Embed Size (px)
Citation preview

JouRAiL OF BACTERIOLOGY, Mar. 1993, p. 1572-15790021-9193/93/061572-08$02.00/0Copyright © 1993, American Society for Microbiology
Vol. 175, No. 6
Structure, Function, and Evolution of the Family ofSuperoxide Dismutase Proteins from
Halophilic ArchaebacteriaPHALGUN JOSHI AND PATRICK P. DENNIS*
Canadian Institute for Advanced Research, Program in Evolutionary Biology, and Department ofBiochemistry, University of British Columbia, 2146 Health Sciences Mall
Vancouver, British Columbia V6T 1Z3, Canada
Received 14 September 1992/Accepted 12 December 1992
The protein sequences of seven members of the superoxide dismutase (SOD) family from halophilicarchaebacteria have been aligned and compared with each other and with the homologous Mn and Fe SODsequences from eubacteria and the methanogenic archaebacterium Methanobacterium thermoautotrophicum.Of 199 common residues in the SOD proteins from halophilic archaebacteria, 125 are conserved in all sevensequences, and 64 of these are encoded by single unique triplets. The 74 remaining positions exhibit a highdegree of variability, and for almost half of these, the encoding triplets are connected by at least twononsynonymous nucleotide substitutions. The majority of nucleotide substitutions within the seven genes arenonsynonymous and result in amino acid replacement in the respective protein; silent third-codon-position(synonymous) substitutions are unexpectedly rare. Halophilic SODs contain 30 specific residues that are notfound at the corresponding positions of the methanogenic or eubacterial SOD proteins. Seven of these arereplacements of highly conserved amino acids in eubacterial SODs that are believed to play an important rolein the three-dimensional structure of the protein. Residues implicated in formation of the active site, catalysis,and metal ion binding are conserved in all Mn and Fe SODs. Molecular phylogenies based on parsimony andneighbor-joining methods coherently group the halophile sequences but surprisingly fail to distinguish betweenthe Mn SOD of Escherichia coli and the Fe SOD of M. thermoautotrophicum as the outgroup. Thesecomparisons indicate that as a group, the SODs of halophilic archaebacteria have many unique andcharacteristic features. At the same time, the patterns of nucleotide substitution and amino acid replacementindicate that these genes and the proteins that they encode continue to be subject to strong and changingselection. This selection may be related to the presence of oxygen radicals and the inter- and intracellularcomposition and concentration of metal cations.
Evolution has produced two unrelated proteins with su-peroxide dismutase (SOD) activity. One is a CuZn enzymeand is confined almost exclusively to the cytoplasm ofeucaryotic organisms. The second contains Fe or Mn asmetal ion cofactor and is found in eubacteria, in the eubac-teria-derived eucaryotic mitochondria, and in archaebacteria(10, 13, 24, 25). These enzymes catalyze the dismutation ofthe reactive superoxide anion (02) to molecular oxygen andperoxide and thus protect organisms from chemical damageand inactivation (4, 5).
Halophilic archaebacteria are a collection of related or-ganisms that evolved from an anaerobic methanogen ances-tor and that now grow in nature either aerobically ormicroaerobically in high-salt environments (7, 27). SODactivity has been detected in at least two representatives ofthe ancestral methanogen group and in numerous halophilicspecies (7, 10, 25). The SOD enzyme from both Halobacte-rium cutirubrum and its immediate relative, H. halobium,has been purified and extensively characterized; the enzymecontains Mn as a metal ion cofactor and is related to the Mn-or Fe-type enzymes of eubacteria (13, 19).The sod gene, encoding the authentic SOD protein, was
cloned from the genome ofH. cutirubrum, and its expressionwas shown to be elevated in the presence of paraquat, acompound that generates superoxide radicals (14, 15). In
* Corresponding author.
addition, a second related gene, slg, was also cloned andcharacterized. Although the slg gene is actively transcribed,its mRNA is not elevated by paraquat, and as yet noenzymatic activity has been associated with the protein thatit encodes (12). The pattern of nucleotide sequence diver-gence between sod and slg is highly unusual; silent third-codon-position substitutions are relatively infrequent, andmost substitutions are consequently nonsynonymous, result-ing in amino acid replacement in the respective proteins.This pattern suggests that the SLG protein and possibly alsothe SOD protein are under intense and perhaps fluctuatingselection for new and divergent function. This selection isalmost certainly influenced by the concentration of thesuperoxide radical and possibly also by the composition andconcentration of intra- and extracellular cations.To extend this analysis, additional genes of the sod family
from other halophilic archaebacteria have been cloned andcharacterized. In this study, we compared the deducedhalophilic protein sequences with those of other Mn and FeSODs and identified amino acid residues that characterizeand distinguish these halophilic SODs.
MATERIALS AND METHODS
The relative rate test (20) was used to estimate the numberof substitutions that have occurred in the sod and the slggenes of H. cutirubrum since their divergence from a com-mon ancestral sequence. This analysis is accomplished by
1572
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

EVOLUTION OF HALOPHILIC SOD PROTEINS 1573
comparison with an outgroup sequence. In this instance, thesod, sequence of Haloferax volcandi and sod sequence ofHaloarcula marismortui were used in two separate tests.The test involves solution of simultaneous equations:
AO = (AB + AC - BC)/2BO = (AB + BC - AC)/2CO = (CA + CB - AB)/2
where A, B, and C represent the two related and theoutgroup gene sequences and AB, AC, and BC are thenumber of nucleotide differences in the pairwise compari-sons of the three sequences. The calculated values of AOand BO estimate the number of substitutions that haveoccurred in sequence A and sequence B since their diver-gence from the common ancestral sequence 0. The value ofCO represents the number of substitutions separating se-quence 0 from sequence C. These relationships are depictedgraphically in Fig. 2.Parsimony analysis was carried out by using the PAUP
analysis package devised by David Swafford (Illinois NaturalHistory Survey, Champaign, Ill.). Neighbor-joining analysiswas carried out by using the application from the Clustal Vanalysis package by Des Higgins (European Molecular Biol-ogy Laboratory, Heidelberg, Germany). The actual opera-tion of the software was performed by Dan Fieldhouse atYork University on a Sun Sparc workstation. For bootstrapresampling (2), 100 and 1,000 repetitions, respectively, werecarried out in the PAUP and neighbor-joining analysis.
