Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1986 hy The American Soeiety of Biological Chemists, Inc.
Vol. 261, No. 15, Issue of May 25, pp. 7001-7010,1986 Printed in U.S.A.
Studies of the DNA Helicase-RNA Primase Unit from Bacteriophage T4 A TRINUCLEOTIDE SEQUENCE ON THE DNA TEMPLATE STARTS RNA PRIMER SYNTHESIS*
(Received for publication, October 28, 1985)
Tai-An ChaS and Bruce M. Alberts From the Department of Biochemistry and Biophysics, University of California, Sun Francisco, Sun Francisco, California 94143
The purified DNA replication proteins encoded by genes 41 and 61 of bacteriophage T4 catalyze efficient RNA primer synthesis on a single-stranded DNA tem- plate. In the presence of additional T4 replication pro- teins, we demonstrate that the template sequences 5’- GTT-3’ and 5’-GCT-3’ serve as necessary and suffi- cient signals for RNA primer-dependent initiation of new DNA chains. These chains start with primers that have the sequences pppApCpNpNpN and pppGpCp- NpNpN, where N can be any one of the four ribonucle- otides. Each primer is initiated from the T (A-start primers) or C (G-start primers) in the center of the recognized template sequence. A subset of the DNA chain starts is observed when one of the four ribonu- cleoside triphosphates used as the substrates for primer synthesis is omitted; the starts observed reveal that both pentaribonucleotide and tetraribonucleotide primers can be used for efficient initiation of new DNA chains, whereas primers that are only 3 nucleotides long are inactive.
It was known previously that, when 61 protein is present in catalytic amounts, the 41 and 61 proteins are both required for observing RNA primer synthesis. However, by raising the concentration of the 61 pro- tein to a much higher level, a substantial amount of RNA-primed DNA synthesis is obtained in the absence of 41 protein. The DNA chains made are initiated by primers that seem to be identical to those made when both 41 and 61 proteins are present; however, only those template sites containing the 5’-GCT-3’ sequence are utilized. The 61 protein is, therefore, the RNA primase, whereas the 41 protein should be viewed as a DNA helicase that is required (presumably via a 41/61 complex) for efficient primase recognition of both the 5‘-GCT-3’ and 5’-GTT-3’ DNA template sequences.
Our understanding of the DNA replication process has advanced rapidly in the past decade. By combining genetic and biochemical approaches, many proteins essential for DNA replication have been identified, purified, and studied in vitro (1, 2, 40). In the bacteriophage T4 replication system, with which this laboratory has been concerned, an accurate and efficient DNA replication process has been reconstituted us- ing purified components (3). These studies reveal that a
* This work was supported by Grant GM24020 from the National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
tutes of Health. Recipient of a postdoctoral fellowship from the National Insti-
mixture of seven proteins (the products of T4 genes 32, 43, 45, 44/62, 41, and 61) constitutes the minimal complex for effective T4 DNA replication fork movement. This 7-protein system produces replication forks that closely resemble those formed by the T4 replication apparatus in vivo, with regard to the efficiency, speed, and fidelity of DNA synthesis (4, 5). The model that emerges from these studies involves a highly sophisticated and elegantly designed “replication machine” at the replication fork (6). While the fork advances, the multiple tasks involved in the DNA replication process are carried out by this machine as a result of the coherent activities of all of the individual protein parts, which move relative to each other without disassembling. This model is attractive from a me- chanistic point of view, because it frees the biological process from the necessity of relying on large numbers of random collisions. Similar protein machines are likely to be assembled in other replication systems, including those of Escherichia coli (7, 8) and the bacteriophages T7 (9) and X (10, l l ) , and to function in many other central biological processes, includ- ing DNA transcription.
Detailed structural studies on the replication apparatus are clearly needed in order to provide further insight into the operation of such protein machines. As one step in this direction, our present work focuses on a DNA helicase-RNA primase subassembly that plays a central part in both leading and lagging strand DNA synthesis in the bacteriophage T4 system. In particular, the template sequence specificity and the protein requirements for the initiation of RNA primer synthesis have been examined. For simulating the process of lagging strand DNA synthesis, we have mixed highly purified T4 replication proteins with an M13 viral DNA template; since no free 3”hydroxyl end is present on this closed circular single-stranded DNA molecule, the initiation of DNA chains is dependent on an RNA-priming process.
In previous studies it was shown that bacteriophage T4 uses the products of T4 genes 41 and 61 for RNA primer synthesis (12-14) and that the major products synthesized are pentaribonucleotides both in vivo (15) and in vitro (12- 14). Whereas primers with a sequence of pppApCpNpNpN are the major products on single-stranded T4 DNA (which contains naturally modified hydroxymethylcytosine residues), additional primers that start with pppG are also found when any cytosine-containing DNA is used as the template (13). The earlier studies also revealed that the third and fourth positions of the primer can be any one of the A, U, C, or G bases; however, no information could be obtained concerning the fifth, 3”terminal nucleotide. Although a large number of discrete sites was found for DNA chain starts, the template sequence required to start a primer remained unknown, and our hypothesis that the 61 protein is the actual RNA primase
7001

7002 DNA Recognition by T4 Priming Proteins
was unproven. In this report, we present experiments that directly address these unresolved issues.
MATERIALS AND METHODS
DNA and Enzymes”M13 phage was isolated from the supernatant of infected E. coli JMlOl cells by two sequential polyethylene glycol precipitations. The phage particles were further purified by centrif- ugation through a CsCl step gradient as described previously (16). Viral DNA was extracted from the purified phage using standard phenol-detergent deproteinization and ethanol precipitation. Analy- sis by agarose gel electrophoresis indicated that 99% of the DNA molecules were single-stranded closed circles. The protein products of T4 genes 43,44/62,45, and 32 were purified from T4-infected cells according to published procedures (17,18). The T4 gene 41 and gene 61 proteins were purified from overproducing E. coli cells carrying the cloned genes on X PL-containing plasmid vectors.’ The plasmid that carries gene 41 was kindly provided by Drs. D. Hinton, L. Silver, and N. Nossal a t National Institutes of Health (19), while the plasmid that carries gene 61 was constructed by Drs. H. Selick and M. Nakanishi in this laboratory.’ All replication proteins were purified to near homogeneity and were free of detectable nuclease activity; their specific activities were tested in an in vitro replication assay described previously (20). 32P-Labeled nucleoside triphosphates were purchased from Amersham Corp. Restriction enzymes were obtained from New England Biolabs.
RNA-primed DNA Synthesis-As mentioned earlier, de m v o ini- tiation of DNA synthesis on a single-stranded closed circular DNA template is dependent on an RNA primer. In the presence of a mixture of six T4 replication proteins (the products of genes 41, 43, 44, 45, 61, and 62), circular duplex molecules are produced in which the new strand is a unit-length molecule that carries a newly synthesized short RNA primer at its 5’-end (20). (For simplicity, we will refer to this reaction as the “standard 6-protein reaction” in the rest of the text.) If the primers are labeled by the incorporation of radioactive ribo- nucleotides, and the circular duplex DNA product molecules are treated with a restriction endonuclease that produces a single cut in each molecule, discrete labeled bands whose lengths correspond to the distance from the sites of RNA primer initiation to the restriction- cutting site are detected by gel electrophoresis under denaturing conditions (see Fig. 1, below).
For 6-protein reactions, the reaction mixture in a 0.1-ml volume contained replication buffer (33 mM Tris-acetate, pH 7.8, 66 mM potassium acetate, 10 mM magnesium acetate, 0.5 mM dithiothreitol, and nuclease-free bovine serum albumin at 112 pg/ml), nucleoside triphosphates (120 p~ each of the four ribonucleoside triphosphates, 1 mM dATP, and 200 p~ each of dCTP, dGTP, and TTP), DNA template (M13 viral DNA at 7.5 pg/ml), and replication proteins (41 protein at 60 pg/ml, 61 protein at 1.3 pg/ml, 43 protein at 2.7 pg/ml, and 45 and 44/62 proteins both at 27 pglml). In order to label the RNA primers, [cY-~’P]CTP at 35 Ci/mmol was included in the reac- tion. The reaction mixture was incubated at 37 “C for 20 min and stopped with Na3EDTA (25 mM) and sodium dodecyl sulfate (0.5%). The DNA was extracted with phenol/chloroform and precipitated with ethanol containing 2 M ammonium acetate to remove unincor- porated nucleotides. After this DNA was treated with a restriction enzyme, the length of its radioactive strands was analyzed by electro- phoresis through a 5% polyacrylamide gel containing 8 M urea fol- lowed by autoradiography. For measuring DNA synthesis rates, [a-32P]dGTP at 2 Ci/mmol replaced the [a-32P]CTP, and small aliquots of solution were taken at various time intervals and acid precipitated onto GF/C filters (Whatman) followed by scintillation counting.
