Embed Size (px)
Citation preview

Original Articles
Sporadic Leber hereditary optic neuropathy in Australia and New Zealand
Christopher Chan, MB BS* David A Mackey, FRACO, MDI Edward Byme, FRACP, MDf
Abstract
Background: Leber hereditary optic neuropathy (LHON) is one of the more common forms of hered- itary optic neuropathy and one of the few mito- chondrial neuropathies. Prior to the advent of mol- ecular DNA testing, the diagnosis depended on the recognition of typical fundal changes, as well as a family history of maternal transmission. Sporadic cases were therefore diagnosed with a level of uncertainty. The aim of this study is to identify the proportion of patients with idiopathic bilateral optic neuropathy/atrophy who are suffering from LHON.
Methods: Requests were sent to all ophthalmolo- gists and neurologist in Australia and New Zealand for blood or hair follicle samples of patients with diagnosis of bilateral optic neuropathy/atrophy of uncertain aetiology for DNA testing by restriction endonuclease analysis.
Results: One hundred and fourty-four samples were received, of which 96 were sporadic cases of idiopathic optic atrophy. Eleven of these sporadic patients were found to harbour pathogenetic mito- chondrial point mutations associated with LHON.
Conclusions: Our results indicated that 11 % of patients with bilateral optic neuropathy/atrophy of
uncertain aetiology are suffering from LHON. Comparing this data with all the known familial cases of LHON, we report that at least 8% of all LHON cases in Australia are sporadic. We conclud- ed that mtDNA testing for LHON in patients with idiopathic optic atrophy should be included in the initial laboratory work-up.
Key words: Idiopathic bilateral optic neuropathy, mitochondrial DNA mutations, sporadic Leber hereditary optic neuropathy.
Leber hereditary optic neuropathy (LHON) is a maternally transmitted hereditary optic neu- ropathy, predominantly affecting young adult males between the ages of 15 and 35 years. It usually presents as an acute or subacute bilat- eral visual loss, which may be simultaneous or sequential, separated by weeks or months.’ The peculiar form of transmission was noted in the earliest description of the disease’ and the pos- sibility of cytoplasmic transmission was pro- posed as early as the 1930s.’ By the early 1970s, mitochondrial transmission of LHON was s ~ g g e s t e d . ~ Mitochondria1 DNA (mtDNA) is the only extra-nuclear DNA found within the cell. It is a 16 569 base-pair, double-stranded, closed circular structure,
* University of Melbourne, Department of Ophthalmoloa. Clinical Neuroscience Centre, St. Vincents Hospital. -f University of Melbourne, Departments of Ophthalmology and Paediatrics. $ Clinical Neuroscience Centre, St Vincents Hospital, Melbourne. Reprints: Dr David A Mackey, University of Melbourne, Department of Ophthalmology, Royal Victorian Eye and Ear Hospital, 32 Gisborne Srreet, East Melbourne,Victoria 3002, Australia.
Sporadic Leber hereditary optic neuropathy in Australia and New Zealand

within the mitochondrial matrix space.5 The mtDNA codes for 13 subunits of the oxidative phosphorylative mechanism (OXPHOS) of the cell, and the two ribosomal RNAs (rRNA) and 22 transfer RNAs (tRNA) required for their synthesis. The 13 mitochondrial-encoded respiratory chain components include seven subunits of Complex I (NADH dehydroge- nase; subunits 1, 2, 3, 4, 4L, 5 and 6),6,' one subunit of Complex I11 (ubiquinolcytochrome c oxido-reductase; subunit cytochrome b), three subunits of Complex IV (cytochrome c oxidase; subunit 1,2 and 3), and two subunits of Complex V (ATP synthase; subunits ATPase 6 and 8).5 The remaining subunits of the respiratory chain and other proteins within the mitochondria are nuclear encoded, and are transported into the mitochondria by way of specific transporters within the mitochondrial membranes.