RESULTS AND DISCUSSION
The deduced amino acid sequences of the SOD family ofproteins from halophilic archaebacteria are aligned in Fig. 1.To facilitate comparison with SOD sequences from othernonhalophilic organisms, the numbering system of Parkerand Blake (17) has been adopted. The three extra residues inthe Haloarcula marismortui sequence near the amino termi-nus are accommodated between positions 2 and 3.The most striking features of the halophilic SOD se-
quences are their high degree of sequence similarity andtheir high proportion of acidic amino acids. In pairwisecomparison, the amino acid sequences are between 76 and100% identical, and 125 of the 199 common residues (62%)are conserved in all seven sequences (see the halophilicconsensus sequence in Fig. 1). In contrast, any one of theseven halophilic proteins exhibits only about 35 to 40%sequence identity with eubacterial and eucaryotic mitochon-drial SODs and about 40 to 45% identity to the SOD ofMethanobacterium thermoautotrophicum. At the DNAlevel, 64 of the 125 codon positions, specifying amino acidsconserved in the halophilic SOD proteins, use single uniquetriplets, whereas the remaining 61 are degenerate (7). Espe-cially interesting is the use of both the UCN and AGY Sercodons at three positions (-1, 19, and 168) where Ser isconserved in all seven halophilic proteins. Also striking arethe 74 positions of amino acid variability. At nearly half ofthese positions, connection between the encoding tripletsrequires two or more nonsynonymous nucleotide substitu-tions. For example, at position 125, aspartic acid (GAC),serine (AGC and TCG), and alanine (GCG) all appear.
Signature amino acid residues in SOD. Comparison of theX-ray diffraction structures of the Bacillus stearotherno-philus Mn SOD and a number of eubacterial and mitochon-
drial Mn and Fe SOD sequences resulted in the identificationof 39 positions of high sequence conservation that appearedto be important in the structure or activity of the protein (17).These residues are listed in Table 1. Four of these positions,His-26, His-81, Asp-175, and His-179, are essential forbinding the metal ion cofactor (22). In addition, residuesHis-30, His-31, Tyr-34, Trp-85, Trp-133, and Tyr-181 havebeen implicated in the formation of a hydrophobic pocketaround the active site and in enzyme catalysis (23). Theremaining residues appear to have structural rather thanfunctional roles. Of these 39 positions, 35 are conserved inthe Fe SOD from M. thermoautotrophicum (25). In thehalophilic SODs, only 26 of these positions are absolutelyconserved; 3 at residues 5, 39, and 131, are partially con-served; 1 at position 130 is deleted, and the remaining 9 aresubstituted with the same amino acid in all seven sequences.None of the 11 positions of partial or complete replacementaffect residues implicated in formation of the active site or inenzyme catalysis.The SODs from halophilic archaebacteria contain 30
unique and conserved amino acid residues that are not foundin any other SODs at the corresponding alignment positions(Table 1). Seven of these represent total replacement ofeubacterial signature residues. In addition, there are ninepositions where the single methanogenic and seven halo-philic SODs exhibit a single amino acid not found in anyknown eubacterial SOD. These comparisons indicate in aqualitative way that the halophilic SOD proteins are almostas different from the SOD of their immediate methanogenrelative as they are from the SODs of their more distanteubacterial cousins. Furthermore, the substitutions specificto the halophiles almost certainly have a substantial effect onthe overall structure of the protein but leave the hydrophobicactive site and the four metal ion-binding residues intact.
Virtually all proteins from halophilic archaebacteria (incomparison with proteins from nonhalophilic organisms)exhibit a high content of acidic amino acids (11). Theseresidues, usually located on the surface of the protein, arethought to sequester water molecules and create a hydrationshell that allows the protein to function within the high-ionic-strength intracellular environment of the cell. For example,in the eubacterial SOD protein, the acidic and basic residuesare equally prevalent, whereas halophilic SODs containmore acidic residues and proportionately fewer basic resi-dues. The pIs of these seven halophilic SODs range from4.16 to 4.20 (Fig. 1). One might predict that of the 30halophile-specific signature residues listed in Table 1, asubstantial proportion would involve replacement of Lys orArg by either Asp or Glu. That is, many of these substitu-tions should reflect the general adaptation of the enzyme tofunction in high salt. Surprisingly, this appears not to be thecase. The 30 halophile-specific substitutions increase thenumber of acidic residues by only three (positions 20, 152,and 201), and an additional acidic residue (position 217) isgenerated within the halophile-specific extension at thecarboxy terminus of the protein. Only position 20 involves alysine/arginine (Escherichia coliiM. thermoautotrophicum)replacement by glutamic acid in the halophilic proteins.
