Embed Size (px)
Citation preview

DOI: 10.1542/neo.11-6-e290 2010;11;e290-e305 NeoReviews
Daniel T. Swarr and V. Reid Sutton Genetics
Skeletal Dysplasias in the Newborn: Diagnostic Evaluation and Developmental
http://neoreviews.aappublications.org/cgi/content/full/neoreviews;11/6/e290located on the World Wide Web at:
The online version of this article, along with updated information and services, is
Online ISSN: 1526-9906. Illinois, 60007. Copyright © 2010 by the American Academy of Pediatrics. All rights reserved. by the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village,it has been published continuously since 2000. NeoReviews is owned, published, and trademarked NeoReviews is the official journal of the American Academy of Pediatrics. A monthly publication,
by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

Skeletal Dysplasias in the Newborn:Diagnostic Evaluation and Developmental GeneticsDaniel T. Swarr, MD,*
V. Reid Sutton, MD†
Author Disclosure
Drs Swarr and Sutton
have disclosed no
financial relationships
relevant to this
article. This
commentary does
contain a discussion
of an unapproved/
investigative use of a
commercial
product/device.
AbstractMany of the genetic disorders of skeletal development lead to significant morbidityand mortality in utero or in the early neonatal period. Due to the large number andheterogeneous nature of these disorders, their diagnosis and management can beoverwhelming. A basic knowledge of skeletal development and a structured, compre-hensive approach to the history, physical examination, and interpretation of radio-graphic studies are crucial. Understanding the power and limitations of prenataldiagnostic technology and genetic testing is essential for accurate counseling andjudicious use of resources. Finally, familiarity with individual disorders and onlineresources aids the neonatologist in coordinating the complex, multidisciplinary carethat these infants demand in the neonatal intensive care unit (NICU) and afterhospital discharge.
Objectives After completing this article, readers should be able to:
1. Define the terms “dysplasia” and “dysotosis” and describe common skeletalabnormalities using accepted terminology.
2. List key elements of the history and physical examination of a neonate in whom adisorder of skeletal development is suspected, describe how to interpret a skeletalsurvey, and collectively use these data to search major atlases and databases.
3. List the most common disorders of skeletal development presenting in the newbornperiod, characteristic features of each disorder, and the molecular basis for eachcondition, if known.
4. Discuss the initial treatment of a neonate presenting with a suspected disorder ofskeletal development.
IntroductionGenetic disorders of skeletal development are a large, extremely heterogeneous group ofconditions that may present anytime from the prenatal period to adulthood. The estimatedincidence of disorders of skeletal development manifesting in the neonatal period is 15.7per 100,000 births. Long-term prognosis ranges from inevitable death shortly after birth
to survival into adulthood with normal intellectual develop-ment. In the neonatal period, respiratory compromise is theleading cause of morbidity and mortality.
Skeletal dysplasias are developmental disorders ofchondro-osseous tissues or an “abnormal organization ofcells into tissue and its morphologic result.” (1) These dis-orders are the result of an insult that occurs after organogen-esis but persists throughout later stages of development intopostnatal life. Dysplasias are referred to as primary if theyresult from mutations in genes expressed in chondro-osseoustissues and secondary if they result from extraosseous factorsthat affect development of bone. Dysplasias continue toaffect the skeleton through late pre- and postnatal life, lead-
*Department of Pediatrics, Baylor College of Medicine & Texas Children’s Hospital, Houston, Tex.†Department of Molecular & Human Genetics, Baylor College of Medicine & Texas Children’s Hospital, Houston, Tex.
Abbreviations
AP: anteroposteriorCNS: central nervous systemNICU: neonatal intensive care unitOI: osteogenesis imperfectaSADDAN: severe achondroplasia with developmental
delay and acanthosis nigricansSEDc: spondyloepiphyseal dysplasia congenitaTD: thanatophoric dysplasia
Article genetics
e290 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

ing to short stature, differing clinical characteristics withage, and in some cases, an increased risk of primarymalignancies of chondro-osseous tissues. In contrast,dysostoses are “malformations of single bones, alone orin combination.” (1) They are typically the result of adiscrete insult during organogenesis. As such, they arestatic lesions without risk of progression or malignantdegeneration and usually are not associated with shortstature. Key definitions important in the diagnosis ofthese disorders are summarized in Table 1.
To date, 372 disorders have been described and clas-sified into 37 groups. (2) As the number of disorders ofskeletal development has increased and understanding oftheir molecular causes has grown, this classification sys-tem has evolved into a complex amalgam of clinical,radiographic, biochemical, and molecular features. Someclassification groups are based purely on molecular cause(eg, FGFR3 group, type II collagen group), others arebased on radiologic findings (eg, metaphyseal dysplasiasgroup, increased bone density group), and yet others arebased on a combination of clinical and radiographicfeatures (eg, mesomelic and rhizomesomelic dysplasias,bent bone dysplasias group).
This bewildering array of disorders and terminologycan make the approach to a newborn who has a suspecteddisorder of skeletal development overwhelming for eventhe most experienced clinician. In this article, we reviewbasic aspects of skeletal development, discuss how toapproach an infant who has a suspected skeletal dysplasiaor dysostosis, summarize common and important disor-ders of skeletal development presenting in the neonatalperiod, and present advances in treatment.
Case PresentationYou are called to evaluate respiratory distress and cyanosisin a term infant who was born to a 35-year-old multipa-
rous woman following a cesarean section for failure toprogress. The mother received late prenatal care, but herserology results were within normal parameters. In thedelivery room, the infant developed mild tachypnea andcyanosis that improved with oxygen administered by nasalcannula. On physical examination, the infant has a weightof 3,830 g (71st percentile), head circumference of 39 cm(97th percentile), and length of 40 cm (�3.7 standarddeviations below the mean). Despite a small chest, his lungfields are clear, and cardiac examination yields normalresults. The most striking findings on examination are shortupper and lower extremities and bilateral club feet. Youtransfer him to the NICU for further evaluation.
Overview of Skeletal DevelopmentDevelopment of the skeleton begins very early in embry-onic life, starting with patterning of the somites duringthe end of the third week and formation of the limb budsduring the fourth week of embryogenesis. Complex mo-lecular pathways mold the somites into the developingaxial skeleton, which ultimately consists of the vertebralcolumn, scapula, ribs, and pelvis. Distinct, yet equallysophisticated developmental signals direct the primordiallimb buds to form the mature appendicular skeleton.Disruptions in these early patterning events classicallyresult in dysostoses (Table 2).
Patterning of the skeleton is followed during the fifthweek of development with the formation of cartilagefrom mesenchyme or embryonic connective tissue. Theskeleton subsequently develops through two primarymechanisms: intramembranous ossification and endo-chondral ossification. In intramembranous ossification,neural crest-derived mesenchymal stem cells condenseand differentiate directly into bone. This process is in-volved primarily in the formation of the flat bones of theskull and proximal portions of the clavicles. The remain-
Table 1. Key DefinitionsTerm Definition
Skeletal dysplasia “An abnormal organization of cells into [chondro-osseous] tissue(s) and its morphologicresult(s)” (1)
Primary skeletal dysplasia A skeletal dysplasia due to mutated genes that are expressed in chondro-osseous tissueSecondary skeletal dysplasia A skeletal dysplasia due to abnormalities of extraosseous factors with secondary effects on
the skeletal systemDysostosis “Malformations of single bones, alone or in combination,” as a result of an insult during
organogenesis (1)Disruptions Secondary malformations of bones as a result of exposure to a noxious agent (such as an
infection or toxin) during a limited period during developmentOsteolysis Postnatal disruption of bone development (includes hereditary and external causes, such as
infection, neuropathies, and toxic exposures)
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e291 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

der of the skeleton is formed through endochondralossification, whereby mesenchymal stem cells initiallycreate a well-organized cartilage structure that subse-quently is converted to bone. A number of disordersresult from disruption of this normal differentiation pro-cess, leading to a phenotype of abnormal skeletal pattern-ing as well as a defect in skeletal growth. Because thesedisorders combine features of dysotosis and dysplasia,they sometimes are referred to as dysostoplasias. Proto-typic examples include campomelic dysplasia and cleido-cranial dysplasia (Table 2).
Newly formed chondrocytes within the immatureskeletal architecture subsequently undergo an elaborateprocess involving proliferation, differentiation, and
growth. Throughout these changes, the chondrocytesare supported by a complex array of organic molecules,referred to as the extracellular matrix. This matrix, whichincludes collagen, proteoglycans, and glycoproteins, notonly provides critical structural support but also interactsdirectly with the signaling pathways modulating chon-drocyte development. A disruption of either this com-plex structural matrix or the intricate molecular signalingpathways regulating chondrocyte growth and develop-ment leads to many of the known skeletal dysplasias.