More importantly, there are certain unique properties of the mtDNA. Firstly, each cell can possess up to thousands of copies of the DNA within its cytoplasm.8 Secondly, the mitochon- drial genetic code is slightly different from the nuclear code and therefore, mitochondria require their own rRNAs and tRNAs for trans- lation of gene products. Thirdly, the mutation rate of mtDNA is much higher than nuclear DNA, possibly due to a combination of increased exposure to oxidative stress, absence of repair mechanisms and lack of protective effect of histones?," Lastly, in common with all cytoplasmic contents, nearly all mtDNA is transmitted through the ovum, and therefore, along the maternal lineage."
Despite our knowledge of the structure and inheritance of mtDNA, it was not until 1988 that Wallace et al., with the aid of the newly discovered DNA amplification technique, the polymerase chain reaction," reported the iden- tification of a point mutation within the mito- chondrial genome associated with LHON. l 3
This G to A transition at nucleotide position 11778 was later found to occur in 50% to 80% of LHON pedigrees ~orldwide.'".'~
Following on from this seminal discovery, Huoponen et a1.I6 and Howell et al.I7 indepen- dently reported the discovery of a G to A tran- sition at mitochondrial nucleotide position 3460, found in 15% to 20% of all LHON pedigrees; while Mackey and Howell'8 and
Johns et al.I9 pointed to a T to C transition at nucleotide position 14484 of the mitochondri- a1 DNA, associated with approximately 15% of LHON cases.
The above three proven point mutations are thought to be responsible for 75% to 80% of all LHON cases worldwide.20 Other, rarer, pro- posed mitochondrial point mutations which are thought to be causative of LHON include the 9804 G to A transition within the cytochrome c oxidase gene,z1 the 13730 G to A transition found in one LHON patient" and the 14459 G to A transition which is found in families with a combination of LHON and early onset dystonia with bilateral striatal necrosis.z3
Other mtDNA point mutations have also been reported to be associated with LHON, but these are probably of little pathogenetic s ignif ican~e.~~,~~ Although we have tested for these proposed mutations, they are not helpful in confirming a diagnosis of LHON and are not included in this report.
Prior to the identification of LHON-associ- ated mtDNA point mutations, the diagnosis of LHON depended on a high level of clinical suspicion and demonstration of maternal transmission within the family. Diagnosis of sporadic cases, therefore, was difficult, as the characteristic fundal changesz5 (dilatation and tortuosity of retinal vessels, telangiectatic microangiopathy and swelling of nerve fibre layer) might be absent."With the availability of mtDNA testing, increasing numbers of spo- radic cases were identified.z7~'8 The reported proportion of sporadic cases within LHON populations varied between different coun- tries. No doubt, part of this may be due to ascertainment bias, difficulty in pedigree trac- ing and availability and completeness of genealogical records. Nevertheless, it is esti- mated that in the United States, 50% to 80% of LHON cases are sporadic,2h while in Europe, the figure is approximately 50%.29 In Australia, where extensive genealogical records are available, truly sporadic cases of LHON were reported to account for only 2% of all cases.3o
The aim of this study is to investigate further the number of sporadic cases of LHON in Australia and New Zealand and to identify the proportion of patients with bilateral optic neu-
8 Australian and New Zealand Journal of Ophthalmology 1996; 24(1)

ropathylatrophy, who harbour at least one of the six pathogenetic, proven or proposed, LHON mtDNA mutations mentioned previ- ously.
