Embed Size (px)
Citation preview

Department of Clinical PharmacologyUniversity of Helsinki
Finland
Studies on Drug Interactions
between CYP3A4 Inhibitors and
Glucocorticoids
by
Tiina Varis
ACADEMIC DISSERTATIONTo be presented, with the permission of the Medical Faculty of the University ofHelsinki, for public examination in the small lecture hall of Haartman Institute,
Haartmaninkatu 3, on December 15th, 2000, at 12 noon.
Helsinki 2000

2
Supervised by:
Professor Pertti Neuvonen, MDDepartment of Clinical PharmacologyUniversity of HelsinkiHelsinki, Finland
Docent Kari Kivistö, MDDepartment of Clinical PharmacologyUniversity of HelsinkiHelsinki, Finland
Reviewed by:
Docent Arja Rautio, MDDepartment of Pharmacology and ToxicologyUniversity of OuluOulu, Finland
Docent Kari Aranko, MDFocus Inhalation OYTurku, Finland
Official opponent:
Docent Risto Huupponen, MDDepartment of Pharmacology and Clinical PharmacologyInstitute of BiomedicineUniversity of TurkuTurku, Finland
ISBN 952-91-2933-5 (nid.)ISBN 952-91-2934-3 (PDF)Helsinki 2000Yliopistopaino

3
To Juha, Antti, and Kristiina

4
CONTENTS
ABBREVIATIONS..................................................................................................................6
LIST OF ORIGINAL PAPERS..............................................................................................7
ABSTRACT .............................................................................................................................8
INTRODUCTION ...................................................................................................................9
REVIEW OF THE LITERATURE......................................................................................10
1. Drug metabolism.............................................................................................................101.1. Principles of drug metabolism.................................................................................101.2. The CYP enzymes...................................................................................................101.3. CYP3A subfamily ...................................................................................................111.4. Drug interactions .....................................................................................................141.5. Drug interactions mediated by inhibition of CYP3A4 ............................................161.6. CYP3A4 inhibitors studied .....................................................................................171.6.1. Itraconazole..........................................................................................................171.6.2. Diltiazem..............................................................................................................181.6.3. Mibefradil.............................................................................................................201.6.4. Grapefruit juice ....................................................................................................211.7. CYP3A4 and P-glycoprotein...................................................................................24
2. Glucocorticoids...............................................................................................................242.1. General ....................................................................................................................242.2. Regulation of cortisol secretion...............................................................................272.3. Pharmacodynamics..................................................................................................272.4. Therapeutic use .......................................................................................................282.5. Adverse effects of glucocorticoid treatment............................................................302.6. Methylprednisolone.................................................................................................302.7. Prednisolone............................................................................................................332.8. Dexamethasone .......................................................................................................35
AIMS OF THE STUDY ........................................................................................................38
MATERIALS AND METHODS ..........................................................................................39
1. Ethical considerations .....................................................................................................392. Study subjects .................................................................................................................393. Study protocol.................................................................................................................39
3.1. Study design ............................................................................................................393.2. Determination of plasma drug and cortisol concentrations .....................................423.3. Pharmacokinetic calculations ..................................................................................433.4. Pharmacodynamic measurements............................................................................443.5. Statistical methods...................................................................................................453.6. Archiving.................................................................................................................45

5
RESULTS .............................................................................................................................. 46
1. Interaction between itraconazole and glucocorticoids .................................................... 461.1. Oral methylprednisolone (Study I) ......................................................................... 461.2. Intravenous methylprednisolone (Study II) ............................................................ 471.3. Oral prednisolone (Study IV) ................................................................................. 481.4. Oral and intravenous dexamethasone (Study VI) ................................................... 49
2. Interaction between diltiazem or mibefradil and oral methylprednisolone (Study III) ... 533. Interaction between grapefruit juice and oral methylprednisolone (Study V) ................ 55
DISCUSSION ........................................................................................................................ 57
1. Methodological considerations....................................................................................... 572. Interaction between CYP3A4 inhibitors and glucocorticoids......................................... 58
2.1. Methylprednisolone ................................................................................................ 582.2. Prednisolone ........................................................................................................... 602.3. Dexamethasone....................................................................................................... 61
3. General discussion.......................................................................................................... 62
SUMMARY AND CONCLUSIONS.................................................................................... 65
ACKNOWLEDGEMENTS.................................................................................................. 67
REFERENCES...................................................................................................................... 69

6
ABBREVIATIONS
ACTH adrenocorticotropin hormoneANOVA analysis of varianceAUC(t-ti) area under the plasma drug concentration-time curve from t to ti hoursCBG corticosteroid-binding globulinCl systemic clearanceCPMP committee for proprietary medicinal productsCmax peak concentrationCRH corticotropin-releasing hormoneCV coefficient of variationCYP cytochrome P450DDD defined daily doseECG electrocardiogramEDTA ethylendiaminetetraacetic acidGRE glucocorticoid regulatory elementHIV human immunodeficiency virusHMG-CoA 3-hydroxy-3-methylglutaryl-coenzyme AHPA-axis hypothalamic-pituitary-adrenal axisHPLC high-performance liquid chromatographyIBW ideal body weightkel elimination rate constantLC/MS/MS liquid chromatography-ionspray-tandem mass spectrometryMDR multiple drug resistanceMI-complex metabolic intermediate complexP-gp P-glycoproteinQT QT interval in electrocardiogramQTc heart rate-corrected QT intervalSD standard deviationSEM standard error of meantmax time of peak concentrationt½ elimination half-lifeVd volume of distribution

7
LIST OF ORIGINAL PAPERS
This thesis is based on the following articles which will be referred to in the textby the Roman numerals I to VI.
I Varis T, Kaukonen K-M, Kivistö KT, Neuvonen PJ. Plasmaconcentrations and effects of oral methylprednisolone are considerablyincreased by itraconazole. Clin Pharmacol Ther 1998;64:363-368
II Varis T, Kivistö KT, Backman JT, Neuvonen PJ. Itraconazole decreasesthe clearance and enhances the effects of intravenously administeredmethylprednisolone in healthy volunteers. Pharmacol Toxicol1999;85:29-32
III Varis T, Backman JT, Kivistö KT, Neuvonen PJ. Diltiazem andmibefradil increase the plasma concentrations and greatly enhance theadrenal-suppressant effect of oral methylprednisolone. Clin PharmacolTher 2000;67:215-221
IV Varis T, Kivistö KT, Neuvonen PJ. The effect of itraconazole on thepharmacokinetics and pharmacodynamics of oral prednisolone. Eur J ClinPharmacol 2000;56:57-60
V Varis T, Kivistö KT, Neuvonen PJ. Grapefruit juice can increase theplasma concentrations of oral methylprednisolone. Eur J Clin Pharmacol2000;56:489-493
VI Varis T, Kivistö KT, Backman JT, Neuvonen PJ. The cytochrome P4503A4 inhibitor itraconazole markedly increases the plasma concentrationsof dexamethasone and enhances its adrenal-suppressant effect. ClinPharmacol Ther, in press
The original publications are reprinted with permission of the copyright holders.

ABSTRACT
8
ABSTRACT
The aim of this thesis was to investigate the effect of CYP3A4 inhibitors on thepharmacokinetics and pharmacodynamics of systemic glucocorticoids; theeffect of itraconazole on oral and intravenous methylprednisolone, on oralprednisolone, and on oral and intravenous dexamethasone; and the effect ofdiltiazem, mibefradil, and grapefruit juice on oral methylprednisolone. Theeffect of route of administration of methylprednisolone and dexamethasone onthe possible interaction with itraconazole was also evaluated.
A total of 8 to 10 healthy subjects were enrolled in each study. Each was adouble-blind, randomised, placebo-controlled, cross-over study, except thegrapefruit juice-methylprednisolone interaction study, with its open, randomisedcross-over design. The pretreatment was followed by a single dose ofglucocorticoid, blood samples were collected and plasma drug and cortisolconcentrations determined.
The use of itraconazole enhanced the adrenal-suppressant effect of bothmethylprednisolone and dexamethasone. Itraconazole increased the AUC oforal and intravenous methylprednisolone and oral and intravenousdexamethasone 3.9-fold (p <0.001), 2.6-fold (p <0.001), 3.7-fold (p <0.001),and 3.3-fold (p <0.001), respectively. In contrast, itraconazole increased theAUC of oral prednisolone by only 24% (p <0.001), and the adrenal-suppressanteffect of prednisolone was slightly enhanced. Both diltiazem and mibefradilincreased the AUC of oral methylprednisolone (2.6-fold and 3.8-fold; p <0.001)and its adrenal-suppressant effect was greatly enhanced. Grapefruit juiceincreased the AUC of oral methylprednisolone by 75% (p <0.001), but itsadrenal-suppressant effect was not significantly affected. The route ofmethylprednisolone and dexamethasone administration may have only a minoreffect on the extent of itraconazole-methylprednisolone and itraconazole-dexamethasone interactions. Despite the differences seen in pharmacokineticinteractions, the extent of the adrenal-suppressant effect was similar after asingle oral or intravenous dose of these glucocorticoids.
In conclusion, these interactions probably result from inhibition of the CYP3A4enzyme both during the first-pass and the elimination phases. Inhibition of theP-glycoprotein may have contributed to the interactions of methylprednisoloneand dexamethasone. Care should be taken if methylprednisolone ordexamethasone is used concomitantly with a potent inhibitor of CYP3A4 over along period. However, the itraconazole-prednisolone and grapefruit juice-methylprednisolone interactions are probably of limited clinical significance.

INTRODUCTION
9
INTRODUCTIONThe cytochrome P450 (CYP) enzymes catalyse the oxidative metabolism ofmany drugs (Pelkonen et al 1998, Dresser et al 2000). Among the human drug-metabolising CYP enzymes, CYP3A4 is the most abundant enzyme both in theliver and intestine (Shimada et al 1994, de Waziers et al 1990).
Because several drugs and also dietary factors can inhibit the catalytic activity ofCYP3A4 (Pelkonen et al 1998, Evans 2000), inhibitors of CYP3A4 can increaseexposure to drugs metabolised mainly via this pathway. Drug-drug interactionscan therefore significantly enhance both the therapeutic effects of the drug andalso the risk of adverse effects (Dresser et al 2000). For example, concomitantuse of itraconazole with commonly used drugs like terfenadine, midazolam, andsimvastatin is associated with potentially serious adverse effects (Pohjola-Sintonen et al 1993, Olkkola et al 1994, Segaert et al 1996). However, enzymeinhibition may also result in therapeutic failure if the drug is administered as aninactive prodrug and its metabolic activation is inhibited (Lin et Lu 1998).
Many important drug-drug interactions have been reported after the drug inquestion has received marketing approval. Some interactions have been seriousenough to cause limitations in the use of the drug or even its withdrawal from themarket. Concern for the safe use of drugs has increased, and the drug interactionpotential of a new investigational compound has to be evaluated beforemarketing approval (Note for guidance on the investigation of drug interactions,CPMP, 1997). In vitro studies are used, for example, to give information aboutspecific CYP enzymes involved in the metabolism of new compounds andpossible drug-drug interactions. However, an interaction observed in an in vitrostudy should be confirmed in relevant, controlled in vivo studies.
Intravenous and oral glucocorticoids have been widely used for many decades invarious clinical conditions for their anti-inflammatory and immunosuppressiveproperties (Hench et al 1949, Swartz and Dluhy 1978, Mies et al 1998). Systemicglucocorticoids are eliminated mainly by metabolism (Vree et al 1999, Begg et al1987, Tomlinson et al 1996), and inhibition of the catalytic activity of CYP3A4by other drugs or dietary constituents can affect their elimination. CYP3A4enzyme, however, probably has a dissimilar role in the metabolism of differentglucocorticoids.
The purpose of the present study was to investigate the potential of CYP3A4inhibitors to affect the pharmacokinetics and adrenal-suppressant effect ofsystemic methylprednisolone, prednisolone, and dexamethasone in controlledclinical studies in healthy volunteers. Furthermore, the effect of the route ofglucocorticoid administration on the interaction potential was evaluated.

REVIEW OF THE LITERATURE
10
REVIEW OF THE LITERATURE
1. Drug metabolism1.1. Principles of drug metabolismMost pharmacologically active compounds are lipophilic because lipophilicityenables them to pass through biological membranes. In order to be excreted fromthe body, these lipophilic compounds must be biotransformed into more water-soluble metabolites (Meyer 1996). Metabolites generated are generallypharmacologically less active than the parent drug or even inactive, but themetabolites may also show enhanced pharmacological activity. Furthermore, themetabolism of drugs can produce harmful reactive and toxic products, which maybe responsible for various forms of toxicity, e.g., hepatotoxicity (Nelson et al1996, Mitchell et al 1974).
One major enzyme system involved in drug biotransformation is the cytochromeP450 (CYP) monooxygenase system. Other important enzymes include esterases,epoxide hydrolase, and conjugating enzymes such as glucuronosyltransferasesand sulfotransferases. These metabolic reactions are divided into functionalisation(phase I) and conjugation (phase II) reactions (Meyer 1996). The phase Ioxidative biotransformations catalysed by the hepatic and extrahepatic CYPenzymes include oxidation, reduction, and hydrolysis. Oxidative metabolismrepresents a major route of elimination for many drugs (Lin and Lu 1998).
During phase II reactions, the parent drug or its phase I metabolite is conjugatedwith an endogenous substrate. The phase II conjugation reactions involveglucuronidation, sulphation, acetylation, and conjugation with glutathione. Theconjugated metabolites are usually highly water-soluble and are readily excretedinto urine and bile, which are the main routes of elimination for most drugs(Meyer 1996).
1.2. The CYP enzymesCYP enzymes are a family of enzymes which are found not only in human beingsbut also in bacteria, fungi, plants, and animals (Nelson et al 1999). CYP enzymesare membrane-bound, localised to the endoplasmic reticulum (Meyer 1996). Theyare responsible for most of the oxidative metabolism of both xenobiotics andendogenous substrates (Dresser et al 2000, Lin and Lu 1998).
The nomenclature of all CYP enzymes is based on their amino acid sequence(Nelson et al 1996). The CYP enzymes within the same family are designated byan Arabic numeral (e.g., CYP3) and share more than 40% identity in the amino

REVIEW OF THE LITERATURE
11
acid sequence. The families are further divided into subfamilies, with theenzymes within a subfamily designated by a capital letter (e.g., CYP3A) andsharing more than 55% identity in the amino acid sequence. Finally, an Arabicnumerical after the letter denotes each individual isoenzyme (e.g., CYP3A4).
Functionally, the CYP enzymes are divided into two major classes, those with aspecific role e.g, in steroid biosynthesis, bile and arachidonic acid metabolism(CYP4, CYP5, and higher) and those primarily involved in the metabolism ofxenobiotics such as drugs, anti-oxidants, and chemicals (CYP1, CYP2, CYP3)(Nelson et al 1999). Many of these enzymes are involved in metabolism of a widerange of different substrates, e.g., endogenous compounds, chemicals, and naturalplant products (Nelson et al 1996). Among the 17 CYP gene families reported inhumans by Nelson in 1999, three families: CYP1, CYP2, and CYP3, areimportant for hepatic drug metabolism (Wrighton et al 1996). The mostprominent CYP isoenzymes with regard to the number of substrate drugs areCYP3A4 and CYP2D6, with a smaller number of drugs metabolised by CYP2C9,CYP2C19, CYP1A2, and CYP2E1 (Wrighton and Stevens 1992, Meyer 1996).CYP enzymes are found mainly in the liver, which plays the major role in humandrug metabolism, but they are present in lower amounts in many extrahepatictissues (Krishna and Klotz 1994).
The expression and catalytic activity of CYP enzymes may vary greatly betweenindividuals, due to genetic, non-genetic endogenous (e.g. diseases), andenvironmental (drugs, diet) factors (Pelkonen et al 1998). Genetic polymorphismgreatly affects the activity of some CYP enzymes, e.g., CYP2C19 and CYP2D6(Wrighton et al 1996, Nakamura et al 1985). Furthermore, some CYP enzymesare inducible by environmental factors such as drugs (Wrighton et al 1996,Pelkonen et al 1998). In addition, catalytic activity of CYP enzymes can beinhibited, or the CYP enzymes can be inactivated. Induction and inhibition ofCYP enzymes increase the intraindividual and interindividual variability of drugmetabolism. Examples of the substrates, inhibitors, and inducers of CYP3A4,CYP2D6, CYP2C9, CYP2C19, CYP1A2, and CYP2E1 isoenzymes are presentedin Table I (page 12).
1.3. CYP3A subfamilyThe CYP3A subfamily is involved in the metabolism of a large number ofendogenous and exogenous compounds, e.g., cortisol, testosterone, cyclosporin,midazolam, nifedipine, and simvastatin (Table I). Because approximately 40-50%of the drugs used in man are metabolised at least partly by CYP3A-mediated

REVIEW OF THE LITERATURE
12
Table I. Examples of substrates, inhibitors, and inducers of the main humanhepatic drug-metabolising CYP enzymes and their relative amounts of the totalhepatic CYP content. * indicates that the enzyme exhibits genetic polymorphism.Modified from Bertz and Granneman (1997) and Pelkonen et al (1998).
CYP enzyme Substrates Inhibitors Inducers
CYP2C19*< 5%
omeprazolemephenytoindiazepam
fluoxetinefluvoxamine
rifampicin
CYP2C8/9*∼ 20%
warfarinphenytoincerivastatin
sulphaphenazolefluconazole
rifampicin
CYP3A4∼ 30%
simvastatinmidazolamtriazolamnifedipinecyclosporincisapridetestosteroneethinylestradiolcortisolterfenadine
ketoconazoleitraconazolediltiazemverapamilgrapefruit juiceerythromycinchlarithromycin
rifampicinphenytoincarbamazepine
CYP1A2∼ 15%
caffeinetheophyllineclozapine
fluvoxamine smokingrifampicin
CYP2E1< 10%
ethanolhalothanechlorzoxazone
disulfiram ethanolisoniazide
CYP2D6*<5%
dextrometorphandebrisoquinecodeinemetoprolol
quinidinefluoxetine
not inducible

REVIEW OF THE LITERATURE
13
oxidation, it appears to be the most important drug-metabolising CYP subfamily(Thummel and Wilkinson 1998).
CYP3A consist of three isoenzymes, CYP3A4, CYP3A5, and CYP3A7(Wrighton and Stevens 1992, de Wildt et al 1999). Among these, CYP3A4 is themost abundant isoenzyme in adult liver, accounting for about 30% of total CYPcontent (Shimada et al 1994). CYP3A4 is shown to be present also e.g, in smallintestine, colon, and stomach (de Waziers et al 1990). CYP3A5 is found in 25-30% of adult livers (Wrighton and Stevens 1992). More recently, however,CYP3A5 protein was detected in 74% of livers examined (Jounaidi et al 1996).CYP3A5 is the major isoenzyme in the lung tissue, in which CYP3A4 isexpressed in only the minority of cases (Kivistö et al 1996a, Anttila et al 1997).Furthermore, CYP3A5 is expressed in the small intestine in 70 to 100% ofindividuals (Lown et al 1994, Kivistö et al 1996b).
CYP3A5 displays 84% similarity in its amino acid sequence to CYP3A4(Aoyama et al 1989). Although the substrate specificity of CYP3A5 is similar tothat of CYP3A4, differences exist in these isoensymes’ catalytic properties(Wrighton and Stevens 1992). In contrast to CYP3A4, CYP3A5 does not appearto metabolise erythromycin, quinidine, or 17α-ethinylestradiol at significant rates(Wrighton et al 1990). Furthermore, testosterone, cortisol, and nifedipine aremetabolised by CYP3A5 at a slower rate than by CYP3A4 (Wrighton et al 1990).
CYP3A7 is the major CYP isoenzyme in the fetal liver, accounting for up to 50%of total CYP content (Kitada et al 1985, Wrighton and VandenBranden 1989). Inthe adult liver, CYP3A7 mRNA is found in about 55 to 85% of samples, althoughat lower levels than CYP3A4 (Schuetz et al 1994, Hakkola et al 1994).
In vitro, the expression and catalytic activity of hepatic CYP3A4 can vary asmuch as 6-fold and 30-fold, respectively (Shimada et al 1994, Yun et al 1993).Despite this large interindividual variation in CYP3A4 activity, CYP3A4 does notappear to be subject to genetic polymorphism, in contrast to CYP2C9, CYP2C19,and CYP2D6 (Aithal et al 1999, Gaedigk 2000). Recent studies have, however,identified a mutation in the CYP3A4 that has been linked to tumour stage andgrade at the time of diagnosis of prostatic cancer (Rebbeck et al 1998)Furthermore, this variation may influence the risk of treatment-related leukemias(Felix et al 1998). A recent study has found this promoter region polymorphismnot to affect the CYP3A4-mediated metabolism of erythromycin or thepharmacokinetics of nifedipine (Ball et al 1999). Sata et al (2000) have identifiedother variant alleles of CYP3A4 designated CYP3A4*2 and CYP3A4*3;CYP3A4*2, which is present at a frequency of 2.7% in a white Finnishpopulation, showed in vitro altered catalytic activity compared with that of wild-type P450 (Sata et al 2000). However, the impact of these mutations on CYP3A4

REVIEW OF THE LITERATURE
14
activity and on the pharmacokinetics of CYP3A4 substrates needs furtherevaluation.
Erythromycin is metabolised about 25% more rapidly by microsomes fromfemale than from male human liver (Hunt et al 1992). Furthermore, midazolam iscleared 20-40% faster by women than by men (Harris et al 1995). Not all studies,however, have found a sex-related difference in CYP3A4 activity (Schmucker etal 1990, Shimada et al 1994). Neither menstrual-cycle phase, smoking, norethanol consumption appear to influence CYP3A4 activity (Kharasch et al 1999,Hunt et al 1992). Sotaniemi et al (1997) have suggested that liver CYP contentdeclines after age 40. However, two other studies have failed to find anyrelationship between age and liver CYP3A activity (Hunt et al 1992, Blanco et al2000). The CYP3A4 activity is decreased in severe liver disease (Pelkonen et al1998), and celiac disease can decrease CYP3A4 activity in gut (Lang et al 1996).
During the last few years research interest has increasingly focused on drugmetabolism in the intestinal wall. This location makes suitable to play animportant role in first-pass metabolism and in limiting the bioavailability of orallyadministered drugs (Hoppu et al 1991, Dresser et al 2000). Many CYP enzymesincluding CYP3A4, CYP3A5, and CYP2D6 appear to be expressed in theintestinal wall (de Waziers et al 1990, Kivistö et al 1996b). After the liver, thesmall intestine contains the highest amount of CYP3A4 (de Waziers et al 1990).Hepatic and intestinal CYP3A4 appear to be regulated independently (Lown et al1994), and the content of intestinal CYP3A4 varies 11-fold and its catalyticactivity 6-fold among individuals (Lown et al 1994). This finding may contributeto the interindividual variability in disposition of CYP3A4 substratesadministered orally. The small intestine is an important place for first-passmetabolism of oral drugs. Interestingly, the intestinal first-pass metabolism ofsome oral drugs, for example, midazolam and cyclosporin, may even exceed thatoccurring in the liver (Thummel et al 1996, Wu et al 1995).
1.4. Drug interactionsDrug interaction can occur by several different mechanisms, and these can beeither harmful or beneficial. Pharmacodynamic interactions cause changes in theeffects of drugs. For example, alcohol enhances the sedative effect ofbenzodiazepines (Seppälä et al 1982). Pharmacokinetic interactions can takeplace at the level of drug absorption, distribution, metabolism, and excretion. Forexample, antacids containing aluminium, calcium, or magnesium and ironproducts reduce the absorption of tetracyclines, resulting in reduced antibacterialeffects (Neuvonen et al 1970). An example of an interaction due to changes in

REVIEW OF THE LITERATURE
15
excretion is reduced excretion of penicillin by probeniside (Kampmann et al1972).
Enzyme induction and inhibition affect drug metabolism. CYP-enzyme inductionusually results from an increased amount of enzyme protein, and a period of timeis needed before enzyme induction becomes clinically significant (Pelkonen et al1998). Enzyme induction increases the metabolism of drugs that are metabolisedby induced enzymes, leading to decreased plasma drug concentrations (Lin andLu 1998). For example, rifampicin can greatly decrease the plasma concentrationsof triazolam and abolishes the effect of triazolam (Villikka et al 1997).
CYP enzyme inhibition usually occurs rapidly, even after a single dose ofinhibitor (Pelkonen et al 1998). Enzyme inhibition can be competitive or non-competitive (Lin and Lu 1998). In case of competitive inhibition, enzymeinhibitor competes the active site of CYP enzyme. Competitive inhibition isprobably the most common mechanism responsible for the drug-drug-interactions. For example, itraconazole and indinavir, a potent humanimmunodeficiency virus (HIV) protease inhibitor, are competitive inhibitors ofCYP3A4 (Back and Tjia 1991, Fitzsimmons and Collins 1997). Competitiveinhibitors are often substrates of the CYP isoenzyme whose activity they inhibit.However, this is not always the case. Quinidine is a substrate for CYP3A4, but isa potent reversible inhibitor of CYP2D6 (Guengerich et al 1986, Nielsen et al1990).
Many drugs, including the macrolide antibiotics troleandomycin anderythromycin, undergo metabolic activation by CYP3A4 to form inhibitorymetabolites (Pessayere et al 1982, Danan et al 1981). These metabolites can formcomplexes with the enzyme called metabolic intermediate (MI) complexes,resulting in a functionally inactive CYP3A4 enzyme. Ethinylestradiol andgrapefruit juice, for example, are classified as mechanism-based inactivators orsuicide substrates, because they cause irreversible inactivation of the enzyme(Guengerich 1988, Lown et al 1997).
In addition to the hepatic, the intestinal CYP3A4 can be induced by such drugs asrifampicin (Kolars et al 1992) and inhibited by known CYP3A4 inhibitors such asketoconazole (Tsunoda et al 1999). The orally administered drug is exposed toCYP3A4-mediated first-pass metabolism, and inhibition of CYP-mediatedmetabolism during the first-pass can result in increased bioavailability. Afterintravenous administration, the drug bypasses the first-pass metabolism, and itssystemic bioavailability is complete. Thus, the effect of enzyme inhibitiondepends on whether the drug is administered orally or intravenously.

REVIEW OF THE LITERATURE
16
Drugs can be classified by wheather their hepatic clearance is enzyme-limited(low; hepatic extraction ratio < 0.3) or flow-limited (high; hepatic extraction ratio> 0.7) (Lin and Lu 1998). If the drug is metabolised mainly in the liver, decreasein metabolic activity by enzyme inhibition results in an almost proportionalincrease in the area under the plasma drug concentration-time curve (AUC) of anorally administered drug, regadless whether the drug is a high- or low-clearancedrug (Lin and Lu 1998). However, after intravenous administration enzymeinhibition significantly affects the AUC of a low-clearance drug, whereas theAUC of high-clearance drug is only slightly affected (the hepatic clearance islimited by the hepatic blood flow) (Lin and Lu 1998).
1.5. Drug interactions mediated by inhibition of CYP3A4The drug-drug interactions mediated through CYP3A4 result either frominduction or from inhibition of this enzyme (Lin and Lu 1998, de Wildt et al1999). Both in vitro and in vivo studies have demonstrated that CYP3A4 isinhibited, for example, by the azole antimycotics ketoconazole and itraconazole(Back and Tjia 1991, Varhe et al 1994, Olkkola et al 1994), the macrolideantibiotics troleandomycin, erythromycin, roxithromycin, and clarithromycin(Gascon et al 1991, Fleming et al 1992, Olkkola et al 1993, Backman et al 1994a,Bailey et al 1996, Yeates et al 1996), the calcium-channel antagonists diltiazem,verapamil, and mibefradil (Backman et al 1994b, Kantola et al 1998a, Welker etal 1998), and by grapefruit juice (Bailey et al 1991, Hukkinen et al 1995, Lilja etal 1998a).
Many drugs from different therapeutic areas are substrates for CYP3A4, which ispresent in high amounts in both the liver and the intestine (Shimada et al 1994, deWaziers et al 1990). Use of a CYP3A4 inhibitor can decrease drug metabolismboth during the first-pass and the elimination phases. Decreased drug eliminationmay enhance a drug’s therapeutic and also its toxic effects. There are severalexamples of an interaction due to inhibition of CYP3A4 being clinicallyimportant. QT-interval prolongation or torsades de pointes ventricular arrhytmiahave been reported with concomitant use of a CYP3A4 inhibitor with terfenadineor cisapride (Pohjola-Sintonen et al 1993, van Haarst et al 1998, Piquette 1999).The risk of rhabdomyolysis appeared to increase with co-administration of aCYP3A4 inhibitor and lovastatin or simvastatin (Neuvonen and Jalava 1996,Neuvonen et al 1998, Lees and Lees 1995). Furthermore, concomitant use ofdrugs such as ketoconazole, itraconazole, or verapamil increases the sedativeeffects of midazolam and triazolam (Olkkola et al 1994, Backman et al 1994b,Varhe et al 1994).

