Embed Size (px)
Citation preview
..............................................................
Signal-dependent regulation ofsplicing via phosphorylationof Sam68Nathalie Matter*†, Peter Herrlich*‡ & Harald Konig*
* Forschungszentrum Karlsruhe GmbH, Institut fur Toxikologie und Genetik, and‡ Universitat Karlsruhe, Institut fur Genetik, Postfach 3640, D-76021 Karlsruhe,Germany.............................................................................................................................................................................
Evolution of human organismal complexity from a relativelysmall number of genes1,2—only approximately twice that of wormor fly—is explained mainly by mechanisms generating multipleproteins from a single gene, the most prevalent of which isalternative pre-messenger-RNA splicing1,3,4. Appropriate spatialand temporal generation of splice variants demands that alterna-tive splicing be subject to extensive regulation, similar to tran-scriptional control. Activation by extracellular cues of severalcellular signalling pathways can indeed regulate alternativesplicing5–8. Here we address the link between signal transductionand splice regulation. We show that the nuclear RNA-bindingprotein Sam68 is a new extracellular signal-regulated kinase(ERK) target. It binds exonic splice-regulatory elements of analternatively spliced exon that is physiologically regulated by theRas signalling pathway, namely exon v5 of CD44. Forcedexpression of Sam68 enhanced ERK-mediated inclusion of thev5-exon sequence in mRNA. This enhancement was impaired bymutation of ERK-phosphorylation sites in Sam68, whereas ERKphosphorylation of Sam68 stimulated splicing of the v5 exonin vitro. Finally, Ras-pathway-induced alternative splicing of theendogenous CD44-v5 exon was abolished by suppression ofSam68 expression. Our data define Sam68 as a prototype regu-lator of alternative splicing whose function depends on proteinmodification in response to extracellular cues.
Alternatively spliced isoforms of CD44 are generated by theinclusion of up to 10 variant exon sequences (v1–v10) in CD44mRNA and are decisive in embryonic development, in immuneresponse and in tumour progression9–12. We previously identifiedRNA elements necessary for signalling-induced alternative splicingin CD44 exon v5 (ref. 5), an exon frequently included in the courseof tumour progression and upon T-cell activation8,12, and weshowed that in T lymphocytes and lymphoma cells alternativesplicing is regulated through these elements by the ERK mitogen-activated protein (MAP) kinase pathway8, an evolutionarily con-served enzyme cascade connecting cell surface receptors toregulatory targets13. Recently, Stoss et al. in collaboration with uslinked Etoile/Sam68-like mammalian protein (SLM-2) to alterna-tive splicing14. SLM-2 belongs to the STAR (signal tranduction andactivation of RNA) protein family15, whose domain organizationsuggests RNA binding and modification in response to signaltransduction. This led to the hypothesis that STAR proteins couldbe involved in coupling signal transduction to alternative pre-mRNA splicing14.
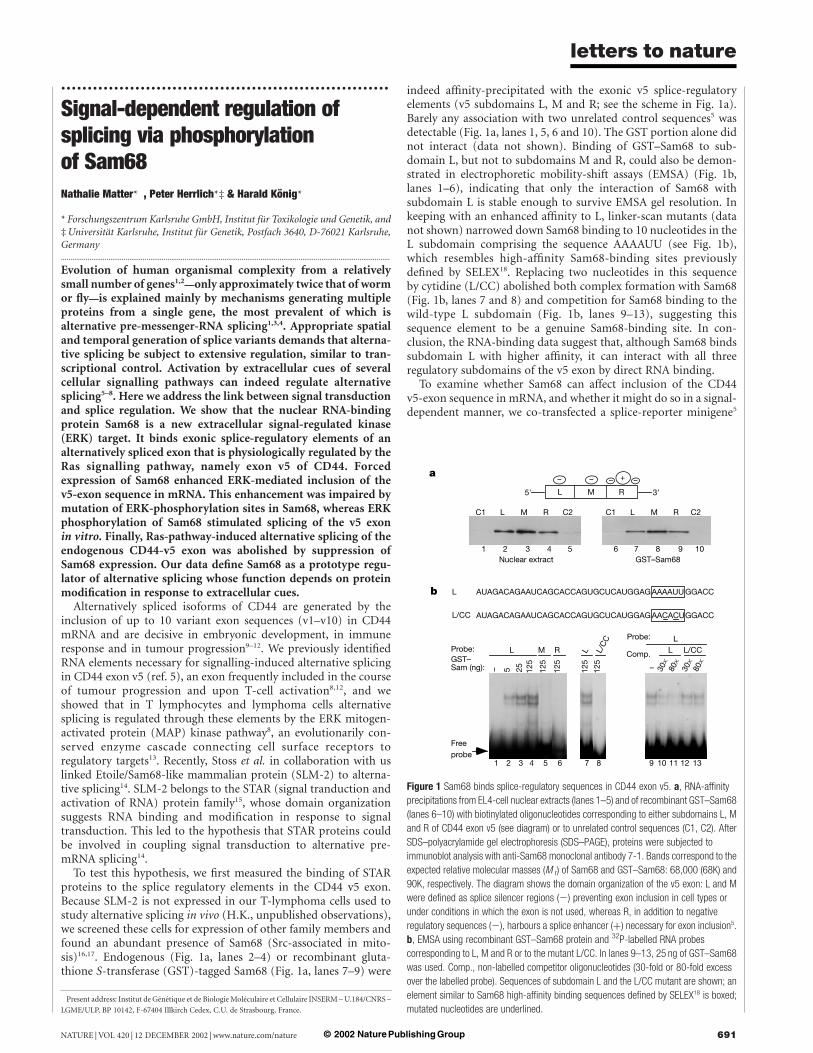