In about 70% of the alignment positions where acidicresidues are found in one or more of the halophilic SODs, anacidic residue is also found in at least one other nonhalo-philic SOD. Of the remaining positions, about half exhibit abasic residue in at least one nonhalophilic sequence. Thus,the high acidity of the halophilic SODs has been achieved byinsertion of Asp and Glu residues most often at positionswhere charged residues can be tolerated in nonhalophilic
VOL. 175, 1993
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

1574 JOSHI AND DENNIS
Hcu Rn SODGRB SODHcu SLGGRB SLGHuo SODIHlo S002HMa SOD
o o o o n s Eo o o o - - -
0 0 0 0 - - Q0 0 0 0 - - Qo o o o -- 00 0 0 0. - - 0
0 0 0 0 - - -
HALOPH IL IC CONSENSUSfth Fe SOD N 0 LEco Rn SOD
I0Y o o o E L P P L P Y D0 0 0 0 - - - - -
H o o o - - - S - - - -H o o o - - - S - - - -- o o o - - 0 - - - - E- o o o - - 0 - - - - EH S N P - - - - - - - -
n s o o o E L . L P YE KE F Y a o o E L P E L P Y P
n SY o o o o T L P S L P Y R
Y D A L E P H- - - - -
- - - -
- - -
- - - - - - -
Y D R L E P HY D A L E P HY D R L E P H
20 30S E Q U L T U H H D r H H Q G Y U O GO-_ - - E--_TEH H QT U- - -
- - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - -
I S E Q M H H D r H H Q . Y U GS R E Q L r H H Q K H H Q R Y U D G
F D K Q r n E H H r K H H Q r Y u N 11
40N A E E r L R E N
- - - - - - - - -
- - - - - - - - - -
L E S - - - - - - - -
.E L R .NR NR L L R K L D E RR N R R L E S L P E F
50 60R E T o o o o o o GoG H A S T R 6R- --00 0 00o o 00 - - - - - - - - -
---000 0o o o o 00 - - - - - - - - -
0000000-- - - - -
- - o o o o o oo E F 6- S - - -
- - R o o o o oo E F 6- S - - --O R o o o o o oo --F G- S - R-
R . o o o o o oo 6. S A. RR E S o o o o ooEo AD U D 1 RRR N r P U E E L Ir K L D Q L P R D K
70L G D_
-
U R NU R N
L i E,1 6 u
80U T HNG SGH L H T LF U Q S
- - - - --
- --
- - --
- -- -
---- c - - Y - - - n - - - E H- - - - - - - - - - - D- - - - - - - - - - - D - - - - N
C- Q D - - - - - - E N
U T H N .6 L H .F U ..L S F H U GG Y U L H L FF U G NL R N N AG GHAR N H S LF U KG
90n s P o o R G G DE
- - - o o D - - 6 ---- o o D -
--- o o E - - - ---- o a E - - - -- o o N G -
* 0
H P o o . G G EG P R D EC G GG E
L K E o o oG T TL
140 150oY D o S H S N r L R N U A
o - - - - - - - - - -
_ P U R K Q - - - - -- o P U R K: Q - - - - -- - o - F - - Q - - - - U- - o - F - - Q - - - - U- - o P C R K Q - - - - P
Y D o . . . L R N U .Y C o Q Rr D R L F I n QL K o o a G D K L A U U S
100 110P o S 6 R L R D R R A D-0 .----U--
o-
----_ _ _ _ _ _
o - - - - - - - -
- o E - E - - - E -
-o E -L --E- - - E-- o E - E - L - - - E E -
P o 1. L R 0P o S G KL R EY E K DQ o o G DL K RA E R D
160U D N HN E G R L U
_ K _ _ Q _ _ _ _- K - _ Q _ _ _ __ K _ _ Q _ _ _ _
U D . H D0 L UU E K H N U N U PT R N Q D S P L n 6
120F 6 S Y E NHU R R E
- - - - - - - -: - -
- - - - - - - - - -
- - - - - - - - -
F G S Y E .U. .EF G S F E R F R K EF 6 S U D N F K R E
170o o o o oG S H P I0 0 0 0 0 0
0 0 0 0 0 0
o o o o o o - - - -
o o o o o o H F R I LE A S G R SF F P
130F E A R R S R R o o S G U R L L U
0 0
--U -- -- o o - - - - - - -
--U -- -- o o - - - - - - -
6--G-- o aA - - - - - -
6-G--o o AR6oo - - - - -
F E. RA oo GU A L L UF S Q R R S R E 6 S G U R U L TF E K R R R S R F G S 6 U R U L U
1I0L R L D U U E N S Y YY D Y 6 P
- - - -
- - - - - -
_- - - - - -
L R L O U U E H S Y Y D Y GL U L D U U E H R Y Y O Y R NL G L D U U E H R Y Y L K F Q N
200U D R F F E U U D U D
- - - - - - - -
S - - - - - -
-S- - - - - - -
.R F F E U D U OU E R F U N U N U KI K E F U N U U N U D
210E PT E R F E
P IAR R 11 Y DP IAR R N Y D- - R R - Y -- - R A - Y -
K R R - E Y -
E U E i R, F EE R R R R F A
Q A A E R F E_ - - - - - -
D U U S L - -
D U U S L - -- - U - L - -- - U -U L- -K S U S H - -
* 0
F ED L o o o oR K K o o o o
PROTEINHcu SODGRB SOOHcu SLGGRB SLGHvo SODIHuo SOD2Hoa SOD
n.U.22386224142220922122224852235222771
HRLO CONSENSUS1th SOD 24066Eco SOD 23042
FIG. 1. Amino acid sequence alignments of SOD proteins. The seven halophilic SOD sequences are aligned according to the numberingsystem of Parker and Blake (17). Abbreviations: Hcu, H. cutirubrum; GRB, Halobacterium sp. strain GRB; Hvo, Haloferax volcanii; Hma,Haloarcula marismortui; Mth, M. thermoautotrophicum; Eco, E. coli. The three extra residues near the amino terminus of the Haloarculamarismortui SOD are accommodated between positions 2 and 3. The H. cutirubrum sequence is given in its entirety; within the halophilicgroup, amino acids identical to the H. cutirubrum residues are indicated by dashes. The halophilic consensus sequence illustrates onlyresidues conserved in all seven sequences; nonconserved residues are indicated by small dots. The halospecific signature residues areindicated by large dots above the halophile consensus sequence. Deletions required to maintain alignments are indicated by open circles. Atthe end of the alignment, the molecular weights (m.w.), the number of acidic and basic amino acid residues, and the pIs of the respectiveproteins, as determined from their primary sequences, are indicated. The M. thermoautotrophicum and E. coli sequences were obtained fromTakao et al. (25) and Takeda and Avila (26).