REVIEW OF THE LITERATURE
17
Drug-drug interactions can also prove useful. The reduction in cyclosporin dosagewould result in extensive cost savings (Jones et al 1997, Martin et al 1999).Concomitant use of ketoconazole with cyclosporin allows the reduction ofcyclosporin dosage by 70 to 85% in renal-, cardiac-, and liver-transplant patients.
1.6. CYP3A4 inhibitors studied
1.6.1. ItraconazoleItraconazole is a triazole antimycotic introduced onto the market in the early1990’s. Itraconazole inhibits the fungal CYP enzyme 14-demethylase, therebyinhibiting the conversion of lanosterol to ergosterol, the primary sterol in fungalmembranes. The azole antifungals are primarily fungistatic (Gupta et al 1994).
Itraconazole is used in treatment and prevention of systemic fungal infections andin oral therapy of superficial dermatomycoses such as onychomycosis (Gupta1994, Huijgens et al 1999, Harari 1999, Venkatakrishnan et al 2000). Therecommended daily dose is 100 to 400 mg for superficial infections, but doses upto 800 mg/day may be needed for treatment of systemic mycoses (Sanchez et al1995). Itraconazole is usually well tolerated; incidence of adverse effects is 7%,rising to 12.5% with therapy longer than one month. The most common adverseeffects are gastrointestinal symptoms, headache, dizziness, and rash. Furthermore,elevation of transaminases has occurred in 0.3 to 5% of patients (Gupta et al1994). Unlike ketoconazole, itraconazole has no significant effect on adrenalfunction and plasma cortisol concentration at doses up to 400 mg daily (Gupta etal 1994, Phillips et al 1987, Queiroz-Telles et al 1997).
Itraconazole is well absorbed when administered with a meal, but its absorption isreduced during fasting (Van Peer et al 1989). The time of the peak concentration(tmax) in plasma ranges from 1.5 to 4 h (Hardin et al 1988). It is highly bound toplasma proteins (Grant and Clissold 1989, Gupta et al 1994). About 95% is boundto plasma protein, mainly albumin, 5% to blood cells, and only 0.2 to 3% remainsfree in plasma (Arredondo at al 1995, Grant and Clissold 1989, Backman et al2000). The volume of distribution (Vd) of itraconazole is about 11 l/kg, whichindicates extensive tissue distribution (Heykants et al 1989). Due to itslipophilicity, concentrations of itraconazole are 3 to 10 times as high in skin, and10 to 20 times as high in liver as in plasma (Heykants et al 1989, Backman et al2000). Itraconazole exhibits non-linear pharmacokinetics at therapeutic doses.The mean AUC increases more than proportionally with dose, suggestingsaturable metabolism (Hardin et al 1988, Van Peer et al 1989). The eliminationhalf-life (t½) of itraconazole after a single dose ranges from 15 h to 25 h and atsteady state from 30 h to 50 h (Heykants et al 1989).

REVIEW OF THE LITERATURE
18
Itraconazole has a significant first-pass metabolism and is eliminated bymetabolism. More than 30 metabolites have been found, but onlyhydroxyitraconazole possesses clinically significant activity (Heykants et al1989). CYP3A4 appears to have an important role in metabolism of itraconazole(Ducharme et al 1995a). About 54% of an oral dose is excreted in faeces and 35%in urine (Heycants et al 1989).
It had been suggested that itraconazole is a selective inhibitor of fungal CYPenzymes and will not inhibit drug metabolism in humans (Grant and Clissold1989). However, it is known today that itraconazole inhibits the metabolism ofmany CYP3A4 substrates and causes many clinically significant drug-druginteractions. Itraconazole has increased the AUC of midazolam and triazolam 11-and 27-fold, respectively and markedly enhanced their hypnotic effects (Olkkolaet al 1994, Varhe et al 1994). In addition, itraconazole has increased the AUC ofbuspirone about 20-fold and increased its pharmacodynamic effects (Kivistö et al1997). Plasma concentrations of lovastatin, lovastatin acid, simvastatin, andsimvastatin acid are greatly increased by itraconazole (Neuvonen and Jalava1996, Kivistö et al 1998, Neuvonen et al 1998). Importantly, concomitant use ofitraconazole and certain HMG-CoA reductase inhibitors may increase risk ofmyalgia, and cases of rhabdomyolysis have been reported (Lees and Lees 1995,Segaert et al 1996). Furthermore, itraconazole inhibits the metabolism ofterfenadine, resulting in increased risk for torsades de pointes ventriculartachycardia (Pohjola-Sintonen et al 1993).
Itraconazole potently inhibits P-glycoprotein (P-gp) in vitro and probably also invivo (Miyama et al 1998, Kurosawa et al 1996, Backman et al 2000). P-gp andCYP3A show substrate overlap (Wacher et al 1995), although the CYP3A4substrate midazolam, for instance, is not a substrate of P-gp (Kim et al 1999). Therelative role of P-gp- and CYP3A-inhibition to the effect of itraconazole on thepharmacokinetics of dual substrates of P-gp and CYP3A is unclear(Venkatakrishnan et al 2000).
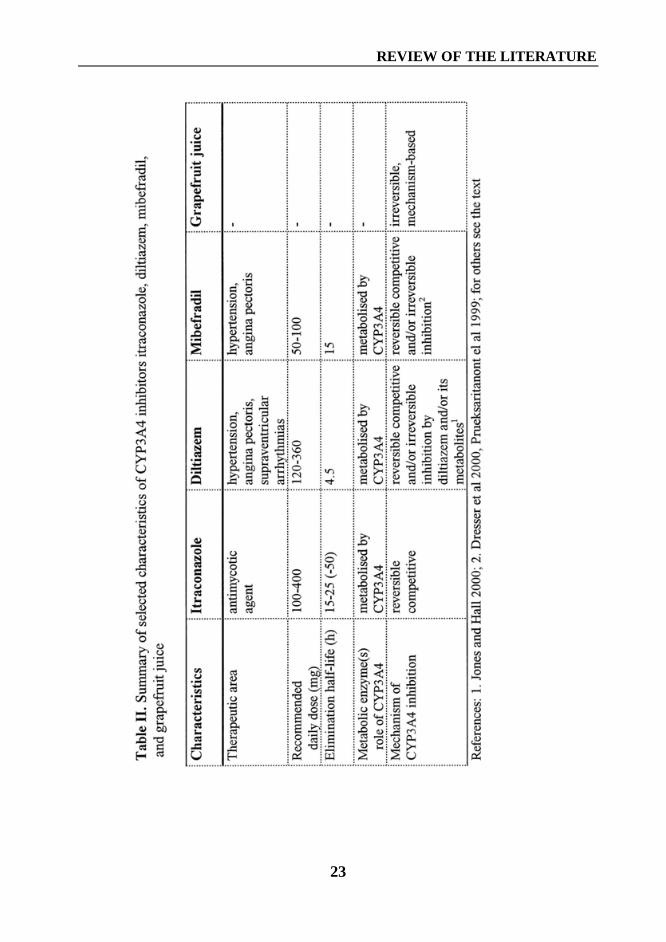
A summary of selected characteristics of itraconazole, diltiazem, mibefradil, andgrapefruit juice is shown in Table II (page 23).
1.6.2. DiltiazemDiltiazem is a benzothiazepine calcium-channel blocker used in treatment ofangina pectoris, hypertension, and supraventricular arrhythmias (Dollery 1999a,Buckley et al 1990). Diltiazem inhibits the influx of calcium into vascular smoothmuscle and myocardial cells through ”slow” calcium channels and therebyreduces the tone of coronary and peripheral arterioles (Jones and Hall 2000). Therecommended oral daily dose of diltiazem ranges from 120 to 360 mg divided

REVIEW OF THE LITERATURE
19
into one to three doses (Buckley et al 1990). Diltiazem is usually well tolerated;the incidence of adverse effects is less than 2% (McGraw et al 1982). The mostcommon adverse effects are related to the vasodilator properties of diltiazem, likeflushing, headache, ankle oedema, and hypotension.
Diltiazem is almost completely absorbed after oral administration; it undergoes asignificant first-pass metabolism, resulting in a bioavailability of only about 40%(Kelly and O’Malley 1992). The peak plasma concentration (Cmax) is reachedwithin 1.5 h with fast-release formulation (Buckley et al 1990).
Diltiazem is about 80 to 90% bound to plasma proteins, mainly albumin (Buckleyet al 1990). The Vd of diltiazem is about 5.0 l/kg (Hermann et al 1983). It islargely eliminated through CYP3A4-mediated metabolism (Pichard et al 1990,Sutton et al 1997). About 70% of the dose is excreted into the urine and 17% intothe faeces (Höglund and Nilsson 1988). Metabolites of diltiazem possesspharmacologic activity, but their role in therapeutic effects is unclear (Jones andHall 2000). The t½ of orally administered diltiazem averages about 4.5 h (range 2-11 h) (Buckley et al 1990). Multiple oral dosing with diltiazem results inprolongation of its elimination, which may be due to inhibition of diltiazemmetabolism by diltiazem itself or by accumulated metabolites (Tsao et al 1990).
The ability of diltiazem to inhibit microsomal drug oxidation was first reported byRenton in 1985. More recently, in a study in human liver microsomes, bothdiltiazem and its metabolites inhibit CYP3A4 (Sutton et al 1997). However, theN-desmethyl and N,N-didesmethyl metabolites were much more potent inhibitorsof CYP3A4 than the parent drug itself (Sutton et al 1997, Jones and Hall 2000).Thus, the metabolites of diltiazem probably contribute to enzyme inhibition anddrug-drug interactions in vivo (Sutton et al 1997, Jones and Hall 2000).
In humans, the plasma concentrations of several CYP3A4 substrates such ascyclosporin, midazolam, triazolam, and simvastatin are increased by diltiazem(Pochet and Pirson 1986, Brockmöller at al 1990, Backman et al 1994b, Varhe etal 1996, Mousa et al 2000). Furthermore, diltiazem increases the concentrationsof carbamazepine, resulting in its enhanced neurotoxicity (Brodie and MacPhee1986, Eimer and Carter 1987). In addition, diltiazem has been shown to increasethe metabolic ratio of debrisoquine, indicating its potential to inhibit the CYP2D6enzyme (Sakai et al 1991). Because diltiazem does not significantly affect hepaticblood flow (Bauer et al 1986), the changes in drug metabolism are most probablydue to the inhibition of CYP enzymes by diltiazem and/or its metabolites.
Diltiazem is also an inhibitor of P-gp (Cornwell et al 1987), and may therebyaffect the pharmacokinetics of substrates of P-gp. In fact, diltiazem can prolongthe t½ and decrease the Cl of digoxin, which is actively secreted by P-gp in renaltubular cells (Rameis et al 1984, de Lannoy and Silverman 1992).

REVIEW OF THE LITERATURE
20
1.6.3. MibefradilMibefradil belongs to a new class of calcium blockers; unlike traditional calciumblockers, it selectively blocks T-type (transient, low-voltage-activated) calciumchannels (Welker et al 1998). Mibefradil can thus serve as an oral treatment forhypertension and stable angina pectoris. It dilates the peripheral and coronaryvessels, which results in a reduction in arterial pressure and peripheral vascularresistance, and an increase in coronary artery blood flow (Welker et al 1998).Mibefradil reduces heart rate dose-dependently and does not cause reflextachycardia, as is commonly seen with dihydropyridine calcium blockers (Welkeret al 1998). The recommended dosage is 50 to 100 mg once daily.
About 99.5% of mibefradil is bound to plasma proteins, mainly to α1-acidglycoprotein (Welker et al 1998). The Vd of mibefradil in the steady state rangesfrom 130 l to 200 l (Dollery 1999b, du Souich et al 2000). Mibefradil exhibitsdose- and time-dependent pharmacokinetics that can be explained by partialsaturation of the metabolism or by inhibition of mibefradil metabolism bymibefradil itself or by accumulated metabolites (du Souich et al 2000). Its t½
averages 15 h, with a range of about 10 to 38 h (Dollery 1999b, Welker et al1998, du Souich et al 2000).
Mibefradil is eliminated largely by metabolism, and excreted mainly in the faeces(75%), to a lesser extent in the urine (25%); less than 3% is excreted unchangedin the urine. The metabolism of mibefradil is mediated by two main pathways,CYP3A4-mediated oxidation and hydrolysis of the ester side-chain. Suchhydrolysis produces Ro 40-5966, which is, however, the major plasma metabolite,possessing about 10% of the pharmacodynamic activity of the parent drug(Welker et al 1998). A total of some 30 metabolites of mibefradil have beendetected (Wiltshire et al 1997).
In vitro studies on human liver microsomes have shown mibefradil to be a potentinhibitor of CYP3A4 and CYP2D6 (Welker et al 1998, Wang et al 1999, Bohetset al 2000). In vivo, mibefradil has increased the total AUC of CYP3A4 substratescyclosporin and triazolam 2.7-fold and 9-fold, respectively, and enhanced thesedative effects of triazolam (Welker et al 1998, Backman et al 1999). Inaddition, mibefradil raises the plasma concentration of terfenadine and the meanQTc interval, resulting in an increased risk for cardiac arrhythmias (Welker et al1998). Mibefradil also considerably increases plasma concentrations ofmetoprolol, a substrate of CYP2D6 (Welker et al 1998). Besides inhibitingCYP3A4, mibefradil also inhibits P-gp in vitro (Wandel et al 2000). However, inhumans mibefradil raises plasma concentrations of digoxin only atsupratherapeutic levels (Siepmann et al 1995, Welker et al 1998).

REVIEW OF THE LITERATURE
21
Mibefradil was withdrawn from the market in 1998, within its first year afterlaunch, because of severe interactions with other drugs (Ellison 1998, Mullins etal 1998, Schmassmann-Suhijar et al 1998, Krähenbühl et al 1998, Anonymous1998).
1.6.4. Grapefruit juiceA finding in 1989 in an ethanol-felodipine interaction study suggested thatgrapefruit juice may increase the bioavailability of felodipine (Bailey et al 1989).This observation was confirmed in a subsequent study, in which the mean AUCof felodipine was increased 284% by grapefruit juice (Bailey et al 1991).
Grapefruit juice has been shown also to raise plasma concentrations of otherdihydropyridine calcium-channel blockers such as nifedipine, nisoldipine, andnitrendipine (Bailey et al 1991, Bailey et al 1993a, Soons et al 1991). Grapefruitjuice has increased the AUC of orally administered midazolam by 52% andenhanced its effects (Kupferschmidt et al 1995). However, Vanakoski et al (1996)found no interaction between grapefruit juice and oral midazolam. In addition,grapefruit juice has increased the plasma concentrations of such drugs ascyclosporin, terfenadine, and cisapride (Ducharme et al 1995b, Benton et al 1996,Kivistö et al 1999). Grapefruit juice appears to affect particularly those drugs withan extensive CYP3A4-mediated first-pass metabolism. It has greatly increased theAUC of of simvastatin (16-fold) and simvastatin acid (7-fold), the AUC oflovastatin (15-fold) and lovastatin acid (5-fold), and the AUC of buspirone (9-fold) (Lilja et al 1998a, Kantola et al 1998b, Lilja et al 1998b).
A study by Lee et al (1996) suggests that grapefruit juice inhibits 11β-hydroxysteroid dehydrogenase that oxidises cortisol to cortisone. However,grapefruit juice does not interact with prednisone and it had no effect on themetabolism of prednisone to prednisolone, a reaction catalysed by 11β-hydroxysteroid dehydrogenase (Hollander et al 1995).
Graprefruit juice reduces intestinal CYP3A4 without affecting hepatic CYP3A4activity. Therefore, the most probable mechanism for grapefruit juice interactionsis inhibition of first-pass metabolism by reduced CYP3A4-mediated metabolismin the intestine (Lown et al 1997, Evans 2000). For example, although grapefruitjuice raises plasma concentrations of oral cyclosporin, felodipine, and midazolam,it has no effect on their clearance after intravenous administration (Ducharme etal 1995b, Lundahl et al 1997, Kupferschmidt et al 1995).
Because grapefruit juice does not reduce the amount of CYP3A4 mRNA, itappears to damage the CYP3A4 enzyme, resulting in accelerated degradation ofthe enzyme (mechanism-based inhibition) (Lown et al 1997, Bailey et al 1998a,

REVIEW OF THE LITERATURE
22
Evans 2000). This ihibition is consistent with the finding that grapefruit juiceaffects felodipine pharmacokinetics even when the juice is ingested 24 h beforethe drug (Lundahl et al 1995). Thus, the return of CYP3A4 activity would need denovo enzyme synthesis (Bailey et al 1998a).
Grapefruit juice-drug interactions can be greater after repeated consumption thanafter a single glass of grapefruit juice (Gross et al 1999, Kivistö et al 1999). TheAUC values of oral cisapride have been increased by about 40% and 145% after asingle glass of regular strength and after repeated doses of double-strengthgrapefruit juice, respectively (Gross et al 1999, Kivistö et al 1999). In addition,repeated consumption of double-strength grapefruit juice has increased the t½ oforal cisapride, buspirone, and atorvastatin (Kivistö et al 1999, Lilja et al 1998b,Lilja et al 1999). Increase in t½ indicates an effect on hepatic CYP3A4.Accordingly, high amounts of grapefruit juice have been reported to affect bothpresystemic and systemic metabolism of oral midazolam, a well-known marker ofCYP3A4 (Veronese et al 1999).
Because of the overlapping substrate specificity of CYP3A4 and P-gp (Wacher etal 1995), it is possible that grapefruit juice affects the intestinal P-gp. Lown et al(1997) found grapefruit juice to reduce the amount of intestinal CYP3A4 withoutaffecting intestinal P-gp expression. Recently grapefruit juice has beendemonstrated in vitro to inhibit vinblastine cellular efflux by P-gp (Takanaga et al1998). Eagling et al (1999) have reported grapefruit juice components to inhibitCYP3A4-mediated metabolism of saquinavir, and to modulate to a limited extentP-gp-mediated transport in vitro. Eagling et al (1999) suggest that the in vivoeffects of grapefruit juice on saquinavir pharmacokinetics most likely result frominhibition of CYP3A4 and only to a minor extent from modulation of P-gp.
It was originally thought that the flavonoid naringin, which is related to thetypical bitter taste of grapefruit juice, and is its most abundant flavonoid, isresponsible for the interaction of grapefruit juice with drugs (Bailey et al 1998a,Evans 2000). Both naringin and its aglucone naringenin inhibit the metabolism offelodipine and nifedipine in vitro (Bailey et al 1998b, Miniscalco et al 1992).Also, furanocumarins can cause a mechanism-based inhibition of CYP enzymesand a furanocumarin called 6’,7’-dihydroxybergamottin is present in grapefruitjuice (Evans 2000). However, in vivo studies show that neither naringin nor 6’,7’-dihydroxybergamottin is the component responsible for grapefruit juice-druginteractions in humans (Bailey et al 1993b and 1998b).
A summary of selected characteristics of itraconazole, diltiazem, mibefradil, andgrapefruit juice is shown in Table II.
REVIEW OF THE LITERATURE
23

REVIEW OF THE LITERATURE
24
1.7. CYP3A4 and P-glycoproteinThe P-gp in humans is a MDR-1 (multiple drug resistance) gene product, whichacts as a transmembrane efflux pump. It has an important role in the developmentof multidrug resistance by tumour cells exposed to cancer chemotherapeuticagents (Sikic et al 1997). P-gp is found in a number of human organs, includinggut, liver, adrenal gland, and kidney (Thiebaut et al 1987).
P-gp has an important role in elimination of drugs into bile and urine, and inlimiting the drug penetration into brain through the blood-brain barrier (Jalava etal 1997, Schinkel et al 1994 and 1995). P-gp and CYP3A4 are colocalised inintestine, where they act together to limit the drug absorption into portalcirculation and to decrease plasma drug concentrations. Both P-gp and CYP3A4are expressed and upregulated co-ordinately in human cancer cells (Schuetz et al1996). In addition, known CYP3A4 inducers like rifampicin also induce P-gp(Schuetz et al 1996) and many inhibitors of CYP3A4 e.g. itraconazole are alsoinhibitors of P-gp (Kurosawa et al 1996, Wacher et al 1995).
Many drugs metabolised by CYP3A4 are also substrates for P-gp (Wacher et al1995). Accordingly, the coadministration of two CYP3A4 substrates can result ininteractions that reflect inhibition of the CYP3A4-mediated metabolism, reducedefflux by P-glycoprotein or a combination of both effects (Kivistö et al 1995).
2. Glucocorticoids2.1. GeneralThe adrenal glands are endocrine organs essential for life. This was established in1849 by Addison, who described a fatal outcome in a patient with adrenaldestruction. The adrenal gland consists of cortex and medulla. The adrenal cortexsecretes corticosteroids, glucocorticoids that regulate carbohydrate and proteinmetabolism, the mineralocorticoid aldosterone that regulates fluid and electrolytebalance, and also precursors of the sex steroids. The main glucocorticoid inhumans is cortisol. (Schimmer and Parker 1996)
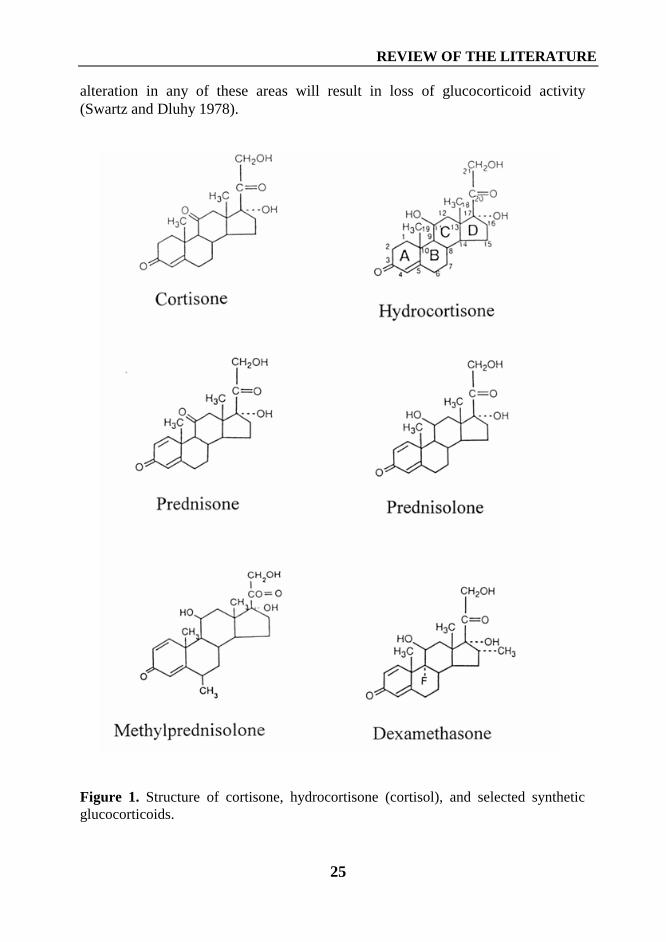
Chemical modifications of the cortisol molecule have generated syntheticglucocorticoid derivates. Because effects of natural and synthetic glucocorticoidsare mediated by the same receptor, the glucocorticoids do not differ in theireffects in humans. However, changes in chemical structure may cause changes inpotencies relative to glucocorticoid and mineralocorticoid activity, alterations inabsorption, protein binding, and metabolism (Schimmer and Parker 1996). Allglucocorticoids, cortisol and its synthetic analogues, are referred as to C21steroids (Figure 1). Some areas of the steroid molecule are critical (Figure 2), and
REVIEW OF THE LITERATURE
25
alteration in any of these areas will result in loss of glucocorticoid activity(Swartz and Dluhy 1978).
Figure 1. Structure of cortisone, hydrocortisone (cortisol), and selected syntheticglucocorticoids.

