Embed Size (px)
Citation preview

Spatio-Temporal Image Correlation Spectroscopy:Extension to Three Dimensions and Application to
Biological Systems
Dominique Guillet
Department of Physics
McGill University, Montreal
Quebec, Canada
April 2012
A Thesis submitted to McGill University
in partial fulfillment of the requirements of the degree of
Master of Science
c© Dominique Guillet, 2012


Contents
Abstract vii
Resume viii
Statement of Originality ix
Acknowledgments x
1 Introduction 11.1 Fluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.2 Fluorescence Microscopy . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.1 Confocal laser scanning microscopy (CLSM) . . . . . . . . . . 41.3 Fluorescence Correlation Spectroscopy . . . . . . . . . . . . . . . . . 61.4 Image Correlation Spectroscopy . . . . . . . . . . . . . . . . . . . . . 7
2 Spatiotemporal Image Correlation Spectroscopy 82.1 Image Correlation Techniques . . . . . . . . . . . . . . . . . . . . . . 8
2.1.1 Image Correlation Spectroscopy . . . . . . . . . . . . . . . . . 92.2 Spatio-Temporal Image Correlation Spectroscopy . . . . . . . . . . . 10
2.2.1 Immobile Population Removal . . . . . . . . . . . . . . . . . . 132.2.2 Experimental Limitations . . . . . . . . . . . . . . . . . . . . 14
3 Vesicle Dynamics during Plant Cell Cytokinesis 183.1 Biological System: Somatic Cytokinesis . . . . . . . . . . . . . . . . . 183.2 Materials and Methods . . . . . . . . . . . . . . . . . . . . . . . . . . 243.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
3.3.1 Vesicle dynamics . . . . . . . . . . . . . . . . . . . . . . . . . 253.3.2 Cell plate growth rate . . . . . . . . . . . . . . . . . . . . . . 29
3.4 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
4 Extension of STICS to 3 Dimensions 364.1 Theory . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 374.2 Computer Simulations . . . . . . . . . . . . . . . . . . . . . . . . . . 40
4.2.1 3D simulator . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
iii

iv CONTENTS
4.2.2 Simulation Results . . . . . . . . . . . . . . . . . . . . . . . . 43
5 Conclusion 55
References 58

List of Figures
1.1 Jablonski diagram . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31.2 Fluorescent spectra of two fluorescent proteins . . . . . . . . . . . . . 41.3 Schematic of a laser scanning confocal microscope . . . . . . . . . . . 51.4 Schematic representation of the image correlation spectroscopy technique 7
2.1 Evolution of the correlation function of a STICS analysis . . . . . . . 122.2 Image from a dividing plant cell with region of analysis overlayed . . 162.3 Evolution of the correlation function as a function of time lag . . . . 172.4 Example of a vector map from a STICS analysis . . . . . . . . . . . . 17
3.1 Sketch of vesicles and vesicle fusion with each other and the cell plate 193.2 Sketch of the different components of plant cell cytokinesis . . . . . . 203.3 Summary of the stages of cell division . . . . . . . . . . . . . . . . . . 233.4 Selected frames from a CLSM fluorescence image time series showing
the reorientation of the cell plate . . . . . . . . . . . . . . . . . . . . 253.5 STICS vector map during accumulation of vesicles . . . . . . . . . . . 263.6 STICS vector maps at different stages of cytokinesis . . . . . . . . . . 283.7 STICS vector map at the ring phragmoplast stage of cell division . . 293.8 Analysis of the growth rate of the cell plate . . . . . . . . . . . . . . 303.9 FRAP experiment on the cell plate of a dividing BY-2 cell . . . . . . 35
4.1 Schematic representation of a 3D image stack time series . . . . . . . 374.2 Contour plots of the 3D autocorrelation function . . . . . . . . . . . . 434.3 Contour plots of the time evolution of the 3D correlation function . . 444.4 3D STICS results for simulations with increasing velocity magnitudes
in the z dimension . . . . . . . . . . . . . . . . . . . . . . . . . . . . 464.5 Spatial spreading of the correlation function with movement . . . . . 474.6 3D STICS results of simulations for increasing values of flowing particle
density . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 484.7 3D STICS results of simulations with velocity components in all three
spatial dimensions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 524.8 3D STICS results of simulations with flowing and filtered immobile
populations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
v

List of Tables
4.1 Adjustable parameters for 3D simulations . . . . . . . . . . . . . . . 424.2 Comparison between STICS and 3D STICS results for simulations of
2D velocities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454.3 Effect of resolution and number of images in z on 3D STICS . . . . . 50
vi

Abstract
The object of this thesis is to present work done using spatio-temporal image correla-
tion spectroscopy (STICS), a technique that uses fluorescence intensity fluctuations in
a microscopy image time series to calculate a complete space-time correlation function
in order to measure transport dynamics in cells. The time evolution of this correla-
tion function gives information on the magnitude and direction of a flow of fluorescent
particles sampled in the image series. First, a new application of STICS to plant cell
biology is shown. In dividing plant cells, delivery of new cell wall material to the
forming cell plate requires intricate coordination of secretory vesicle trafficking and
delivery. In this work, STICS is used to measure vesicle dynamics during plant cell
division. It was discovered that vesicle transport to the plane of division occurs in
three phases, each with its characteristic flow patterns and range of velocities, which
directly reflect the rate of growth of the forming cell plate. The second part of this
thesis presents the extension of the STICS technique to a third spatial dimension. The
development of this new technique, called 3D STICS, allows the study of transport
dynamics in three dimensions, which is more relevant in tissues and non adherent cells
which are inherently 3D. Computer simulations were performed to test the accuracy
and precision of the technique under a range of parameters such as particle density of
immobile and moving populations; and number of images, velocity and resolution in
the third spatial dimension. A comparison between values of velocities in a 2D plane
recovered using STICS and its new 3D version is also presented.
vii

Resume
L’objet de cette these est de presenter des travaux faits a l’aide de la spectroscopie
par correlation spatiotemporelle d’images (STICS), une technique qui utilise les fluc-
tuations d’intensite dans une serie d’images capturees a l’aide d’un microsope par
fluorescence pour calculer la fonction complete de correlation spatiotemporelle, et
ainsi mesurer la dynamique du transport de proteines a l’interier de cellules vivantes.
L’evoultion temporelle de cette fonction de correlation donne de l’information sur
la direction et la vitesse d’un flot de particules fluorescentes presentes dans la serie
d’images. Tout d’abord, une nouvelle application de la technique en biologie vegetale
est presentee. Lors de la division cellulaire vegetale, le transport du materiel mem-
branaire necessaire a la formation de la plaque cellulaire requiert une grande precision
dans la coordination du transport et de la livraison des vesicules de secretion. Dans
cette these, STICS est utilisee pour mesurer la dynamique de ces vesicules pendant la
division cellulaire vegetale. Les resultats obtenus revelent l’existence de trois phases
dans le transport des vesicules de secretion au site de division cellulaire, chacune
presentant une echelle de vitesse et des motifs de mouvement characteristiques qui se
refletent dans le taux de croissance de la plaque cellulaire. Dans un deuxieme temps, le
developpement de STICS pour inclure l’analyse de la troisieme dimension spatiale est
presente. Cette nouvelle technique, appelee STICS 3D, permet l’etude de dynamiques
en trois dimensions, ce qui est plus pertinent que la version deux-dimensionnelle pour
les tissus et les cellules non adherentes, qui ont un environnement intrinsequement
3D. Des simulation par ordinateur ont ete effectuees pour determiner l’exactitude,
la precision et les limites de la technique pour un eventail de parametres comme la
vitesse, le nombre d’images et la resoultion dans la troisieme dimension spatiale ainsi
que la densite des populations immobiles et en mouvement. Une comparaison en-
tre les resultats obtenus avec STICS et la nouvelle version 3D de la technique est
egalement presentee.
viii

Statement of Originality
The author claims the following aspects of the thesis constitute original scholarship
and an advancement of knowledge:
Chapter 3 STICS measurements of vesicle dynamics during plant cell cytokinesis.
This chapter presents the first application of spatio-temporal image correla-
tion spectroscopy (STICS) on dividing plant cells and is the first comprehen-
sive characterization of endogenous secretory vesicle dynamics during plant cell
cytokinesis. Because of the interdisciplinary nature of this project, the work
presented was done in collaboration with Prof. Anja Geitmann’s group, from
Universite de Montreal. Chloe van Oostende performed cell culture, imaging,
FRAP experiments and analysis with FluMOS.
Chapter 4 Extension of STICS to 3 Dimensions. This chapter presents the devel-
opment of a new technique, 3D spatio-temporal image correlation spectroscopy
(3D STICS), which is an extension of STICS. This new technique allows the
study of transport dynamics in three dimensions, which is more relevant in tis-
sues and non adherent cells which are inherently 3D. This chapter presents the
theoretical basis of the technique as well as its characterization under various
simulation parameters.
ix

Acknowledgments
First I would like to thank my supervisor, Prof. Paul Wiseman, for giving me the
opportunity to be a part of his research group. It is thanks to him that I have ex-
perienced stimulating collaborations and leading-edge research. He has my gratitude
for giving me a chance as a simple undergraduate one summer and accepting me in
his group ever since.
I also want to acknowledge my research collaborators, without whom part of this
thesis would have never existed. First I need to thank Prof. Anja Geitmann, from
Universite de Montreal, for allowing me to work with her group on this interesting
project of dividing plant cells. I also have to recognize the incredible work of her
postdoc, Chloe van Oostende, who was a big part of this project, taking care of the
cell culture and imaging, and analyses of cells with FRAP and FluMOS. I have to
thank her for the numerous meetings and long - but always enjoyable! - discussions
that were necessary to obtain the results we have now.
I also need to thank many people from my own research group. Antoine Godin
helped me considerably with the coding of 3D STICS and the 3D simulator in MatLab.
I also have to thank Laurent Potvin-Trottier and Benjamin Rappaz for the stimulating
discussions around cups of coffee in the morning and for the training they gave me
in cell culture. Finally I have to thank all other Wiseman group members, present or
past, alongside whom I have worked. They have all contributed in their own way to
make my time in the group an enjoyable and priceless experience.
The final acknowledgements must go to my family, and especially to my parents,
Christiane and Jean-Pierre. They have instilled in me, ever since I was very young, a
thirst for knowledge and a wonderment of life. They have given me the confidence to
believe in myself and the drive to seek out challenges. They have always supported
me and have given me their unconditional love, for which I am forever grateful.
x

Chapter 1
Introduction
Despite being the smallest living organisms, cells are arguably amongst the most
important subject of study for human society since they form the basis of every
living thing. They interact together through extremely complex pathways using an
incredible variety of molecules to perform tasks like signalling, transport and cell
division, to name only a few. In spite of abundant research in the field since their
discovery hundreds of years ago, much still remains to be understood about the
functions and mechanisms of cells.
The emergence of new microscopy techniques to study the many different aspects
of cells and cell functions have advanced the field of biophysics in the last decade.
However, new quantitative analysis techniques also had to be developed to keep up
with the increasing quantity of data generated and to probe the system under study
at various spatial and time scales. Fluorescence fluctuation techniques are among
the most useful and versatile tools to have been established in recent years. One
such technique is called spatio-temporal image correlation spectroscopy (STICS), a
technique that extracts information from fluorescence fluctuations in an image time
series to map the movement of fluorescently labeled particles inside a living cell.
This thesis presents the application of STICS to a new biological system: the
dynamics of secretory vesicle in dividing plant cells. These vesicles are crucial to
1

2 CHAPTER 1. INTRODUCTION
the formation of the new cell wall: their transport to the proper location in the cell
at the right time is of utmost importance to the normal development of the plant.
As well, the extension of the technique to a third spatial dimension is presented, by
application to 3D image time series. This will allow the study of protein dynamics
in cells in a 3D environment, which is a more biologically relevant environment for
tissues.
This works will review, in Chapter 1, the fundamental concepts and experimental
techniques that are essential to the work done in this thesis. Then, Chapter 2 will
introduce the STICS technique: the theoretical basis behind it along with its strengths
and limitations. Chapter 3 will show the application of STICS to dividing plant cells
and the results obtained. This will be followed by Chapter 4, where the extension
of the technique to three spatial dimensions is presented. The final chapter will give
a summary of the work and discuss potential future research projects based on this
work.
1.1 Fluorescence
The field of live cell biology took a giant leap forward when the naturally occurring
green fluorescent protein (GFP) was extracted and purified for the first time from the
jellyfish Aequorea victoria [1]. Indeed, this was a breakthrough because it became
possible to have the cell synthesize a protein of interest attached with a GFP fluo-
rescent maker by genetically splicing the GFP codon within the genetic code of the
protein. This revolutionized live cell imaging. Since its discovery, many mutants have
been made that fluoresce at different wavelengths, making fluorescent proteins a ver-
satile tool for fluorescence microscopy, one of the most widely used imaging technique
in the field of biology.
Fluorescence occurs when a molecule in a singlet excited state rapidly returns to
the ground singlet state via the emission of a photon of a longer wavelength than