The final step in the formation of bone is mineraliza-tion. Osteoblasts deposit an osteoid matrix, which be-comes impregnated with hydroxyapatite crystals. Thiscalcified material provides bone with its characteristic
Table 2. Overview of Skeletal DevelopmentDevelopmentalStage Timing Example Pathway/Disorder
Patterning Third through sixthweeks
Notch signaling occurs in oscillations that pattern somite formation.Mutation of the Notch ligand, dll3, leads to autosomal recessivespondylocostal dysostosis, a disorder characterized by multipleabnormalities of the axial skeletal system (hemivertebrae; fused andblock vertebrae; and abnormal, fused ribs).
Condensation andDifferentiation
Fifth week onwards The Sox family of transcription factors (eg, SOX5, SOX6, and SOX9) playsan essential role in the differentiation of precursor cells intochondrocytes in endochondral bone. Campomelic dysplasia is due tohaploinsufficiency of SOX9, which leads to bowing and angulation oflong bones, hypoplasia of the scapula and pelvis, abnormalities of theribs and vertebral column, and craniofacial abnormalities. This conditionusually is fatal in the neonatal period due to a small rib cage, narrowairways, and hypoplastic lungs. Because of the importance of this genein sexual differentiation, 75% of 46,XY patients also have complete orpartial sex reversal.
Growth Fifth week throughadolescence
PTHrP is a secreted molecule that maintains chondrocytes in thenonhypertrophic proliferating state. It is signals through the PTH/PTHrPreceptor (PTHR) and is regulated indirectly by the Indian hedgehog gene.Abnormalities in this pathway lead to a spectrum of disorders, includingBlomstrand chondrodysplasia, Jansen-type metaphyseal chondrodysplasia,and enchondromatosis. For example, homozygous loss-of-functionmutations in the receptor for PTHrP lead to excessive chondrocytehypertrophy and inadequate proliferation. The result is Blomstrandchondrodysplasia, a severe short-limb dwarfism characterized byaccelerated bone maturation and generalized sclerosis of bone. It isusually fatal in the neonatal period.
Mineralization andHomeostasis
Eighth week throughadulthood
Mutations in the gene encoding tissue-nonspecific alkaline phosphatase(TNAP) result in hypophosphatasia. This disorder can manifest in one ofsix recognized clinical forms: perinatal lethal, perinatal benign, infantile,childhood, adult, and odontohypophosphatasia. Fetuses affected by theperinatal lethal form have markedly impaired bone mineralization inutero, with characteristic osteochondral spurs on the arms and legs.Death usually results at or shortly after birth from respiratory failure dueto hypoplastic lungs and rachitic changes of the chest. The infantileform may not be clinically evident at birth but subsequently manifestswith similar respiratory complications, craniosynostosis, and symptomatichypercalcemia.
genetics skeletal dysplasias
e292 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

hard, durable structure. A delicate balance betweenbone-forming cells (osteoblasts) and bone-degradingcells (osteoclasts) continues throughout life, reshapingand remodeling the skeleton. A variety of skeletal disor-ders result from the disruption of normal mineralizationand homeostasis (Table 2).
Initial Diagnostic ApproachThe evaluation and initial treatment of the neonate whohas a suspected disorder of skeletal development is amultidisciplinary endeavor, requiring, at minimum, theexpertise of neonatologists, medical geneticists, and ra-diologists. After addressing any respiratory compromiseand clinically stabilizing the infant, initial evaluationshould begin with a detailed history and physical exami-nation. A comprehensive prenatal history should be elic-ited, looking for potential infectious, environmental, or
teratogenic disruptions of skeletaldevelopment. A family history alsomay be informative because manyof the disorders of skeletal develop-ment are transmitted in an autoso-mal dominant pattern. Theoreti-cally, affected individuals transmitthe trait to 50% of their offspringwith this mode of inheritance. Inactuality, the numbers are lowerdue to germline mosaicism and in-complete penetrance. More often,there is no family history of thedisorder due to de novo mutations.More than 80% of cases of achon-droplasia, for example, are due tode novo mutations in FGFR3, andthere is a well-described associationbetween these new mutations andadvanced paternal age.
A head-to-toe physical examina-tion should be performed, focusingon physical features and other birthdefects that may aid in determininga specific diagnosis (Table 3). Thehead shape, fontanelles, and suturewidth are examined, looking forthe presence of a craniosynostosis.In addition, any dysmorphic facialfeatures should be noted. The oralcavity is examined for cleft palate,gingival frenulae, and abnormaldentition. Consultation with anophthalmologist may be indicated
to examine for retinitis pigmentosa, cataracts, colobo-mas, or myopia. Abnormalities of the skin, hair, and nailsshould be noted and the joints examined carefully for anyabnormalities, such as hyperlaxity or contractures. Innoting any asymmetry or deformity of the extremities, itis important to look specifically for syndactyly or anabnormal number of digits. Careful cardiac and neuro-logic examinations should be completed to look forevidence of congenital heart disease or central nervoussystem (CNS) dysfunction, respectively. Finally, a com-plete skeletal survey should be performed and consulta-tion with a medical geneticist and pediatric radiologistobtained.
RadiologyA skeletal survey performed for the assessment of apotential abnormality of skeletal development should
Table 3. Notable Physical Examination Findings inNeonatal Skeletal Dysplasias
Finding Dysplasia
SkullMacrocephalyLarge fontanelles, wide suturesCloverleaf deformity
Achondroplasia, hypochondroplasiaCleidocranial dysplasiaThanatophoric dysplasia
TeethNatal teethDentinogenesis imperfectaHypoplasia of dental cementumSupernumerary teeth
Ellis-van Creveld dysplasiaOsteogenesis imperfectaHypophosphatasiaCleidocranial dysplasia
Oral cavityCleft palate
Gingival frenulae
Type II collagenopathies, campomelicdysplasia
Ellis-van Creveld dysplasia
EyesCataractsSevere myopia
Chondrodysplasia punctataType II collagenopathies
SkinRedundant skin foldsIchthyosiform erythrodermaAcanthosis nigricans
Achondroplasia, hypochondroplasiaChondrodysplasia punctataSevere achondroplasia (SAADAN),
hypochondroplasia
NailsHypoplastic nails Ellis-van Creveld syndrome, chondrodysplasia
punctata-brachytelephalangic typeLimbsAsymmetry
Fractures
Chondrodysplasia punctata-Conradi-Hunermann type
Osteogenesis imperfecta, hypophosphatasia
Adapted from Mortier. (3)
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e293 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

include the following views: anteroposterior (AP) andlateral skull; AP and lateral thoracolumbar spine; and APimages of chest, pelvis, upper and lower limbs, andhands/feet. Additional films may be obtained to assessspecific abnormalities or if asymmetry exists.
A brief review of all of the radiographs can identify theportion(s) of the skeleton that appear to be affected mostseverely. Common terms used to refer to regions of theaxial skeleton appear in Table 4. Abnormalities of theappendicular skeleton are referred to by the region mostaffected within individual long bones (epiphyseal, me-taphyseal, diaphyseal) as well as along the proximo-distalaxis. Shortening that predominantly involves the proxi-mal, middle, or distal region is referred to as rhizomelic,mesomelic, or acromelic, respectively.
In the neonate, epiphyseal dysplasias typically mani-fest as delayed epiphyseal ossification. The following os-sification centers are expected to be seen radiographicallyin a term or near-term neonate: humeral head, distalfemoral, proximal tibial, calcaneus, and talus. Radio-graphic findings of metaphyseal dysplasias include irreg-ular, broad, and cupped (or lucent) metaphyses. Theterm “overtubulated” (metaphyseal flaring resulting in asharp transition between the metaphysis and diaphysis) isused occasionally to describe the radiographic findings ofmetaphyseal dysplasias. Diaphyseal dysplasias present asrelatively wide diaphyses with narrow metaphyses andoften are referred to as “undertubulated” bones. In-volvement of the spine (most often platyspondyly in theneonate) frequently is seen in conjunction with abnor-malities of the long bones. In such cases, the categoriza-tion is spondyloepiphyseal, spondylometaphyseal, orspondyloepimetaphyseal dysplasia. This basic informa-tion of radiographic and clinical categorization providesthe starting point for a query of major reference booksand electronic databases.
For examination of individual bones, Offiah and Hall
(4) have proposed a helpful mnemonic of “the five S’s”:Structure, Shape, Size, Sum, and Soft tissues. Peculiar orunusual features of individual bones may provide helpfuldiagnostic clues. It is important to look for increased ordecreased bone density, abnormal bony growths, andany bones that appear to be of an unusual shape. The sizeof individual bones, particularly with respect to the re-mainder of the skeleton, should be considered. Finally, itis important to note whether there is an abnormal num-ber or fusion of any bones and whether there appears tobe any soft-tissue involvement.
Additional Diagnostic TestingDepending on the specific disorder of skeletal develop-ment, it may be possible to establish a diagnosis with aclinical history, physical examination, and skeletal surveyalone. In other cases, additional evaluation may be re-quired. Histologic examination of bone and cartilagemay be very informative, although it can be obtainedonly through bone biopsy. Therefore, it often is reservedfor autopsy cases or for infants and children undergoinga surgical procedure for another indication. Biochemicalanalysis may be helpful in specific cases, such as examina-tion of type I collagen in osteogenesis imperfecta. Fi-nally, DNA testing now is available for many disorders ofskeletal development and generally is preferred because itis less invasive and can provide a definitive diagnosis. Theutility of these tests varies greatly between disorders, andinterpretation of results can be difficult, so consultationwith a medical geneticist is advised.