REVIEW OF THE LITERATURE
26
Figure 2. Groups/areas essential for anti-inflammatory activity of glucocorticoids(Adapted from Swartz and Dluhy 1978)
Synthetic corticosteroids are grouped according to their relative potencies in Na+
retention and anti-inflammatory effects in relation to cortisol (Table III). Basedon these potencies, synthetic corticosteroids are divided into glucocorticoids andmineralocorticoids, although some steroids traditionally classified asglucocorticoids, such as prednisolone, also possess some mineralocorticoidactivity. Synthetic glucocorticoids with long plasma half-lives also possess longbiological half-lives, reflecting the duration of the activity at tissue level (TableIII) (Swartz and Dluhy 1978, Schimmer and Parker 1996).
Table III. Relative potencies of some natural and synthetic glucocorticoids(Modified from Swartz and Dluhy 1978, Schimmer and Parker 1996)
Compound Equivalentpotency (mg)
Anti-inflammatory
potency
Na+-retainingpotency
Biologicalhalf-life (h)
Cortisol 20 1 1 8-12Cortisone 25 0.8 0.8 8-12
Prednisone 5 4 0.8 18-36
Prednisolone 5 4 0.8 18-36
Methylprednisolone 4 5 0.5 18-36
Dexamethasone 0.75 25 0 36-54

REVIEW OF THE LITERATURE
27
2.2. Regulation of cortisol secretionThe main glucocorticoid of human beings is cortisol (hydrocortisone). Thesecretion of cortisol follows a circadian diurnal variation with peakconcentrations in the morning and the lowest concentrations occurringapproximately at midnight (Jusko et al 1975, Meibohm et al 1999). Esteban et al(1991) suggest that cortisol secretion rather than metabolism is mainlyresponsible for the changes in plasma cortisol.
Hypothalamus, pituitary, and adrenal cortex are collectively called thehypothalamic-pituitary-adrenal (HPA) axis. This system is responsible for thediurnal rhythm in basal cortisol secretion and the increase in cortisol secretion instress, e.g., in infection or surgery (Naito et al 1992). The adrenocorticotropinhormone (ACTH), which is secreted by the anterior pituitary gland, stimulates thesecretion of cortisol. The secretion of ACTH is regulated by a hypothalamichormone, corticotropin-releasing hormone (CRH). Cortisol has negative feedbackeffects on the hypothalamus and anterior pituitary gland to reduce formation ofCRH and ACTH and thereby can control its own secretion.
2.3. PharmacodynamicsIntracellular action of glucocorticoids. Glucocorticoids enter the target cell andbind the glucocorticoid receptor in cytoplasm. This binding activates the receptorthat moves into the nucleus and binds to specific glucocorticoid regulatoryelements (GREs). Glucocorticoids mainly increase the expression of the targetgenes. They can, however, also suppress gene transcription. Glucocorticoids havewidespread actions affecting many cells and tissues (Schimmer and Parker 1996,Berne and Levy 1998).
Carbohydrate, protein, and lipid metabolism. In the liver, glucocorticoidsenhance the formation of glucose from amino acids and glycerol, and thedeposition of glucose as glycogen. In peripheral tissues, glucocorticoids increaseprotein breakdown in muscle, bone, connective tissue, and skin. Furthermore,glucocorticoids antagonise the effect of insulin on glucose metabolism and inhibitinsulin-stimulated glucose uptake in cells. The net effect of glucocorticoids is anincrease in blood glucose concentrations. Glucocorticoids facilitate the effect oflipolytic substances, such as growth hormone, resulting in an increase in free fattyacids. An excess of glucocorticoids results in the typical distribution of fat in theback and neck, face, and abdomen, with loss of fat in the extremities.
Effects on muscle, bone, and connective tissue. Glucocorticoids reduce musclemass and strength and inhibit bone formation by several mechanisms. Theyreduce synthesis of the type I collagen essential for bone matrix. Glucocorticoids

REVIEW OF THE LITERATURE
28
reduce the availability of calcium for bone mineralisation. In addition,glucocorticoids increase bone reabsorption. Finally, the net effect is a decrease inbone density (osteoporosis). Glucocorticoids inhibit the synthesis of collagen,resulting in thinning of the skin and capillary walls.
Effects on the vascular system. Glucocorticoids enhance vascular reactivity toother vasoactive substances, e.g., angiotensin II. Glucocorticoids, even thoselacking mineralocorticoid activity, may cause hypertension, the mechanism ofwhich is still unclear.
Effects on the central nervous system. Glucocorticoids modulate behaviour andmood. Lack of cortisol in Addison’s disease may cause apathy and depression. Anexcess of glucocorticoids may cause insomnia, decrease memory skills, lower thethreshold to seizure activity, or even cause psychoses.
Effects on inflammatory and immune responses. Glucocorticoids reduce thenumber of lymphocytes and reduce the mass of lymphoid tissue. In addition,glucocorticoids inhibit such processes as the synthesis and release of arachinodicacid, a precursor for many mediators of immune responses. The hormone reducesthe number and function of thymus-derived lymphocytes (T-cells), therebyinhibiting cell-mediated immunity. The production of e.g., interleukin-1, -2, -6,and tumour necrosis factor α is reduced. The suppression of T-cell activation alsosuppresses the activation of B-lymphocytes. The net effects of these complexactions are diminished inflammatory and immune responses.
2.4. Therapeutic useThe finding of Hench et al in 1949 that cortisone has beneficial effects intreatment of rheumatoid arthritis generated great interest in cortisol and itssynthetic analogues. In addition to being replacement therapy for adrenalinsufficiency, glucocorticoids are widely used because of their anti-inflammatoryand immunosuppressive properties in treatment of nonendocrine diseases.
Systemic glucocorticoids are included in the standard immunosuppressive drugregimen for organ-transplant patients (Mies et al 1998, Sarna et al 1997, Pericoand Remuzzi 1997). They are used in treatment of diseases of connective tissue,e.g., rheumatoid arthritis, polymyositis, and systemic lupus erythematosus(LaRochelle et al 1993, Laan et al 1999) and in treatment of acute exacerbation ofasthma (Rowe et al 2000). Many antineoplastic combinations used to treat acuteleukaemia, lymphomas, and myeloma include glucocorticoids (Gaynon andCarrel 1999, Cain et al 1998, Oken 1997). Furthermore, renal diseases, e.g.,nephrotic syndrome secondary to minimal change disease usually respond well tothe glucocorticoids (Friedman and Chesney 1982). In addition, glucocorticoidsare used for the management of nausea and vomiting induced by cancer
REVIEW OF THE LITERATURE
29
chemotherapy and for management of brain tumours (Ater et al 1997, Perez1998).
Usually the appropriate dose has to be determined case by case. The dosage ofglucocorticoid varies, depending on nature and severity of the disease. Prolongedtherapy with doses that exceed the normal physiological level of glucocorticoidrequire careful consideration. If possible, the total daily dose should be given as asingle morning dose, and an alternate day therapy has been recommended tominimise the catabolic actions and HPA-axis suppression of glucocorticoids(Melby 1974, Swartz and Dluhy 1978).
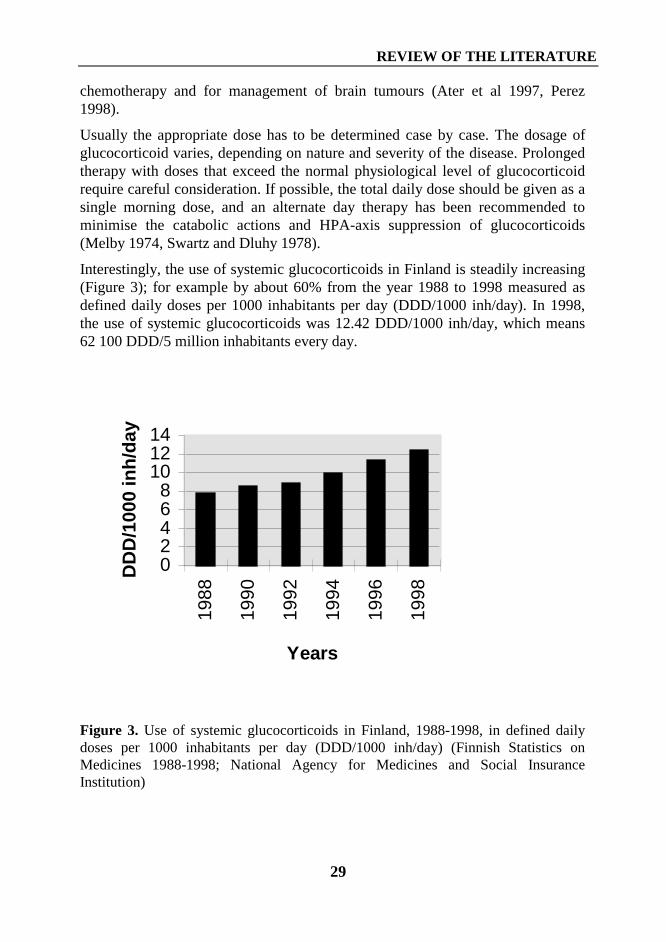
Interestingly, the use of systemic glucocorticoids in Finland is steadily increasing(Figure 3); for example by about 60% from the year 1988 to 1998 measured asdefined daily doses per 1000 inhabitants per day (DDD/1000 inh/day). In 1998,the use of systemic glucocorticoids was 12.42 DDD/1000 inh/day, which means62 100 DDD/5 million inhabitants every day.
02468
101214
1988
1990
1992
1994
1996
1998
Years
DD
D/1
000
inh
/day
Figure 3. Use of systemic glucocorticoids in Finland, 1988-1998, in defined dailydoses per 1000 inhabitants per day (DDD/1000 inh/day) (Finnish Statistics onMedicines 1988-1998; National Agency for Medicines and Social InsuranceInstitution)

REVIEW OF THE LITERATURE
30
2.5. Adverse effects of glucocorticoid treatmentGlucocorticoid adverse effects can be described as an extension of theirpharmacodynamic and physiological effects. Synthetic glucocorticoids cansuppress endogenous cortisol secretion and abolish the daily circadian rhythm ofcortisol (Salassa et al 1953, Melby 1974, Slayter et al 1996). The failure of theadrenal gland to response to stress (infection, surgery) is one major andunpredictable complication of glucocorticoid therapy (Fraser et al 1952, Salassaet al 1953, Melby 1974, Henzen et al 2000). Treatment with high doses ofglucocorticoids even for less than a week and daily doses as low as 5 mgprednisone equivalent or less can, in some sensitive individuals, impair theadrenal response (Streck and Lockwood 1979, Schlaghecke et al 1992, Henzen etal 2000). Neither the dose of glucocorticoid nor the duration of the treatmentappears to predict the adrenal insufficiency (Livanou et al 1967, Schlaghecke et al1992, Henzen et al 2000).
Glucocorticoids increase the susceptibility to bacterial, viral, and fungalinfections (Twycross 1994). Severe, even fatal varicella cases have beenassociated with corticosteroid use (Kasper and Howe 1990, Dowell and Bresee1993). Glucocorticoids can cause myopathy, which may be severe enough todisturb daily activities and even impair respiratory function (Batchelor et al1997). Osteoporosis and vertebral compression fractures are seriouscomplications of glucocorticoid therapy (Adachi et al 1993). It has been estimatedthat the incidence of fractures in patients receiving chronic glucocorticoid therapyis about 30% to 50% (Adachi et al 1993). In addition, glucocorticoids can causehyperglycaemia and increase insulin requirements of diabetes patients (David etal 1980, Twycross 1994). Furthermore, glucocorticoids can cause electrolyteabnormalities, hypertension, psychiatric disorders, cataract, a typical moon-face,striae, and truncal obesity (Melby 1974, Swartz and Dluhy 1978, Twycross 1994,Jenkins 1999). In children treatment with glucocorticoids can result in growthretardation (Sarna et al 1997).
2.6. MethylprednisolonePharmacokinetics. Methylprednisolone is rapidly absorbed following oraladministration with its Cmax reached within 1 to 3 h (Antal et al 1983, Al-Habetand Rogers 1989). The mean absolute bioavailability of methylprednisolone ishigh, ranging from 80% to 90% (Antal et al 1983, Al-Habet and Rogers 1989,Patel et al 1993). However, Patel et al (1993) reported large interindividualvariability in absolute bioavailability, ranging from 50% to completebioavailability.

REVIEW OF THE LITERATURE
31
Methylprednisolone is about 80% bound to plasma protein with only a littlebound to the corticosteroid-binding protein, transcortin (Szefler et al 1986, Konget al 1989). The mean Vd of methylprednisolone averages 1.2 to 1.5 l/kg (Al-Habet and Rogers 1989, Kong et al 1989, Szefler et al 1986). Ferry et al (1994)suggest that methylprednisolone exhibits dose- and time-dependentpharmacokinetics. Others have, however, shown the pharmacokinetics to belinear in the dose ranges 0 to 40 mg (Kong et al 1989), 20 to 80 mg (Al-Habet andRogers 1989), and 540 to 1026 mg (Assael et al 1982). The mean t½ ofmethylprednisolone averages 1.9 to 3.3 h (Szefler et al 1986, Ferry et al 1994, Al-Habet and Rogers 1989). The systemic plasma clearance (Cl) ofmethylprednisolone averages 350 to 450 ml/h/kg (Al-Habet and Rogers 1989,Patel et al 1993).
Due to its lipophilicity and low water-solubility, methylprednisolone itself can notbe administered intravenously. Methylprednisolone sodium succinate is a prodrugand is hydrolysed in the body rapidly and almost completely with a half-life ofabout 4 minutes to active methylprednisolone alcohol (Al-Habet and Rogers1989).
Methylprednisolone undergoes reduction of the ketone group at the C20 position,yielding the inactive metabolites 20α- and 20β-hydroxymethylprednisolone, andfurther oxidation of the C20,C21 side chain (Stjernholm and Katz 1975, Vree et al1999). The 6α-methyl group has been suggested to prevent the hydroxylation atC6 position. However, Vree et al (1999) found 6β-hydroxy-6α-methylprednisolone and a compound that might be 6α-hydroxy-6β-methylprednisolone in the urine of patients receiving methylprednisolone.Oxidation at C11 of methylprednisolone produces a minor metabolite,methylprednisone; this reaction is reversible. In addition, Rodchenkov et al(1987) found 6,7-dehydro metabolites in urine, indicating that formation of a 6,7-double band may be possible. The suggested metabolic pathways ofmethylprednisolone are presented in Figure 4. The specific CYP enzymesinvolved in the various metabolic pathways of methylprednisolone have not beendetermined.
After radio-labelled doses of methylprednisolone, a total of 75% of the label wasdetected in urine and 20% in bile (Slaunwhite and Sandberg 1961). Furthermore,during the first 24 h after administration of methylprednisolone sodium succinate,about 30% of the dose was excreted in the urine: 10% as unmetabolised ester,12% as methylprednisolone, 8% as 20α- and 1.0% as 20β-hydroxymethylprednisolone, and less than 1.0% as methylprednisone (Lawson etal 1992). In urine, glucuronides, sulphates and unconjugated metabolites werepresent (Slaunwhite and Sandberg 1961).

REVIEW OF THE LITERATURE
32
Figure 4. Metabolic pathways of methylprednisolone reported in the literature(Adapted from Vree et al 1999).
Women appear to eliminate methylprednisolone more rapidly than do men;however, the clinical importance of this observation is unclear (Lew et al 1993).The Cl of methylprednisolone is slower in elderly (range 69-82 years) than inyounger (range 24-37 years) men (Tornetore et al 1994). This pharmacokineticalteration may increase the risk for adverse effects among elderly patients(Tornetore et al 1994). Furthermore, the Cl of methylprednisolone was lower inthe obese than in non-obese subjects (Dunn et al 1991). The metabolism ofmethylprednisolone may be impaired in severe liver disease; however, patientswith renal impairment appears to need no dose adjustment of methylprednisolone(Dollery 1999c).
Pharmacokinetic drug interactions. Enzyme inducers phenobarbital, phenytoin,and carbamazepine induce metabolism of methylprednisolone. Phenobarbitalincreased the Cl of methylprednisolone by 208%, phenytoin by 478%, andcarbamazepine by 342%, suggesting clinically significant interactions(Stjernholm and Katz 1975, Bartoszek et al 1987).
The effect of some CYP3A4 inhibitors on intravenous methylprednisolone hasbeen evaluated. Troleandomycin decreased the Cl of intravenousmethylprednisolone by 64% and increased its mean t½ from 2.5 to 4.6 h (Szefleret al 1980). Erythromycin 250 mg 4 times daily for one week decreased the Cl ofmethylprednisolone by 46% and increased its t½ by 51%, from 2.4 to 3.5 h(LaForce et al 1983). In addition, ketoconazole increased the methylprednisoloneAUC by 135% and reduced its Cl by 66% after intravenous administration (Glynn

REVIEW OF THE LITERATURE
33
et al 1986). Kandrotas et al (1987) recommend a 50% lower dose ofmethylprednisolone during concomitant use of ketoconazole andmethylprednisolone.
Cimetidine did not affect methylprednisolone disposition (Green et al 1984).Furthermore, cyclosporin appears not to affect methylprednisolone dispositionfollowing intravenous administration of methylprednisolone (Tornatore et al1993). Oral contraceptives decreased the Cl of intravenous methylprednisolone,increased total AUC by 49%, and t½ from 1.7 to 2.2 h (Slayter et al 1996).
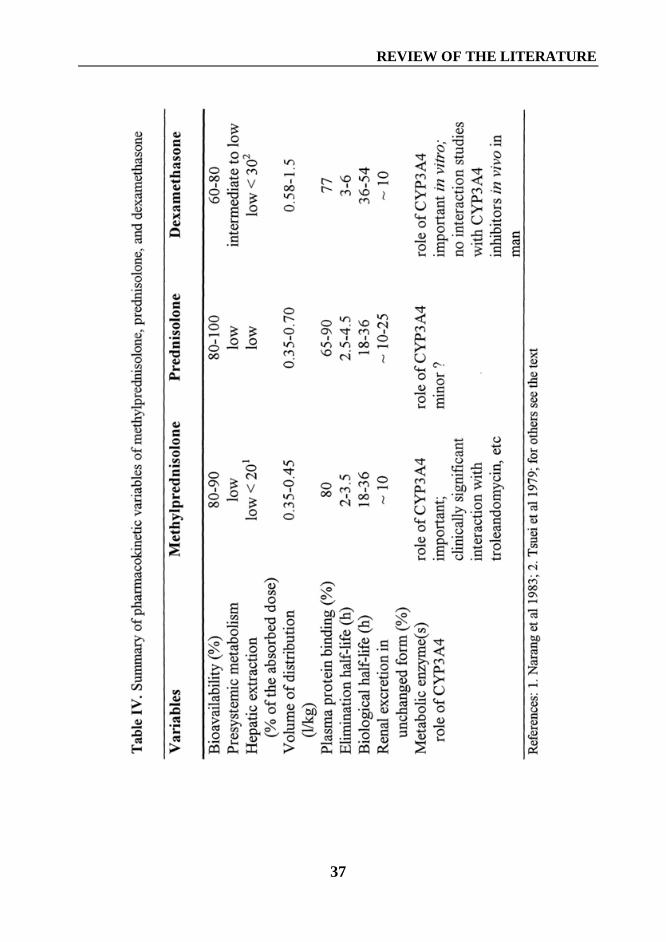
A summary of pharmacokinetic variables of methylprednisolone, prednisolone,and dexamethasone is shown in Table IV (page 37).
2.7. PrednisolonePharmacokinetics. Prednisolone is rapidly absorbed after oral administration andreaches the Cmax within approximately 1 to 2 h (Leclercq and Copinschi 1974,Pickup 1979). The mean absolute bioavailability of prednisolone ranged from80% to about 100% (Petereit and Meikle 1977, Al-Habet and Rogers 1980). Theprotein binding of prednisolone varies from 65% to 90% (Pickup 1979).Prednisolone is reversibly bound to albumin and to transcortin. The degree ofprotein binding of prednisolone is concentration-dependent, since binding totranscortin is saturable (Pickup 1979).
The Vd of prednisolone increases with dose from about 0.35 l/kg to 0.7 l/kg (Begget al 1987). In addition, the Cl of prednisolone is dose-dependent. After a 5 mgand 40 mg dose of prednisolone the Cl has been about 7 l/h and 12 l/h,respectively (Rose et al 1981a). These dose-dependent changes may result from aconcentration-dependent change in protein binding. Because both Vd and the Clincrease, there may be none (Tanner et al 1979, Bergrem et al 1983) or only aminor dose-dependent change in the t½ of prednisolone (Rose et al 1981a). Themean t½ of prednisolone has ranged from 2.6 h to 4.5 h (Al-Habet and Rogers1980, Rose et al 1981a, Bergrem et al 1983).
The oxidation of prednisolone at C11 position produces prednisone, a syntheticglucocorticoid in an inactive form. This metabolic reaction is interconversible andfavours the formation of prednisolone. The concentrations of prednisolone are 4to 10 times as high as those of prednisone after administration of eitherprednisone or prednisolone (Rose et al 1981a). Prednisolone can be hydroxylatedat the 6β position like cortisol, and the urinary 6β-hydroxyprednisolone makes up8% to 10% of the administered prednisolone dose and some 2% to 5% is excretedas prednisone in the urine (Rose et al 1981a, Frey et al 1987, Zürcher et al 1989).About 7% to 24% of the prednisolone dose is excreted in the urine as the parent

REVIEW OF THE LITERATURE
34
drug (Rose et al 1981a, Frey et al 1987, Zürcher et al 1989). Furthermore, bothprednisone and prednisolone can undergo reduction of the ketone group at C20(Gray et al 1956). After radiolabelled doses of prednisolone, more than 90% ofthe label is detected in urine and less than 5% in bile (Slaunwhite and Sandberg1961).
The specific CYP enzymes involved in the metabolic pathways of prednisolonehave not been determined. Based on the fact that 6β-hydroxylation of cortisol iscatalysed by CYP3A4 and CYP3A5 (Ged et al 1989, Wrighton et al 1990), theseenzymes have also been assumed to be responsible for the 6β-hydroxylation ofprednisolone, although this is a relatively minor metabolic pathway ofprednisolone.
The Cl of prednisolone appears to be slightly, about 20%, higher in females thanin males (Meffin et al 1984), the clinical significance of which remains unclear.The Cl of prednisolone is 35% higher in obese subjects than in non-obesesubjects, and dosage should reflect total body weight (Milsap et al 1984). Its Clmay be slightly decreased in chronic active hepatitis (Powell and Axelsen 1972);however, in a study by Kawai et al (1985), liver disease does not affect itspharmacokinetics. Smoking does not affect the pharmacokinetics of prednisolone(Rose et al 1981b). In addition, results concerning the autoinduction ofprednisolone metabolism during long-term therapy are contradictory (Begg et al1987).
Pharmacokinetic drug interactions. Rifampicin, phenobarbital, carbamazepine,and phenytoin induce the metabolism and increase the Cl of prednisolone(McAllistar et al 1983, Brooks et al 1976, Bartoszek et al 1987, Petereit andMeikle 1977). Concomitant use of rifampicin or phenobarbital with prednisolonecan worsen the symptoms of the disease being treated, e.g., nephrotic syndromeor rheumatoid arthritis (Hendrickse et al 1979, Brooks et al 1976).
Lanzoprazole and omeprazole, following prednisone administration, do not affectthe pharmacokinetics of prednisolone (Cavanaugh and Karol 1996). Thepharmacokinetics of prednisolone are altered only slightly after cimetidine andranitidine, and this interaction appears not to be clinically significant (Sirgo et al1985). Neither azathioprine nor interleukin-10 affect the pharmacokinetics ofprednisolone (Frey et al 1981, Chakraborty et al 1999). In addition, indomethacinand naproxen have increased free prednisolone concentration by 30 to 60%without affecting total prednisolone concentrations (Rae et al 1982). Oralcontraceptive steroids have increased the t½ of prednisolone about 1.5- to 2.5-fold, and careful monitoring is recommended when oral contraceptives andprednisolone are used concomitantly (Legler and Benet 1986).

REVIEW OF THE LITERATURE
35
The CYP3A4 inhibitor ketoconazole (200 mg/day) has increased plasmaconcentrations of prednisolone by about 50% following oral prednisone orintravenous prednisolone administration (Zürcher et al 1989). Two other studies,however, failed to find any effect with 200 mg/day ketoconazole on either thepharmacokinetics or pharmacodynamics of prednisolone (Ludwig et al 1989,Yamashita et al 1991). Furthermore, neither troleandomycin nor grapefruit juiceaffected its pharmacokinetics (Szefler et al 1982, Hollander et al 1995). However,administration of diltiazem increased the AUC of prednisolone by about 20%after ingestion of prednisone (Imani et al 1999). Whether cyclosporin affects theelimination of prednisolone is unclear. Two reports have suggested cyclosporininhibits the elimination of prednisolone (Öst 1984, Langhoff et al 1985). Incontrast, some investigators have found cyclosporin not to alter its metabolism(Frey et al 1987, Rocci et al 1988).
2.8. DexamethasonePharmacokinetics. Dexamethasone is rapibly absorbed after oral administration.The Cmax of oral dexamethasone is reached between 1.2 and 2.7 h (Rose et al1981b, Loew et al 1986, O’Sullivan et al 1997). Although mean absolutebioavailability of dexamethasone has ranged from 61 to 78% (Duggan et al 1975,Rose et al 1981b, O’Sullivan et al 1997), large interindividual variation in theabsolute bioavailability can exist ,e.g., from 51% to 89% in healthy volunteersand from 34% to 79% in depressed patients (O’Sullivan et al 1997).
Dexamethasone is about 77% bound to plasma protein, mainly albumin, and itappears neither to bind transcortin nor to compete with cortisol for its bindingprotein (Peets et al 1969). Dexamethasone bound to albumin is unaffected byincreasing dexamethasone concentrations (Peets et al 1969). The mean Vd ofdexamethasone averages 0.58 to 1.5 l/kg (Rose et al 1981b, Workman et al 1986,O’Sullivan et al 1997). Loew et al (1986) found the AUC of dexamethasone toincrease less than proportionally to dose after oral 0.5- to 1.5-mg doses. However,no dose-dependency was observed when dexamethasone was administered tohealthy subjects at a 15-mg dose and at high 1.5 mg/kg doses or to cancer patientsat 40- to 200-mg doses (Rohdewald et al 1987, Brady et al 1987). The t½ ofdexamethasone averages 3.1 to 6.1 h and its Cl 110 to 180 ml/h/kg (Rose et al1981b, O’Sullivan et al 1997).
Dexamethasone sodium phosphate is a prodrug. After intravenous administration,the hydrolysis of the phosphate ester to dexamethasone is rapid, with half-life of 5min, and the Cmax of dexamethasone is reached after 10 min (Rohdewald et al1987). Conversion of dexamethasone phosphate to dexamethasone alcoholappears to be about 90% (Hare et 1975).

REVIEW OF THE LITERATURE
36
The 6β-hydroxylation is the major metabolic pathway of dexamethasone(Minagawa et al 1986), and hydroxylation both at the 6α and at the 6β positionhave shown to be CYP3A4-mediated in human liver microsomes in vitro (Gentileet al 1996). About 9 to 10% of the dose was excreted as unchangeddexamethasone in the urine (Duggan et al 1975, Miyabo et al 1981), withglucuronides present in small amounts (Minagawa et al 1986, Tomlinson et al1996). After an intravenous dose of labelled dexamethasone, about 64% wasexcreted in the urine within 24 h, and unconjugated metabolites formed thelargest fraction, about 30% of the dose (Haque et al 1972).
The t½ of dexamethasone has been reported to be prolonged in liver disease andshortened in chronic renal failure (Kawai et al 1985). However, Workman et al(1986) found chronic renal failure not to affect the pharmacokinetics ofdexamethasone. The t½ of oral dexamethasone was longer in men than in women(6.4 h vs. 4.8 h), and the gender difference appears to be weight-related(O’Sullivan et al 1997). In addition, smoking did not affect the pharmacokineticsof dexamethasone (Rose et al 1981b).
Pharmacokinetic drug interactions. Phenobarbital has increased the Cl ofdexamethasone by 88% and decreased its t½ by 44% (Brooks et al 1972).Phenytoin increased the Cl and reduces the systemic bioavailability ofdexamethasone (Chalk 1984). Carbamazepine 800 mg daily raises the dose ofdexamethasone needed to suppress plasma cortisol 2- to 4-fold, when measuredas a part of the dexamethasone suppression test (Kobberling and v zur Muhlen1973).
Treatment with ephedrine has decreased the t½ of dexamethasone by 36% andincreased its Cl by 42%, but theophylline appears not to interact withdexamethasone (Brooks et al 1977). Cimetidine does not affect thepharmacokinetics of intravenous dexamethasone (Peden et al 1984). There appearto be no controlled studies concerning the pharmacokinetic interactions betweenCYP3A4 inhibitors and dexamethasone in humans.
A summary of pharmacokinetic variables of methylprednisolone, prednisolone,and dexamethasone is shown in Table IV.
REVIEW OF THE LITERATURE
37

AIMS OF THE STUDY
38
AIMS OF THE STUDY
Synthetic glucocorticoids are widely used because of their anti-inflammatory andimmunosuppressive properties in many different types of formulations. Thisstudy, however, will focus on systemic oral and/or intravenous glucocorticoidsmethylprednisolone, prednisolone, and dexamethasone. It has been shown thatthese glucocorticoids are eliminated mainly by metabolism, and CYP3A4 mayplay a different role in their biotransformation. Many drugs currently on themarket are known to inhibit CYP3A4-mediated metabolism in vitro and in vivo.CYP3A4 is also present in the gut, and inhibition of drug metabolism can occureven in the intestinal mucosa, a fact which may be important in regard to orallyadministered drugs.
Glucocorticoids have a very wide therapeutic margin, and high doses can begiven intravenously, e.g., in an emergency. However, for continuous use at dosesas low as possible, intermediate-acting glucocorticoids and alternate-day therapyhave been recommended to minimise the catabolic action and HPA-axissuppression of glucocorticoids. Thus, pharmacokinetic interaction ofglucocorticoids with other drugs, resulting in an increased and prolongedexposure to glucocorticoids, can be clinically important. Increased knowledge ofpotential drug-drug interactions of systemic glucocorticoids would result in moreproper and safe use of these drugs.
The specific aims of this thesis were to study:
1. The effect of itraconazole on the pharmacokinetics and adrenal-suppressanteffect of oral and intravenous methylprednisolone (Studies I and II), with specialattention to the route of methylprednisolone administration.
2. The effect of itraconazole on the pharmacokinetics and adrenal-suppressanteffect of oral prednisolone (Study IV)
3. The effect of itraconazole on the pharmacokinetics and adrenal-suppressanteffect of oral and intravenous dexamethasone (Study VI), with special attentionto the route of dexamethasone administration.
4. The effect of diltiazem and mibefradil on the pharmacokinetics and adrenal-suppressant effect of oral methylprednisolone (Study III)
5. The effect of grapefruit juice on the pharmacokinetics and adrenal-suppressanteffect of oral methylprednisolone (Study V).