Methods Letters were sent to all ophthalmologists and neurologists in Australia and New Zealand, requesting blood or hair follicles samples from patients with bilateral optic neuropathy/atro- phy of uncertain aetiology. The blood samples were collected on Guthrie card^,^' more often used for neonatal screening of inherited dis- ease~.~* The Guthrie cards or hair samples were sent to our laboratory and DNA extrac- tion performed. Extraction of mtDNA from hair follicles has been previously de~cribed.’~ For DNA extraction from Guthrie cards, we punched three 4 mm Guthrie blood spots with a sterile puncher into a 1.5 mL microfuge tube. The puncher was then washed with 70% ethanol and flamed, to remove any lingering DNA. Blood was eluted from the filter paper in 50 mM Pis-HC1 pH 8.5, 1 mM EDTA, 0.5% Tween 20 and 200 mg/pL of proteinase K. The mixture was incubated overnight at 37°C in a temperature-controlled water bath. After the
Table 1. LHON mutations
incubation, the microfuge tube was placed in boiling water for 10 minutes, to denature any residual proteins, followed by cooling on ice. A phenol-chloroform extraction was then per- formed. DNA was precipitated with 0.1 vol- ume of 3 M sodium acetate and 2.5 volume of 100% cold ethanol at -80°C for one hour. After centrifugation (1 3 000 g for 15 minutes), the precipitant was washed with 70% cold ethanol and further centrifuged (13 000 g for 15 min- utes). The pellet was air dried and resuspend- ed in filtered T E buffer (10 mM Eis-HCL, 1 mM EDTA pH 8.0). Then 10 pL of this was used for PCR amplification. Amplification of the desired mtDNA fragments was performed in a reaction mixture containing 10 pL of tem- plate DNA, 1 x PCR buffer (5 mM KCL, 1 mM Eis-HCL pH 9.0 and 0.01% Triton X- loo), 2.5 mM MgC12, 200 mM of equimolar mixture of dNTPs, 20 pmol of each synthetic oligonucleotide primer (Table 1) and 1.25 units of Tuq polymerase, in a final reaction vol- ume of 50pL. A control blank was also pre- pared at the same time, substituting template DNA with sterilised, double-distilled water. The reaction mixture was subjected to an ini- tial denaturation of three minutes at 94”C, followed by 35 cycles of denaturation (one
H-strand L-strand Restriction Normal Mutated Mutation Gene acid primer Sequence primer Sequence endonuclease fragments fragments
change ~~ _______-
14459 ND6 Ala ,Val 14560- 5’GATTGTT 14439- SGATACTC Acil’” 11,19,110 11,129 (G ‘A) (Complex I) 14578 AGCGGTGT 14458 CTCAATAG
14484 ND6 Met *Val 14519- 5’TITGGGG 14463- S’TAGTATA Sau3AIt 21,54 75 -
(H1457) GGTC3’ (L1443M) CCACC3’
(T + C) (Complex I) 14538 GAGGTTAT 14483 TCCAAAGA
____ (334) ATGGG3’ (336) CAACGA3’
13730 ND5 Gly P Glu 13733 - 5’TGCGGGG 13630 - S’ACAGGTC EcoRVt 29 103 132 (C + A) (Complex 1) 13761 GAAATGTT 13647 AACCTCGC
(H1373M) GTTAGTAA (L1363) TTC3’ TGAGAT3’ ___________ __- -~
11778 ND4 Arg + HIS 11918 - S’GTAGGAG 11240 - 5’CTCATCG SfaN I 155,548 703 (G + A) (Complex I) I1942 AGTGATAT 11266 CACTAATT
TTGATCAG (L1124) TACACTCA M a e 111s 37,255,411 37,124,131,411 G3’ CAAC3’
(P2)
~~
9804 cox I11 Ala >Thr 10159- 5’CGAAGCC 8907- 5’CTTCTTA MaeIIl$ 480,796 382,414,480 (G +A) (ComplexIV) 10182 GCACTCGT 8929 CCACAAGG
rER2) AAGGGGTG IATP6L) CACACCTA G3’ C3’
~~~
115,184 299 3460 ND 1 Ala .Thr 3553 - S’CAGTCAG 3215- 5’GAGGGG AcyIt (G -+A) (Complex I) 3573 AGGTTCAA 3295 GGTTCATA
(ND1 1B) TTCCTC3’ (NDI 1A) GTAGAAG3’
Three mismatch primers (L1443M, 336 and H1373M) were used to create restriction sites for the screening of point mutations 14459,14484 and 13730. The altered nucleotide bases were underlined. * New England Biolab; t Promega; $ Borhringer Mannheim.