SODs without adversely affecting enzyme activity. These tween two gene sequences initially degenerates more rapidlysites are presumed to be on the surface of the protein than the amino acid identity between the two correspondingmolecule and well removed from the site of catalysis. proteins. Only later, when a substantial number of first- and
Nucleotide sequence divergence. When orthologous or par- second-position changes accumulate, does the amino acidalogous gene sequences initially begin to diverge during identity fall below the nucleotide identity. Typical of thisevolution, mutational differences usually accumulate most situation are the paralogous p-vac and c-vac (plasmid-encod-rapidly in the third codon position and most substitutions are ed and chromosome-encoded gas vacuole) genes of H.synonymous. As a consequence, nucleotide identity be- halobium and the orthologous c-vac gene of Haloferax
J. BACrERIOL.
190D R G S F
A - - D -A - - D -
R - - D -
. R G FU R P D YR R P D Y
ACIDIC BASIC38 73S 736 635 639 a38 a41 9
p14. 174.164. 174. 174. 74.204. 16
35 23 S. 2825 23 6. 78
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
EVOLUTION OF HALOPHILIC SOD PROTEINS 1575
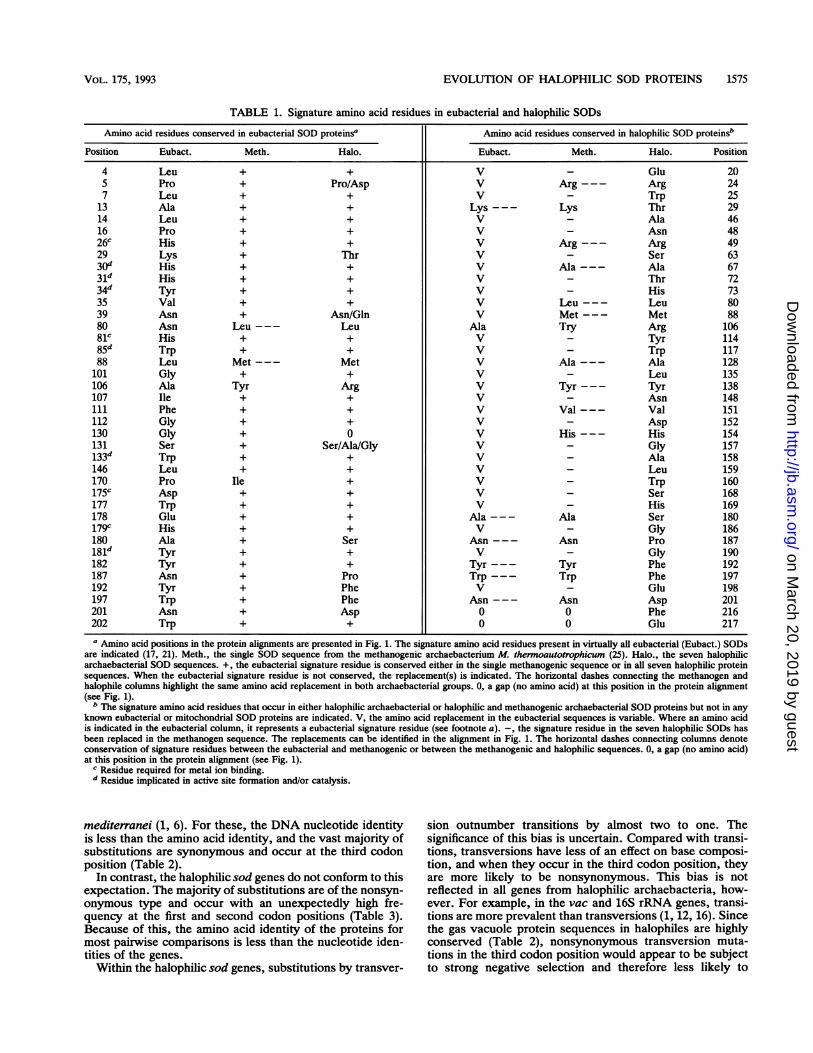
TABLE 1. Signature amino acid residues in eubacterial and halophilic SODs
Amino acid residues conserved in eubacterial SOD proteinsa Amino acid residues conserved in halophilic SOD proteinsb
Position Eubact. Meth. Halo. Eubact. Meth. Halo. Position
4 Leu + + V - Glu 205 Pro + Pro/Asp V Arg--- Arg 247 Leu + + V - Trp 25
13 Ala + + Lys--- Lys Thr 2914 Leu + + V - Ala 4616 Pro + + V - Asn 4826C His + + V Arg--- Arg 4929 Lys + Thr V - Ser 6330d His + + V Ala--- Ala 6731d His + + V - Thr 7234d Tyr + + V _ His 7335 Val + + V Leu--- Leu 8039 Asn + Asn/Gln V Met--- Met 8880 Asn Leu--- Leu Ala Try Arg 10681C His + + V - Tyr 11485d Trp + + V - Trp 11788 Leu Met --- Met V Ala --- Ala 128
101 Gly + + V - Leu 135106 Ala Tyr Arg V Tyr --- Tyr 138107 Ile + + V - Asn 148111 Phe + + V Val --- Val 151112 Gly + + V - Asp 152130 Gly + 0 V His--- His 154131 Ser + Ser/Ala/Gly V - Gly 157133d Trp + + V - Ala 158146 Leu + + V - Leu 159170 Pro Ile + V - Trp 160175c Asp + + V - Ser 168177 Trp + + V - His 169178 Glu + + Ala --- Ala Ser 180179c His + + V - Gly 186180 Ala + Ser Asn --- Asn Pro 187181d Tyr + + V - Gly 190182 Tyr + + Tyr--- Tyr Phe 192187 Asn + Pro Trp --- Trp Phe 197192 Tyr + Phe V - Glu 198197 Trp + Phe Asn --- Asn Asp 201201 Asn + Asp 0 0 Phe 216202 Trp + + 0 0 Glu 217a Amino acid positions in the protein alignments are presented in Fig. 1. The signature amino acid residues present in virtually all eubacterial (Eubact.) SODs
are indicated (17, 21). Meth., the single SOD sequence from the methanogenic archaebacterium M. thermoautotrophicum (25). Halo., the seven halophilicarchaebacterial SOD sequences. +, the eubacterial signature residue is conserved either in the single methanogenic sequence or in all seven halophilic proteinsequences. When the eubacterial signature residue is not conserved, the replacement(s) is indicated. The horizontal dashes connecting the methanogen andhalophile columns highlight the same amino acid replacement in both archaebacterial groups. 0, a gap (no amino acid) at this position in the protein alignment(see Fig. 1).