Prenatal Diagnosis of Fetal SkeletalDysplasiasDue to improvements in fetal imaging technology, itnow is possible to recognize disorders of skeletal devel-opment in the prenatal period. Suspicion for a fetalskeletal dysplasia should be raised when routine ultra-sonographic measurements of femora or humeri fall be-low the 5th percentile or other skeletal abnormalitiesare apparent. Any fetus meeting these criteria should bereferred to a high-risk perinatal center that has expertisein fetal skeletal dysplasias for complete ultrasonographicevaluation. Additional imaging studies may includethree-dimensional ultrasonography for better definitionof craniofacial abnormalities and fetal magnetic reso-nance imaging if there is a concern for spinal defects.Despite these sophisticated imaging modalities, estab-lishing a definitive diagnosis in the prenatal period re-mains very difficult. This dilemma presents a majorchallenge for both obstetricians and neonatologists in
Table 4. Terminology: AxialSkeletonSkull: Cranio-/cranialFace: Facio-/facialMandible: Mandibulo-Clavicle: Cleido-Ribs: Costo-Spine: Spondylo-/vertebralPelvis: Ischio-/ilio-/pubic
Adapted from Offiah and Hal. (4)
genetics skeletal dysplasias
e294 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from
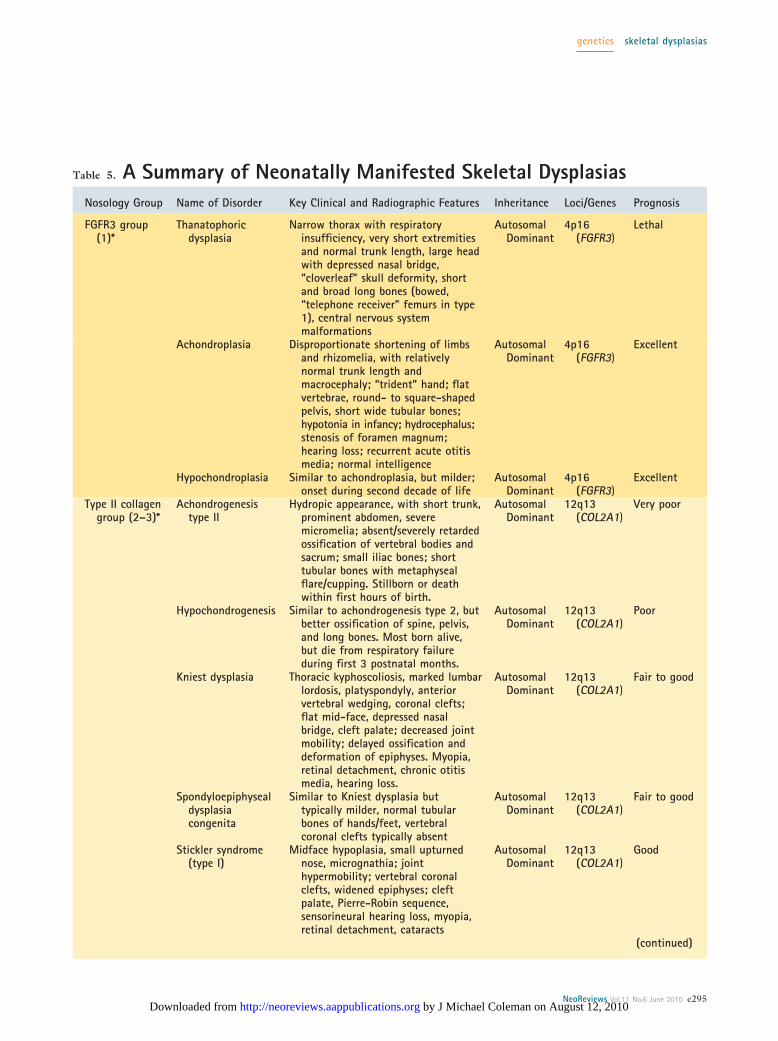
Table 5. A Summary of Neonatally Manifested Skeletal DysplasiasNosology Group Name of Disorder Key Clinical and Radiographic Features Inheritance Loci/Genes Prognosis
FGFR3 group(1)*
Thanatophoricdysplasia
Narrow thorax with respiratoryinsufficiency, very short extremitiesand normal trunk length, large headwith depressed nasal bridge,“cloverleaf” skull deformity, shortand broad long bones (bowed,“telephone receiver” femurs in type1), central nervous systemmalformations
AutosomalDominant
4p16(FGFR3)
Lethal
Achondroplasia Disproportionate shortening of limbsand rhizomelia, with relativelynormal trunk length andmacrocephaly; “trident” hand; flatvertebrae, round- to square-shapedpelvis, short wide tubular bones;hypotonia in infancy; hydrocephalus;stenosis of foramen magnum;hearing loss; recurrent acute otitismedia; normal intelligence
AutosomalDominant
4p16(FGFR3)
Excellent
Hypochondroplasia Similar to achondroplasia, but milder;onset during second decade of life
AutosomalDominant
4p16(FGFR3)
Excellent
Type II collagengroup (2–3)*
Achondrogenesistype II
Hydropic appearance, with short trunk,prominent abdomen, severemicromelia; absent/severely retardedossification of vertebral bodies andsacrum; small iliac bones; shorttubular bones with metaphysealflare/cupping. Stillborn or deathwithin first hours of birth.
AutosomalDominant
12q13(COL2A1)
Very poor
Hypochondrogenesis Similar to achondrogenesis type 2, butbetter ossification of spine, pelvis,and long bones. Most born alive,but die from respiratory failureduring first 3 postnatal months.
AutosomalDominant
12q13(COL2A1)
Poor
Kniest dysplasia Thoracic kyphoscoliosis, marked lumbarlordosis, platyspondyly, anteriorvertebral wedging, coronal clefts;flat mid-face, depressed nasalbridge, cleft palate; decreased jointmobility; delayed ossification anddeformation of epiphyses. Myopia,retinal detachment, chronic otitismedia, hearing loss.
AutosomalDominant
12q13(COL2A1)
Fair to good
Spondyloepiphysealdysplasiacongenita
Similar to Kniest dysplasia buttypically milder, normal tubularbones of hands/feet, vertebralcoronal clefts typically absent
AutosomalDominant
12q13(COL2A1)
Fair to good
Stickler syndrome(type I)
Midface hypoplasia, small upturnednose, micrognathia; jointhypermobility; vertebral coronalclefts, widened epiphyses; cleftpalate, Pierre-Robin sequence,sensorineural hearing loss, myopia,retinal detachment, cataracts
AutosomalDominant
12q13(COL2A1)
Good
(continued)
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e295 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

counseling the families of affected fetuses and optimallyplanning for their postnatal care.
In some cases, prenatal DNA testing may be em-ployed to help establish a definitive fetal diagnosis. Forfamilies that have a history of a specific disorder ofskeletal development that has been confirmed with mo-lecular methods, mutation analysis may be performed onDNA obtained by chorionic villus sampling or amnio-centesis. In the absence of a family history, large genesizes, “private” mutations, and disorders of unknowncause may hinder timely molecular diagnosis. In manycases, however, it still may be possible to perform suc-cessful mutation analysis when a known, common muta-tion exists (as with FGFR3 mutations in thanatophoricdysplasia and achondroplasia). The interested reader is
referred to Krakow and associates (5) for an excellent andmore complete discussion of this topic.
Selected Disorders of Skeletal DevelopmentWith Neonatal OnsetA summary of some of the more common and clinicallyimportant disorders of skeletal development presentingin the perinatal and neonatal periods appears in Table 5.
Type II CollagenopathiesAchondrogenesis type II, hypochondrogenesis, Kniestdysplasia, spondyloepiphyseal dysplasia congenita (SEDc),and Stickler syndrome form a spectrum of disorders dueto mutations in the COL2A1 gene that may presentduring the neonatal period. These disorders are charac-
Table 5. A Summary of Neonatally Manifested SkeletalDysplasias—Continued
Nosology Group Name of Disorder Key Clinical and Radiographic Features Inheritance Loci/Genes Prognosis
Short-ribdysplasiagroup (7)*
Ellis-van Creveldsyndrome
Narrow, shallow thorax; pre- andpostaxial polydactyly, fusion of themetacarpals or phalanges, dysplasiaof the pelvis, rhizomelia; congenitalheart disease; hypoplastic nails;dental anomalies
AutosomalRecessive
4p16(EVC1,EVC2)
Good in absence ofcardiopulmonaryabnormalities
Asphyxiatingthoracic dysplasia(Jeune)
Narrow, shallow thorax withrespiratory insufficiency;metaphyseal irregularities, shorthands/feet, postaxial polydactyly,short middle and distal phalanges,cone-shaped epiphyses; progressiverenal disease, pancreatic and hepaticfibrosis, Hirschsprung disease,multiple gingival frenulae,hydrocephalus
AutosomalRecessive
3q24–26(IFT80),15q13
Fair
Decreased bonedensity group(24)*
Osteogenesisimperfecta
See Table 6
Defectivemineralizationgroup (25)*
Hypophosphatasia,perinatal lethal
Severe undermineralization of bone;marked shortening of extremities;osteochondral spurs on arms/legs;respiratory failure, apnea, seizures
AutosomalRecessive
1p36(TNAP)
Poor
Defectivemineralizationgroup (25)*
Hypophosphatasia,infantile forms
May appear normal at birth, butsymptoms almost always manifest infirst 6 postnatal months. Markedundermineralization of bone onradiography, rachitic changes ofmetaphyses, craniosynostosis,respiratory insufficiency,symptomatic hypercalcemia, shortstature, premature loss of deciduousteeth
AutosomalRecessive
1p36(TNAP)
Variable
*Numbers in parentheses refer to the group number as they appear in the International Nosology Classification System. (6)
genetics skeletal dysplasias
e296 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from
terized by disproportionate involvement of the spine andepiphyses of long bones (spondyloepiphyseal involve-ment).