_ _ _ _ _ _ _ _ ~ ~ ._____---~ _____ __--
Sporadic Leber hereditary optic neuropathy in Australia and New Zealand 9

minute at 94"C), annealing (one minute at 55"C), extension (2.5 minutes at 72°C) and a final cycle of extension for 7.5 minutes at 72°C. Presence of the amplified PCR frag- ments was confirmed by electrophoresis of 10pL of the PCR product through a 3.0% (w/v) agarose gel containing 0.05 pg/mL ethidium bromide, in TAE buffer (0.04 M Tris-acetate, 0.002 M EDTA). Bands were visualised by UV light. Untreated PCR prod- uct (1 0 pL) was digested with the appropriate restriction enzymes (Table 1) over four hours according to manufacturers recommendations. All digested products were electrophoresed against low molecular weight marker (Promega) on a 3.0% (w/v) agarose gel as described above. Digested bands were again visualised by UV light. Samples testing positive for any of the six LHON mutations were repeated, with further samples prepared from the original Guthrie cards or hair follicles. If confirmed, the sample was sent away for further confirmatory mtDNA testing by sequencing.
Results Over a period of one-and-a-half years, 144 samples from cases of idiopathic optic atrophy, previously investigated by the referring physi- cians, were sent for mtDNA screening of LHON mutations. Many of these were from relatively young patients with limited visual recovery. Of these, 21 samples were collected from individuals belonging to eight of the 17
multiple affected families with LHON in Australia and New Zealand, documented by one of us (DAM) previously (Table 2)?','' These samples were sent for screening either because a confirmatory testing for mtDNA sta- tus of the individual was requested, or the patient was unaware that they belonged to a family with LHON. Seventeen samples belonging to seven new families (Table 2) have positive family histories of optic neuropathy/atrophy and therefore, were not sporadic cases. They were excluded from our final analysis. Five of these families have the 11778 mutation, one family has the 14484 mutation, and the remaining family presum- ably has an unknown mutation. Nine individu- als belonging to three families were subse- quently diagnosed with autosomal dominant optic neuropathy. One sample was received from an adopted patient, whom we later iden- tified, on molecular DNA basis, to be a mem- ber of one of the known multigeneration fami- lies in Australia.
By excluding the above samples, we were left with 96 presumably sporadic cases of bilateral optic neuropathy/atrophy of uncertain aetiology. Eleven patients screened were positive for LHON mutations. Of these, seven males and one female harbour the 11778 mutation, one male and one female have the 14484 mutation and one female has the 3460 mutation. Screening results of these patients are summarised in Figure 1.
85 patients do not harbour any of the six primary LHON mutations. 1 I patients were screened positive for LHON mutations. 8 patients have the I1 778 mutation, two have the 14484 mutation and one has the 3460 mutation.
Figure 1
10 Australian and New Zealand Journal of Ophthalmology 1996; 24(1)
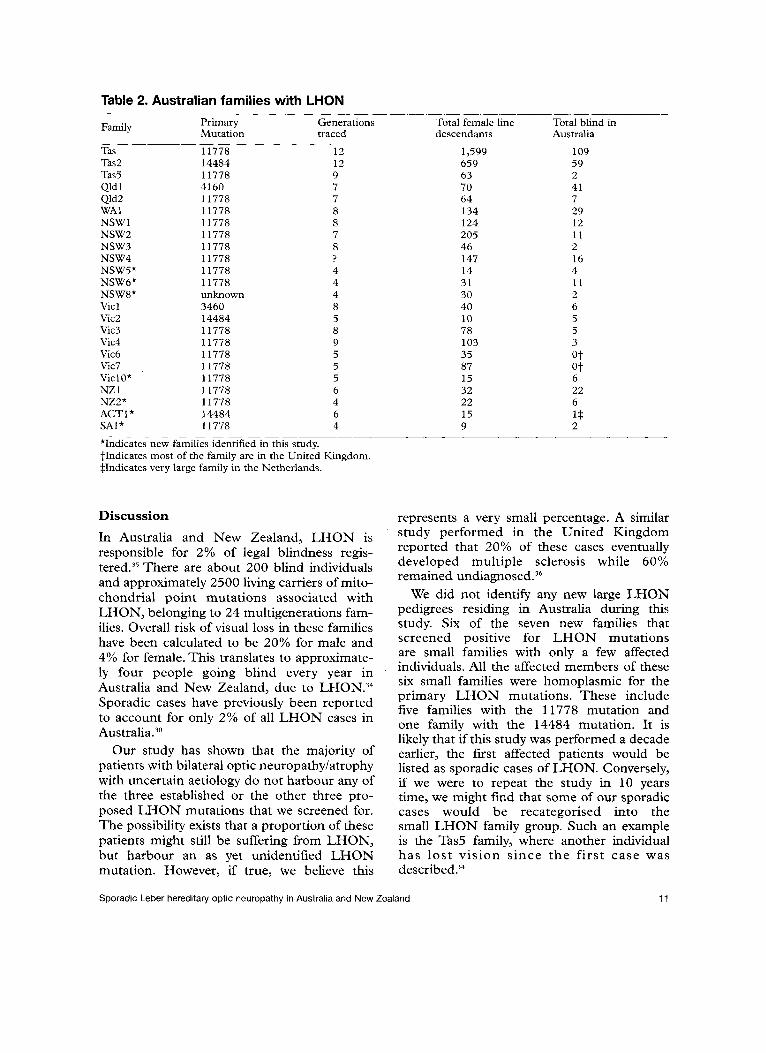
Table 2. Australian families with LHON _____ ______ ~~ _ _