b The signature amino acid residues that occur in either halophilic archaebacterial or halophilic and methanogenic archaebacterial SOD proteins but not in anyknown eubacterial or mitochondrial SOD proteins are indicated. V, the amino acid replacement in the eubacterial sequences is variable. Where an amino acidis indicated in the eubacterial column, it represents a eubacterial signature residue (see footnote a). -, the signature residue in the seven halophilic SODs hasbeen replaced in the methanogen sequence. The replacements can be identified in the alignment in Fig. 1. The horizontal dashes connecting columns denoteconservation of signature residues between the eubacterial and methanogenic or between the methanogenic and halophilic sequences. 0, a gap (no amino acid)at this position in the protein alignment (see Fig. 1).
C Residue required for metal ion binding."Residue implicated in active site formation and/or catalysis.
mediterranei (1, 6). For these, the DNA nucleotide identityis less than the amino acid identity, and the vast majority ofsubstitutions are synonymous and occur at the third codonposition (Table 2).
In contrast, the halophilic sod genes do not conform to thisexpectation. The majority of substitutions are of the nonsyn-onymous type and occur with an unexpectedly high fre-quency at the first and second codon positions (Table 3).Because of this, the amino acid identity of the proteins formost pairwise comparisons is less than the nucleotide iden-tities of the genes.
Within the halophilic sod genes, substitutions by transver-
sion outnumber transitions by almost two to one. Thesignificance of this bias is uncertain. Compared with transi-tions, transversions have less of an effect on base composi-tion, and when they occur in the third codon position, theyare more likely to be nonsynonymous. This bias is notreflected in all genes from halophilic archaebacteria, how-ever. For example, in the vac and 16S rRNA genes, transi-tions are more prevalent than transversions (1, 12, 16). Sincethe gas vacuole protein sequences in halophiles are highlyconserved (Table 2), nonsynonymous transversion muta-tions in the third codon position would appear to be subjectto strong negative selection and therefore less likely to
VOL. 175, 1993
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

1576 JOSHI AND DENNIS
TABLE 2. Comparison of paralogous and orthologous gasvacuole genesa
Comparison
Species and gene Halobacterium Haloferaxhalobium mediterraneip-vac c-vac
Halobactenum halobium c-vac 84.7/97.42/0/3321/14
Haloferax mediterranei c-vac 86.4/96.1 85.5/98.73/1/27 1/1/3320/11 20/13
a Each entry in the matrix consists of three rows. The first row indicates theDNA (nucleotide) sequence identity/protein (amino acid) sequence identity,each expressed as a percent. The second row indicates the distribution ofnucleotide substitutions in the comparison between first/second/third codonpositions. The third row indicates the number of transition substitution/transversion substitutions.
become fixed in the population. Many of the 16S rRNAsubstitutions occur at compensatory positions within regionsof RNA secondary structure. Transition mutations allowthese compensatory changes to proceed through stableG. U base pair intermediates and therefore could accountfor the transitional bias.When this high proportion of nonsynonymous substitu-
tions was first observed between the paralogous sod and sIggenes of H. cutirubrum, it was suggested that the causemight be intense selection at the molecular level for newprotein function. Since the sod gene encodes the authenticSOD activity, it was imagined that the superfulous sig genewas being used to generate a new and different enzymaticactivity. That is, the sod gene was being conserved, whereasthe sIg gene was being subjected to frequent nucleotidesubstitution.
(i)
C
0
AKB
(ii)
Hvosodl
(52.)/ 4(24.)Hcu
Hcu sod819
J. BA=RIOL.
(il'' Hmasod
(e0
Hcu HcuBig sod
FIG. 2. Relative rates of nucleotide substitutions in the sod andsig genes of H. cutirubrum. The relative rate test is described inMaterials and Methods. (i) The generalized situation in which A andB are contemporary gene sequences; 0 is the common ancestralsequence of A and B, and C is the outgroup sequence. The lengthsof the three branches connecting at 0 are proportional to the numberof nucleotide substitutions (in parentheses) separating 0 from A, B,and C. These lengths are calculated by using the three equationslisted in Materials and Methods. The distances (in nucleotidesubstitution) AB, BC, and AC can be obtained from Table 3. (ii) Forthe H. cutirubrum (Hcu) sig (A), H. cutirubrum sod (B), andHaloferax volcandi (Hvo) sod, (C) calculations, AB, BC, and AC are77, 114, and 142, respectively. (iii) For the H. cutirubrum sig (A), H.cutirubrum sod (B), and Haloarcula marismortui (Hma) sod (C)calculations, AB, BC, and AC are 77, 134, and 137, respectively.