DNA Sequencing and High Resolution Gel Electrophoresis-Two approaches were employed to map the primer site sequence at the nucleotide level. In the first approach, standard Maxam-Gilbert se- quencing techniques (21) were used to determine the 5’-end sequence of the RNA-primed DNA fragments that were detected by the dena-
’ J. Barry, T. Cha, and H. Selick, unpublished experiments. H. Selick and M. Nakanishi, unpublished results. A T4 DNA
fragment (spanning nucleotide positions 16978 to 18610) containing gene 61 was joined to a synthetic T7 bacteriophage gene 10 ribosomal binding site sequence attached immediately upstream. This modified gene was cloned into the X PL-containing pUC9 expression vector. E. coli cells carrying this plasmid, after heat induction, overproduce 61 protein to a level of 10-20% of the total cellular protein.
turing gel. Since the conditions used in Maxam-Gilbert reactions degrade the RNA primer, radioactive label had to be introduced at the 5’-end of the DNA. The reaction mixture was, therefore, treated with alkali to remove the RNA primers and the single-stranded DNA molecules produced then labeled with [-p3’P]ATP (5000 Ci/mmol) using polynucleotide kinase. The labeled DNA was next annealed to the template DNA and cut by treatment with a restriction endonu- clease. The labeled DNA fragments were then separated by gel electrophoresis, purified from gel slices, and subjected to the standard DNA sequencing reactions. The 5’-end sequence determined in this way was used to locate the primer site sequence on the template according to base complementarity by reference to the known M13 DNA sequence (22); these DNA fragments were five nucleotides shorter than the corresponding RNA-labeled fragments from which they were derived. This approach is similar to the system developed to analyze T7 RNA primer sites by Tabor and Richardson (23).
The second approach used to position primers made use of the high resolving power of electrophoresis through a long and thin polyacrylamide gel. Under the conditions we used (a 60 X 20 X 0.03- cm polyacrylamide gel containing 8 M urea, with the xylene cyanole dye run to a point 15 cm from the bottom using 75 watts of constant power), the lengths of DNA fragments between 100 and 300 nucleo- tides long could be resolved with a single loading by reference to appropriate markers. The primer site sequence on the template could then be read from the known M13 sequence, since the lengths of these fragments correspond to the distance from the primer start to the restriction cleavage site. Two types of size markers were employed in these gel analyses: for coarse calibration, restriction fragments generated from HueIII-digested 6x174 DNA and M13 DNA were used, whereas for high resolution gel analysis a sequencing ladder was generated from the Maxam-Gilbert “G plus A reaction” performed on a 310-nucleotide-long fragment isolated from HueIII-cut 4x174 DNA.
RESULTS
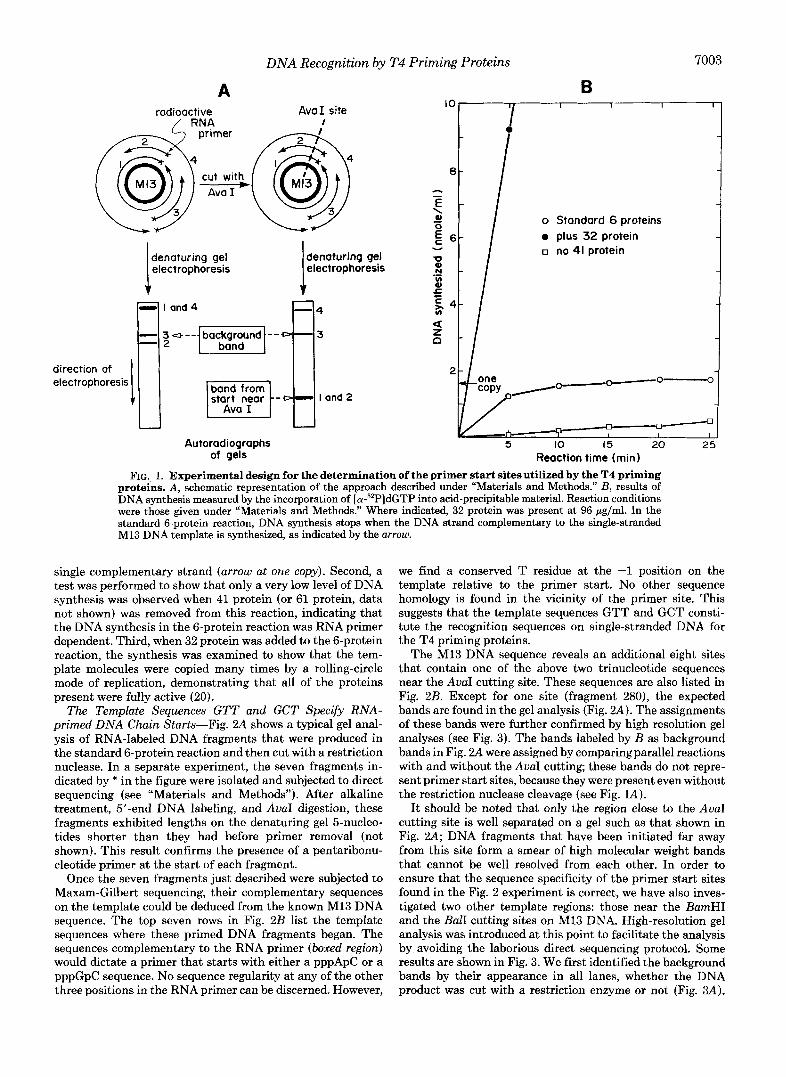
Fig. lA presents a schematic representation of the method described under “Materials and Methods” for localizing primer start sites. Briefly, pentaribonucleotide primers are synthesized at multiple sites on an M13 DNA template in a standard 6-protein reaction. The T4 DNA polymerase (gene 43 protein) catalyzes DNA synthesis that begins at the 3’- hydroxyl end of these primers and, aided by the polymerase accessory proteins (44/62 and 45 proteins), proceeds around the circular template to produce a population of nicked duplex circular molecules. The RNA primers are radioactively labeled by the incorporation of [a-32P]CTP present in the reaction. On the autoradiograph obtained after polyacrylamide gel elec- trophoresis under denaturing conditions, the unit-length product strands will produce a single predominant band cor- responding to the unit length of the template. In order to separate the RNA-primed DNA fragments with different start sites, a restriction endonuclease that produces a single cut in the duplex product molecules (AuaI in this example) is used to cut the DNA circles prior to gel electrophoresis. The lengths of the resolved DNA fragments then identify the various positions of primer starts relative to the restriction site.
As shown in Fig. lA, if DNA synthesis terminates at specific pause sites before the template is completely copied, DNA bands will appear on the gel in addition to the ones generated by the restriction cutting. These “background bands” are identified by being present even without restriction enzyme treatment. Such background bands would be expected because of the DNA polymerase pause sites created by short helical regions in the template, as described previously (24, 25).