1.1. FLUORESCENCE 3
that of the photon that was originally absorbed by the molecule to excite the elec-
tron [2]. Fluorescence can be explained with Jablonski diagrams, named after Alexan-
der Jablonski, who introduced them as a representation of electronic energy levels [3].
Figure 1.1: Jablonski diagram showing the different energy levels
Figure 1.1 is an example of such a diagram. Initially, the molecule is in the ground
state S0. Upon absorbing a photon of excitation light, electrons can be promoted to
the first excited singlet state S1. This process occurs very rapidly, on the femtosecond
time scale. After absorption, the molecule is in a high vibrational state of the singlet
excited state. It rapidly relaxes to the lowest vibrational state level of S1 through a
non-radiative decay process, on the time scale of 10−12 s. When the molecule drops
from the lowest vibrational excited state back to the ground state, a photon is emitted
as fluorescence emission on the nanosecond time scale. Some energy is lost by the
molecule during the vibrational relaxation, so the absorbed and emitted photons dif-
fer in wavelength. This shift of the emission to longer wavelength is called the Stokes
shift. It is essential for fluorescence microscopy, as it allows the simultaneous exci-
tation and detection of fluorescent molecules by using optical filters to block out the
excitation light and detect the weaker fluorescence emission. Each fluorescent protein
4 CHAPTER 1. INTRODUCTION
has its own characteristic absorption-emission spectrum. This allows the observation
of more than one fluorescent population at the same time, if they have sufficiently
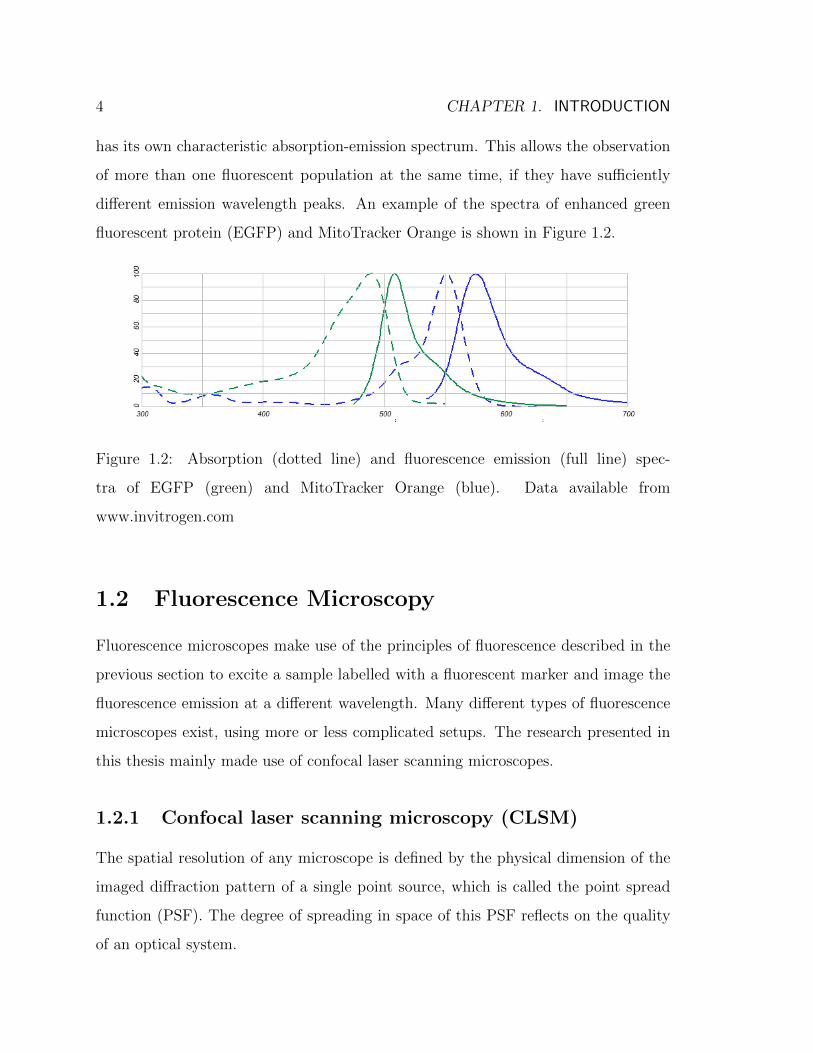
different emission wavelength peaks. An example of the spectra of enhanced green
fluorescent protein (EGFP) and MitoTracker Orange is shown in Figure 1.2.
Figure 1.2: Absorption (dotted line) and fluorescence emission (full line) spec-
tra of EGFP (green) and MitoTracker Orange (blue). Data available from
www.invitrogen.com
1.2 Fluorescence Microscopy
Fluorescence microscopes make use of the principles of fluorescence described in the
previous section to excite a sample labelled with a fluorescent marker and image the
fluorescence emission at a different wavelength. Many different types of fluorescence
microscopes exist, using more or less complicated setups. The research presented in
this thesis mainly made use of confocal laser scanning microscopes.
1.2.1 Confocal laser scanning microscopy (CLSM)
The spatial resolution of any microscope is defined by the physical dimension of the
imaged diffraction pattern of a single point source, which is called the point spread
function (PSF). The degree of spreading in space of this PSF reflects on the quality
of an optical system.

1.2. FLUORESCENCE MICROSCOPY 5
Figure 1.3: Schematic of a laser scanning confocal microscope. Image reproduced
from [5]
The principal advantage of the confocal microscope, invented in 1957 by Marvin
Minsky [4], is to use a pin-hole to spatially filter out-of-focus light for samples thicker
than the PSF. This allows the imaging of thin cross-sections of a sample since flu-
orescence is only detected from a small illumination volume defined by the spatial
extent of the PSF. This also reduces blurring due to scattered light and improves the
signal-to-noise ratio. Furthermore, a confocal laser scanning microscope can raster
scan the sample point-by-point with a focused laser beam and reconstruct the fluores-
cence image using a computer. A schematic of a laser scanning confocal microscope
is shown in Fig 1.3.
In a CLSM, the laser beam is usually scanned across a sample using a set of
mirrors. Lenses collimate the beam, which passes through a dichroic mirror before
being focused by an objective lens into a small focal volume on or within the sample.
By scanning the focal point across the sample, the fluorophores in the focus are ex-
cited sequentially as the beam is scanned across the sample. Fluorescence emitted is
collected back through the same objective (epi mode) or the dichroic mirror, which
reflects the fluorescence emission into a CCD or PMT detector. Fluorophores slightly
below or above the focal plane may also be excited. This creates background fluores-
cence which reduces the signal-to-noise ratio. This background can be decreased with

6 CHAPTER 1. INTRODUCTION
a series of pinholes. The size of the PSF at the focal point is reduced by bringing
the excitation beam through a first pinhole. The emission signal passes through a
second pinhole in order to block the out-of-focus components along the edge of the
beam (see Figure 1.3) [6].
1.3 Fluorescence Correlation Spectroscopy
Many quantitative fluorescence techniques have been developed based on fluorescence
microscopy. One of the earliest fluorescence fluctuation techniques introduced is called
fluorescence correlation spectroscopy (FCS). Such time signal-correlation techniques
were first applied in the early 1970’s at Cornell University [7]. FCS essentially looks
at the intensity fluctuations arising from fluorescent particles moving into and out of
a small stationnary laser beam focus within a sample. FCS records these fluorescence
intensity fluctuations as a time series that is then used to calculate a temporal au-
tocorrelation function (see Figure 1.4). Information about the transport dynamics,
number of molecules and chemical reaction rates, for example, can be extracted from
the magnitude and characteristic time scale of the intensity fluctuations. Although
it has high time resolution, it is important to note that FCS measurements are made
only at a single point in space. Furthermore, results depend strongly on the size and
shape of the focal volume. The time scales involved in FCS range from microseconds
to milliseconds, making it possible to probe reactions and dynamics on short time
scales within the sample. However, extended exposure times can photobleach the
fluorescent labels under study and thus prevent the study of long timescale dynam-
ics. Many extensions of FCS have been developed over the years to tackle different
challenges [8]. The main technique of interest for this thesis is an imaging analog of
FCS called image correlation spectroscopy.

1.4. IMAGE CORRELATION SPECTROSCOPY 7
Figure 1.4: Schematic representation of the image correlation spectroscopy technique.
A) The fluorescence intensity is collected from the focal volume. Intensity fluctua-
tions arise from fluorophores moving in and out of the volume. B) The fluorescence
intensity is recorded as a function of time. The temporal autocorrelation function
will be calculated from this time series and reveal information on the dynamics of the
fluorescent molecules in the system.
1.4 Image Correlation Spectroscopy
Image correlation spectroscopy (ICS) differs from FCS in that it analyzes intensity
fluctuations sampled in space across an image recorded using a fluorescence micro-
scope instead of sampling in time from a fixed position in space. It can be thought
of as its spatial equivalent. The technique was first developed to look at aggregation
states and protein surface densities in cells [9]. The technique was later extended to
temporal ICS by calculating the time autocorrelation function from an image time
series [10]. Using both spatial and temporal correlation functions, ICS can recover
information about degree of aggregation, concentration, diffusion coefficients and dy-
namics of proteins. A more rigorous introduction to ICS will be presented in the next
chapter.

Chapter 2
Spatiotemporal Image Correlation
Spectroscopy
This thesis focuses on work done using spatio-temporal image correlation spectroscopy
(STICS), a technique developed in 2005 by Hebert et al. [11] as an extension of image
correlation spectroscopy (ICS) which was briefly introduced in the previous chapter.
STICS calculates a full space-time correlation function from the intensity fluctuations
in a fluorescence microscopy image time series. The time evolution of this correlation
function gives information on the magnitude and direction of a flow of fluorescent
particles sampled in the image series. This chapter will first present an introduction
to ICS, followed by a more in-depth review of STICS.
2.1 Image Correlation Techniques
All image correlation techniques are based on one common principle: calculating
the correlation function of intensity fluctuations recorded within a microscopy image
time series. They differ in the way they treat this data. Consider the generalized
correlation function,
8

2.1. IMAGE CORRELATION TECHNIQUES 9
rab(ξ, η, τ) =〈δia(x, y, t)δib(x+ ξ, y + η, t+ τ)〉〈ia(x, y, t)〉t〈ib(x, y, t+ τ)〉t+τ
(2.1)
where ξ and η are spatial lag variables and τ is the temporal lag variable. δi(x, y, t)
represents the intensity fluctuation of a pixel at position (x,y) and at time t and is
given by
δi(x, y, t) = i(x, y, t)− 〈i〉t (2.2)
where i(x, y, t) is the intensity at pixel (x, y) recorded in the image at time t. 〈i〉tis the average intensity of that image. The angular brackets in the numerator of
Eq. 2.1 represent an ensemble average of pixel intensity fluctuations over all pairs of
images separated by τ . The subcripts a and b refer to the general case of two detection
channels. By making different approximations to this generalized correlation function,
image correlation techniques can extract various information contained within the
image time series.
The correlation techniques described in this chapter make use of the microscope
point-spread function (PSF) to correlate fluorescence intensity fluctuations over space,
time, or both. Indeed, each image from a fluorescence microscope is the result of the
spatial convolution of the PSF with the location of all fluorescent point-emitters.
For spatial correlation, it is important for the signal from each point-source to be
oversampled: the convolution causes the integrated emission to be spread over a
number of adjacent pixels in the image, to an extent dependent on the size of the
PSF and the pixel size.
2.1.1 Image Correlation Spectroscopy
Taking τ = 0 in Eq. 2.1 reduces the problem to calculating and fitting the spatial
autocorrelation function for each separate frame. This corresponds to the original
technique called image correlation spectroscopy (ICS). The correlation function of
the image frame at time t is well approximated as a two-dimensional Gaussian:

10 CHAPTER 2. SPATIOTEMPORAL IMAGE CORRELATION SPECTROSCOPY
r(ξ, η, 0)t =〈δi(x, y, t)δi(x+ ξ, y + η, t)〉
〈i(x, y, t)〉2t(2.3)
This can be fitted to a 2D Gaussian as a function of the spatial lag variables η and
ξ:
r(ξ, η, 0)t = g(0, 0, 0)t exp (−ξ2 + η2
ω20
) + g∞t (2.4)
where the fitting parameters are g∞t, the offset introduced by the finite size of
the region of analysis; ω0, the fit waist radius and g(0, 0, 0)t, the zero spatial-lags
amplitude of the image frame at time t.
From the spatial correlation function, the mean number density of independent
fluorescent particles in the focal volume (〈np〉) can be calculated and is inversely
related to the peak amplitude [9]:
limξ,η→0r(ξ, η, 0)t = g(0, 0, 0)t =1
〈np〉(2.5)
2.2 Spatio-Temporal Image Correlation Spectroscopy
The main technique of interest for this thesis, spatio-temporal image correlation spec-
troscopy (STICS), calculates the complete spatio-temporal correlation function be-
tween pairs of images within an image time series. In other words, it correlates in
space and time the fluorescence fluctuations recorded in an image time series. The
evolution of this function as a function of time lag directly reflects the underlying
transport dynamics of the fluorescently labeled particles in the imaged system. Since
the technique was first developed, it has been successful in measuring the directed
flow of α5 integrin and alpha-actinin proteins in living CHO cells [11] [5], to probe
the integrin-actin linkage during cell migration [12] and to measure vesicle dynamics
in growing plant pollen tubes [13], to name only a few examples.