To test this hypothesis, we first measured the binding of STARproteins to the splice regulatory elements in the CD44 v5 exon.Because SLM-2 is not expressed in our T-lymphoma cells used tostudy alternative splicing in vivo (H.K., unpublished observations),we screened these cells for expression of other family members andfound an abundant presence of Sam68 (Src-associated in mito-sis)16,17. Endogenous (Fig. 1a, lanes 2–4) or recombinant gluta-thione S-transferase (GST)-tagged Sam68 (Fig. 1a, lanes 7–9) were
indeed affinity-precipitated with the exonic v5 splice-regulatoryelements (v5 subdomains L, M and R; see the scheme in Fig. 1a).Barely any association with two unrelated control sequences5 wasdetectable (Fig. 1a, lanes 1, 5, 6 and 10). The GST portion alone didnot interact (data not shown). Binding of GST–Sam68 to sub-domain L, but not to subdomains M and R, could also be demon-strated in electrophoretic mobility-shift assays (EMSA) (Fig. 1b,lanes 1–6), indicating that only the interaction of Sam68 withsubdomain L is stable enough to survive EMSA gel resolution. Inkeeping with an enhanced affinity to L, linker-scan mutants (datanot shown) narrowed down Sam68 binding to 10 nucleotides in theL subdomain comprising the sequence AAAAUU (see Fig. 1b),which resembles high-affinity Sam68-binding sites previouslydefined by SELEX18. Replacing two nucleotides in this sequenceby cytidine (L/CC) abolished both complex formation with Sam68(Fig. 1b, lanes 7 and 8) and competition for Sam68 binding to thewild-type L subdomain (Fig. 1b, lanes 9–13), suggesting thissequence element to be a genuine Sam68-binding site. In con-clusion, the RNA-binding data suggest that, although Sam68 bindssubdomain L with higher affinity, it can interact with all threeregulatory subdomains of the v5 exon by direct RNA binding.
To examine whether Sam68 can affect inclusion of the CD44v5-exon sequence in mRNA, and whether it might do so in a signal-dependent manner, we co-transfected a splice-reporter minigene5
Figure 1 Sam68 binds splice-regulatory sequences in CD44 exon v5. a, RNA-affinity
precipitations from EL4-cell nuclear extracts (lanes 1–5) and of recombinant GST–Sam68
(lanes 6–10) with biotinylated oligonucleotides corresponding to either subdomains L, M
and R of CD44 exon v5 (see diagram) or to unrelated control sequences (C1, C2). After
SDS–polyacrylamide gel electrophoresis (SDS–PAGE), proteins were subjected to
immunoblot analysis with anti-Sam68 monoclonal antibody 7-1. Bands correspond to the
expected relative molecular masses (M r) of Sam68 and GST–Sam68: 68,000 (68K) and
90K, respectively. The diagram shows the domain organization of the v5 exon: L and M
were defined as splice silencer regions (2) preventing exon inclusion in cell types or
under conditions in which the exon is not used, whereas R, in addition to negative
regulatory sequences (2), harbours a splice enhancer (þ) necessary for exon inclusion5.
b, EMSA using recombinant GST–Sam68 protein and 32P-labelled RNA probes
corresponding to L, M and R or to the mutant L/CC. In lanes 9–13, 25 ng of GST–Sam68
was used. Comp., non-labelled competitor oligonucleotides (30-fold or 80-fold excess
over the labelled probe). Sequences of subdomain L and the L/CC mutant are shown; an
element similar to Sam68 high-affinity binding sequences defined by SELEX18 is boxed;
mutated nucleotides are underlined.† Present address: Institut de Genetique et de Biologie Moleculaire et Cellulaire INSERM – U.184/CNRS –
LGME/ULP, BP 10142, F-67404 Illkirch Cedex, C.U. de Strasbourg, France.