The control assays shown in Fig. 1B were routinely per- formed to test new batches of DNA and proteins, in order to ensure reproducible conditions. Three reactions were moni- tored. First, in the standard 6-protein reaction the level of incorporated deoxyribonucleotides was examined to ensure that the synthesis rapidly reached a plateau where the major- ity of the template molecules had been used to synthesize a
DNA Recognition by T4 Priming Proteins 7003
A B radioactive AvaI site
IO I I I
I ‘I primer I @L@ 0
, [ 2 F,l I I
o Standard 6 proteins 0 plus 32 protein 0 no 41 protein
denaturing gel denaturing gel electrophoresis electrophoresis
- I and 4 4
- 3 4 ” background ”-0 3
direction of electrophoresis /o-- 0 ° C
start near - I and 2
1-0-= I I I
Autoradiographs 5 IO 15 20 2: of gels Reaction time (min)
FIG. 1. Experimental design for the determination of the primer start sites utilized by the T4 priming proteins. A, schematic representation of the approach described under “Materials and Methods.” B, results of DNA synthesis measured by the incorporation of [cY-~’P]~GTP into acid-precipitable material. Reaction conditions were those given under “Materials and Methods.” Where indicated, 32 protein was present at 96 gg/ml. In the standard 6-protein reaction, DNA synthesis stops when the DNA strand complementary to the single-stranded M13 DNA template is synthesized, as indicated by the arrow.
single complementary strand (arrow at one copy). Second, a test was performed to show that only a very low level of DNA synthesis was observed when 41 protein (or 61 protein, data not shown) was removed from this reaction, indicating that the DNA synthesis in the 6-protein reaction was RNA primer dependent. Third, when 32 protein was added to the 6-protein reaction, the synthesis was examined to show that the tem- plate molecules were copied many times by a rolling-circle mode of replication, demonstrating that all of the proteins present were fully active (20).
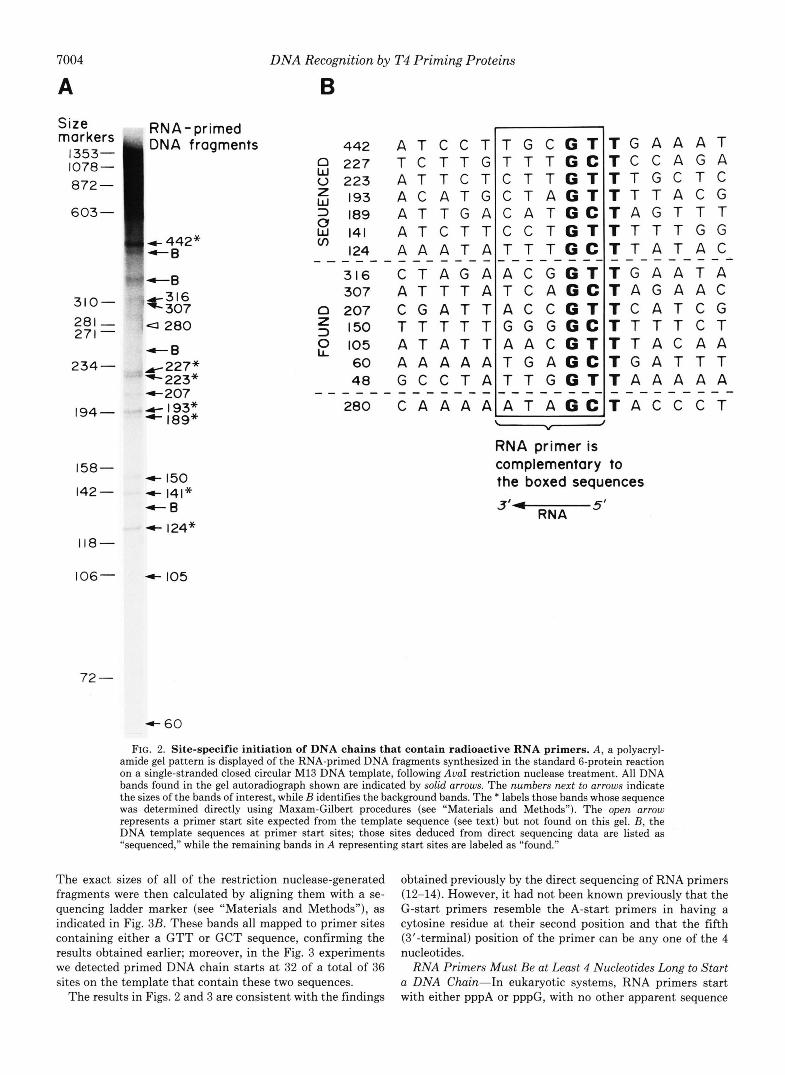
The Template Sequences GTT and GCT Specify R N A - primed D N A Chain Starts-Fig. 2A shows a typical gel anal- ysis of RNA-labeled DNA fragments that were produced in the standard 6-protein reaction and then cut with a restriction nuclease. In a separate experiment, the seven fragments in- dicated by * in the figure were isolated and subjected to direct sequencing (see “Materials and Methods”). After alkaline treatment, 5’-end DNA labeling, and AuaI digestion, these fragments exhibited lengths on the denaturing gel 5-nucleo- tides shorter than they had before primer removal (not shown). This result confirms the presence of a pentaribonu- cleotide primer at the start of each fragment.
Once the seven fragments just described were subjected to Maxam-Gilbert sequencing, their complementary sequences on the template could be deduced from the known M13 DNA sequence. The top seven rows in Fig. 2B list the template sequences where these primed DNA fragments began. The sequences complementary to the RNA primer (boxed region) would dictate a primer that starts with either a pppApC or a pppGpC sequence. No sequence regularity at any of the other three positions in the RNA primer can be discerned. However,
we find a conserved T residue at the -1 position on the template relative to the primer start. No other sequence homology is found in the vicinity of the primer site. This suggests that the template sequences GTT and GCT consti- tute the recognition sequences on single-stranded DNA for the T4 priming proteins.
The M13 DNA sequence reveals an additional eight sites that contain one of the above two trinucleotide sequences near the AuaI cutting site. These sequences are also listed in Fig. 2B. Except for one site (fragment 280), the expected bands are found in the gel analysis (Fig. 2 A ) . The assignments of these bands were further confirmed by high resolution gel analyses (see Fig. 3). The bands labeled by B as background bands in Fig. 2A were assigned by comparing parallel reactions with and without the AuaI cutting; these bands do not repre- sent primer start sites, because they were present even without the restriction nuclease cleavage (see Fig. L4).
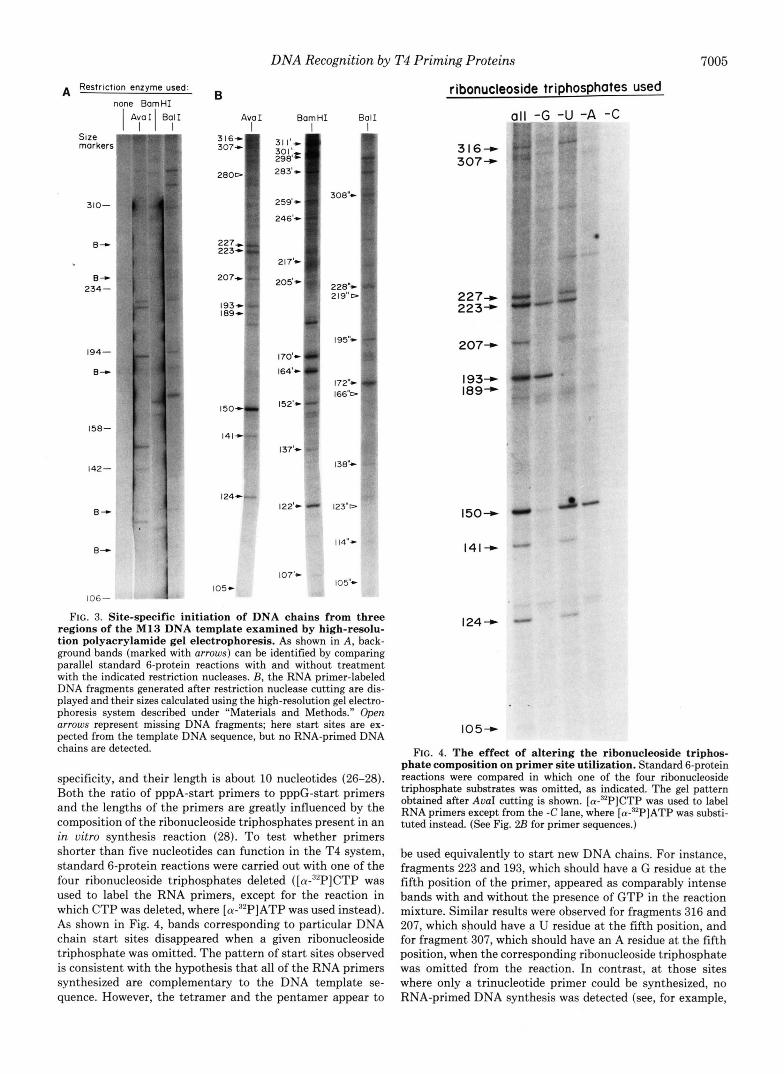
It should be noted that only the region close to the AuaI cutting site is well separated on a gel such as that shown in Fig. 2 . 4 ; DNA fragments that have been initiated far away from this site form a smear of high molecular weight bands that cannot be well resolved from each other. In order to ensure that the sequence specificity of the primer start sites found in the Fig. 2 experiment is correct, we have also inves- tigated two other template regions: those near the BamHI and the BalI cutting sites on M13 DNA. High-resolution gel analysis was introduced at this point to facilitate the analysis by avoiding the laborious direct sequencing protocol. Some results are shown in Fig. 3. We first identified the background bands by their appearance in all lanes, whether the DNA product was cut with a restriction enzyme or not (Fig. 3A).
