Embed Size (px)
Citation preview
40
60708090
RNR3
Fol
d In
crea
se
= 1hr. +MMS = 2.5 hrs +MMS
= No MMS
Ant
i-TB
P Fo
ld C
hang
e
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
Ant
i-Crt
1p F
old
Cha
nge
0.00.51.01.52.02.53.03.54.04.5
50
0
302010
BY4741 dhh1∆ ccr4∆ pop2∆ not5∆
BY4741 dhh1∆ ccr4∆ pop2∆ not5∆ BY4741 dhh1∆ ccr4∆ pop2∆ not5∆
= - MMS = +MMS
= - MMS = +MMS
A. B.
C. D.
Kruk et al. Fig S1
= - MMS = +MMS
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Ant
i-Taf
1 Fo
ld C
hang
e
BY4741 dhh1∆ ccr4∆ pop2∆ not5∆
Northern blot mRNA
0.0
0.2
0.4
0.6
0.8
1.0
TBP
%IP
(IP/
Inpu
t)
E.
B(-85)
C(+448)
D(+1100)
E(+1500)
F(+2420)
= - MMS = +MMS

Full-length Rpb1 ∆CTD-Rpb1WCE IP WCE
myc Rpb3
Dhh1-myc 1 2 3 4 5 6 7 8 9 10
1
IP
untagged Ccr4-mycCcr4-mycrpb4∆
Dhh1
Rpb1
WCE WCEIP IPWT rpb4∆
Ccr4-myc
Rpb1
WCE WCE WCEIP IPIP
IP: Rpb1
IP Antibody
Full-length ∆CTD
WCE IP WCE IP
Not2-myc
FL ∆CTD
A. B.
D. E.
C.Full-length Rpb1 ∆CTD-Rpb1
*
2 3 4 5 6
11 12 13 14 15 16 17 18 19 20 21
Rpb1
FL Rpb1 ∆CTD Rpb1
Rpb1
IP: Rpb1
IP: anti-myc IP: anti-myc
WCE IP IP WCEmyc
Rpb3
IP IP
WCE IP
CTD-lessPlasmidR P B 1
CTD-lessPlasmid
~191kD~168kD
GenomicCopy
Fig. S2

TAP-Ccr4complex
-
EC70
TAP-Not4complex
Not1
TAP-Ccr4
Not5
Pop2
Caf40
Not2
Caf120Caf130
Not3
Dhh1Not4
EC70/Ccr4-Not
A. B.
150 nuc runoffproduct
70 nuc
% ru
noff
prod
uct
time (minutes)
0102030405060708090
100
0 5 10 15 20 25 30
C.
time (minutes) -10 0
+Ccr4-NotBSA
0
dRNAPII
dRNAPIIdRNAPII +Ccr4-Not
Fig S3

510152025303540
2 4 6 8 10 12 14 16
1 kb runoffproduct
70 nuc
time (minutes) -10 0 0
time (minutes) 0 0
% ru
noff
prod
uct
time (minutes)
5’
yRNAPII, template and UpG
Ccr4-Notor BSA Add 100 uM (D) or 10 uM (E) GTP
100 uM ATP, CTP5 uM UTP, αP32UTP
5’Run-off
yRNAPIIyRNAPII +Ccr4-Not
5’
yRNAPII, template UpG
20’ 10’
NTPs, -GTP Ccr4-Not or BSA Add 100 uM GTP
50 uM UTP
Aliquot 1
(-10’)
Run off
A.
B.
C.
D.
+Ccr4-Not
1 kb runoffproduct
+Ccr4-Not
BSA
BSA
0
20
40
60
80
100
120
0 2 4 6 8 10 12 14 16 18
% ru
noff
prod
uct
time (minutes)
yRNAPIIyRNAPII +Ccr4-Not
EC70 formationPreincubation
Fig S4
E.
0
20
40
60
80
100
120
0 2 4 6 8 10 12 14 16 18
% ru
noff
prod
uct
time (minutes)
time (minutes) 0 0
+Ccr4-NotBSA
yRNAPIIyRNAPII +Ccr4-Not