letters to nature
NATURE | VOL 420 | 12 DECEMBER 2002 | www.nature.com/nature 691© 2002 Nature Publishing Group
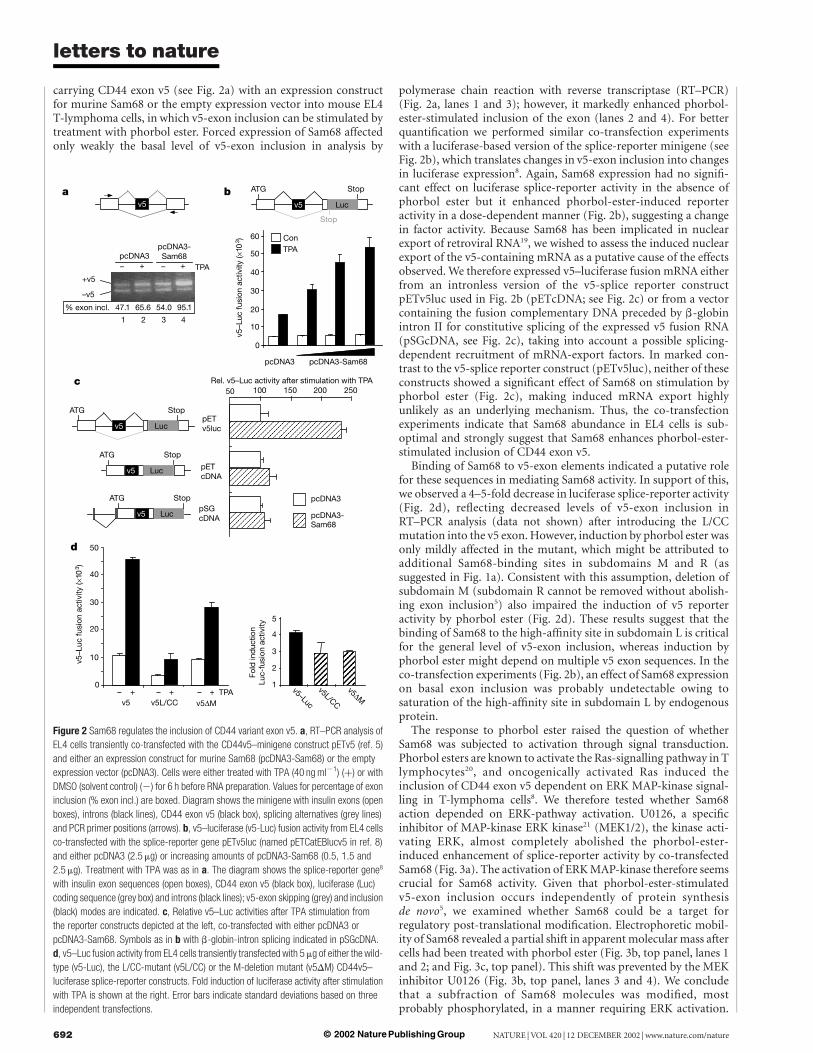
carrying CD44 exon v5 (see Fig. 2a) with an expression constructfor murine Sam68 or the empty expression vector into mouse EL4T-lymphoma cells, in which v5-exon inclusion can be stimulated bytreatment with phorbol ester. Forced expression of Sam68 affectedonly weakly the basal level of v5-exon inclusion in analysis by
polymerase chain reaction with reverse transcriptase (RT–PCR)(Fig. 2a, lanes 1 and 3); however, it markedly enhanced phorbol-ester-stimulated inclusion of the exon (lanes 2 and 4). For betterquantification we performed similar co-transfection experimentswith a luciferase-based version of the splice-reporter minigene (seeFig. 2b), which translates changes in v5-exon inclusion into changesin luciferase expression8. Again, Sam68 expression had no signifi-cant effect on luciferase splice-reporter activity in the absence ofphorbol ester but it enhanced phorbol-ester-induced reporteractivity in a dose-dependent manner (Fig. 2b), suggesting a changein factor activity. Because Sam68 has been implicated in nuclearexport of retroviral RNA19, we wished to assess the induced nuclearexport of the v5-containing mRNA as a putative cause of the effectsobserved. We therefore expressed v5–luciferase fusion mRNA eitherfrom an intronless version of the v5-splice reporter constructpETv5luc used in Fig. 2b (pETcDNA; see Fig. 2c) or from a vectorcontaining the fusion complementary DNA preceded by b-globinintron II for constitutive splicing of the expressed v5 fusion RNA(pSGcDNA, see Fig. 2c), taking into account a possible splicing-dependent recruitment of mRNA-export factors. In marked con-trast to the v5-splice reporter construct (pETv5luc), neither of theseconstructs showed a significant effect of Sam68 on stimulation byphorbol ester (Fig. 2c), making induced mRNA export highlyunlikely as an underlying mechanism. Thus, the co-transfectionexperiments indicate that Sam68 abundance in EL4 cells is sub-optimal and strongly suggest that Sam68 enhances phorbol-ester-stimulated inclusion of CD44 exon v5.