7004 DNA Recognition by T4 Priming Proteins
B Size markers 1353- IO78 - 072 - 603-
RNA-primed DNA fragments
c 442" -6
158-
142 - 4 141" 4 150
+R 4 124"
118-
442
E 2 2 7
A T C C T T G C G T T C T T G T T T G C
0 2 2 3 A T T C T C T T G T 6 193 A C A T G C T A G T 2 189 A T T G A C A T G C S 141 A T C T T C C T G T
124 A A A T A T T T G C
316 C T A G A A C G G T 307 A T T T A T C A G C
0 2 0 7 C G A T T A C C G T 5 150 T T T T T G G G G C 0 105 A T A T T A A C G T
60 A A A A A T G A G C 48 G C C T A T T G G T
280 C A A A A A T A G C - RNA primer is
0 """"_""""""
LI
"_"""""""""
T G A A A T T C C A G A T T G C T C T T T A C G T A G T T T T T T T G G T T A T A C
T G A A T A T A G A A C T C A T C G T T T T C T T T A C A A T G A T T T T A A A A A
T A C C C T
"""" i
""""-
complementary to the boxed sequences 3" 5'
RNA
106- + 105
72 -
FIG. 2. Site-specific initiation of DNA chains that contain radioactive RNA primers. A, a polyacryl- amide gel pattern is displayed of the RNA-primed DNA fragments synthesized in the standard 6-protein reaction on a single-stranded closed circular M13 DNA template, following AuaI restriction nuclease treatment. All DNA bands found in the gel autoradiograph shown are indicated by solid arrows. The numbers next to arrows indicate the sizes of the bands of interest, while B identifies the background bands. The * labels those bands whose sequence was determined directly using Maxam-Gilbert procedures (see "Materials and Methods"). The open arrow represents a primer start site expected from the template sequence (see text) but not found on this gel. B, the DNA template sequences at primer start sites; those sites deduced from direct sequencing data are listed as "sequenced," while the remaining bands in A representing start sites are labeled as "found."
The exact sizes of all of the restriction nuclease-generated fragments were then calculated by aligning them with a se- quencing ladder marker (see "Materials and Methods"), as indicated in Fig. 3B. These bands all mapped to primer sites containing either a GTT or GCT sequence, confirming the results obtained earlier; moreover, in the Fig. 3 experiments we detected primed DNA chain starts at 32 of a total of 36 sites on the template that contain these two sequences.
The results in Figs. 2 and 3 are consistent with the findings
obtained previously by the direct sequencing of RNA primers (12-14). However, it had not been known previously that the G-start primers resemble the A-start primers in having a cytosine residue at their second position and that the fifth (3'-terminal) position of the primer can be any one of the 4 nucleotides.
RNA Primers Must Be at Least 4 Nucleotides Long to Start a DNA Chain-In eukaryotic systems, RNA primers start with either pppA or pppG, with no other apparent sequence
DNA Recognition by T4 Priming Proteins 7005
A Restriction enzyme used: none BomHI
I 1 ATII Bo11
2 Z e r s .. i '
310-
B"
B" 234-
194-
B"
158-
142-
B"
B"
106-
B
Avo I BomHI B o l l
I 207
193 I89
I50
141
I24
105-
170" 1)1 164'-
+. 122'- -
107'-
.* :. .
228% 4% 219°C-
195"- - ..e
172'#- rl 166%
I 3 8"-
123"D
114"-
I 0 5"-
FIG. 3. Site-specific initiation of DNA chains from three regions of the M13 DNA template examined by high-resolu- tion polyacrylamide gel electrophoresis. As shown in A, back- ground bands (marked with arrows) can be identified by comparing parallel standard 6-protein reactions with and without treatment with the indicated restriction nucleases. B, the RNA primer-labeled DNA fragments generated after restriction nuclease cutting are dis- played and their sizes calculated using the high-resolution gel electro- phoresis system described under "Materials and Methods." Open arrows represent missing DNA fragments; here start sites are ex- pected from the template DNA sequence, but no RNA-primed DNA chains are detected.
specificity, and their length is about 10 nucleotides (26-28). Both the ratio of pppA-start primers to pppG-start primers and the lengths of the primers are greatly influenced by the composition of the ribonucleoside triphosphates present in an in uitro synthesis reaction (28). To test whether primers shorter than five nucleotides can function in the T4 system, standard 6-protein reactions were carried out with one of the four ribonucleoside triphosphates deleted ( [cx-~~PICTP was used to label the RNA primers, except for the reaction in which CTP was deleted, where [cx-~~PIATP was used instead). As shown in Fig. 4, bands corresponding to particular DNA chain start sites disappeared when a given ribonucleoside triphosphate was omitted. The pattern of start sites observed is consistent with the hypothesis that all of the RNA primers synthesized are complementary to the DNA template se- quence. However, the tetramer and the pentamer appear to
ribonucleoside triphosphates used
all -G -U -A -C
316+ 307+
227- 223*
207-
189- I93+
150-
141-
124-
105+
FIG. 4. The effect of altering the ribonucleoside triphos- phate composition on primer site utilization. Standard 6-protein reactions were compared in which one of the four ribonucleoside triphosphate substrates was omitted, as indicated. The gel pattern obtained after AvaI cutting is shown. [cY-~'P]CTP was used to label RNA primers except from the -C lane, where [cY-~*P]ATP was substi- tuted instead. (See Fig. 2B for primer sequences.)
