Embed Size (px)
Citation preview

Supporting Information:
Electronic effects on the substitution reactions of benzhydrols and fluorenyl alcohols. Determination of mechanism and effects of antiaromaticityStephen R. D. George, Timothy E. Elton and Jason B. Harper*
Table of Contents
Hammett plots for the methanolysis of the species 7 and 3 using parameters 2
Hammett plot for the methanolysis of species 3 using + parameters showing the effect
of varying the effective substituent constant for 3h 3
General reduction of the ketones 9a,b using lithium aluminium hydride 4
General reaction of benzaldehyde with in situ generated aryl lithium from the aryl
bromides 10c-e 5
General method for the esterification of benzoic acids 8
General Suzuki coupling to give methyl biphenyl benzoates 9
Preparation of compounds S6 and S7 10
General deprotection of methyl benzoates 11
Formation of the substituted biphenyl-2-carboxylic acids 15g,h 13
Preparation of 2-(4'-methoxy)benzoic acid 15e 14
General method for the ring closure of the benzoic acids 15 to the fluorenones 16 16
Preparation of compounds 16i and 3i 19
General method for the formation of the fluorenols 3 21
General method for the formation of the methyl ethers 6 and 8 25
Kinetic analysis, including all rate data, for the methanolysis of species 3 and 7 31
1H and 13C NMR spectra for all novel compounds 34
1
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry.This journal is © The Royal Society of Chemistry 2015

y = -5.055x + 0.232 R² = 0.974
-3.0
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
-0.3 -0.2 -0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6
log
(k X
/ k
H)
σ
Figure S1 Hammett plot (using values) for the methanolysis of the benzhydrols 7 at 23.8ºC with errors calculated through standard means.1
y = -4.909x + 0.343 R² = 0.949
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
-0.3 -0.2 -0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6
log
(k X
/ k
H)
σ
Figure S2 Hammett plot (using values) for the methanolysis of the fluorenols 3 at 60.0ºC with errors calculated through standard means.1
2
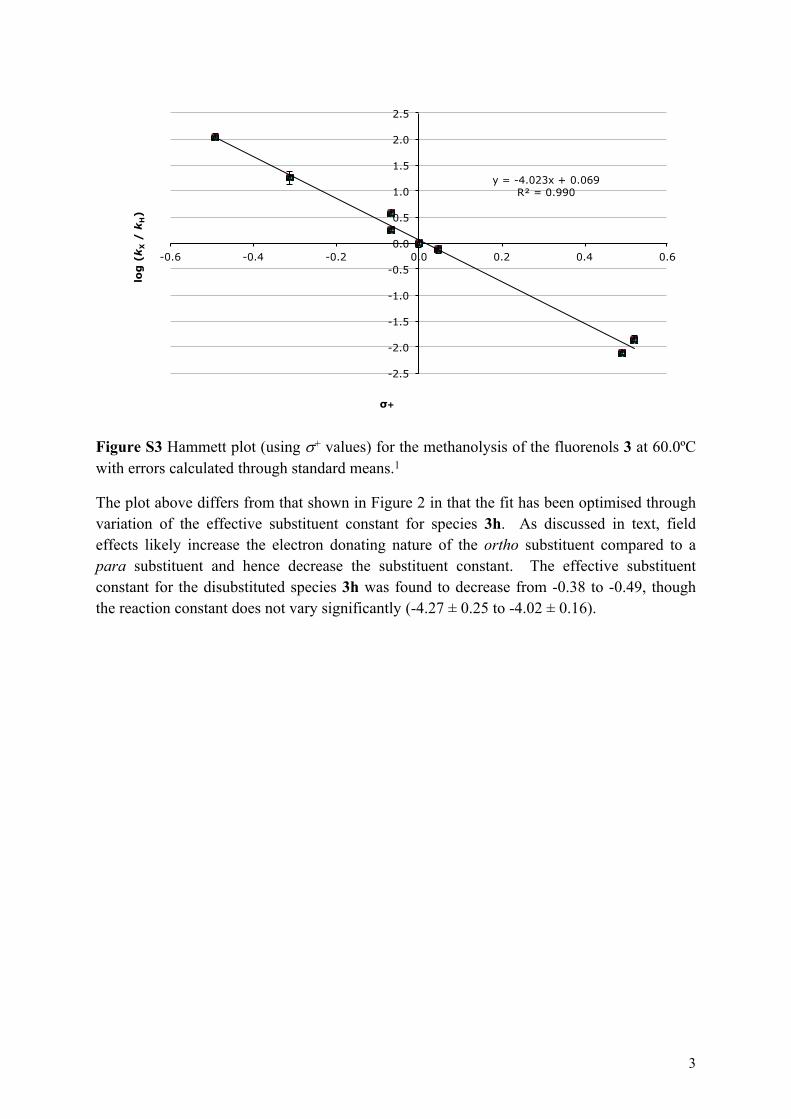
y = -4.023x + 0.069 R² = 0.990
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
-0.6 -0.4 -0.2 0.0 0.2 0.4 0.6
log
(k X
/ k
H)
σ+
Figure S3 Hammett plot (using + values) for the methanolysis of the fluorenols 3 at 60.0ºC with errors calculated through standard means.1
The plot above differs from that shown in Figure 2 in that the fit has been optimised through variation of the effective substituent constant for species 3h. As discussed in text, field effects likely increase the electron donating nature of the ortho substituent compared to a para substituent and hence decrease the substituent constant. The effective substituent constant for the disubstituted species 3h was found to decrease from -0.38 to -0.49, though the reaction constant does not vary significantly (-4.27 ± 0.25 to -4.02 ± 0.16).
3

General reduction of the ketones 9a,b using lithium aluminium hydride
To an ice-cooled solution of the appropriate ketone 9 (2.77 mmol) in anhydrous
tetrahydrofuran (25 mL) under dynamic nitrogen was added lithium aluminium hydride (158
mg, 4.16 mmol) in one portion at 0ºC. After the addition was complete, the reaction mixture
was stirred at 0oC for one hour before being very carefully quenched with water until no
further bubbling was noted (ca. 3 mL). The resulting white suspension was filtered and the
residue was washed with ether (30 mL). The filtrate was dried over magnesium sulfate,
filtered and concentrated under reduced pressure to yield the products as white solids that
were used without further purification.
Benzyhydrol 7a
0.487 g, 95%. M.p. 64.3-66.6ºC (Lit.2 68ºC). 1H NMR (300 MHz,
Chloroform-d) δ 7.76 – 7.58 (m, 5H, ArH), 7.49 – 7.30 (m, 5H, ArH), 5.60
(s, 1H, CHOH). 1H NMR (500 MHz, Methanol- d4) δ 7.35 (d, J = 7.5 Hz,
4H, Ar), 7.29 (t, J = 7.5 Hz, 4H, Ar), 7.21 (t, J = 7.5 Hz, 2H, Ar), 5.76 (s, 1H, CHOH). The
NMR data is in good agreement with literature sources.2
4-Methylbenzhydrol 7b
0.79 g, 3.79 mmol, 79%. M.p. 52oC (Lit.3 52ºC). 1H NMR (300 MHz,
Chloroform-d) δ 7.45 – 7.30 (m, 7H, ArH), 7.27 – 7.20 (m, 2H, ArH),
5.81 (s, 1H, CHOH), 2.35 (s, 3H, ArCH3). The NMR data is in good
agreement with literature sources.4
4
OH
7b
OH
7a

General reaction of benzaldehyde with in situ generated aryl lithium from the aryl
bromides 10c-e
To a stirred solution of an appropriately substituted aryl bromide 10c-e (1.92 mmol) in
tetrahydrofuran (15 ml) at -78ºC, nbutyl lithium (1.50 ml, 2.40 mmol) was slowly added. The
temperature was maintained and the mixture stirred for 1 hour before benzaldehyde (0.24 ml,
2.30 mmol) was added. The solution was allowed to warm to ambient temperature and the
reaction mixture was stirred for 4 hours. Water (5 ml) was added and the resultant mixture
was extracted with ethyl acetate (3 x 10 ml). The organic extracts were combined and dried
over magnesium sulfate and excess solvent removed under reduced pressure to give a crude
oil which was purified using flash column chromatography, eluting with 5% ethyl acetate /
95% hexane to give a yellow oil. The oil was dissolved in hexane (15 ml) and cooled to -20ºC
for 48 hours. The supernatant solution was decanted to give the products.
3-Methylbenzhydrol 7c
Obtained as a white solid. 0.284 g, 49%. M.p. 62ºC (Lit.5 61ºC). 1H
NMR (300 MHz, Chloroform-d) δ 7.47 – 7.17 (m, 9H, ArH), 7.13 – 7.08
(m, 1H, ArH), 5.85 (s, 1H, CHOH), 2.43 – 2.30 (s, 3H, ArCH3). The
NMR data is in good agreement with literature sources.6
4-Trifluoromethylbenzhydrol 7d
Obtained as a white solid (0.42 g, 87%). 1H NMR (300 MHz,
Chloroform-d) δ 7.60 (d, J = 8.4 Hz, 2H, ArH), 7.52 (d, J = 8.4 Hz,
5
OH
CF37d
OH
7c