MATERIALS AND METHODS
39
MATERIALS AND METHODS
1. Ethical considerationsAll study protocols were approved by independent Ethics Committee of theDepartment of Clinical Pharmacology, University of Helsinki, and reviewed by theNational Agency of Medicines in Finland. Before entering the study all subjectsreceived both oral and written information and gave their written consent.
2. Study subjectsAll subjects participating in Studies I to VI were healthy volunteers (age range 19-42 years; weight 45-90 kg). Altogether 43 study subjects (25 females, 18 males)participated in Studies I to IV. A total of ten subjects participated in more than onestudy; seven subjects in two, two subjects in three and one subject in four times.None of the subjects were smokers or used continuous medication, except that inStudies I to III eight female subjects used oral contraceptive steroids. Use of otherdrugs was prohibited starting 2 weeks before each study day. Furthermore,participation in another trial and blood donation within one month before the studyperiod were exclusion criteria. Each subject underwent a medical screening 1 to 3weeks before participation in a study. This screening included clinicalexamination, medical history, and routine laboratory tests, and in Study III astandard electrocardiogram (ECG) to ascertain that each was in good health.
3. Study protocol3.1. Study designIn Studies I to IV and VI the subjects were pretreated with a potential enzymeinhibitor or placebo and in Study V with double-strength grapefruit juice or water.A randomised crossover design was used in two or three phases with a washoutperiod of 4 weeks. All studies were double-blind, except Study V, which was anopen study.
The pretreatment drugs and matched placebos were supplied, packed, and labelledaccording to a randomisation list for each subject by the Pharmacy of the HelsinkiUniversity Central Hospital. Pretreatment drugs and matched placebos wereidentical in order to maintain the the double-blind nature of the studies. The studydrugs (glucocorticoids) were also supplied by the Pharmacy of the HelsinkiUniversity Central Hospital. Glucocorticoids were given under the supervision ofthe investigator in the morning at 9:00 after an overnight fast. The oral study drugs
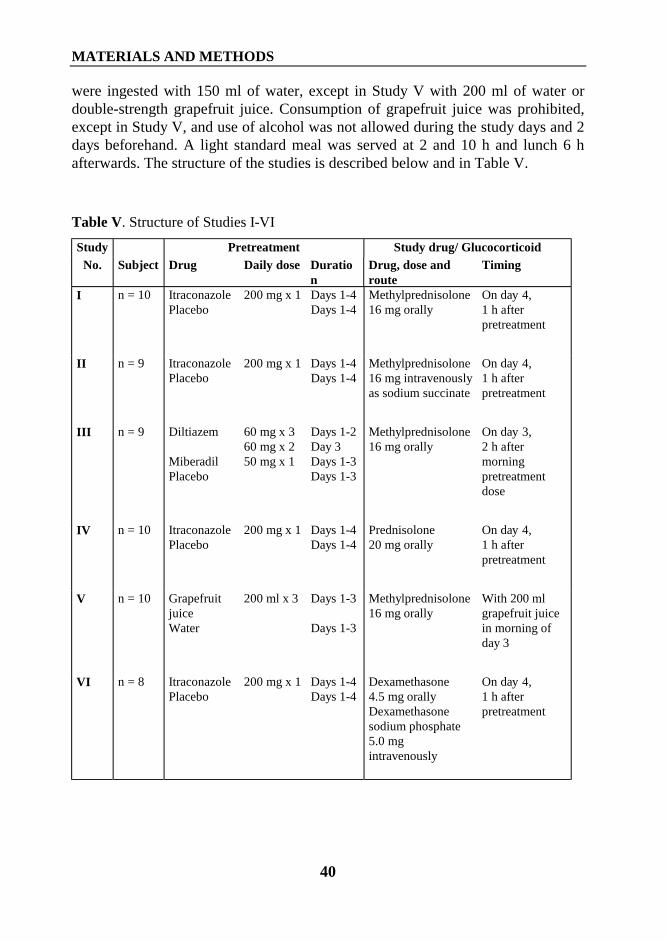
MATERIALS AND METHODS
40
were ingested with 150 ml of water, except in Study V with 200 ml of water ordouble-strength grapefruit juice. Consumption of grapefruit juice was prohibited,except in Study V, and use of alcohol was not allowed during the study days and 2days beforehand. A light standard meal was served at 2 and 10 h and lunch 6 hafterwards. The structure of the studies is described below and in Table V.
Table V. Structure of Studies I-VI
Study Pretreatment Study drug/ GlucocorticoidNo. Subject Drug Daily dose Duratio
nDrug, dose androute
Timing
I n = 10 ItraconazolePlacebo
200 mg x 1 Days 1-4Days 1-4
Methylprednisolone16 mg orally
On day 4,1 h afterpretreatment
II n = 9 ItraconazolePlacebo
200 mg x 1 Days 1-4Days 1-4
Methylprednisolone16 mg intravenouslyas sodium succinate
On day 4,1 h afterpretreatment
III n = 9 Diltiazem
MiberadilPlacebo
60 mg x 360 mg x 250 mg x 1
Days 1-2Day 3Days 1-3Days 1-3
Methylprednisolone16 mg orally
On day 3,2 h aftermorningpretreatmentdose
IV n = 10 ItraconazolePlacebo
200 mg x 1 Days 1-4Days 1-4
Prednisolone20 mg orally
On day 4,1 h afterpretreatment
V n = 10 GrapefruitjuiceWater
200 ml x 3 Days 1-3
Days 1-3
Methylprednisolone16 mg orally
With 200 mlgrapefruit juicein morning ofday 3
VI n = 8 ItraconazolePlacebo
200 mg x 1 Days 1-4Days 1-4
Dexamethasone4.5 mg orallyDexamethasonesodium phosphate5.0 mgintravenously
On day 4,1 h afterpretreatment

MATERIALS AND METHODS
41
Study I. Ten subjects (7 females/3 males) each received 200 mg itraconazole orplacebo orally once daily for 4 days. On day 4, a dose of 16 mgmethylprednisolone was ingested at 9:00 a.m., one hour after theitraconazole/placebo. Blood samples were drawn before the methylprednisoloneadministration and ½, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, and 24 h later. Plasma wasseparated within 30 min and stored at -40° C until analysis. Methylprednisolone,cortisol, itraconazole, and hydroxyitraconazole concentrations were determined.
Study II. Nine subjects (4 females/5 males) each received 200 mg itraconazole orplacebo orally once daily for 4 days. On day 4, an intravenous dose of 16 mgmethylprednisolone as sodium succinate was given within 2 min at 9:00 a.m., onehour after itraconazole/placebo. Blood samples were drawn beforemethylprednisolone administration and 3 min and ½, 1, 2, 3, 4, 5, 6, 8, 10, 12, and24 h later. The sampling tubes were immediately placed into ice water, and plasmawas separated within 30 min by centrifugation at +4°C. The samples were stored at-80°C until analysis. Methylprednisolone, cortisol, itraconazole, andhydroxyitraconazole concentrations were determined.
Study III. Nine subjects (8 females/1 male) each received 60 mg diltiazem 3 timesa day (at 7:00 a.m., 3:00 p.m., and 9:00 p.m.), 50 mg mibefradil once a day (at 7:00a.m. and corresponding placebo at 3:00 p.m. and 9:00 p.m.), or placebo three timesa day, all of these orally and all for 2 days. On the next day (day 3), only twopretreatment doses were given (at 7:00 a.m. and 3:00 p.m), and a dose of 16 mgmethylprednisolone was ingested at 9:00 a.m., 2 h after diltiazem, mibefradil, orplacebo. Blood samples were drawn before methylprednisolone administration andat ½, 1, 1½, 2, 3, 4, 5, 6, 8, 10, 12, 23, and 47 h. Plasma was separated within 30min and stored at -80° C until analysis. Methylprednisolone, cortisol, diltiazem,and mibefradil concentrations were determined. Each subject underwent ascreening including ECG, blood pressure, and heart rate on the first and third dayof each study period.
Study IV. Ten subjects (5 females/5 males) each received 200 mg itraconazole orplacebo orally once daily for 4 days. On day 4, a dose of 20 mg prednisolone wasingested at 9:00 a.m., one h after itraconazole/placebo. Blood samples were drawnbefore prednisolone administration and at ½, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 23, and47 h. Plasma was separated within 30 min and stored at -40 °C until analysis.Prednisolone, cortisol, itraconazole, and hydroxyitraconazole concentrations weredetermined.
Study V. Ten subjects (2 females/8 males) each drank 200 ml double-strengthgrapefruit juice or 200 ml water 3 times a day for 2 days. On day 3, a dose of 16mg methylprednisolone was ingested at 9:00 a.m. with 200 ml double-strengthgrapefruit juice or water. In addition, 200 ml grapefruit juice or water was given ½

MATERIALS AND METHODS
42
and 1½ h after methylprednisolone administration. Blood samples were drawnbefore methylprednisolone administration and at ½, 1, 1½, 2, 3, 4, 5, 6, 8, 10, 12,23, and 47 h. Plasma was separated within 30 min and stored at -40 °C untilanalysis. Methylprednisolone and cortisol concentrations were determined.
Study VI. Eight subjects (7 females/1 male) each ingested either 200 mgitraconazole (during 2 phases) or placebo (during 2 phases) once daily for 4 days.On the fourth day, at 9:00 a.m., one h after itraconazole/placebo, each subjectreceived 4.5 mg dexamethasone orally or a dose of 5.0 mg dexamethasone sodiumphosphate (corresponds to 3.85 mg dexamethasone) intravenously during both theitraconazole and placebo phases. Dexamethasone sodium phosphate was given asan intravenous injection over 2 min. Blood samples were drawn beforedexamethasone administration and at 1, 2, 3, 4, 6, 8, 10, 12, 23, 47, and 71 h. Afterintravenous administration, an additional sample was drawn 0.25 h afterdexamethasone injection. Plasma was separated within 30 min and stored at -70°Cuntil analysis. Dexamethasone, cortisol, itraconazole, and hydroxyitraconazoleconcentrations were determined.
3.2. Determination of plasma drug and cortisol concentrationsAll plasma drug and cortisol concentrations were determined in the AnalyticalLaboratory of the Department of Clinical Pharmacology, University of Helsinki.
Methylprednisolone. In Studies I, II, and III, plasma methylprednisoloneconcentrations were determined by high-performance liquid chromatography(HPLC) with ultraviolet detection (Ebling et al 1984, Kong et al 1988).Dexamethasone served as the internal standard. The quantification limit was 3ng/ml in Studies I and II and 2 ng/ml in Study III. The interday coefficient ofvariation (CV) for methylprednisolone ranged from 5.8% to 11.9% at < 20 ng/mlconcentrations and 1.2% to 4.9% at higher methylprednisolone concentrations.
In Study V, the plasma methylprednisolone concentrations were determined byliquid chromatography-ionspray-tandem mass spectrometry with use of the PESCIEX API 3000 LC/MS/MS system (Sciex Division of MDS Inc, Toronto,Canada) (Kong et al 1988, Dodds et al 1997). Dexamethasone served as theinternal standard. The quantification limit was 0.5 ng/ml and the interday CV 6.3%at 4.3 ng/ml, 2.3% at 25 ng/ml, and 3.6% at 118 ng/ml (n = 4 at all concentrations).
Prednisolone. Plasma prednisolone concentrations were determined by HPLCwith ultraviolet detection (McWhinney et al 1996). Dexamethasone served as theinternal standard. The quantification limit was 5 ng/ml and the interday CV 8.5%at 11 ng/ml, 2.8% at 49 ng/ml, and 3.8% at 227 ng/ml (n = 6 at all concentrations).

MATERIALS AND METHODS
43
Dexamethasone. Plasma dexamethasone concentrations were determined byHPLC with ultraviolet detection (McWhinney et al 1996). Methylprednisoloneserved as the internal standard. The quantification limit was 2 ng/ml and theinterday CV 9.0% at 3.7 ng/ml (n = 11), 1.6% at 36 ng/ml (n = 14), and 2.2% at158 ng/ml (n = 14).
Itraconazole and hydroxyitraconazole. Itraconazole and hydroxyitraconazoleplasma concentrations were determined by HPLC with fluorescence detection(Allenmark et al 1990, Remmel et al 1988). R51012 (Janssen Pharma, Beerse,Belgium) served as the internal standard. The quantification limit was 10 ng/ml forboth compounds. The interday CV for itraconazole ranged from 0.5% to 5.6% andthat for hydroxyitraconazole from 0.7% to 9.3% at relevant itraconazoleconcentrations.
Diltiazem. Plasma diltiazem concentrations were determined by HPLC withultraviolet detection (Dube et al 1988, Hynning et al 1988). Verapamil served asthe internal standard. The quantification limit was 10 ng/ml, and the interday CVfor diltiazem was 4.0% at 24.7 ng/ml (n = 6) and 3.4% at 232 ng/ml (n = 6).
Mibefradil. Plasma mibefradil concentrations were determined by HPLC(Skerjanec and Tam 1995) with fluorescence detection. An external standardserved in this assay. The quantification limit was 10 ng/ml. The intraday CV formibefradil was 1.5% at 15.8 ng/ml (n = 10) and 2.0% at 50.9 ng/ml (n = 10).
Cortisol. In Studies I to IV and VI plasma cortisol concentrations were determinedby HPLC with ultraviolet detection (Ebling et al 1984, Kong et al 1988,McWhinney et al 1996). Dexamethasone served as the internal standard. Thequantification limit was 3 ng/ml in Studies I and II, 2 ng/ml in III and IV, and 5ng/ml in VI. The interday CV for cortisol ranged from 3.7% to 14.5% at < 20ng/ml concentrations and from 1.6% to 6.5% at higher cortisol concentrations.
In Study V, the plasma cortisol concentrations were determined by liquidchromatography-ionspray-tandem mass spectrometry with use of the PE SCIEXAPI 3000 LC/MS/MS system (Sciex) with dexamethasone as the internal standard.The quantification limit was 0.5 ng/ml. The interday CV was 1.2% at 4.1 ng/ml,6.1% at 25 ng/ml, and 4.3% at 123 ng/ml (n = 5 at all concentrations).
3.3. Pharmacokinetic calculationsThe peak concentration in plasma (Cmax) and time to Cmax (tmax) were obtaineddirectly from the data. The elimination rate constant (kel) was determined forcalculation of elimination half-life (t½) and total area under the plasmaconcentration-time curve [AUC(0-∞)]. The terminal log-linear phase of the plasmaconcentration-time curve was visually identified for each subject. The elimination

MATERIALS AND METHODS
44
rate constant (kel) was calculated by regression analysis of this log-linear part of theplasma concentration-time curve. The t½ was calculated from the equation
t½ = ln2 / kel
The area under the plasma concentration-time curve (AUC) values were calculatedby use of the linear trapezoidal rule (Studies I, II and IV) or by log trapezoidal rule(Studies III, V, and VI). To determine the AUC to infinity, the extrapolation toinfinity was done by dividing the last measured concentration by the correspondingkel value. AUC values were calculated, referring to the time from 0 to 24 h after theadministration of the study drug in Study I. Furthermore, AUC values from 12 to24 h (Study II), from 12 to 23 h (Studies III and VI), and from 23 to 47 h (StudyVI) were calculated as a posthoc analysis of AUC. Plasma concentrations forcalculation of the AUC(12-24), AUC(12-23), and AUC(23-47) values wereestimated with use of kel values when the concentrations were below thequantification limit. Furthermore, for itraconazole, diltiazem, and mibefradil, theAUC values [AUC(0-24) or AUC(0-23)] after administration of the study drugwere calculated by linear trapezoidal rule.
In Studies II and VI, after intravenous administration of methylprednisolone anddexamethasone the systemic clearance (Cl) and volume of distribution (Vd) werecalculated from the equations
Cl = Dose / AUC(0-∞)
Vd = Cl / kel
In Study VI the bioavailability of oral dexamethasone (F) was calculated (Gibaldiand Perreir 1982) from the equation
F = [AUC(0-∞)po·Doseiv] / [AUC(0-∞)iv·Dosepo]
For the pharmacokinetic calculations, the conversion of intravenousdexamethasone sodium phosphate to dexamethasone alcohol was expected to be90% (Hare et al 1975). The pharmacokinetic program, MK model, version 5(Biosoft, Cambridge, UK) were used for calculation of the pharmacokineticparameters.
3.4. Pharmacodynamic measurementsIn all studies, endogenous cortisol secretion and the adrenal-suppressant effect ofglucocorticoids were evaluated by measuring morning plasma cortisolconcentrations before the administration of the glucocorticoid and in the morning