be used equivalently to start new DNA chains. For instance, fragments 223 and 193, which should have a G residue at the fifth position of the primer, appeared as comparably intense bands with and without the presence of GTP in the reaction mixture. Similar results were observed for fragments 316 and 207, which should have a U residue at the fifth position, and for fragment 307, which should have an A residue at the fifth position, when the corresponding ribonucleoside triphosphate was omitted from the reaction. In contrast, at those sites where only a trinucleotide primer could be synthesized, no RNA-primed DNA synthesis was detected (see, for example,
7006 DNA Recognition by T4 Priming Proteins
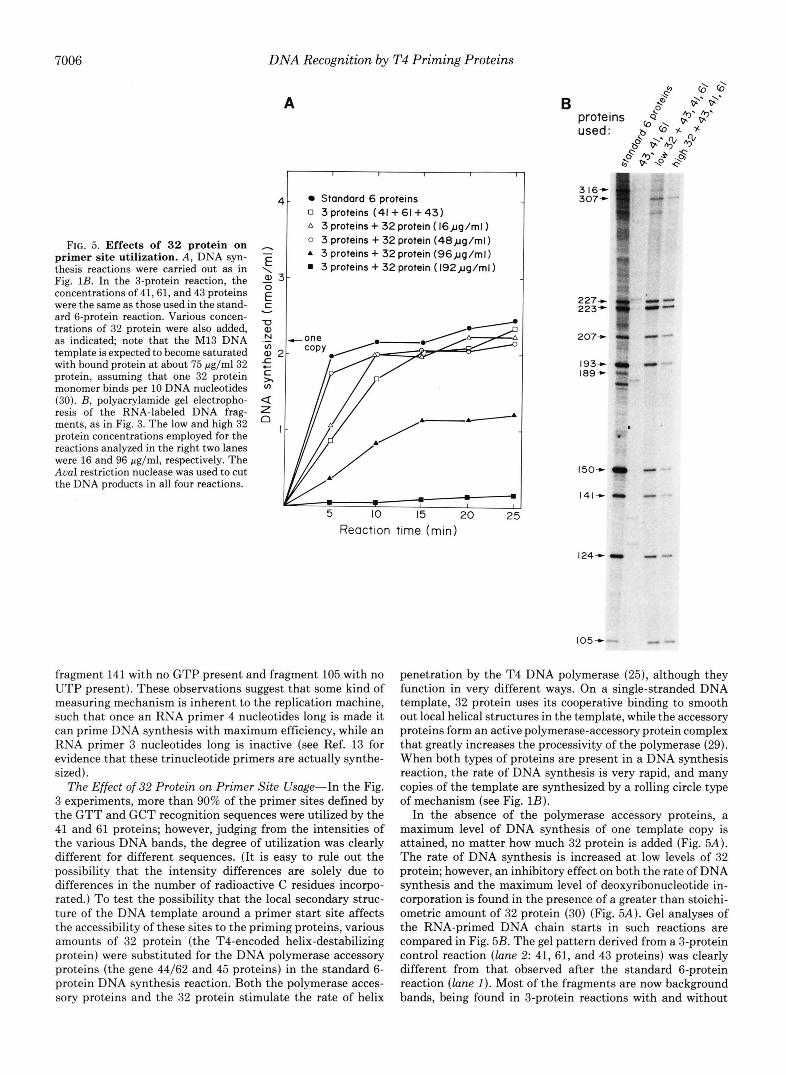
FIG. 5. Effects of 32 protein on primer site utilization. A , DNA syn- thesis reactions were carried out as in Fig. 1B. In the 3-protein reaction, the concentrations of 41,61, and 43 proteins were the same as those used in the stand- ard 6-protein reaction. Various concen- trations of 32 protein were also added, as indicated; note that the M13 DNA template is expected to become saturated with bound protein at about 75 pg/ml32 protein, assuming that one 32 protein monomer binds per 10 DNA nucleotides (30). B, polyacrylamide gel electropho- resis of the RNA-labeled DNA frag- ments, as in Fig. 3. The low and high 32 protein concentrations employed for the reactions analyzed in the right two lanes were 16 and 96 pg/ml, respectively. The AuaI restriction nuclease was used to cut the DNA products in all four reactions.
A
4. Standard 6 proteins 0 3proteins ( 4 1 + 6 1 + 4 3 ) A 3 proteins + 32 protein ( 1 6 ~ ~ g / m l ) o 3 proteins + 32 protein ( 4 8 ~ ~ g / m l )
h -
- 0, 3- 3 proteins + 32 protein ( 1 9 2 ~ ~ g / m l ) E rl. 3 proteins + 32 protein ( 9 6 ~ ~ g / m l )
\
E v c
3 16- 307'
227" 223"
207-3
189- 193+
150-
141-
Reaction time (min)
124-
105-
fragment 141 with no GTP present and fragment 105 with no UTP present). These observations suggest that some kind of measuring mechanism is inherent to the replication machine, such that once an RNA primer 4 nucleotides long is made it can prime DNA synthesis with maximum efficiency, while an RNA primer 3 nucleotides long is inactive (see Ref. 13 for evidence that these trinucleotide primers are actually synthe- sized).
The Effect of 32 Protein on Primer Site Usage-In the Fig. 3 experiments, more than 90% of the primer sites defined by the GTT and GCT recognition sequences were utilized by the 41 and 61 proteins; however, judging from the intensities of the various DNA bands, the degree of utilization was clearly different for different sequences. (It is easy to rule out the possibility that the intensity differences are solely due to differences in the number of radioactive C residues incorpo- rated.) To test the possibility that the local secondary struc- ture of the DNA template around a primer start site affects the accessibility of these sites to the priming proteins, various amounts of 32 protein (the T4-encoded helix-destabilizing protein) were substituted for the DNA polymerase accessory proteins (the gene 44/62 and 45 proteins) in the standard 6- protein DNA synthesis reaction. Both the polymerase acces- sory proteins and the 32 protein stimulate the rate of helix
penetration by the T4 DNA polymerase (25), although they function in very different ways. On a single-stranded DNA template, 32 protein uses its cooperative binding to smooth out local helical structures in the template, while the accessory proteins form an active polymerase-accessory protein complex that greatly increases the processivity of the polymerase (29). When both types of proteins are present in a DNA synthesis reaction, the rate of DNA synthesis is very rapid, and many copies of the template are synthesized by a rolling circle type of mechanism (see Fig. 1B).
In the absence of the polymerase accessory proteins, a maximum level of DNA synthesis of one template copy is attained, no matter how much 32 protein is added (Fig. 5A). The rate of DNA synthesis is increased at low levels of 32 protein; however, an inhibitory effect on both the rate of DNA synthesis and the maximum level of deoxyribonucleotide in- corporation is found in the presence of a greater than stoichi- ometric amount of 32 protein (30) (Fig. 5A). Gel analyses of the RNA-primed DNA chain starts in such reactions are compared in Fig. 5B. The gel pattern derived from a 3-protein control reaction (lane 2: 41, 61, and 43 proteins) was clearly different from that observed after the standard 6-protein reaction (lane 1). Most of the fragments are now background bands, being found in 3-protein reactions with and without
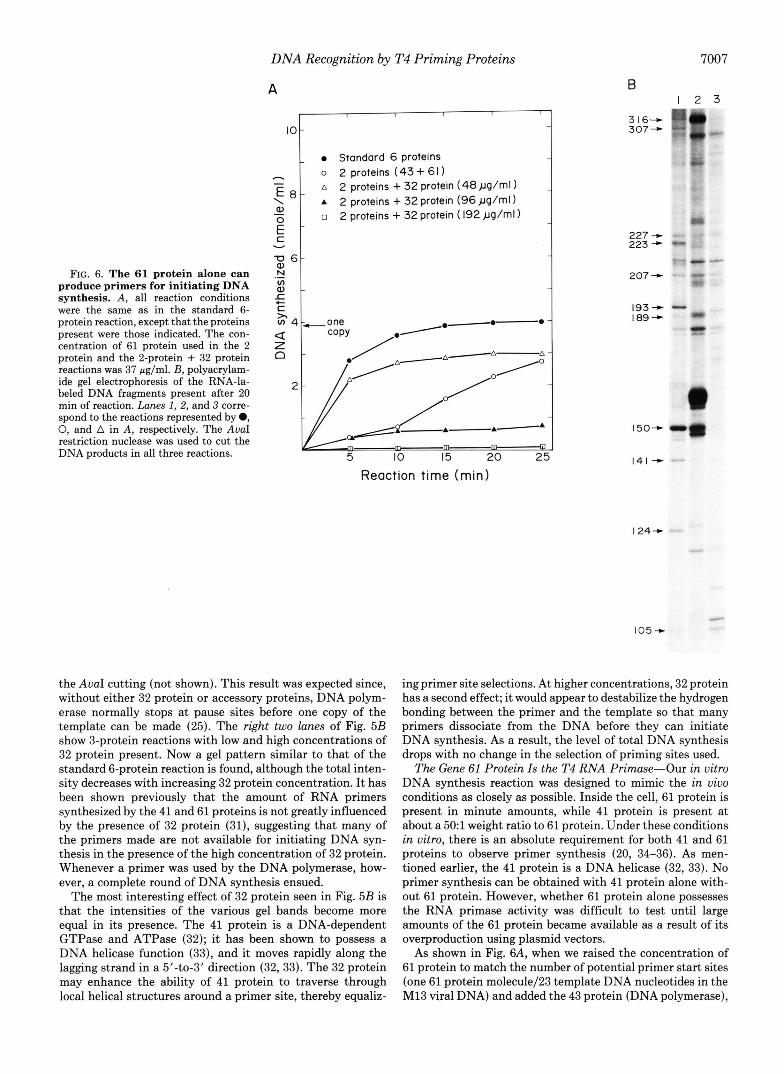
FIG. 6. The 61 protein alone can produce primers for initiating DNA synthesis. A , all reaction conditions were the same as in the standard 6- protein reaction, except that the proteins present were those indicated. The con- centration of 61 protein used in the 2 protein and the 2-protein + 32 protein reactions was 37 pg/ml. E, polyacrylam- ide gel electrophoresis of the RNA-la- heled DNA fragments present after 20 min of reaction. Lunes I , 2, and 3 corre- spond to the reactions represented by 0, 0, and A in A, respectively. The AvaI restriction nuclease was used to cut the DNA products in all three reactions.