2H, ArH), 7.37 - 7.28 (m, 5H, ArH), 5.89 (s, 1H, CHOH), 2.28 (s, 1H, OH). The NMR data is
in good agreement with literature sources.7
3-Methoxybenzhydrol 7e
Obtained as a clear oil (0.22 g, 53%). 1H NMR (300 MHz, Chloroform-
d) δ 7.47 – 7.19 (m, 6H, ArH), 7.06 – 6.93 (m, 2H, ArH), 6.84 (ddd, J =
8.1, 2.4, 1.2 Hz, 1H, ArH), 5.80 (s, 1H, CHOH), 3.80 (s, 3H, ArOCH3).
The NMR data is in good agreement with literature sources.8
6
OHO
7e

Overall scheme for the synthesis of the substituted fluorenols 3
Scheme 1 - a) cat. H2SO4, MeOH, Δ. b) ArB(OH)2, SPhos, Pd(OAc)2, Na2CO3, ACN/H2O, Δ. c) Tf2O, NEt3, DCM, 0ºC. d)
NaOH, EtOH, Δ. e) KMnO4, Py/H2O, Δ. f), ArB(OH)2, Pd(PPh3)4, Cs2CO3, THF, Δ. g) i) SOCl2, ii) AlCl3, DCM, 0ºC. h)
LiAlH4, THF, 0ºC. i) NaBH4, MeOH, 0ºC.
7
Br
O
OH
OH
O
OHO
O
R2R4
OH
CO2Me
R3
R1
O
OH
R2R4R3
R1
O
R2
R4R3
R1OH
R2
R4R3
R1
O
Br
a, b a, c, b
d
b, e
g
h e, b, i
over 2 steps R5
R5
R5 R5
I
O
Of, d
11 13
1412
15
1633i
S1-6

General method for the esterification of benzoic acids
The desired substituted benzoic acid (0.05 mol) was added to methanol (125 ml) and
concentrated sulfuric acid was then added (2 ml). The reaction mixture was then heated at
reflux for 5 hours. The yellow reaction mixture was cooled to ambient temperature, and
saturated aqueous sodium carbonate (200 ml) was added. The resultant mixture was extracted
with ethyl acetate (3 x 50 ml), the combined organic layers were washed with water (50 ml)
and brine (50 ml) and dried over magnesium sulfate. Excess solvent was then removed under
reduced pressure to yield the desired product which was used without further purification
Methyl-2-bromobenzoate S1
Obtained as a clear yellow oil. 9.75 g, 92%. 1H NMR (300 MHz,
Chloroform-d) δ 7.85 – 7.72 (m, 1H, ArH), 7.71 – 7.59 (m, 1H, ArH), 7.43 –
7.21 (m, 2H, ArH), 3.93 (s, 3H, CO2CH3). The NMR data is in good
agreement with literature sources.9
Methyl 4-methyl-2-hydroxybenzoate S2
Obtained as a dark orange oil. 7.62 g, 92%. 1H NMR (300 MHz,
Chloroform-d) δ 10.70 (s, 1H, ArOH), 7.70 (d, J = 7.9 Hz, 1H, ArH), 6.79
(s, 1H, ArH), 6.69 (d, J = 7.9 Hz, 1H, ArH), 3.92 (s, 3H, CO2CH3), 2.34 (s,
3H, ArCH3). The NMR data is in good agreement with literature sources.10
8
Br
O
O
S1
OH
O
O
S2

General Suzuki coupling to give methyl biphenyl benzoates
The desired substituted methyl benzoate (4.65 mmol) was dissolved in acetonitrile (15 ml)
and water (7 ml) followed by the required boronic acid coupling partner (6.97 mmol),
palladium acetate (10.4 mg, 0.0465 mmol), 2-dicyclohexylphosphino-2',6'-
dimethoxybiphenyl (SPhos) (38.2 mg, 0.0930 mmol) and sodium carbonate (2.73 g, 25.7
mmol). The reaction mixture was heated at reflux overnight, allowed to cool and eluted
through a silica gel pad (25 g) with dichloromethane (300 ml). The filtrate was dried over
magnesium sulfate and excess solvent was removed under reduced pressure to give a crude
brown oil. The crude product was purified using flash column chromatography eluting with
2.5% ethyl acetate / 97.5% hexane to give the methyl biphenyl benzoates as a clear oil.
Methyl 2-(4'-methylphenyl)benzoate S3
0.598 g, 57%. 1H NMR (300 MHz, Chloroform-d) δ 7.83 (ddd, J = 7.6, 1.4,
0.7 Hz, 1H, ArH), 7.60 – 7.48 (m, 1H, ArH), 7.48 – 7.35 (m, 2H, ArH),
7.24 (s, 4H, ArH), 3.69 (d, 3H, CO2CH3), 2.43 (s, 3H, ArCH3). The NMR
data is in good agreement with literature sources.11
Methyl 2-(4'-Trifluoromethylphenyl)benzoate S4
1.06 g, 81%. 1H NMR (300 MHz, Chloroform-d) δ 7.94 (ddd, J = 7.7,
1.5, 0.5 Hz, 1H, ArH), 7.68 (ddt, J = 8.1, 1.5, 0.7 Hz, 2H, ArH), 7.59
(td, J = 7.5, 1.5 Hz, 1H), 7.55 – 7.39 (m, 3H, ArH), 7.37 (ddd, J = 7.6,
1.4, 0.5 Hz, 1H, ArH), 3.69 (s, 3H, CO2CH3). The NMR data is in good
agreement with literature sources.12
9
O
O
S3
O
O
CF3S4

Methyl 4-methyl-2-(phenyl)benzoate S5
1.96 g, 90%. 1H NMR (300 MHz, Chloroform-d) δ 7.79 (d, J = 8.9 Hz, 1H,
ArH), 7.42 (t, J = 7.3 Hz, 2H, ArH), 7.37 (t, J = 7.3 Hz, 1H, ArH), 7.32 (d,
J = 6.8 Hz, 2H, ArH), 7.24 (d, J = 7.7 Hz, 2H, ArH), 7.20 (s, 1H, ArH),
3.65 (s, 3H, CO2CH3), 2.45 (s, 3H, ArCH3). The NMR data is in good agreement with
literature sources.13
2-(4'-Methoxyphenyl)toluene S6
This compound was synthesised from 4-bromoansiole 12 (0.58 mL, 4.65
mmol) and otolyl boronic acid (0.759 mg, 5.58 mmol) as per the
methodology utilised in the formation of the methyl diphenyl benzoates
with the exception of the elution solvent during purification, where only neat hexane was
used. The compound was isolated as a colourless oil (0.857 g, 93%). 1H NMR (300 MHz,
Chloroform-d) δ 7.30 - 7.24 (m, 6H, ArH), 6.98 (d, J = 8.6 Hz, ArH), 3.89 (s, 3H, ArOCH3),
2.31 (s, 3H, ArCH3). The NMR data is in good agreement with literature sources.14
Methyl 4-methyl-2-(trifluoromethylsulfonyloxy)benzoate S7
To a solution of triethylamine (3.36 mL, 24.1 mmol) and methyl
4-methyl-2-hydroxybenzoate S2 (2.00 g, 12.0 mmol) in anhydrous
dichloromethane (200 mL) was cautiously added triflic anhydride (3.64
mL, 21.7 mmol) at 0oC under dynamic nitrogen over 2 mins. After being stirred for two hours
at 0ºC and one hour at room temperature, the reaction was quenched with 2 M sulfuric acid
(200 mL) and the organic phase separated. Any organic species in the aqueous phase were
10
OTf
O
O
S7
OS6
O
O
S5

extracted with further dichloromethane (2 x 200 mL). The combined dichloromethane layers
were then dried over magnesium sulfate, filtered and the organic components adsorbed onto
silica gel (15 g). This silica placed on a fresh pad of silica gel (30 g) in a filter funnel and the
product S6 was eluted with hexane:ethyl acetate (9:1) as a clear light yellow oil (3.26 g,
91%). 1H NMR (300 MHz, Chloroform-d) δ 7.98 (d, J = 8.2 Hz, 1H, ArH), 7.27 (d, J = 8.2
Hz, 1H, ArH), 7.09 (s, 1H, ArH), 3.95 (s, 3H, CO2CH3), 2.45 (s, 3H, ArCH3). The NMR data
is in good agreement with literature sources.15
General deprotection of methyl benzoates
The desired methyl benzoate (2.64 mmol) was dissolved in ethanol (15 ml) and sodium
hydroxide (1.05 g, 20.6 mmol) was added. The resultant solution was heated at reflux for 5
hours and allowed to cool before the solvent was removed under reduced pressure. The
residue was then redissolved in ethyl acetate (25 ml) and extracted with 3 M aqueous sodium
hydroxide (3 x 15 ml). The aqueous layers were then acidified with 5 M hydrochloric acid
(40 ml) and the mixture extracted with ethyl acetate (3 x 25 ml). The organic layers were
combined, washed with water (20 ml) and bicarbonate (25 ml) and dried over magnesium
sulfate. The solvent was removed under reduced pressure to the desired products which were
used without further purification.
2-(4'-Methylphenyl)benzoic acid 15c
Obtained as a yellow solid. 0.396 g, 43%. M.p. 187.7ºC (Lit.16 171ºC). 1H
NMR (600 MHz, Chloroform-d) δ 7.96 (dd, J = 7.8, 1.4 Hz, 1H), 7.57 (td,
J = 7.6, 1.4 Hz, 1H), 7.43 (td, J = 7.6, 1.2 Hz, 1H), 7.39 (dd, J = 7.7, 1.2
11
OH
O
15c

