Embed Size (px)
Citation preview

ISSN 0012�5008, Doklady Chemistry, 2011, Vol. 439, Part 1, pp. 194–199. © Pleiades Publishing, Ltd., 2011.Original Russian Text © K.K. Kalninsh, E.F. Panarin, 2011, published in Doklady Akademii Nauk, 2011, Vol. 439, No. 2, pp. 205–210.
194
The elementary step of the polycondensation ofaromatic diamines and dianhydrides proceeds throughplanar reaction complexes [1] with an extremely lowenergy (less than 1 eV) caused by a specific hydrogenbond in the electronically excited state [2]. The energyof this stage, which has the meaning of the activationenergy Ea, is rather sensitive to the solvent nature anddecreases with an increase in the polarity of amedium. The solvation effect can be theoreticallyestimated by the self�consistent reaction field(SCRF) method. The original computational pro�cedure, implemented in the Gaussian09W programpackage, calculates the energy of a polar moleculein a solvent reaction field.
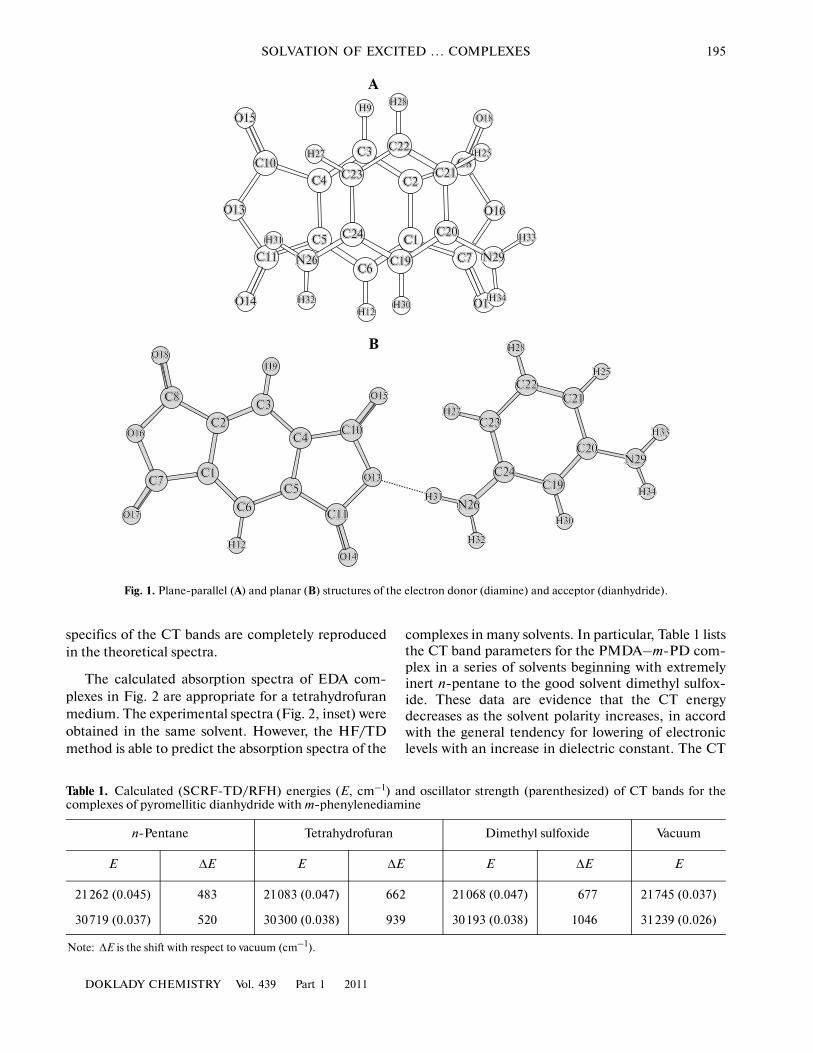
In this paper, we report one of the first attempts toestimate the solvent effect on the excited electron�transfer reaction states, for the complexes of pyromel�litic and dichloropyromellitic dianhydrides (PMDAand DCPMDA, respectively) with m� and p�phe�nylenediamines (m�PD and p�PD, respectively) as anexample, by the SCRF iteration procedure [3] and,thus, to quantitatively describe the solvation effects. Inaddition, absorption spectra of diamine–dianhydridecharge�transfer (CT) complexes were simulated withinclusion of the dielectric medium effect. Figure 1shows the structures of two types of electron donor–acceptor (EDA) diamine–dianhydride complexes: theplane�parallel structure (A) active in optical absorp�tion or emission spectra and planar structure (B)determining the occurrence of thermochemical reac�tions. The latter complexes are characterized byclearly pronounced conjugation between the electrondonor and acceptor through the shared hydrogenatom.