DNA Recognition by T4 Priming Proteins
A I I I I
IO -
- Standard 6 proteins CI o 2 proteins (43+ 61) -
A 2 proteins + 32 protein (48pg/rnl)
O 2 proteins + 32 protein ( 192 pg/rnl) \ E
E - - A 2 proteins + 32 protein (96 pg/ml)
0) - c Y
f Q) I L )r
-0-0
5 IO 15 20 2
Reaction time (min)
the AvuI cutting (not shown). This result was expected since, without either 32 protein or accessory proteins, DNA polym- erase normally stops at pause sites before one copy of the template can be made (25). The right two lunes of Fig. 5B show 3-protein reactions with low and high concentrations of 32 protein present. Now a gel pattern similar to that of the standard 6-protein reaction is found, although the total inten- sity decreases with increasing 32 protein concentration. It has been shown previously that the amount of RNA primers synthesized by the 41 and 61 proteins is not greatly influenced by the presence of 32 protein (31), suggesting that many of the primers made are not available for initiating DNA syn- thesis in the presence of the high concentration of 32 protein. Whenever a primer was used by the DNA polymerase, how- ever, a complete round of DNA synthesis ensued.
The most interesting effect of 32 protein seen in Fig. 5B is that the intensities of the various gel bands become more equal in its presence. The 41 protein is a DNA-dependent GTPase and ATPase (32); it has been shown to possess a DNA helicase function (33), and it moves rapidly along the lagging strand in a 5’40-3’ direction (32,33). The 32 protein may enhance the ability of 41 protein to traverse through local helical structures around a primer site, thereby equaliz-
7007
B I 2 3 ”
3 16” 307 -
227 ”t 223 +
. .”- 207 +
I93 --t 189-
ing primer site selections. At higher concentrations, 32 protein has a second effect; it would appear to destabilize the hydrogen bonding between the primer and the template so that many primers dissociate from the DNA before they can initiate DNA synthesis. As a result, the level of total DNA synthesis drops with no change in the selection of priming sites used.
The Gene 61 Protein Is the T4 RNA Primase-Our in vitro DNA synthesis reaction was designed to mimic the in vivo conditions as closely as possible. Inside the cell, 61 protein is present in minute amounts, while 41 protein is present at about a 501 weight ratio to 61 protein. Under these conditions in vitro, there is an absolute requirement for both 41 and 61 proteins to observe primer synthesis (20, 34-36). As men- tioned earlier, the 41 protein is a DNA helicase (32, 33). No primer synthesis can be obtained with 41 protein alone with- out 61 protein. However, whether 61 protein alone possesses the RNA primase activity was difficult to test until large amounts of the 61 protein became available as a result of its overproduction using plasmid vectors.
As shown in Fig. 6A, when we raised the concentration of 61 protein to match the number of potential primer start sites (one 61 protein molecule/23 template DNA nucleotides in the M13 viral DNA) and added the 43 protein (DNA polymerase),
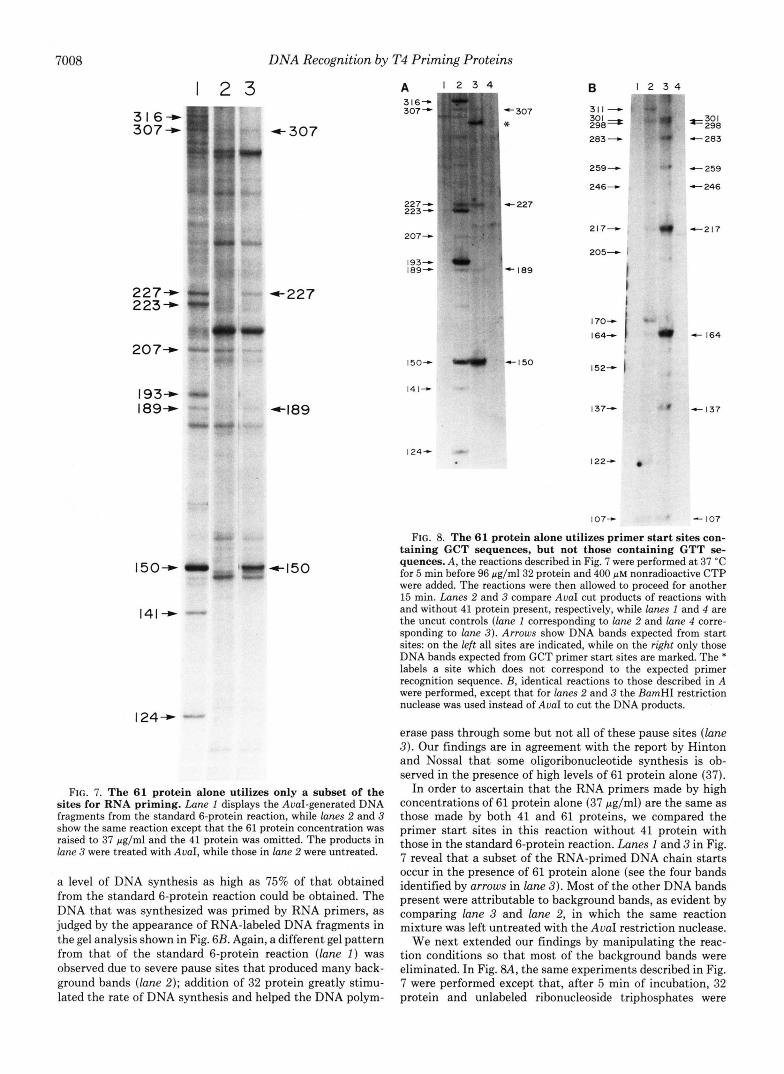
7008 DNA Recognition by T4 Priming Proteins
I 2 3 316- 307- + 307
mr .-
m d
A 316- 307 ”
I 2 3 4
* *
227- 223-
207-
193- 189-
150- - 141 - 124- - -
r-227
e189
FIG. 7. The 61 protein alone utilizes only a subset of the sites for RNA priming. Lane I displays the AuaI-generated DNA fragments from the standard 6-protein reaction, while lanes 2 and 3 show the same reaction except that the 61 protein concentration was raised to 37 pg/ml and the 41 protein was omitted. The products in lane 3 were treated with AuaI, while those in lane 2 were untreated.
a level of DNA synthesis as high as 75% of that obtained from the standard 6-protein reaction could be obtained. The DNA that was synthesized was primed by RNA primers, as judged by the appearance of RNA-labeled DNA fragments in the gel analysis shown in Fig. 6B. Again, a different gel pattern from that of the standard 6-protein reaction (lane 1 ) was observed due to severe pause sites that produced many back- ground bands (lane 2) ; addition of 32 protein greatly stimu- lated the rate of DNA synthesis and helped the DNA polym-
227 + 223-
207”
193- 189”
1504
141-
124-
307
+ 227
- 189
+I50
217-
205+
170”
164”
152”
I 3 7 4
122”
107+
e217
c 164
* I37
- IO7
FIG. 8. The 61 protein alone utilizes primer start sites con- taining GCT sequences, but not those containing GTT se- quences. A, the reactions described in Fig. 7 were performed at 37 “C for 5 min before 96 pg/ml32 protein and 400 p~ nonradioactive CTP were added. The reactions were then allowed to proceed for another 15 min. Lanes 2 and 3 compare AuaI cut products of reactions with and without 41 protein present, respectively, while lanes I and 4 are the uncut controls (lane I corresponding to lane 2 and lane 4 corre- sponding to lane 3) . Arrows show DNA bands expected from start sites: on the left all sites are indicated, while on the right only those DNA bands expected from GCT primer start sites are marked. The * labels a site which does not correspond to the expected primer recognition sequence. B, identical reactions to those described in A were performed, except that for lanes 2 and 3 the BamHI restriction nuclease was used instead of AuaI to cut the DNA products.
erase pass through some but not all of these pause sites (lane 3) . Our findings are in agreement with the report by Hinton and Nossal that some oligoribonucleotide synthesis is ob- served in the presence of high levels of 61 protein alone (37).
