ofthe vertebral bodies andirregular vertebral endplates, especially in the upper lumbarregion.
FIG 6 Typicalfacialfeatures ofa neonatewith Stickler's syndrome. Note the flat nasalbridge, small nose, anteverted nares, andprominent eyes.
FIG 5 Photograph ofa child with Stickler'ssyndrome in early infancy. Note in particularthe marked micrognathia. He also had a cleft
palate and initialfeeding difficulties.
Stickler's syndrome
I K Temple
FIG 7 Facialfeatures ofan infant agedfour months. Theflat nasal bridge and small nose are marked. Note thepresence ofepicanthic folds.
FIG 8 Flattening ofthe midface with micrognathia isobviousfrom this side view ofthe same neonate as shown infig 6.
features can be severe and very worrying to theparents of affected children. However, many ofthese features, especially the micrognathia, improvewith age, so that the face can look normal in adult-hood (figs 5, 9, 10, and 11).
HypodontiaDental maleruption and enamel hypoplasia havebeen described.4
DeafnessSensorineural deafness originally described byStickler and Pugh2 can be severe and progressive.9Glue ear associated with cleft palate can exacerbatethe problem but is remedial to surgery. In somepatients a mild conductive element remains owing toossicular defects.
OTHER FEATURESMitral valve prolapse has been reported in 45% of57 patients reviewed by Liberfarb and Goldblatt.12Intelligence is usually normal unless affected byearly hypoxia secondary to the Pierre Robinsequence or deafness.
Management
(1) All patients presenting with the Pierre Robinsequence or at a 50% risk should have their eyes
F s
FIG 9 The same child as shown in fig 5.
122

Stickler's syndrome
examined for myopia and retinal degeneration inearly infancy and at regular intervals thereafter.(2) Deteriorating visual acuity is usually the result ofa complication rather than progressive myopia andwarrants careful examination for cataract or retinalchanges.
w:4r/.. ..
FIG 10 The same child as seen in figs 5 and 9 aged threeyears. There has been good mandibular growth and thesepictures emphasise the change in facial appearance thatoccurs with time.
(3) Blindness through retinal detachment can belargely prevented by yearly follow up by anophthalmologist until the age of 30 years. Treatmentafter detachment is difficult.(4) In families presenting with dominant myopia thediagnosis of Stickler's syndrome should always beconsidered and the 'exclusion list' followed (table 1).(5) All children diagnosed must have an audiologicalexamination to exclude deafness.(6) Severe midfacial hypoplasia may require plasticsurgery with nasal reconstruction.(7) Mitral valve prolapse should be looked for and,if present, appropriate prophylactic antibiotics givenbefore surgery.(8) Genetic counselling is important for other familymembers at risk and the 'exclusion list' should befollowed before reassuring relatives that they areunaffected (table 1).
TABLE 1 An exclusion check list to follow in patients with apositive family history of Stickler's syndrome. The variablemanifestations of the condition make complete reassuranceof subjects at risk difficult.
(1) Ophthalmology Visual acuity-severe myopia and blindnessSlit lamp examination-cataracts and vitreousFundoscopy-retinopathy
(2) Audiology Deafness(3) Radiology AP and lateral spine
AP pelvisLateral skullAP handAP knee
(4) Palate examination-clefting(5) Early photographs
FIG 11 Mother and child with Stickler'ssyndrome showing the relatively normalfacialappearance in adulthood. The mother ismyopic and had a cleft palate. She hadfouraffected children. The flat nasal bridge andmidfacial hypoplasia are more marked in herfour year old child.
123
Inheritance Differential diagnosis
Inheritance is autosomal dominant with variable The following dominant syndromes should beexpression. excluded.
Incidence
The incidence of Stickler's syndrome is unknownbut the condition is not rare and had tended to beunderdiagnosed in the past. The diagnosis should beconsidered in all families with dominant cleft palateor myopia.
Aetiology
A candidate gene for Stickler's syndrome has beenproposed and linkage established with the type IIcollagen gene, COL2A1, on chromosome 12. Lodscores of 3-96 at 0=0 have recently been reported byFrancomano et al. 13 Type II collagen is made up ofthree al(II) collagen chains and is the major colla-gen of vitreous, nucleus pulposus, and cartilage. Astructural defect in this protein could thereforeexplain the connective tissue defects found inStickler's syndrome in at least some families.
It remains a possibility that clinical heterogeneityis the result of several gene loci and linkage infurther families is awaited. Gene tracking will alsohelp to show whether there is a distinction betweenStickler's syndrome and other connective tissuedysplasia syndromes.