The presence of additional sequences from the halophilicsod gene family sheds some light on this situation. Clearly,not just the sod and sig genes of H. cutirubrum and Halo-bacterium sp. strain GRB but all of the genes in the collec-tion when compared pairwise appear to exhibit this unusualpattern of divergence. When the relative rate test (20) wasapplied to the paralogous sod and sig genes ofH. cutirubrum(or Halobacterium sp. strain GRB), using either the Halof-erax volcanii sod, gene or the Haloarcula marismortui sodgene as the outgroup, it was quite clear that both sod and sighave accumulated substitutions at a substantial rate (Fig. 2).Relative to the sod gene of Haloarcula marismortui, the sodand sig genes of H. cutirubrum have accumulated mutations
TABLE 3. Comparison of halophilic sod genes and proteinsa
Comparison
Species and gene Halobacterium cutirubrum Halobacterium sp. strain GRB Haloferax volcanusod sig sod sig sod, sod2
Halobacterium cutirubrum sig 87.2/82.525/21/3131/46
Halobacterium sp. strain GRBsod 95.5/95.5 87.3/82.0
0/1/2 25/22/292/1 31/45
slg 86.7/81.0 99.2/98.5 86.7/80.525/24/31 0/3/2 85/25/3033/47 3/2 33/47
Haloferax volcaniisod, 81.0/80.0 76.3/72.0 80.7/79.5 76.0/70.5
25/28/61 41/30/71 25/29/62 41/33/7034/80 42/100 35/81 45/99
sod2 80.9/80.4 77.1/72.4 80.6/79.9 76.7/70.9 99.5/10026/29/59 39/29/69 26/30/60 39/32/68 1/1/134/80 42/95 35/81 45/94 0/3
Haloferax marismortui sod 77.7/74.5 77.2/76.0 78.0/75.0 76.3/74.5 76.3/78.0 76.2/78.436/35/63 37/30/70 36/34/62 37/33/72 29/26/87 30/27/8562/72 57/80 61/71 60/82 45/97 45/97
a Matrix entries are as described in the footnote to Table 2.
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

EVOLUTION OF HALOPHILIC SOD PROTEINS 1577
A. NUCLEOTIDE PARSIMONY- nth sod
1.00 , Hcu sod0.ss | GRB sod
owed~~~~~~10
---I l Hcu s I
" r ~~~~~~HvosodlLHvo sod2
HMO sod
B. AMINO ACID PARSIMONY1Mth SOD
Hcu SODGRB SODHcu SLGOGRB SLGHuo SOD0
fHuo S002
Hma SOD
C. NUCLEOTIDE NEIGHBOR JOINING
1.00 Hcu 319
1.00 GRB sig
1.00 Hcu sod0.92,
OGRB sod
,."0 rHuo sodirI Huo sod2
I Hma sodI
.- - - - 11th sod1/5 meale
D. AMINO ACID NEIGHBOR JOINING
1.00 Hcu SLG
o.84 GRB SLG
1.00 Hcu SOD
GRB SODr-I 1.00 1Huo S001I Hvo S0021 0.62
l Hoo SOD
---- - th sod1/5 scale
FIG. 3. Phylogenetic relationships among members of the halophilic sod gene family or the proteins that they encode. Phylogenetic treeswere constructed by using the maximum parsimony (A and B) or neighbor-joining (C and D) method. The neighbor-joining method containsa correction for multiple substitutions occurring at a single position (8, 9). The lengths of the branches in the neighbor-joining trees reflectevolutionary distance. The stippled branch leading to the outgroup is drawn at one-fifth scale. Trees A and C are based on DNA sequencealignments, and trees B and D are based on protein sequence alignments. The numbers preceding each branch are bootstrap consistencyvalues and indicate the proportions of replications that group all of the taxonomic units within the branch and exclude all other taxonomicunits represented in the tree. These consistency values were computed from 100 or more bootstrap resamplings. Abbreviations are as in Fig.1.
at essentially the same rate, whereas relative to the sod,gene of Haloferax volcanji, the sIg gene has accumulatedsubstitutions at twice the rate of the sod gene. This findingimplies that both the sod and sIg genes and probably all ofthe genes in this halophilic family have been subjected tointense but variable selective pressures that has resulted infrequent amino acid replacements in each of the respectiveproteins. These replacements are confined to 74 of the 199common amino acid positions (Fig. 1). For the nearlyidentical paralogous sod, and sod2 genes of Haloferax vol-canii, the forces and processes maintaining sequence homo-geneity are not understood.
Phylogenetic analysis. Two methods, maximum parsimony(3) and neighbor joining (18), were used to analyze thephylogenetic relationships among the halophilic sod genesand among the proteins that they encode (Fig. 3). Thenucleotide and protein alignments used for the analysis werecolinear and are depicted in the accompanying paper (7) andin Fig. 1. The use of the sod gene sequence from M.thermoautotrophicum as an outgroup allows for the position-ing of the ancestral halophilic gene (or root) within the tree.These phylogenetic analyses have been evaluated for
significance at each branch point by using the commonlyemployed bootstrap resampling technique (2). The consis-tency values indicated are the proportions in 100 or moreresamplings for exclusive grouping of the taxonomic units(genes or proteins) within that branch of the tree, separatefrom all the other taxonomic units represented on the tree. A
value greater than 0.95 is considered significant, whereasvalues less than 0.95 are not necessarily significant.The nucleotide parsimony, amino acid parsimony, and
nucleotide neighbor-joining trees exhibit identical topolo-gies; the sequences from Halobacterium spp. form a coher-ent group with the two sod-genes (or proteins) on one branchand the two sig genes (or proteins) on another. The singlesod gene (or protein) from Haloarcula marismortui branchesearly and the two nearly identical sod genes (or proteins)from Haloferax volcanii branch later from the lineage lead-ing to the Halobacterium spp. The bootstrap consistencyvalues for most of the branch points are greater than 0.95.Only the amino acid neighbor-joining tree exhibits a slightlyaltered topology. Here, the Haloferax volcanji proteinbranches early from the lineage leading to Haloarcula mans-mortui rather than the lineage leading to Halobactenium spp.The bootstrap consistency for this grouping is 0.62.