2.2. SPATIO-TEMPORAL IMAGE CORRELATION SPECTROSCOPY 11
Starting from the general spatiotemporal correlation function defined in Eq. 2.1,
we can define a discrete approximation to the function. Indeed, since the correlation
function is applied to an image time series, time is a discrete variable with increments
δt, the time between each image frame. Let us consider a = b, the case of a one-
channel detection. In terms of the discrete time lag variable s, the correlation function
becomes
raa(ξ, η, s) =1
N − s
N−s∑t=1
〈δia(x, y, t)δia(x+ ξ, y + η, t+ s)〉〈ia〉t〈ia〉t+s
(2.6)
where N is the total number of images in the time series, the spatial lag variables
ξ and η are now pixel shifts in x and y, and s is the discrete time-lag representing the
number of frames separating the images being correlated. The real discrete time-lag,
∆t ∼ τ , is simply the product of the frame lag s and the sampling time per image
frame δt: ∆t = sδt. Each calculated function is averaged over the number of the
N − s pairs of images in the time series.
In practice, the correlation functions are calculated using Fast Fourier Transforms
to reduce computation time and deal with the discrete nature of the function:
r(ξ, η, s)t =FFT−1[FFT (i(x, y, t))FFT (i(x+ ξ, y + η, t+ s))∗]
〈i(x, y, t)〉〈i(x, y, t+ s)〉(2.7)
The STICS algorithm begins by calculating the time lag 0 autocorrelation function
for each frame in the image time series. This is essentially the calculation for ICS,
except that the function is averaged over every frame. Then, for every increment
of the time-lag s, the average correlation function of pairs of images in the series is
calculated. For each lag-time, the result is a 2D correlation function that can be fitted
to a 2D Gaussian of the form:
r′(ξ, η, s) = g(x(s), y(s), s) exp(−(ξ − x(s))2 + (η − y(s))2
ω20
) + g∞(s) (2.8)

12 CHAPTER 2. SPATIOTEMPORAL IMAGE CORRELATION SPECTROSCOPY
where g(x(s), y(s), s) is the peak intensity, x(s) and y(s) are the x and y coordi-
nates of the peak position, ω0 is the e−2 waist radius and g∞ is the spatial lag offset
of the correlation function. Following the evolution of the fit values as a function
of time lag can reveal information about the dynamics of the fluorescent population.
Indeed, the peak’s position is directly related to the flow of the particles. Therefore,
if the fluorescent population is undergoing directed flow, the correlation function’s
peak position will shift according to its magnitude and direction. On the other hand,
tracking the width of the peak will give information about the diffusion coefficient of
the particles. Some idealized cases are shown in Figure 2.1 calculated from computer
simulations of an image series.
Figure 2.1: STICS analysis of computer simulated images for different dynamics: i)
Directed flow. The peak moves in the direction opposite to the flow. ii) Diffusion. The
peak stays centered and progressively widens. iii) Directed flow and diffusion. The
peak divides in two parts, each presenting the behaviour of either diffusion (central
peak) or flow (translating peak). Image adapted from [11].
Once we have extracted the fit values of x(s) and y(s), we can recover the mag-
nitude and direction of the flow by calculating the velocity vectors vx and vy, given
that we know the pixel size and the time δt between each frame:

2.2. SPATIO-TEMPORAL IMAGE CORRELATION SPECTROSCOPY 13
x(δt) = −vxδty(δt) = −vyδt (2.9)
The negative sign in Eq. 2.9 comes from the fact that we multiply the FFT of the
first image with the complex conjugate of the second one in Eq. 2.7, which introduces
a minus sign.
2.2.1 Immobile Population Removal
In biological systems, the fluorescent population under study will not always be ho-
mogeneous: there will often be immobile or slow-moving populations present that can
represent an important fraction of the total fluorescence signal. This population will
correlate strongly in space and time and, for small time lags, can mask the smaller
correlation peak of a translating population in the vicinity of zero spatial lags. This
is problematic since it makes it difficult to track the peak of the correlation function
arising form the flowing population.
An approach has been developed to solve this problem [5]. It consists of removing
the DC component of each pixel’s time trace by filtering in Fourier space. This
method defines the immobile or slow-moving population as the component that does
not fluctuate over the time scale of the series. It removes the DC component by
imposing a cutoff frequency and calculating the corrected intensities. This must
be done before analyzing the image time series with STICS. The corrected pixel
intensities are given by:
i′(x, y, t) = F−1f Ft(i(x, y, t))×H 1T
(f) (2.10)
where T is the total time of the series, F−1f is the inverse Fourier transform with
respect to frequency, Ft is the Fourier transform with respect to time, and H 1T
(f) is
the Heaviside function which is zero for f < 1/T and one for f ≥ 1/T .
It is important to note that this method strongly depends on the time sampling
of the image time series. The cutoff frequency that is used to remove the immobile

14 CHAPTER 2. SPATIOTEMPORAL IMAGE CORRELATION SPECTROSCOPY
component is defined by the Heaviside function H 1T
(f) and depends on the total time
T of the series. Filtering an image time series that is shorter in time will result in a
higher cutoff frequency, meaning more signal - and thus more information - will be
lost. It is therefore important to keep this in mind when comparing STICS results
for different image time series on which the Fourier filter was first applied. If there si
a distribution of velocities, the comparison will only be meaningful if the same time
sampling was used for all image time series.
2.2.2 Experimental Limitations
With correlation techniques, there is a lower limit on the sampling required in order
to have enough averaging of the correlation function to obtain reliable and meaningful
results. The accuracy and precision of ICS was investigated [14] and it was found that
the smallest usable region of interest (ROI) for ICS analysis was a region of 16x16
pixels2 area for typical imaging with a high NA objective lens. The same limit is
applicable to STICS. However, it suffers an additional restriction: since it calculates
the velocity of particles by tracking the translating peak of the correlation function,
the latter needs to stay in the ROI for a minimum of one time lag. This imposes an
upper limit to the velocities that can be recovered using this technique. Taking the
x-axis as an example, the maximal velocity that can be measured is
vmaxx =Nx × δx2× δt
(2.11)
where Nx is the size of the region in pixels, δx is the spatial resolution in µm per
pixel and δt is the time between image frames. The same equation is applicable to
the y axis. When analyzing data, the ROI chosen would usually have a size of a
16x16 pixels2 area, in order to obtain relatively finely sampled vector maps across a
cell. Therefore, the imaging rate has to be adjusted to fit the flow rate of the system
under study. A system that reaches high velocities will have to be imaged at a much
higher time resolution than a slower system.

2.2. SPATIO-TEMPORAL IMAGE CORRELATION SPECTROSCOPY 15
The STICS analysis is performed on image time series using a software first written
in Matlab by David Kolin of the Wiseman lab in 2007 and improved by various group
members since then. Some parameters have to be set in order to perform the analysis.
The user has to choose the maximum time lag τ over which the fluctuations are
correlated. The size of the analysis ROI’s also has to be set, with a minimum size of
16x16 pixels2 area as mentioned previously. The spacing between each ROI can also
be adjusted. If a spacing smaller than the subregion size is chosen, the subregions will
overlap. If they do, the velocity vectors from adjacent regions are not independent
of each other: hence there should be a certain continuity from one region to another.
The user can set a vector mismatch threshold to filter out vectors that deviate from
their neighbour by a certain amount.
The software ensures the reliability of the results obtained by implementing other
parameters that will stop the analysis for a specific subregion if a threshold value is
reached. For example, the user can set a beam radius threshold in µm that sets the
maximum e−2 radius allowed for the correlation functions. Another parameter is the
correlation local maxima, which is the ratio of the noise correlation peak amplitudes
(off center) to the amplitude of the correlation function from the fluctuation signal.
This ensures that the noise correlation peaks are not larger than the signal correlation
peak.
An example of the STICS technique applied to real experimental data is presented
here. Figure 2.2 shows the first image of the series with the region chosen to perform
the STICS analysis overlayed. The data presented is taken from a dividing plant
cell with secretory vesicles labeled with Knolle-GFP. This biological system and its
analysis with STICS will be the subject of the next chapter.
Figure 2.3B shows the time evolution of the correlation function for the suared
16x16 pixels2 ROI in Figure 2.3A. Figure 2.4 shows the resulting flow vectors for this
region and a few neighbouring ones, which were shown in Figure 2.2, overlayed on the
first image of the time series. These vectors give quantitative information on vesicle
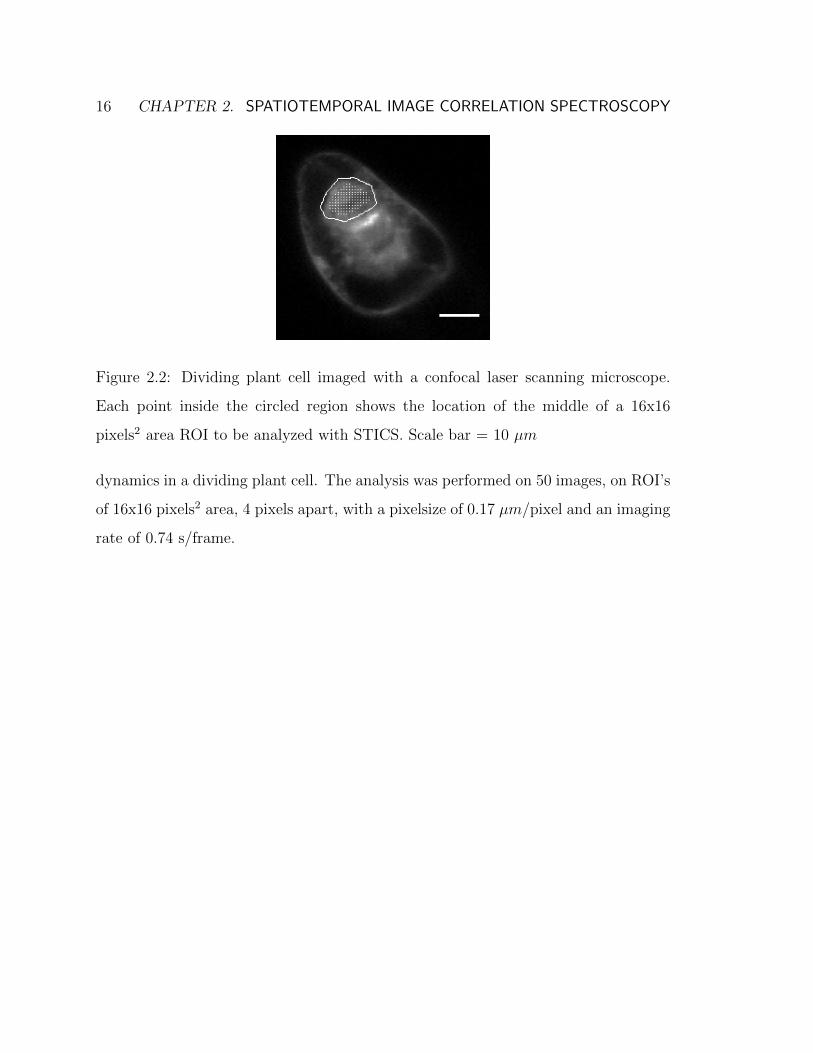
16 CHAPTER 2. SPATIOTEMPORAL IMAGE CORRELATION SPECTROSCOPY
Figure 2.2: Dividing plant cell imaged with a confocal laser scanning microscope.
Each point inside the circled region shows the location of the middle of a 16x16
pixels2 area ROI to be analyzed with STICS. Scale bar = 10 µm
dynamics in a dividing plant cell. The analysis was performed on 50 images, on ROI’s
of 16x16 pixels2 area, 4 pixels apart, with a pixelsize of 0.17 µm/pixel and an imaging
rate of 0.74 s/frame.

2.2. SPATIO-TEMPORAL IMAGE CORRELATION SPECTROSCOPY 17
Figure 2.3: A) Close-up of the circled region in Figure 2.2. The square shows the
location of a ROI on which STICS is applied. Scale bar = 10 µm. B) Evolution of
the correlation function as a function of time lag shown for the squared region. For
each time lag, the correlation function is fitted to a 2D Gaussian function and the
time evolution of the peak’s location gives the magnitude and direction of the flow of
vesicles. Scale bar = 1 µm.
Figure 2.4: Resulting vector map for the STICS analysis of the ROI’s shown in
FIgure 2.2, which gives information on vesicle dynamics in a dividing BY-2 plant cell.
Scale bar = 10 µm.