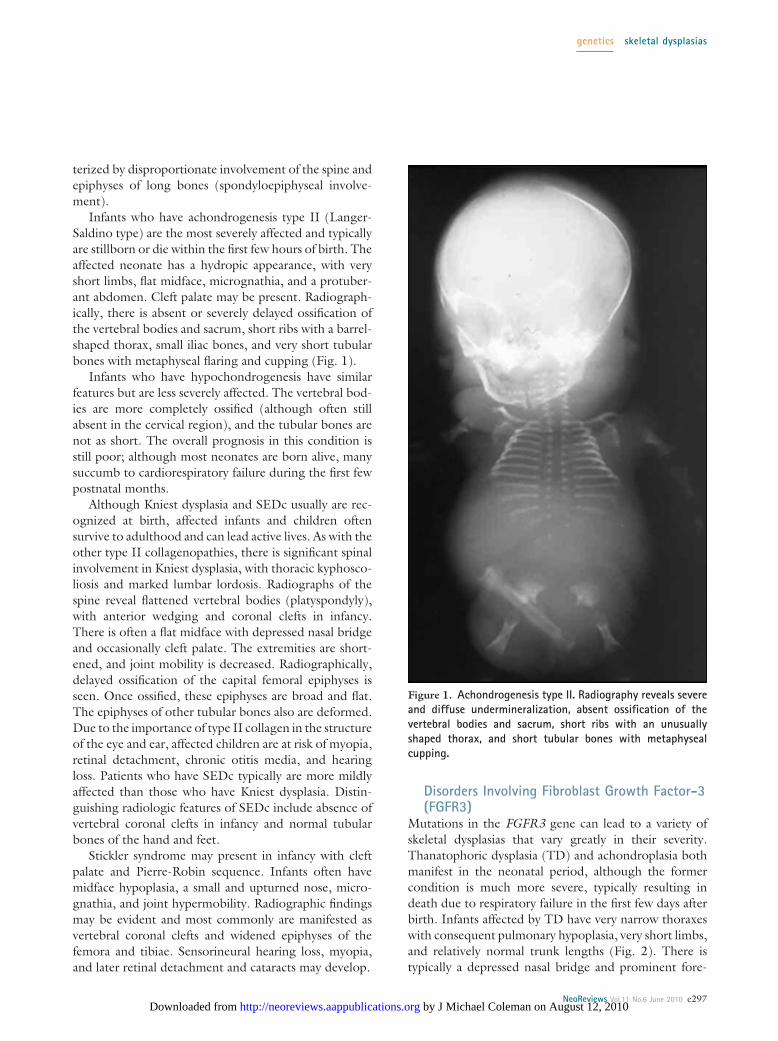
Infants who have achondrogenesis type II (Langer-Saldino type) are the most severely affected and typicallyare stillborn or die within the first few hours of birth. Theaffected neonate has a hydropic appearance, with veryshort limbs, flat midface, micrognathia, and a protuber-ant abdomen. Cleft palate may be present. Radiograph-ically, there is absent or severely delayed ossification ofthe vertebral bodies and sacrum, short ribs with a barrel-shaped thorax, small iliac bones, and very short tubularbones with metaphyseal flaring and cupping (Fig. 1).
Infants who have hypochondrogenesis have similarfeatures but are less severely affected. The vertebral bod-ies are more completely ossified (although often stillabsent in the cervical region), and the tubular bones arenot as short. The overall prognosis in this condition isstill poor; although most neonates are born alive, manysuccumb to cardiorespiratory failure during the first fewpostnatal months.
Although Kniest dysplasia and SEDc usually are rec-ognized at birth, affected infants and children oftensurvive to adulthood and can lead active lives. As with theother type II collagenopathies, there is significant spinalinvolvement in Kniest dysplasia, with thoracic kyphosco-liosis and marked lumbar lordosis. Radiographs of thespine reveal flattened vertebral bodies (platyspondyly),with anterior wedging and coronal clefts in infancy.There is often a flat midface with depressed nasal bridgeand occasionally cleft palate. The extremities are short-ened, and joint mobility is decreased. Radiographically,delayed ossification of the capital femoral epiphyses isseen. Once ossified, these epiphyses are broad and flat.The epiphyses of other tubular bones also are deformed.Due to the importance of type II collagen in the structureof the eye and ear, affected children are at risk of myopia,retinal detachment, chronic otitis media, and hearingloss. Patients who have SEDc typically are more mildlyaffected than those who have Kniest dysplasia. Distin-guishing radiologic features of SEDc include absence ofvertebral coronal clefts in infancy and normal tubularbones of the hand and feet.
Stickler syndrome may present in infancy with cleftpalate and Pierre-Robin sequence. Infants often havemidface hypoplasia, a small and upturned nose, micro-gnathia, and joint hypermobility. Radiographic findingsmay be evident and most commonly are manifested asvertebral coronal clefts and widened epiphyses of thefemora and tibiae. Sensorineural hearing loss, myopia,and later retinal detachment and cataracts may develop.
Disorders Involving Fibroblast Growth Factor-3(FGFR3)
Mutations in the FGFR3 gene can lead to a variety ofskeletal dysplasias that vary greatly in their severity.Thanatophoric dysplasia (TD) and achondroplasia bothmanifest in the neonatal period, although the formercondition is much more severe, typically resulting indeath due to respiratory failure in the first few days afterbirth. Infants affected by TD have very narrow thoraxeswith consequent pulmonary hypoplasia, very short limbs,and relatively normal trunk lengths (Fig. 2). There istypically a depressed nasal bridge and prominent fore-
Figure 1. Achondrogenesis type II. Radiography reveals severeand diffuse undermineralization, absent ossification of thevertebral bodies and sacrum, short ribs with an unusuallyshaped thorax, and short tubular bones with metaphysealcupping.
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e297 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from
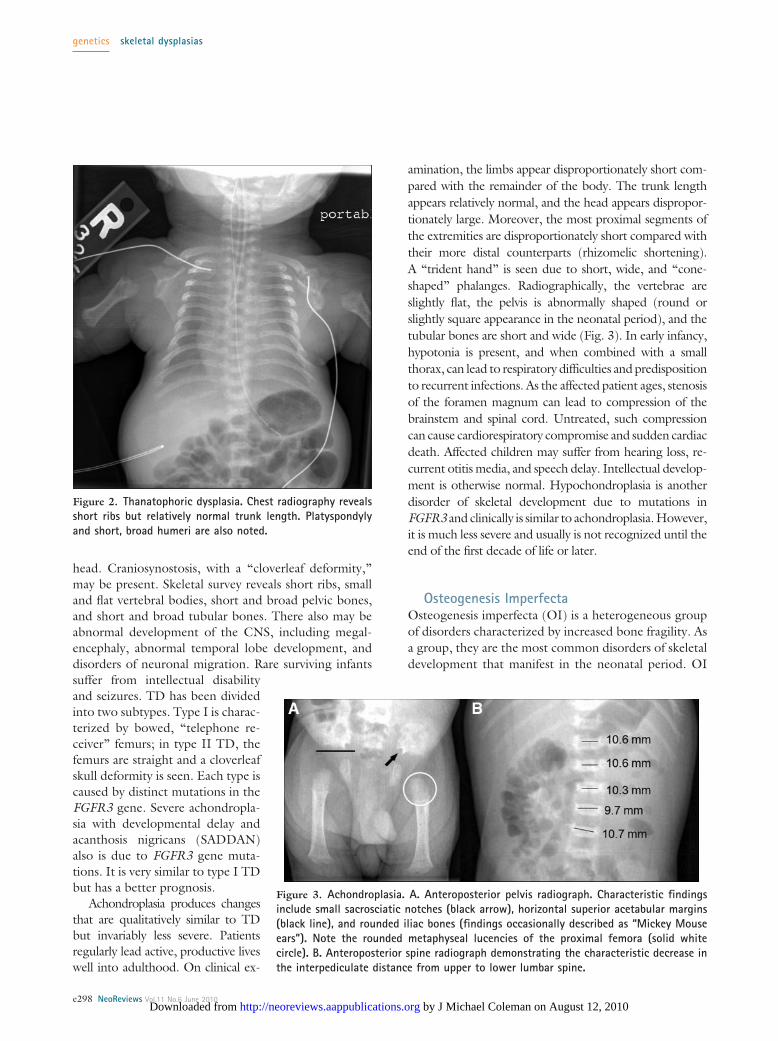
head. Craniosynostosis, with a “cloverleaf deformity,”may be present. Skeletal survey reveals short ribs, smalland flat vertebral bodies, short and broad pelvic bones,and short and broad tubular bones. There also may beabnormal development of the CNS, including megal-encephaly, abnormal temporal lobe development, anddisorders of neuronal migration. Rare surviving infantssuffer from intellectual disabilityand seizures. TD has been dividedinto two subtypes. Type I is charac-terized by bowed, “telephone re-ceiver” femurs; in type II TD, thefemurs are straight and a cloverleafskull deformity is seen. Each type iscaused by distinct mutations in theFGFR3 gene. Severe achondropla-sia with developmental delay andacanthosis nigricans (SADDAN)also is due to FGFR3 gene muta-tions. It is very similar to type I TDbut has a better prognosis.