Hz, 1H), 7.29 – 7.20 (m, 5H), 2.42 (s, 3H). The NMR data is in good agreement with
literature sources.17
2-(4'-Trifluoromethylphenyl)benzoic acid 15d
Obtained as a yellow solid. 1.26 g, 3.55 mmol, 94%. M.p. 190.6ºC
(Lit.17 165-166ºC). 1H NMR (600 MHz, Chloroform-d) δ 7.97 (dd, J =
7.8, 1.4 Hz, 2H), 7.65 – 7.56 (m, 6H), 7.52 – 7.42 (m, 6H), 7.34 (dd, J =
7.6, 1.2 Hz, 2H), 2.07 (d, J = 2.5 Hz, 9H). The NMR data is in good
agreement with literature sources.17
4-Methyl-2-phenylbenzoic acid 15f
Obtained as a white solid. 0.534 g, 95%. M.p. 165.8-167.2ºC (Lit.18 167ºC).
1H NMR (600 MHz, Acetone-d6) δ 7.80 (d, J = 7.9 Hz, 1H, ArH), 7.42 -
7.32 (m, 6H, ArH), 7.30 (d, J = 8.5 Hz, 1H, ArH), 7.22 (s, 1H, ArH), 2.44
(s, 3H, ArCH3). The NMR data is in good agreement with literature
sources.19
12
OH
O
CF315d
OH
O
15f

Formation of the substituted biphenyl-2-carboxylic acids 15g,h
2-Iodobenzoate 14 (2.00 g, 7.63 mmol), caesium carbonate (7.46 g, 22.9 mmol), and either
otolyl boronic acid (1.14 g, 8.40 mmol) or 2,5-dimethylphenylboronic acid (1.26 g, 8.4 mmol)
and palladium tetrakis(triphenylphosphine) (0.441 g, 382 μmol) were heated to reflux in
anhydrous tetrahydrofuran (50 mL) whilst under dynamic nitrogen for 48 hours. After
cooling to room temperature, the reaction was quenched with water (50 mL). At this point
concentrated sulfuric acid was added cautiously at 0ºC until no more bubbling could be noted
and all the residual caesium carbonate was reacted. The aqueous layer was extracted with
dichloromethane (3 x 50 mL), the organic layers were combined and the solvent was
removed under reduced pressure. Potassium hydroxide (5.03 g, 89.6 mmol) and ethanol (100
mL) were added to the residue and the resulting mixture was heated to reflux for 2 hours.
After the reaction mixture was cooled, the ethanol was removed under reduced pressure and
the resultant dark solid was extracted with 3 M aqueous potassium hydroxide solution (200
mL), which was then filtered through celite to remove insoluble impurities. The celite was
further washed with 3 M aqueous potassium hydroxide (2 x 100 mL) and the combined clear
aqueous solutions carefully acidified to pH 1 using concentrated hydrochloric acid at 0ºC.
The milky suspension was extracted with ethyl acetate (2 x 300 mL). The combined organic
layers were dried over magnesium sulfate, filtered and the solvent removed under reduced
pressure to yield the products as beige solids.
2′,5'-Dimethylbiphenyl-2-carboxylic acid 15g
1.34 g, 78%. M.p. 95-98ºC. 1H NMR (300 MHz, Chloroform-d) δ 8.06 (m,
1H, ArH), 7.58 (t, J = 7.5 Hz, 1H, ArH), 7.44 (t, J = 7.6 Hz, 1H, ArH), 7.31
– 7.20 (m, 1H, ArH), 7.18 – 7.03 (m, 2H, ArH), 6.94 (s, 1H, 6'-ArH), 2.35
(s, 3H, ArCH3), 2.05 (s, 3H, ArCH3). 13C NMR (101 MHz, CDCl3) δ
13
O
OH
15g

173.92 (ArCO2H), 145.08 (ArC), 142.39 (ArC), 136.04 (ArC), 133.77 (ArC), 133.66 (ArC),
132.78 (ArC), 132.19 (ArC), 130.87 (ArC), 130.64 (ArC), 130.58 (ArC), 129.50 (ArC),
128.53 (ArC), 22.39 (ArCH3), 20.95 (ArCH3). IR (solid press, cm-1) 738 (sh, s), 812 (sh, s),
930 (br, m), 1088 (sh, w), 1138 (sh, m), 1295 (sh, s), 1405 (sh, m), 1596 (sh, m) , 1674 (sh, s),
2859 (br, m). Found HR-MS (ESI) m/z: 249.0882 ([M+Na+], 100), 250.0912 ([M+Na+], 16);
C15H14O2Na+ requires m/z: 249.0886 ([M+Na+], 100), 250.0920 ([M+Na+], 18).
2′-Methylbiphenyl-2-carboxylic acid 15h
0.89 g, 50%. M.p. 98-101ºC (Lit.17 104-106ºC). 1H NMR (500 MHz,
Chloroform-d) δ 8.07 (d, J = 7.9 Hz, 1H, ArH), 7.60 (t, J = 7.4 Hz, 1H, ArH),
7.46 (t, J = 7.7 Hz, 1H, ArH), 7.26 (m, 4H, ArH), 7.11 (d, J = 7.3 Hz, 1H,
ArH), 2.11 (s, 3H, ArCH3). The NMR data is in good agreement with
literature sources17.
2-(4'-Methoxy)benzoic acid 15e
A suspension of 2-(4'-methoxyphenyl)toluene (0.223 mg, 1.12 mmol)
and potassium permanganate (0.889 mg, 5.62 mmol) in a 1:1 mixture of
pyridine (5 mL) and water (5 mL) was heated to reflux overnight. After
cooling to room temperature, solid sodium metabisulfite was added to
the reaction mixture until no further decolourisation was noted. The white suspension was
then cautiously poured into a biphasic mixture of 3 M hydrochloric acid (50 mL) and ethyl
acetate (50 mL). After complete dissolution had occurred, the organic layer was separated
and further washed with 3 M hydrochloric acid (3 x 50 mL). The organic layer was then dried
over magnesium sulfate, filtered and concentrated under reduced pressure to yield the product
14
O
OH
15h
O
OH
O
15e

as a white solid (0.255 g, 99%). M.p. 139-142ºC (Lit.17 141-143ºC). 1H NMR (300 MHz,
Acetone-d6) δ 11.29 (s, 1H, CO2H), 7.98 (d, J = 7.6 Hz, 1H, ArH), 7.58 (m, 1H, ArH), 7.47 -
7.26 (m, 4H, ArH), 6.97 (d, J = 8.1 Hz, 2H, ArH), 3.87 (s, 3H, ArOCH3). The NMR data is in
good agreement with literature sources.20
15

General method for the ring closure of the benzoic acids 15 to the fluorenones 16
The benzoic acid of interest (2.02 mmol) was dissolved in thionyl chloride (8 ml) and stirred
for 4 hours. Excess thionyl chloride was removed via trap to trap distillation and
dichloromethane (20 ml) added to the remaining brown solid. To the stirred solution
aluminium trichloride (0.490 g, 6.06 mmol) was added, causing a deep red colouration. The
reaction mixture was stirred for a further 2 hours and water (5 ml) added before the mixture
was filtered and extracted with a further portion of dichloromethane (3 x 10 ml). The organic
extracts were combined and dried over magnesium sulfate and excess solvent removed under
reduced pressure to give the products as orange / yellow solids.
3-Methylfluorenone 16b
0.364 g, 93%. M.p. 63-64ºC (Lit.21 63-64ºC) 1H NMR (600 MHz,
Chloroform-d) δ 7.66 (d, J = 7.1 Hz, 1H, ArH), 7.57 (d, J = 7.6 Hz, 1H,
ArH), 7.52 - 7.45 (m, 2H, ArH), 7.34 (s, 1H, ArH), 7.32 - 7.27 (m, 1H,
ArH), 7.10 (d, J = 7.6 Hz, 1H, ArH), 2.44 (s, 3H, ArCH3). The NMR data is in good
agreement with literature sources.22
2-Methylfluorenone 16c
0.215 g, 55%. M.p. 93ºC (Lit.23 92ºC). 1H NMR (400 MHz, Chloroform-
d) δ 7.65 (dt, J = 7.3, 1.0 Hz, 1H, ArH), 7.54 – 7.39 (m, 4H, ArH), 7.35 –
7.22 (m, 2H, ArH), 2.40 (s, 3H, ArCH3). The NMR data is in good
agreement with literature sources.22
16
O
16c
O
16b