MATERIALS AND METHODS
45
at 23 or 24, 47, and 71 h after glucocorticoid administration. In addition, in StudiesIII and V, the AUC of cortisol from 0 to 12 h after methylprednisoloneadministration [AUC(0-12)] was calculated by use of the linear trapezoidal rule.
3.5. Statistical methodsAll individual subject data, medical history, results from the clinical examination,chemical laboratory tests, ECG, and plasma drug and cortisol concentrations areconsidered as source data. The individual data were recorded into a database.However, plasma drug and cortisol concentrations were sent as an electronicdatabase.
Results are expressed as mean values with standard deviation (SD) and as meanvalues with standard error of mean (SEM) in figures. Each tmax value is presentedas median with range. The pharmacokinetic variables and the 0-, 23- or 24-, 47-,and 71-hour plasma cortisol concentrations between the phases were comparedwith Student’s 2-tailed t-test for paired data. In Study III, the statistical analysiswas done with two-way analysis of variance (ANOVA) with the Tukey test. In allstudies, the Wilcoxon test was used for analysis of tmax. The 95% confidenceintervals (CI) and ranges were presented for selected variables.
Pearson’s correlation coefficient was used to evaluate the linear correlations.Spearman rank-order correlation coefficient was used to investigate the possiblerelationship between the morning plasma cortisol concentration and the AUC(12-23) and AUC(0-∞) of methylprednisolone (Study III). In Study VI, nonlinearregression was used to evaluate the relationship between the AUC(23-47) ofdexamethasone and morning plasma cortisol concentrations at 47 and 71 h.
In all studies, the level of statistical significance was p <0.05. The number ofsubjects in each study was 8-10, which was considered to be sufficient in apharmacokinetic cross-over study to discover an about 30 to 50% change in theAUCs, sufficient large to be clinically significant. The statistical analyses weredone with the statistical program Systat for Windows, version 6.0.1 (SPSS Inc.,Chicago, Il, USA).
3.6. ArchivingAll study-related documents, e.g., protocols, original signed informed consents,source data are archived at the Department of Clinical Pharmacology, University ofHelsinki.
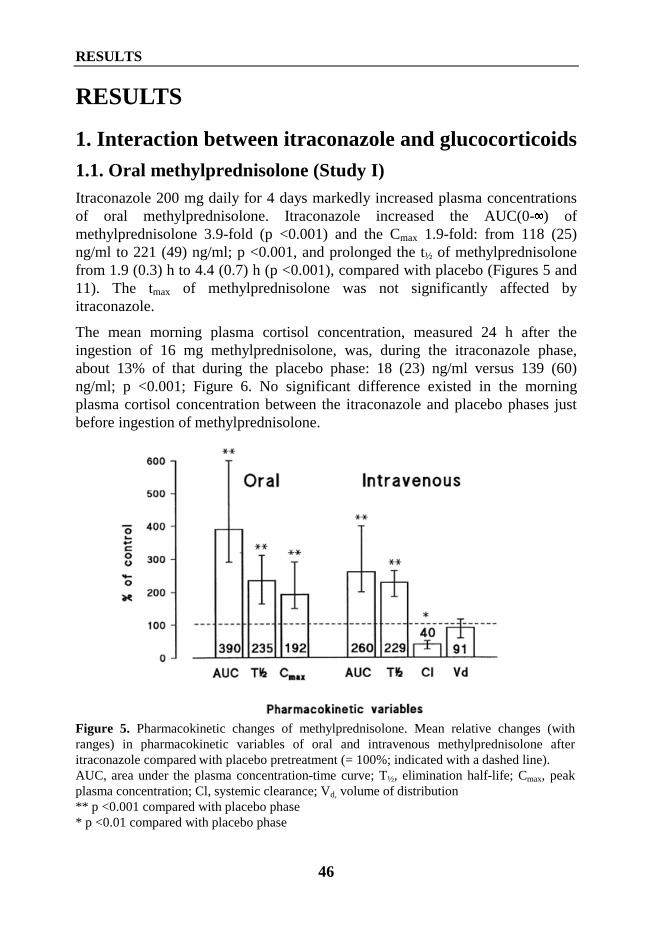
RESULTS
46
RESULTS
1. Interaction between itraconazole and glucocorticoids1.1. Oral methylprednisolone (Study I)Itraconazole 200 mg daily for 4 days markedly increased plasma concentrationsof oral methylprednisolone. Itraconazole increased the AUC(0-�) ofmethylprednisolone 3.9-fold (p <0.001) and the Cmax 1.9-fold: from 118 (25)ng/ml to 221 (49) ng/ml; p <0.001, and prolonged the t½ of methylprednisolonefrom 1.9 (0.3) h to 4.4 (0.7) h (p <0.001), compared with placebo (Figures 5 and11). The tmax of methylprednisolone was not significantly affected byitraconazole.
The mean morning plasma cortisol concentration, measured 24 h after theingestion of 16 mg methylprednisolone, was, during the itraconazole phase,about 13% of that during the placebo phase: 18 (23) ng/ml versus 139 (60)ng/ml; p <0.001; Figure 6. No significant difference existed in the morningplasma cortisol concentration between the itraconazole and placebo phases justbefore ingestion of methylprednisolone.
Figure 5. Pharmacokinetic changes of methylprednisolone. Mean relative changes (withranges) in pharmacokinetic variables of oral and intravenous methylprednisolone afteritraconazole compared with placebo pretreatment (= 100%; indicated with a dashed line).AUC, area under the plasma concentration-time curve; T½, elimination half-life; Cmax, peakplasma concentration; Cl, systemic clearance; Vd, volume of distribution** p <0.001 compared with placebo phase* p <0.01 compared with placebo phase
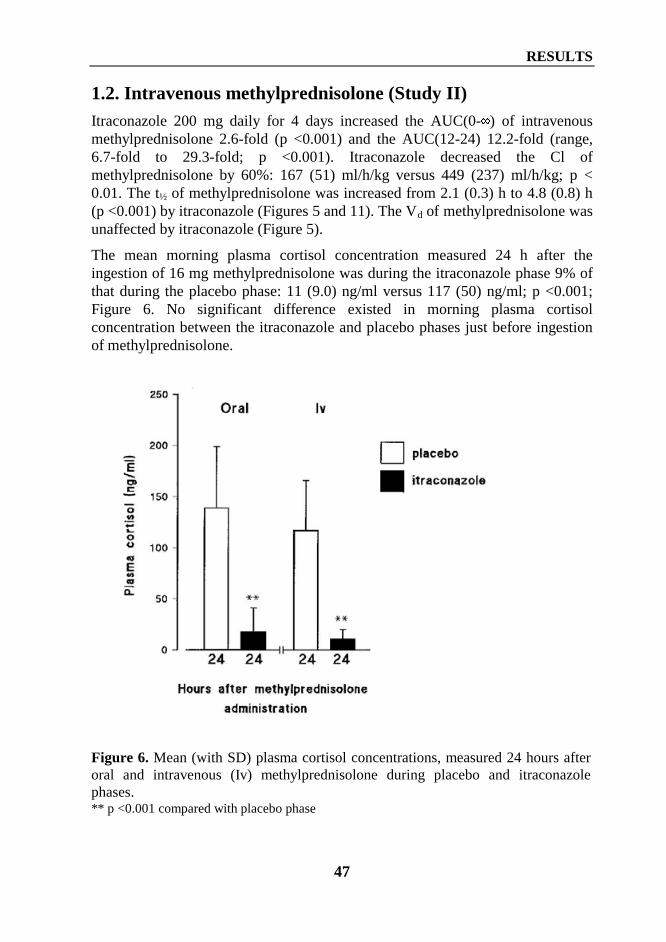
RESULTS
47
1.2. Intravenous methylprednisolone (Study II)Itraconazole 200 mg daily for 4 days increased the AUC(0-�) of intravenousmethylprednisolone 2.6-fold (p <0.001) and the AUC(12-24) 12.2-fold (range,6.7-fold to 29.3-fold; p <0.001). Itraconazole decreased the Cl ofmethylprednisolone by 60%: 167 (51) ml/h/kg versus 449 (237) ml/h/kg; p <0.01. The t½ of methylprednisolone was increased from 2.1 (0.3) h to 4.8 (0.8) h(p <0.001) by itraconazole (Figures 5 and 11). The Vd of methylprednisolone wasunaffected by itraconazole (Figure 5).
The mean morning plasma cortisol concentration measured 24 h after theingestion of 16 mg methylprednisolone was during the itraconazole phase 9% ofthat during the placebo phase: 11 (9.0) ng/ml versus 117 (50) ng/ml; p <0.001;Figure 6. No significant difference existed in morning plasma cortisolconcentration between the itraconazole and placebo phases just before ingestionof methylprednisolone.
Figure 6. Mean (with SD) plasma cortisol concentrations, measured 24 hours afteroral and intravenous (Iv) methylprednisolone during placebo and itraconazolephases.** p <0.001 compared with placebo phase
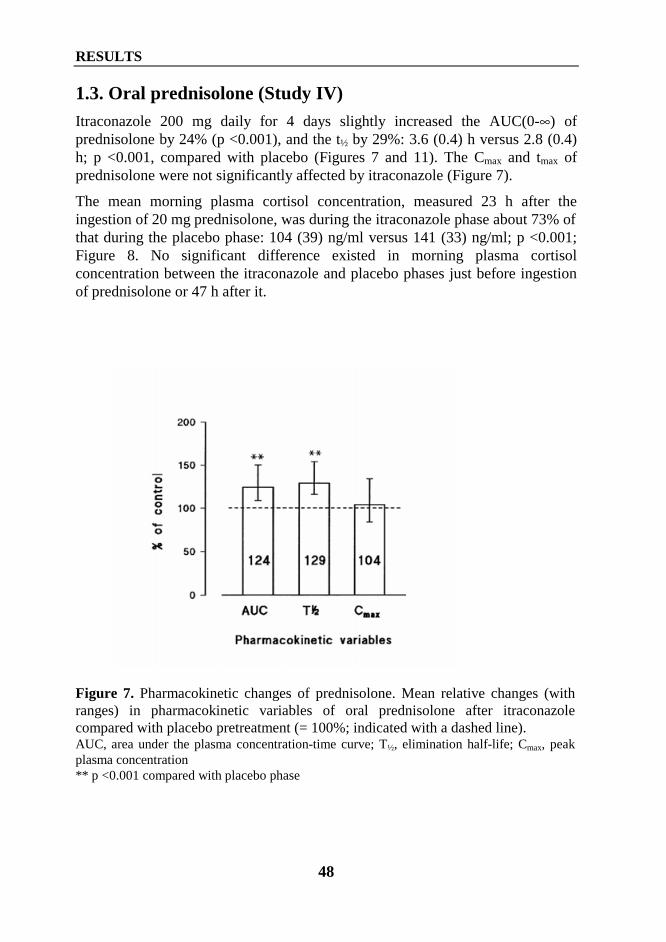
RESULTS
48
1.3. Oral prednisolone (Study IV)Itraconazole 200 mg daily for 4 days slightly increased the AUC(0-∞) ofprednisolone by 24% (p <0.001), and the t½ by 29%: 3.6 (0.4) h versus 2.8 (0.4)h; p <0.001, compared with placebo (Figures 7 and 11). The Cmax and tmax ofprednisolone were not significantly affected by itraconazole (Figure 7).
The mean morning plasma cortisol concentration, measured 23 h after theingestion of 20 mg prednisolone, was during the itraconazole phase about 73% ofthat during the placebo phase: 104 (39) ng/ml versus 141 (33) ng/ml; p <0.001;Figure 8. No significant difference existed in morning plasma cortisolconcentration between the itraconazole and placebo phases just before ingestionof prednisolone or 47 h after it.
Figure 7. Pharmacokinetic changes of prednisolone. Mean relative changes (withranges) in pharmacokinetic variables of oral prednisolone after itraconazolecompared with placebo pretreatment (= 100%; indicated with a dashed line).AUC, area under the plasma concentration-time curve; T½, elimination half-life; Cmax, peakplasma concentration** p <0.001 compared with placebo phase
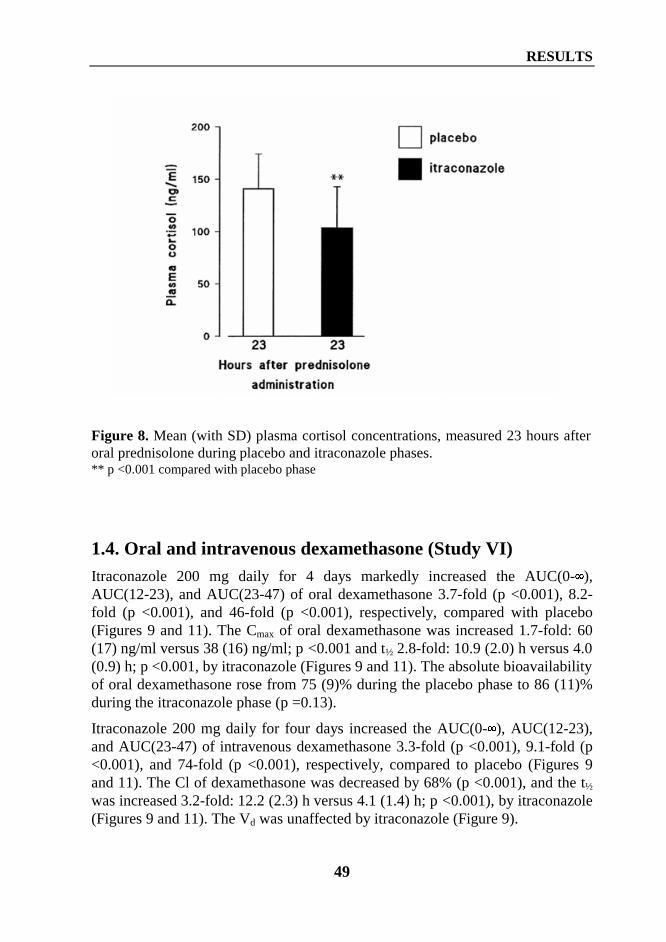
RESULTS
49
Figure 8. Mean (with SD) plasma cortisol concentrations, measured 23 hours afteroral prednisolone during placebo and itraconazole phases.** p <0.001 compared with placebo phase
1.4. Oral and intravenous dexamethasone (Study VI)Itraconazole 200 mg daily for 4 days markedly increased the AUC(0-�),AUC(12-23), and AUC(23-47) of oral dexamethasone 3.7-fold (p <0.001), 8.2-fold (p <0.001), and 46-fold (p <0.001), respectively, compared with placebo(Figures 9 and 11). The Cmax of oral dexamethasone was increased 1.7-fold: 60(17) ng/ml versus 38 (16) ng/ml; p <0.001 and t½ 2.8-fold: 10.9 (2.0) h versus 4.0(0.9) h; p <0.001, by itraconazole (Figures 9 and 11). The absolute bioavailabilityof oral dexamethasone rose from 75 (9)% during the placebo phase to 86 (11)%during the itraconazole phase (p =0.13).
Itraconazole 200 mg daily for four days increased the AUC(0-�), AUC(12-23),and AUC(23-47) of intravenous dexamethasone 3.3-fold (p <0.001), 9.1-fold (p<0.001), and 74-fold (p <0.001), respectively, compared to placebo (Figures 9and 11). The Cl of dexamethasone was decreased by 68% (p <0.001), and the t½
was increased 3.2-fold: 12.2 (2.3) h versus 4.1 (1.4) h; p <0.001), by itraconazole(Figures 9 and 11). The Vd was unaffected by itraconazole (Figure 9).
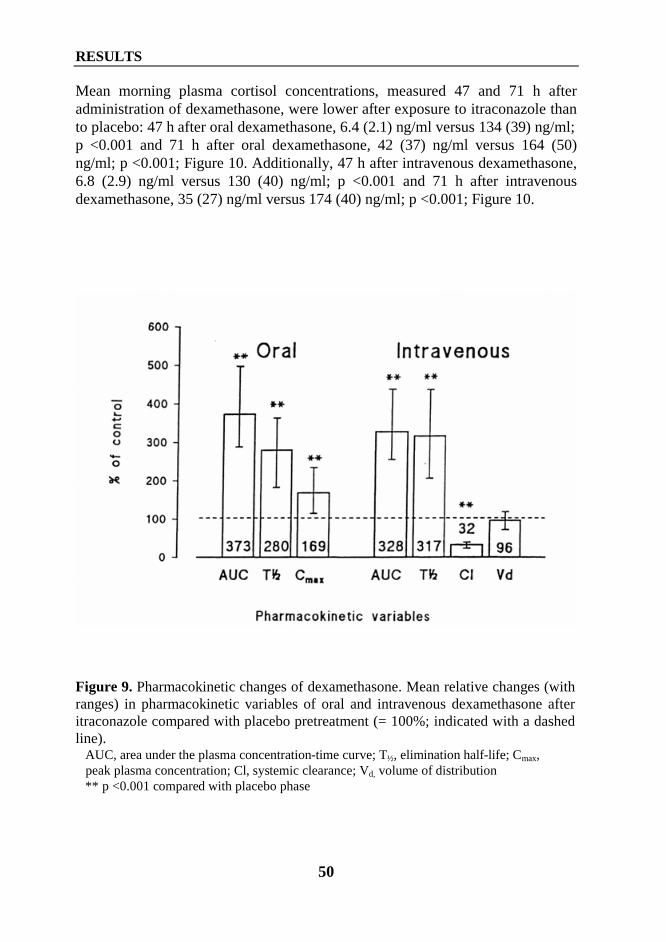
RESULTS
50
Mean morning plasma cortisol concentrations, measured 47 and 71 h afteradministration of dexamethasone, were lower after exposure to itraconazole thanto placebo: 47 h after oral dexamethasone, 6.4 (2.1) ng/ml versus 134 (39) ng/ml;p <0.001 and 71 h after oral dexamethasone, 42 (37) ng/ml versus 164 (50)ng/ml; p <0.001; Figure 10. Additionally, 47 h after intravenous dexamethasone,6.8 (2.9) ng/ml versus 130 (40) ng/ml; p <0.001 and 71 h after intravenousdexamethasone, 35 (27) ng/ml versus 174 (40) ng/ml; p <0.001; Figure 10.
Figure 9. Pharmacokinetic changes of dexamethasone. Mean relative changes (withranges) in pharmacokinetic variables of oral and intravenous dexamethasone afteritraconazole compared with placebo pretreatment (= 100%; indicated with a dashedline). AUC, area under the plasma concentration-time curve; T½, elimination half-life; Cmax, peak plasma concentration; Cl, systemic clearance; Vd, volume of distribution ** p <0.001 compared with placebo phase
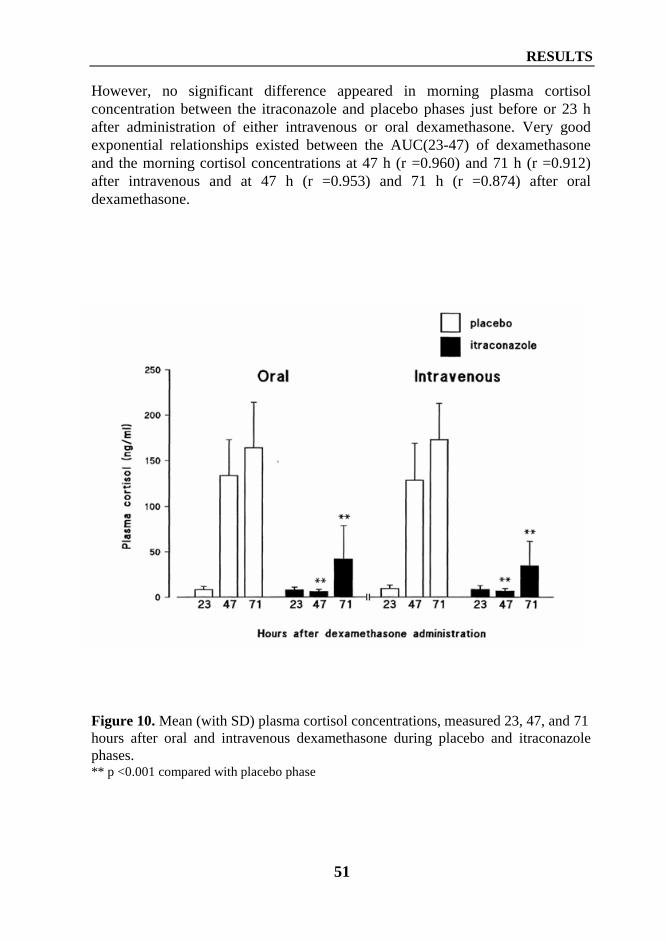
RESULTS
51
However, no significant difference appeared in morning plasma cortisolconcentration between the itraconazole and placebo phases just before or 23 hafter administration of either intravenous or oral dexamethasone. Very goodexponential relationships existed between the AUC(23-47) of dexamethasoneand the morning cortisol concentrations at 47 h (r =0.960) and 71 h (r =0.912)after intravenous and at 47 h (r =0.953) and 71 h (r =0.874) after oraldexamethasone.
Figure 10. Mean (with SD) plasma cortisol concentrations, measured 23, 47, and 71hours after oral and intravenous dexamethasone during placebo and itraconazolephases.** p <0.001 compared with placebo phase
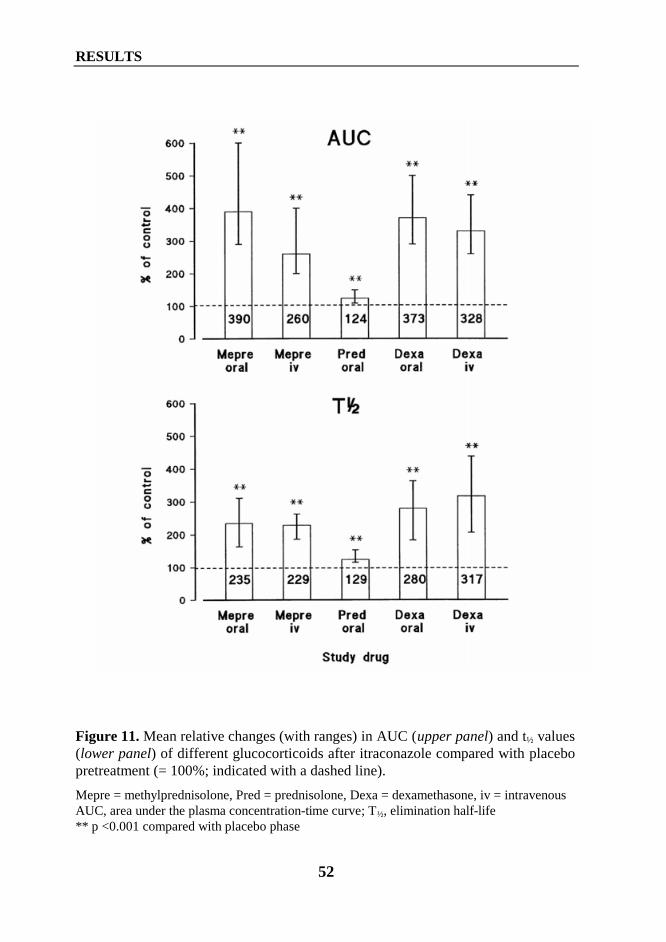
RESULTS
52
Figure 11. Mean relative changes (with ranges) in AUC (upper panel) and t½ values(lower panel) of different glucocorticoids after itraconazole compared with placebopretreatment (= 100%; indicated with a dashed line).
Mepre = methylprednisolone, Pred = prednisolone, Dexa = dexamethasone, iv = intravenousAUC, area under the plasma concentration-time curve; T½, elimination half-life** p <0.001 compared with placebo phase
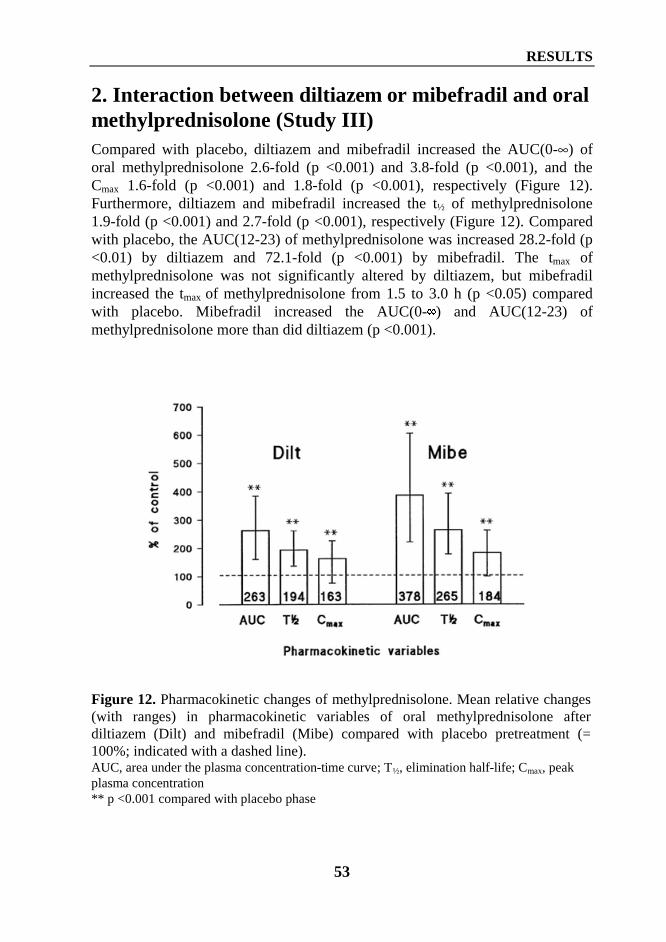
RESULTS
53
2. Interaction between diltiazem or mibefradil and oralmethylprednisolone (Study III)Compared with placebo, diltiazem and mibefradil increased the AUC(0-∞) oforal methylprednisolone 2.6-fold (p <0.001) and 3.8-fold (p <0.001), and theCmax 1.6-fold (p <0.001) and 1.8-fold (p <0.001), respectively (Figure 12).Furthermore, diltiazem and mibefradil increased the t½ of methylprednisolone1.9-fold (p <0.001) and 2.7-fold (p <0.001), respectively (Figure 12). Comparedwith placebo, the AUC(12-23) of methylprednisolone was increased 28.2-fold (p<0.01) by diltiazem and 72.1-fold (p <0.001) by mibefradil. The tmax ofmethylprednisolone was not significantly altered by diltiazem, but mibefradilincreased the tmax of methylprednisolone from 1.5 to 3.0 h (p <0.05) comparedwith placebo. Mibefradil increased the AUC(0-�) and AUC(12-23) ofmethylprednisolone more than did diltiazem (p <0.001).
Figure 12. Pharmacokinetic changes of methylprednisolone. Mean relative changes(with ranges) in pharmacokinetic variables of oral methylprednisolone afterdiltiazem (Dilt) and mibefradil (Mibe) compared with placebo pretreatment (=100%; indicated with a dashed line).AUC, area under the plasma concentration-time curve; T½, elimination half-life; Cmax, peakplasma concentration** p <0.001 compared with placebo phase
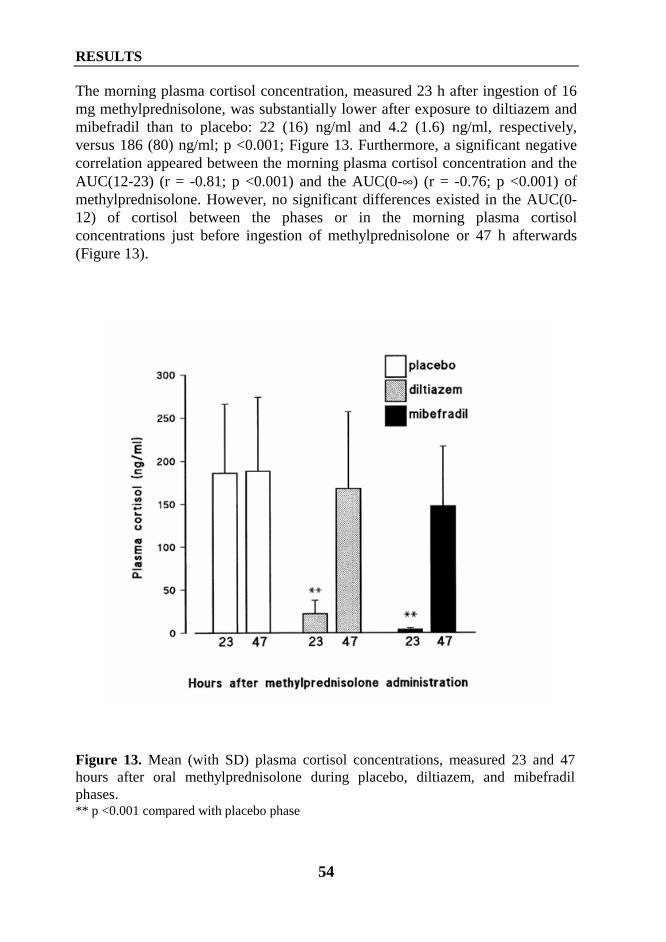
RESULTS
54
The morning plasma cortisol concentration, measured 23 h after ingestion of 16mg methylprednisolone, was substantially lower after exposure to diltiazem andmibefradil than to placebo: 22 (16) ng/ml and 4.2 (1.6) ng/ml, respectively,versus 186 (80) ng/ml; p <0.001; Figure 13. Furthermore, a significant negativecorrelation appeared between the morning plasma cortisol concentration and theAUC(12-23) (r = -0.81; p <0.001) and the AUC(0-∞) (r = -0.76; p <0.001) ofmethylprednisolone. However, no significant differences existed in the AUC(0-12) of cortisol between the phases or in the morning plasma cortisolconcentrations just before ingestion of methylprednisolone or 47 h afterwards(Figure 13).
Figure 13. Mean (with SD) plasma cortisol concentrations, measured 23 and 47hours after oral methylprednisolone during placebo, diltiazem, and mibefradilphases.** p <0.001 compared with placebo phase
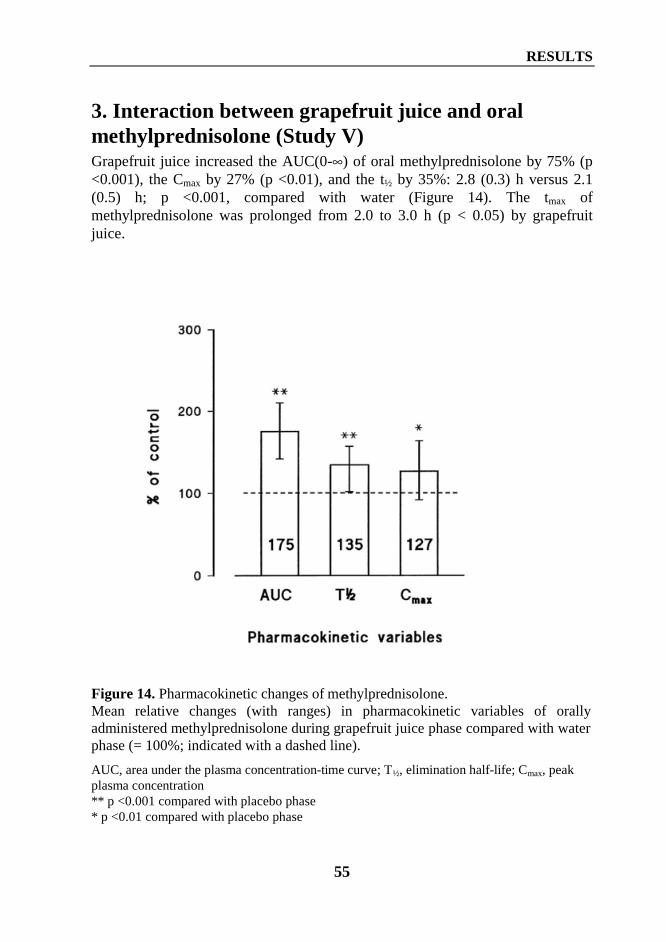
RESULTS
55
3. Interaction between grapefruit juice and oralmethylprednisolone (Study V)Grapefruit juice increased the AUC(0-∞) of oral methylprednisolone by 75% (p<0.001), the Cmax by 27% (p <0.01), and the t½ by 35%: 2.8 (0.3) h versus 2.1(0.5) h; p <0.001, compared with water (Figure 14). The tmax ofmethylprednisolone was prolonged from 2.0 to 3.0 h (p < 0.05) by grapefruitjuice.
Figure 14. Pharmacokinetic changes of methylprednisolone.Mean relative changes (with ranges) in pharmacokinetic variables of orallyadministered methylprednisolone during grapefruit juice phase compared with waterphase (= 100%; indicated with a dashed line).
AUC, area under the plasma concentration-time curve; T½, elimination half-life; Cmax, peakplasma concentration** p <0.001 compared with placebo phase* p <0.01 compared with placebo phase
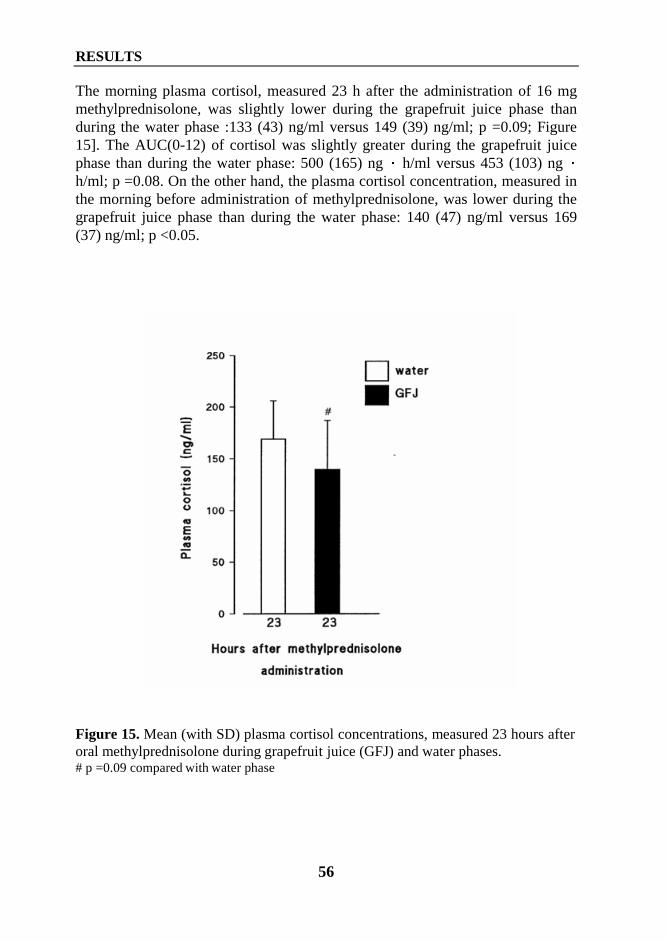
RESULTS
56
The morning plasma cortisol, measured 23 h after the administration of 16 mgmethylprednisolone, was slightly lower during the grapefruit juice phase thanduring the water phase :133 (43) ng/ml versus 149 (39) ng/ml; p =0.09; Figure15]. The AUC(0-12) of cortisol was slightly greater during the grapefruit juicephase than during the water phase: 500 (165) ng � h/ml versus 453 (103) ng �h/ml; p =0.08. On the other hand, the plasma cortisol concentration, measured inthe morning before administration of methylprednisolone, was lower during thegrapefruit juice phase than during the water phase: 140 (47) ng/ml versus 169(37) ng/ml; p <0.05.
Figure 15. Mean (with SD) plasma cortisol concentrations, measured 23 hours afteroral methylprednisolone during grapefruit juice (GFJ) and water phases.# p =0.09 compared with water phase

DISCUSSION
57
DISCUSSION
1. Methodological considerationsThe present study evaluated the effect of CYP3A4 inhibitors on syntheticglucocorticoids after their systemic administration. In addition tomethylprednisolone, prednisolone, and dexamethasone, also betamethasone andprednisone are in clinical use in Finland. Betamethasone and prednisone were notevaluated in the present study. Prednisone, however, is considered a prodrug andis further metabolised to prednisolone.
All studies were carried out in healthy subjects, and after their pretreatment withCYP3A4 inhibitor, a single dose of glucocorticoid was administered. This shouldbe considered in the extrapolation of the pharmacokinetic and pharmacodynamicresults to the patient using systemic glucocorticoids as a long-term treatment ofchronic disease. The doses of 20 mg prednisolone and of 16 mgmethylprednisolone used in the present study are equivalent regarding theirpotency to glucocorticoid activity (Table III, page 26). However, an equivalentdose of dexamethasone would be 3.0 mg. Thus, dexamethasone was used at higher(3.85 mg and 4.5 mg) doses regarding the potency than were methylprednisoloneand prednisolone.
The susceptibility of glucocorticoids to interact with CYP3A4 inhibitors wasstudied with potential CYP3A4 inhibitors currently on the market and in clinicaluse. Mibefradil was, however, withdrawn from the market in 1998 during theclinical phase of Study III. Each subject received oral and written information andgave additional written consent.
The CYP3A4-enzyme inhibitors of the present study possess different inhibitorymechanisms. Itraconazole, for example, is a competitive inhibitor of CYP3A4, andgrapefruit juice and maybe also mibefradil are non-competitive mechanism-basedinactivators of CYP3A4 (Back and Tjia 1991, Lown et al 1997, Prueksaritanont elal 1999). The pretreatment period ranged from 3 to 4 days, because even such ashort treatment is sufficient to reveal a clinically significant inhibition ofCYP3A4. The t½ of mibefradil ranges from 10 h to 38 h, and it may be suggestedthat the steady-state concentration was not reached during the 3-day treatment. Inaddition, diltiazem has been shown dose-dependently to increase the plasmaconcentrations of nifedipine (Tateishi et al 1989). Because the steady-stateconcentration of the CYP3A4 inhibitors was perhups not reached during the 3 to 4days, the interactions observed may become even more pronounced afterpretreatment for longer period or with a higher dose.

DISCUSSION
58
The adrenal-suppressant effect of glucocorticoids was evaluated by measuringmorning plasma cortisol concentrations. The normal range for the morning plasmacortisol concentration taken between 8:00 a.m. and 10:00 a.m. varies between 50and 250 ng/ml according to the laboratory of Helsinki University Central Hospital.In the present study blood samples for plasma cortisol determination were taken at8:00 a.m. or at 9:00 a.m.. Cortisol secretion is pulsatile and exhibits a diurnalpattern with the highest concentration occurring early in the morning. Morningcortisol measurement has been used as an index of adrenal function, because itreflects the peak endogenous activation of the HPA-axis (Grinspoon and Biller1994). According to Grinspoon and Biller (1994), patients with a morning cortisolconcentration above 190 ng/ml do not need further testing for adrenalinsufficiency; although a low morning cortisol concentration may reflect the nadirbetween pulses of cortisol secretion, a cortisol concentration of less than 30 ng/mlis considered diagnostic for adrenal insufficiency. In the present series ofinteraction studies, cortisol concentrations were not measured during the night-time, which could have provided additional information.
2. Interaction between CYP3A4 inhibitors andglucocorticoids2.1. MethylprednisoloneItraconazole markedly affected the pharmacokinetics both of oral and ofintravenous methylprednisolone. Itraconazole increased the AUC of oralmethylprednisolone 3.9- fold, the AUC of intravenous methylprednisolone 2.6-fold, and decreased the Cl of methylprednisolone by 60%. The effect ofitraconazole on the AUC of methylprednisolone was about 50% greater after oralthan after intravenous administration. This finding can be explained by theinhibition of the first-pass metabolism after oral administration, resulting inincreased bioavailability of methylprednisolone. This absolute bioavailability,however, could not be calculated, because the effects of itraconazole on the oraland intravenous methylprednisolone were evaluated in separate studies.
Diltiazem and mibefradil significantly affected the pharmacokinetics of oralmethylprednisolone. The extent to which mibefradil affected the pharmacokineticsof oral methylprednisolone was as great as that seen after itraconazole in Study I.This finding is in good agreement with that of an in vitro study in whichmibefradil was as potent inhibitor of CYP3A4 as was itraconazole (Wang et al1999). Thus, these in vitro results predicted well the interactions observed in vivoin the present studies. Furthermore, the effect of diltiazem on thepharmacokinetics of methylprednisolone was smaller than that of mibefradil.