Achondroplasia produces changesthat are qualitatively similar to TDbut invariably less severe. Patientsregularly lead active, productive liveswell into adulthood. On clinical ex-
amination, the limbs appear disproportionately short com-pared with the remainder of the body. The trunk lengthappears relatively normal, and the head appears dispropor-tionately large. Moreover, the most proximal segments ofthe extremities are disproportionately short compared withtheir more distal counterparts (rhizomelic shortening).A “trident hand” is seen due to short, wide, and “cone-shaped” phalanges. Radiographically, the vertebrae areslightly flat, the pelvis is abnormally shaped (round orslightly square appearance in the neonatal period), and thetubular bones are short and wide (Fig. 3). In early infancy,hypotonia is present, and when combined with a smallthorax, can lead to respiratory difficulties and predispositionto recurrent infections. As the affected patient ages, stenosisof the foramen magnum can lead to compression of thebrainstem and spinal cord. Untreated, such compressioncan cause cardiorespiratory compromise and sudden cardiacdeath. Affected children may suffer from hearing loss, re-current otitis media, and speech delay. Intellectual develop-ment is otherwise normal. Hypochondroplasia is anotherdisorder of skeletal development due to mutations inFGFR3 and clinically is similar to achondroplasia. However,it is much less severe and usually is not recognized until theend of the first decade of life or later.
Osteogenesis ImperfectaOsteogenesis imperfecta (OI) is a heterogeneous groupof disorders characterized by increased bone fragility. Asa group, they are the most common disorders of skeletaldevelopment that manifest in the neonatal period. OI
Figure 2. Thanatophoric dysplasia. Chest radiography revealsshort ribs but relatively normal trunk length. Platyspondylyand short, broad humeri are also noted.
Figure 3. Achondroplasia. A. Anteroposterior pelvis radiograph. Characteristic findingsinclude small sacrosciatic notches (black arrow), horizontal superior acetabular margins(black line), and rounded iliac bones (findings occasionally described as “Mickey Mouseears”). Note the rounded metaphyseal lucencies of the proximal femora (solid whitecircle). B. Anteroposterior spine radiograph demonstrating the characteristic decrease inthe interpediculate distance from upper to lower lumbar spine.
genetics skeletal dysplasias
e298 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from
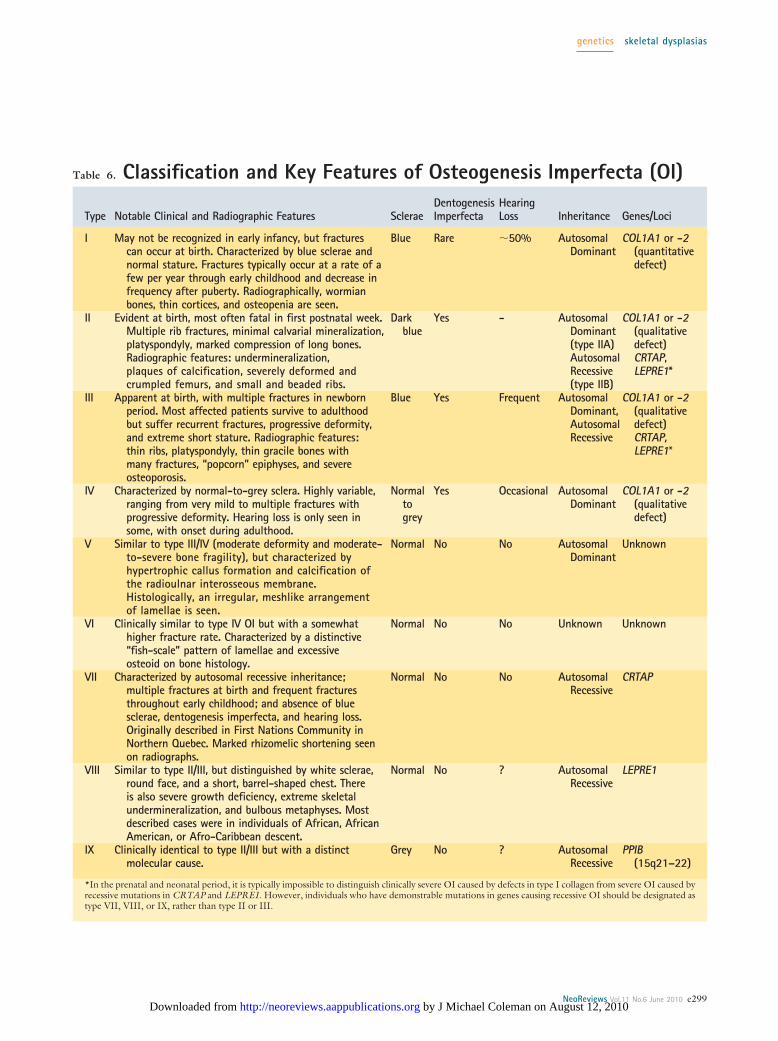
Table 6. Classification and Key Features of Osteogenesis Imperfecta (OI)
Type Notable Clinical and Radiographic Features ScleraeDentogenesisImperfecta
HearingLoss Inheritance Genes/Loci
I May not be recognized in early infancy, but fracturescan occur at birth. Characterized by blue sclerae andnormal stature. Fractures typically occur at a rate of afew per year through early childhood and decrease infrequency after puberty. Radiographically, wormianbones, thin cortices, and osteopenia are seen.
Blue Rare �50% AutosomalDominant
COL1A1 or -2(quantitativedefect)
II Evident at birth, most often fatal in first postnatal week.Multiple rib fractures, minimal calvarial mineralization,platyspondyly, marked compression of long bones.Radiographic features: undermineralization,plaques of calcification, severely deformed andcrumpled femurs, and small and beaded ribs.
Darkblue
Yes - AutosomalDominant(type IIA)AutosomalRecessive(type IIB)
COL1A1 or -2(qualitativedefect)CRTAP,LEPRE1*
III Apparent at birth, with multiple fractures in newbornperiod. Most affected patients survive to adulthoodbut suffer recurrent fractures, progressive deformity,and extreme short stature. Radiographic features:thin ribs, platyspondyly, thin gracile bones withmany fractures, “popcorn” epiphyses, and severeosteoporosis.
Blue Yes Frequent AutosomalDominant,AutosomalRecessive
COL1A1 or -2(qualitativedefect)CRTAP,LEPRE1*
IV Characterized by normal-to-grey sclera. Highly variable,ranging from very mild to multiple fractures withprogressive deformity. Hearing loss is only seen insome, with onset during adulthood.
Normaltogrey
Yes Occasional AutosomalDominant
COL1A1 or -2(qualitativedefect)
V Similar to type III/IV (moderate deformity and moderate-to-severe bone fragility), but characterized byhypertrophic callus formation and calcification ofthe radioulnar interosseous membrane.Histologically, an irregular, meshlike arrangementof lamellae is seen.
Normal No No AutosomalDominant
Unknown
VI Clinically similar to type IV OI but with a somewhathigher fracture rate. Characterized by a distinctive“fish-scale” pattern of lamellae and excessiveosteoid on bone histology.
Normal No No Unknown Unknown
VII Characterized by autosomal recessive inheritance;multiple fractures at birth and frequent fracturesthroughout early childhood; and absence of bluesclerae, dentogenesis imperfecta, and hearing loss.Originally described in First Nations Community inNorthern Quebec. Marked rhizomelic shortening seenon radiographs.
Normal No No AutosomalRecessive
CRTAP
VIII Similar to type II/III, but distinguished by white sclerae,round face, and a short, barrel-shaped chest. Thereis also severe growth deficiency, extreme skeletalundermineralization, and bulbous metaphyses. Mostdescribed cases were in individuals of African, AfricanAmerican, or Afro-Caribbean descent.
Normal No ? AutosomalRecessive
LEPRE1
IX Clinically identical to type II/III but with a distinctmolecular cause.