In all of these analyses, the M. thermoautotrophicumprotein was clearly and unambiguously identified as theoutgroup. This protein is apparently an Fe-containing SOD(25), whereas the halophilic proteins are most likely allMn-containing enzymes (13). The difference in metal ioncertainly accounts for some of the amino acid differencesbetween the methanogenic and halophilic proteins. Whenthe E. coli Mn-sod gene and protein sequences were in-cluded, the parsimony analysis grouped the halophiles to-gether but was unable to reliably distinguish between E. colior M. thermoautotrophicum as the outgroup (not shown).
VOL. 175, 1993
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

1578 JOSHI AND DENNIS
GENE GRIN OFDUPLICRTION INOUCIBILITY
R
RNCESTRIALSTRTE
i--- Hcu sod
B -GAB sod____.PSD 1
< ~~~Hcu 3 19
___S2 GRO sag
RACESTRIARL Ho sodlSTATE Si- ----- P0
Hvo sod2
Hea sod
\ DELETION OFINDUCIBLE GENE
FIG. 4. Consensus tree illustrating phylogenetic relationshipsamong the halophilic sod genes (or proteins). The consensus treewas obtained from the four constructed trees illustrated in Fig. 3.Speciation events are designated S1, S2, and S3. The commence-ment of paralogous gene sequence divergence is indicated by PSD1and PSD2. (A) Positions of gene duplication and acquisition ofinducibility by paraquat. Double lines indicate the presence ofparalogous genes of identical coding sequence; solid lines representnoninducible genes; stippled lines represent paraquat induciblegenes. (B) The alternative scenario in which the halophilic ancestoralready possesses duplicated sod genes.
This finding reinforces the impression that the halophilicproteins as a group are unique and different from all otherSOD proteins (Table 1).The consensus tree. The consensus derived from these
phylogenetic analyses is illustrated in Fig. 4. The branchpoints represent either speciation events, designated Si, S2,and S3, or divergence of paralogous gene sequences, desig-nated PSD1 and PSD2. The first speciation event, Si,separates the lineage leading to Haloarcula spp. from otherhalophiles. The second event, S2, splits Haloferax spp. fromHalobacterium spp., and the third, more recent event, S3,splits H. cutirubrum from Halobacterium sp. strain GRB.The first paralogous sequence divergence event, PSD1,
represents the commencement of divergence between theparalogous sod and sig genes within the Halobacteriumbranch. The majority of the differences that have accumu-lated between these two sequences predate S3, the specia-tion of H. cutirubrum and Halobacterium sp. strain GRB.The second event, PSD2, is meant to depict the minordifferences that exist between the sod, and sod2 genes ofHaloferax volcani.
Other features characteristic of halophilic sod genes canbe superimposed on this diagram. One is sod gene copynumber, and another is response to paraquat. Both themethanogen ancestor represented by M. thermoautotrophi-cum and the early-branching Haloarcula mansmortui haveonly a single sod gene (7, 25). This finding suggests that thehalophilic ancestor may also have had only a single sod gene
in its genome. If this scenario is correct, the position forduplication would be early in the branch leading to thegenera Halobactenum and Haloferax. In the extant speciesexamined that possess two sod-related genes, one gene isinducible by paraquat and the other gene is not inducible byparaquat (7, 12). This finding implies that shortly afterduplication and prior to the separation ofHalobacterium andHaloferax, one of the duplicated genes acquired the propertyof inducibility. The Haloarcula marismortui branch retainsthe ancestral single-copy noninducible state.There is a second alternative scenario that is equally
likely. The ancestral halophile might have already possessedduplicated sod genes, one of which was inducible and theother of which was noninducible by paraquat. To reach thecurrent state would require simply the loss of the paraquat-inducible gene from the branch leading to Haloarcula ma-is-mortui. This possibility is depicted in Fig. 3B. Other, morecomplex explanations are not considered here.Although partially satisfying, these models for the evolu-
tion of sod-related sequences within halophilic archaebacte-ria fail to explain a number of observations. The first is theabsence of any detectable sequence similarity in the 5'flanking regions of the sod-related genes that were inducedby paraquat (see Fig. 4 in the accompanying paper [7]). Thisis especially enigmatic since the 5' flanking sequences of theuninducible genes that possibly have a deeper evolutionaryorigin (Fig. 3A) nonetheless exhibit easily identifiable se-quence similarity. Second, the sod, and sod2 genes of Hf.volcanji, although virtually identical within their codingregions, exhibit no detectable similarity in their flankingregions. This must mean that coding sequence homogeneityis maintained by concerted evolution and probably involvesrecombination- or gene conversion-type events; these eventsapparently do not involve or include either the 5' or 3'flanking regions.
In general, the flanking regions of homologous genes,excluding conserved regulatory elements, accumulate nucle-otide substitutions more rapidly than do coding regions. Thisis in general true for the halophilic sod gene family. Com-parison between genera indicate that only the noninduciblesod genes exhibit similarity and that this similarity is con-fined to a 50- to 55-nucleotide long region that contains thetranscriptional promoter (7). Even here, however, sequenceidentity is substantially less than in the coding regions. Thisexpected pattern is reversed within H. cutirubrum andHalobactenum sp. strain GRB. Although the sample size issomewhat restricted, substitutions appear to be almost two-fold more frequent in the coding than in the noncodingregion.
In summary, nucleotide sequence divergence of the sodgene family from halophilic archaebacteria exhibits manyunusual and remarkable features (7). These features indicatequite clearly that evolutionary processes are not uniform andpredictable. Rather, they are complex and involve subtle butintense selection that is presumably exerted at the level ofprotein structure and function; somehow, this selectioninfluences processes at the level of DNA that are only nowbecoming apparent from sequence data analysis. Theseprocesses include duplication, generation of variability bymutation, fixation or elimination of mutations by drift orselection, and recombination or gene conversion.
