Embed Size (px)
Citation preview
Syndrome of the month
Journal of Medical Genetics 1987, 24, 193-196
Craniofrontonasal dysplasiaI D YOUNGFrom the Department of Child Health, Leicester Royal Infirmary, Leicester LE2 7LX.
The term 'craniofrontonasal dysplasia' (CFND) wasintroduced by Cohen' in 1979 when describing a 14year old girl with coronal craniosynostosis, hyper-telorism, limitation of shoulder movement, anddigital abnormalities. The child's mother was alsoaffected. In the same volume of Birth Defects,Slover and Sujansky2 reported similar findings inthree female sibs, both of whose parents hadhypertelorism, as did the paternal grandmother.These papers established CFND as a distinct
entity. Subsequent reports have expanded the pheno-type to include numerous trunk and limbabnormalities.-36 This condition is probably not
Received for publication 14 November 1986.Accepted for publication 19 November 1986.
exceedingly rare. Approximately 25 cases from 10families have been well documented, as reviewedrecently by Sax and Flannery4 and Kumar et al,5 andthe abstract by Reich et a16 provides brief details of afurther 21 cases. The author is aware of three casesin Leicestershire which has a population of 850 000.
Clinical features
There is evidence that CFND may show consider-able variation in severity even within a family. Thefollowing observations are based on a review ofpublished and personally encountered patientsshowing marked dysmorphism and in whom thediagnosis is undoubted.
{:1 1i:,)/u
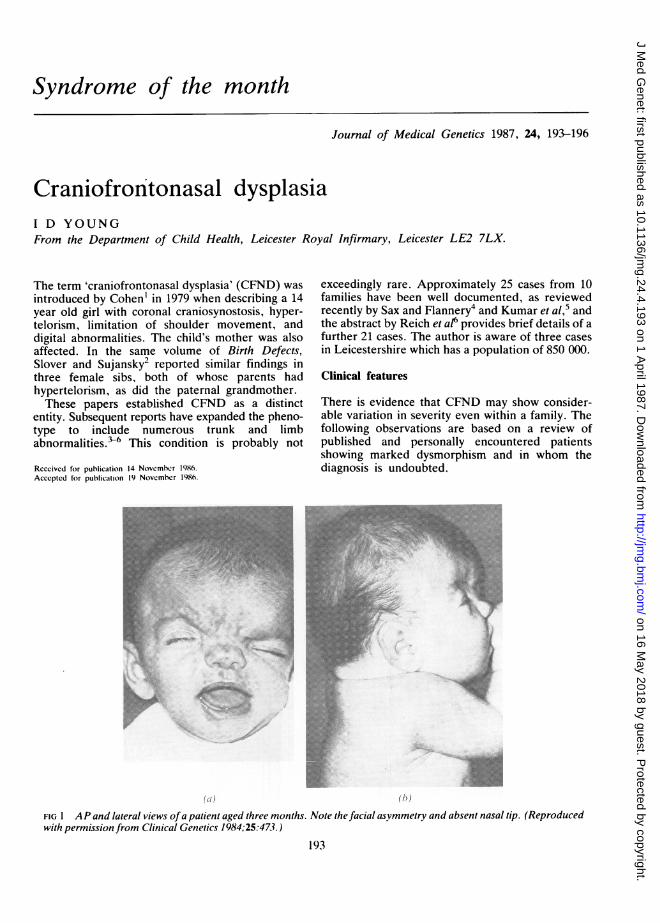
FIG 1 A P and lateral views ofa patient aged three months. Note the facial asymmetry and absent nasal tip. (Reproducedwith permission from Clinical Genetics 1984;25:473.)