Grey No ? AutosomalRecessive
PPIB(15q21–22)
*In the prenatal and neonatal period, it is typically impossible to distinguish clinically severe OI caused by defects in type I collagen from severe OI caused byrecessive mutations in CRTAP and LEPRE1. However, individuals who have demonstrable mutations in genes causing recessive OI should be designated astype VII, VIII, or IX, rather than type II or III.
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e299 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from
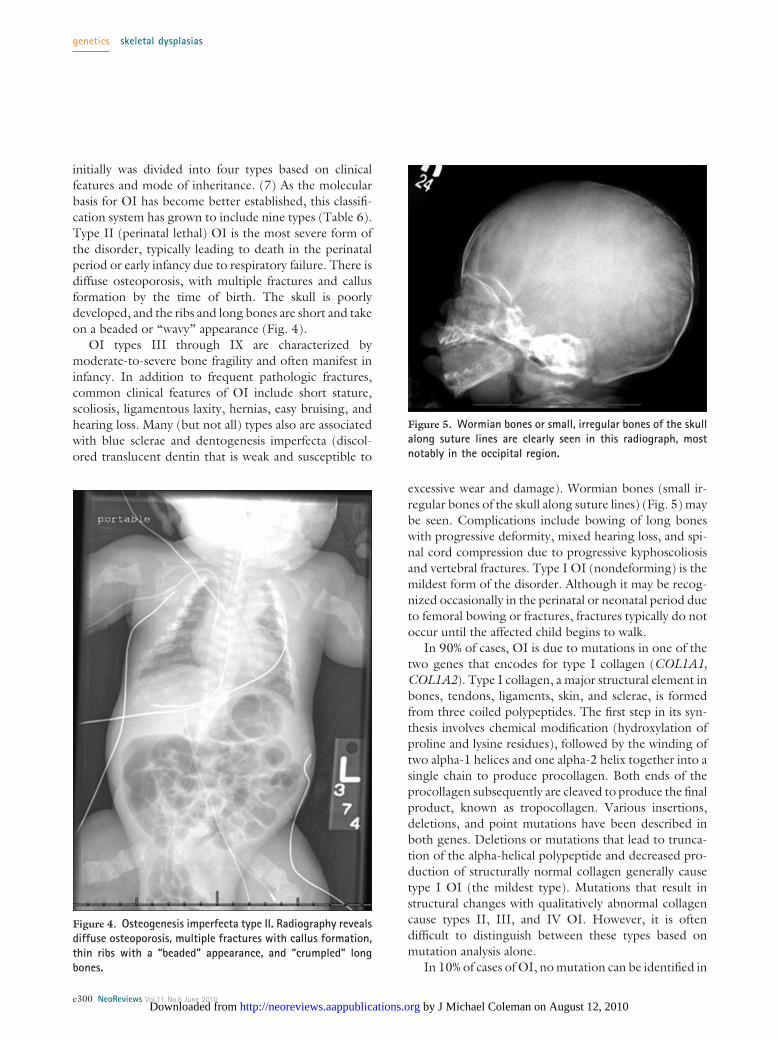
initially was divided into four types based on clinicalfeatures and mode of inheritance. (7) As the molecularbasis for OI has become better established, this classifi-cation system has grown to include nine types (Table 6).Type II (perinatal lethal) OI is the most severe form ofthe disorder, typically leading to death in the perinatalperiod or early infancy due to respiratory failure. There isdiffuse osteoporosis, with multiple fractures and callusformation by the time of birth. The skull is poorlydeveloped, and the ribs and long bones are short and takeon a beaded or “wavy” appearance (Fig. 4).
OI types III through IX are characterized bymoderate-to-severe bone fragility and often manifest ininfancy. In addition to frequent pathologic fractures,common clinical features of OI include short stature,scoliosis, ligamentous laxity, hernias, easy bruising, andhearing loss. Many (but not all) types also are associatedwith blue sclerae and dentogenesis imperfecta (discol-ored translucent dentin that is weak and susceptible to
excessive wear and damage). Wormian bones (small ir-regular bones of the skull along suture lines) (Fig. 5) maybe seen. Complications include bowing of long boneswith progressive deformity, mixed hearing loss, and spi-nal cord compression due to progressive kyphoscoliosisand vertebral fractures. Type I OI (nondeforming) is themildest form of the disorder. Although it may be recog-nized occasionally in the perinatal or neonatal period dueto femoral bowing or fractures, fractures typically do notoccur until the affected child begins to walk.
In 90% of cases, OI is due to mutations in one of thetwo genes that encodes for type I collagen (COL1A1,COL1A2). Type I collagen, a major structural element inbones, tendons, ligaments, skin, and sclerae, is formedfrom three coiled polypeptides. The first step in its syn-thesis involves chemical modification (hydroxylation ofproline and lysine residues), followed by the winding oftwo alpha-1 helices and one alpha-2 helix together into asingle chain to produce procollagen. Both ends of theprocollagen subsequently are cleaved to produce the finalproduct, known as tropocollagen. Various insertions,deletions, and point mutations have been described inboth genes. Deletions or mutations that lead to trunca-tion of the alpha-helical polypeptide and decreased pro-duction of structurally normal collagen generally causetype I OI (the mildest type). Mutations that result instructural changes with qualitatively abnormal collagencause types II, III, and IV OI. However, it is oftendifficult to distinguish between these types based onmutation analysis alone.
In 10% of cases of OI, no mutation can be identified in
Figure 4. Osteogenesis imperfecta type II. Radiography revealsdiffuse osteoporosis, multiple fractures with callus formation,thin ribs with a “beaded” appearance, and “crumpled” longbones.
Figure 5. Wormian bones or small, irregular bones of the skullalong suture lines are clearly seen in this radiograph, mostnotably in the occipital region.
genetics skeletal dysplasias
e300 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from
either COL1A gene. Recently, three genes have beendescribed that when mutated, can lead to severe OItransmitted in an autosomal recessive pattern. Thesegenes encode for the proteins cartilage-associated pro-tein (CRTAP), leprecan-like 1 (LEPRE1) and peptidyl-prolyl isomerase B (PPIB), which form an interactivecomplex. It is not clear whether the underlying mecha-nism is defective formation of this complex of proteinsor defective hydroxylation of the proline 936 residueof type I collagen. Up to 5% of cases of type II OImay be due to mutations in these genes. Type VII OI,characterized by moderate deformation, white sclera,absence of dentogenesis imperfecta, fish-scale pattern
of bone lamellation, and an autosomal recessive pat-tern of inheritance, also has been mapped to theCRTAP gene region. Homozygous (recessive) muta-tions in PPIB have been identified in type IX OI. Finally,it has been proposed that patients who have OI due tomutations in CRTAP or LEPRE1 that appear clinicallydistinct from type II or type VII OI should be classified astype VIII.
Because the dominant and recessive forms of OI arenot easily distinguished clinically, DNA testing is neces-sary to provide accurate diagnostic, management, andrecurrence risk information. We recommend startingwith sequencing of COL1A1 and COL1A2, and if this isnormal, proceeding to sequencing of CRTAP, LEPRE1,and PPIB.
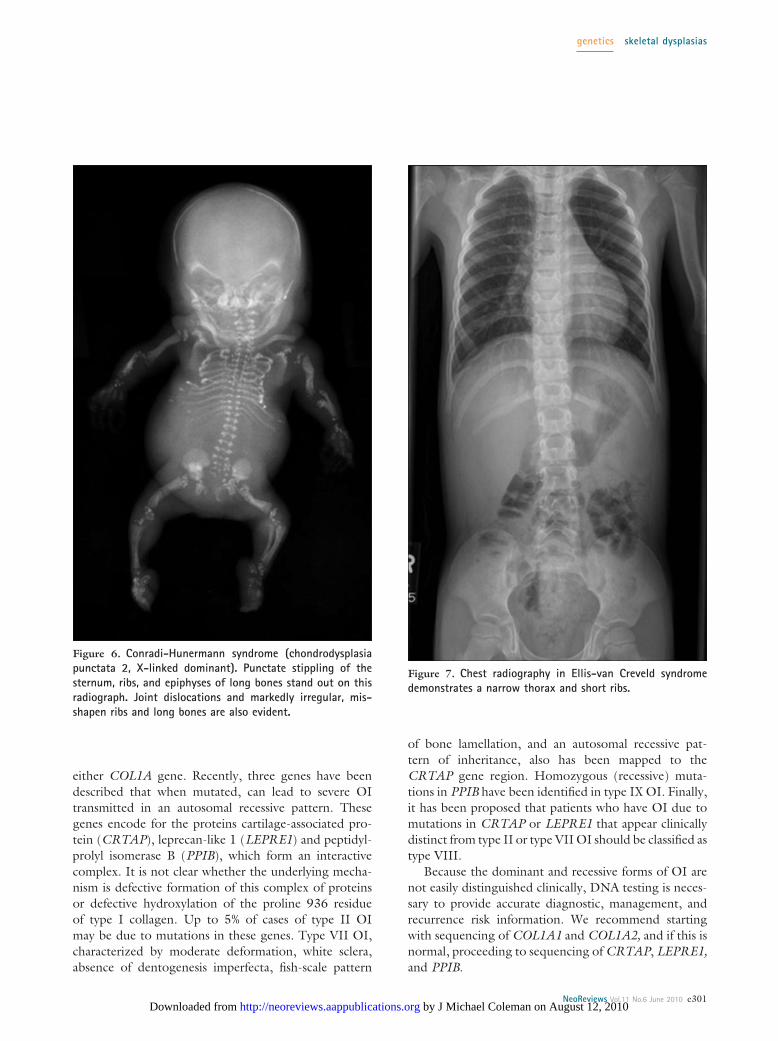
Figure 6. Conradi-Hunermann syndrome (chondrodysplasiapunctata 2, X-linked dominant). Punctate stippling of thesternum, ribs, and epiphyses of long bones stand out on thisradiograph. Joint dislocations and markedly irregular, mis-shapen ribs and long bones are also evident.