2-Trifluoromethylfluorenone 16d
0.181 g, 16%. M.p. 131ºC (Lit.21 136-137ºC). 1H NMR (400 MHz,
Chloroform-d) δ 8.25 (dd, J = 2.1, 0.6 Hz, 1H, ArH), 8.12 (dd, J = 8.0,
2.1 Hz, 1H, ArH), 7.75 (dt, J = 7.4, 0.9 Hz, 1H, ArH), 7.67 – 7.53 (m,
3H, ArH), 7.41 (td, J = 7.4, 1.2 Hz, 1H, ArH). The NMR data is in good agreement with
literature sources.24
2-Methoxyfluorenone 16e
0.325 g, 77%. M.p. 75-77ºC (Lit.21 77-78ºC). 1H NMR (600 MHz,
Chloroform-d) δ 7.60 (d, J = 7.6 Hz, 1H, ArH), 7.47 - 7.37 (m, 3H,
ArH), 7.22 - 7.16 (m, 3H, ArH), 6.98 (dd, J = 8.2, 3.5 Hz, 1H, ArH),
3.85 (s, 3H, OCH3). The NMR data is in good agreement with literature sources.22
1,4-Dimethylfluorenone 16g
0.432 g, 94%. M.p. 79-82ºC (Lit.25 80-82ºC). 1H NMR (600 MHz,
Chloroform-d) δ 7.69 (d, J = 7.3 Hz, 1H, ArH), 7.64 (d, J = 7.6 Hz, 1H,
ArH), 7.50 (t, J = 7.6 Hz, 1H, ArH), 7.31 (t, J = 7.3 Hz, 1H, 2/3ArH), 7.15
(d, J = 7.6 Hz, 1H, ArH), 6.98 (d, J = 7.6 Hz, 1H, ArH), 2.63 (s, 3H, 4-CH3),
2.59 (s, 3H, 1-CH3). The NMR data is in good agreement with literature sources.22
17
O
16g
O
CF3
16d
O
O
16e

4-Methylfluorenone 16h
0.389 g, 91%. M.p. 74-78ºC (Lit.26 78ºC). 1H NMR (600 MHz, Chloroform-
d) δ 7.69 (d, J = 7.3 Hz, 1H, ArH), 7.62 (d, J = 7.5 Hz, 1H, ArH), 7.53 (d, J
= 7.2 Hz, 1H, ArH), 7.49 (t, J = 7.5 Hz, 1H, ArH), 7.33 – 7.23 (m, 2H,
ArH), 7.19 (t, J = 7.4 Hz, 1H, ArH), 2.59 (s, 3H, ArCH3). The NMR data is
in good agreement with literature sources.27
18
O
16h

Methyl 9-fluorenone-3-carboxylate 16i
A suspension of 3-methylfluorenone (0.218 mg, 1.12 mmol) and
potassium permanganate (0.889 mg, 5.62 mmol) in a 1:1 mixture of
pyridine (5 mL) and water (5 mL) was heated to reflux overnight.
After cooling to room temperature, solid sodium metabisulfite was added to the reaction
mixture until no further decolourisation was noted. The white suspension was then cautiously
poured into a biphasic mixture of 3 M hydrochloric acid (50 mL) and ethyl acetate (50 mL).
After complete dissolution had occurred, the organic layer was separated and further washed
with 3 M hydrochloric acid (3 x 50 mL). The organic layer was then dried over magnesium
sulfate, filtered and concentrated under reduced pressure. The resulting white solid was
immediately added to methanol (125 ml) and concentrated sulfuric acid was then added (2
ml). The reaction mixture was then heated at reflux for 5 hours. The yellow reaction mixture
was cooled to ambient temperature, and saturated aqueous sodium carbonate (200 ml) was
added. The resultant mixture was extracted with ethyl acetate (3 x 50 ml), the combined
organic layers were washed with water (50 ml) and brine (50 ml) and dried over magnesium
sulfate. Excess solvent was then removed under reduced pressure to yield the desired product
as a yellow solid. 0.241 g, 90%. M.p. 144-146ºC (Lit.28 145ºC). 1H NMR (500 MHz,
Chloroform-d) δ 8.11 (s, 1H, ArH), 7.96 (d, J = 7.7 Hz, 1H, ArH), 7.66 (t, J = 6.4 Hz, 1H,
ArH), 7.58 - 7.49 (m, 1H, ArH), 7.32 (t, J = 7.1 Hz, 1H, ArH), 3.97 (s, 3H, CO2CH3). The
NMR data is in good agreement with literature sources.28
19
O
CO2Me16i

Methyl 9-fluorenol-3-carboxylate 3i
To a solution of methyl 9-fluorenone-3-carboxylate 16i (0.150 mg,
0.630 mmol) in anhydrous methanol (10 mL) was slowly added
sodium borohydride (48 mg, 1.26 mmol). After completion of the
reaction as assessed using TLC (ca. 20 minutes), the reaction mixture was poured into water
(40 mL) which was then acidified to pH 1 using concentrated sulfuric acid. The organic
components were then extracted into ethyl acetate (2 x 50 mL), the combined extracts were
dried over magnesium sulfate and filtered. Evaporation of the filtrate under reduced pressure
yielded the product as a light white solid (0.146 g, 97%). M.p. 129-132ºC. 1H NMR (500
MHz, Chloroform-d) δ 8.31 (s, 1H, ArH), 7.98 (d, J = 7.8 Hz, 1H, ArH), 7.84 (d, J = 7.3 Hz,
1H, ArH), 7.74 (d, J = 7.7 Hz, 1H, ArH), 7.66 (d, J = 7.6 Hz, 1H, ArH), 7.43 (t, J = 7.6 Hz,
1H, ArH), 7.37 (t, J = 7.6 Hz, 1H, ArH), 5.63 (s, 1H, ArCHOHAr), 3.92 (s, 3H, ArCO2CH3).
13C NMR (101 MHz, Chloroform-d) δ 168.51 (ArCO2CH3), 151.83 (ArC), 147.04 (ArC),
141.72 (ArC), 140.45 (ArC), 132.21 (ArC), 130.75 (ArC), 130.74 (ArC), 129.80 (ArC),
126.65 (ArC), 126.37 (ArC), 122.46 (ArC), 121.80 (ArC), 76.39 (ArCHOHAr), 53.68
(ArCO2CH3). IR (solid press, cm-1) 752 (sh, s), 1030 (sh, s), 1096 (sh, m), 1183 (sh, m), 1253
(sh, s), 1419 (sh, m), 1684 (sh, s), 3459 (sh, m). Found HR-MS (ESI) m/z: 241.0856 ([M+H+],
100), 242.0889 ([M+H+], 10); C15H14OH+ requires m/z: 241.0859 ([M+H+], 100), 242.0889
([M+H+], 16).
20
OH
CO2Me3i

General method for the formation of the fluorenols 3
These compounds were formed as white solids in an analogous manner to the preparation of
benzhydrol 7a from the appropriate fluorenone 16 (2.77 mmol, limiting reagent).
Fluorenol 3a
0.498 g, 99%. M.p. 149-151ºC (Lit.29 156-158ºC). 1H NMR (300 MHz,
Chloroform-d) δ 7.74 – 7.58 (m, 4H, Ar), 7.51 – 7.29 (m, 4H, Ar), 5.57 (s,
1H, ArCHOHAr). 1H NMR (300 MHz, Methanol-d4) δ 7.71 (d, J = 7.1 Hz,
2H, Ar), 7.62 (d, J = 6.6 Hz, 2H, Ar), 7.45 – 7.26 (m, 4H, Ar), 5.55 (s, 1H, CHOH). The
NMR data is in good agreement with literature sources.30
3-Methylfluorenol 3b
0.522 g, 96%. M.p. 143-144ºC (Lit.31 143.5-144.5ºC) 1H NMR (300 MHz,
Chloroform-d) δ 7.63 (d, J = 7.7 Hz, 2H, ArH), 7.52 (d, J = 7.6 Hz, 1H,
ArH), 7.47 (s, 1H, ArH), 7.38 (t, J = 7.2 Hz, 1H, ArH), 7.31 (t, J = 7.2 Hz,
1H, ArH), 7.13 (d, J = 7.2 Hz, 1H, ArH), 5.55 (s 1H, CHOH), 2.43 (s, 3H, ArCH3). 13C NMR
(101 MHz, Chloroform-d) δ 147.54 (ArC), 144.30 (ArC), 141.57 (ArC), 141.46 (ArC),
140.35 (ArC), 130.37 (ArC), 129.99 (ArC), 129.08 (ArC), 126.54 (ArC), 126.28 (ArC),
122.03 (ArC), 121.23 (ArC), 76.34 (ArCHOHAr), 23.06 (ArCH3). IR (solid press, cm-1) 738
(sh, s), 835 (sh, m), 1022 (sh, s), 1163 (sh, m), 1199 (sh, m), 1297 (sh, m), 1447 (sh, m), 1613
(sh, w), 3299 (br, m). Found HR-MS (ESI) m/z: 219.0781 ([M+Na+], 100), 220.0813
([M+Na+], 16); C14H12O2Na+ requires m/z: 219.0780 ([M+Na+], 100), 220.0814 ([M+Na+],
18).
21
OH
3a
OH
3b