ACKNOWLEDGMENTS
This work was supported by a research grant from the MedicalResearch Council of Canada, of which P.P.D. is a principal inves-
J. BACTERIOL.
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

EVOLUTION OF HALOPHILIC SOD PROTEINS 1579
tigator. P.P.D. is a fellow of the Canadian Institute of AdvancedResearch, Program in Evolutionary Biology.We thank Dan Fieldhouse of York University for help in the
neighbor-joining analysis.
REFERENCES1. Englert, C., M. Horne, and F. Pfeifer. 1990. Expression of the
major gas vesicle protein gene in the halophilic archaebacteriumHaloferax mediterranei is modulated by salt. Mol. Gen. Genet.222:225-232.
2. Felsenstein, J. 1985. Confidence limits on phylogenies: an ap-proach using the bootstrap. Evolution 39:783-791.
3. Fitch, W. M. 1971. Toward defining the course of evolution;minimal change for a specific tree typology. Syst. Zool. 20:406-416.
4. Fridovich, I. 1978. The biology of oxygen radicals. Science201:875-880.
5. Fridovich, I. 1986. Biological effects of the superoxide radical.Arch. Biochem. Biophys. 247:1-8.
6. Horne, M., C. Englert, and F. Pfeifer. 1988. Two genes encodinggas vacuole proteins in halobacterium halobium. Mol. Gen.Genet. 213:459-464.
7. Joshi, P., and P. P. Dennis. 1993. Characterization of paralogousand orthologous members of the superoxide dismutase genefamily from genera of the halophilic bacteria. J. Bacteriol.175:1561-1571.
8. Kimura, M. 1980. A simple method for estimating evolutionaryrates of base substitutions through comparative studies ofnucleotide sequences. J. Mol. Evol. 16:111-120.
9. Kimura, M. 1983. The neutral theory of molecular evolution.Cambridge University Press, Cambridge.
10. Kirby, T. W., J. R. Lancaster, and I. Fridovich. 1981. Isolationand characterization of the iron-containing superoxide dismu-tase of Methanobacterium bryantii. Arch. Biochem. Biophys.210:140-148.
11. Lanyi, J. 1974. Salt dependence of proteins from extremelyhalophilic bacteria. Bacteriol. Rev. 38:272-290.
12. May, B., and P. P. Dennis. 1990. Unusual evolution of a
superoxide dismutase-like gene from the extreme halophilicarchaebacterium Halobacterium cutirubrum. J. Bacteriol. 172:3725-3729.
13. May, B. P., and P. P. Dennis. 1987. Superoxide dismutase fromthe extremely halophilic archaebacterium Halobacterium cutir-ubrum. J. Bacteriol. 169:1417-1422.
14. May, B. P., and P. P. Dennis. 1989. Evolution and regulation of
the gene encoding superoxide dismutase from the archaebacte-rium Halobacterium cutirubrum. J. Biol. Chem. 264:12253-12258.
15. May, B. P., P. Tam, and P. P. Dennis. 1989. The expression ofthe superoxide dismutase gene in Halobacterium cutirubrumand Halobacterium volcani. Can. J. Microbiol. 35:171-175.
16. Mylvaganam, S., and P. P. Dennis. 1992. Sequence heterogene-ity between the two genes encoding 16S rRNA from thehalophilic archaebacterium Haloarcula marismortui. Genetics130:399-410.
17. Parker, M. W., and C. C. Blake. 1988. Iron- and manganese-superoxide dismutases can be distinguished by analysis of theirprimary structures. FEBS Lett. 229:377-382.
18. Saitou, N., and M. Nei. 1987. The neighbour joining method; anew method for reconstructing phylogenetic trees. Mol. Biol.Evol. 4:406-425.
19. Salin, M. L., and D. Oesterhelt. 1988. Purification of a manga-nese-containing superoxide dismutase from Halobactenumhalobium. Arch. Biochem. Biophys. 260:806-810.
20. Sarich, V. M., and A. C. Wilson. 1973. Generation time andgenomic evolution in primates. Science 179:1144-1147.
21. Smith, M., and R. Doolittle. 1992. A comparison of evolutionaryrates of the two major kinds of superoxide dismutase. J. Mol.Evol. 34:175-184.
22. Stallings, W. C., K. A. Pattridge, R. K. Strong, and M. L.Ludwig. 1984. Manganese and iron superoxide dismutases arestructural homologs. J. Biol. Chem. 259:10695-10699.
23. Stallings, W. C., K. A. Pattridge, R. K. Strong, and M. L.Ludwig. 1985. The structure of manganese superoxide dismu-tase from Thermus thermophilus HB8 at 2.4-A resolution. J.Biol. Chem. 260:16424-16432.
24. Steinman, H. M. 1982. Superoxide dismutases: protein chemis-try and structure-function relationships, p. 11-68. In L. W.Oberly (ed.), Superoxide dismutases, vol. 1. CRC Press, BocaRaton, Fla.
25. Takao, M., A. Yasui, and A. Oikawa. 1991. Unique character-istics of superoxide dismutase of a strictly anaerobic archaebac-terium Methanobacterium thermoautotrophicum. J. Biol.Chem. 266:14151-14154.
26. Takeda, Y., and H. Avila. 1986. Structure and gene expressionof the E. coli Mn-superoxide dismutase gene. Nucleic AcidsRes. 14:4577-4589.
27. Woese, C. R. 1987. Bacterial evolution. Microbiol. Rev. 51:221-271.
VOL. 175, 1993
on March 20, 2019 by guest
http://jb.asm.org/
Dow
nloaded from







![TOP 10 RouterOS configuration mistakes - MikroTik · PDF filePresenter –Andis Arins andis[at]router.lv 2 MikroTik Consultant at MikroTik/ Microsoftcertified trainer Member of the](https://img.pdfslide.us/doc/110x75/5a79d4447f8b9ab05f8cc617/top-10-routeros-configuration-mistakes-mikrotik-andis-arins-andisatrouterlv.jpg)





