Figure 7. Chest radiography in Ellis-van Creveld syndromedemonstrates a narrow thorax and short ribs.
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e301 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

Chondrodysplasia Punctata GroupThe chondrodysplasia punctata syndromes are character-ized by small calcifications in cartilaginous epiphyses(punctuate epiphyses, stippled epiphyses) (Fig. 6) thatare the result of dyssynchronous ossification. In additionto inherited osteochondrodysplasias, a wide range ofother conditions, including peroxisomal disorders (Zell-weger syndrome), chromosomal abnormalities (trisomy18 and 21, Turner syndrome), single-gene disorders(Cornelia de Lange syndrome), metabolic disorders(GM-1 gangliosidosis, galactosialidosis), infections (con-genital rubella), or toxic exposures (maternal diabetes,warfarin embryopathy) can lead to stippled epiphyses.The pattern and location of the stippling often help toestablish a specific diagnosis, which is critical to accuratecounseling and management. For example, Greenbergdysplasia almost always leads to death in utero or at birth,whereas the tibial-metacarpal type of chondrodysplasiapunctata has a very good prognosis, with normal devel-opment and survival to adulthood. Interestingly, in all ofthese conditions, the epiphyseal stippling disappears byabout 3 to 5 years of age, making subsequent diagnosismuch more difficult.
Asphyxiating Thoracic Dystrophy (JeuneSyndrome)
Asphyxiating thoracic dystrophy is characterized by avery narrow and shallow thorax, which can lead to severerespiratory compromise. Patients have short extremitieswith metaphyseal irregularities, short hands and feet withpostaxial polydactyly, and shortmiddle and distal phalanges withcone-shaped epiphyses. Other as-sociated medical problems includeprogressive renal disease, pancreaticand hepatic fibrosis, Hirschsprungdisease, and hydrocephalus. Respi-ratory distress and recurrent respi-ratory infections are the greatestcause of morbidity and mortalityduring the neonatal period and in-fancy. The disorder is transmitted inan autosomal recessive pattern, andtwo genetic loci have been identi-fied to date. A subset of cases aredue to mutations in intraflagellertransport protein (IFT80), locatedin 3q24-q26. Other cases havebeen mapped to the 15q13 region,but a causal gene has yet to be iden-
tified. It can be difficult to distinguish this disorder fromEllis-van Creveld syndrome.
Chondroectodermal Dysplasia (Ellis-vanCreveld Syndrome)
Ellis-van Creveld syndrome, originally described in theAmish population, has many similarities with Jeune syn-drome. Skeletal changes include narrow thorax (Fig. 7),pre- or postaxial polydactyly, fusion of the metacarpals orphalanges (Fig. 8), dysplasia of the pelvis, and progres-sive shortening of the extremities in a proximal-to-distalpattern. Multiple gingival frenulae are common. In con-trast to Jeune syndrome, however, Ellis-van Creveldsyndrome commonly is associated with congenital heartdisease, hypoplastic nails (Fig. 8), and dental anomalies(eg, natal teeth, partial odontia, enamel hypoplasia, dys-plastic teeth). The most commonly seen cardiac lesionsare atrial septal defect or single atrium.
ManagementThe care of children who have disorders of skeletaldevelopment presenting in the neonatal period is a com-plex, multidisciplinary effort best coordinated by a centerwith expertise in these conditions. Neonatologists play acrucial role in the stabilization and initial identification ofaffected infants because many have a variety of medicalcomplications shortly after birth, including respiratorydistress due to pulmonary hypoplasia and restrictive lungdisease. Medical geneticists and radiologists play criticalroles in establishing a definitive diagnosis, which is cru-cial to provide neonatologists and families with prognos-
Figure 8. Hands in Ellis-van Creveld syndrome A. Note the short, broad fingers anddeep-set, hypoplastic, ridged fingernails. B. Radiography reveals cone-shaped epiphyses ofthe phalanges (white arrow), and fusion of the fourth and fifth metacarpals and thecapitate and hamate bones.
genetics skeletal dysplasias
e302 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from
tic information to direct the overall course and intensityof care. Pediatric orthopedists, physical medicine andrehabilitation specialists, and physical and occupationaltherapists are needed to monitor the physical develop-ment of the children and address specific deformities.Depending on the disorder, a host of specialists may beinvolved in the affected child’s care, including but notlimited to pulmonology (eg, pulmonary hypoplasia, re-strictive lung disease), cardiology, dentistry, nephrology,ophthalmology (eg, cataracts, retinal detachment, myo-pia), otorhinolaryngology, and neurosurgery (eg, steno-sis of the foramen magnum, hydrocephalus). Equallyimportant, psychologists, social workers, and family sup-
port groups take critical parts in the psychosocial care ofpatients and their families.
Few pharmacologic treatments are available for themanagement of disorders of skeletal development. Intra-venous infusion of pamidronate (a bisphosphonate) hasbeen reported to increased bone mineral density, de-crease biochemical markers of bone reabsorption, andpossibly decrease fracture risk in OI. This is presently thestandard of care for types III and IV OI. Human growthhormone has been employed on an experimental basis toincrease final adult height in several skeletal dysplasias,but its use remains controversial.
Case Continued: Evaluating a SuspectedSkeletal DysplasiaAfter transfer the NICU, the patient is stabilized withnasal continuous positive airway pressure. A skeletal surveyis performed (Figs. 9 and 10) and a genetics consultationobtained. The most notable features on review of the skeletal
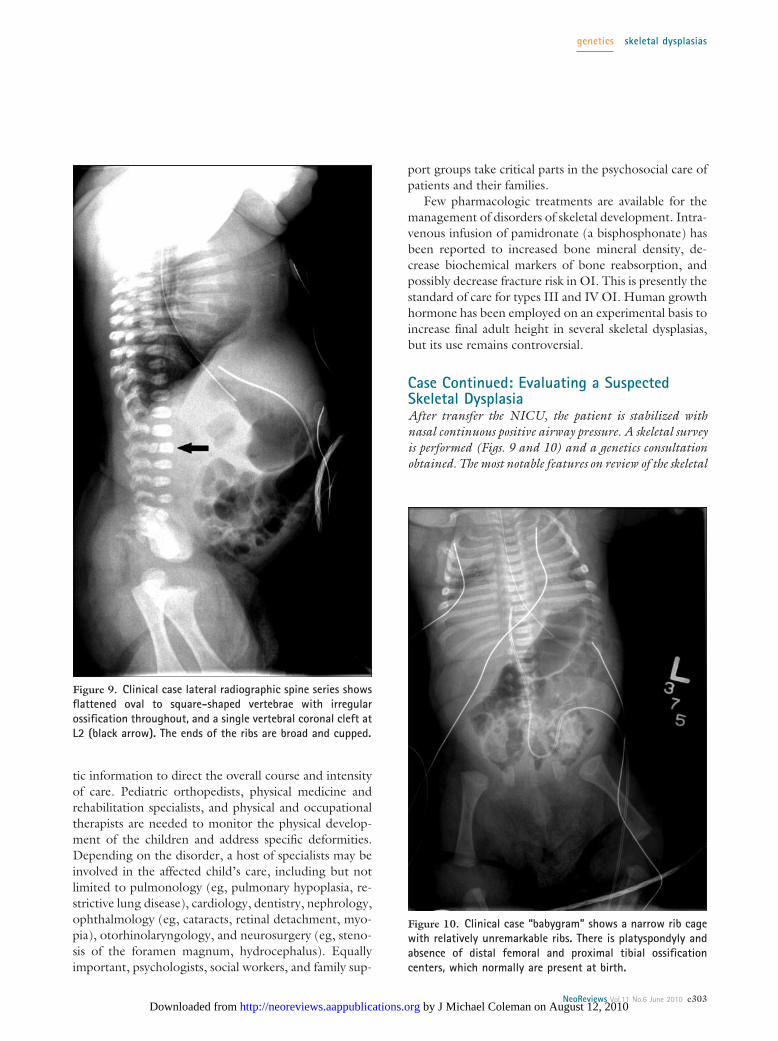
Figure 9. Clinical case lateral radiographic spine series showsflattened oval to square-shaped vertebrae with irregularossification throughout, and a single vertebral coronal cleft atL2 (black arrow). The ends of the ribs are broad and cupped.