193
on 16 May 2018 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.24.4.193 on 1 April 1987. D
ownloaded from
I D Young
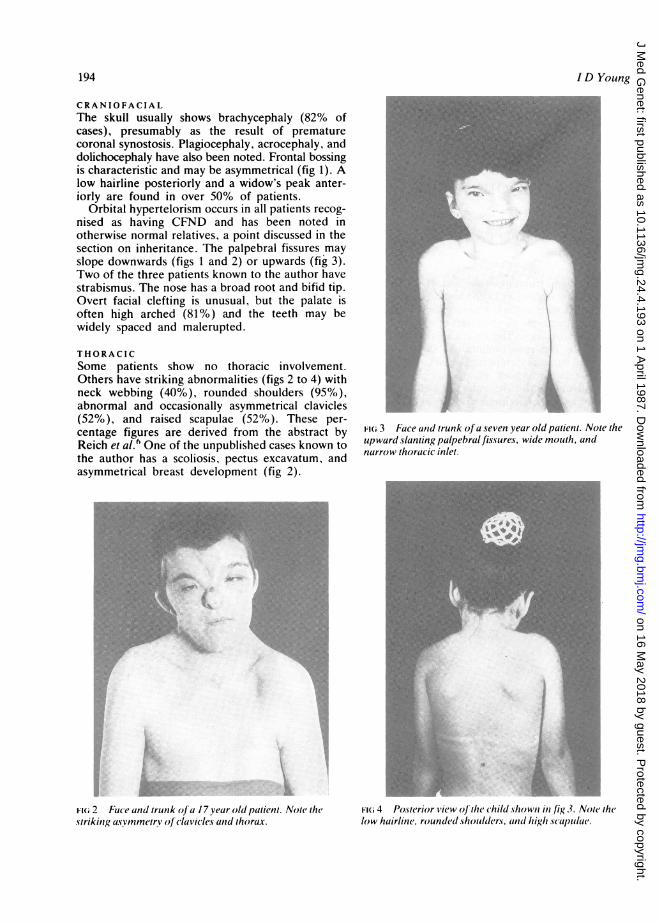
CRANIOFACIALThe skull usually shows brachycephaly (82% ofcases), presumably as the result of prematurecoronal synostosis. Plagiocephaly, acrocephaly, anddolichocephaly have also been noted. Frontal bossingis characteristic and may be asymmetrical (fig 1). Alow hairline posteriorly and a widow's peak anter-iorly are found in over 50% of patients.
Orbital hypertelorism occurs in all patients recog-nised as having CFND and has been noted inotherwise normal relatives, a point discussed in thesection on inheritance. The palpebral fissures mayslope downwards (figs 1 and 2) or upwards (fig 3).Two of the three patients known to the author havestrabismus. The nose has a broad root and bifid tip.Overt facial clefting is unusual, but the palate isoften high arched (81%) and the teeth may bewidely spaced and malerupted.
THORACICSome patients show no thoracic involvement.Others have striking abnormalities (figs 2 to 4) withneck webbing (40%), rounded shoulders (95%),abnormal and occasionally asymmetrical clavicles(52%), and raised scapulae (52%). These per-centage figures are derived from the abstract byReich et al.6 One of the unpublished cases known tothe author has a scoliosis, pectus excavatum, andasymmetrical breast development (fig 2).
...~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~....
Fii 2 uce and trunk olJ a l7year old paluin Noe ihestrikinga, nsymmetrv ofc(fclavic acaiid thorax.
FIG 3 Face anid trunk o (a seven year old patient. Note theupward slanting palpebral fissures, wide mouth, andnarrow thoracic inlet.