0
5
10
15
20
25
30
0 2 4 6 8 10 12 14 16 18
yPol II
time (minutes)
% ru
noff
prod
uct
yPol II : Ccr4-Not :: 1: 2
yPol II : Ccr4-Not :: 1: 6
yPol II : Ccr4-Not :: 1: 4
A.Kruk_Figure S5
B.
yPol IICcr4-Not4 complex
200
116 97
66
45
+ + + +- - + +
UV-crosslinking - + + -
Rpb1
Ccr4-Not specificband
Rpb2
Autoradiograph
5’
Preincubation of yRNAP II with template and Upg
20’ 10’
NTPs, with O-me GTPadded, G-less transcript formation
Incubationwith Ccr4-Not compexor buffer
10’
Crosslink with300 nm UV
Treat with RNAseand DNAse
15’SDS-PAGE,
silve
r stai
nig
exposu
re to phosp
hoimag
er

WT ccr4∆ dst1∆ dhh1∆ not4∆
Rel
ativ
e R
NA
PII D
ensi
ty in
Gal
acto
se
Kruk-Fig. S6
GAL1P YLR454W
+1 1KB 2KB 4KB 6KB 8KB
A.
B.
0.0
0.2
0.4
0.6
0.8
1.0
1.2 +11KB2KB4KB6KB8KB

Supplemental TABLE 1. Strain used in the study
Strain Genotype BY4741 MATa, his3∆1, leu2∆0, met15∆0, ura3∆0 JR1178 BY4741 with dhh1∆::kanMX JR1179 BY4741 with ccr4∆::kanMX JR1180 BY4741 with pop2∆::kanMX JR1181 BY4741 with not5∆::kanMX JR1182 BY4741 with rpb4∆::kanMX JR1188 BY4741 with not3∆::kanMX JR1189 BY4741 with not4∆::kanMX JR1358 BY4741 with chd1∆::kanMX Euroscarf BY4741 with ctk1∆::kanMX JR1376 BY4741 with cdc73∆::kanMX JR1377 BY4741 with paf1∆::kanMX MY16 MATα, leu2::PET56, trp1∆1, ura3-52, gal2, gcn4∆1
Collart and Struhl (1993) JR1192 BY4741 with Dhh1-myc::HIS3 JR1193 BY4741 with Ccr4-myc::HIS3 JR1194 BY4741 with Ccr4-myc::HIS3, rpb4∆::kanMX JR1195 BY4741 with Pop2-myc::HIS3 JR1196 BY4741 with Not2-myc::HIS3 JR1220 MATa, Dhh1-myc::HIS3, rpb4::kanMX, trp1∆63,
met15∆0, leu2∆0, ura3∆0 JR1222 BY4741 with Not5-myc::HIS3 JR1328 BY4741 with 3HA-kanMX-GAL1P-YLR454W JR1329 JR1328 with dst1∆::URA3 JR1330 JR1328 with with ccr4∆::URA3 JR1331 JR1328 dhh1∆::LEU2 JR1332 JR1328 with not4∆::URA3 JR1408 BY4741 with Not4-TAP::HIS3, dst1∆::URA3 JR1409 BY4741 with Ccr4-TAP::HIS3, dst1∆::URA3