Binding of Sam68 to v5-exon elements indicated a putative rolefor these sequences in mediating Sam68 activity. In support of this,we observed a 4–5-fold decrease in luciferase splice-reporter activity(Fig. 2d), reflecting decreased levels of v5-exon inclusion inRT–PCR analysis (data not shown) after introducing the L/CCmutation into the v5 exon. However, induction by phorbol ester wasonly mildly affected in the mutant, which might be attributed toadditional Sam68-binding sites in subdomains M and R (assuggested in Fig. 1a). Consistent with this assumption, deletion ofsubdomain M (subdomain R cannot be removed without abolish-ing exon inclusion5) also impaired the induction of v5 reporteractivity by phorbol ester (Fig. 2d). These results suggest that thebinding of Sam68 to the high-affinity site in subdomain L is criticalfor the general level of v5-exon inclusion, whereas induction byphorbol ester might depend on multiple v5 exon sequences. In theco-transfection experiments (Fig. 2b), an effect of Sam68 expressionon basal exon inclusion was probably undetectable owing tosaturation of the high-affinity site in subdomain L by endogenousprotein.
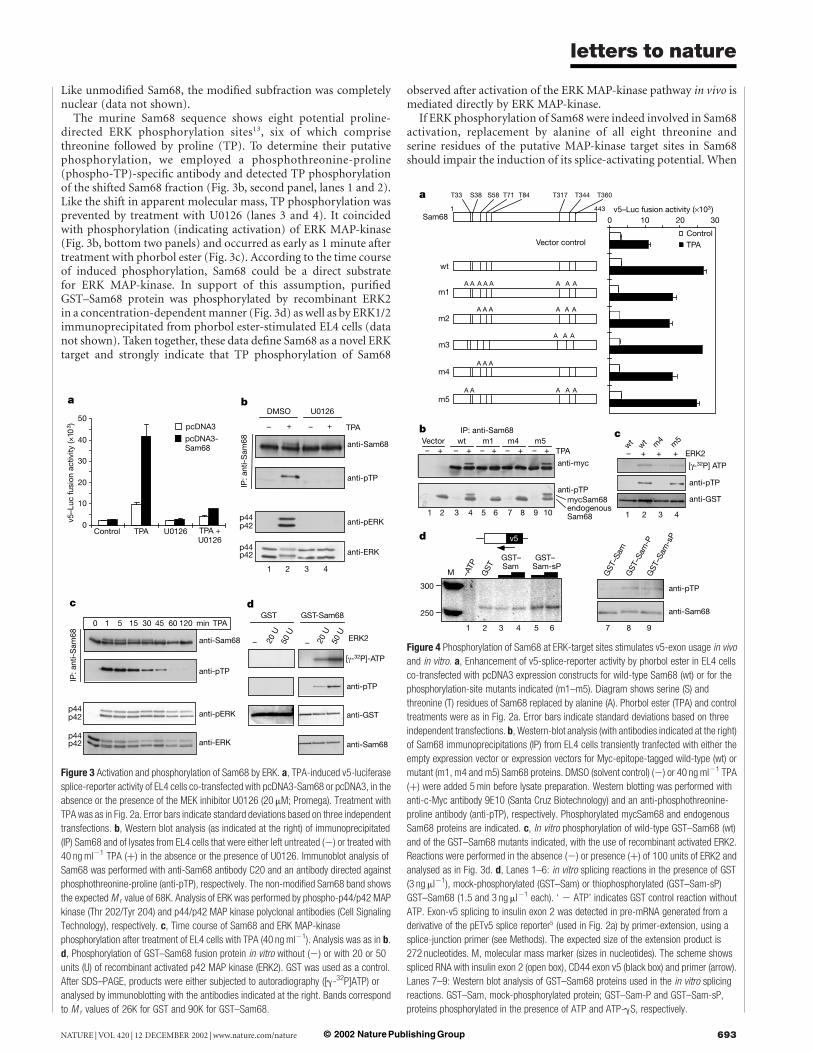
The response to phorbol ester raised the question of whetherSam68 was subjected to activation through signal transduction.Phorbol esters are known to activate the Ras-signalling pathway in Tlymphocytes20, and oncogenically activated Ras induced theinclusion of CD44 exon v5 dependent on ERK MAP-kinase signal-ling in T-lymphoma cells8. We therefore tested whether Sam68action depended on ERK-pathway activation. U0126, a specificinhibitor of MAP-kinase ERK kinase21 (MEK1/2), the kinase acti-vating ERK, almost completely abolished the phorbol-ester-induced enhancement of splice-reporter activity by co-transfectedSam68 (Fig. 3a). The activation of ERK MAP-kinase therefore seemscrucial for Sam68 activity. Given that phorbol-ester-stimulatedv5-exon inclusion occurs independently of protein synthesisde novo5, we examined whether Sam68 could be a target forregulatory post-translational modification. Electrophoretic mobil-ity of Sam68 revealed a partial shift in apparent molecular mass aftercells had been treated with phorbol ester (Fig. 3b, top panel, lanes 1and 2; and Fig. 3c, top panel). This shift was prevented by the MEKinhibitor U0126 (Fig. 3b, top panel, lanes 3 and 4). We concludethat a subfraction of Sam68 molecules was modified, mostprobably phosphorylated, in a manner requiring ERK activation.