DISCUSSION
59
However, in some subjects diltiazem affected the AUC of methylprednisolone asalmost as greatly as mibefradil.
Troleandomycin, a potent inhibitor of CYP3A4, has shown to decrease the Cl ofmethylprednisolone by 64% (Szefler et al 1980). Furthermore, ketoconazole,which is an even more potent inhibitor of CYP3A4 in vitro than is itraconazole,increased the AUC of methylprednisolone about 2.4-fold and decreased its Cl by66% (Glynn et al 1986). Erythromycin decreased the Cl of methylprednisolone by46%, and in a recent study, clarithromycin reduced the apparent oral Cl ofmethylprednisolone by 65% (LaForce et al 1983, Fost et al 1999). In summary, ourresults for itraconazole, diltiazem, and mibefradil are in concordance with those ofprevious studies on the effect of other potent CYP3A4 inhibitors on thepharmacokinetics of methylprednisolone. Furthermore, in a congress report,itraconazole for 4 days increased the AUC of oral methylprednisolone (48 mg)about 2.5-fold, confirming the results of the present study (Lebrun-Vignes et al1998).
In this study, high amounts of grapefruit juice increased the AUC of oralmethylprednisolone by 75%. Because grapefruit juice reduced intestinal CYP3A4without affecting hepatic CYP3A4 activity (Lown et al 1997), it may be suggestedthat the increase in AUC of oral methylprednisolone mainly resulted frominhibition of its first-pass metabolism. However, recent studies have shown highand repeated consumption of grapefruit juice to increase also the t½ of such drugsas oral buspirone and cisapride (Lilja et al 1998b, Kivistö et al 1999). In thepresent study, grapefruit juice slightly but significantly increased the t½ of oralmethylprednisolone, suggesting that it inhibited also systemic (hepatic) CYP3A4-mediated methylprednisolone metabolism. Grapefruit juice alters thepharmacokinetics of methylprednisolone considerably less than that of drugs suchas simvastatin, lovastatin, and buspirone (Lilja et al 1998a, Kantola et al 1998b,Lilja et al 1998b). This may be because of the rather small first-pass metabolismof methylprednisolone compared to that of simvastatin, lovastatin, and buspirone.
The increased and prolonged exposure to methylprednisolone during theitraconazole, the diltiazem, and the mibefradil phases was associated with muchlower morning plasma cortisol concentrations at 23/24 h than those seen duringthe placebo phase. Thus, concomitant use of methylprednisolone with itraconazoleor with these calcium-channel blockers enhances the adrenal-suppressant effect ofmethylprednisolone by completely suppressing the peak cortisol concentrationseen in the placebo phase.
The concentration-effect relationship is log-linear, and despite the fact thatitraconazole increased the AUC of methylprednisolone by some 50% more afteroral than after intravenous administration, the extent of adrenal-suppressant effectduring the itraconazole phases was about the same. Furthermore, a negative

DISCUSSION
60
correlation appeared between the night-time exposure to methylprednisolone[AUC(12-23)] and the morning plasma cortisol concentrations (Study III). It isknown that endogenous cortisol is secreted almost immediately after synthesis(Berne and Levy 1998). Therefore, increases in t½ and the night-timeconcentrations of methylprednisolone may effectively suppress the peak cortisolconcentrations occurring early in the morning.
It can be concluded that these interactions between itraconazole, diltiazem, ormibefradil and methylprednisolone are clinically significant and enhance theadrenal-suppressant and perhups also other effects of methylprednisolone. In thepresent study, the adrenal-suppressant effect of methylprednisolone tended toincrease during the grapefruit juice phase compared with that of the water phase.Despite the statistically significant increase in the AUC, Cmax, and t½ of oralmethylprednisolone caused by grapefruit juice, the clinical significance of thisinteraction appears to be minor. However, it might enhance the effects ofmethylprednisolone in a few subjects.
2.2. PrednisoloneDespite the statistically significant increase in the AUC and t½ of oralprednisolone caused by itraconazole, the interaction was quantitatively minor andis probably of limited clinical significance. This observation is in agreement withearlier findings that prednisolone does not significantly interact withtroleandomycin, ketoconazole, or clarithromycin, all potent inhibitors of CYP3A4(Szefler et al 1982, Ludwig et al 1989, Yamashita et al 1991, Frost et al 1999). Inaddition, the pharmacodynamics of prednisolone, evaluated by its suppressiveeffects on serum cortisol, blood basophil, and helper T-lymphocyte values, werenot affected during the ketoconazole phase (Ludwig et al 1989, Yamashita et al1991). In the present study, the adrenal-suppressant effect slightly increasedduring the itraconazole phase compared with that of the placebo phase. Theclinical significance of this change is probably minor.
However, in contrast to other reports, Zürcher et al (1989) found ketoconazole toraised prednisolone plasma concentration by 50% after both oral prednisone andintravenous prednisolone, and Imani et al (1999) reported diltiazem to raise theAUC of prednisolone slightly, by about 20%, after prednisone administration.Imani et al (1999) suggested that diltiazem may in some patients enhance theeffects of prednisone. Furthermore, Zürcher et al (1989) suggested that the enzymeresponsible for the interconversion of prednisolone ↔ prednisone was unaffectedby ketoconazole. It may be assumed that the effect of itraconazole, another azoleantimycotic agent and potent inhibitor of CYP3A4, on plasma concentrations ofprednisolone is also similar after both prednisolone and prednisone administration.

DISCUSSION
61
A congress report by Lebrun-Vignes et al (1998) confirms that itraconazole forfour days does not affect the pharmacokinetics prednisolone after oral prednisone(60 mg).
In conclusion, the results of the present study strongly suggest that CYP3A4 doesnot play a major role in the metabolic pathways of prednisolone. The concomitantuse of prednisolone or prednisone with itraconazole or with other potent CYP3A4inhibitors should not markedly alter the effects of these glucocorticoids.
2.3. DexamethasoneItraconazole greatly affected the pharmacokinetics both of oral and of intravenousdexamethasone. Itraconazole increased the AUC of oral dexamethasone 3.7-fold,the AUC of intravenous dexamethasone 3.3-fold, and decreased the Cl ofdexamethasone by 68%. The effect of itraconazole on the AUC of dexamethasonewas similar after oral and after intravenous administration. Furthermore, theabsolute bioavailability of oral dexamethasone was slightly elevated, from 75% to86%, by itraconazole. It is known that a significant increase in the AUC byenzyme inhibition both after oral and after intravenous administration togetherwith small change in the absolute bioavailability are typical of a drug with lowhepatic clearance (Lin and Lu 1998).
The extent of itraconazole-dexamethasone interaction was approximately as greatas the extent of itraconazole-methylprednisolone interaction. Thus, thesusceptibility of dexamethasone and of methylprednisolone to interact withitraconazole is similar. There seem to be no previous studies on the effects ofCYP3A4 inhibitors on the pharmacokinetics of oral and intravenousdexamethasone. However, CYP3A4 has been shown to be important inmetabolism of dexamethasone in vitro (Gentile et al 1996). The present resultsstrongly suggest that this is the case also in vivo in human beings.
The increased exposure to dexamethasone during the itraconazole phases wasassociated with much reduced morning plasma cortisol concentrations at 47 and71 h than those during the placebo phases. The extent of the adrenal-suppressanteffect of dexamethasone during the itraconazole phase was about the same,regardless of the route of dexamethasone administration.
In conclusion, concomitant use of itraconazole or probably also other potentCYP3A4 inhibitors woth dexamethasone greatly enhances the adrenal-suppressantand perhaps also other effects of dexamethasone.

DISCUSSION
62
3. General discussion
Itraconazole, diltiazem, and mibefradil are known to inhibit CYP3A4-mediateddrug metabolism. Therefore, it can be assumed that inhibition of CYP3A4-mediated metabolism during first-pass and elimination phases is the most probablemechanism explaining the observed interactions in the present study. However,itraconazole, diltiazem, and mibefradil are also inhibitors of P-gp (Miyama et al1998, Cornwell et al 1987, Wandel et al 2000). In addition, grapefruit juice hasbeen demonstrated to inhibit the vinblastine cellular efflux by P-gp and tomodulate to a limited extent P-gp mediated transport of saquinavir in vitro(Takanaga et al 1998, Eagling et al 1999). Eagling et al (1997) suggest, however,that the in vivo effects of grapefruit juice most likely result from inhibition ofCYP3A4 and only to a minor extent from modulation of P-gp. In contrast toprednisolone (Saitoh et al 1998), methylprednisolone and dexamethasone havebeen shown to be substrates for P-gp (Saitoh et al 1998, Ueda et al 1992);therefore, inhibition of this transmembrane efflux pump may have contributed tothe observed interactions. However, present studies do not allow the separation ofthe role of CYP3A4 and P- gp in these interactions.
All glucocorticoids are readily eliminated by metabolism. Although the role ofindividual CYP enzymes in the metabolic pathways of various glucocorticoidsremains unclear, CYP3A4 probably has a dissimilar role in the metabolism ofeach glucocorticoid. Based on the studies presented here, itraconazole, a potentinhibitor of CYP3A4, greatly interacted with both methylprednisolone anddexamethasone, but only slightly affected the pharmacokinetics of prednisolone.This strongly suggests that the metabolic pathways of these glucocorticoids aredissimilar and that, in contrast to prednisolone, CYP3A4 has an important role inthe metabolism of methylprednisolone and dexamethasone.
Induction of CYP3A4 by glucocorticoids may counteract the effects of CYP3A4inhibitors on these glucocorticoids during long-term use. Dexamethasone has beenshown to induce CYP3A4 in vitro (Pichard et al 1992), and in cancer patients toincrease the Cl of cyclophosphamide, a substrate for CYP3A4 (Yule et al 1996,Busse et al 1997). However, in controlled studies, dexamethasone does not affectthe AUC of triazolam and midazolam, both known substrates for CYP3A4(Villikka et al 1998, Palmer et al 2000). Thus, dexamethasone seems not to be apotent inducer of CYP3A4-mediated metabolism in human beings.
The concomitant use of the CYP3A4 inhibitors itraconazole, diltiazem, ormibefradil with methylprednisolone, and of itraconazole with dexamethasonemarkedly enhanced the adrenal-suppressant effect of these glucocorticoids. Theprologned suppression was due to the increased effect of methylprednisolone ordexamethasone, because neither itraconazole, diltiazen nor mibefradil

DISCUSSION
63
significantly affected the plasma cortisol concentrations measured in the morningbefore glucocorticoid administration. The marked decrease in elimination ofmethylprednisolone and dexamethasone is particularly important, because itappears also to prolong the biological half-lives of these steroids. Thus, not onlythe exposure to methylprednisolone and dexamethasone increased, but also the”biological profile” of these steroids changed. Furthermore, the increasedexposure to methylprednisolone, especially during night-time, enhanced itsadrenal-suppressant effect by suppressing the peak cortisol concentration evidentduring the placebo phase. This may be important in patients receiving low dosesof methylprednisolone once daily.
Present studies were performed in healthy volunteers after administration of asingle dose of glucocorticoid. It can be assumed, however, that the great effect ofCYP3A4 inhibitors on the pharmacokinetics and pharmacodynamics ofmethylprednisolone or dexamethasone can be seen also in patients receiving long-term glucocorticoid treatment. During long-term treatment, the increased andprolonged exposure to glucocorticoids caused by other concomitant medicationmay alter the clinical response to glucocorticoid. Besides plasma cortisolconcentratios and the addrenal-suppressant effect of glucocorticoid, no otherpharmacodynamic parameters were assessed in the studies of this thesis. However,there are several lines of evidence in the literature to suggest that concomitant useof a potent inhibitor of CYP3A4 with glucocorticoid, or increased exposure to aglucocorticoid may enhance many different glucocorticoid-related effects.
In one case report, addition of itraconazole to a transplant patient´simmunosuppressive regimen which includied methylprednisolone led to myopathyand diabetes, suggesting a clinically important interaction between itraconazoleand methylprednisolone (Linthoudt et al 1996). Similar, or even more severeitraconazole-methylprednisolone interactions are known to have occurred duringtheir combined clinical use. Furthermore, combination of troleandomycin andmethylprednisolone had a beneficial ”steroid-sparing” effect on difficult asthma(Siracusa et al 1993). However, this combination led to increased risk ofglucocorticoid-related adverse effects (e.g., reduction in bone mineral content,increase in plasma glucose levels), and even one case of fatal varicella has beenreported, suggesting enhanced immunosuppression (Harris et al 1989, Siracusa etal 1993, Lantner et al 1990). Diltiazem and verapamil can improve the outcome oforgan-transplant patients treated with methylprednisone (Wagner et al 1987,Kunzendorf et al 1991, Dawidson et al 1989, Mies et al 1998), which may in partbe explained by the increase in the AUC of methylprednisolone caused bydiltiazem. Furthermore, methylprednisolone exposure rather than themethylprednisolone dose predicts adrenal suppression and growth inhibition inchildren with liver and renal transplants (Sarna et al 1997). In addition, the

DISCUSSION
64
diabetogenic effect of dexamethasone appears to be dose-dependent (Matsumotoet al 1996).
The pharmacokinetic interaction between itraconazole and dexamethasone andbetween itraconazole and methylprednisolone was slightly greater after oral thanafter intravenous administration of these two steroids. This finding may favourintravenous rather than oral use of methylprednisolone and dexamethasone.However, the extent of the adrenal-suppressant effect was similar after their oraland their intravenous administration. The extent of interaction observed after oraladministration might be reduced by giving itraconazole some 12 h afterglucocorticoid, as shown in a recent study of ketoconazole with budesonide(Seidegård 2000).

SUMMARY AND CONCLUSIONS
65
SUMMARY AND CONCLUSIONS
One of the most important drug-metabolising enzymes in human beings isCYP3A4. Many drugs and also grapefruit juice can inhibit the catalytic activityof CYP3A4, which may result in a clinically significant increase in exposure todrugs metabolised mainly by this enzyme.
The systemic glucocorticoids methylprednisolone, prednisolone, anddexamethasone have been used for many years for various clinical indicationsand are eliminated mainly by metabolism. Although the role of individual CYPenzymes in the metabolic pathways of these glucocorticoids seems not to beclear, CYP3A4 probably plays a dissimilar role in their metabolism.
The aim of this series of studies was to investigate the effect of CYP3A4inhibitors on the pharmacokinetics and pharmacodynamics of systemicglucocorticoids: the effect of itraconazole on oral (Study I) and intravenous(Study II) methylprednisolone, on oral prednisolone (Study IV), and on oral andintravenous dexamethasone (Study VI). In addition, the effect of diltiazem ormibefradil (Study III), and grapefruit juice (Study V) on oral methylprednisolonewas investigated. The effect of route of administration of methylprednisolone anddexamethasone on their possible interaction with itraconazole was also evaluated(Studies I, II, and VI).
The studies were double-blind, randomised, placebo-controlled, and cross-over,except for the grapefruit juice-methylprednisolone interaction study, which hadan open, randomised, cross-over design. All studies were carried out in 8 to 10healthy subjects. A single dose of glucocorticoid was administered after or duringthe administration of a CYP3A4 inhibitor for 3 to 4 days. Blood samples werecollected, and plasma drug and cortisol concentrations determined by specifichigh-performance liquid chromatographic and mass spectrometric methods.
Itraconazole increased the AUC of oral and intravenous methylprednisolone 3.9-fold (p <0.001) and 2.6-fold (p <0.001). The AUC of oral and intravenousdexamethasone was increased 3.7-fold (p <0.001) and 3.3-fold (p <0.001) byitraconazole. The use of itraconazole enhanced the adrenal-suppressant effect ofboth methylprednisolone and dexamethasone. In contrast, itraconazole increasedthe AUC of oral prednisolone by only 24% (p <0.001) and had a minor effect onthe adrenal-suppressant effect of prednisolone. Both diltiazem and mibefradilincreased the AUC of oral methylprednisolone (2.6-fold and 3.8-fold; p <0.001)and greatly enhanced the adrenal-suppressant effect of methylprednisolone.Grapefruit juice increased the AUC of oral methylprednisolone by 75% (p<0.001); however, it did not significantly affect the adrenal-suppressant effect ofmethylprednisolone. The route of methylprednisolone or dexamethasone

SUMMARY AND CONCLUSIONS
66
administration may have a only minor effect on the extent of itraconazole-methylprednisolone and itraconazole-dexamethasone interactions.
The following conclusions can be drawn on the basis of Studies I-VI:
1. Interactions between the antifungal agent itraconazole and methylprednisoloneor dexamethasone are clinically important. Itraconazole and probably also otherpotent CYP3A4 inhibitors should be used with caution in patients receivingdexamethasone or methylprednisolone, particularly during long-termconcomitant use.
2. The interaction between itraconazole and prednisolone is unlikely to beclinically significant.
3. The interactions between the calcium-channel blockers diltiazem or mibefradiland methylprednisolone are clinically important. Diltiazem and mibefradilshould be used with caution in patients receiving methylprednisolone,particularly during long-term concomitant use.
4. The clinical significance of the interaction between grapefruit juice and oralmethylprednisolone is probably minor. However, high repeated doses ofgrapefruit juice may enhance the effects of oral methylprednisolone in somesensitive subjects.
5. The route of methylprednisolone or dexamethasone administration may have aminor effect on the extent of itraconazole-methylprednisolone and itraconazole-dexamethasone interactions. Despite the differences seen in pharmacokineticinteractions, the extent of the adrenal-suppressant effect was similar after oral orintravenous dose of these glucocorticoids.
6. All of these interactions are probably due to inhibition of the CYP3A4 enzymeduring both the first-pass and the elimination phases. Inhibition of the P-glycoprotein may have contributed to the observed interactions ofmethylprednisolone and of dexamethasone.
7. The metabolism and interaction potential of different glucocorticoids aredissimilar. In contrast to prednisolone and also prednisone, CYP3A4 appears tobe important in the metabolism of methylprednisolone and dexamethasone.Therefore, the pharmacokinetics of methylprednisolone and dexamethasone aresensitive to itraconazole and probably also to other potent CYP3A4 inhibitors.However, interactions of itraconazole and other CYP3A4 inhibitors withprednisolone and probably with prednisone are likely to be of limited clinicalsignificance.

ACKNOWLEDGEMENTS
67
ACKNOWLEDGEMENTS
These studies were carried out at the Department of Clinical Pharmacology,University of Helsinki, during the years 1997 to 1999. I am grateful to all thosewho have contributed to my work during these years.
I express my deepest gratitude to my supervisers:
Professor Pertti Neuvonen, Head of the Department of ClinicalPharmacology, University of Helsinki, for introducing me to the field ofdrug-drug interactions, for his encouraging supervision, his time, and alsofor his providing excellent working facilities. His amazing energy combinedwith his wide knowledge in the field of clinical pharmacology creates astrong foundation for high quality research.
Docent Kari Kivistö, for his continuous interest, his time, and for hisscientific guidance during these years. His enthusiasm for scientific workand wide expertise in the field of clinical pharmacology has alwaysimpressed me.
Docent Arja Rautio and Docent Kari Aranko are gratefully acknowledged fortheir valuable review and comments on the present work.
I express my warm thanks to Kerttu Mårtensson, Lisbeth Partanen, EijaMäkinen-Pulli, Mikko Neuvonen, and Jouko Laitila for their guidance and skilfuldetermination of plasma drug and cortisol concentrations. Airi Järvinen is alsowarmly acknowledged.
I thank all my colleagues at the Department of Clinical Pharmacology, includingJanne Backman, Maija Kaukonen, Kirsti Villikka, Jari Lilja, Outi Lapatto-Reiniluoto, Kari Raaska, Harri Luurila, Jun-Sheng Wang, Xia Wen, MikkoNiemi, Elina Saarenmaa, Mika Jokinen, Carl Kyrklund, Matti Kivikko, KatiAaltonen, and Laura Juntti-Patinen, for their friendship and collaboration. I alsowant to thank all the people who join us in the ”first-pass group” meetings. Veryspecial thanks belong to Tuija Itkonen for her joyful company during these yearsand for her help in many practical matters.
Docent Kalle Hoppu and the staff of the Poison Information Centre are alsowarmly acknowledged.
Sincere thanks to Carol Norris for editing of the English language of this thesis.
Financial support by the Helsinki University Central Hospital Research Fund, theNational Technology Agency of Finland (TEKES), the Finnish Medical Society

ACKNOWLEDGEMENTS
68
Duodecim, and the Clinical Drug Research Graduate School are gratefullyacknowledged.
Haluan osoittaa lämpimät kiitokseni myös vanhemmilleni, jotka ovat tukeneetminua tutkimustyöni aikana ja osoittaneet jatkuvaa kiinnostusta sitä kohtaan.Lisäksi kiitos myös käytännön avusta, jota olen teiltä saanut erityisesti kuluneenkesän ja syksyn aikana.
Finally, I express my deepest and most loving thanks to my husband Juha for hisunselfish support and love during these years. My gratitude and love go to ourchildren Antti and Kristiina, who are the most important challenge of my life,more important even than science.
Karkkila, November 2000
Tiina Varis

REFERENCES
69
REFERENCESAdachi JD, Bensen WG, Hodsman AB. Corticosteroid-induced osteoporosis. Semin Arthritis
Rheum 1993;22:375-384Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome
P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet1999;353(9154):717-719
Al-Habet S, Rogers HJ. Pharmacokinetics of intravenous and oral prednisolone. Br J ClinPharmacol 1980;10:503-508
Al-Habet SM, Rogers HJ. Methylprednisolone pharmacokinetics after intravenous and oraladministration. Br J Clin Pharmacol 1989;27:285-290
Allenmark S, Edebo A, Lindgren K. Determination of itraconazole in serum with high-performance liquid chromatography and fluorescence detection. J Chromatogr1990;532:203-206
Anonymous. Roche Laboratories announced withdrawal of Posicor from the market. FDATalk Paper June 8, 1998
Antal EJ, Wright CE 3d, Gillespie WR, Albert KS. Influence of route of administration onthe pharmacokinetics of methylprednisolone. J Pharmacokinet Biopharm 1983;11:561-576
Anttila S, Hukkanen J, Hakkola J, Stjernvall T, Beaune P, Edwards RJ, Boobis AR, PelkonenO, Raunio H. Expression and localization of CYP3A4 and CYP3A5 in human lung. Am JRespir Cell Mol Biol 1997;16:242-249
Arredondo G, Calvo R, Marcos F, Martinez-Jorda R, Suarez E. Protein binding ofitraconazole and fluconazole in patients with cancer. Int J Clin Pharmacol Ther1995;33:449-452
Assael BM, Banfi G, Appiani AC, Edefonti A, Jusko WJ. Disposition of pulse dosemethylprednisolone in adult and paediatric patients with the nephrotic syndrome. Eur JClin Pharmacol 1982;23:429-433
Ater JL, van Eys J, Woo SY, Moore B 3rd, Copeland DR, Bruner J. MOPP chemotherapywithout irradiation as primary postsurgical therapy for brain tumors in infants and youngchildren. J Neurooncol 1997;32:243-252
Aoyama T, Yamano S, Waxman DJ, Lapenson DP, Meyer UA, Fischer V, Tyndale R, InabaT, Kalow W, Gelboin HV, Gonzalez FJ. Cytochrome P-450 hPCN3, a novel cytochromeP-450 IIIA gene product that is differentially expressed in adult human liver. cDNA anddeduced amino acid sequence and distinct specificities of cDNA-expressed hPCN1 andhPCN3 for the metabolism of steroid hormones and cyclosporine. J Biol Chem1989;264(18):10388-10395
Back DJ, Tjia JF. Comparative effects of the antimycotic drugs ketoconazole, fluconazole,itraconazole and terbinafine on the metabolism of cyclosporin by human liver microsomes.Br J Clin Pharmacol 1991;32:624-626
Backman JT, Aranko K, Himberg J-J, Olkkola KT. A pharmacokinetic interaction betweenroxithromycin and midazolam. Eur J Clin Pharmacol 1994a;46:551-555
Backman JT, Olkkola KT, Aranko K, Himberg J-J, Neuvonen PJ. Dose of midazolam shouldbe reduced during diltiazem and verapamil treatments. Br J Clin Pharmacol1994b;37:221-225

REFERENCES
70
Backman JT, Wang JS, Wen X, Kivistö KT, Neuvonen PJ. Mibefradil but not isradipinesubstantially elevates the plasma concentrations of the CYP3A4 substrate triazolam. ClinPharmacol Ther 1999;66:401-407
Backman JT, Kivistö KT, Wang JS, Neuvonen PJ. Antifungals. In: Levy RH, Thummel KE,Trager WF, Hansten PD, Eichelbaum M, editors. Metabolic drug interactions. LippincottWilliams & Wilkins. 2000: 623-646
Bailey DG, Spence JD, Edgar B, Bayliff CD, Arnold JM. Ethanol enhances the hemodynamiceffects of felodipine. Clin Invest Med 1989;12:357-362
Bailey DG, Spence JD, Munoz C, Arnold JM. Interaction of citrus juices with felodipine andnifedipine. Lancet 1991;337:268-269
Bailey DG, Arnold JM, Strong HA, Munoz C, Spence JD. Effect of grapefruit juice andnaringin on nisoldipine pharmacokinetics. Clin Pharmacol Ther 1993a;54:589-594
Bailey DG, Arnold JM, Munoz C, Spence JD. Grapefruit juice--felodipine interaction:mechanism, predictability, and effect of naringin. Clin Pharmacol Ther 1993b;53:637-42
Bailey DG, Bend JR, Arnold JM, Tran LT, Spence JD. Erythromycin-felodipine interaction:magnitude, mechanism, and comparison with grapefruit juice. Clin Pharmacol Ther1996;60:25-33
Bailey DG, Malcolm J, Arnold O, Spence JD. Grapefruit juice-drug interactions. Br J ClinPharmacol 1998a;46:101-110
Bailey DG, Kreeft JH, Munoz C, Freeman DJ, Bend JR. Grapefruit juice-felodipineinteraction: effect of naringin and 6',7'-dihydroxybergamottin in humans. Clin PharmacolTher 1998b;64:248-256
Ball SE, Scatina J, Kao J, Ferron GM, Fruncillo R, Mayer P, Weinryb I, Guida M, HopkinsPJ, Warner N, Hall J. Population distribution and effects on drug metabolism of a geneticvariant in the 5' promoter region of CYP3A4. Clin Pharmacol Ther 1999;66:288-294
Bartoszek M, Brenner AM, Szefler SJ. Prednisolone and methylprednisolone kinetics inchildren receiving anticonvulsant therapy. Clin Pharmacol Ther 1987;42:424-432.
Batchelor TT, Taylor LP, Thaler HT, Posner JB, DeAngelis LM. Steroid myopathy in cancerpatients. Neurology 1997;48:1234-1238
Bauer LA, Stenwall M, Horn JR, Davis R, Opheim K, Greene L. Changes in antipyrine andindocyanine green kinetics during nifedipine, verapamil, and diltiazem therapy. ClinPharmacol Ther 1986;40:239-242
Begg EJ, Atkinson HC, Gianarakis N. The pharmacokinetics of corticosteroid agents. Med JAust 1987;146:37-41
Benton RE, Honig PK, Zamani K, Cantilena LR, Woosley RL. Grapefruit juice altersterfenadine pharmacokinetics, resulting in prolongation of repolarization on theelectrocardiogram. Clin Pharmacol Ther 1996;59:383-388
Bergrem H, Grøttum P, Rugstad HE. Pharmacokinetics and protein binding of prednisoloneafter oral and intravenous administration. Eur J Clin Pharmacol 1983;24:415-419
Berne RM, Levy MN. Physiology. 4th ed.:Mosby, Inc. 1998:930-949Bertz RJ, Granneman GR. Use of in vitro and in vivo data to estimate the likelihood of
metabolic pharmacokinetic interactions. Clin Pharmacokinet 1997;32:210-258Blanco JG, Harrison PL, Evans WE, Relling MV. Human cytochrome P450 maximal
activities In pediatric versus adult liver. Drug Metab Dispos 2000;28:379-382Bohets H, Lavrijsen K, Hendrickx J, van Houdt J, van Genechten V, Verboven P,
Meuldermans W, Heykants J. Identification of the cytochrome P450 enzymes involved inthe metabolism of cisapride: in vitro studies of potential co-medication interactions. Br JPharmacol 2000;129:1655-1667