Supplemental figure legends
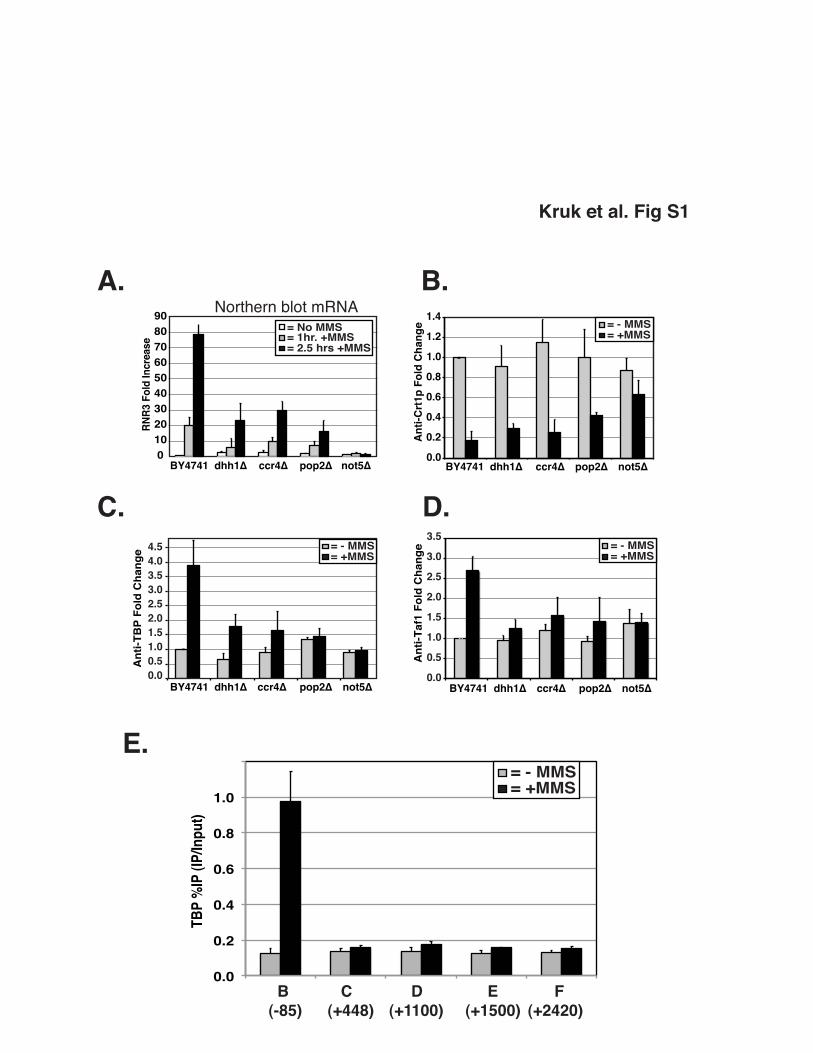
Figure S1. Ccr4-Not is needed for optimal expression of RNR3. (A.) Quantification
of northern blot analysis of RNR3 expression in Ccr4-Not mutants. Cells were grown
overnight and untreated or treated with MMS for 1hr and 2.5hrs. Blots were probed with
RNR3 and loading control, ScR1. RNR3 signal was corrected based on ScR1 signal.
Bars represent average and standard deviation of at least three independent
experiments. (B - D.) ChIP analysis of Crt1 release, TBP recruitment, and TAF1
recruitment. Cells were grown overnight and treated for 2.5hrs with MMS or untreated
prior to crosslinking. Crt1 release was detected at the DRE (Region A in Figure 1),
while TBP and Taf1 recruitment are at the RNR3 promoter (Region B in Figure 1). Bars
represent average and standard deviation of at least three independent experiments.
Crt1 release is checkpoint-dependent, and this step is used to monitor the integrity of
the DNA damage checkpoint. Deletion of DHH1, CCR4 and POP2 does not significantly
affect the release of Crt1, suggesting that the DNA damage-signaling pathway is intact
in these mutants. However, deletion of NOT5 significantly affects Crt1 release from the
promoter. We have also observed that other Ccr4-Not mutants that have severe growth
defects, such as cells with NOT2 mutations, also have impaired DNA damage signaling
(not shown). Thus, most experiments were conducted in mutants with intact
checkpoints. (E) Localization of TBP across RNR3. TBP is recruited only to the
promoter. ChIP DNA in this panel was analyzed by qPCR.