Figure 10. Clinical case “babygram” shows a narrow rib cagewith relatively unremarkable ribs. There is platyspondyly andabsence of distal femoral and proximal tibial ossificationcenters, which normally are present at birth.
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e303 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

survey are vertebral abnormalities and an absence of epiph-yses normally present at birth. Vertebrae are flattened ovalto square-shaped, with irregular ossification throughout.A single vertebral coronal cleft is apparent at L2 (a featureusually not seen in this particular diagnosis). Distal femo-ral and proximal tibial epiphyseal ossification centers areabsent (normally present at birth). The rib cage appearssomewhat narrow, but the ribs themselves are grossly nor-mal. Summarizing these preliminary data, the infant ap-pears to be affected by a disorder of skeletal developmentwith prominent spondyloepiphyseal involvement.
This specific pattern of skeletal involvement can be usedto search for spondyloepiphyseal dysplasias presenting in thenewborn period in “The Nosology and Classification ofGenetic Skeletal Disorders”; major atlases, such as BoneDysplasias and Taybi & Lachman’s Radiology of Syn-dromes, Metabolic Disorders, and Skeletal Dysplasias;public electronic databases, such as Online Mendelian In-heritance in Man; and commercial databases, such as theLondon Dysmorphology Database and the RadiologicalElectronic Atlas of Malformation Syndromes and Skele-tal Dysplasia. In this case, consultation with experts inradiology and medical genetics establishes a diagnosis of atype II collagenopathy, most likely spondyloepiphyseal dys-plasia congenita, on clinical and radiologic grounds.
Genetic testing is conducted to confirm the diagnosis.A query of www.genetests.org can be performed to identifyclinical laboratories offering testing for this and otherskeletal dysplasias. In this case, sequence analysis of theentire coding region of COL2A1 is performed, which re-veals a 1546G�A missense mutation in exon 22, convert-ing a conserved glycine in the triple helical domain to serine(Gly316Ser). A similar mutation (Gly316Asp) had beenreported previously in a patient who had achondrogenesistype II. (8)
The patient has a prolonged NICU stay due to respira-tory failure from pulmonary hypoplasia, which requireslong-term mechanical ventilation and tracheostomy place-ment. A gastrostomy tube also is placed due to inabilityto feed orally. Further evaluation reveals moderate-to-profound hearing loss bilaterally, mild myopia, and cervi-cal spine instability requiring use of a Minerva stabilizingbrace. The patient is discharged from the hospital at ap-proximately 9 weeks of age.
ACKNOWLEDGMENTS. We would like to thank SeemaJilani, MD, Jessica Swarr, PA-C, and Kelly Wade, MD,for their critical reviews of this manuscript.
References1. Spranger JW, Brill PW, Poznanski AK, et al. Bone Dysplasias: AnAtlas of Genetic Disorders of Skeletal Development. New York, NY:Oxford University Press; 20022. Superti-Furga A, Unger S. Nosology and classification of geneticskeletal disorders: 2006 revision. Am J Med Genet A. 2007;143:1–183. Mortier GR. The diagnosis of skeletal dysplasias: a multidisci-plinary approach. Eur J Radiol. 2001;40:161–1674. Offiah AC, Hall CM. Radiological diagnosis of the constitu-tional disorders of bone. As easy as A, B, C? Pediatr Radiol.2003;33:153–1615. Krakow D, Lachman RS, Rimoin DL. Guidelines for the pre-natal diagnosis of fetal skeletal dysplasias. Genet Med. 2009;11:127–1336. Superti-Furga A, Bonafe L, Rimoin DL. Molecular-pathogeneticclassification of genetic disorders of the skeleton. Am J Med Genet.2001;106:282–2937. Sillence DO, Rimoin DL, Danks DM. Clinical variability inosteogenesis imperfecta-variable expressivity or genetic heteroge-neity. Birth Defects Orig Artic Ser. 1979;15(5B):113–1298. Faivre L, M. Le Merrer, Douvier S, et al. Recurrence of achon-drogenesis type II within the same family: evidence for germlinemosaicism. Am J Med Genet A. 2004;126A:308–312
Suggested ReadingByers PH, Krakow D, Nunes ME, Pepin M. Genetic evaluation of
suspected osteogenesis imperfecta (OI). Genet Med. 2006;8:383–388
Kornak U, Mundlos S. Genetic disorders of the skeleton: a devel-opmental approach. Am J Hum Genet. 2003;73:447–474
Lindsay R. Modeling the benefits of pamidronate in children withosteogenesis imperfecta. J Clin Invest. 2002;110:1239–1241
Marlowe A, Pepin MG, Byers PH. Testing for osteogenesis imper-fecta in cases of suspected non-accidental injury. J Med Genet.2002;39:382–386
Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007;2:40Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;
363:1377–1385Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteo-
genesis imperfecta. J Med Genet. 1979;16:101–116Spranger J, Winterpacht A, Zabel B. The type II collagenopathies: a
spectrum of chondrodysplasias. Eur J Pediatr. 1994;153:56–65Trotter TL, Hall JG. Health supervision for children with achon-
droplasia. Pediatrics. 2005;116:771–783Tuysuz B, Baris S, Aksoy F, Madazli R, Ungur S, Sever L. Clinical
variability of asphyxiating thoracic dystrophy (Jeune) syndrome:evaluation and classification of 13 patients. Am J Med Genet A.2009;149A:1727–1733
American Board of Pediatrics Neonatal-PerinatalMedicine Content Specification• Recognize the clinical features and know
how to diagnose and manage skeletaldysplasias, such as achondrogenesis,achondroplasia, chondrodermal dysplasia,epiphyseal dysostosis, osteogenesisimperfecta, hypophosphatasia, etc.
genetics skeletal dysplasias
e304 NeoReviews Vol.11 No.6 June 2010 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

NeoReviews Quiz
1. Development of the skeleton begins early in embryonic life, starting with patterning of the somites andformation of the limb buds in the third and fourth weeks of embryogenesis. Of the following, abnormalitiesin early patterning events during skeletal development are most likely to result in:
A. Disruptions.B. Dysostosis.C. Osteolysis.D. Primary dysplasia.E. Secondary dysplasia.
2. When evaluating a neonate who has a skeletal disorder, it is important to perform a detailed clinicalexamination, focusing on physical features and other birth defects that may aid in determining a specificdiagnosis. Of the following, large fontanelles and wide sutures are most notable for:
A. Campomelic dysplasia.B. Chondrodysplasia punctata.C. Cleidocranial dysplasia.D. Osteogenesis imperfecta.E. Spondylometaphyseal dysplasia.
3. For families with a history of a specific disorder of skeletal development that has been confirmed withmolecular methods, mutation analysis may be performed on DNA obtained by chorionic villus sampling oramniocentesis. Of the following, the diagnosis of campomelic dysplasia is most likely with a mutation inthe gene:
A. COL2A1.B. FGFR3.C. IFT80.D. SOX9.E. TNAP.
4. You are caring for a 4-day-old female infant whom you suspect has asphyxiating thoracic dystrophy (Jeunesyndrome) based on the findings that include a small chest, short hands, and short feet with postaxialpolydactyly. The infant is receiving mechanical ventilation, intravenous fluids, and partial enteral feedings.The nurse informs you that the infant has not passed meconium. Of the following, the most likely cause offailure to pass meconium in this infant is:
A. Anal stenosis.B. Hirschsprung disease.C. Ileal atresia.D. Meconium plug.E. Mid-gut volvulus.
5. You are caring for a term newborn in whom you suspect achondroplasia based on the findings that includerhizomelic shortening of extremities, “trident” hands with “cone-shaped” phalanges, and macrocephaly. Youorder a radiographic skeletal survey. Of the following, the most specific finding for achondroplasia onimaging of the spine is:
A. Decreasing lumbar interpedicular distance.B. Hemivertebrae.C. Spina bifida occulta.D. Thoracolumbar scoliosis.E. Vertebral coronal clefts.
genetics skeletal dysplasias
NeoReviews Vol.11 No.6 June 2010 e305 by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from

DOI: 10.1542/neo.11-6-e290 2010;11;e290-e305 NeoReviews
Daniel T. Swarr and V. Reid Sutton Genetics
Skeletal Dysplasias in the Newborn: Diagnostic Evaluation and Developmental
& ServicesUpdated Information
s;11/6/e290http://neoreviews.aappublications.org/cgi/content/full/neoreviewincluding high-resolution figures, can be found at:
Permissions & Licensing
http://neoreviews.aappublications.org/misc/Permissions.shtmltables) or in its entirety can be found online at: Information about reproducing this article in parts (figures,
Reprints http://neoreviews.aappublications.org/misc/reprints.shtml
Information about ordering reprints can be found online:
by J Michael Coleman on August 12, 2010 http://neoreviews.aappublications.orgDownloaded from





