REFERENCES
71
Brady ME, Sartiano GP, Rosenblum SL, Zaglama NE, Bauguess CT. The pharmacokineticsof single high doses of dexamethasone in cancer patients. Eur J Clin Pharmacol1987;32:593-596
Brockmöller J, Neumayer HH, Wagner K, Weber W, Heinemeyer G, Kewitz H, Roots I.Pharmacokinetic interaction between cyclosporin and diltiazem. Eur J Clin Pharmacol1990;38:237-242
Brodie MJ, MacPhee GJ. Carbamazepine neurotoxicity precipitated by diltiazem. BMJ1986;292(6529):1170-1171
Brooks PM, Buchanan WW, Grove M, Downie WW. Effects of enzyme induction onmetabolism of prednisolone. Clinical and laboratory study. Ann Rheum Dis 1976;35:339-343
Brooks SM, Werk EE, Ackerman SJ, Sullivan I, Thrasher K. Adverse effects ofphenobarbital on corticosteroid metabolism in patients with bronchial asthma. N Engl JMed 1972;286:1125-1128
Brooks SM, Sholiton LJ, Werk EE Jr, Altenau P. The effects of ephedrine and theophyllineon dexamethasone metabolism in bronchial asthma. J Clin Pharmacol 1977;17:308-318.
Buckley MM, Grant SM, Goa KL, McTavish D, Sorkin EM. Diltiazem. A reappraisal of itspharmacological properties and therapeutic use. Drugs 1990;39:757-806
Busse D, Busch FW, Bohnenstengel F, Eichelbaum M, Fischer P, Opalinska J, SchumacherK, Schweizer E, Kroemer HK. Dose escalation of cyclophosphamide in patients withbreast cancer: consequences for pharmacokinetics and metabolism. J Clin Oncol1997;15:1885-1896
Cain MS, Burton GV, Holcombe RF. Fatal leukoencephalopathy in a patient with non-Hodgkin's lymphoma treated with CHOP chemotherapy and high-dose steroids. Am J MedSci 1998;315:202-207
Cavanaugh JH, Karol MD. Lack of pharmacokinetic interaction after administration oflansoprazole or omeprazole with prednisone. J Clin Pharmacol 1996;36:1064-1071
Chakraborty A, Blum RA, Mis SM, Cutler DL, Jusko WJ. Pharmacokinetic and adrenalinteractions of IL-10 and prednisone in healthy volunteers. J Clin Pharmacol1999;39:624-635
Chalk JB, Ridgeway K, Brophy T, Yelland JD, Eadie MJ. Phenytoin impairs thebioavailability of dexamethasone in neurological and neurosurgical patients. J NeurolNeurosurg Psychiatry 1984;47:1087-1090
Cornwell MM, Pastan I, Gottesman MM. Certain calcium channel blockers bind specificallyto multidrug-resistant human KB carcinoma membrane vesicles and inhibit drug binding toP-glycoprotein. J Biol Chem 1987;262:2166-2170
Danan G, Descatoire V, Pessayre D. Self-induction by erythromycin of its owntransformation into a metabolite forming an inactive complex with reduced cytochrome P-450. J Pharmacol Exp Ther 1981;218:509-514
David DS, Cheigh JS, Braun DW Jr, Fotino M, Stenzel KH, Rubin AL. HLA-A28 andsteroid-induced diabetes in renal transplant patients. JAMA 1980;243:532-533
Dawidson I, Rooth P, Fry WR, Sandor Z, Willms C, Coorpender L, Alway C, Reisch J.Prevention of acute cyclosporine-induced renal blood flow inhibition and improvedimmunosuppression with verapamil. Transplantation 1989;48:575-580
Dodds HM, Taylor PJ, Cannell GR, Pond SM. A high-performance liquid chromatography-electrospray-tandem mass spectrometry analysis of cortisol and metabolites in placentalperfusate. Anal Biochem 1997;247:342-347

REFERENCES
72
Dollery C, editor. Therapeutic drugs. 2nd ed. Edinburgh, Great Britain: Churchill Livingstone,1999a;1:D139-143
Dollery C, editor. Therapeutic drugs. 2nd ed. Edinburgh, Great Britain: Churchill Livingstone,1999b;2:M164-171
Dollery C, editor. Therapeutic drugs. 2nd ed. Edinburgh, Great Britain: Churchill Livingstone,1999c;2:M125-129
Dowell SF, Bresee JS. Severe varicella associated with steroid use. Pediatrics 1993;92:223-228
Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences andclinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet 2000;38:41-57
Dube LM, Mousseau N, McGilveray IJ. High-performance liquid chromatographicdetermination of diltiazem and four of its metabolites in plasma: evaluation of theirstability. J Chromatogr 1988;430:103-111
Ducharme MP, Slaughter RL, Warbasse LH, Chandrasekar PH, Van de Velde V, MannensG, Edwards DJ. Itraconazole and hydroxyitraconazole serum concentrations are reducedmore than tenfold by phenytoin. Clin Pharmacol Ther 1995a;58:617-624
Ducharme MP, Warbasse LH, Edwards DJ. Disposition of intravenous and oral cyclosporineafter administration with grapefruit juice. Clin Pharmacol Ther 1995b;57:485-491
Duggan DE, Yeh KC, Matalia N, Ditzler CA, McMahon FG. Bioavailability of oraldexamethasone. Clin Pharmacol Ther 1975;18:205-209
Dunn TE, Ludwig EA, Slaughter RL, Camara DS, Jusko WJ. Pharmacokinetics andpharmacodynamics of methylprednisolone in obesity. Clin Pharmacol Ther 1991;49:536-549
Eagling VA, Profit L, Back DJ. Inhibition of the CYP3A4-mediated metabolism and P-glycoprotein-mediated transport of the HIV-1 protease inhibitor saquinavir by grapefruitjuice components. Br J Clin Pharmacol 1999;48:543-552
Ebling WF, Szefler SJ, Jusko WJ. Analysis of cortisol, methylprednisolone, andmethylprednisolone hemisuccinate. Absence of effects of troleandomycin on esterhydrolysis. J Chromatogr 1984;305:271-280
Eimer M, Carter BL. Elevated serum carbamazepine concentrations following diltiazeminitiation. Drug Intell Clin Pharm 1987;21:340-342
Ellison RH. Voluntary withdrawal of Posicor from the market. Press release, RocheLaboratories. June 8, 1998
Esteban NV, Loughlin T, Yergey AL, Zawadzki JK, Booth JD, Winterer JC, Loriaux DL.Daily cortisol production rate in man determined by stable isotope dilution/massspectrometry. J Clin Endocrinol Metab 1991;72:39-45
Evans AM. Influence of dietary components on the gastrointestinal metabolism and transportof drugs. Ther Drug Monit 2000;22:13113-6
Felix CA, Walker AH, Lange BJ, Williams TM, Winick NJ, Cheung NK, Lovett BD, NowellPC, Blair IA, Rebbeck TR. Association of CYP3A4 genotype with treatment-relatedleukemia. Proc Natl Acad Sci U S A 1998;95:13176-13181
Ferry JJ, Della-Coletta AA, Weber DJ, VanderLugt JT. Pilot study of the pharmacokineticsof methylprednisolone after single and multiple intravenous doses of methylprednisolonesodium succinate and methylprednisolone suleptanate to healthy volunteers. J ClinPharmacol 1994;34:1109-1115
Finnish statistics on medicines 1988-1998. National Agency for Medicines and SocialInsurance Institution.

REFERENCES
73
Fleming CM, Branch RA, Wilkinson GR, Guengerich FP. Human liver microsomal N-hydroxylation of dapsone by cytochrome P-4503A4. Mol Pharmacol 1992;41:975-980
Fost DA, Leung DY, Martin RJ, Brown EE, Szefler SJ, Spahn JD. Inhibition ofmethylprednisolone elimination in the presence of clarithromycin therapy. J Allergy ClinImmunol 1999;103:1031-1035
Fraser CG, Preuss FS, Bigford WD. Adrenal atrophy and irreversible shock associated withcortisone therapy. JAMA 1952;149:1542-1543.
Frey FJ, Lozada F, Guentert T, Frey BM. A single dose of azathioprine does not affect thepharmacokinetics of prednisolone following oral prednisone. Eur J Clin Pharmacol1981;19:209-212
Frey FJ, Schnetzer A, Horber FF, Frey BM. Evidence that cyclosporine does not affect themetabolism of prednisolone after renal transplantation. Transplantation 1987;43:494-498
Friedman AL, Chesney RW. Glucocorticoids in renal disease. Theoretical basis,consequences and efficacy of use in the pediatric patient. Am J Nephrol 1982;2:330-341
Fitzsimmons ME, Collins JM. Selective biotransformation of the human immunodeficiencyvirus protease inhibitor saquinavir by human small-intestinal cytochrome P4503A4:potential contribution to high first-pass metabolism. Drug Metab Dispos 1997;25:256-266
Gaedigk A. Interethnic differences of drug-metabolizing enzymes. Int J Clin Pharmacol Ther2000;38:61-68
Gascon M-P, Dayer P. In vitro forecasting of drugs which may interfere with thebiotransformation of midazolam. Eur J Clin Pharmacol 1991;41:573-578
Gaynon PS, Carrel AL. Glucocorticosteroid therapy in childhood acute lymphoblasticleukemia. Adv Exp Med Biol 1999;457:593-605
Ged C, Rouillon JM, Pichard L, Combalbert J, Bressot N, Bories P, Michel H, Beaune P,Maurel P. The increase in urinary excretion of 6 beta-hydroxycortisol as a marker ofhuman hepatic cytochrome P450IIIA induction. Br J Clin Pharmacol 1989;28:373-387
Gentile DM, Tomlinson ES, Maggs JL, Park BK, Back DJ. Dexamethasone metabolism byhuman liver in vitro. Metabolite identification and inhibition of 6-hydroxylation. JPharmacol Exp Ther 1996;277:105-112
Gibaldi M, Perrier D. Pharmacokinetics. 2nd ed. New York: Marcel Dekker; 1982Glynn AM, Slaughter RL, Brass C, D'Ambrosio R, Jusko WJ. Effects of ketoconazole on
methylprednisolone pharmacokinetics and cortisol secretion. Clin Pharmacol Ther1986;39:654-659
Grant SM, Clissold SP. Itraconazole. A review of its pharmacodynamic and pharmacokineticproperties, and therapeutic use in superficial and systemic mycoses. Drugs 1989;37:310-344
Gray CH, Green MA, Holness NJ, Lunnon JB. Urinary metabolic products of prednisone andprednisolone. J Endocrinol 1956;14:146-154
Green AW, Ebling WF, Gardner MJ, Jusko WJ. Cimetidine-methylprednisolone-theophyllinemetabolic interaction. Am J Med 1984;77:1115-1118
Grinspoon SK, Biller BM. Clinical review 62: Laboratory assessment of adrenalinsufficiency. J Clin Endocrinol Metab 1994;79:923-931
Gross AS, Goh YD, Addison RS, Shenfield GM. Influence of grapefruit juice on cisapridepharmacokinetics. Clin Pharmacol Ther 1999;65:395-401
Guengerich FP, Müller-Enoch D, Blair IA. Oxidation of quinidine by human livercytochrome P-450. Mol Pharmacol 1986;30:287-295
Guengerich FP. Oxidation of 17 alpha-ethynylestradiol by human liver cytochrome P-450.Mol Pharmacol 1988;33:500-508

REFERENCES
74
Gupta AK, Sauder DN, Shear NH. Antifungal agents: an overview. Part II. J Am AcadDermatol 1994;30:911-33
van Haarst AD, van 't Klooster GA, van Gerven JM, Schoemaker RC, van Oene JC,Burggraaf J, Coene MC, Cohen AF. The influence of cisapride and clarithromycin on QTintervals in healthy volunteers. Clin Pharmacol Ther 1998;64:542-546
Hakkola J, Pasanen M, Purkunen R, Saarikoski S, Pelkonen O, Mäenpää J, Rane A, RaunioH. Expression of xenobiotic-metabolizing cytochrome P450 forms in human adult andfetal liver. Biochem Pharmacol 1994;48:59-64
Harari S. Current strategies in the treatment of invasive Aspergillus infections inimmunocompromised patients. Drugs 1999;58:621-631
Hardin TC, Graybill JR, Fetchick R, Woestenborghs R, Rinaldi MG, Kuhn JG.Pharmacokinetics of itraconazole following oral administration to normal volunteers.Antimicrob Agents Chemother 1988;32:1310-1313
Hare LE, Yeh KC, Ditzler CA, McMahon FG, Duggan DE. Bioavailability ofdexamethasone. II. Dexamethasone phosphate. Clin Pharmacol Ther 1975;18:330-337
Harris R, German D. The incidence of corticosteroid side effects in chronic steroid-dependent asthmatics on TAO (troleandomycin) and methylprednisolone. Ann Allergy1989;63:110-111
Harris RZ, Benet LZ, Schwartz JB. Gender effects in pharmacokinetics andpharmacodynamics. Drugs 1995;50:222-239
Haque N, Thrasher K, Werk EE Jr, Knowles HC Jr, Sholiton LJ. Studies on dexamethasonemetabolism in man: effect of diphenylhydantoin. J Clin Endocrinol Metab 1972;34:44-50
Hench PS, Kendall EC, Slocumb CH, Pollery HF. The effect of a hormone of the adrenalcortex and of pituitary adrenocorticotrophic hormone on rheumatoid arthritis. Proc StaffMeetings Mayo Clinic 1949;24:181-197
Hendrickse W, McKiernan J, Pickup M, Lowe J. Rifampicin-induced non-responsiveness tocorticosteroid treatment in nephrotic syndrome. BMJ 1979;1(6159):306
Henzen C, Suter A, Lerch E, Urbinelli R, Schorno XH, Briner VA. Suppression and recoveryof adrenal response after short-term, high-dose glucocorticoid treatment. Lancet2000;355:542-545
Hermann P, Rodger SD, Remones G, Thenot JP, London DR, Morselli PL.Pharmacokinetics of diltiazem after intravenous and oral administration. Eur J ClinPharmacol 1983;24:349-352
Heykants J, Van Peer A, Van de Velde V, Van Rooy P, Meuldermans W, Lavrijsen K,Woestenborghs R, Van Cutsem J, Cauwenbergh G. The clinical pharmacokinetics ofitraconazole: an overview. Mycoses 1989;32 Suppl 1:67-87
Hollander AA, van Rooij J, Lentjes GW, Arbouw F, van Bree JB, Schoemaker RC, van EsLA, van der Woude FJ, Cohen AF. The effect of grapefruit juice on cyclosporine andprednisone metabolism in transplant patients. Clin Pharmacol Ther 1995;57:318-324
Hoppu K, Koskimies O, Holmberg C, Hirvisalo EL. Evidence for pre-hepatic metabolism oforal cyclosporine in children. Br J Clin Pharmacol 1991;32:477-481
Huijgens PC, Simoons-Smit AM, van Loenen AC, Prooy E, van Tinteren H, OssenkoppeleGJ, Jonkhoff AR. Fluconazole versus itraconazole for the prevention of fungal infectionsin haemato-oncology. J Clin Pathol 1999;52:376-380
Hukkinen SK, Varhe A, Olkkola KT, Neuvonen PJ. Plasma concentrations of triazolam areincreased by concomitant ingestion of grapefruit juice. Clin Pharmacol Ther 1995;58:127-131.

REFERENCES
75
Hunt CM, Westerkam WR, Stave GM. Effect of age and gender on the activity of humanhepatic CYP3A. Biochem Pharmacol 1992;44:275-283
Hynning P-Å, Anderson P, Bondesson U, Boréus LO. Liquid-chromatographic quantificationcompared with gas-chromatographic-mass-spectrometric determination of verapamil andnorverapamil in plasma. Clin Chem 1988;34:2502-2503
Höglund P, Nilsson LG. Physiological disposition of intravenously administered 14C-labeleddiltiazem in healthy volunteers. Ther Drug Monit 1988;10:401-409
Imani S, Jusko WJ, Steiner R. Diltiazem retards the metabolism of oral prednisone witheffects on T-cell markers. Pediatr Transplant 1999;3:126-130
Jalava K-M, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin.Ther Drug Monit 1997;19:609-613
Jenkins S. My route to a steroid psychosis. BMJ 1999;318:67Jones DR, Hall DS. Calcium Channel Blockers. In: Levy RH, Thummel KE, Trager WF,
Hansten PD, Eichelbaum M, editors. Metabolic drug interactions. Lippincott Williams &Wilkins. 2000: 333-345
Jones TE. The use of other drugs to allow a lower dosage of cyclosporin to be used.Therapeutic and pharmacoeconomic considerations. Clin Pharmacokinet 1997;32:357-367
Jounaidi Y, Hyrailles V, Gervot L, Maurel P. Detection of CYP3A5 allelic variant: acandidate for the polymorphic expression of the protein? Biochem Biophys Res Commun1996;221:466-470
Jusko WJ, Slaunwhite WR Jr, Aceto T Jr. Partial pharmacodynamic model for the circadian-episodic secretion of cortisol in man. J Clin Endocrinol Metab 1975;40:278-289
Kampmann J, Hansen JM, Siersboek-Nielsen K, Laursen H. Effect of some drugs onpenicillin half-life in blood. Clin Pharmacol Ther 1972;13:516-519
Kandrotas RJ, Slaughter RL, Brass C, Jusko WJ. Ketoconazole effects onmethylprednisolone disposition and their joint suppression of endogenous cortisol. ClinPharmacol Ther 1987;42:465-470
Kantola T, Kivistö KT, Neuvonen PJ. Erythromycin and verapamil considerably increaseserum simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther1998a;64:177-182
Kantola T, Kivistö KT, Neuvonen PJ. Grapefruit juice greatly increases serum concentrationsof lovastatin and lovastatin acid. Clin Pharmacol Ther 1998b;63:397-402
Kasper WJ, Howe PM. Fatal varicella after a single course of corticosteroids. Pediatr InfectDis J 1990;9:729-732
Kates RE. Calcium antagonists. Pharmacokinetic properties. Drugs 1983;25:113-124Kawai S, Ichikawa Y, Homma M. Differences in metabolic properties among cortisol,
prednisolone, and dexamethasone in liver and renal diseases: accelerated metabolism ofdexamethasone in renal failure. J Clin Endocrinol Metab 1985;60:848-854
Kelly JG, O'Malley K. Clinical pharmacokinetics of calcium antagonists. An update. ClinPharmacokinet 1992 ;22:416-433
Kharasch ED, Mautz D, Senn T, Lentz G, Cox K. Menstrual cycle variability in midazolampharmacokinetics. J Clin Pharmacol 1999;39:275-280
Kim RB, Wandel C, Leake B, Cvetkovic M, Fromm MF, Dempsey PJ, Roden MM, Belas F,Chaudhary AK, Roden DM, Wood AJ, Wilkinson GR. Interrelationship betweensubstrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res 1999;16:408-414

REFERENCES
76
Kitada M, Kamataki T, Itahashi K, Rikihisa T, Kato R, Kanakubo Y. Purification andproperties of cytochrome P-450 from homogenates of human fetal livers. Arch BiochemBiophys 1985;241:275-280
Kivistö KT, Kroemer HK, Eichelbaum M. The role of human cytochrome P450 enzymes inthe metabolism of anticancer agents: implications for drug interactions. Br J ClinPharmacol 1995;40:523-530
Kivistö KT, Griese EU, Fritz P, Linder A, Hakkola J, Raunio H, Beaune P, Kroemer HK.Expression of cytochrome P 450 3A enzymes in human lung: a combined RT-PCR andimmunohistochemical analysis of normal tissue and lung tumours. Naunyn SchmiedebergsArch Pharmacol 1996a;353:207-212
Kivistö KT, Bookjans G, Fromm MF, Griese EU, Münzel P, Kroemer HK. Expression ofCYP3A4, CYP3A5 and CYP3A7 in human duodenal tissue. Br J Clin Pharmacol1996b;42:387-389
Kivistö KT, Lamberg TS, Kantola T, Neuvonen PJ. Plasma buspirone concentrations aregreatly increased by erythromycin and itraconazole. Clin Pharmacol Ther 1997;62:348-354
Kivistö KT, Kantola T, Neuvonen PJ. Different effects of itraconazole on thepharmacokinetics of fluvastatin and lovastatin. Br J Clin Pharmacol 1998;46:49-53
Kivistö KT, Lilja JJ, Backman JT, Neuvonen PJ. Repeated consumption of grapefruit juiceconsiderably increases plasma concentrations of cisapride. Clin Pharmacol Ther1999;66:448-453
Kobberling J, Muhlen A von zur. The influence of diphenylhydantoin and carbamazepine onthe circadian rhythm of free urinary corticoids and on the suppressibility of the basal andthe "impulsive" activity by dexamethasone. Acta Endocrinol 1973;72:308-318
Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB. Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Invest1992;90:1871-1878
Kong AN, Slaughter RL, Jusko WJ. Simultaneous analysis of methylprednisolonehemisuccinate, cortisol and methylprednisolone by normal-phase high-performance liquidchromatography in human plasma. J Chromatogr 1988;432:308-314
Kong AN, Ludwig EA, Slaughter RL, DiStefano PM, DeMasi J, Middleton E Jr, Jusko WJ.Pharmacokinetics and pharmacodynamic modeling of direct suppression effects ofmethylprednisolone on serum cortisol and blood histamine in human subjects. ClinPharmacol Ther 1989;46:616-628
Krishna DR, Klotz U. Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet1994;26:144-160
Krähenbühl S, Menafoglio A, Giostra E, Gallino A. Serious interaction between mibefradiland tacrolimus. Transplantation 1998;66:1113-1115
Kunzendorf U, Walz G, Brockmoeller J, Neumayer HH, Jochimsen F, Roots I, Offermann G,Strom TB. Effects of diltiazem upon metabolism and immunosuppressive action ofcyclosporine in kidney graft recipients. Transplantation 1991;52:280-284
Kupferschmidt HH, Ha HR, Ziegler WH, Meier PJ, Krähenbühl S. Interaction betweengrapefruit juice and midazolam in humans. Clin Pharmacol Ther 1995;58:20-28
Kurosawa M, Okabe M, Hara N, Kawamura K, Suzuki S, Sakurada K, Asaka M. Reversaleffect of itraconazole on adriamycin and etoposide resistance in human leukemia cells.Ann Hematol 1996;72:17-21
Laan RF, Jansen TL, van Riel PL. Glucocorticosteroids in the management of rheumatoidarthritis. Rheumatology 1999;38:6-12

REFERENCES
77
LaForce CF, Szefler SJ, Miller MF, Ebling W, Brenner M. Inhibition of methylprednisoloneelimination in the presence of erythromycin therapy. J Allergy Clin Immunol 1983;72:34-39
Lang CC, Brown RM, Kinirons MT, Deathridge MA, Guengerich FP, Kelleher D, O'BriainDS, Ghishan FK, Wood AJ. Decreased intestinal CYP3A in celiac disease: reversal aftersuccessful gluten-free diet: a potential source of interindividual variability in first-passdrug metabolism. Clin Pharmacol Ther 1996;59:41-46
Langhoff E, Madsen S, Flachs H, Olgaard K, Ladefoged J, Hvidberg EF. Inhibition ofprednisolone metabolism by cyclosporine in kidney-transplanted patients. Transplantation1985;39:107-109
de Lannoy IA, Silverman M. The MDR1 gene product, P-glycoprotein, mediates thetransport of the cardiac glycoside, digoxin. Biochem Biophys Res Commun 1992;189:551-557
Lantner R, Rockoff JB, DeMasi J, Boran-Ragotzy R, Middleton E Jr. Fatal varicella in acorticosteroid-dependent asthmatic receiving troleandomycin. Allergy Proc 1990;11:83-87
LaRochelle GE Jr, LaRochelle AG, Ratner RE, Borenstein DG. Recovery of thehypothalamic-pituitary-adrenal (HPA) axis in patients with rheumatic diseases receivinglow-dose prednisone. Am J Med 1993;95:258-264
Lawson GJ, Chakraborty J, Dumasia MC, Baylis EM. Methylprednisolone hemisuccinateand metabolites in urine from patients receiving high-dose corticosteroid therapy. TherDrug Monit 1992;14:20-26
Lebrun-Vignes B, Chosidow O, Diquet B, Levron JC, Herson S. Effect of itraconazole on thepharmacokinetics of prednisolone and methylprednisolone in healthy subjects. (Abstract).J Dermatol Science 1998;16(Suppl 1): 212
Lee DA, Taylor GM, Walker JG, James VH. The effect of concurrent administration ofantacids on prednisolone absorption. Br J Clin Pharmacol 1979;8:92-94
Lee YS, Lorenzo BJ, Koufis T, Reidenberg MM. Grapefruit juice and its flavonoids inhibit11 beta-hydroxysteroid dehydrogenase. Clin Pharmacol Ther 1996;59:62-71
Lees RS, Lees AM. Rhabdomyolysis from the coadministration of lovastatin and theantifungal agent itraconazole. N Engl J Med 1995;333:664-665
Legler UF, Benet LZ. Marked alterations in dose-dependent prednisolone kinetics in womentaking oral contraceptives. Clin Pharmacol Ther 1986;39:425-429
Leclercq R, Copinschi G. Patterns of plasma levels of prednisolone after oral administrationin man. J Pharmacokinet Biopharm 1974;2:175-187
Lew KH, Ludwig EA, Milad MA, Donovan K, Middleton E Jr, Ferry JJ, Jusko WJ. Gender-based effects on methylprednisolone pharmacokinetics and pharmacodynamics. ClinPharmacol Ther 1993;54:402-414
Lilja JJ, Kivistö KT, Neuvonen PJ. Grapefruit juice-simvastatin interaction: effect on serumconcentrations of simvastatin, simvastatin acid, and HMG-CoA reductase inhibitors. ClinPharmacol Ther 1998a;64:477-483
Lilja JJ, Kivistö KT, Backman JT, Lamberg TS, Neuvonen PJ. Grapefruit juice substantiallyincreases plasma concentrations of buspirone. Clin Pharmacol Ther 1998b;64:655-660
Lilja JJ, Kivistö KT, Neuvonen PJ. Grapefruit juice increases serum concentrations ofatorvastatin and has no effect on pravastatin. Clin Pharmacol Ther 1999;66:118-127
Lin JH, Lu AY. Inhibition and induction of cytochrome P450 and the clinical implications.Clin Pharmacokinet 1998;35:361-390

REFERENCES
78
Linthoudt H, Van Raemdonck D, Lerut T, Demedts M, Verleden G. The association ofitraconazole and methylprednisolone may give rise to important steroid-related sideeffects. J Heart Lung Transplant 1996;15:1165
Livanou T, Ferriman D, James VHT. Recovery of hypothalamo-pituitary-adrenal functionafter corticosteroid therapy. Lancet 1967;ii:856-859
Loew D, Schuster O, Graul EH. Dose-dependent pharmacokinetics of dexamethasone. Eur JClin Pharmacol 1986;30:225-230
Lown KS, Kolars JC, Thummel KE, Barnett JL, Kunze KL, Wrighton SA, Watkins PB.Interpatient heterogeneity in expression of CYP3A4 and CYP3A5 in small bowel. Lack ofprediction by the erythromycin breath test. Drug Metab Dispos 1994;22:947-955
Lown KS, Bailey DG, Fontana RJ, Janardan SK, Adair CH, Fortlage LA, Brown MB, GuoW, Watkins PB. Grapefruit juice increases felodipine oral availability in humans bydecreasing intestinal CYP3A protein expression. J Clin Invest 1997;99:2545-2553
Ludwig EA, Slaughter RL, Savliwala M, Brass C, Jusko WJ. Steroid-specific effects ofketoconazole on corticosteroid disposition: unaltered prednisolone elimination. DICP1989;23:858-861
Lundahl J, Regårdh CG, Edgar B, Johnsson G. Relationship between time of intake ofgrapefruit juice and its effect on pharmacokinetics and pharmacodynamics of felodipine inhealthy subjects. Eur J Clin Pharmacol 1995;49:61-67
Lundahl J, Regårdh CG, Edgar B, Johnsson G. Effects of grapefruit juice ingestion--pharmacokinetics and haemodynamics of intravenously and orally administered felodipinein healthy men. Eur J Clin Pharmacol 1997;52:139-145
Martin JE, Daoud AJ, Schroeder TJ, First MR. The clinical and economic potential ofcyclosporin drug interactions. Pharmacoeconomics 1999;15:317-337
Matsumoto K, Yamasaki H, Akazawa S, Sakamaki H, Ishibashi M, Abiru N, Uotani S,Matsuo H, Yamaguchi Y, Tokuyama K, Nagataki S. High-dose but not low-dosedexamethasone impairs glucose tolerance by inducing compensatory failure of pancreaticbeta-cells in normal men. J Clin Endocrinol Metab 1996;81:2621-2626
McAllister WA, Thompson PJ, Al-Habet SM, Rogers HJ. Rifampicin reduces effectivenessand bioavailability of prednisolone. BMJ 1983;286(6369):923-925
McGraw BF, Walker SD, Hemberger JA, Gitomer SL, Nakama M. Clinical experience withdiltiazem in Japan. Pharmacotherapy 1982;2:156-161
McWhinney BC, Ward G, Hickman PE. Improved HPLC method for simultaneous analysisof cortisol, 11-deoxycortisol, prednisolone, methylprednisolone, and dexamethasone inserum and urine. Clin Chem 1996;42: 979-981
Meffin PJ, Brooks PM, Sallustio BC. Alterations in prednisolone disposition as a result oftime of administration, gender and dose. Br J Clin Pharmacol 1984;17:395-404
Meibohm B, Derendorf H, Möllmann H, Fröhlich P, Tromm A, Wagner M, Homrighausen S,Krieg M, Hochhaus G. Mechanism-based PK/PD model for the lymphocytopenia inducedby endogenous and exogenous corticosteroids. Int J Clin Pharmacol Ther 1999;37:367-376
Melby JC. Drug spotlight program: systemic corticosteroid therapy: pharmacology andendocrinologic considerations. Ann Intern Med 1974;81:505-512
Meyer UA. Overview of enzymes of drug metabolism. J Pharmacokinet Biopharm1996;24:449-459
Mies S, Massarollo PC, Figueira ER, Leitao RM, Raia S. Lower incidence of liver graftrejection in patients on diltiazem plus cyclosporine therapy. Transplant Proc1998;30:1437-1438