2-Methylfluorenol 3c
48.9 mg, 23%. M.p. 148ºC (Lit.32 145ºC). 1H NMR (400 MHz,
Chloroform-d) δ 7.65 (dt, J = 7.3, 1.0 Hz, 1H, ArH), 7.54 – 7.39 (m, 4H,
ArH), 7.35 – 7.22 (m, 2H, ArH), 5.45 (s, 1H, CHOH), 2.40 (s, 3H,
ArCH3). 13C NMR (101 MHz, Chloroform-d) δ 147.30 (ArC), 146.97 (ArC), 141.55 (ArC),
139.25 (ArC), 138.75 (ArC), 131.17 (ArC), 130.43 (ArC), 128.75 (ArC), 127.30 (ArC),
126.49 (ArC), 121.16 (ArC), 121.08 (ArC), 76.53 (ArCHOHAr), 23.01 (ArCH3). IR (solid
press, cm-1) 668 (sh, s), 820 (sh, s), 1022 (sh, s), 1124 (sh, m), 1178 (sh, m), 1294 (sh, m),
1454 (sh, m), 3301 (br, m). Found HR-MS (ESI) m/z: 219.0777 ([M+Na+], 100), 220.0809
([M+Na+], 16); C14H12O2Na+ requires m/z: 219.0780 ([M+Na+], 100), 220.0814 ([M+Na+],
18).
2-Trifluoromethylfluorenol 3d
63.5 mg, 32%. M.p. 148ºC. 1H NMR (300 MHz, Chloroform-d) δ 8.24
(dt, J = 2.1, 0.7 Hz, 1H, ArH), 8.01 (ddd, J = 8.2, 2.1, 0.6 Hz, 1H,
ArH), 7.77 – 7.65 (m, 3H, ArH), 7.43 (pd, J = 7.5, 1.2 Hz, 2H, ArH),
5.66 (s, 1H, CHOH). 13C NMR (101 MHz, Chloroform-d) δ 147.79 (ArC), 147.23 (ArC),
145.10 (ArC), 143.54 (ArC), 139.94 (ArC), 130.82 (ArC), 130.26 (ArC), 128.34 (ArC),
126.75 (ArC), 123.97 (ArC), 122.11 (ArC), 121.04 (ArC), 76.47 (ArCHOHAr). IR (solid
press, cm-1) 665 (sh, m), 741 (sh, s), 800 (sh, s), 1040 (sh, m), 1121 (sh, w), 3178 (br, m).
22
OH
3c
OH
CF33d

2-Methoxyfluorenol 3e
0.567 g, 96%. M.p. 158-161ºC (Lit.33 160ºC) 1H NMR (300 MHz,
Chloroform-d) δ 7.64 (d, J = 8.3 Hz, 2H, ArH), 7.58 (d, J = 7.2 Hz, 1H,
ArH), 7.33 (t, J = 7.6 Hz, 3H, ArH), 7.27 - 7.20 (m, 2H, ArH), 6.95 (dd,
J = 8.3, 2.4 Hz, 1H, ArH), 5.53 (d, J = 7.9 Hz, 1H, CHOH), 4.70 (d, J = 7.9 Hz, 1H, CHOH).
13C NMR (101 MHz, Acetone-d6) δ 161.52 (ArC), 150.33 (ArC), 147.97 (ArC), 141.46
(ArC), 133.97 (ArC), 129.82 (ArC), 127.63 (ArC), 126.28 (ArC), 121.98 (ArC), 120.32
(ArC), 115.62 (ArC), 112.12 (ArC), 75.79 (ArCHOHAr), 56.33 (ArOCH3). IR (solid press,
cm-1) 741 (sh, s), 814 (sh, m), 1042 (sh, m), 1142 (sh, m), 1181 (sh, m), 1242 (sh, s), 1301 (sh,
s), 1461 (sh, s) , 1620 (sh, w), 3226 (br, m). Found HR-MS (ESI) m/z: 235.0728 ([M+Na+],
100), 236.0763 ([M+Na+], 16); C14H12O2Na+ requires m/z: 235.0730 ([M+Na+], 100),
236.0763 ([M+Na+], 18).
1,4-Dimethylfluorenol 3g
0.567 g, 97%. M.p. 168-170ºC. 1H NMR (400 MHz, Chloroform-d) δ 7.80
(d, J = 7.7 Hz, 1H, ArH), 7.69 (d, J = 7.4 Hz, 1H, ArH), 7.42 (t, J = 7.7 Hz,
1H, ArH), 7.34 (t, J = 7.4 Hz, 1H, ArH), 7.10 (d, J = 7.6 Hz, 1H, ArH), 7.03
(d, J = 7.6 Hz, 1H, ArH), 5.62 (s, 1H, ArCHOHAr), 2.65 (s, 3H, ArCH3),
2.55 (s, 3H, ArCH3). 13C NMR (101 MHz, Chloroform-d) δ 147.38 (ArC), 144.75 (ArC),
142.58 (ArC), 139.35 (ArC), 134.96 (ArC), 133.05 (ArC), 131.86 (ArC), 130.62 (ArC),
130.41 (ArC), 128.48 (ArC), 126.46 (ArC), 124.52 (ArC), 76.08 (ArCHOHAr), 21.91
(ArCH3), 19.37 (ArCH3). IR (solid press, cm-1) 731 (sh, s), 804 (sh, m), 933 (sh, w), 1050 (sh,
s), 1190 (sh, m), 1305 (sh, m), 1453 (sh, m), 1500 (sh, w) , 2917 (sh, w), 3264 (br, w). Found
23
OH
O
3e
OH
3g

HR-MS (ESI) m/z: 233.0933 ([M+Na+], 100), 234.0966 ([M+Na+], 16); C15H14ONa+ requires
m/z: 233.0937 ([M+Na+], 100), 234.0970 ([M+Na+], 16).
4-Methylfluorenol 3h
0.521 g, 96%. M.p. 144-148ºC (Lit.34 159-160ºC). 1H NMR (500 MHz,
Chloroform-d) δ 7.81 (d, J = 7.6 Hz, 1H, ArH), 7.70 (d, J = 7.2 Hz, 1H,
ArH), 7.58 – 7.51 (d, J = 7.2 Hz, 1H, ArH), 7.47 – 7.40 (m, 1H, ArH), 7.35
(m, 1H, ArH), 7.26 (t, J = 7.6 Hz, 1H, ArH), 7.22 – 7.16 (m, 1H, ArH), 5.57
(s, 1H, CHOH), 2.69 (s, 3H, ArCH3). 13C NMR (101 MHz, Chloroform-d) δ 147.48 (ArC),
147.46 (ArC), 142.37 (ArC), 139.38 (ArC), 134.59 (ArC), 132.81 (ArC), 130.44 (ArC),
128.96 (ArC), 128.58 (ArC), 126.48 (ArC), 124.55 (ArC), 123.98 (ArC), 76.46
(ArCHOHAr), 22.19 (ArCH3). IR (solid press, cm-1) 733 (sh, s), 809 (sh, w), 1050 (sh, m),
1196 (sh, m), 1299 (sh, m), 1453 (sh, m), 1703 (sh, w), 3280 (br, m). Found HR-MS (ESI)
m/z: 219.0776 ([M+Na+], 100), 220.0808 ([M+Na+], 14); C14H12O2Na+ requires m/z:
219.0780 ([M+Na+], 100), 220.0814 ([M+Na+], 16).
24
OH
3h