| - i _. i l rz9 | EE * a
:.: a | S: 1E D Es
L : _ g[. :s. - | ES | s is>@ t W r'.S I ' ::s. I *
! E '' Sg w | Etla l F &t *e 11 1fg \:.''ffY.>. &' illZ 111 liltg w lL 1|1 Ew . .X X XZ9 :: . _ X1 011 liEis I _ | | :|, IW.joF' w 1 W W
FI(i 4 Po.sterior Svic w oJ the chilcl hown in /is,} .Z. Not( thelow huirline. resuncled *holxider.s tind hip,Xh .Scupul{(
194
on 16 May 2018 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.24.4.193 on 1 April 1987. D
ownloaded from
Craniofrontonasal dysplasia
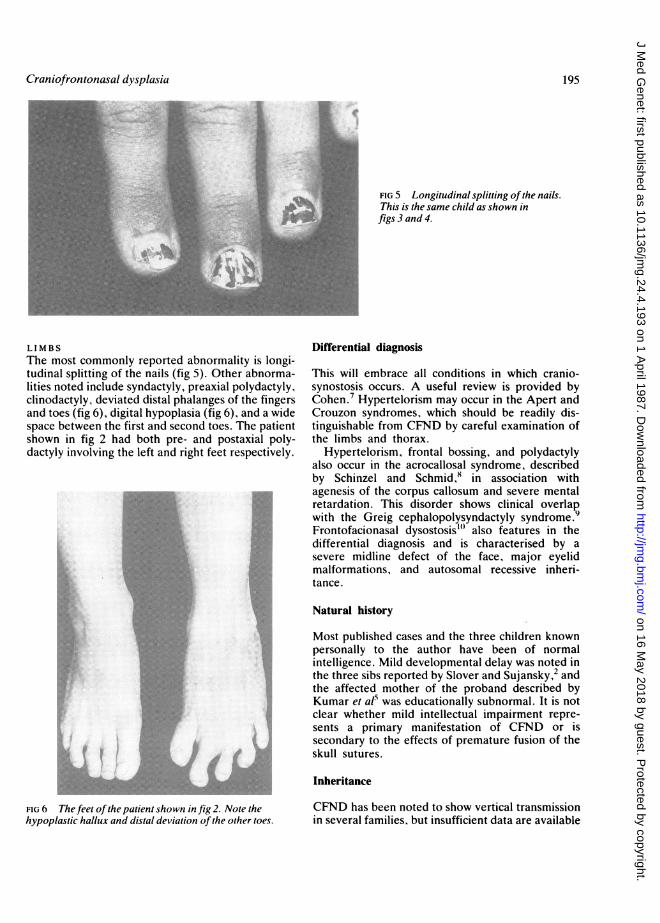
FIG 5 Longitudinal splitting ofthe nails.This is the same child as shown infigs3and4.
LIMBSThe most commonly reported abnormality is longi-tudinal splitting of the nails (fig 5). Other abnorma-lities noted include syndactyly, preaxial polydactyly,clinodactyly, deviated distal phalanges of the fingersand toes (fig 6), digital hypoplasia (fig 6), and a widespace between the first and second toes. The patientshown in fig 2 had both pre- and postaxial poly-dactyly involving the left and right feet respectively.
.~~~~~~~~~~~~~~~~~~-I ieS
iSI.S @.r~~~~~~~~~~~~L:..
Differential diagnosis
This will embrace all conditions in which cranio-synostosis occurs. A useful review is provided byCohen.7 Hypertelorism may occur in the Apert andCrouzon syndromes, which should be readily dis-tinguishable from CFND by careful examination ofthe limbs and thorax.
Hypertelorism, frontal bossing, and polydactylyalso occur in the acrocallosal syndrome, describedby Schinzel and Schmid,8 in association withagenesis of the corpus callosum and severe mentalretardation. This disorder shows clinical overlapwith the Greig cephalopolysyndactyly syndrome.9Frontofacionasal dysostosis"' also features in thedifferential diagnosis and is characterised by asevere midline defect of the face, major eyelidmalformations, and autosomal recessive inheri-tance.
Natural history
Most published cases and the three children knownpersonally to the author have been of normalintelligence. Mild developmental delay was noted inthe three sibs reported by Slover and Sujansky,2 andthe affected mother of the proband described byKumar et al" was educationally subnormal. It is notclear whether mild intellectual impairment repre-sents a primary manifestation of CFND or issecondary to the effects of premature fusion of theskull sutures.