Primary Generations Total female line Total blind in Mutation traced descendants Australia
Family
Tas 11778 12 1,599 109 Tas2 14484 12 659 59 Tas5 11778 9 63 2 Qld 1 4160 7 70 41 Qld2 11778 7 64 7 MA1 11778 8 134 29 NSWl 11778 8 124 12 NSW2 11778 7 205 11 NSW3 11778 8 46 2 NSW4 11778 ? 147 16 NSW5* 11778 4 14 4 NSW6* 11778 4 31 11 NSW8* unknown 4 30 2 Vicl 3460 8 40 6 Vic2 14484 5 10 5 Vic3 11778 8 78 5 Vic4 11778 9 103 3 Vic6 11778 5 35 ot Vic7 11778 5 87 @t
______ ___
ViclO* 11778 5 15 6 NZ 1 11778 6 32 22 NZ2* 11778 4 22 6 ACT1* 14484 6 15 1* SAl* 11778 4 9 2
*Indicates new families identified in this study. tIndicates most of the family are in the United I n g d o m . $Indicates very large family in the Netherlands.
__ ___
Discussion
In Australia and New Zealand, LHON is responsible for 2% of legal blindness regis- t e ~ e d . ~ ~ There are about 200 blind individuals and approximately 2500 living carriers of mito- chondrial point mutations associated with LHON, belonging to 24 multigenerations fam- ilies. Overall risk of visual loss in these families have been calculated to be 20% for male and 4% for female. This translates to approximate- ly four people going blind every year in '
Australia and New Zealand, due to LHON.34 Sporadic cases have previously been reported to account for only 2% of all LHON cases in A ~ s t r a l i a . ~ ~
Our study has shown that the majority of patients with bilateral optic neuropathy/atrophy with uncertain aetiology do not harbour any of the three established or the other three pro- posed LHON mutations that we screened for. The possibility exists that a proportion of these patients might still be suffering from LHON, but harbour an as yet unidentified LHON mutation. However, if true, we believe this
represents a very small percentage. A similar study performed in the United Kingdom reported that 20% of these cases eventually developed multiple sclerosis while 60% remained ~ndiagnosed.~~
We did not identify any new large LHON pedigrees residing in Australia during this study. Six of the seven new families that screened positive for LHON mutations are small families with only a few affected individuals. All the affected members of these six small families were homoplasmic for the primary LHON mutations. These include five families with the 11778 mutation and one family with the 14484 mutation. It is likely that if this study was performed a decade earlier, the first affected patients would be listed as sporadic cases of LHON. Conversely, if we were to repeat the study in 10 years time, we might find that some of our sporadic cases would be recategorised into the small LHON family group. Such an example is the Tas5 family, where another individual has lost vision since the first case was described?4
Sporadic Leber hereditary optic neuropathy in Australia and New Zealand 11

Table 3. Sporadic LHON in Australia and New Zealand ____ ~ _ _ _ _ _ _ _ _ _ ____ ____
Primary Generations Total female line Total blind in Mutation traced descendants Australia
Family ___
Tas3* 3460 9 83 1 Qld3 Qld4 WA2 WA3 NSW7 NZ3 NZ4 NZ5 SA2 Vic5* Vic9 Vicll
11778 11778 11778 11778 3460 11778 11778 11778 14484 11778 11778 14484
4 5 ? 7 ? ? ? ? 2 6 ? ?