Figure 2 Sam68 regulates the inclusion of CD44 variant exon v5. a, RT–PCR analysis of
EL4 cells transiently co-transfected with the CD44v5–minigene construct pETv5 (ref. 5)
and either an expression construct for murine Sam68 (pcDNA3-Sam68) or the empty
expression vector (pcDNA3). Cells were either treated with TPA (40 ng ml21) (þ) or with
DMSO (solvent control) (2) for 6 h before RNA preparation. Values for percentage of exon
inclusion (% exon incl.) are boxed. Diagram shows the minigene with insulin exons (open
boxes), introns (black lines), CD44 exon v5 (black box), splicing alternatives (grey lines)
and PCR primer positions (arrows). b, v5–luciferase (v5-Luc) fusion activity from EL4 cells
co-transfected with the splice-reporter gene pETv5luc (named pETCatEBlucv5 in ref. 8)
and either pcDNA3 (2.5 mg) or increasing amounts of pcDNA3-Sam68 (0.5, 1.5 and
2.5 mg). Treatment with TPA was as in a. The diagram shows the splice-reporter gene8
with insulin exon sequences (open boxes), CD44 exon v5 (black box), luciferase (Luc)
coding sequence (grey box) and introns (black lines); v5-exon skipping (grey) and inclusion
(black) modes are indicated. c, Relative v5–Luc activities after TPA stimulation from
the reporter constructs depicted at the left, co-transfected with either pcDNA3 or
pcDNA3-Sam68. Symbols as in b with b-globin-intron splicing indicated in pSGcDNA.
d, v5–Luc fusion activity from EL4 cells transiently transfected with 5 mg of either the wild-
type (v5-Luc), the L/CC-mutant (v5L/CC) or the M-deletion mutant (v5DM) CD44v5–
luciferase splice-reporter constructs. Fold induction of luciferase activity after stimulation
with TPA is shown at the right. Error bars indicate standard deviations based on three
independent transfections.
letters to nature
NATURE | VOL 420 | 12 DECEMBER 2002 | www.nature.com/nature692 © 2002 Nature Publishing Group
Like unmodified Sam68, the modified subfraction was completelynuclear (data not shown).
The murine Sam68 sequence shows eight potential proline-directed ERK phosphorylation sites13, six of which comprisethreonine followed by proline (TP). To determine their putativephosphorylation, we employed a phosphothreonine-proline(phospho-TP)-specific antibody and detected TP phosphorylationof the shifted Sam68 fraction (Fig. 3b, second panel, lanes 1 and 2).Like the shift in apparent molecular mass, TP phosphorylation wasprevented by treatment with U0126 (lanes 3 and 4). It coincidedwith phosphorylation (indicating activation) of ERK MAP-kinase(Fig. 3b, bottom two panels) and occurred as early as 1 minute aftertreatment with phorbol ester (Fig. 3c). According to the time courseof induced phosphorylation, Sam68 could be a direct substratefor ERK MAP-kinase. In support of this assumption, purifiedGST–Sam68 protein was phosphorylated by recombinant ERK2in a concentration-dependent manner (Fig. 3d) as well as by ERK1/2immunoprecipitated from phorbol ester-stimulated EL4 cells (datanot shown). Taken together, these data define Sam68 as a novel ERKtarget and strongly indicate that TP phosphorylation of Sam68
observed after activation of the ERK MAP-kinase pathway in vivo ismediated directly by ERK MAP-kinase.
If ERK phosphorylation of Sam68 were indeed involved in Sam68activation, replacement by alanine of all eight threonine andserine residues of the putative MAP-kinase target sites in Sam68should impair the induction of its splice-activating potential. When
Figure 4 Phosphorylation of Sam68 at ERK-target sites stimulates v5-exon usage in vivo
and in vitro. a, Enhancement of v5-splice-reporter activity by phorbol ester in EL4 cells
co-transfected with pcDNA3 expression constructs for wild-type Sam68 (wt) or for the
phosphorylation-site mutants indicated (m1–m5). Diagram shows serine (S) and
threonine (T) residues of Sam68 replaced by alanine (A). Phorbol ester (TPA) and control
treatments were as in Fig. 2a. Error bars indicate standard deviations based on three
independent transfections. b, Western-blot analysis (with antibodies indicated at the right)
of Sam68 immunoprecipitations (IP) from EL4 cells transiently tranfected with either the
empty expression vector or expression vectors for Myc-epitope-tagged wild-type (wt) or
mutant (m1, m4 and m5) Sam68 proteins. DMSO (solvent control) (2) or 40 ng ml21 TPA
(þ) were added 5 min before lysate preparation. Western blotting was performed with
anti-c-Myc antibody 9E10 (Santa Cruz Biotechnology) and an anti-phosphothreonine-
proline antibody (anti-pTP), respectively. Phosphorylated mycSam68 and endogenous
Sam68 proteins are indicated. c, In vitro phosphorylation of wild-type GST–Sam68 (wt)
and of the GST–Sam68 mutants indicated, with the use of recombinant activated ERK2.