Figure S2. Ccr4-Not interaction with RNAPII is not dependent on the CTD or
RPB4. (A.) Schematic of simultaneous expression of full-length (genomic) and CTD-
less (plasmid) Rpb1 subunit within yeast cells. Full-length and CTD-less Rpb1 can be
detected by western blotting using an N-terminal Rpb1 antibody (y-80, SCBT), migrating
at approximately 191kD and 168kD, respectively. (B.) Protein extracts prepared from
strains transformed with plasmid carrying either a full-length version of RPB1 or a CTD-
less version of RPB1. Rpb1 was detected using y-80. The amount of full length Rpb1 to
CTD-less Rpb1 in the cells was approximately 3:1. Extracts were IPʼed using anti-myc
(lanes 2 and 5) as a control and show no Rpb1 protein binding non-specifically to IgG or
the resin. Anti-Rpb3 IP confirms both the full-length (lane 3) and CTD-less Rpb1 (lane
6) can be incorporated into the RNAPII complex. The amount of CTD-less Rpb1 versus
full length Rpb1 brought down by Rpb3 reflected their relative expression in cells. (C.)
Dhh1-myc (left) and Not2-myc (right) strains were transformed with plasmids as
described above. Protein extracts were immunoprecipitated with anti-myc followed by
western blot probing for the presence of RNAPII using an antibody specific to the N-
terminus of Rpb1 (y-80). The amount of CTD-less versus full length Rpb1 copurifying
with Dhh1 and Not2 is the same as that brought down with Rpb3 antibody and reflects
the relative expression of each in cells. (D.) Protein extracts prepared from wild type
and rpb4∆ strain were immunoprecipitated with Rpb1 antibody (8WG16), followed by
western blotting with Dhh1 antibody. Anti-Rpb1 was used as an IP and loading control.
(E.) Protein extracts prepared from untagged and Ccr4-myc strains, either with or

without RPB4 deleted, were immunoprecipitated with Rpb1 antibody (8WG16) followed
by detection of Ccr4-myc using anti-myc. Anti-Rpb1 was used as an IP and loading
control. The asterisk marks a version of Rpb1 with a proteolyzed CTD, which is
observed in some WCE preparations. It is not observed in the IP lanes because the
8WG16 antibody used in the immunoprecipitation step recognizes the CTD.
Figure S3. Ccr4-Not purified through a Ccr4-TAP subunit binds ECs in vitro and
Ccr4-Not does not stimulate elongation of dRNAPII complexes. (A) Silver stained
SDS-PAGE gel showing the composition of the Ccr4-Not complex purified from a TAP-
Ccr4 strain. (B) Analysis of interaction of Ccr4-Not with elongation complexes. RNAPII
elongation complexes (EC70) were formed as described in the legend of Figure 3 and in
the materials and methods section and incubated for 10 min with increasing amounts of
purified Ccr4-Not complex from either TAP-Ccr4 strain or TAP-Not4 (approximately 0.5
pmol, 1 pmol and 1.5 pmol of Ccr4-Not complex was added in each case). RNAPII only
lane contains 1 µg of BSA. (C) Ccr4-Not complex does not stimulate the resumption of
transcription of stalled Drosophila RNAPII elongation complexes. Drosophila elongation
complexes were incubated with 1.5 pmol of Ccr4-Not complex or 1 µg BSA for 10
minutes. 100 µM GTP and 50 µM UTP were added to generate a 150 base run-off
transcript. Percentage of run-off product was calculated and plotted as a function of
time.
Figure S4. Ccr4-Not predominantly stimulates the resumption of transcription
from pauses. (A) Outline of the in vitro elongation system using a template generating

a 1kb run off product. (B) The stalled elongation complexes were incubated with 1.5
pmol of Ccr4-Not complex or 1 µg BSA for 10 minutes. Samples were collected at
different times after addition of 100 µM GTP and 50 µM UTP. Percentage of run-off
product was calculated and plotted as a function of time. The stimulation is observed on
the longer run-off template if RNAPII is arrested at a G-tract. (C) Outline of the in vitro
elongation system to analyze the affects of Ccr4-Not on elongation of unarrested
complexes. RNAPII is incubated with UpG and the tailed template (EC70 1kb) to form
initiated complexes, followed by the addition of 1.5 pmol of Ccr4-Not complex or 1 µg
BSA for 10 minutes. All nucleotides were then added and samples were collected at
different time points. (D) Analysis of transcripts following addition of 100 uM GTP. (E)
Analysis of transcripts produced under limiting (10 uM) GTP concentrations.
Figure S5. (A) Dose dependent stimulation of elongation by Ccr4-Not. Transcription
elongation assays on the 1Kb run off template was conducted as described in
Supplemental Figure 4B on stalled elongation complexes. Different ratios of Ccr4-Not to
RNAPII were analyzed. The amount of run-of products is plotted on the Y-axis. (B)
Experimental scheme for the crosslinking assays. Samples were treated, or not, with UV
light where indicated. The migration of molecular weight markers is indicated on the left.
Figure S6. (A) Schematic of the GAL1p-454W reporter gene. (B) Alternative analysis of
the data from Figure 6 in the manuscript. In this panel, the level of RNAPII in the
mutants at each location was normalized to the value in wild type, which was set at 1.0.
This method of quantification does not correct for the amount of RNAPII loaded onto the
promoter (+1); thus, does not correct for any effects of the mutation on initiation.