In order to ascertain that the RNA primers made by high concentrations of 61 protein alone (37 pg/ml) are the same as those made by both 41 and 61 proteins, we compared the primer start sites in this reaction without 41 protein with those in the standard 6-protein reaction. Lanes 1 and 3 in Fig. 7 reveal that a subset of the RNA-primed DNA chain starts occur in the presence of 61 protein alone (see the four bands identified by arrows in lane 3) . Most of the other DNA bands present were attributable to background bands, as evident by comparing lane 3 and lane 2, in which the same reaction mixture was left untreated with the AvaI restriction nuclease.
We next extended our findings by manipulating the reac- tion conditions so that most of the background bands were eliminated. In Fig. 8A, the same experiments described in Fig. 7 were performed except that, after 5 min of incubation, 32 protein and unlabeled ribonucleoside triphosphates were

DNA Recognition by
TABLE I Ribonucleoside triphosphate dependence of the DNA synthesis
stimulated by high concentrations of 61 protein in the absence of 41 protein
The incorporation of [cY-~’P]~GTP into acid-precipitable material is shown. The reaction conditions were as described under “Materials and Methods” for the standard 6-protein reaction, except that the proteins and rNTPs present in the reactions were as indicated, with the concentration of the 61 or the 41 protein being that listed in the first column. The 43+44/62+45 protein in brackets constitute the DNA polymerase “holoenzyme.” All incubations were for 20 min at 37 “C. An incorporation level of 100% corresponds to one copy of the strand complementary to the single-stranded M13 DNA template (see Fig. 6).
Proteins present rNTPs Present inz$rt$on Standard 6-protein control [43+44/62+45]+61(37 pg/ml) [43+44/62+45]+61 (37 pg/ml) [43+44/62+45]+61(37 pg/ml) [43+44/62+45]+61 (37 pg/ml) [43+44/62+45]+61 (37 pg/ml) [43+44/62+45]+61 (1.3 pg/ml) [43+44/62+45]+41 (65 pglml)
A, G, C, U ( A, G, C, U A, G, C (no LJ) G, C, U (no A) A, G, U (no C ) A, C, U (no G) A, G, C, U A, G, C, U
% 100) 74 71 61 14 12 12 10
added to the reaction for a further 15 min of incubation. During this time, 32 protein catalyzed a strand displacement reaction, allowing DNA replication to proceed beyond one copy of the template. (Because excess unlabeled ribonucleo- side triphosphates were introduced, the RNA primers synthe- sized at later times of the reaction were not significantly radioactively labeled.) After stopping the DNA synthesis re- action, excess M13 DNA template was added and allowed to anneal to the nacent DNA chains before restriction nuclease cutting. As seen in Fig. 8A (lune 4 ) , the background bands were strongly reduced. The four bands identified by their sizes in lune 3 appeared after AuuI nuclease cutting, confirming the results of Fig. 7. It is interesting that one major DNA band (labeled by the * in lune 3) , which was also present in Fig. 7, is not complementary to either of the specific template rec- ognition sequences at its 5‘-end; it, therefore, appears to start from a unique DNA structure that is only recognizable by 61 protein alone.
All four of the DNA fragments identified above were initi- ated from primer sites containing the GCT recognition se- quence. Fig. 8B shows an identical experiment as that re- ported in 8A, except that the BumHI restriction nuclease was employed instead of AuuI, so that more template sites could be analyzed. Again, all fragments identified in lune 3 (Fig. 8B) originate from primer sites containing the GCT sequence. We conclude that the 61 protein is the RNA primase in the T4 replication system; by itself, it can utilize a specific subset of the same RNA primer start sites that are recognized much more efficiently when 41 protein is present.
The above conclusions are supported by an analysis of the ribonucleoside triphosphate requirements for 61 protein-de- pendent DNA synthesis on an M13 DNA template when no 41 protein is present. As shown in Table I, under these conditions RNA-primed DNA synthesis becomes absolutely dependent on the presence of GTP and CTP, while omission of ATP or UTP has very little effect. These results are different from those obtained in the presence of 41 protein, where omission of GTP allows nearly full DNA synthesis because of the extensive use of A-start primers (12, 13).
DISCUSSION
The main features of the present work can be summarized as follows. 1) The two template DNA sequences GTT and
T4 Priming Proteins 7009
GCT are necessary and sufficient to specify RNA primer synthesis by the T4 RNA priming proteins. The RNA primers begin by the template-directed synthesis of a pppApC or a pppGpC from the center of these trinucleotide sequences. 2) The utilization of various primer sites by the priming proteins is apparently governed in part by the different secondary structures around different primer sites in the template mol- ecules, and in the presence of 32 protein most primer site sequences are utilized to nearly the same extent. 3) The 61 protein is the RNA primase in the T4 replication system; however, by itself it only utilizes those primer start sites on the template that contain the GCT sequence. The 41 protein is a DNA helicase that greatly increases the efficiency with which these GCT sites are used, and it is absolutely required for 61 protein recognition of the GTT primer start sites. This recognition is likely to be mediated through specific protein- protein interactions that create a helicase-primase complex.
Compared with eukaryotic systems, in which primers with variable lengths are synthesized with either pppA or pppG at their 5’-ends (26-28), the synthesis of RNA primers in the T4 replication system is under tight control. In addition to the sequence-specific template recognition, the major RNA products synthesized by the priming proteins both in vivo and in vitro are pentaribonucleotides. Thus, a measuring mecha- nism must exist in the interaction between the priming pro- teins and the template. The mechanism determining the length of the primer cannot be dictated by the template sequence alone, since the 3‘-side of the primer exhibits no nucleotide sequence specificity. In addition, we find that a tetranucleotide primer (but not a trinucleotide primer) can be efficiently utilized by the polymerase to initiate DNA synthe- sis. We suggest that the stringency of primer length determi- nation is imposed by a unique structure formed when the priming proteins locate a primer start site on the template.
In this study, we have observed a T residue that is conserved at the position one nucleotide upstream from the site on the template where primer synthesis is initiated. A similar obser- vation was reported earlier for the bacteriophage T7 replica- tion system (23); in their studies of the template recognition sequence for RNA primer synthesis by the T7 gene 4 protein using methods similar to ours, Tabor and Richardson reported a conserved C residue at the -1 position relative to the primer start. It is worth noting that the sequence specificity found for the primer start sites can also explain our previous finding that the synthetic random co-polymer poly(d1,dT) works as a good template for RNA primer synthesis by the 41 and 61 proteins (13, 36).
The requirement for a T nucleotide at the -1 position relative to a primer start can account for the observed spacing of potential primer sites found earlier (12,34). However, there is a large difference between the frequency of appearance of the trinucleotide recognition sequence on the template and the frequency of the DNA chain initiation events that is reflected in the lengths of Okazaki fragments. The C residues are modified in normal T4 DNA; this prevents the recognition of the GCT sequence by the priming proteins (13), leaving only the GTT primer start site that occurs about once every 55 nucleotides. But the average Okazaki fragment synthesized on a T4 DNA template is about 1500 nucleotides long (38). Several models have been proposed to account for the length of the Okazaki fragment synthesized at a replication fork (3, 39). An initial “trombone model” (3, 39) suggested that the size of an Okazaki fragment might be determined by the length of the previously synthesized Okazaki fragment. How- ever, the length of each Okazaki fragment could alternatively be determined by the distance traveled by the 41 protein

7010 DNA Recognition by T4 Priming Proteins
during the time required for the lagging strand DNA polym- erase molecule to complete the previous Okazaki fragment (39). Both models require a high frequency of primer sites on the template, SO that nearby start sites will be available whenever a new primer needs to be made.