Quantum�chemical calculations were performedby the RHF and ROHF/D95 ab initio methods with
the Gaussian09W program package [3]. Details of cal�culations of molecules in the ground (S0) and excited(T1) states were described in [4]. The absorption spec�tra of the EDA complexes were calculated by the time�dependent (TD) Hartree–Fock method.
A satisfactory correspondence between the calcu�lated and experimental absorption spectra in the rangeof CT electronic transitions (Fig. 2) was obtained bythe TD�RHF/D95 method for the radical ion struc�ture with the plane�parallel arrangement of the donorand acceptor molecules at a distance of ~3.3 Å fromeach other. It has been precisely this distance that hasbeen found in crystalline EDA complexes by X�raycrystallography. Mutual orientation of the donor andacceptor in the complex, which has a considerableeffect on the spectral characteristics, was chosen tak�ing into account data of the semiempirical ZINDO/Smethod, which requires minimal CPU time for mod�eling of the complex structure.
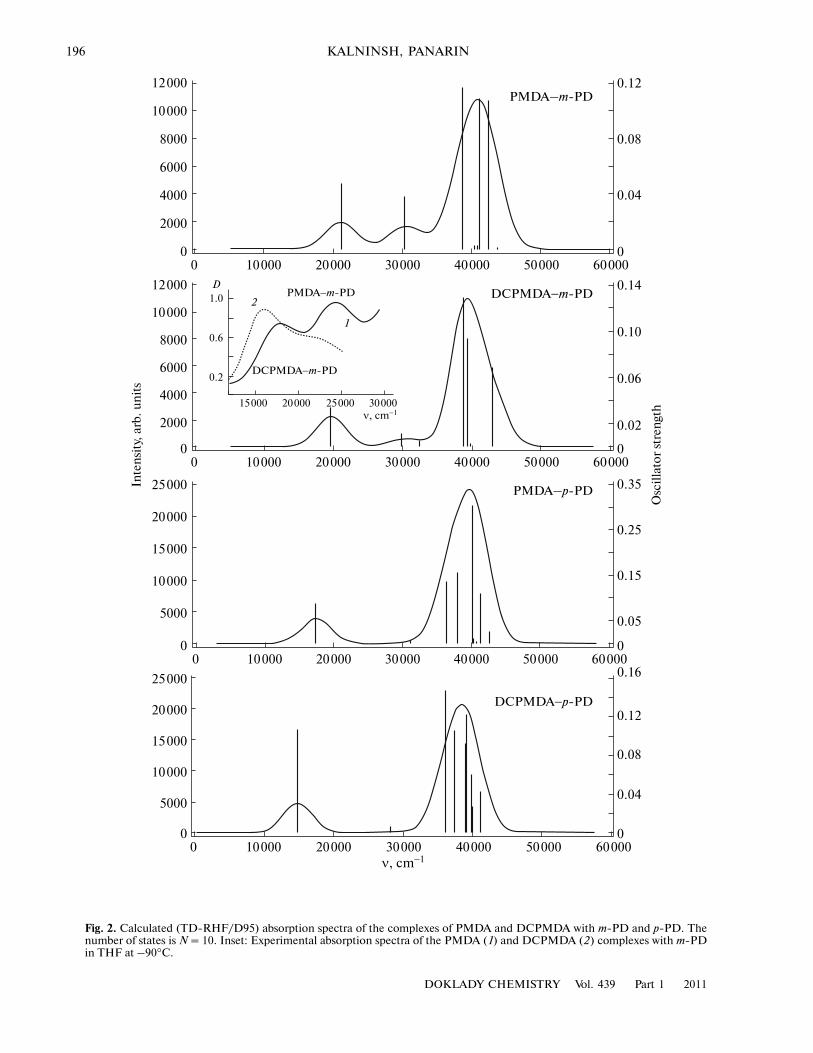
Figure 2 shows that the visible spectrum shows CTbands of medium intensity due to transitions betweenthe frontier orbitals. Two bands in the calculated andexperimental spectra (inset) of the m�phenylenedi�amine complexes correspond to transitions from thetwo upper closely spaced (splitting, 0.9 eV) occupiedorbitals. According to the calculation, the 0.25 eV(2000 cm–1) higher electron affinity of DCPMDA, ascompared with PMDA, leads to the low�frequencyshift of the CT band (by 1500 cm–1, while the experi�mental shift is 2000 cm–1). In the calculated absorp�tion spectra of the p�PD complexes, the doublet of CTbands degenerate into single lines at 17 450 (0.09) and14 850 (0.09) cm–1 (the oscillator strength is parenthe�sized) for PMDA and DCPMDA, respectively, whichis caused by a large difference in energy between twoupper occupied MOs of p�PD (2.3 eV). The secondCT band is expected to be in a high�frequency region(above 30 000 cm–1) and is masked by the properabsorption of the complex components. The observed
CHEMISTRY
Solvation of ExcitedDonor–Acceptor Diamine–Dianhydride Complexes
K. K. Kalninsh and Corresponding Member of the RAS E. F. Panarin
Received March 25, 2011
DOI: 10.1134/S0012500811070056
Institute of Polymers, Russian Academy of Sciences,Bol’shoi pr. 31, St. Petersburg, 199004 Russia
DOKLADY CHEMISTRY Vol. 439 Part 1 2011
SOLVATION OF EXCITED ... COMPLEXES 195
specifics of the CT bands are completely reproducedin the theoretical spectra.