Chapter 3
Vesicle Dynamics during Plant Cell
Cytokinesis
This chapter will discuss a new application of STICS to study the phenomenon of cell
division in plant cells. Here, STICS was used to measure the dynamics of secretory
vesicles carrying new cell wall material in plant cells undergoing somatic cytokinesis.
The high density and small size of the vesicles make this a perfect candidate for STICS
measurement. This chapter will first introduce the biological process studied, the
experimental techniques are then presented, followed by the results and a discussion
on their meaning.
3.1 Biological System: Somatic Cytokinesis
In eukaryotic cells, the cell cycle is divided into two main phases: interphase and
mitosis. Interphase is the longest of the two, and is itself divided in three stages: G1,
S and G2. G1 and G2 are two growth phases, while S in the stage at which the DNA
replicates in the cell. Mitosis, or M phase, is the relatively brief but complex period
when nuclear division occurs (karyokinesis). It is divided into five stages: prophase,
metaphase, anaphase, telophase and finally cytokinesis, in which the cytoplasm of
18

3.1. BIOLOGICAL SYSTEM: SOMATIC CYTOKINESIS 19
the cell is divided in two to form two daughter cells [15].
In plant cells, cytokinesis occurs when a cell plate forms in the middle of the cell,
expands in a centrifugal direction and attaches to the parent cell wall, thus physically
dividing the cytoplasm of the cell. The material components of the cell plate, like
pectins, hemicelluloses, callose and cellulose synthesizing enzyme complexes [16], are
delivered by secretory vesicles produced by Golgi stacks in the cell that fuse with the
cell plate. Figure 3.1 represents vesicles produced by the Golgi apparatus delivering
material components to the cell plate and fusing with each other and with the cell
plate during its formation.
Figure 3.1: Sketch representing vesicles produced by the Golgi apparatus delivering
material components to the cell plate and fusing with each other and with the cell
plate during its formation. Adapted from [5]
The vesicles need to be transported rapidly and efficiently to precise locations in
the cell at specific times in order for cell division to occur normally. Indeed, failure to
do so will result in abnormal growth patterns or aberrant deposition of the cell wall
components, or in the extreme, a complete absence of both processes [17]. Knowledge
about the dynamics between the production of the vesicles by Golgi stacks and their
fusion with the cell plate - the vesicle movements within the cytoplasmic space and
their method of delivery to the cell plate - is scarce. Measurement of the vesicles

20 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
dynamics is the subject of investigation of the work presented in this chapter.
The new cell plate forms in the equatorial plane of a cytoskeletal array known as
the phragmoplast. This consists of opposing actin filaments and microtubules with
their (+) end facing the plane of division. The major function of the microtubules is
to deliver the Golgi-derived vesicles to the site of cell plate assembly in the center of
the structure, while the main role of the actin filaments is to guide the cell plate to
the proper site of fusion with the cell wall. [17]
Another distinct structure of plant cell cytokinesis is the cell plate assembly ma-
trix (CPAM). It is a transient, membrane-less cytoplasmic domain that encompasses
the vesicles fusing to form the cell plate. It is composed of various scaffolding, enzy-
matic, structural and regulatory proteins necessary for the formation of the cell plate
and these are transported to the assembly region by microtubules from the phragmo-
plast. [18] [19] The cell plate arises within the CPAM and undergoes a maturation
process throughout its formation, which takes typically approximately 30 minutes [20].
A sketch of the components of plant cytokinesis during cell plate formation is shown
in Figure 3.2.
Figure 3.2: Sketch of the different components of plant cell cytokinesis in the telophase
stage of cell division. Phragmoplast microtubules direct vesicles from Golgi stacks to
the site of fusion with the cell plate, which is expanding outwards.
The Golgi stacks that produce the secretory vesicles are known to accumulate

3.1. BIOLOGICAL SYSTEM: SOMATIC CYTOKINESIS 21
at the equatorial plane in a belt-shaped arrangement. Although the Golgi stacks
are close to the phragmoplast, they are excluded from it [21]. The mean distance
between the bulk of the Golgi stacks and the destination sites of the vesicles in the
equatorial plane is usually several micrometers and about 5-10 µm in the BY-2 cells
studied. Golgi stack dynamics and the eventual fusion of vesicles in the equatorial
plane have been extensively investigated by optical as well as transmission electron
microscopy [22] [23]. It is interesting to note that taking into account the average
diameter of a new cell wall (∼ 9 µm), the mean number of Golgi stacks per cell (∼
68 during mitosis [21]), the dimension of Golgi-derived vesicles (∼ 50 nm [20]) and
the fact that about 30% of the cell plate membrane is recycled by vesicles during
maturation [20], each Golgi stack must produce on average 310 vesicles to form the
new cell wall. It is believed that most vesicles are transported to the site of cell plate
assembly in 6 to 8 minutes (from metaphase to the early stage of karyokinesis) [24].
Such estimates show that there must be a high degree of vesicle trafficking during
that period of time.
The process of cell division occurs in different stages, which are summarized in Fig-
ure 3.3. It involves a highly complex set of carefully controlled membrane transforma-
tion and maturation events. The first stage is the late mitotic anaphase, known as the
phragmoplast initials stage. At this stage, vesicle clouds accumulate within multiple
CPAM’s in the central region of the cell. Some fuse together, forming dumbbell-
shaped cell plate intermediates. At the early stage of karyokinesis, known as the
solid phragmoplast stage, the cell plate is in the form of a tubulo-vesicular network
and is surrounded by a cocoon-like CPAM. It expands outwards during this stage.
Afterwards comes the mid-telophase stage of karyokinesis, known as the transitional
phragmoplast stage. Microtubules disassemble at the central region of the cell plate
and new ones form at the cell plate margins, directing the vesicles to these expanding
regions. The central domain of the cell plate is in the form of a tubular network, free
of CPAM, while its peripheral region is surrounded by a ring-shaped CPAM and is

22 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
still expanding outwards. The last stage, occuring during the late telophase stage of
cytokinesis, is called the ring phragmoplast stage. The cell plate has matured into a
planar fenestrated sheet and grows until it attaches to the cell wall [23]. Complete
cell division occurs in approximately 30 minutes in the commonly used BY-2 plant
cell line that was used for the cytokinesis studies presented in this thesis.

3.1. BIOLOGICAL SYSTEM: SOMATIC CYTOKINESIS 23
Figure 3.3: Summary of the stages of plant cell division. A) G2-M stage (premitotic).
B) Phragmoplast initials stage. C) Solid phragmoplast stage. D) Transitional phrag-
moplast stage. E) Ring phragmoplast stage. F) G1 stage in the daughter cells. (chr:
chromosome, cpam: cell plate assembly matrix, cw: cell wall; db: dumbbell-shaped
intermediate, gs: golgi stack, mt: microtubule, N: nucleus, ne: nuclear envelope,
pfs: planar fenestrated sheet cell plate, pgz: peripheral cell plate growth zone, pm:
plasma membrane, ppb: pre-prophase band, tn: tubular network cell plate, tvn:
tubulo-vesicular network cell plate, v: golgi-derived vesicle) Adapted from [23].

24 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
3.2 Materials and Methods
The work presented in this chapter was done in collaboration with Prof. Anja Geit-
mann’s group from the Institut de recherche en biologie vegetale, affiliated with the
Universite de Montreal. Cell culture and imaging was done by Chloe van Oostende
in the Geitmann lab. STICS analysis were performed by Dominique Guillet. Other
image analysis techniques (FRAP and FluMOS) were performed by Thomas Triplet
and Chloe van Oostende.
The tobacco BY-2 cell line was cultured in the dark (25 C, impeller velocity of 150
revolutions per minute (rpm)) [25]. Cells were subcultured weekly by transferring
1 mL of a 7-day-old culture into 50 mL of fresh medium. Stable transformations
of BY-2 cells were performed using Agrobacterium tumefaciens strain LBA4404 fol-
lowing established protocols [26]. Secretory vesicles were labeled with Knolle-GFP.
Stably transfected BY-2 cell lines were maintained independently by culturing them
every week in BY-2 medium in a 6-well plate with gentle shaking. All lines were
selected and subcultured in BY-2 medium containing carbenicillin at 150 mg/L, to
kill agrobacteria.
Confocal laser scanning microscopy (CLSM) imaging was performed with a Zeiss
LSM 510 META/LSM 5 LIVE/Axiovert 200 M system. The microscope was fitted
with a Plan Apochromat 100x/1.4 oil differential interference contrast (DIC) objec-
tive. We excited GFP using the 488 nm diode laser (100 mW) with an emission filter
LP 505. Live imaging was performed using 19% of the diode laser power. Image res-
olution was based on 1024x1024 pixels, with a pixel size of 0.17 µm and an imaging
time of 0.74 s/frame.
Unless specified otherwise, STICS analysis were performed on subsets of 50 images,
on ROI subregions of 16x16 pixels with 4 pixel spacing between adjacent ROI’s (which
means they overlapped by 12 pixels). To obtain statistics on vesicle velocities, 17
CLSM fluorescence image series of dividing BY-2 cells of length ranging 100 to 2500
images were analyzed. For STICS analysis, they were divided in subsets of either 50

3.3. RESULTS 25
or 100 images corresponding to different stages of cell division, resulting in a total of
over 50 different vector maps. The vector maps shown in this chapter are overlayed
on the first image of the time series analyzed.
3.3 Results
3.3.1 Vesicle dynamics
Knolle is a syntaxin protein specifically expressed between the mitosis phase of the
cell cycle and the end of cytokinesis. Its function is required for the formation of the
cell plate and cytokinetic vesicle fusion [27]. It is sorted in the Golgi and delivered
in vesicles to the cell plate. Several image time series of dividing cells expressing
Knolle-GFP were analyzed at different stages of the cell plate formation using the
STICS technique.
Figure 3.4: Frames from a fluorescence image time series showing the formation of
the cell plate and its reorientation over time. A) Vesicles accumulate at the center.
B) Fusion of vesicles at the site of cell plate formation. C) Reorientation of the cell
plate towards its target on the cell wall. D) The cell plate has extended to the plasma
membrane. Scale bar = 10 µm
The process of cytokinesis is relatively long, taking up to 30 minutes to complete.
Therefore, it is difficult to image the same cell over the whole process of cell plate
formation without causing significant photobleaching of the fluorescent probe. Fur-
thermore, it is not unusual for the plane of cell division to shift and reorient during

26 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
the growth of the cell plate. Figure 3.4 shows a typical example of such shifting
behaviour in a cell undergoing cytokinesis. Therefore, it is important, when apply-
ing the STICS technique, to choose a analysis time window that is shorter than the
time-scale of these large-scale morphological changes.
Many image time sub-series were analyzed using STICS. Figures 3.5 to 3.7 show the
output STICS vector maps of analysis done on image time series at different moments
of the cell division process. They were chosen because they are representative of the
general tendencies of vesicle dynamics over the course of cell plate formation that
were observed in our analysis and that will be described for each vector map.
Figure 3.5: STICS vector map of vesicle dynamics calculated over 16x16 pixel ROI’s
4 pixels apart, correlated over 50 images in time, early in the division process of the
cell. There is clear movement of vesicles towards the cell plate, as well as towards the
plasma membrane, in the direction of cell plate growth. Scale bar = 10 µm.
Figure 3.5 shows the results of the STICS analysis applied on a subset of 50 images
collected during the accumulation of vesicles at the site of fusion. No cell plate is

3.3. RESULTS 27
perceptible at this point in time. There is very clear vesicle movement towards the
center of the cell, where the cell plate formation will initiate.
Figure 3.6A shows the results of the STICS analysis applied on a subset of 50
images collected at an early stage of cell division, as can be seen from the fact that
the cell plate is still of a rather small size (about 9 µm) and has not extended very
far. The flow vectors show clear movement of the vesicles towards the cell plate, as
well as movement directed towards the plasma membrane, in the direction of growth
of the cell plate. The few vectors that can be seen pointing away from the cell plate
may be due to the recycling of cell plate membrane by vesicles.
Figure 3.6B shows the results of the STICS analysis applied on a 50 image subset
from the same image series as Figure 3.6A, 148 s later. During this time, the ”right”
side of the cell plate has almost reached the plasma membrane and as a result vesicle
trafficking on that side has dramatically decreased, as can be seen on the vector map.
It also shows vesicle dynamics towards the ”left” side of the cell plate, which is still
expanding. Although the mean velocity in Figure 3.6B is higher than in Figure 3.6A
(3.16 µm/min compared to 2.49 µm/min), the velocity of vesicles being brought to
the expanding side of the cell plate, and thus responsible for cell plate growth, is
comparable to that of the earlier time. The cell plate spans approximatively 18 µm
at this time.

28 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
Figure 3.6: STICS vector maps measured for different subsets of 50 images taken from
the same image series A) Early stage of cell division, with the cell plate measuring ∼ 9
µm. There is clear movement of vesicles towards the cell plate, as well as towards the
plasma membrane, in the direction of cell division. B) Same cell, 148 seconds later,
with the cell plate now spanning ∼ 18 µm. Vesicle dynamics are now concentrated
more towards the ”left” side of the cell plate, which is still extending. Scale bars =
10 µm.
Figure 3.7 shows the results of the STICS analysis applied on a 50 image subset
from the same image series as Figure 3.6, 389 s later. Because vesicle dynamics had
notably slowed down at that point, the time window for analysis was increased to 100
images to be able to detect the slower movements. At that moment, the ’left’ side of
the cell plate spanned 23 µm and had almost reached the parent plasma membrane.
Most vesicle dynamics have died down: the overall velocity has decreased and the
number of vectors detected above noise is much smaller. It is important to note also
that because of this decrease in the number of vectors generated under lower S/N
ratio, ’noise’ vectors are less filtered and appear in the resulting vector maps as some

3.3. RESULTS 29
of the highest velocity vectors.
Figure 3.7: STICS vector map of vesicle dynamics towards the end of cell plate
growth, with the cell plate almost spanning the whole cell diameter at ∼ 23 µm.
Vesicle dynamics have greatly slowed down. The number of detected vectors as well
as the overall velocity has notably decreased. Scale bar = 10 µm.
3.3.2 Cell plate growth rate
As a parallel investigation, live microscopy image time series were analysed using
Geitmann lab’s fluorescence morphological operators software (FluMOS) by to mea-
sure cell plate growth rate in BY-2 cells. This image analysis software was designed
as a fully automated pipeline of arithmetic, morphological and thresholding operators
and is capable of extracting the cell plate and computing its length from a series of
fluorescence images. The detection rate achieved with this program is over 95%, thus
requiring very little manual correction. This allows processing of high-throughput mi-

30 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
Figure 3.8: Measurements of cell plate growth using FluMOS. A) Example of a typ-
ical curve of cell plate diameter as a function of time. The primary and secondary
centrifugal growths can clearly be observed, as well as the early accumulation of vesi-
cles at 5 µm diameter. B) Expansion rate of the cell plate diameter. Between 5
and 15 µm (primary centrifugal growth), the cell plate grows at a higher rate than
when it has reached 15 µm (secondary centrifugal growth). Measurements come from
FluMOS analysis of 14 different cells, and the error bars represent the standard error
on these measurements.
croscopy imaging data resulting in reliable statistical analysis of the cell plate growth.
Analysis with FLuMOS revealed that cell plate growth in dividing BY-2 cells
occurs in 3 phases (see Figure 3.8A). The first one is an accumulation of vesicles on
a region of 5 µm diameter. This is followed by a phase of fast primary centrifugal
growth (PCG), at a rate of 1.2 ± 0.45 µm/s, until it reaches 15 µm (15.4 ± 1.89 µm).
Afterwards, the plane of cell division sometimes shifts and reorients itself towards its
target on the plasma membrane. The cell plate then slows down during the secondary
centrifugal growth (SCG) until all sides are fused with the parental plasma membrane.
The rate of growth averages 0.34 µm/min, but the variability of this last phase is high.
Apparently, whatever the final diameter of the cell plate (between 20 to 35 µm), the