In synthesis on an M13 DNA template, 90% of the predicted primer sites were used by the priming proteins in this study, though their degree of utilization differed. These variations were not affected by the changes in the composition of the ribonucleoside triphosphates present in the reactions, but were greatly influenced by the presence of 32 protein. Helix destabilization due to the cooperative binding of 32 protein to single-stranded DNA has been shown to have a profound effect on many cellular processes. In the present case, it appears to help RNA priming events by removing helical secondary structures from the DNA template, making all sites nearly equally available for primer synthesis.
The failure of earlier attempts to observe primer synthesis by 61 protein alone was due to the small amount of the 61 protein available for these tests (35, 36). However, the solo action of 61 protein only makes primers from sites containing the GCT sequence. In order to utilize primer sites containing both GTT and GCT sequences, the 61 protein seems to require a conformational change induced by its association with 41 protein. These results strongly suggest that a 41/61 helicase- primase complex forms during the priming event. Along these lines, it is very interesting to recall the E. coli DNA replication system, in which the 61 protein is replaced by dnaG protein, while the 41 protein is apparently replaced by the dnaB protein (1, 41). In contrast, in the T7 bacteriophage system a single polypeptide chain possesses both helicase and primase activities (9, 23).
Inside the T4-infected cell, the GCT sites on the DNA template would normally be prevented from serving as primer start sites by hydroxymethylation of the C residue. Thus, even at high concentrations, 61 protein alone should be incapable of RNA primer synthesis. Nature has, therefore, designed a system that limits the synthesis of RNA primers to those sites on the DNA that contain a lagging strand DNA helicase. Such helicases seem to be put onto DNA only at a DNA replication origin inside the cell, and they are highly processive proteins that are likely to remain with the same fork until DNA synthesis is completed. Because these proteins thereby serve as markers for a lagging strand of a replication fork, coupling the RNA primase to them guarantees that RNA primers will be made only on those regions of single-stranded DNA inside a cell that are appropriate places for starting an Okazaki fragment.
REFERENCES
1. Kornberg, A. (1980) DNA Replication, Freeman Publications, San
2. Nossal, N. G. (1983) Annu. Reu. Biuchem. 52,581-615 3. Alberts, B. M., Barry, J., Bedinger, P., Formosa, T., Jongeneel,
C. V., and Kreuzer, K. N. (1983) Cold Spring Harbor Symp. Quant. Biol. 47,655-668
4. Sinha, N. K., Morris, C. F., and Alberts, B. M. (1980) J. Biol. Chem. 255,4290-4303
5. Hibner, U., and Alberts, B. M. (1980) Nature 285,300-305 6. Alberts, B. M. (1984) Cold Spring Harbor Symp. Quant. Biol. 4 9 ,
Francisco, CA
1-12
7. Ogawa, T., Baker, T. A., van der Ende, A., and Kornberg, A.
8. van der Ende, A., Baker, T. A., Ogawa, T., and Kornberg, A.
9. Richardson, C. C. (1983) Cell 33,315-317
(1985) Proc. Natl. Acad. Sei. U. S. A. 82,3562-3566
(1985) Proc. Natl. Acad. Sei. U. S. A. 8 2 , 3954-3958
10. LeBowitz, J. H., Zylicz, M., Georgopoulos, C., and McMacken, R. (1985) Proc. Natl. Acad. Sei. U. S. A. 82,398&3992
11. Dodson, M., Roberts, J., McMacken, R., and Echols, H. (1985) Proc. Natl. Acad. Sei. U. S. A. 82, 4678-4682
12. Liu, C. C., and Alberts, B. M. (1980) Proc. Natl. Acad. Sei. U. S.
13. Liu, C-C., and Alberts, B. M. (1981) J. Bwl. Chem. 2 5 6 , 2821-
14. Nossal, N. G. (1980) J. Biol. Chem. 255, 2176-2182 15. Kurosawa, Y., and Okazaki, T. (1979) J. Mol. Biol. 135,841-861 16. Yamamoto, K. R., Alberts, B. M., Benzinger, R., Lawhorne, L.,
17. Morris, C. F., Hama-Inaba, H., Mace, D., Sinha, N. K., and
18. Bittner, M., Burke, R. L., and Alberts, B. M. (1979) J. Biol. Chem.
19. Hinton, D. M., Silver, L. L., and Nossal, N. G. (1985) J. Biol.
20. Liu, C. C., Burke, R. L., Hibner, U., Barry, J., and Alberts, B. M.
21. Maxam, A. M., and Gilbert, W. (1980) Methods Enzymol. 6 5 ,
22. van Wezenbeek, P. M. G. F., Hulsebos, T. J. M., and Schoen-
23. Tabor, S., and Richardson, C. C. (1981) Proc. NatL Acad. Sci. U.
24. Wuang, C-C., and Hearst, J. E. (1980) Anal. Biochem. 103 , 127- 139
25. Huang, C-C., Hearst, J. E., and Alberts, B. M. (1981) J. Biol. Chem. 256,4087-4094
26. Reichard, P., and Eliasson, R. (1978) Cotd Spring Harbor Symp. Quant. Bwl. 43,271-277
27. Tseng, B. Y., Erickson, J. M., and Goulian, M. (1979) J. Mol. Biol. 129,531-546
28. Yamaguchi, M., Hendrickson, E. A., and DePamphilis, M. L. (1985) J. Bwl. Chem. 260 , 6254-6263
29. Newport, J. W., Kowalczykowski, S. C., Lonberg, N., Paul, L. S., and von Hippel, P. H. (1980) ICN-UCLA Symp. Mol. Cell. Biol.
A. 77,5698-5702
2829
and Treiber, G. (1970) Virology 40, 734-744
Alberts, B. M. (1979) J. Biol. Chem. 254,67874196
254,9565-9572
Chem. 260,12851-12857
(1979) Cold Spring Harbor Symp. Quant. Biol. 43 , 469-487
499-560
makers, J. G. G. (1980) Gene 11,129-148
S. A. 78 , 205-209
19,485-505 30. Alberts, B. M., and Frey, L. (1970) Nautre 227,1313-1318 31. Burke, R. L., Alberts, B. M., and Hosoda, J. (1980) J. Bwl. Chem.
32. Liu, C-C., and Alberts, B. M. (1981) J. Biol. Chem. 2 5 6 , 2813- 2820
33. Venkatesan, M., Silver, L. L., and Nossal, N. G. (1982) J. Bwl. Chem. 257,12426-12434
34. Alberts, B. M., Barry, J., Bedinger, P., Burke, R. L., Hibner, U., Liu, C. C., and Sheridan, R. (1980) ICN-UCLA Symp. Mol. Cell. Bwl. 19,449-473
35. Silver, L. L., andNossal, N. G. (1982) J. Biol. Chem. 257, 11696- 11705
36. Burke, R. L., Mum, M., Barry, J., and Alberts, B. M. (1985) J.
37. Hinton, D. M., and Nossal, N. G. (1985) J. Bwl. Chem. 2 6 0 ,
38. Okazaki, R., Okazaki, T., Sakabe, K., Sugimoto, 0. K., and Sugino, A. (1968) Proc. Natl. Acad. Sci. U. S. A. 59,598-605
39. Nossal, N. G., and Alberts, B. M. (1983) in Bacteriophage T4 (Mathews, C. K., Kutter, E. M., Mosig, G., and Berget, P. B., eds) pp. 71-81, American Society for Microbiology, Washing- ton, D. C.
40. Kornberg, A. (1982) Supplement to DNA Replication, Freeman Publications, San Francisco, CA
41. LeBowitz, J. H., and McMacken, R. (1986). J. Biol. Chem. 2 6 1 , 4738-4748
255,11484-11493
BWl. Chem. 260,1711-1722
12858-12865
![Characterization of DNA Helicase II from a uvrD252 Mutant of · Purification of DNA helicase HI. To overproduce DNA helicase II, 6 liters of SK8118 (SK707 [uvrD+] containing pBWK58[uvrD+]](https://img.pdfslide.us/doc/110x75/5ff8a53c2b681343f2207317/characterization-of-dna-helicase-ii-from-a-uvrd252-mutant-of-purification-of-dna.jpg)




