The calculated absorption spectra of EDA com�plexes in Fig. 2 are appropriate for a tetrahydrofuranmedium. The experimental spectra (Fig. 2, inset) wereobtained in the same solvent. However, the HF/TDmethod is able to predict the absorption spectra of the
complexes in many solvents. In particular, Table 1 liststhe CT band parameters for the PMDA–m�PD com�plex in a series of solvents beginning with extremelyinert n�pentane to the good solvent dimethyl sulfox�ide. These data are evidence that the CT energydecreases as the solvent polarity increases, in accordwith the general tendency for lowering of electroniclevels with an increase in dielectric constant. The CT
������
���������
��������������� ���
������
� � � ������
���������
���������
���������
���
������
������
���������
��������� ���������
������
������
������������
������ ������
�� �� ��
������
���������
���
� � �
������������
������
���������
���������
������
������
���
������
���
������
������������
� � �
���������
������
���������
���������
������
���������
���������
������
������
������
������
���
���������
������
�� �� �� ������
������
���
������
������
���
� � �
A
B
Fig. 1. Plane�parallel (A) and planar (B) structures of the electron donor (diamine) and acceptor (dianhydride).
Table 1. Calculated (SCRF�TD/RFH) energies (E, cm–1) and oscillator strength (parenthesized) of CT bands for thecomplexes of pyromellitic dianhydride with m�phenylenediamine
n�Pentane Tetrahydrofuran Dimethyl sulfoxide Vacuum
E ΔE E ΔE E ΔE E
21262 (0.045) 483 21083 (0.047) 662 21068 (0.047) 677 21745 (0.037)
30719 (0.037) 520 30300 (0.038) 939 30193 (0.038) 1046 31239 (0.026)
Note: ΔE is the shift with respect to vacuum (cm–1).
196
DOKLADY CHEMISTRY Vol. 439 Part 1 2011
KALNINSH, PANARIN
12000
10000
8000
6000
4000
2000
00 10000 20000 30000 40000 50000 60000
0.12
0.08
0.04
0
PMDA–m�PD
12000
10000
8000
6000
4000
2000
00 10000 20000 30000 40000 50000 60000
0.14
0.06
0.02
0
DCPMDA−m�PD
0.10
PMDA−m�PD
DCPMDA−m�PD
1.0
0.6
0.2
15000 20000 25000 30000
25000
20000
00 10000 20000 30000 40000 50000 60000
0.35
0.25
0.05
0
PMDA–p�PD
15000
10000
5000
0.15
25000
20000
00 10000 20000 30000 40000 50000 60000
0.16
0.12
0
DCPMDA−p�PD
15000
10000
5000
0.08
D
ν, cm−1
ν, cm−1
Inte
nsi
ty,
arb.
un
its
Osc
illa
tor
stre
ngt
h
1
2
0.04
Fig. 2. Calculated (TD�RHF/D95) absorption spectra of the complexes of PMDA and DCPMDA with m�PD and p�PD. Thenumber of states is N = 10. Inset: Experimental absorption spectra of the PMDA (1) and DCPMDA (2) complexes with m�PDin THF at –90°C.