3.4. DISCUSSION 31
second phase is less variable (34%) than the third one (47%) (see Figure 3.8B). These
results were obtained by analyzing 14 different cells, and the errors on these values
are the standard errors on the measurements.
3.4 Discussion
Secretory vesicles are directly responsible for the formation of the cell plate, by car-
rying membrane components to the site of cell plate formation and fusing together
to form the cell plate. Therefore, the speed and number of vesicles delivered defines
the diameter and growth rate of the cell plate. A slower growth rate of the cell plate
should correspond with an decrease in vesicle trafficking in the cytoplasm of the cell.
The different phases of cell plate growth that were observed in the last section should
therefore agree with vesicle dynamics detected with STICS.
Figure 3.5 shows rather slow dynamics, at an average velocity of 0.794 µm/min,
but a high number of velocity vectors detected above noise. In terms of stages of cy-
tokinesis discussed earlier, this corresponds to the phragmoplast initials stage, where
vesicle clouds accumulate within multiple CPAM’s in the central region of the cell.
Morphologically, this reflects the first stage of cell plate growth that was detectable:
an accumulation of vesicles, without any distinct cell plate growth.
Figure 3.6A illustrates elevated vesicle dynamics. Flow vectors can be seen sur-
rounding the entirety of the cell plate, which is expanding but still relatively small, at
a diameter of 9 µm. This corresponds to the solid phragmoplast stage of cell division,
where the cell plate is surrounded by a cocoon-like CPAM and is expanding outwards.
The large quantity and high velocity of vesicles transported to the cell plate cause
it to expand very rapidly, growing approximately 9 µm in 148 s (Figure 3.6B). This
corresponds to the primary centrifugal growth of the cell plate observed in Figure 3.8.
In Figure 3.6B, the main change is that the flow vectors are now concentrated on
the side of the cell plate that is still growing. This is consistent with the transitional

32 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
phragmoplast stage of cell division. At this point, microtubules disassemble at the
central region of the cell plate and new ones form at the cell plate margins, directing
the vesicles to these expanding regions.
Vesicle dynamics then attenuate, as can be seen by the reduced number of vectors
in Figure 3.7 and by the lower velocity range in that figure. This also becomes clear by
comparing cell plate growth: in 389 s, it has only expanded 5 µm. This is consistent
with the secondary centrifugal growth of the cell plate that was distinguished in
Figure 3.8.
It is important to note that the vector maps presented in this chapter are rep-
resentative of the results obtained with STICS on all cells analyzed, even though
these results come from the analysis of the same cell at different stages of cell divi-
sion. They still serve as a good example of vesicle dynamics over the course of plant
cell cytokinesis. Indeed, this specific cell was imaged for the whole duration of the
formation of its cell plate, allowing a good visualization and comprehension of the
evolution of the characteristics of vesicle dynamics during cell division in terms of
flow pattern, range of velocities and density of vesicle trafficking. The other cells
analyzed exhibited similar vesicle dynamics characteristics at corresponding stages of
cell division.
To further confirm these results, fluorescence recovery after photobleaching (FRAP)
experiments were performed by Chloe van Oostende, from the Geitmann lab. In
FRAP, a high intensity laser pulse is used to photobleach the target fluorescent par-
ticles in a small (∼ µm) region of the sample, and the rate at which the fluorescence
intensity recovers in time after the bleach pulse across this region is interpreted as
the rate of lateral transport of the labeled species back into the bleached area [28].
A parameter that can be calculated from FRAP experiments is the mobile fraction
(Mf) of fluorescent species [29]. It is calculated by comparing the fluorescence in the
bleached region after full recovery (F∞), just after bleaching (F0) and before bleaching
(Fi), using the following equation:

3.4. DISCUSSION 33
Mf = (F∞ − F0)/(Fi − F0) (3.1)
Figure 3.9A, the whole length of the cell plate was bleached and the recovery along
different regions was calculated. It is important to note that at this point, neither of
the two sides have reached the parental plasma membrane. Therefore, they should
have similar dynamics and show a similar recovery of fluorescence. This is what is
seen here: very limited recovery is seen in the central region of the cell plate, while
the two sides show a dramatically higher recovery value. Six cells were bleached and
analyzed in a similar way. The curve shown here is a typical representation of the
results obtained.
Figure 3.9B presents average results from FRAP experiments done at different
stages of cell plate formation. It appears that at the beginning of the formation of
the cell plate (CP beginning), vesicles are delivered faster than afterwards. At that
stage, the average mobile fraction (Mf) was 85%. For later stages, a distinction was
made between the expanding sides of the cell plate (growing) and its center. The
growing extremities presented a higher recovery (Mf=51%) than the central region
(Mf=36%) of the cell plate (paired T-test, two-tailed P value < 0.001). Each bar
represents the average result of the analysis of nine cells, and the error bars are
the standard error on the measurements. This is in agreement with the STICS and
FluMOS results presented and discussed above.
This work represents the first comprehensive characterization of secretory vesi-
cle dynamics during plant cell cytokinesis. This process is extremely well studied
in many regards. Studies have used methods like electron tomography and elec-
tron microscopy improved by using high-pressure freezing [23] to study the geometry
and ultra-structure of a dividing plant cell. Studies also investigated the effects of
treatment of the cell with various drugs acting as inhibitors to different components
of plant cell division [30] [31]. Vesicle pathways have also been studied using syn-
thetic vesicles and polystyrene beads, which are bigger and stiffer than endogenous

34 CHAPTER 3. VESICLE DYNAMICS DURING PLANT CELL CYTOKINESIS
ones [32] [33]. However, none of these techniques combine live imaging of dividing
plant cells and analysis of the dynamics of endogenous fluorescent secretory vesicles.
The results obtained in this chapter are unprecedented and bring important insights
on the delivery of vesicles to the forming cell plate during plant cell cytokinesis.
The vesicle dynamics measured in this chapter were obtained with STICS, a tech-
nique that is restricted to the study of dynamics in a two dimensional plane. However,
vesicle transport and delivery occur in a 3D environment. Some information is there-
fore sure to be lost in the analysis of vesicle dynamics in the 2D focal plane of the cell
plate. The analysis of data in 3D space would provide additional information on this
process and would require the development of a technique able to analyze particles as
small and dense as vesicles. A first step in this direction is taken in the next chapter.
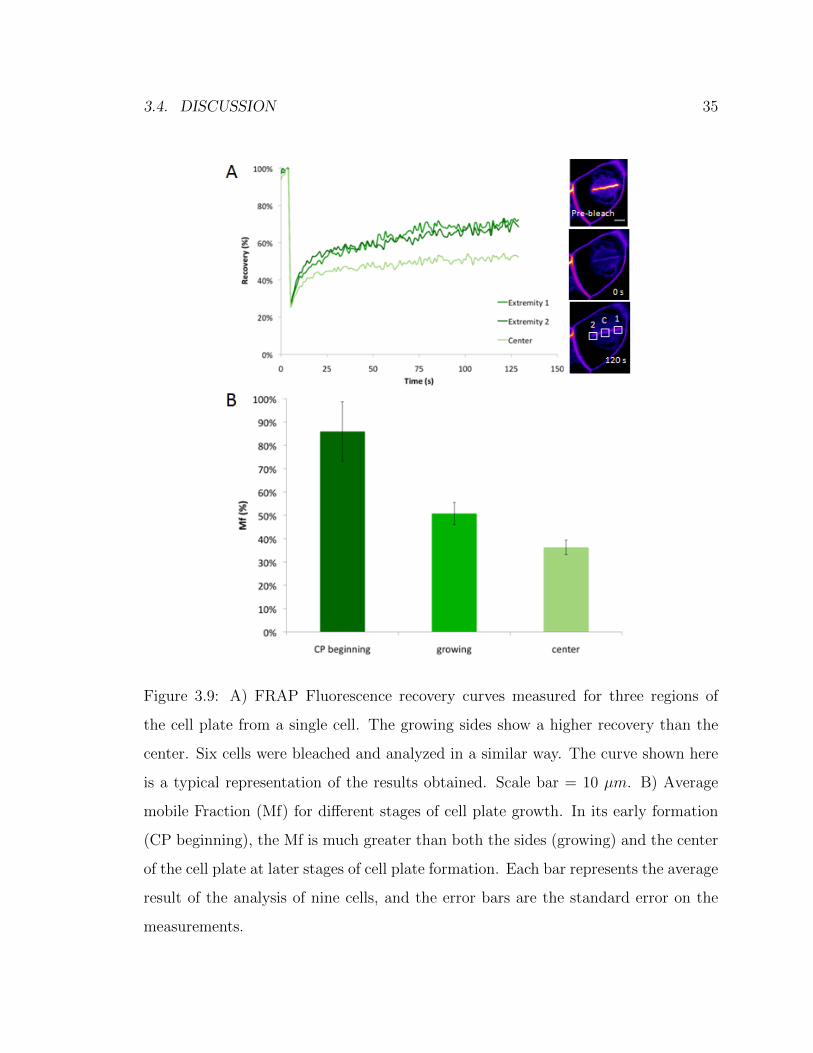
3.4. DISCUSSION 35
Figure 3.9: A) FRAP Fluorescence recovery curves measured for three regions of
the cell plate from a single cell. The growing sides show a higher recovery than the
center. Six cells were bleached and analyzed in a similar way. The curve shown here
is a typical representation of the results obtained. Scale bar = 10 µm. B) Average
mobile Fraction (Mf) for different stages of cell plate growth. In its early formation
(CP beginning), the Mf is much greater than both the sides (growing) and the center
of the cell plate at later stages of cell plate formation. Each bar represents the average
result of the analysis of nine cells, and the error bars are the standard error on the
measurements.

Chapter 4
Extension of STICS to 3 Dimensions
The STICS technique for 2D and time measurement was introduced in Chapter 2 and
applied to study secretory vesicle transport in BY-2 plant cells undergoing cytokinesis
in Chapter 3, and has been used to study dynamics in a variety of biological systems
since it was first developed [11] [12] [13]. It has been shown to be a robust and
reliable technique, given sufficient spatial and time sampling. However, STICS has
one important limitation: it is restricted to the study of dynamics in a two dimensional
plane set by the spatial resolution of the microscope, whereas the environment of
tissues and non adherent cells is inherently 3D. It is therefore crucial to be able
to study dynamics in three dimensions in order to completely understand biological
processes not restricted to membranes in these cells [34] [35]. Vesicle transport in
dividing plant cells as studied in Chapter 3 also occurs in three dimensions, although
it was only measured here in the 2D focal plane of the cell plate. The work presented
could therefore benefit from an analysis method that can study dynamics in three
dimensions. A few techniques have tried to solve that problem, namely 3D Single
Particle Tracking [36], but they are usually limited in terms of low density labeling
for spatial sampling and tracking a small number of molecules at once. STICS, as
was discussed previously, works very well in high density situations and does not
need to individually resolve each protein. An extension of the technique to a third
36

4.1. THEORY 37
spatial dimension, called 3D STICS, was developed to overcome its limitation to two
dimensions.
This chapter will first present the theory behind the development of 3D STICS,
followed by computer simulations performed to map the detection and spatial sam-
pling of the technique for ranges of parameters such as particle density of immobile
and moving populations; total number of images, particle velocity and image spacing
in the third spatial dimension.
4.1 Theory
The central idea behind 3D STICS is to extend calculation of the spatio-temporal
correlation function to times series of 3D image stacks. A 3D stack is composed of
many 2D (x and y) images separated by a constant spacing in the third (z) spatial
dimension. The z voxel (volume element in 3D) resolution is set by the spacing, in
µm, between each 2D image in z. A schematic representation of a 3D image time
series is shown in Figure 4.1.
Figure 4.1: Schematic representation of a 3D image time series, with k images in the
third spatial dimension, z, and n images in time.
The spatial resolution in x, y and z is set by the 3D PSF of the microscope. Each
2D image from a fluorescence microscope is the integrated intensity distribution from
the spatial convolution of the PSF with the location of all fluorescent point-emitters.