Initiation defects are observed, but the build up of RNAPII in the body of the gene is still
evident.

SupplementalinformationKruketal.
Supplemental experimental procedures
RNA isolation and Northern Blotting.
RNA isolation and northern blotting was carried out as previously described 1. 15µg of
total RNA was separated on 1% formaldehyde gel and transferred to nitrocellulose
membrane (Hybond-N+; Amersham Pharmacia Biotech, Piscataway, NJ) via capillary
action. After UV crosslinking and a ≥4 hr prehybridization at 65˚C, radioactively-labeled
gene-specific probes were added. Signal was detected using Phosphor Screen
(Molecular Dynamics), scanned with the Typhoon system (Molecular Dynamics), and
quantified using ImageQuant.
GAL-YLR454 assay and Chromatin Immunoprecipitation.
Cells in the GAL1 shutdown experiment (Figure 1I) were grown overnight in YPD+ 2%
galactose, and then a portion of culture was removed for crosslinking. The remaining
cells were centrifuged and resuspended in 2% dextrose medium. Cells were
crosslinked at indicated times indicated in the figure. For the GAL1p-YLR454W
experiments, cultures were grown overnight in YPD+ 2% galactose. An aliquot was
removed and 2% dextrose was added directly to the media. Samples were crosslinked
with formaldehyde for 15 minutes at indicated time points. RNAPII present at the
promoter under galactose-inducing conditions is the maximal amount of RNAPII
initiating transcription in each strain; therefore, after normalizing each region to wild
type, the density of RNAPII at the promoter was set to 1. This assumes a processivity

SupplementalinformationKruketal.
of 100% for wild type cells 2. Whole-cell extracts were prepared by glass bead disruption
in FA-lysis buffer and sheared into fragments averaging 200 to 600 bp in size using a
Bioruptor (Diagenode, Philadelphia, PA). Chromatin was immunoprecipitated (IP) with
the antibodies indicated below. After purification, the precipitated and input DNAs were
analyzed by semi-quantitative PCR or real-time PCR. PCR products were analyzed by
electrophoresis and ethidium bromide staining, scanned with the Typhoon system
(Molecular Dynamics), and quantified by using ImageQuant. Real-time PCR was
performed with SYBR® Green detection (Quanta Biosciences, Gaithersburg, MD) using
a StepOne Plus qPCR thermocycler (Applied Biosystems, Foster City, CA). Percent IP
was calculated using the following formula: (IP signal/input signal) x 100. The following
antibodies were used in ChIP: 2µL of αmyc (9E10 ascites; Covance, Emeryville, CA),
2µL of αDhh1 (αDhh1; our lab), 2µL of αRNAPII (8WG16; Covance, Princeton, NJ), 1uL
of αCrt1 N-terminal (αCrt1; our lab), 1uL αTATA-binding protein (TBP) (αTBP; our lab),
1ul anti-TAF1 (our lab) and 1uL αRpb3 (αRpb3 ascites; Neoclone, Madison, WI).
Co-immunoprecipitation and Western blotting.
Extracts were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE),
transferred to nitrocellulose membranes (Whatman Protran®). Membranes were
blocked for at least 1 hr with 5% dry milk in TBST (50 mM Tris, pH 7.4, 150 mM NaCl,
0.05% Tween-20). Antibodies diluted in TBST+ 2% milk. Proteins were detected using
HRP-secondary antibodies and enhanced chemiluminescence. The following antibodies
were used in Western blotting: 1:5000 of anti-myc (9E10 ascites; Covance, Emeryville,
CA), 1:3000 of anti-Dhh1 (anti-Dhh1; our lab), 1:1000 of anti-Rpb1 N-terminal (y-80;