? ? ? 106 ? ? ? ? 3 32 ? ?
1 1 1 1 1 1 1 1 1 1 1 1
*Indicates previous reported sporadic cases
Of the 11 cases of LHON confirmed by mol- ecular testing, we failed to identify any individ- ual harbouring either the 14459,13730 or 9804 mutations. 73% of our cases have the 11778 mutation, 18% have the 14484 mutation and 9% have the 3460 mutation. These figures are similar to those reported in the world literature.
In addition, our present study has indicated that the incidence of sporadic cases of LMON might have been underestimated. The 1 1 cases of sporadic LHON added to the two previous- ly reported cases, increases the proportion of sporadic cases of LHON in Australia and New Zealand to 8% (Table 3).
The results also showed that 11% of patients suffering from bilateral optic neuropathy/ atrophy of uncertain aetiology and without a family history of a similar ailment, harbour one of the primary LHON mutations and are, in fact, suffering from visual loss secondary to LHON. This figure is lower than the 20% of sporadic LHON cases reported from a similar group of patients with bilateral simultaneous optic neu- ropathy in the United Kingdom.96
The male to female ratio of confirmed spo- radic LHON cases is 2.67: 1. This ratio is lower than the reported ratio of 5:1 in established LHON pedigrees in A~stralia?~ but is within range of those reported in the United Kingdom.37 The explanation for male predominance in LHON cases is still unexplained. Previous study on the possibility of nuclear-cytoplasmic interac- tions in the phenotypic expression of LHON have been un~evealing.~~ A genetic factor residing
in the X chromosome has been suggested to interact with primary LHON mutations to pro- duce such predominan~e.~~ However, investiga- tions have failed to identrfy any linkage of the disease to locus markers on the X chromosome.4o It is likely that additional factors, be they genetic, endogenous or environmental, interact with the primary LHON mutations to produce the male predominance, as well as the delay in onset of the disease and the reduced penetrance, since not all individuals with the primary mutations develop LHON.
In conclusion, we recommend that all patients who present with idopathic bilateral optic neuropathylatrophy, even if there were no family history, should undergo mtDNA testing for LHON mutations. We predict that about 10% of these would be confirmed sporadic LHON cases. As some members of LHON families are unaware of their family history, the likelihood of a positive LHON screen is even higher.
Acknowledgements We would like to thank all the staff at the Melbourne Neuromuscular Research center for their help and the NHMRC for financial support. We would especially like to thank Dr B Jean-Francois for her technical advice and Dr R Blok for her invaluble comment on this manuscript.
We also thank Prof N Howell from the University of Texas Medical branch, Galveston, Texas, USA, for performing confirmatory sequencing of our positive LHON samples.
12 Australian and New Zealand Journal of Ophthalmology 1996; 24(1)

We thank all the ophthalmologists and neurologists who have participated in this study: Dr R Agnello, Dr C Banks, Dr P Beaumont, Dr C Benson, Dr S Best, Prof F Billson, Dr S Birchley, Dr J Bond, Dr J Bowbyes, Dr S Bower, Dr R Buttery, Dr D Colville, Prof I Constable, Dr S Crowe, Dr J Dixon, Dr N Downie, Dr M Edwards, Dr J Elder, Dr D Farlow, Dr C Ferraris, Dr M Flaherty, Dr E Finkelstein, Dr T Gin, Dr J Grigg, Dr Hadden, Dr A Hall, Dr C Harding, Dr G Harley, Dr A Harper, Dr B Harrisberg, Dr M Hennessey, Dr R Higgins, Dr D Hor, D r T Hodson, Dr R Kearney, Dr C Keith, Dr J King, Dr L Kowal, Dr J L a Nauze, Dr B Lansdell, Dr M Loane, Dr P Lockie Dr D Lowe, Dr P McCartney, Assoc Prof H Maclean, Dr C McKenzie, Dr Malone, Dr W Marshman, Dr J Mazzetti, Assoc Prof P Mitchell, Dr I Murrell, Dr S Nicholls, Dr M Noble, Dr J O’Day, DrV Phakey, Dr R Rawson, Dr R Rischbieth, Dr R Roberts, Dr D Roden, Dr J Rogers, Dr J Runciman, Dr N Saad, Dr R Sachdev, Dr R Sellens, Dr K Snow, Dr G Suthers, Dr D Tamblyn, Dr M Toohey, Dr M Treplin, Dr R Westmore, Dr J Whitford, Dr G Wise, Dr D Workman and Dr E Wysocki.