38 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
The degree of spreading in space of this PSF reflects on the quality of the optical
system. It is important to note that in the z optical axis, the PSF has a larger
spreading in space than in the other two dimensions for standard objective lenses.
Furthermore, even though 3D STICS calculations approximates the 3D PSF profile
as Gaussian, this is not really the case: the PSF deviates from Gaussian in z. The
spatial resolution is therefore lower in z than it is in x and y [37] [38].
In Chapter 2, we defined in Equation 2.1 a generalized fluorescence intensity fluc-
tuation correlation function for the two spatial lag variables ξ and η and one time lag
variable τ . This function can be extended to three dimensions in space by adding a
third spatial lag variable χ. The equation becomes
rab(ξ, η, χ, τ) =〈δia(x, y, z, t)δib(x+ ξ, y + η, z + χ, t+ τ)〉〈ia(x, y, z, t)〉t〈ib(x, y, z, t+ τ)〉t+τ
(4.1)
We can define a discrete approximation to the function similar to Eq. 2.6. In the
third spatial dimension, z, the discretization comes from the voxel spacing in z in the
image stack. In this case, time is a discrete variable with increments ∆T , the time
taken to image each z stack of 2D images. In terms of the discrete time lag variable
s, the correlation function becomes
raa(ξ, η, χ, s) =1
N − s
N−s∑t=1
〈δia(x, y, z, t)δia(x+ ξ, y + η, z + χ, t+ s)〉〈ia〉t〈ia〉t+s
(4.2)
where N is the total number of image stacks in the time series, the spatial lag
variables ξ, η and χ are voxel shifts in x, y and z, and s is the discrete time-lag
representing the number of image stacks separating the two z stacks being correlated.
The real time-lag, ∆T ∼ τ , is the product of the discrete time lag s, the sampling
time per image frame δt and the number of 2D images in a a stack n: ∆T = nsδt.
In practice, similarly to the 2D version of STICS, the correlation functions are
calculated using Fast Fourier Transforms for computational efficiency:

4.1. THEORY 39
r(ξ, η, χ, s)t =FFT−1[FFT (i(x, y, z, t))FFT (i(x+ ξ, y + η, z + χ, t+ s))∗]
〈i(x, y, z, t)〉〈i(x, y, z, t+ s)〉(4.3)
The 3D STICS algorithm begins by calculating the time lag 0 autocorrelation
function for each image stack in the 3D image time series. Then, for every increment
of the time lag s, the average correlation function of pairs of image stacks in the series
is calculated. For each time lag, the result is a 3D correlation function that can be
fitted to a 3D Gaussian of the form:
r′(ξ, η, χ, s) = g(x(s), y(s), z(s), s) exp(−((ξ − x(s))2 + (η − y(s))2
ω20x,y
+(χ− z(s))2
ω20z
))+g∞(s)
(4.4)
where g(x(s), y(s), z(s), s) is the peak intensity, x(s), y(s) and z(s) are the x, y
and z coordinates of the peak position, ω0x,y is the e−2 waist radius in the x and y
axes, ω0z is the e−2 waist radius in the z axis and g∞ is the long spatial lag offset of
the correlation function. It is important to note that the value of ω0z is greater than
ω0x,y . As discussed previously, the 3D PSF has a larger spreading in z than in the
other dimensions. With CLSM’s, it can be greater than two times the value of ω0x,y
depending on whether the objective lens is overfilled [38].
As in the 2D version of STICS, following the evolution of the fit values as a function
of time lag reveals information about the dynamics of the fluorescent population. If
the fluorescent population under study is undergoing directed flow, the correlation
peak will shift in x, y and z according to its magnitude and direction if the dynamics
are sampled appropriately.

40 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
4.2 Computer Simulations
4.2.1 3D simulator
Computer simulations with set inputs were employed to test the collection parameter
space of 3D STICS to investigate the feasibility of the approach. In order to do
this, an existing simulator limited to simulating 2D image time series, programmed
in MatLab by David Kolin of the Wiseman group, was extended to simulate three
dimensional image time series. This 2D simulator worked in the following way: using
a set input density supplied by the user, point particles are given random positions
inside a 2D matrix. The convolution of this matrix with a 2D Gaussian of set e−2
spatial radius is then performed to model the integration of fluorescence from a PSF
with Gaussian profile. Particle positions are recalculated for every image in the time
series, by moving each frame according to the dynamics set by the user input diffusion
coefficient and velocity [39].
The same basic principles apply to the 3D simulator that was developed. Using
the set input density (in particles/µm3) supplied by the user, point particles are
given random positions inside a 3D matrix. The convolution of this matrix with a
3D Gaussian of set e−2 spatial radius in the x and y axes and a larger e−2 spatial
radius in the z axis is performed to model the integration of fluorescence from a 3D
PSF with asymmetric Gaussian intensity profile. The equation for the convolution is
C3D = P3D ∗G3D, where C3D is the resulting 3D convolved matrix, PM is the 3D
particle position matrix and G3D is the 3D Gaussian matrix defined as
G3D = exp (−(2(x− x0)2
ω20x
+2(y − y0)2
ω20y
+2(z − z0)2
ω20z
)) (4.5)
Particle positions are recalculated for every 2D image in the time series, by moving
each frame according to the dynamics set by the user input diffusion coefficient and
velocity in the following way:

4.2. COMPUTER SIMULATIONS 41
x′ = x+ vxδt+ arand√
2Dxδt/px (4.6)
where x’ is the new particle position, x is the previous particle position, δt is the set
sampling time per image frame, vx is the set magnitude of the velocity in the x axis,
arand is a random number taken from a normal distribution with mean 0 and variance
1, Dx is the set diffusion coefficient in the x axis and px is the set voxel resolution
in the x axis. Similar equations hold for the y and z axes. Recalculating particle
positions for every 2D image models the fact that during real microscopy acquisition,
particles in a sample will have moved in the time taken to record one image, although
this does not model faster dynamics on the image frame time scale. Furthermore,
to create a z stack, each 2D image is set to sample the convolved matrix at the
appropriate value of the stack in the z dimension, effectively modeling the focal plane
of a CLSM moving in the z axis during acquisition of 3D-time data. The adjustable
parameters for 3D simulations are shown in Table 4.1. The default values represent
the typical values used for simulations, unless specified otherwise. The PSF size in
the z dimension was taken to be twice as large as the PSF in the x and y dimensions
to model the 3D PSF of a CLSM. Values of pixel size and sampling time used are
also typical of confocal laser-scanning microscopy. Most simulations presented in this
chapter are done with velocity components in the x and y dimensions set to 0 and
set to 0.01 µm/s in the z dimension, in order to characterize the capabilities of the
technique in this new dimension.
To give a general idea of the size and shape of the correlation function in the three
spatial dimensions, contour plots of correlation functions calculated from computer
simulations are presented in Figures 4.2 and 4.3. Both simulations were performed
with a PSF size of 0.4 µm in x and y and of 0.8 µm in z. Figure 4.2 shows contour
plots in the x-y (A) and x-z (B) planes of the autocorrelation function of a 3D stack of
64 images in z of 64x64 pixels, with a pixel size of 0.1 µm and a z spacing of 0.1 µm.
The effect on the 3D correlation function of the larger PSF size in the z dimension

42 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
Simulation Parameter Default Value
Number of pixels in x dimension 64 pixels
Number of pixels in y dimension 64 pixels
Number of images in z dimension 32 pixels
Spacing between images in z 0.4 µm
Number of image stacks 10
Particle density 35 particles/µm3
Pixel size 0.1 µm
PSF size in x and y 0.4 µm
PSF size in z 0.8 µm
Time between 2D images 0.1 s
Velocity in x dimension 0 µm/s
Velocity in y dimension 0 µm/s
Velocity in z dimension 0.01 µm/s
Table 4.1: Adjustable parameters for 3D simulations. Default values represent the
values used for simulations, unless specified otherwise.

4.2. COMPUTER SIMULATIONS 43
can clearly be seen. Figure 4.3 shows contour plots in the x-y (A), x-z (B) and y-z (C)
planes of the time evolution of the correlation function for 5 time lags. The 3D stacks
were composed of 32 images in z of 64x64 pixels, with a pixel size of 0.1 µm and a z
spacing of 0.4 µm. Velocity components were set to 0 µm/s, 0.1 µm/s and 0.1 µm/s
in the x, y and z dimensions respectively. The correlation function translates in the
y and z dimensions, while it stays constant in the x dimension. Also, the correlation
function appears of a similar size in all dimensions only because the z voxel number
(number of z images) and resolution (z spacing) is larger.
Figure 4.2: Multi-level contour plots of the 3D autocorrelation function of a 3D stack
of 64 images in z of 64x64 pixels, with a pixel size of 0.1 µm and a z spacing of 0.1
µm, in the A) x-y plane and B) x-z plane. The effect on the 3D correlation function
of the larger PSF size in the z dimension can clearly be seen.
4.2.2 Simulation Results
The first set of simulations presented was done as a verification that 3D STICS
has the same capacity at recovering velocities in a 2D plane as the original STICS
technique. To do this, computer simulations were performed with increasing set
velocities in x and y. The velocity magnitude in z was set to 0. 3D STICS was
applied on the resulting 3D image time series and velocity magnitudes in x, y and z
were recovered. Recovered velocity magnitudes in z were negligibly close to 0 for all

44 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
Figure 4.3: Contour plots of the time evolution of the 3D correlation function for 5
time lags in the A) x-y plane, B) x-z plane and C) y-z plane. The 3D stacks were
composed of 32 images in z of 64x64 pixels, with a pixel size of 0.1 µm and a z spacing
of 0.4 µm. Velocity components were set to vx = 0 µm/s, vy = 0.1 µm/s and vz =
0.1 µm/s.
simulations performed, as expected. To compare resulting velocity magnitudes in x
and y to STICS results, a 2D image was randomly chosen in the first z stack and an
image time series was created with this frame and the corresponding ones at similar
values of depth in the z axis in the following z stacks. This effectively created a 2D
image time series with an adjusted sampling time per image frame ∆T corresponding
to the time taken to image a z stack:
∆T = Nzδt (4.7)
where Nz is the number of 2D images in a z stack and δt is the real sampling time
per image frame. STICS was applied on this 2D image time series and the resulting
recovered velocity magnitudes in x and y were compared to those obtained with 3D
STICS. This was done for increasing values of velocities, keeping all other parameters
constant. The results are presented in Table 4.2. It shows that 3D STICS can recover
the correct values of velocity magnitude as well as the original technique for the case
of simple 2D flows.
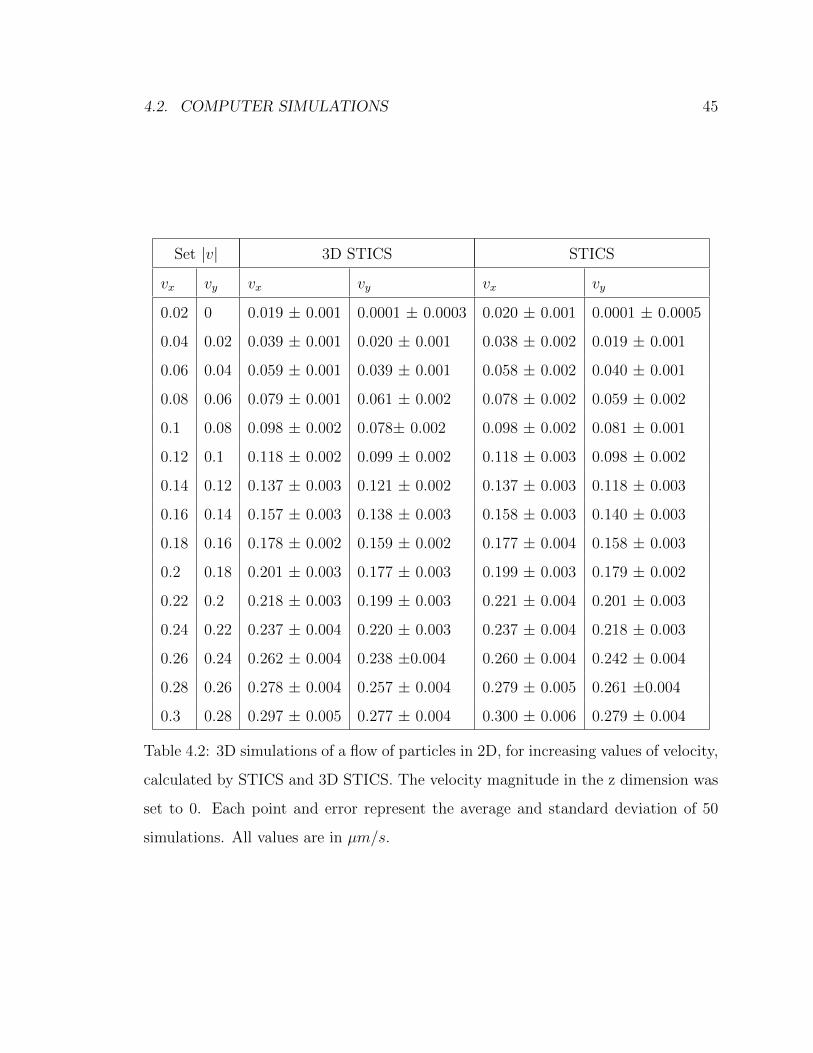
4.2. COMPUTER SIMULATIONS 45
Set |v| 3D STICS STICS
vx vy vx vy vx vy
0.02 0 0.019 ± 0.001 0.0001 ± 0.0003 0.020 ± 0.001 0.0001 ± 0.0005
0.04 0.02 0.039 ± 0.001 0.020 ± 0.001 0.038 ± 0.002 0.019 ± 0.001
0.06 0.04 0.059 ± 0.001 0.039 ± 0.001 0.058 ± 0.002 0.040 ± 0.001
0.08 0.06 0.079 ± 0.001 0.061 ± 0.002 0.078 ± 0.002 0.059 ± 0.002
0.1 0.08 0.098 ± 0.002 0.078± 0.002 0.098 ± 0.002 0.081 ± 0.001
0.12 0.1 0.118 ± 0.002 0.099 ± 0.002 0.118 ± 0.003 0.098 ± 0.002
0.14 0.12 0.137 ± 0.003 0.121 ± 0.002 0.137 ± 0.003 0.118 ± 0.003
0.16 0.14 0.157 ± 0.003 0.138 ± 0.003 0.158 ± 0.003 0.140 ± 0.003
0.18 0.16 0.178 ± 0.002 0.159 ± 0.002 0.177 ± 0.004 0.158 ± 0.003
0.2 0.18 0.201 ± 0.003 0.177 ± 0.003 0.199 ± 0.003 0.179 ± 0.002
0.22 0.2 0.218 ± 0.003 0.199 ± 0.003 0.221 ± 0.004 0.201 ± 0.003
0.24 0.22 0.237 ± 0.004 0.220 ± 0.003 0.237 ± 0.004 0.218 ± 0.003
0.26 0.24 0.262 ± 0.004 0.238 ±0.004 0.260 ± 0.004 0.242 ± 0.004
0.28 0.26 0.278 ± 0.004 0.257 ± 0.004 0.279 ± 0.005 0.261 ±0.004
0.3 0.28 0.297 ± 0.005 0.277 ± 0.004 0.300 ± 0.006 0.279 ± 0.004
Table 4.2: 3D simulations of a flow of particles in 2D, for increasing values of velocity,
calculated by STICS and 3D STICS. The velocity magnitude in the z dimension was
set to 0. Each point and error represent the average and standard deviation of 50
simulations. All values are in µm/s.