DOKLADY CHEMISTRY Vol. 439 Part 1 2011
SOLVATION OF EXCITED ... COMPLEXES 197
band shift, depending on the solvent polarity, is smalland does not exceed 1000 cm–1.
The planar complexes (Fig. 1, B) in the excitedstate are radical ion pairs with a large dipole momentμ (Table 2) directed along the major axis of the com�plex. The dipole moment clearly depends on the sol�vent polarity and increases with an increase in thedielectric constant (ε). The full electron transfer fromthe diamine to the dianhydride is supported by thecharge distribution and spin population on the atomsof the molecules constituting the complex. In particu�lar, the calculation of the triplet excited state of thePMDA–p�PD complex shows that the overall chargeon the atoms of the diamine molecule is +1.000, andthe total spin population index (F) is 1.000. These dataare convincing evidence of the radical cation nature ofthe diamine, which acts as the electron donor in theEDA complex. Analogously, the dianhydride moleculein the excited state becomes a radical anion andacquires the charge q = –1.000 and the spin popula�tion F = 1.000.
In the solvent reaction field, the dipole moment ofthe diamine–dianhydride reaction complex notice�ably increases and reaches the maximum of 38.1 D forthe DCPMDA–m�PD complex. In a polar medium,the complex “swells,” which is indicated by anincrease in its size and the length of the hydrogen bond
between its components (Table 2). This result is con�sistent with the data by Mataga [5], obtained whenstudying transient femtosecond absorption spectra. Itwas demonstrated that, in a polar medium, loose exci�plexes are formed, which dissociate into isolated ions,whereas tight ion pairs that appear in a nonpolar ofslightly polar solvent are prone to rapid recombinationand formation of the reaction product. The thermallygenerated excited states studied in this work are similarin many respects to photo exciplexes, so that they canbe considered jointly. An important interdisciplinaryquestion concerning the energy gap between theexcited and ground states is solved with confidence forthermally excited states (thermo exciplexes). How�ever, in the case of photo exciplexes, it is difficult toestimate the ground state energy [5] since this state isof dissociative character and escapes direct opticaldetection. In this context, the computationally pre�dicted noticeable decrease in the electron transferactivation energy, which is more than 7 kcal/mol forthe DCPMDA–p�PD complex, with an increase inthe solvent polarity (Table 2) is of special interest. Thelow activation energy Ea = 2.6 kcal/mol for this com�plex in polar dimethyl sulfoxide points to the sponta�neous electron transfer, which does not require anythermal activation.
Table 2. Calculated (SCRF�ROHF/D95) activation energy of electron transfer between diamine and dianhydride (Ea,kcal/mol), hydrogen bond length (R(N–H···OCC), Å, and dipole moment of the complex μ, D) in the excited triplet state
Complex Acceptor–donor Solvent ε R μ Ea
PMDA–m�PD DMSO 46.83 1.984 37.6 24.00
PMDA–p�PD
n�Pentane 1.84 1.771 30.9 16.39
Benzene 2.27 1.793 31.9 15.78
THF 7.43 1.870 35.2 12.97
DMSO 46.83 1.903 36.7 11.71
DCPMDA–m�PD DMSO 46.83 2.013 38.1 15.81
DCPMDA–p�PD
n�Pentane 1.84 1.576 29.0 9.75
Benzene 2.27 1.600 29.8 8.77
THF 7.43 1.767 34.5 4.10
DMSO 46.83 1.802 36.2 2.63
Note: THF is tetrahydrofuran, and DMSO is dimethyl sulfoxide.

198
DOKLADY CHEMISTRY Vol. 439 Part 1 2011
KALNINSH, PANARIN
The spin populations F on the atoms of the reactioncomplex as a function of the solvent type were calcu�lated by the SCRF�ROHF method (Fig. 3). As is seen,the decrease in polarity is accompanied by an increasein F on the C10 and N26 atoms, which is evidence ofan increase in the chemical activity of these atomsinvolved in the reaction product formation. The pro�cess of recombination of radical sites leading to theappearance of the new C10–N26 chemical bond isfavored by the low polarity of the solvent, in agreementwith photophysical data [5] on the increase in the for�mation rate of compact ion pairs in slightly polarmedia.