Reactions were performed in the absence (2) or presence (þ) of 100 units of ERK2 and
analysed as in Fig. 3d. d, Lanes 1–6: in vitro splicing reactions in the presence of GST
(3 ng ml21), mock-phosphorylated (GST–Sam) or thiophosphorylated (GST–Sam-sP)
GST–Sam68 (1.5 and 3 ng ml21 each). ‘ 2 ATP’ indicates GST control reaction without
ATP. Exon-v5 splicing to insulin exon 2 was detected in pre-mRNA generated from a
derivative of the pETv5 splice reporter5 (used in Fig. 2a) by primer-extension, using a
splice-junction primer (see Methods). The expected size of the extension product is
272 nucleotides. M, molecular mass marker (sizes in nucleotides). The scheme shows
spliced RNA with insulin exon 2 (open box), CD44 exon v5 (black box) and primer (arrow).
Lanes 7–9: Western blot analysis of GST–Sam68 proteins used in the in vitro splicing
reactions. GST–Sam, mock-phosphorylated protein; GST–Sam-P and GST–Sam-sP,
proteins phosphorylated in the presence of ATP and ATP-gS, respectively.
Figure 3 Activation and phosphorylation of Sam68 by ERK. a, TPA-induced v5-luciferase
splice-reporter activity of EL4 cells co-transfected with pcDNA3-Sam68 or pcDNA3, in the
absence or the presence of the MEK inhibitor U0126 (20 mM; Promega). Treatment with
TPA was as in Fig. 2a. Error bars indicate standard deviations based on three independent
transfections. b, Western blot analysis (as indicated at the right) of immunoprecipitated
(IP) Sam68 and of lysates from EL4 cells that were either left untreated (2) or treated with
40 ng ml21 TPA (þ) in the absence or the presence of U0126. Immunoblot analysis of
Sam68 was performed with anti-Sam68 antibody C20 and an antibody directed against
phosphothreonine-proline (anti-pTP), respectively. The non-modified Sam68 band shows
the expected M r value of 68K. Analysis of ERK was performed by phospho-p44/p42 MAP
kinase (Thr 202/Tyr 204) and p44/p42 MAP kinase polyclonal antibodies (Cell Signaling
Technology), respectively. c, Time course of Sam68 and ERK MAP-kinase
phosphorylation after treatment of EL4 cells with TPA (40 ng ml21). Analysis was as in b.
d, Phosphorylation of GST–Sam68 fusion protein in vitro without (2) or with 20 or 50
units (U) of recombinant activated p42 MAP kinase (ERK2). GST was used as a control.
After SDS–PAGE, products were either subjected to autoradiography ([g-32P]ATP) or
analysed by immunoblotting with the antibodies indicated at the right. Bands correspond
to M r values of 26K for GST and 90K for GST–Sam68.
letters to nature
NATURE | VOL 420 | 12 DECEMBER 2002 | www.nature.com/nature 693© 2002 Nature Publishing Group

co-transfected with the CD44-v5 splice-reporter construct, theeight-amino-acid exchange mutant was significantly impaired inits ability to mediate phorbol-ester-induced exon inclusion (Fig. 4a,mutant m1). By swapping Sam68 portions comprising mutated andnon-mutated ERK target sites, we found that Sam68 mutated inSer 58, Thr 71 and Thr 84 showed the same extent of impairmentin induced exon inclusion as did Sam68 mutated in all S/TP sites(Fig. 4a, mutant m4). Conversely, mutants in which these residueswere unchanged behaved like the wild-type Sam68 protein (Fig. 4a,mutants m3 and m5). Single mutations of residues Ser 58, Thr 71 orThr 84 showed no significant effects. Binding of RNA by multiplephosphorylation-site mutants was comparable to that of the wild-type protein (data not shown), excluding reduced binding ofRNA as a cause of their impaired function. Myc-epitope-taggedversions of the total S/TP ! A mutant (m1) as well as of the theS58A-T71A-T84A-triple mutant (m4) expressed in EL4 cells nolonger produced the phorbol-ester-induced shift in apparent mol-ecular mass (Fig. 4b, upper panel, lanes 3–8) nor the additional(mycSam68) TP-phosphorylated band (lower panel, lanes 3–8),whereas the complementary mutant (m5) did (lanes 9 and 10). Inagreement with this result, incorporation of 32PO4 into recombi-nant GST–Sam68 proteins by incubation with ERK2 and[g-32P]ATP in vitro was severely reduced, and overall TP phos-phorylation was not detectable when using the triple mutant (m4)(Fig. 4c, lanes 2 and 3). Incorporation of 32PO4 into m5 mutantprotein was affected to a smaller extent, and phospho-TP antibodystaining was comparable to that of wild-type GST–Sam68 (Fig. 4c,lanes 2 and 4). Thus, Ser 58, Thr 71 and Thr 84 are the predominantERK-phosphorylation sites. Proline-directed phosphorylation ofthese sites is, at least in part, required for induced alternative
splicing of exon v5 after activation of the ERK MAP-kinase pathway.Residual activity of the phosphorylation-site mutants after treat-ment with phorbol ester could be explained by the recruitment ofERK through the mutants into complexes containing endogenousSam68. Alternatively, additional ERK targets recruited by Sam68 orphosphorylated dependent on ERK recruitment through Sam68might participate in splice regulation.