46 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
The second set of simulations was done to verify that 3D STICS achieves its goal of
recovering velocities in the third spatial dimension and to test the range of velocities
at which it obtains precise and accurate results. Setting velocity magnitudes in x and
y to 0, 3D STICS was applied on simulations of flowing populations with increasing
values of velocities in the z dimension, keeping all other parameters constant. The
results are shown in Figure 4.4.
Figure 4.4: 3D STICS results for computer simulations with increasing values of ve-
locity magnitudes in the z dimension. Velocity magnitudes in the x and y dimensions
were set to 0. Each point and error bars represent the average and standard deviation
of 50 simulations.
As can be seen, the technique performs rather well for smaller values of velocities,
but results start to deviate at higher values of velocities. This is because as the
velocity magnitude increases, the displacement of particles in the z dimension within
a z stack in the time taken to image it becomes non negligible. With the parameters

4.2. COMPUTER SIMULATIONS 47
used for the simulations shown in Table 4.1, an image stack takes 3.2 seconds to
image. At a velocity of 0.5 µm/s, the particles move 1.6 µm in the time it takes to
image a single stack. As the correlation function is calculated from such an image
time series, it will be more spread out in the axis of movement. Figure 4.5 shows the
autocorrelation function of a 3D stack composed of 32 images of 64x64 pixels, with a
pixel size of 0.1 µm and a z spacing of 0.4 µm, without flow (A) and with a flow in the
z direction of 0.5 µm/s (B). The spreading of the correlation function is due to the
movement of particles. For higher time lags, the correlation function in Figure 4.5B
will quickly move past the edge of the correlation window, at which point part of it
will reappear on the other side. The fit values of such a correlation function to a 3D
Gaussian will be incorrectly calculated, resulting in an incorrect peak position. This
effect is lessened as the imaging rate is increased. Current high-speed microscopes,
which can record hundreds of images in a second, will not be as affected by this error,
although there will always be a sampling limit.
Figure 4.5: Spatial spreading of the correlation function. A) x-z plane of the autocor-
relation function of a 3D stack composed of 32 images of 64x64 pixels, with a pixel
size of 0.1 µm and a z spacing of 0.4 µm, without flow. B) x-z plane of the autocor-
relation function of a similar 3D stack with particles flowing in the z dimension at a
rate of 0.5 µm/s. The spreading of the correlation function is due to the movement
of particles.
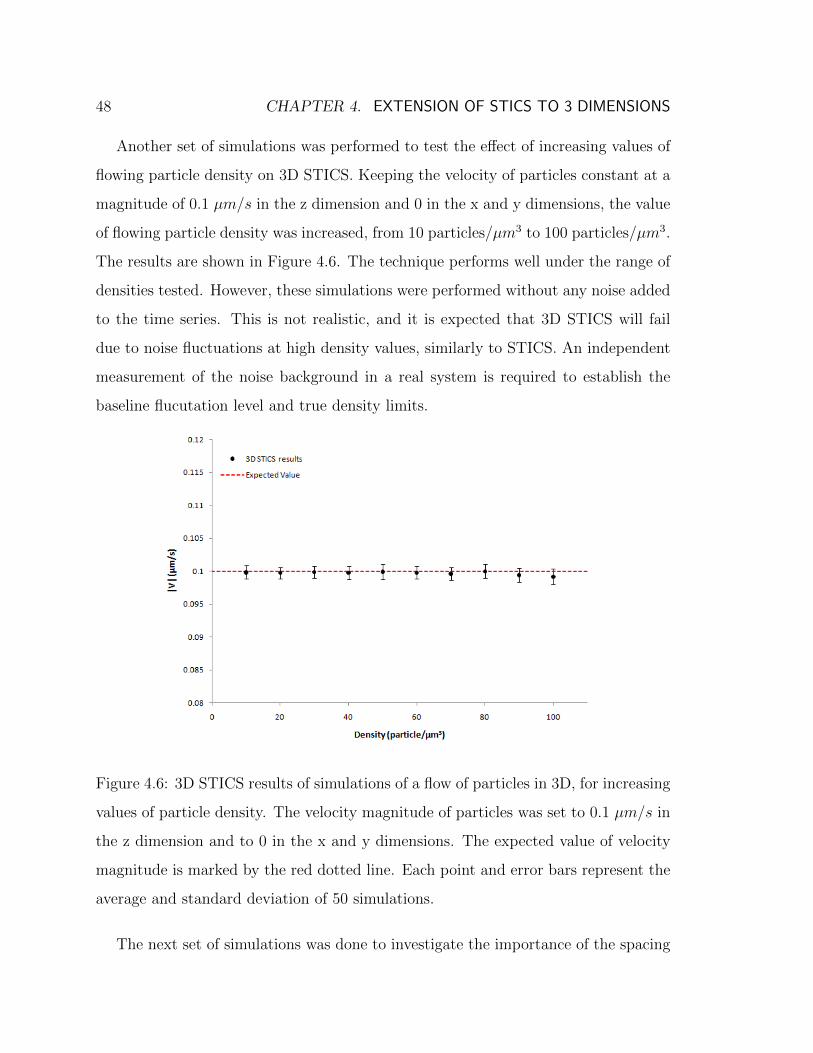
48 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
Another set of simulations was performed to test the effect of increasing values of
flowing particle density on 3D STICS. Keeping the velocity of particles constant at a
magnitude of 0.1 µm/s in the z dimension and 0 in the x and y dimensions, the value
of flowing particle density was increased, from 10 particles/µm3 to 100 particles/µm3.
The results are shown in Figure 4.6. The technique performs well under the range of
densities tested. However, these simulations were performed without any noise added
to the time series. This is not realistic, and it is expected that 3D STICS will fail
due to noise fluctuations at high density values, similarly to STICS. An independent
measurement of the noise background in a real system is required to establish the
baseline flucutation level and true density limits.
Figure 4.6: 3D STICS results of simulations of a flow of particles in 3D, for increasing
values of particle density. The velocity magnitude of particles was set to 0.1 µm/s in
the z dimension and to 0 in the x and y dimensions. The expected value of velocity
magnitude is marked by the red dotted line. Each point and error bars represent the
average and standard deviation of 50 simulations.
The next set of simulations was done to investigate the importance of the spacing

4.2. COMPUTER SIMULATIONS 49
between images in a 3D stack and the number of images used to create the z stacks.
With the original version of STICS, a criterion for accurate and precise results was
sufficient spatial resolution and sampling. There is a lower limit on the sampling
required in order to have enough averaging of the correlation function to obtain
reliable and meaningful results. It was found that the smallest usable region of interest
(ROI) for ICS and STICS analysis was a region of 16x16 pixels2 area for typical
imaging with a high NA objective lens [14]. A similar condition should exist on
the sampling required in the z dimension. Also, as was discussed in Chapter 2,
it is important for spatial correlation for the signal from each point-source to be
oversampled: the spatial convolution of the PSF with the location of all fluorescent
point-emitters causes the integrated emission to be spread over a number of adjacent
pixels in the image, to an extent dependent on the size of the PSF and the pixel
size. The same principle applies to 3D STICS for the third spatial dimension. To get
oversampling in the z dimension, it is important to image 3D stacks with a z spacing
smaller than the diameter of the PSF in z.
Keeping the velocity of particles constant at a magnitude of 0.1 µm/s in the z
dimension and 0 in the x and y dimensions, the z spacing was varied from 0.2 µm to
2 µm and tested for z stacks of 16, 32 and 48 images. The results are summarized in
Table 4.3, where ∆z is the z spacing between images in µm and ∆N z is the number of
images in a z stack. Each value shown is the average of the results of 50 simulations
and the error is the standard deviation on these results. These show that 3D STICS
fails to be accurate for large z spacing, as expected, because the signal from each
point-source is not oversampled enough in z. The technique also has lower accuracy
for z stacks of 16 images because there is less averaging of the correlation function at
that size: half the number of fluctuations are expected to be sampled compared to z
stacks of 32 images.
To illustrate the effect of sampling in three spatial dimensions and how it is dif-
ferent in z because of the larger PSF size, simulations of particles flowing with equal

50 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
HHHHHHH
HH∆z
∆N z16 32 48
0.2 0.083±0.004 0.098±0.002 0.102±0.001
0.4 0.092±0.003 0.099±0.001 0.1016±0.0006
0.6 0.100±0.004 0.100±0.001 0.1001±0.0009
0.8 0.102±0.004 0.098±0.001 0.1017±0.0005
1.0 0.095±0.006 0.102±0.002 0.0996±0.0009
1.2 0.10±0.01 0.09±0.01 0.104±0.005
1.4 0.10±0.02 0.09±0.01 0.108±0.008
1.6 0.12±0.03 0.091±0.008 0.08± 0.01
1.8 0.12±0.03 0.094±0.006 0.08±0.01
2.0 0.14±0.04 0.096±0.004 0.09±0.01
Table 4.3: 3D STICS simulation analysis results for the measured z-velocity (set value
vz = 0.1 µm/s). ∆z is the spacing between images in µm and ∆N z is the number
of images in a stack. The velocity magnitude of particles was set at 0 in the x and y
dimensions. Each value and error represent the average and standard deviation of 50
simulations.

4.2. COMPUTER SIMULATIONS 51
velocity components in all three dimensions were performed. The velocity of flow-
ing particles was increased from vx = vy = vz = 0.05 µm/s to vx = vy = vz = 0.5
µm/s, in 3D stacks of 32 images, 32x32 pixels, with a pixel size of 0.4 µm and a z
spacing of 0.4 µm. The only difference in the resulting recovered velocity components
should therefore come from the difference in sampling in z compared to x and y. The
principle behind STICS and 3D STICS is to sample fluctuations in space and time,
with the PSF acting as the correlator. In 2D, the sampling of spatial fluctuations
depends on how many beam areas are sampled per ROI. In 3D, the number of PSF
fluctuation volumes sampled will depend on the direction, since it is the PSF is lager
in z. However, there will also be a temporal component: the particles will not cross
the PSF as fast in z as in x and y. The results presented in Figure 4.7 clearly show
the effect of the larger PSF size in z. For smaller velocity values, 3D STICS can
recover the set velocity component in z as well as in x and y, indicating that the lower
sampling in z is compensated by the fact that particles take more time to cross the
PSF in that direction. However, for higher velocity values, this effect is lessened by
the fast flow of the particles, and recovered velocity components in z start to deviate
due to lower sampling. Each point and error bars in the figure represent the average
and standard deviation of 50 simulations.
The final set of simulations performed was done to investigate velocity recovery in
the presence of an immobile population. As mentioned in Chapter 2, there will often
be immobile or slow-moving populations present that can represent an important
fraction of the total fluorescence signal. This population will correlate strongly in
space and time and, for small time lags, can mask the smaller correlation peak of a
translating population in the vicinity of zero spatial lags, making it difficult to track
the correlation function’s peak location in order to calculate velocities. Therefore, a
filter is needed to remove the contribution of the immobile population. In this 3D
case, this can be done in a similar way as with STICS, by removing the DC component
of each pixel’s time trace by filtering in Fourier space. The corrected pixel intensities

52 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
Figure 4.7: 3D STICS results of simulations with increasing and equal velocity com-
ponents in all three spatial dimensions in 3D stacks of 32 images, 32x32 pixels, with
a pixel size of 0.4 µm and a z spacing of 0.4 µm. The effect of the larger PSF size
in z (0.8 µm compared to 0.4 µm) can clearly be seen. Each point and error bars
represent the average and standard deviation of 50 simulations.
are given by:
i′(x, y, z, t) = F−1f Ft(i(x, y, z, t))×H 1T
(f) (4.8)
where T is the total time of the series, F−1f is the inverse Fourier transform with
respect to frequency, Ft is the Fourier transform with respect to time, and H 1T
(f) is
the Heaviside function which is zero for f < 1/T and one for f ≥ 1/T .
The results shown in Figure 4.8 present the velocities recovered by 3D STICS for
simulated image time series containing an immobile population of increasing density
and a flowing population of set constant density. The immobile population density
was increased from 10 particles/µm3 to 100 particles/µm3. The velocity of the moving

4.2. COMPUTER SIMULATIONS 53
particles was set at a constant magnitude of 0.1 µm/s in the z dimension and 0 in
the x and y dimensions. The density of the flowing population is kept constant at
a value of 35 particles/µm3. Once the image series is filtered, 3D STICS is able to
recover the flowing population’s velocity, with no significant change even when 75%
of the total population is immobile.
Figure 4.8: 3D STICS results of simulations with flowing and filtered immobile pop-
ulations, for increasing values of immobile population particle density. The velocity
magnitude of flowing particles was kept at a constant value of 0.1 µm/s in the z di-
mension and at a value of 0 in the x and y dimensions. The expected value of velocity
magnitude is marked by the red dotted line. Each point and error bars represent the
average and standard deviation of 50 simulations.
From the computer simulations shown above, it is clear that 3D STICS performs
well under parameters consistent with current CLSM microscopes. Results could even
be improved by using faster imaging systems such as spinning-disk microscopes, ca-
pable of imaging hundreds of images in a second. However, faster imaging rates can
result in significant photobleaching, which could be incorporated into future simula-

54 CHAPTER 4. EXTENSION OF STICS TO 3 DIMENSIONS
tions. 3D STICS was only tested so far with computer-simulated image time series.
Before the technique can be applied in real studies, it will have to be tested with
real control experiments and the results obtained compared to those of an estab-
lished technique, like 3D single particle tracking at labeling densities accessible to
both. There is a great number of potential avenues to be explored with 3D STICS.
Other biological processes that were previously studied with STICS could be ideal
test subjects for the new 3D version of the technique, such as vesicle transport and
delivery dynamics in dividing plant cells. Intracellular endosomal delivery and traf-
ficking would be another candidate system for this method. Although some details
of 3D STICS remain to be explored, the work presented in this chapter represents
a stepping stone in the development of a new 3D correlation technique. With this
powerful tool, more biologically relevant and complex systems will be accessible and
perhaps reveal new details on molecular transport mechanisms that are essential to
cell function.