Thus, typical reagents in polycondensation, such asaromatic diamines and dianhydrides, act as moder�ately strong electron donors and acceptors and aresimultaneously involved in a strong intermolecularhydrogen bond. In the electronically excited state,
these two fundamental interactions ensure the forma�tion of an extended conjugation chain. An importantfeature of this chain is the interplay of electron andproton transfers in the low�energy electronic state thatcan be thermally populated. The SCRF�CPCM cal�culation of the energy and electronic characteristicswith the Gaussian09W program package revealed largedipole moments (up to 38 D), the radical ion characterof the reaction complex, and a strong dependence ofthese characteristics on the solvent nature.
REFERENCES
1. Kalnin’sh, K.K. and Panarin, E.F., Dokl. Chem., 2010,vol. 434, part 1, pp. 241–244 [Dokl. Akad. Nauk, 2010,vol. 434, no. 3, pp. 348–351].
2. Kalnin’sh, K.K., Opt. Spektrosk., 2007, vol. 103, no. 4,pp. 569–587.
� � �
������
���
������ ������
������
�� �� �� ������
��� ������
���������
��� ������ ������
������
���������
������
���������
���������
���������
������������
������������
���� � �
���������
������
������������������
���������������
12345678910111213141516171819202122232425262728293031323334
ССССССССHССHOOOOOOССССССNNHHHHHHHH
0.1510.1480.0000.1720.1740.0000.0340.0330.0000.0510.0490.0000.0000.0540.0540.0000.0400.0400.0900.0660.2160.0610.0900.1880.1280.1600.0000.0000.0000.0000.0000.0000.0000.000
0.143
0.0010.1790.1820.0000.0270.0260.0000.0590.0550.0000.0000.0590.0600.0000.0360.0350.0940.0640.2130.0570.0920.1890.1210.1690.0000.0000.0000.0000.0000.0000.0000.000
0.1370.1300.1120.0030.1890.1920.0000.0180.0150.0000.0810.0630.0000.0000.0670.0750.0000.0290.0250.1020.0610.2050.0470.0970.1890.1050.1930.0000.0000.0000.0000.0000.0000.0000.000
0.1300.0990.0060.1920.1910.0000.0170.0120.0000.0930.0610.0000.0000.0660.0830.0000.0280.0210.1050.0600.2020.0440.0990.1890.1000.2020.0000.0000.0000.0000.0000.0000.0000.000
Ben� n�Pen�THFDMSO
1.54 Å
(b)(а)
Fig. 3. (a) Calculated (ROHF/D95) structure of the electronically excited reaction complex of PMDA with p�PD in the T1 statein DMSO, THF, benzene and n�pentane (SCRF optimization) and (b) spin populations F in the triplet state (atoms with F > 0.09are labeled with an asterisk).
tanezene

DOKLADY CHEMISTRY Vol. 439 Part 1 2011
SOLVATION OF EXCITED ... COMPLEXES 199
3. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scu�seria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G.,Barone, V., Mennucci, B., Petersson, G.A., Nakat�suji, H., Caricato, M., Li, X., Hratchian, H.P., Izmay�lov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L.,Hada, M., Ehara, M., Toyota, K., Fukuda, R., Haseg�awa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O.,Nakai, H., Vreven, T., Montgomery, J.A., Jr., Peralta, J.E.,Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E.,Kudin, K.N., Staroverov, V.N., Kobayashi, R., Nor�mand, J., Raghavachari, K., Rendell, A., Burant, J.C.,Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Mil�lam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V.,
Adamo, C., Jaramillo, J., Gomperts, R., Strat�mann, R.E., Yazyev, O., Austin, A.J., Cammi, R.,Pomelli, C., Ochterski, J.W., Martin, R.L., Moro�kuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P.,Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O.,Foresman, J.B., Ortiz, J.V., Cioslowski, J., and Fox, D.J.,Gaussian 09, Revision A.1, Wallingford (CT): Gaussian,Inc., 2009.
4. Kalnin’sh, K.K. and Semenov, S.G., Zh. Prikl. Khim.(S.�Peterburg), 2003, vol. 76, no. 10, pp. 1585–1600.
5. Mataga, N. and Miyasaka, H., Adv. Chem. Phys., 2007,vol. 107, part 2, pp. 431–496.








![1,3‐Diamine‐Derived Bifunctional Organocatalyst Prepared ... · 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor ... [12f] Camphor is one of ... These are](https://img.pdfslide.us/doc/110x75/5b0406ee7f8b9a89208d0264/13diaminederived-bifunctional-organocatalyst-prepared-3-diamine-derived.jpg)




