MARSHALL'S SYNDROMEIn 1958, Marshall14 described the association ofcataract, myopia and fluid vitreous, deafness, andmarked midfacial hypoplasia. Since then Zellwegeret al,15 Keith et al, and O'Donnell et a116 havereported further families and included short stature,cleft palate, and spondyloepiphyseal dysplasiaindistinguishable from that in Stickler's syndrome.Every sign described in Marshall's syndrome hasbeen seen in families with Stickler's syndrome, withone exception, namely that thickening of thecalvarium and dural calcification have been seen inMarshall's syndrome and not Stickler's syndrome.However, when Ayme and Preus17 sought to
resolve this question they performed computerisedcluster analysis on 17 fully documented patients withejther Marshall's or Stickler's syndrome and foundthat two phenotypically separate groups correspond-ing to the two syndromes emerged. This mightsupport the presence of separate mutations or couldsimply reflect the fact that gene expression dependson other inherited factors which alter the featuresthat predominate in different families.
WEISSENBACHER-ZWEYMULLER SYNDROMEIn 1964 the authors17a described a newborn male
TABLE 2 Differential diagnosis in Stickler's syndrome.
Stick Marsh WZ Kniest Med Sed Wag Cerv NS
EyeMyopia + + + + - + + +Retinal
degeneration + + + + - + + +Cataract + + - - - - + -
JointsEpiphyseal
dysplasia + + + + + + - - +Flaredmetaphyses + + + + - -- - +
Platyspondyly + + + + + + - - +
OrofacialFlat midface + + + + - + - + +Cleft palate + + + + - + - + +Deafness + + + + - + - - +
Otherdifferentiating Thick - Rhizomelic - Truncal - - Autfeatures calvarium, dwarfism dwarfism rec
duralcalcification
Stick=Stickler's syndrome, Marsh=Marshall's syndrome, WZ=Weissenbacher-Zweymuller syndrome, Kniest=Kniest's syndrome, MED=multiple epiphysealdysplasia, SED=spondyloepiphyseal dysplasia, WAG=Wagner's syndrome, Cerv=Cervenka's syndrome, NS=Nance-Sweeny syndrome. +=symptom reportedin syndrome. -=symptom not reported.
124 I K Temple

Stickler's syndrome
with the Pierre Robin sequence and chondrodys-plasia. He had rhizomelic shortening of the limbsand x rays showed a dumb bell appearance of thefemora and humeri. Follow up of this and otherpatients'8 showed normal growth of limbs andmandible with regression of the x ray findings andnormal intelligence. Later, Kelly et a18 described aneonate with these characteristic changes who hadfirst degree relatives with Stickler's syndrome. It isnow widely believed that the Weissenbacher-Zweymuller syndrome is the same condition asStickler's syndrome, the neonatal x ray findings re-presenting a more severe form of the metaphysealflaring which caused the 'prominent joints' inStickler's original article.Three other neonates described by Winter et al7
with Weissenbacher-Zweymuller syndrome hadmidfacial hypoplasia and deafness characteristic ofMarshall's syndrome, providing further evidencethat all three syndromes result from the samemutant gene.
KNIEST S SYNDROMEDescribed in 1952 by Kniest,19 this syndrome ischaracterised by short trunked dwarfism withkyphoscoliosis, deafness, myopia, and depressednasal bridge. Cleft palate and detached retina canalso occur. X rays show broad metaphyses andirregular epiphyses of long bones and platyspondylywith thoracolumbar kyphoscoliosis.
It is only likely to be confused with Stickler'ssyndrome in the neonatal period. Unlike the normalgrowth in Weissenbacher-Zweymuller syndrome,deformity increases with age and stature is markedlyreduced, making follow up a most important factorin differentiating the two conditions.The dumb bell appearance of the long bones seen
in metatropic dwarfism is much more pronouncedthan in Stickler's syndrome.
MULTIPLE EPIPHYSEAL DYSPLASIASimilar joint changes are seen in both conditions butmultiple epiphyseal dysplasia is not associated withthe non-skeletal manifestations of Stickler's syn-drome. Spinal changes are very mild.
SPONDYLOEPIPHYSEAL DYSPLASIA CONGENITAThis is differentiated by extreme short stature,usually prenatal in origin. Myopia, retinal detach-ment, and flat facies are features of the syndromeand occasionally the epiphyseal changes can be con-fused but tend to be more severe, particularly in thespine and hip where there is invariably severe coxavara.
CERVENKA'S SYNDROME
The combination of myopia, retinal detachment,
cleft palate, and flat facies was described byCervenka and reviewed by Cohen et al.20 Otherfamilies have since been reported and thought tohave Stickler's syndrome.2'
WAGNER'S SYNDROMEThis refers to a dominantly inherited ocularsyndrome of myopia, cataract, and vitreoretinaldegeneration progressing to retinal detachment.The findings cannot be distinguished from thoseseen in Stickler's syndrome. In 1979, Liberfarb et a16looked at 15 index cases and their families anddiscovered on close inspection the presence of non-ocular manifestations which had been previouslyoverlooked. They suggested that Wagner's andStickler's syndromes were the same condition.However, over 250 subjects have been describedwith only eye signs and it cannot be excluded thatWagner's syndrome is a separate entity.
The following recessive syndromes should beconsidered.
NANCE-SWEENY SYNDROMEInsley and Astley22 described sibs with deafness,marked flattening of the midface, cleft palate, andgeneralised skeletal anomalies with short longbones, flaring of metaphyses, and large epiphyses.Vertebral changes were also present resulting inprogressive spinal curvature.