REFERENCES
79
Milsap RL, Plaisance KI, Jusko WJ. Prednisolone disposition in obese men. Clin PharmacolTher 1984;36:824-831
Minagawa K, Kasuya Y, Baba S, Knapp G, Skelly JP. Identification and quantification of 6beta-hydroxydexamethasone as a major urinary metabolite of dexamethasone in man.Steroids 1986;47:175-188
Miniscalco A, Lundahl J, Regardh CG, Edgar B, Eriksson UG. Inhibition of dihydropyridinemetabolism in rat and human liver microsomes by flavonoids found in grapefruit juice. JPharmacol Exp Ther 1992;261:1195-1199
Mitchell JR, Thorgeirsson SS, Potter WZ, Jollow DJ, Keiser H. Acetaminophen-inducedhepatic injury: protective role of glutathione in man and rationale for therapy. ClinPharmacol Ther 1974;16:676-684
Miyabo S, Nakamura T, Kuwazima S, Kishida S. A comparison of the bioavailability andpotency of dexamethasone phosphate and sulphate in man. Eur J Clin Pharmacol1981;20:277-282
Miyama T, Takanaga H, Matsuo H, Yamano K, Yamamoto K, Iga T, Naito M, Tsuruo T,Ishizuka H, Kawahara Y, Sawada Y. P-glycoprotein-mediated transport of itraconazoleacross the blood-brain barrier. Antimicrob Agents Chemother 1998;42:1738-1744
Mousa O, Brater DC, Sundblad KJ, Hall SD. The interaction of diltiazem with simvastatin.Clin Pharmacol Ther 2000;67:267-274
Mullins ME, Horowitz BZ, Linden DH, Smith GW, Norton RL, Stump J. Life-threateninginteraction of mibefradil and beta-blockers with dihydropyridine calcium channelblockers. JAMA 1998;280:157-158
Naito Y, Tamai S, Shingu K, Shindo K, Matsui T, Segawa H, Nakai Y, Mori K. Responses ofplasma adrenocorticotropic hormone, cortisol, and cytokines during and after upperabdominal surgery. Anesthesiology 1992;77:426-431
Nakamura K, Goto F, Ray WA, McAllister CB, Jacqz E, Wilkinson GR, Branch RA.Interethnic differences in genetic polymorphism of debrisoquin and mephenytoinhydroxylation between Japanese and Caucasian populations. Clin Pharmacol Ther1985;38:402-408
Narang PK, Wilder R, Chatterji DC, Yeager RL, Gallelli JF. Systemic bioavailability andpharmacokinetics of methylprednisolone in patients with rheumatoid arthritis following'high-dose' pulse administration. Biopharm Drug Dispos 1983;4:233-248
Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, WatermanMR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW. P450 superfamily:update on new sequences, gene mapping, accession numbers and nomenclature.Pharmacogenetics 1996;6:1-42
Nelson DR. Cytochrome P450 and the individuality of species. Arch Biochem Biophys1999;369:1-10
Neuvonen PJ, Gothoni G, Hackman R, Björksten K. Interference of iron with the absorptionof tetracyclines in man. Br Med J 1970;4:532-534
Neuvonen PJ, Jalava K-M. Itraconazole drastically increases plasma concentrations oflovastatin and lovastatin acid. Clin Pharmacol Ther 1996;60:54-61
Neuvonen PJ, Kantola T, Kivistö KT. Simvastatin but not pravastatin is very susceptible tointeraction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther 1998;63:332-341
Nielsen MD, Brøsen K, Gram LF. A dose-effect study of the in vivo inhibitory effect ofquinidine on sparteine oxidation in man. Br J Clin Pharmacol 1990;29:299-304
Note for guidance on the investigation of drug interactions, CPMP, 1997

REFERENCES
80
Oken MM. Multiple myeloma: prognosis and standard treatment. Cancer Invest 1997;15:57-64
Olkkola KT, Aranko K, Luurila H, Hiller A, Saarnivaara L, Himberg J-J, Neuvonen PJ. Apotentially hazardous interaction between erythromycin and midazolam. Clin PharmacolTher 1993;53:298-305
Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receivingthe systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther1994;55:481-485
O'Sullivan BT, Cutler DJ, Hunt GE, Walters C, Johnson GF, Caterson ID. Pharmacokineticsof dexamethasone and its relationship to dexamethasone suppression test outcome indepressed patients and healthy control subjects. Biol Psychiatry 1997;41:574-584
Palmer JL, Barrington P, Collyer S. An evaluation of the effects of oral dexamethasone 2 mgand 5 mg once daily for 7 days on CYP3A4 activity. Br J Clin Pharmacol 2000;49:504P
Patel PM, Selby PJ, Graham MA, Viner C, Newell DR, McElwain TJ. Pharmacokinetics ofhigh dose methylprednisolone and use in hematological malignancies. Hematol Oncol1993;11:89-96
Peden NR, Rewhorn I, Champion MC, Mussani R, Ooi TC. Cortisol and dexamethasoneelimination during treatment with cimetidine. Br J Clin Pharmacol 1984;18:101-103
Peets EA, Staub M, Symchowicz S. Plasma binding of betamethasone-3H, dexamethasone-3Hand cortisol -14C. A comparative study. Biochem Pharmacol 1969;18:1655-1663
Pelkonen O, Mäenpää J, Taavitsainen P, Rautio A, Raunio H. Inhibition and induction ofhuman cytochrome P450 (CYP) enzymes. Xenobiotica 1998;28:1203-1253
Perez EA. Use of dexamethasone with 5-HT3-receptor antagonists for chemotherapy-inducednausea and vomiting. Cancer J Sci Am 1998;4:72-77
Perico N, Remuzzi G. Prevention of transplant rejection: current treatment guidelines andfuture developments. Drugs 1997;54:533-570
Pessayre D, Larrey D, Vitaux J, Breil P, Belghiti J, Benhamou JP. Formation of an inactivecytochrome P-450 Fe(II)-metabolite complex after administration of troleandomycin inhumans. Biochem Pharmacol 1982;31:1699-1704
Petereit LB, Meikle AW. Effectiveness of prednisolone during phenytoin therapy. ClinPharmacol Ther 1977;22:912-916
Phillips P, Graybill JR, Fetchick R, Dunn JF. Adrenal response to corticotropin duringtherapy with itraconazole. Antimicrob Agents Chemother 1987;31:647-649
Pichard L, Gillet G, Fabre I, Dalet-Beluche I, Bonfils C, Thenot JP, Maurel P. Identificationof the rabbit and human cytochromes P-450IIIA as the major enzymes involved in the N-demethylation of diltiazem. Drug Metab Dispos 1990;18:711-719
Pichard L, Fabre I, Daujat M, Domergue J, Joyeux H, Maurel P. Effect of corticosteroids onthe expression of cytochromes P450 and on cyclosporin A oxidase activity in primarycultures of human hepatocytes. Mol Pharmacol 1992;41:1047-1055
Pickup ME. Clinical pharmacokinetics of prednisone and prednisolone. Clin Pharmacokinet1979;4:111-128
Piquette RK. Torsade de pointes induced by cisapride/clarithromycin interaction. AnnPharmacother 1999;33:22-26
Pochet JM, Pirson Y. Cyclosporin-diltiazem interaction. Lancet 1986;1(8487):979Pohjola-Sintonen S, Viitasalo M, Toivonen L, Neuvonen P. Itraconazole prevents
terfenadine metabolism and increases risk of torsades de pointes ventricular tachycardia.Eur J Clin Pharmacol 1993;45:191-193

REFERENCES
81
Powell LW, Axelsen E. Corticosteroids in liver disease: studies on the biological conversionof prednisone to prednisolone and plasma protein binding. Gut 1972;13:690-696
Prueksaritanont T, Ma B, Tang C, Meng Y, Assang C, Lu P, Reider PJ, Lin JH, Baillie TA.Metabolic interactions between mibefradil and HMG-CoA reductase inhibitors: an in vitroinvestigation with human liver preparations. Br J Clin Pharmacol 1999;47:291-298
Queiroz-Telles F, Purim KS, Boguszewski CL, Afonso FC, Graf H. Adrenal response tocorticotrophin and testosterone during long-term therapy with itraconazole in patients withchromoblastomycosis. J Antimicrob Chemother 1997;40:899-902
Rae SA, Williams IA, English J, Baylis EM. Alteration of plasma prednisolone levels byindomethacin and naproxen. Br J Clin Pharmacol 1982;14:459-461
Rameis H, Magometschnigg D, Ganzinger U. The diltiazem-digoxin interaction. ClinPharmacol Ther 1984;36:183-189
Rebbeck TR, Jaffe JM, Walker AH, Wein AJ, Malkowicz SB. Modification of clinicalpresentation of prostate tumors by a novel genetic variant in CYP3A4. J Natl Cancer Inst1998;90:1225-1229
Remmel RP, Dombrovskis D, Canafax DM. Assay of itraconazole in leukemic patient plasmaby reversed-phase small-bore liquid chromatography. J Chromatogr 1988;432:388-394
Renton KW. Inhibition of hepatic microsomal drug metabolism by the calcium channelblockers diltiazem and verapamil. Biochem Pharmacol 1985;34:2549-2553
Rocci ML Jr, Tietze KJ, Lee J, Harris H, Danzeisen J, Burke JF. The effect of cyclosporineon the pharmacokinetics of prednisolone in renal transplant patients. Transplantation1988;45:656-660
Rodchenkov GM, Uralets VP, Semenov VA. Determination of methylprednisolonemetabolites in human urine by gas chromatography-mass spectrometry. J Chromatogr1987;423:15-22
Rohdewald P, Möllmann H, Barth J, Rehder J, Derendorf H. Pharmacokinetics ofdexamethasone and its phosphate ester. Biopharm Drug Dispos 1987;8:205-212
Rose JQ, Yurchak AM, Jusko WJ. Dose dependent pharmacokinetics of prednisone andprednisolone in man. J Pharmacokinet Biopharm 1981a;9:389-417
Rose JQ, Yurchak AM, Meikle AW, Jusko WJ. Effect of smoking on prednisone,prednisolone, and dexamethasone pharmacokinetics. J Pharmacokinet Biopharm1981b;9:1-14
Rowe BH, Spooner CH, Ducharme FM, Bretzlaff JA, Bota GW. Corticosteroids forpreventing relapse following acute exacerbations of asthma. Cochrane Database Syst Rev2000;2:CD000195
Saitoh H, Hatakeyama M, Eguchi O, Oda M, Takada M. Involvement of intestinal P-glycoprotein in the restricted absorption of methylprednisolone from rat small intestine. JPharm Sci 1998;87:73-75
Sakai H, Kobayashi S, Hamada K, Iida S, Akita H, Tanaka E, Uchida E, Yasuhara H. Theeffects of diltiazem on hepatic drug metabolizing enzymes in man using antipyrine,trimethadione and debrisoquine as model substrates. Br J Clin Pharmacol 1991;31:353-355
Salassa RM, Bennet WA, Keating FR Jr, Sprague RG. Postoperative adrenal corticalinsufficiency occurrence in patients previously treated with cortisone. JAMA1953;152:1509-1515.
Sanchez C, Mauri E, Dalmau D, Quintana S, Aparicio A, Garau J. Treatment of cerebralAspergillosis with itraconazole: do high doses improve the prognosis? Clin Infect Dis1995;21:1485-1487

REFERENCES
82
Sarna S, Hoppu K, Neuvonen PJ, Laine J, Holmberg C. Methylprednisolone exposure, ratherthan dose, predicts adrenal suppression and growth inhibition in children with liver andrenal transplants. J Clin Endocrinol Metab 1997;82:75-77
Sata F, Sapone A, Elizondo G, Stocker P, Miller VP, Zheng W, Raunio H, Crespi CL,Gonzalez FJ. CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12:evidence for an allelic variant with altered catalytic activity. Clin Pharmacol Ther2000;67:48-56
Schimmer BP, Parker KL. Adrenocorticotropic hormone; adrenocortical steroids and theirsynthetic analoqs; inhibitors of the synthesis and actions of adrenocortical hormones. InGoodman & Gilman’s. The pharmacological basis of therapeutics. 1996:1459-1485
Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA,van der Valk MA, Robanus-Maandag EC, te Riele HP, Berns AJM, Borst P. Disruption ofthe mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier andto increased sensitivity to drugs. Cell 1994;77:491-502
Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone,digoxin, and cyclosporin A. J Clin Invest 1995;96:1698-1705
Schlaghecke R, Kornely E, Santen RT, Ridderskamp P. The effect of long-termglucocorticoid therapy on pituitary-adrenal responses to exogenous corticotropin-releasinghormone. N Engl J Med 1992;326:226-230
Schmassmann-Suhijar D, Bullingham R, Gasser R, Schmutz J, Haefeli WE.Rhabdomyolysis due to interaction of simvastatin with mibefradil. Lancet 1998;351:1929-30
Schmucker DL, Woodhouse KW, Wang RK, Wynne H, James OF, McManus M, Kremers P.Effects of age and gender on in vitro properties of human liver microsomalmonooxygenases. Clin Pharmacol Ther 1990;48:365-374
Schuetz JD, Beach DL, Guzelian PS. Selective expression of cytochrome P450 CYP3AmRNAs in embryonic and adult human liver. Pharmacogenetics 1994;4:11-20
Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein andcytochrome P4503A coordinately up-regulate these proteins in human colon carcinomacells. Mol Pharmacol 1996;49:311-318
Segaert MF, De Soete C, Vandewiele I, Verbanck J. Drug-interaction-inducedrhabdomyolysis. Nephrol Dial Transplant 1996;11:1846-1847
Seidegård J. Reduction of the inhibitory effect of ketoconazole on budesonidepharmacokinetics by separation of their time of administration. Clin Pharmacol Ther2000;68:13-17
Seppälä T, Aranko K, Mattila MJ, Shrotriya RC. Effects of alcohol on buspirone andlorazepam actions. Clin Pharmacol Ther 1982;32:201-207
Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations inhuman liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogensand toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. JPharmacol Exp Ther 1994;270:414-423
Siepmann M, Kleinbloesem C, Kirch W. The interaction of the calcium antagonist RO 40-5967 with digoxin. Br J Clin Pharmacol 1995;39:491-496
Sikic BI, Fisher GA, Lum BL, Halsey J, Beketic-Oreskovic L, Chen G. Modulation andprevention of multidrug resistance by inhibitors of P-glycoprotein. Cancer ChemotherPharmacol 1997;40 Suppl:S13-19

REFERENCES
83
Siracusa A, Brugnami G, Fiordi T, Areni S, Severini C, Marabini A. Troleandomycin in thetreatment of difficult asthma. J Allergy Clin Immunol 1993;92:677-682
Sirgo MA, Rocci ML Jr, Ferguson RK, Eshelman FN, Vlasses PH. Effects of cimetidine andranitidine on the conversion of prednisone to prednisolone. Clin Pharmacol Ther1985;37:534-538
Skerjanec A, Tam YK. High-performance liquid chromatographic analysis of mibefradil indog plasma and urine. J Chromatogr B Biomed Appl 1995;669:377-382
Slaunwhite WR, Sandberg AA. Disposition of radioactive 17α-hydroxy-progesterone, 6α-methyl-17α-acetoxy-progesterone and 6α-methylprednisolone in human subjects. J ClinEndocrinol Metab 1961;21:753-764
Slayter KL, Ludwig EA, Lew KH, Middleton E Jr, Ferry JJ, Jusko WJ. Oral contraceptiveeffects on methylprednisolone pharmacokinetics and pharmacodynamics. Clin PharmacolTher 1996;59:312-321
Soons PA, Vogels BA, Roosemalen MC, Schoemaker HC, Uchida E, Edgar B, Lundahl J,Cohen AF, Breimer DD. Grapefruit juice and cimetidine inhibit stereoselectivemetabolism of nitrendipine in humans. Clin Pharmacol Ther 1991;50:394-403
Sotaniemi EA, Arranto AJ, Pelkonen O, Pasanen M. Age and cytochrome P450-linked drugmetabolism in humans: an analysis of 226 subjects with equal histopathologic conditions.Clin Pharmacol Ther 1997;61:331-339
du Souich P, Besner JG, Clozel JP, Welker HA, Lefebvre M, Caille G. Nonlinear kineticsand pharmacologic response to mibefradil. Clin Pharmacol Ther 2000;67:249-257
Stjernholm MR, Katz FH. Effects of diphenylhydantoin, phenobarbital, and diazepam on themetabolism of methylprednisolone and its sodium succinate. J Clin Endocrinol Metab1975;41:887-893
Streck WF, Lockwood DH. Pituitary adrenal recovery following short-term suppression withcorticosteroids. Am J Med 1979;66:910-914
Sutton D, Butler AM, Nadin L, Murray M. Role of CYP3A4 in human hepatic diltiazem N-demethylation: inhibition of CYP3A4 activity by oxidized diltiazem metabolites. JPharmacol Exp Ther 1997;282:294-300
Swartz SL, Dluhy RG. Corticosteroids: clinical pharmacology and therapeutic use. Drugs1978;16:238-525
Szefler SJ, Rose JQ, Ellis EF, Spector SL, Green AW, Jusko WJ. The effect oftroleandomycin on methylprednisolone elimination. J Allergy Clin Immunol 1980;66:447-51
Szefler SJ, Ellis EF, Brenner M, Rose JQ, Spector SL, Yurchak AM, Andrews F, Jusko WJ.Steroid-specific and anticonvulsant interaction aspects of troleandomycin-steroid therapy.J Allergy Clin Immunol 1982;69:455-460
Szefler SJ, Ebling WF, Georgitis JW, Jusko WJ. Methylprednisolone versus prednisolonepharmacokinetics in relation to dose in adults. Eur J Clin Pharmacol 1986;30:323-329
Tanner A, Bochner F, Caffin J, Halliday J, Powell L. Dose-dependent prednisolone kinetics.Clin Pharmacol Ther 1979;25:571-578
Takanaga H, Ohnishi A, Matsuo H, Sawada Y. Inhibition of vinblastine efflux mediated byP-glycoprotein by grapefruit juice components in caco-2 cells. Biol Pharm Bull1998;21:1062-1066
Tateishi T, Ohashi K, Sudo T, Sakamoto K, Toyosaki N, Hosoda S, Toyo-oka T, Kumagai Y,Sugimoto K, Fujimura A, et al. Dose dependent effect of diltiazem on thepharmacokinetics of nifedipine. J Clin Pharmacol 1989;29:994-997

REFERENCES
84
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellularlocalization of the multidrug-resistance gene product P-glycoprotein in normal humantissues. Proc Natl Acad Sci U S A 1987;84:7735-7738
Thummel KE, O'Shea D, Paine MF, Shen DD, Kunze KL, Perkins JD, Wilkinson GR. Oralfirst-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther 1996;59:491-502
Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving humanCYP3A. Annu Rev Pharmacol Toxicol 1998;38:389-430
Tomlinson ES, Maggs JL, Park BK, Back DJ. Dexamethasone as an in vivo probe forCYP3A4. Exp Toxic Pathol 1996;48:395-396
Tornatore KM, Walshe JJ, Reed KA, Holdsworth MT, Venuto RC. Comparativemethylprednisolone pharmacokinetics in renal transplant patients receiving double- ortriple-drug immunosuppression. Ann Pharmacother 1993;27:545-549
Tornatore KM, Logue G, Venuto RC, Davis PJ. Pharmacokinetics of methylprednisolone inelderly and young healthy males. J Am Geriatr Soc 1994;42:1118-1122
Tsao S-C, Dickinson TH, Abernethy DR. Metabolite inhibition of parent drugbiotransformation. Studies of diltiazem. Drug Metab Dispos 1990;18:180-182
Tsuei SE, Moore RG, Ashley JJ, McBride WG. Disposition of synethetic glucocorticoids. I.Pharmacokinetics of dexamethasone in healthy adults. J Pharmacokinet Biopharm1979;7:249-264
Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal andhepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect ofketoconazole. Clin Pharmacol Ther 1999;66:461-471
Twycross R. The risks and benefits of corticosteroids in advanced cancer. Drug Saf1994;11:163-178
Ueda K, Okamura N, Hirai M, Tanigawara Y, Saeki T, Kioka N, Komano T, Hori R. HumanP-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone.J Biol Chem 1992;267:24248-24252
Vanakoski J, Mattila MJ, Seppälä T. Grapefruit juice does not enhance the effects ofmidazolam and triazolam in man. Eur J Clin Pharmacol 1996;50:501-508
Van Peer A, Woestenborghs R, Heykants J, Gasparini R, Gauwenbergh G. The effects offood and dose on the oral systemic availability of itraconazole in healthy subjects. Eur JClin Pharmacol 1989;36:423-426
Varhe A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patientsreceiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther1994;56:601-607
Varhe A, Olkkola KT, Neuvonen PJ. Diltiazem enhances the effects of triazolam byinhibiting its metabolism. Clin Pharmacol Ther 1996;59:369-375
Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents onoxidative drug metabolism: clinical relevance. Clin Pharmacokinet 2000;38:111-180
Veronese ML, Gillen LP, Dorval EP, Pequignot EC, Waldman SA, Greenberg HE. Inhibitionof hepatic and intestinal CYP3A4 by ”high dose” grapefruit juice (GFJ) (Abstract). J ClinPharmacol 1999;39:980
Villikka K, Kivistö KT, Backman JT, Olkkola KT, Neuvonen PJ. Triazolam is ineffective inpatients taking rifampin. Clin Pharmacol Ther 1997;61:8-14
Villikka K, Kivistö KT, Neuvonen PJ. The effect of dexamethasone on the pharmacokineticsof triazolam. Pharmacol Toxicol 1998;83:135-138

REFERENCES
85
Vree TB, Maljers L, Van den Borg N, Nibbering NM, Verwey-van Wissen CP, LagerwerfAJ, Maes RA, Jongen PJ. High-performance liquid-chromatographic-atmospheric-pressurechemical-ionization ion-trap mass-spectrometric identification of isomeric C6-hydroxy andC20-hydroxy metabolites of methylprednisolone in the urine of patients receiving high-dose pulse therapy. J Pharm Pharmacol 1999;51:1155-1166
van der Vring JA, Bernink PJ, van der Wall EE, van Velhuisen DJ, Braun S, Kobrin I.Evaluating the safety of mibefradil, a selective T-type calcium antagonist, in patients withchronic congestive heart failure. Clin Ther 1996;18:1191-1206
Wacher VJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution ofcytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity incancer chemotherapy. Mol Carcinog 1995;13:129-134
Wagner K, Albrecht S, Neumayer HH. Prevention of posttransplant acute tubular necrosis bythe calcium antagonist diltiazem: a prospective randomized study. Am J Nephrol1987;7:287-291
Wandel C, Kim RB, Guengerich FP, Wood AJ. Mibefradil is a P-glycoprotein substrate and apotent inhibitor of both P-glycoprotein and CYP3A In vitro. Drug Metab Dispos2000;28:895-898
Wang JS, Wen X, Backman JT, Taavitsainen P, Neuvonen PJ, Kivistö KT. Midazolamalpha-hydroxylation by human liver microsomes in vitro: inhibition by calcium channelblockers, itraconazole and ketoconazole. Pharmacol Toxicol 1999;85:157-161
de Waziers I, Cugnenc PH, Yang CS, Leroux JP, Beaune PH. Cytochrome P 450isoenzymes, epoxide hydrolase and glutathione transferases in rat and human hepatic andextrahepatic tissues. J Pharmacol Exp Ther 1990 ;253:387-394
Welker HA, Wiltshire H, Bullingham R. Clinical pharmacokinetics of mibefradil. ClinPharmacokinet 1998;35:405-423
de Wildt SN, Kearns GL, Leeder JS, van den Anker JN. Cytochrome P450 3A: ontogeny anddrug disposition. Clin Pharmacokinet 1999;37:485-505
Wiltshire HR, Sutton BM, Heeps G, Betty AM, Angus DW, Harris SR, Worth E, WelkerHA. Metabolism of the calcium antagonist, mibefradil (POSICOR, Ro 40-5967). Part III.Comparative pharmacokinetics of mibefradil and its major metabolites in rat, marmoset,cynomolgus monkey and man. Xenobiotica 1997;27:557-571
Workman RJ, Vaughn WK, Stone WJ. Dexamethasone suppression testing in chronic renalfailure: pharmacokinetics of dexamethasone and demonstration of a normal hypothalamic-pituitary-adrenal axis. J Clin Endocrinol Metab 1986;63:741-746
Wrighton SA, Vandenbranden M. Isolation and characterization of human fetal livercytochrome P450HLp2: a third member of the P450III gene family. Arch BiochemBiophys 1989;268(1):144-151
Wrighton SA, Brian WR, Sari MA, Iwasaki M, Guengerich FP, Raucy JL, Molowa DT,Vandenbranden M. Studies on the expression and metabolic capabilities of human livercytochrome P450IIIA5 (HLp3). Mol Pharmacol 1990;38:207-213
Wrighton SA, Stevens JC. The human hepatic cytochromes P450 involved in drugmetabolism. Crit Rev Toxicol 1992;22:1-21
Wrighton SA, VandenBranden M, Ring BJ. The human drug metabolizing cytochromesP450. J Pharmacokinet Biopharm 1996;24:461-473
Wu CY, Benet LZ, Hebert MF, Gupta SK, Rowland M, Gomez DY, Wacher VJ.Differentiation of absorption and first-pass gut and hepatic metabolism in humans: studieswith cyclosporine. Clin Pharmacol Ther 1995;58:492-497

REFERENCES
86
Yamashita SK, Ludwig EA, Middleton E Jr, Jusko WJ. Lack of pharmacokinetic andpharmacodynamic interactions between ketoconazole and prednisolone. Clin PharmacolTher 1991;49:558-570
Yeates RA, Laufen H, Zimmermann T. Interaction between midazolam and clarithromycin:comparison with azithromycin. Int J Clin Pharmacol Ther 1996;34:400-405
Yun C-H, Okerholm RA, Guengerich FP. Oxidation of the antihistaminic drug terfenadine inhuman liver microsomes. Role of cytochrome P-450 3A(4) in N-dealkylation and C-hydroxylation. Drug Metab Dispos 1993;21:403-409
Yule SM, Boddy AV, Cole M, Price L, Wyllie R, Tasso MJ, Pearson AD, Idle JR.Cyclophosphamide pharmacokinetics in children. Br J Clin Pharmacol 1996;41:13-19
Zürcher RM, Frey BM, Frey FJ. Impact of ketoconazole on the metabolism of prednisolone.Clin Pharmacol Ther 1989;45:366-372
Öst L. Effects of cyclosporin on prednisolone metabolism. Lancet 1984;1(8374):451





