General method for the formation of the methyl ethers 6 and 8
The appropriate alcohol (271 μmol) was added to methanol (5 mL) followed by concentrated
sulfuric acid (0.5 mL). The reaction mixture was then stirred at the temperature specified for
a time as noted below. The reaction mixture was added to water (10 mL) and extracted thrice
with ether (3 x 15 mL). The combined organic layers were dried over magnesium sulfate,
filtered and the solvent removed under reduced pressure to yield the products as clear oils
(unless otherwise noted), which required no further purification (unless otherwise noted). It is
worth mentioning that the methyl ethers 6d and 6i were not isolated as the half-lives of these
reactions precluded their formation in a reasonable time frame.
9-Methoxy-3-methylfluorene 6b
Reflux, 16 hours. 51.7 mg, 91%. 1H NMR (500 MHz, Chloroform-d) δ
7.65 (d, J = 7.5 Hz, 1H, ArH), 7.60 (d, J = 7.5 Hz, 1H, ArH), 7.51-7.47 (m,
2H, ArH), 7.39 (t, J = 7.5 Hz, 1H, ArH), 7.31 (t, J = 7.0 Hz, 1H, ArH),
7.14 (d, J = 7.5 Hz, 1H, ArH), 5.58 (s, 1H, ArCHOCH3Ar), 3.06 (s, 3H, ArCHOCH3Ar), 2.44
(s, 3H, ArCH3). 13C NMR (101 MHz, Chloroform-d) δ 144.41 (ArC), 142.62 (ArC), 142.51
(ArC), 141.06 (ArC), 140.33 (ArC), 130.35 (ArC), 129.78 (ArC), 128.81 (ArC), 126.88
(ArC), 126.67 (ArC), 122.06 (ArC), 121.22 (ArC), 82.59 (ArCHOCH3Ar), 53.54
(ArCHOCH3Ar), 23.07 (ArCH3). IR (solid press, cm-1) 741 (sh, s), 768 (sh, s), 934 (sh, w),
1072 (sh, s), 1204 (sh, m), 1450 (sh, s), 1613 (sh, w), 2926 (br, w). Found HR-MS (ESI) m/z:
211.0753 ([M+H+], 100), 212.0788 ([M+H+], 24); C16H16ONa+ requires m/z: 211.1117
([M+H+], 100), 212.1151 ([M+H+], 16).
25
O
6b

9-Methoxy-2-methylfluorene 6c
Reflux, 16 hours. 53.4 mg, 94%. 1H NMR (500 MHz, Chloroform-d) δ
7.66 (d, J = 7.4 Hz, 1H, ArH), 7.63 (d, J = 7.5 Hz, 1H, ArH), 7.59 (d, J =
7.6 Hz, 1H, ArH), 7.46 (s, 1H, ArH), 7.41 (d, J = 7.3 Hz, 1H, ArH), 7.32
(d, J = 7.5 Hz, 1H, ArH), 7.24 (d, J = 7.5 Hz, 1H, ArH), 5.60 (s, 1H, ArCHOCH3Ar), 3.12 (s,
3H, ArCHOCH3Ar), 2.47 (s, 3H, ArCH3). 13C NMR (101 MHz, Chloroform-d) δ 142.83
(ArC), 142.46 (ArC), 141.17 (ArC), 138.39 (ArC), 137.52 (ArC), 129.75 (ArC), 128.96
(ArC), 127.04 (ArC), 126.21 (ArC), 125.45 (ArC), 119.71 (ArC), 119.64 (ArC), 81.37
(ArCHOCH3Ar), 52.36 (ArCHOCH3Ar), 21.64 (ArCH3). IR (solid press, cm-1) 764 (sh, s),
821 (sh, m), 939 (br, w), 1070 (sh, s), 1126 (sh, m), 1453 (sh, m), 1606 (sh, w), 2925 (br, w).
Found HR-MS (ESI) m/z: 233.0935 ([M+Na+], 100), 234.0967 ([M+Na+], 16); C15H14ONa+
requires m/z: 233.097 ([M+Na+], 100), 234.0970 ([M+Na+], 18).
2,9-Dimethoxyfluorene 6e
Reflux, 72 hours. 58.4 mg, 95%.1H NMR (500 MHz, Chloroform-d) δ
7.63-7.57 (m, 3H, ArH), 7.39 (t, J = 7.5 Hz, 1H, ArH), 7.30-7.25 (m,
1H, ArH), 7.21-7.19 (m, 1H, ArH), 6.98-6.94 (m, 1H, ArH), 5.59 (s,
1H, ArCHOCH3Ar), 3.90 (s, 3H, ArCHOCH3Ar), 3.08 (s, 3H, ArOCH3). 13C NMR (101
MHz, Chloroform-d) δ 159.90 (ArC), 144.47 (ArC), 142.06 (ArC), 141.10 (ArC), 133.83
(ArC), 129.02 (ArC), 126.36 (ArC), 125.36 (ArC), 120.74 (ArC), 119.16 (ArC), 114.99
(ArC), 110.93 (ArC), 81.29 (ArCHOCH3Ar), 55.59 (ArCHOCH3Ar), 52.09 (ArOCH3). IR
(solid press, cm-1) 740 (sh, s), 821 (sh, m), 938 (sh, m), 1068 (sh, s), 1193 (sh, s), 1259 (sh, s),
1457 (sh, m), 1607 (sh, m) , 2932 (br, m). Found HR-MS (ESI) m/z: 249.0886 ([M+Na+],
26
O
6e
O
O
6c

100), 250.0914 ([M+Na+], 16); C15H14O2Na+ requires m/z: 249.0886 ([M+Na+], 100),
250.0920 ([M+Na+], 18).
9-Methoxy-1,4-dimethylfluorene 6g
Reflux, 16 hours. White solid.58.4 mg, 96%. M.p. 79-81ºC. 1H NMR (500
MHz, Chloroform-d) δ 7.83 (d, J = 7.6 Hz, 1H, ArH), 7.66 (d, J = 7.3 Hz,
1H, ArH), 7.44 (t, J = 7.5 Hz, 1H, ArH), 7.35 (t, J = 7.3 Hz, 1H, ArH), 7.12
(d, J = 7.6 Hz, 1H, ArH), 7.03 (d, J = 7.5 Hz, 1H, ArH), 5.68 (s, 1H, ArCHOCH3Ar), 2.92 (s,
3H, ArCHOCH3Ar), 2.67 (s, 3H, ArCH3), 2.50 (s, 3H, ArCH3). 13C NMR (101 MHz,
Chloroform-d) δ 144.20 (ArC), 143.71 (ArC), 141.52 (ArC), 135.20 (ArC), 133.02 (ArC),
131.77 (ArC), 130.26 (ArC), 130.20 (ArC), 128.13 (ArC), 126.64 (ArC), 124.44 (ArC),
121.40 (ArC), 82.02 (ArCHOCH3Ar), 52.11 (ArCHOCH3Ar), 21.99 (ArCH3), 19.17
(ArCH3). IR (liquid film, cm-1) 744 (sh, s), 807 (sh, m), 1024 (sh, w), 1085 (sh, s), 1196 (sh,
m), 1455 (sh, s), 1501 (sh, w), 2925 (br, s). Found HR-MS (ESI) m/z: 247.1089 ([M+Na+],
100), 248.1120 ([M+Na+], 20); C16H16ONa+ requires m/z: 247.1093 ([M+Na+], 100),
248.1127 ([M+Na+], 20).
9-Methoxy-4-methylfluorene 6h
Reflux, 16 hours. White solid. 53.9 mg, 95%. M.p. 85-88ºC. 1H NMR (400
MHz, Chloroform-d) δ 7.85 (d, J = 7.6 Hz, 1H, ArH), 7.68 (d, J = 7.3 Hz,
1H, ArH), 7.51 (d, J = 7.2 Hz, 1H, ArH), 7.46 (t, J = 7.5 Hz, 1H, ArH), 7.37
(t, J = 7.4 Hz, 1H, ArH), 7.31 – 7.24 (m, 1H, ArH), 7.22 (d, J = 7.4 Hz, 1H, ArH), 5.62 (s,
1H, ArCHOCH3Ar), 3.06 (s, 3H, ArCHOCH3Ar) 2.71 (s, 3H, ArCH3). 1H NMR (400 MHz,
27
O
6g
O
6h