Recently Miny and Lenz23 described two similarsibs again with spinal deformity as an importantclinical feature. Winter et al7 referred to this syn-drome as the Nance-Sweeny syndrome after anoriginal description of a 52 year old man,24 butcomparison with childhood reports is difficult.
In all these reports myopia is not a feature.
I would like to thank Dr Michael Baraitser for hishelp and constructive discussions about this paperand for permission to use photographs of hispatients; Dr Christine Hall for her expert adviceregarding the radiological findings in Stickler's syn-drome; Mr Nicholas Geddes in the medical illustra-tion department; Mr Barry Jones for photographsof his patients; and the families for their permissionto print them. Dr I K Temple is funded by theDuchenne Muscular Dystrophy Group.
References
Stickler GB, Belau PG, Farrel FJ, et al. Hereditary progressivearthro-ophthalmology. Mayo Clin Proc 1965;40:433-55.
2 Stickler GB, Pugh D. Hereditary progressive arthro-ophthalmology. Mayo Clin Proc 1967;42:495-500.Opitz JM, France T, Herrmann J, et al. The Stickler syndrome.N Engi J Med 1972;286:546-7.
4 Herrmann J, France T, Spranger J, et al. The Stickler syndrome
125

126
[hereditary arthro-ophthalmology]. Birth Defects 1975;X1(2):76-103.
5 Baraitser M. MarshallStickler syndrome. J Med Genet 1982;19:139-40.
6 Liberfarb RM, Hirose T, Holmes LB. The Wagner-Sticklersyndrome. A genetic study. Birth Defects 1979;15(5B):145-54.
7 Winter RM, Baraitser M, Laurence KM, et al. TheWeissenbacher-Zweymuller, Stickler and Marshall syndromes:further evidence for their indentity. Am J Med Genet1983;16:189-99.
8 Kelly TE, Wells HH, Tuck KB. The Weissenbacher-Zweymuller syndrome. Possible neonatal expression of theStickler syndrome. Am J Med Genet 1982;11:113-9.
9 Keith CG, Dobbs RH, Shaw DG, et al. Abnormal facies,myopia and short stature. Arch Dis Child 1972;47:787-93.
10 Van Balen ATM, Falger ELF. Hereditary hyaloideoretinaldegeneration and palatoschisis. Arch Ophthalmol 1970;83:152-62.Frandsen E. Hereditary hyaloideoretinal degeneration[Wagner] in a Danish family. Acta Ophthalmol (Copenh)1966;44:223-32.
12 Liberfarb RM, Goldblatt A. Prevalence of mitral valveprolapse in the Stickler syndrome. Am J Med Genet1986;24:387-92.
13 Francomano CA, Maumenee I, Liberfarb R, Pyeritz RE.Cosegregation of Stickler syndrome and type II collagen genealleles. HGM9. Cytogenet Cell Genet 1987;46:578A.
14 Marshall D. Ectodermal dysplasia. Report of a kindred withocular deformities and hearing defect. Am J Ophthalmol1958;45:143-56.
`5 Zellweger H, Smith JK, Grutzer P. The Marshall syndrome:report of a new family. J Pediatr 1974;84:868-71.
I K Temple
16 O'Donnell JJ, Sirkin S, Hall BD. Generalised osseousabnormalities in the Marshall syndrome. Birth Defects1976;12(5):299-314.
17 Ayme S, Preus M. The Marshall and Stickler syndromes:objective rejection of lumping. J Med Genet 1984;21:34-8.
17a Weissenbacher G, Zweymuller E. Coincidental occurrence ofPierre Robin and fetal chondrodysplasia. MonatsschrKinderheilkd 1964;112:315-7.
18 Haller JO, Berdon WE, Robinow M, et al. The Weissenbacher-Zweymuller syndrome of micrognathia and rhizomelicchondrodysplasia at birth with subsequent normal growth. AJR1975;125:93643.
19 Kniest W. Zur Abgrenzung der Dysostosis enchondrallis vonder Chondrodystrophie. Z Kinderheilkd 1952;70:633-40.
20 Cohen MM, Knobloch WH, Gorlin RJ. A dominantly inheritedsyndrome of hyaloideoretinal degeneration, cleft palate andmaxillary hypoplasia. Birth Defects 1971;7:83-6.
21 Hall J. Stickler syndrome. Birth Defects. 1974;10(8):157-71.22 Insley J, Astley R. A bone dysplasia with deafness. Br J Radiol
1974;47:244-51.23 Miny P, Lenz W. Autosomal recessive deafness with skeletal
dysplasia and facial appearance of Marshall syndrome. Am JMed Genet 1985;21:317-24.
24 Nance WE, Sweeney A. Recessively inherited chondro-dysplasia. Birth Defects 1970;6(4):25-7.
Correspondence to Dr I K Temple, MothercareDepartment of Genetics, Institute of Child Health,30 Guilford Street, London WC1N 1EH.





