SupplementalinformationKruketal.
Santa Cruz Biotechnologies, Santa Cruz, CA), 1:1000 of anti-RNAPII (8WG16;
Covance, Emeryville, CA).
Extract preparation and co-immunoprecipitation was carried out as described in
detail in previous publications 1,3. One ml of 2 mg/ml whole cell extract in Buffer B (20
mM HEPES-KOH, pH 7.5, 10 mM magnesium acetate, 150 mM potassium acetate, 10
mM EGTA and 20% glycerol. Prior to use, DTT (5 mM) and the following protease
inhibitors: 3 µg/ml each of pepstatin A, leupeptin, aprotinin, bestatin, antipain, and
chymostatin; 2 mM benzamidine-HCl were added) was incubated for 2-3 hrs at 4˚C with
antibody and then 50 ul of a 50:50 slurry of Protein A Sepharose CL-4B (GE Healthcare,
Piscataway, NJ) was added and the incubation was carried out overnight with end-over-
end rotation. The beads were pelleted and washed 4 times with Buffer B containing 0.2
M KoAc. Immune complexes were eluted in SDS-PAGE buffer and analyzed by western
blotting. The following antibodies were used: 2µL anti-myc (9E10 ascites; Covance,
Emeryville, CA), 2µL anti-Dhh1 (anti-Dhh1; our lab), 2µL anti-Rpb3 ascites fluid
(Neoclone, Madison, WI).
Protein Purification
Both TAP-Not4 and TAP-Ccr4 complexes were purified from strains containing a
deletion of DST1. The protocol for TAP purifications was modified from that described in
another publication 4. Cells were broken in buffer containing 250 mM KCl to liberate the
complex from the pellet. All subsequent steps of purification were carried out in buffer
containing 150mm NaCl. The proteins were dialyzed, concentrated by microfiltration

SupplementalinformationKruketal.
and stored in a buffer containing 25 mM HEPES pH 7.6, 150 mM KCl, 12 mM MgCl2,
10% glycerol, 0.5 mM EDTA. Yeast RNAPII was purified using a Rpb4-TAP strain as
described in a previous publication 5. The concentrations of all proteins were estimated
by comparing intensities of their bands to that of known amounts of BSA on a silver
stained SDS-PAGE gel.
Preparation of elongation complexes and gel shift assays
Assays were adapted from those used to analyze Drosophila elongation complexes 6,7.
DNA templates were generated by PCR amplification from a plasmid containing a G-
less cassette, followed by DNA sequence with all four nucleotides. The forward primer
contains a Bgl II site. PCR products were digested with Bgl II, dephosphorylated with λ
phosphatase (NEB) and ligated with a 5ʼ phosphorylated oligonucleotide. The resulting
template contains a 11 nucleotide 5ʼ overhang with either 150 bp or 1kb duplex DNA,
depending on the PCR primers used to generate the substrate. The template was gel
purified before use. Transcription reactions (15 ul) contained 50 mM HEPES pH 7.6,
100 mM KCl, 1 mM MnCl2, 12% glycerol, 0.5 mM DTT, 0.5 mM UpG, 20 units of
RNasin (Promega, Madison, WI), 100 ng of template and ~100 ng (~0.25 pmol) of
purified yeast RNAPII. The template was preincubated with RNAPII for 5 minutes in the
transcription buffer, and then transcription was initiated by adding a 5 μl NTP mix,
yielding final concentrations of 0.1 mM ATP, 0.1 mM CTP, 5 μM UTP, 5 uM 3ʼO-methyl
GTP and 4 uCi/reaction of [a-32 P] UTP. Each reaction was incubated at 30°C for 20
minutes. The final KCl concentration in the transcription reaction was 75 mM.