Methanol-d4) δ 7.84 (d, J = 7.7 Hz, 1H, ArH), 7.63 – 7.58 (m, 1H, ArH), 7.45 – 7.39 (m, 2H,
ArH), 7.33 (td, J = 7.4, 1.0 Hz, 1H, ArH), 7.26 – 7.16 (m, 2H, ArH), 5.49 (s, 1H,
ArCHOCH3Ar), 3.07 (s, 3H, ArCHOCH3Ar), 2.67 (s, 3H, ArCH3). 13C NMR (101 MHz,
Chloroform-d) δ 144.27 (ArC), 143.45 (ArC), 140.45 (ArC), 134.60 (ArC), 132.79 (ArC),
130.38 (ArC), 128.66 (ArC), 128.29 (ArC), 126.81 (ArC), 124.58 (ArC), 124.36 (ArC), 82.52
(ArCHOCH3Ar), 53.31 (ArCHOCH3Ar), 22.33 (ArCH3). IR (solid press, cm-1) 738 (sh, s),
814 (sh, w), 1084 (sh, m), 1199 (sh, w), 1453 (sh, m), 1605 (sh, w), 1711 (sh, w), 2931 (br,
w). Found HR-MS (ESI) m/z: 233.0936 ([M+Na+], 100), 234.0967 ([M+Na+], 18);
C15H14ONa+ requires m/z: 233.0937 ([M+Na+], 100), 234.0970 ([M+Na+], 16).
1,1-Diphenylmethyl methyl ether 8a
Reflux, 2 hours. 52.1 mg, 97%. 1H NMR (300 MHz, Chloroform-d) δ 7.56
- 7.21 (m, 10H, ArH), 5.32 (s, 1H, ArCHOCH3Ar), 3.46 (s, 3H, OCH3). 1H
NMR (600 MHz, Methanol-d4) δ 7.45 – 7.28 (m, 8H, ArH), 7.27 – 7.19
(m, 2H, ArH), 5.29 (s, 1H ArCHOCH3Ar), 3.35 (s, 3H, OCH3). 13C NMR (151 MHz,
Chloroform-d) δ 142.08 (ArC), 128.40 (ArC), 127.46 (ArC), 126.92 (ArC), 85.44
(ArCHOCH3Ar), 57.03 (ArCHOCH3Ar). The NMR data is in good agreement with literature
sources.35
1-Phenyl-1-(4-methylphenyl)methyl methyl ether 8b
Reflux, 2 hours. 53.0 mg, 92%. 1H NMR (400 MHz, Chloroform-d) δ
7.44 – 7.36 (m, 4H, ArH), 7.33 - 7.28 (m, 3H, ArH), 7.21 (d, J = 7.6 Hz,
2H, ArH), 5.31 (s, 1H, ArCHOCH3Ar), 3.42 (s, 3H, OCH3). 13C NMR
(151 MHz, Chloroform-d) δ 142.39 (ArC), 139.19 (ArC), 137.15 (ArC), 129.15 (ArC),
128.42 (ArC), 127.41 (ArC), 126.97 (ArC), 126.89 (ArC), 85.33 (ArCHOCH3Ar), 56.99
28
O
8a
O
8b

(ArCHOCH3Ar). 21.17 (ArCH3). The NMR data is in good agreement with literature
sources.36
1-Phenyl-1-(3-methylphenyl)methyl methyl ether 8c
Reflux, 2 hours. 51.6 mg, 90%. 1H NMR (400 MHz, Chloroform-d) δ
7.44-7.35 (m, 4H, ArH), 7.33 – 7.18 (m, 4H, ArH), 7.12 (m, 1H, ArH),
5.27 (s, 1H, ArCHOCH3Ar), 3.44 (s, 3H, OCH3), 2.39 (s, 3H, ArCH3).
13C NMR (151 MHz, Chloroform-d) δ 143.64 (ArC), 143.47 (ArC), 139.50 (ArC), 129.85
(ArC), 129.75 (ArC), 129.71 (ArC), 128.99 (ArC), 128.86 (ArC), 128.35 (ArC), 128.49
(ArC), 86.95 (ArCHOCH3Ar), 58.48 (ArCHOCH3Ar). 22.95 (ArCH3). The NMR data is in
good agreement with literature sources.36
1-Phenyl-1-(4-trifluoromethylphenyl)methyl methyl ether 8d
Reflux, 2 hours. 64.1 mg, 89%. 1H NMR (400 MHz, Chloroform-d) δ
7.61 (d, J = 8.3 Hz, 2H, ArH), 7.50 (d, J = 8.3 Hz, 2H, ArH), 7.40 –
7.28 (m, 5H, ArH), 5.31 (s, 1H, ArCHOCH3Ar), 3.42 (s, 3H, OCH3).
13C NMR (151 MHz, Chloroform-d) δ 147.64 (ArC), 142.61 (ArC), 130.05 (ArC), 129.34
(ArC), 128.43 (ArC), 128.40 (ArC), 126.9 (ArC), 126.8 (q, J = 3.6 Hz, ArCF3), 124.2 (ArC)
86.22 (ArCHOCH3Ar), 58.52 (ArCHOCH3Ar). IR (solid press, cm-1) 700 (sh, w), 749 (sh, w),
808 (sh, w), 1095 (sh, m), 1123 (sh, m), 1324 (sh, s), 2934 (br, m).
29
O
8c
O
8dCF3

1-Phenyl-1-(3-methoxyphenyl)methyl methyl ether 8e
Reflux, 2 hours. 61.8 mg, quantitative. 1H NMR (500 MHz,
Chloroform-d) δ 7.40 – 7.31 (m, 4H, ArH), 7.29 - 7.24 (m, 3H, ArH),
6.98 – 6.93 (m, 2H, ArH), 6.81 (d, J = 8.2 Hz, 2H, ArH), 5.24 (s, 1H,
ArCHOCH3Ar), 3.81 (s, 1H, ArCHOCH3Ar), 3.42 (s, 3H, ArOCH3). 13C NMR (151 MHz,
Chloroform-d) δ 159.73 (ArC), 143.71 (ArC), 141.99 (ArC), 129.41 (ArC), 128.40 (ArC),
127.50 (ArC), 126.89 (ArC), 119.37 (ArC), 112.89 (ArC), 112.39 (ArC), 85.33
(ArCHOCH3Ar), 56.99 (ArCHOCH3Ar). 55.20 (ArOCH3). The NMR data is in good
agreement with literature sources.37
9-Methoxyfluorene 6a
Reflux, 48 hours. 51.8 mg, 97%. 1H NMR (600 MHz, Chloroform-d) δ 7.68
(d, J = 7.5 Hz, 2H, ArH), 7.65 – 7.58 (m, 2H, ArH), 7.49 – 7.37 (m, 2H,
ArH), 7.33 (td, J = 7.5, 1.2 Hz, 2H, ArH), 5.61 (s, 1H, ArCHOCH3Ar), 3.07
(s, 3H, OCH3). 1H NMR (600 MHz, Methanol-d4) δ 7.72 (d, J = 7.4 Hz, 2H, ArH), 7.58 (d, J
= 7.4 Hz, 2H, ArH), 7.40 (t, J = 7.4 Hz, 2H, ArH), 7.32 (t, J = 7.4 Hz, 2H, ArH), 5.52 (s, 1H,
ArCHOCH3Ar), 3.12 (s, 3H, OCH3). 13C NMR (151 MHz, Chloroform-d) δ 142.57 (ArC),
141.01 (ArC), 129.00 (ArC), 127.51 (ArC), 125.48 (ArC), 119.95 (ArC), 81.42
(ArCHOCH3Ar), 52.29 (ArCHOCH3Ar). The NMR data is in good agreement with literature
sources.38
30
O
6a
O
8e
O

Kinetic analysis, including rate data, for the methanolysis of species 3 and 7
A fresh solution of concentrated sulfuric acid (35.4 mg, 361 μmol) in deuterated methanol
(0.6 mL) was generated. Of this, a portion (0.5 mL) was added to an NMR tube that already
contained the desired amount (ca. 30 μmol) of the respective alcohols. The reaction was held
at the desired temperature either in a NMR spectrometer (for alcohols 7a-d, 3a-c and 3e-h) or
in a temperature controlled water bath at 23.8ºC (for 7e) or 60.0ºC (for alcohols 3d and 3i)
and 1H NMR spectra were taken at appropriate intervals. The extent of reaction at a given
time was determined through comparing the integration due to signals due to the starting
material (at the chemical shifts noted below in Table S2) relative to the integration of the
product signals (at the chemical shifts noted below). This allowed calculation of observed
first order rate constants, which are tabulated below (Table S1).
31