Chapter 5
Conclusion
New applications developments to the spatio-temporal image correlation spectroscopy
(STICS) technique were presented in this thesis. Chapter 3 illustrated its application
for measurements of secretory vesicle dynamics carrying new cell wall material inside
dividing tobacco BY-2 cells. Due to their small size and high density, quantitative
information on the dynamics of these vesicles had always been difficult to obtain.
The STICS technique was successful in measuring velocity maps for the secretory
vesicles from fluorescence microscopy image time series. The values of the velocity
flow obtained varied depending on the stage of cell division. In parallel, the growth
rate of the cell plate was measured using fluorescence morphological operators soft-
ware (FluMOS). It was discovered that the cell plate passed through three specific
phases: an accumulation of vesicles at the site of fusion, followed by a period of high
growth rate until the cell plate reached an approximate diameter of 15 µm, and fi-
nally another slower period of growth until the cell plate reached and attached to
the parental plasma membrane. These stages match well with the dynamics of the
vesicles that were characterized using STICS: slow velocities as the vesicles accumu-
late at the center, rapid and active trafficking as the cell plate first starts expanding,
and slower dynamics focused on the growing extremities as it finishes its matura-
tion. This study sheds new light on the rate of vesicle transport to the cell plate and
55

56 CHAPTER 5. CONCLUSION
how it evolves throughout cell plate formation. Previous studies have used electron
tomography [23], drugs, inhibitors [30], beads and synthetic vesicles to study this
process [32], but none combined live imaging of dividing plant cells and analysis of
the dynamics of endogenous fluorescent secretory vesicles. This work represents the
first comprehensive characterization of secretory vesicle dynamics during plant cell
cytokinesis.
Chapter 4 presented the extension of STICS to three spatial dimensions. Although
the original STICS technique has proven useful for a number of membrane biophysics
applications, it is restricted to the study of dynamics in two dimensions, which is
not always biologically relevant. To overcome this restriction, an extension of the
technique to a third spatial dimension was developed. Its capabilities and limits
were extensively tested using computer simulations. The new technique was first
compared to its 2D predecessor by testing its ability to calculate velocities in a 2D
plane. Then, its efficacy at recovering velocities in the third spatial dimension was
tested. The effects of the densities of the immobile and moving population, as well as
the number of images and resolution in the third spatial dimension was investigated.
The simulations done in this chapter revealed that the technique performs well under
parameters easily achieved with current high-speed microscopes. However, before
it can be applied to study biological systems, the technique remains to be tested
on controlled experimental data and compared with the results of an established
technique such as 3D single particle tracking.
There is a great number of potential avenues to be explored with 3D STICS. One of
them ties in with Chapter 3 of this thesis: the technique could be used to study vesicle
dynamics in three dimensions in plant cells. Vesicles and other organelles do of course
have three degrees of translational freedom in the three dimensional space of the cell.
Therefore, in order to obtain a more complete picture of intracellular trafficking, the
acquisition of 3D image time series would be needed in adherent cell tissue culture.
Other biological systems that were previously studied with STICS could be ideal

57
test subjects for the new 3D version of the technique, such as a study of adhesion
complexes in cells migrating inside a 3D matrix, which is closer to the tissue context
for cell migration [34] [35]. Although some details of 3D STICS remain to be explored,
the work presented in this thesis represents a stepping stone in the development of a
new 3D correlation technique. With this new powerful tool, more biologically relevant
and complex systems will be accessible to researchers and perhaps reveal new details
on molecular transport mechanisms that are essential to cell function.

Bibliography
[1] O. Shimomura, F. Johnson, and Y. Saiga, “Extraction, purification and prop-erties of aequorin, a bioluminescent protein from the luminous hydromedusan,aequorea,” J. Cell. Comp. Physiol., vol. 59, 1962.
[2] B. Valeur, Molecular Fluorescence: Principles and Applications. Wiley, 2002.
[3] A. Jablonski, “ber den Mechanismus der Photolumineszenz von Farbstoffphos-phoren,” Zeitschrift Fr Physik, vol. 94, pp. 38–46, 1935.
[4] M. Monsky, “Memoirs on inventing the confocal scanning microscope,” Scanning,vol. 10(4), pp. 128–138, 1988.
[5] B. Hebert, Spatio-temporal Image Correlation Spectroscopy: Development andImplementation in Living Cells. PhD thesis, Physics Department, McGill Uni-versity, 2006.
[6] S. Paddock, “Confocal laser scanning microscopy,” BioTechniques, vol. 27,pp. 992–1004, 1999.
[7] D. Magde, E. Elson, and W. W. Webb, “Thermodynamic fluctuations in a re-acting system: measurement by fluorescence correlation spectroscopy,” PhysicalReview Letters, vol. 29(11), pp. 705–708, 1972.
[8] E. Haustein and P. Schwille, “Fluorescence correlation spectroscopy: novel vari-ation of an established technique,” Annu Rev Biophys Biomol Struct, vol. 36,pp. 151–169, 2007.
[9] N. Petersen, P. Hddelius, P. W. Wiseman, O. Seger, and K.-E. Magnusson,“Quatitation of membrane receptor distributions by image correlation spec-troscopy: concept and application,” Biophysical Journal, vol. 65, pp. 1135–1146,1993.
[10] P. W. Wiseman and N. O. Petersen, “Image correlation spectroscopy: optimiza-tion for ultrasensitive detection of preexisting platelet-derived growth factor-betareceptor oligomers on intact cells,” Biophysical Journal, vol. 76(2), pp. 963–977,1999.
58

BIBLIOGRAPHY 59
[11] B. Hebert, S. Constantino, and P. W. Wiseman, “Spatiotemporal image cor-relation spectroscopy (STICS) theory, verification, and application to proteinvelocity mapping in living CHO cells,” Biophysical Journal, vol. 88, pp. 3601–3614, 2005.
[12] C. Brown, B. Hebert, D. Kolin, J. Zareno, A. Horwitz, and P. W. Wiseman,“Probing the integrin-actin linkage using high-resolution protein velocity map-ping,” Journal of Cell Science, vol. 119, pp. 5204–5214, 2006.
[13] J. Bove, B. Vaillancourt, J. Kroeger, P. K. Hepler, P. W. Wiseman, and A. Geit-mann, “Magnitude and direction of vesicle dynamics in growing pollen tubesusing spatiotemporal image correlation spectroscopy (STICS) and fluorescencerecovery after photobleaching (FRAP),” Plant Physiology, vol. 147(4), pp. 1646–1658, 2008.
[14] S. Constantino, J. Comean, D. Kolin, and P. W. Wiseman, “Accuracy and dy-namic range of spatial image correlation spectroscopy and cross-correlation spec-troscopy,” Biophysical Journal, vol. 89, pp. 1251–1260, 2005.
[15] G. M. Cooper and R. E. Hausman, The Cell: a molecular approach (5th Edition).Sinauer Associates Inc., 2009.
[16] D. Verma, “Cytokinesis and building of the cell plate in plants,” Annu Rev PlantPhysiol Plant Mol Biol, vol. 52, pp. 751–784, 2001.
[17] T. M. Molchan, A. H. Valster, and P. K. Hepler, “Actomyosin promotes cellplate alignment and late lateral expansion in Tradescantia stamen hair cells,” JCell Biol, vol. 130, pp. 1345–1357, 1995.
[18] A. L. Samuels, T. H. J. Giddings, and S. L. A., “Cytokinesis in tobacco BY-2and root tip cells: a new model of cell plate formation in higher plants,” Planta,vol. 214(5), pp. 683–693, 2002.
[19] M. S. Otegui and S. L. A., “Electron tomographic analysis of post-meiotic cytoki-nesis during pollen development in Arabidopsis thaliana,” Planta, vol. 218(4),pp. 501–515, 2004.
[20] J. M. Seguı-Simarro, J. R. Austin, E. A. White, and L. A. Staehelin, “Electrontomographic analysis of post-meiotic cytokinesis during pollen development inArabidopsis thaliana,” Plant Cell, vol. 16, pp. 836–856, 2004.
[21] J. M. Seguı-Simarro and L. A. Staehelin, “Cell cycle-dependent changes in Golgistacks, vacuoles, clathrin-coated vesicles and multivesicular bodies in meristem-atic cells of Arabidopsis thaliana: quantitative and spatial analysis,” Planta,vol. 223, pp. 223–236, 2006.
[22] A. Nebenfur, “Organelle dynamics during cell division,” Plant Cell Monogr,vol. 9, pp. 195–206, 2007.

60 BIBLIOGRAPHY
[23] M. S. Seguı-Simarro, Otegui, J. M., J. R. Austin, and L. A. Staehelin, “Electrontomographic analysis of post-meiotic cytokinesis during pollen development inArabidopsis thaliana,” Plant Cell Monogr, vol. 9, pp. 251–287, 2008.
[24] K. Ueda, S. Sakaguchi, F. Kumagai, S. Hasezawa, H. Quader, and U. Kristen,“Development and disintegration of phragmoplasts in living cultured cells ofa GFP::TUA6 transgenic Arabidopsis thaliana plant,” Protoplasma, vol. 220,pp. 111–118, 2003.
[25] T. Nagata and F. Kumagai, “Plant cell biology through the window of the highlysynchronized tobacco BY-2 cell line,” Methods Cell Sci, vol. 21(2-3), pp. 123–127,1999.
[26] P. Dhonukshe and T. W. J. J. Gadella, “Alteration of microtubule dynamicinstability during preprophase band formation revealed by Yellow FluorescenceProtein-CLIP170 microtubule plus-end labeling,” Plant Cell, vol. 15(3), pp. 597–611, 2003.
[27] M. H. Lauber, I. Waizenegger, T. Steinmann, H. Schwarz, U. Mayer, I. Hwang,W. Lukowitz, and G. Jurgens, “The Arabidopsis KNOLLE protein is acytokinesis-specific syntaxin,” J Cell Biol, vol. 139(6), pp. 1485–1493, 1997.
[28] M. Edidin, Y. Zagyansky, and T. J. Lardner, “Measurement of membrane proteinlateral diffusion in single cells,” Science, vol. 191, pp. 466–468, 1976.
[29] A. J. Reits and J. J. Neefjes, “From fixed to FRAP: measuring protein mobilityand activity in living cells,” Nature Cell Biol, vol. 3, pp. E145–E147, 2001.
[30] T. Higaki, N. Kutsuna, T. Sano, and S. Hasezawa, “Quantitative analysis ofchanges in actin microfilament contribution to cell plate development in plantcytokinesis,” BMC Plant Biol, vol. 8, p. 80, 2008.
[31] N. Kutsuna, F. Kumagai, M. H. Sato, and S. Hasezawa, “Quantitative analysisof changes in actin microfilament contribution to cell plate development in plantcytokinesis,” Plant Cell Physiol, vol. 44(10), pp. 1045–1054, 2003.
[32] A. Esseling-Ozdoba, J. W. Vos, A. A. M. van Lammeren, and A. M. C. Emons,“Synthetic lipid (DOPG) vesicles accumulate in the cell plate region but do notfuse,” Plant Physiol, vol. 147, pp. 1699–1709, 2008.
[33] A. Esseling-Ozdoba, R. A. Kik, A. A. M. van Lammeren, J. M. Kleijn, andE. A. M. C., “Flexibility contra stiffness: the phragmoplast as a physical barrierfor beads but not for vesicles,” Plant Physiol, vol. 152(2), pp. 1065–1072, 2010.
[34] D. J. Webb and A. F. Horwitz, “New dimensions in cell migration,” Nature CellBiol, vol. 5, pp. 690–692, 2003.

BIBLIOGRAPHY 61
[35] T. Jacks and R. A. Weinberg, “Taking the study of cancer cell survival to a newdimension,” Cell, vol. 111, pp. 923–925, 2002.
[36] I. M. Peters, B. G. De Grooth, J. M. Schins, C. G. Figdor, and J. Greve,“Three-dimensional single-particle tracking with nanometer resolution,” Rev SciInstrum, vol. 69, pp. 2762–2766, 1998.
[37] R. W. Cole, T. Jinadasa, and C. M. Brown, “Measuring and interpreting pointspread functions to determine confocal microscope resolution and ensure qualitycontrol,” Nature Protocols, vol. 6, pp. 1929–1941, 2011.
[38] S. T. Hess and W. W. Webb, “Focal volume optics and experimental artifacts inconfocal fluorescence correlation spectrsocopy,” Biophysical Journal, vol. 83(4),pp. 2300–2317, 2002.
[39] D. L. Kolin, k-Space Image Correlation Spectroscopy: Theory, Verification, andApplications. PhD thesis, Department of Chemistry, McGill University, 2008.





