SupplementalinformationKruketal.
Elongation complexes with Drosophila RNAPII were generated by incubation at room
temperature. Elongation complexes with Pyrococcus furiosus archaeal polymerase (a
kind gift of Katsu Murakami, Penn State University) were generated using the same
conditions as described above, except for that ECs were formed at 75°C for 20 minutes.
The reaction mixture was cooled to room temperature prior to the addition of Ccr4-Not
or carrier protein. Purified Ccr4-Not complex was added to the stalled elongation
complexes formed as described above, and allowed to bind to the yRNAPII, dRNAPII or
archaeal polymerase elongation complexes for 5 minutes. 1 ug of yeast RNA was
added to the reactions to reduce nonspecific binding of proteins to the nascent
transcript. The samples were run on 4% native gel in a buffer containing 50 mM Tris pH
8.5, 0.38 M Glycine, 2 mM EDTA and 5 mM MgCl2 at 4˚C at 200V for 5 hours. The
gels were dried and analyzed using the Typhoon phosphorimaging system (Molecular
Dynamics).
In Vitro Run-On Assay
Transcription reactions (15 ul) contained 50 mM HEPES pH 7.6, 100 mM KCl, 1 mM
MnCl2, 12% glycerol, 0.5 mM DTT, 0.5 mM UpG, 20 units of RNasin (Promega,
Madison, WI), 100 ng of template and ~100 ng (~0.25 pmol) of purified RNAPII. The
template was preincubated with RNAPII for 5 minutes in the transcription buffer, and
then transcription was initiated by adding a 5 μl NTP mix, yielding final concentrations of
0.1 mM ATP, 0.1 mM CTP, 5 μM UTP and 4 uCi/reaction of [a-32 P] UTP. Each

SupplementalinformationKruketal.
reaction was incubated at 30°C for 20 minutes. Following incubation with Ccr4-Not
complex or equivalent amounts of BSA for 10 minutes, 50 uM UTP and 100 uM GTP
were added and samples were removed at the indicated time points. Transcription
reactions were terminated and RNA was purified by phenol chloroform extraction and
ethanol precipitation. The products were analyzed on 8M urea-containing denaturing
gels. The gels were dried and analyzed using the Typhoon phosphorimaging system. In
order to analyze the affect of Ccr4-Not complex on rate of elongation, yRNAPII was
incubated with UpG and 1Kb run off template for 5 minutes, followed by the addition of
the Ccr4-Not complex. Transcription was initiated by addition of all nucleotides (0.1 mM
ATP, 0.1 mM CTP, 0.1 mM GTP, 5 uM UTP, and 1 uCi/reaction of [a-32 P] UTP).
Assays were conducted where the amount of GTP was reduced to 10 uM as indicated.
Samples were removed at regular intervals, and processed as described above.
References:
1. Reese, J.C. & Green, M.R. Functional analysis of TFIID components using conditional mutants. Methods Enzymol 370, 415-30 (2003).
2. Mason, P.B. & Struhl, K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell 17, 831-40 (2005).
3. Reese, J.C., Apone, L., Walker, S.S., Griffin, L.A. & Green, M.R. Yeast TAFIIS in a multisubunit complex required for activated transcription. Nature 371, 523-7 (1994).
4. Rigaut, G. et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17, 1030-2 (1999).
5. Suh, M.H. et al. Fcp1 directly recognizes the C-terminal domain (CTD) and interacts with a site on RNA polymerase II distinct from the CTD. Proc Natl Acad Sci U S A 102, 17314-9 (2005).
6. Zhang, Z., Fu, J. & Gilmour, D.S. CTD-dependent dismantling of the RNA polymerase II elongation complex by the pre-mRNA 3'-end processing factor, Pcf11. Genes Dev 19, 1572-80 (2005).

SupplementalinformationKruketal.
7. Zhang, Z., Wu, C.H. & Gilmour, D.S. Analysis of polymerase II elongation complexes by native gel electrophoresis. Evidence for a novel carboxyl-terminal domain-mediated termination mechanism. J Biol Chem 279, 23223-8 (2004).





