Compound Temp / oC
Amount of alcohol / mg
Observed rate constant (kobs) / s-1
Average kobs/ s-1
5.0 6.45 x 10-5
4.8 6.71 x 10-523.84.8 6.50 x 10-5
6.55(14) x 10-5
5.6 5.53 x 10-3
6.3 5.07 x 10-3Benzhydrol 7a
60.05.0 5.47 x 10-3
5.36(23) x 10-3
5.9 1.27 x 10-3
5.5 1.49 x 10-34-Methylbenzhydrol 7b 23.85.7 1..79 x 10-3
1.42(16) x 10-3
5.9 1.37 x 10-4
5.7 1.70 x 10-43-Methylbenzhydrol 7c 23.86.1 1.40 x 10-4
1.47(26) x 10-4
5.9 4.01 x 10-53-Methoxybenzhydrol 7d5.9 5.19 x 10-523.85.9 4.25 x 10-5
4.12(1.06) x 10-5
5.4 1.93 x 10-7
5.5 1.99 x 10-74-Trifluoromethylbenzhydrol 7e
23.85.9 2.11 x 10-7
2.06(12) x 10-7
5.2 3.03 x 10-5
60.0 5.3 3.24 x 10-5Fluorenol 3a5.0 3.45 x 10-5
3.24(21) x 10-5
5.6 2.04 x 10-5
5.6 1.92 x 10-523.85.7 1.77 x 10-5
1.91(14) x 10-5
5.3 3.26 x 10-3
6.0 3.65 x 10-31,4-Dimethylfluorenol 3g
60.05.7 3.52 x 10-3
3.48(20) x 10-3
5.6 5.52 x 10-4
5.2 3.91 x 10-43-Methylfluorenol 3b 60.07.2 4.89 x 10-4
5.67(1.13) x 10-4
6.0 1.13 x 10-4
5.5 1.14 x 10-44-Methylfluorenol 3h 60.05.2 1.25 x 10-4
1.17(06) x 10-4
5.1 6.30 x 10-5
5.4 6.23 x 10-52-Methylfluorenol 3c 60.06.2 5.73 x 10-5
6.12(33) x 10-5
6.5 2.31 x 10-5
5.2 2.51 x 10-52-Methoxyfluorenol 3e 60.05.4 2.40 x 10-5
2.41(10) x 10-5
5.7 4.64 x 10-7
5.5 4.24 x 10-72-Trifluoromethyl fluorenol 3d
60.05.7 4.59 x 10-7
4.48(20) x 10-7
6.8 2.45 x 10-7
7.1 2.54 x 10-73-Methylcarboxylate fluorenol 3i
60.07.1 2.39 x 10-7
2.46(08) x 10-7
a Due to speed of the reaction at this temperature, the sample was first dissolved in 400 μL of hot deuterated methanol before the addition of the acid catalyst in a further 100 μL of deuterated methanol at room temperature. A dummy sample was locked in the NMR spectrometer before the sample was added and shimmed at 60ºC. The sample was routinely already at 60% extent of conversion, but this had no bearing on our ability to monitor the reaction as it proceeded to completion.
32
Table S1 - The amounts of starting material used, the resulting observed first-order rate constants and the observed first order rate constants for the methanolysis
reactions of the alcohols noted and the uncertainties the averages reported as half the range of the data.

Compound δStarting Material 7 / ppm δProduct 6 or 8 / ppm
Benzhydrol 7a 5.77 5.284-Methylbenzhydrol 7b 5.74 5.253-Methylbenzhydrol 7c 5.74 5.253-Methoxybenzhydrol 7d - 5.364-Trifluoromethylbenzhydrol 7e 5.86 5.39Fluorenol 3a 5.55 5.543-Methylfluorenol 3b 5.50 5.472-Methylfluorenol 3c 5.46 5.402-Trifluoromethylfluorenol 3d 8.24 8.192-Methoxyfluorenol 3e 5.49 5.471,4-Dimethylfluorenol 3g 5.58 5.554-Methylfluorenol 3h 5.50 5.483-Methylcarboxylatefluorenol 3i 5.60 5.59
Table S2 – The signals followed in the 1H NMR spectrum for all of the methanolysis reactions. Only 3-methoxybenzhydrol required reference to the solvent peak due to an
overlap of the starting material signal with the broad acidified methanol signal.
33

1H and 13C NMR spectra
2′,5'-Dimethylbiphenyl-2-carboxylic acid 15g
34
O
OH
15g

3- Methylfluorenol 3b
35
OH
3b

2- Methylfluorenol 3c
36
OH
3c

2-Trifluoromethylfluorenol 3d
37
OH
CF33d

2-Methoxyfluorenol 3e
38
OH
O
3e
1,4-Dimethylfluorenol 3g
39
OH
3g

4-Methylfluorenol 3h
40
OH
3h

Methyl 9-fluorenol-3-carboxylate 3i
41
OH
CO2Me3i
1-Phenyl-1-(4-trifluoromethylphenyl)methyl methyl ether 8d
42
O
8dCF3

9-Methoxy-3-methylfluorene 6b
43
O
6b

9-Methoxy- 2-methylfluorene
6c
44
O
6c
2,9-
Dimethoxyfluorene 6e
45
46
O
6e
O

9-Methoxy-1,4-Dimethylfluorene 6g
47
O
6g

9-Methoxy-4-methylfluorene 6h
48
O
6h

References
1. D. C. Baird, Experimentation: An Introduction to Measurement Theory and Experiment Design, Pearson Education, New York, 1994.
2. C. S. Marvel and N. A. Hansen, Org. Synth., 1928, 8, 24-25.3. A. G. Davies, J. Kenyon, B. J. Lyons and T. A. Rohan, J. Chem. Soc., 1954, 3474-
3479.4. K. Suzuki, T. Arao, S. Ishii, Y. Maeda, K. Kondo and T. Aoyama, Tetrahedron Lett.,
2006, 47, 5789-5792.5. V. J. Shiner and C. J. Verbanic, J. Am. Chem. Soc., 1957, 79, 369-373.6. X. Jia, L. Fang, A. Lin, Y. Pan and C. Zhu, Synlett, 2009, 495-499.7. C.-T. Lee and B. H. Lipshutz, Org. Lett., 2008, 10, 4187-4190.8. M. Kuriyama, N. Ishiyama, R. Shimazawa and O. Onomura, Tetrahedron, 2010, 66,
6814-6819.9. S.-i. Hirashima, T. Nobuta, N. Tada, T. Miura and A. Itoh, Org. Lett., 2010, 12, 3645-
3647.10. J. L. Seidel, W. W. Epstein and D. W. Davidson, J. Chem. Ecol., 1990, 16, 1791-
1816.11. M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133,
19505-19511.12. M. Amatore and C. Gosmini, Angew. Chem., Int. Ed., 2008, 47, 2089-2092.13. J. J. Mousseau, F. Vallee, M. M. Lorion and A. B. Charette, J. Am. Chem. Soc., 2010,
132, 14412-14414.14. L. Ackermann, C. J. Gschrei, A. Althammer and M. Riederer, Chem. Commun., 2006,
1419-1421.15. D. J. Drake and J. M. Wardleworth, Zeneca Limited, UK; Zeneca Pharma S.A. . 1999,
p. 138 pp.16. J. R. Merchant and V. B. Desai, Curr. Sci., 1960, 29, 476.17. D. N. Korolev and N. A. Bumagin, Tetrahedron Lett., 2006, 47, 4225-4229.18. N. C. Deno, J. Am. Chem. Soc., 1950, 72, 4057-4059.19. Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574-
2577.20. C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132, 14006-14008.21. J.-C. Wan, J.-M. Huang, Y.-H. Jhan and J.-C. Hsieh, Org. Lett., 2013, 15, 2742-2745.22. T.-P. Liu, Y.-X. Liao, C.-H. Xing and Q.-S. Hu, Org. Lett., 2011, 13, 2452-2455.23. J. M. Fu, B. P. Zhao, M. J. Sharp and V. Snieckus, J. Org. Chem., 1991, 56, 1683-
1685.24. J. Song, F. Wei, W. Sun, K. Li, Y. Tian, C. Liu, Y. Li and L. Xie, Org. Lett., 2015,
17, 2106-2109.25. J. Gruber, R. W. C. Li, L. H. J. M. C. Aguiar, A. R. V. Benvenho, R. Lessmann and I.
A. Huemmelgen, J. Mater. Chem., 2005, 15, 517-522.26. K. Alder, J. Haydn, K. Heimbach, K. Neufang, G. Hansen and W. Gerhard, Liebigs
Ann. Chem., 1954, 586, 110-137.27. T. Fukuyama, S. Maetani, K. Miyagawa and I. Ryu, Org. Lett., 2014, 16, 3216-3219.28. J. W. Lockner, D. D. Dixon, R. Risgaard and P. S. Baran, Org. Lett., 2011, 13, 5628-
5631.29. Z. Hou, K. Takamine, O. Aoki, H. Shiraishi, Y. Fujiwara and H. Taniguchi, J. Org.
Chem., 1988, 53, 6077-6084.30. K. Hattori, H. Sajiki and K. Hirota, Tetrahedron, 2001, 57, 4817-4824.31. J. D. Dickinson and C. Eaborn, J. Chem. Soc., 1959, 2337-2340.
49

32. E. D. Bergmann, G. Berthier, E. Fischer, Y. Hirshberg, D. Lavie, E. Loewenthal and B. Pullman, Bull. Soc. Chim. Fr., 1952, 78-83.
33. C. L. Arcus and M. M. Coombs, J. Chem. Soc., 1954, 3977-3980.34. B. R. T. Keene and P. Tissington, J. C. Soc., 1965, 3032-3037.35. T. B. Phan, C. Nolte, S. Kobayashi, A. R. Ofial and H. Mayr, J. Am. Chem. Soc.,
2009, 131, 11392-11401.36. M. Shi, Y. Okamoto and S. Takamuku, Bulletin of the Chemical Society of Japan,
1990, 63, 2731-2733.37. J. Clayden and M. Julia, J. Chem. Soc., Chem. Commun., 1993, 1682-1683.38. V. S. Sridevi, W. K. Leong and Y. Zhu, Organometallics, 2006, 25, 283-288.
50





