References 1.
2.
3.
4.
5.
6.
7.
8.
Nikoskelainen EK. Clinical picture of LHON. Clin Neurosci 1994;2:115-20. Leber T. Uber hereditare und congenital-angelegte Sehnervenleiden. Graefe’s Arch Ophthalmol
Imai Y, Moriwaki D. A probable case of cytoplasmic inheritance in man: a critique of Lebers disease. J Genet
Erickson RP. Lebers optic atrophy: a possible example of mitochondrial inheritance. Am J Hum Genet
Anderson S, Bankier AT, Barrel1 BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organisation of the human mitochondrial genome. Nature
Chomyn A, Mariottini P, Cleeter MW, Ragan CL, Matsuno-Yagi A, et al. Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory chain NADH dehydrogenase. Nature 1985;3 14592-7. Chomyn A, Cleeter MW, Ragan CL, &ley M, Doolittle RF, Attardi G. URF6, last unidentified reading kame of human mtDNA, codes for an NADH dehydrogenase subunit. Science 1986;234:6 14- 18. Shuster RC, Rubenstein AJ, Wallace DC. Mitochondrial DNA in anucleate human blood cells. Biochem Biophys Res Commun 1988; 155: 1360-5.
187 1;2:249-91.
1936;33: 163-7.
1972;24:348-9.
198 1;290:457-65.
9. Brown WM, George M, Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA
10. Cann RL, Wilson AC. Length mutations in human mitochondrial DNA. Genetics 1983;104:699-711.
11. Giles RE, Blanc H, Cann HM and Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci USA. 1980;77:6715-19.
12. Saiki RK, Scharf SJ, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of beta-globin sequences and restriction site analysis for diagnosis of sickle cell anemia. Science.
13. Wallace DC, Singh G, Lott MT, Hodge JA, SchurrTG, Lezza AMS, et al. Mitochondrial DNA mutation asso- ciated with Lebers hereditary optic neuropathy. Science
14. Newman NJ, Lott MT, Wallace DC. The clinical char- acteristics of pedigrees of Lebers hereditary optic neu- ropathy with the 11778 mutation. Am J Ophthalmol
15. Mashimay, HiidaY, OguchiY, Kudoh J, Shimizu N. High frequency of mutations at position 11778 in mitochondr- ial ND4 gene in Japanese families with Lebers hereditary optic neuropathy. Hum Genet 1993;92:101-2.
16. Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus M-L. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet 1991;48:1147-53.
17. Howell N, Bindoff L, McCullough DA, Kubacka I, Poulton J, Mackey D, Taylor K, Turnbull DM. Leber hereditary optic neuropathy: identification of the same mitochondrial ND1 mutation in six pedigrees. Am J Hum Genet 1991;49:939-50.
18. Mackey D, Howell N. A variant of Leber hereditary optic neuropathy characterized by recovery of vision and by an unusual mitochondrial genetic etiology. Am J Hum Genet 19923 1: 121 8-28.
19. Johns DR, Neufeld MJ, Raymond DP. An ND-6 mito- chondrial DNA mutation associated with Leber hered- itary optic neuropathy. Biochem Biophys Res C o r n
20. Brown MD, Wallace DC. Spectrum of mitochondrial DNA mutations in Lebers hereditary optic neuropathy. Clin Neurosci 1994;2:138-45.
21. Johns DR, Neufeld MJ. Cytochrome c oxidase muta- tions in Leber hereditary optic neuropathy. Biochem Biophys Res Comm 1993;196;2:810-15.
22. Howell N, Halvorson S, Burns J, McCullough DA, Paulton J. When does bilateral optic atrophy become Leber hereditary optic neuropathy? Am J Hum Genet
23. Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with mater- nally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA. 1994;91:6206-10.