To test whether Sam68 and its phosphorylation by ERK can affectv5 splicing directly, we set up in vitro splicing reactions supple-mented with either non-phosphorylated or ERK-phosphorylatedrecombinant GST–Sam68 protein. Because the phosphorylation ofSam68 by ERK is very rapidly reverted in nuclear extracts (data notshown) and splicing is much less efficient in vitro than in vivo(making long incubation times necessary), we reasoned that Sam68phosphorylation must be stabilized to see putative splice activationin vitro. Because phosphatase inhibitors block the catalytic steps ofsplicing22, we used ATP-gS to stably phosphorylate Sam68 by ERK.Thiophosphorylation of GST–Sam68 led to a similar shift inapparent molecular mass and reactivity to phospho-TP-specificantibody as observed with the ATP-phosphorylated protein(Fig. 4d, lanes 7–9). It prevented dephosphorylation but did notchange protein stability in nuclear extracts (data not shown). Theextensive pre-mRNA length and low efficiency of v5 splicing led usto detect spliced transcripts by primer extension with a splice-junction primer (see the scheme in Fig. 4d) specifically recognizingthe spliced RNA (Fig. 4d, compare lanes 1 and 2). In contrast tomock-phosphorylated GST–Sam68 preparations, thiophosphory-lated GST–Sam68 markedly stimulated v5 splicing compared withthe GST control (Fig. 4d, lanes 2–6), suggesting direct spliceregulation by Sam68 phosphorylation.
Thus, the data indicate that Sam68 can act as splice factor forCD44-v5 alternative splicing whose activity is regulated by ERKphosphorylation. To investigate whether Sam68 is crucial to spliceregulation of endogenous CD44 by Ras-pathway activation, wedownregulated Sam68 expression in EL4 cells by morpholinoantisense technology23,24. Sam68-protein levels were specificallydecreased by more than 80% (as quantified by western blotting)with the cognate Sam68-morpholino sequence, but not when anonspecific control sequence was used (Fig. 5a). When analysingthe expression of exon-v5-containing endogenous CD44 mRNA,the phorbol-ester-dependent induction observed in the controlmorpholino-treated cells (Fig. 5b, lanes 1 and 2) was almostundetectable in the Sam68-morpholino ‘knockdown’ cells (lanes 3and 4). Basal v5 expression was not affected, possibly owing tooccupancy of the high-affinity binding site in the L portion of theexon by residual Sam68 protein. Similarly to murine primary T cellsand other T-lymphoma cells5,8, treatment of EL4 cells with phorbolester stimulated the expression of CD44 mRNAs containing pri-marily single variant exons, of which v1, v3, v5 and v7 are mostreliably detectable. Interestingly, induction of v1, v3 and v7expression by phorbol ester was not affected by ‘knocking down’Sam68 expression (Fig. 5b). Thus, the data suggest exon specificityof Sam68 action and indicate a crucial role for Sam68 in Ras-pathway-induced alternative splicing of exon v5 in endogenousCD44.
The results define a first splice regulator protein that is linked tosignal transduction and modulated directly by ERK and is thereforeunder the control of Ras and growth-factor signalling. We postulatethat splice regulation will become just as complex as transcriptionalcontrol to account for the temporal and spatial requirements ofgene expression, which needs to exploit a limited number of genes.The novel ERK substrate, Sam68, acts exon-specifically and does notseem to change affinity to RNA after phosphorylation in vitro. Itmight act as regulatory component in the splice–silencer complexformed over the v5-exon sequence5,25 by controlling protein–protein contacts to factors involved in splice-site recognition and/or in spliceosome assembly. Our data now open the subject of how
Figure 5 Sam68 is required for phorbol-ester regulation of v5-exon usage in endogenous
CD44. a, Downregulation of Sam68 expression by antisense Sam68-morpholino
sequence. A cognate Sam68-morpholino oligonucleotide (samMO) or a nonspecific
control-morpholino sequence (cMO) were delivered to EL4 cells and Sam68 protein levels
were analysed by western blotting. The blot was re-probed with hnRNP A1 antibody to
check for protein loading. Bands correspond to M r values of 68K for Sam68 and 34K for
hnRNP A1. b, RT–PCR analysis of cells in a that were either mock-treated (2) or treated
with TPA (40 ng ml21) for 6 h. Diagram shows CD44 gene organization with constant-
exon regions (open boxes), variant exons (closed boxes) and primer positions (arrows).