Inheritance
CFND has been noted to show vertical transmissionin several families, but insufficient data are available
195
FIG 6 The feet ofthe patient shown in fig 2. Note thehypoplastic hallux and distal deviation ofthe other toes.
on 16 May 2018 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.24.4.193 on 1 April 1987. D
ownloaded from

I D Young
to establish with certainty the exact mode ofinheritance. Several theories have been proposed toexplain the marked excess of affected females overaffected males. These include metabolicinterference," sex linked dominance with malelethality,'2 and segregation distortion analogous tothe T locus in mice.4Examples of male to male transmission have been
reported6 making both sex linked and cytoplasmicinheritance untenable unless CFND is aetiologicallyheterogeneous. It has been suggested that affectedmales may show only very mild features of thecondition, for example, borderline hypertelorism,and that appropriate males in these families shouldbe carefully assessed both clinically andradiologically3 (R J Gorlin, 1986, personal com-munication).For practical purposes any patient with CFND
should be alerted to a risk of at least 50% foroffspring, particularly if the fetus is female. In viewof the severe cosmetic problems associated with thisdisorder some families might feel that prenataldiagnosis would be justified using ultrasonographyor fetoscopy or both, with the option of terminationif severely affected.
The author is grateful to Dr J R Moore for referringall three patients, to Mrs Penny Marston for typingthe manuscript, and to the Editor of ClinicalGenetics for permission to reproduce fig 1.
References
Cohen MM. Craniofrontonasal dysplasia. Birth Defects1979;XV(5B):85-9.
2 Slover R, Sujansky E. Frontonasal dysplasia with coronalcraniosynostosis in 3 sibs. Birth Defects 1979;XV(5B):75-83.
3 Pruzansky S, Costaras M, Rollnick BR. Radiocephalometricfindings in a family with craniofrontonasal dysplasia. BirthDefects 1982;18(1):121-38.
4Sax CM, Flannery DB. Craniofrontonasal dysplasia: clinical andgenetic analysis. Clin Genet 1986;29:508-15.
5Kumar D, Clark JW, Blank CE, Patton MA. A family withcraniofrontonasal dysplasia and fragile site 12q13 segregatingindependently. Clin Genet 1986;29:530-7.
6 Reich EW, Wishnick MM, McCarthy JG, Risch N. Cranio-frontal dysplasia: clinical delineation. Am J Hum Genet1985;37:72A.
7Cohen MM. Craniosynostosis and syndromes with craniosyno-stosis: incidence, genetics, penetrance, variability and newsyndrome updating. Birth Defects 1979;XV(5B):13-63.
8 Schinzel A, Schmid W. Hallux duplication, post-axial poly-dactyly, absence of the corpus callosum, severe mental retarda-tion and additional anomalies in 2 unrelated patients: a newsyndrome. Am J Med Genet 1980;6:241-9.
9Baraitser M, Winter RM, Brett EM. Greig cephalopolysyn-dactyly: report of 13 affected individuals in 3 families. ClinGenet 1983;24:257-65.
10 Gollop R. Fronto-facio-nasal dysostosis. A new autosomalrecessive syndrome. Am J Med Genet 1981;10:409-12.Rollnick B, Day D, Tissot R, Kaye C. A pedigree: possibleevidence for the metabolic interference hypothesis. Am J HumGenet 1981;33:823-6.
12 Young ID, Moore JR. Craniofrontonasal dysplasia - a distinctentity with lethality in the male? Clin Genet 1984;25:473-4.
Correspondence and requests for reprints to Dr I DYoung, Department of Child Health, Clinical Scien-ces Building, Leicester Royal Infirmary, PO Box 65,Leicester LE2 7LX.
196
on 16 May 2018 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.24.4.193 on 1 April 1987. D
ownloaded from





