24. Howell N. Primary LHON mutations: trying to sepa- rate Fruyt from Chaf. Clin Neurosci 1994;2:130-7.
25. Smith JL, Hoyt WF, Susac JO. Ocular fundus in Lebers optic neuropathy. Arch Ophthalmol 1973;90:349-54.
26. Newman NJ. Lebers hereditary optic neuropathy. New genetic considerations. Arch Neurol 1993;50:540-8.
1979;76: 1967-7 1.
1985;230: 1350-4.
19885242: 1427-30.
199 1; 1 1 1~750-62.
1992; 187: 155 1-7.
1993;53:959-63.
Sporadic Leber hereditary optic neuropathy in Australia and New Zealand 13

27. Oostra RJ, Bolhuis PA, Bleeker-Wagemakers EM. Mitochondria1 DNA analysis as a diagnostic tool in sin- gleton cases of Lebers hereditary optic neuropathy. Ophthalmic Paed Genet 1993;14:109-15.
28. Swartz N, Savino PJ. Is all nondefinable optic atrophy Lebers herediary optic neuropathy? Surv Ophthalmol 1994;39:146-50.
29. Obermaier-Kusser B, Lorenz B, Schubring S, Paprotta A, Zerres K, Meitinger T, et al. Features of mtDNA mutation patterns in European pedigrees and sporadic cases with Leber hereditary optic neuropathy. Am J Hum Genet 1994;55:1063-6.
30. Mackey DA. Epidemiology of Lebers hereditary optic neuropathy in Australia. Clin Neurosci 1994;2: 162-4.
31. Mackey DA, Nasioulas S, Forrest S. Finger prick blood testing in Leber hereditary optic neuropathy. Br J Ophthalmol 1993;77:3 1 1 - 12.
32. McCabe, ER, Huang SZ, Seltzer WK, Law ML. DNA microextraction from dried blood spots on filter paper blotters: potential applications to newborn screening. Hum Genet 1987;75:2 13- 16.
33. Kotsimbos N, Jean-Francois BMJ, Huizing M, Kapsa RMI, Lertrit P, Siregar NC, et al. Rapid and noninvasive screening of patients with mitochondrial myopathy. Hum Genet 1994;4:132-5.
34. Mackey DA, Buttery RG. Leber hereditary optic neu- ropathy in Australia. Aust NZ J Ophthalmol 1992;20: !77-84.
35. Yeates FM. Causes of binocular legal blindness in an Australian metropolitan community. Aust J Ophthalmol
36. Morrissey SP, Borruat FX, Miller DH, Moseley IF, Sweeney MG, Govan GG, Kelly MA, et al. Bilateral simultaneous optic neuropathy in adults: clinical, imag- ing, serological, and genetic studies. J Neurol Neurosurg
37. Riordan-Eva P, Sanders M.D, Govan GG, Sweeney MG, Da Costa J, Harding AE. The clinical fea- tures of Lebers hereditary optic neuropathy defined by the presence of a pathogenic mitochondria1 DNA muta- tion. Brain 1995;118:319-37.
38. Govan GG, Smith PR, Kellar-Wood H, Schapira AHV, Harding AE. HLA class I1 genotypes in Lebers hereditary optic neuropathy. J Neurol Sci
39. Bu XD, Rotter JI. X chromosome-linked and mitochondrial gene control of Leber hereditary optic neuropathy: Evidence from segregation analysis for dependence on X chromosome inactivation. Proc Natl Acad Sci USA 1991;88:81
40. Juvonen V, Vilkki J, Aula P, Nikoskelainen E, Savontaus ML. Reevaluation of the linkage of an optic atrophy susceptibility gene to X-chromosomal markers in Finnish families with Leber hereditary optic neuroretinopathy (LHON). Am J Hum Genet 1993;53:289-92.
1983;11:321-3.
PSYC 1995;58:70-4.
1994;126: 193-6.
98-202.
14 Australian and New Zealand Journal of Ophthalmology 1996; 24(1)





