Exon-specific PCRs were performed with 50
primers5 specific for the CD44 variant (v)
exons indicated and a common 30
primer5 (see the diagram). The additional band in v5
PCR (lane 2) represents CD44v5/v6 and v5/v7 mRNA as analysed by sequencing.
Total CD44, using a 50
constant region primer5 (c), and glyceraldehyde3phosphate
dehydrogenase mRNA levels were monitored to control for CD44 transcriptional
activation and cDNA amounts, respectively. M, 100-bp ladder (indicated sizes in base pairs).
letters to nature
NATURE | VOL 420 | 12 DECEMBER 2002 | www.nature.com/nature694 © 2002 Nature Publishing Group

exon choice is controlled by the signalling-induced modification ofsplice factors in normal physiology and disease. A
MethodsCell culture and transfectionsWe cultured EL4 cells in DMEM with 10% fetal calf serum, 100 U ml21 penicillin and100 mg ml21 streptomycin. Co-transfections (107 cells) were performed with DEAE-Dextran, using 1 mg of reporter plasmid and the indicated amounts of Sam68 expressionplasmid or empty vector. For analysis of mycSam68 modification, 2.5 £ 107 cells weretransfected with 20 mg of plasmid DNA by electroporation.
Plasmid constructionpcDNA3-Sam68 was generated by cloning the murine Sam68 cDNA, amplified from theplasmid pcDNA3-mycmSam68wt using Pwo polymerase and the primers 5 0 -CGGGATCCCCGATGCAGCGCCGGGACGATCCTGCCTCG-3
0and 5
0-
CGGAATTCTTAATAACGTCCATATGGATGCTCTC-3 0 , into the BamHI and EcoRI sitesof pcDNA3.1 þ (Invitrogen). Corresponding expression constructs for phosphorylation-site mutants were generated by replacing serines and threonines with alanine through site-directed mutagenesis (Quickchange kit; Stratagene). pETv5L/CCluc and pETv5DMlucwere generated by site-directed mutagenesis of pETv5luc and MluI deletion ofpETCatEBlucv5DM8, respectively. To obtain pETcDNA and pSGcDNA, insulin-v5-luciferase cDNA was cloned into the HindIII and BglII sites of pETCatEB8 and of amodified version of pSG5 (Stratagene), respectively. pSKv5DAvr/Spe resulted frominsertion of an 1.2-kilobase PvuII/EcoNI fragment of pETv5 (ref. 5) containing insulinexon2, CD44 exon v5 and the intervening intron into the SmaI site of pBluescript SK II(Stratagene), followed by a reduction of the intron size to 248 base pairs (bp) by AvrII/SpeI-fragment deletion.
RNA-binding assaysRNA-affinity precipitations were performed as described25 by using 40 mg of nuclearextract and 0.8 mg of recombinant GST–Sam68 protein, respectively. Oligonucleotidesequences for L, M, R and C1 (named ‘blue’ in ref. 25) are given elsewhere25; C2, 5 0 -CGACGCGUAUCAUGGUUAUGG CAGCACUGCAUACGCGUCG-3
0. EMSA reactions
were performed in 25 ml of binding buffer (10 mM HEPES pH 7.9, 100 mM KCl, 0.025%Nonidet P40, 10% glycerol, 1 mM dithiothreitol (DTT), 1 mM phenylmethanesulphonylfluoride (PMSF)) in the presence of 3 mg of yeast tRNA and 0.05 pmol of labelled RNAoligonucleotide (20,000 c.p.m.) at room temperature for 15 min. Reactions were resolvedon 4% native polyacrylamide gels in 0.5 £ TBE. Probe sequences were as described25,lacking biotinylation.
Cell lysate preparation and immunoprecipitationAfter being washed once with PBS, cells (2 £ 107) were lysed in 800 ml of RIPA buffer(50 mM Tris-HCl pH 8.0, 125 mM NaCl, 0.5% IGEPAL CA-630 (Sigma), 0.5% sodiumdeoxycholate, 0.1% SDS) supplemented with 1 mM DTT, 1 mM PMSF, 2 mg ml21
aprotinin, 2 mg ml21 leupeptin, 2 mg ml21 pepstatin, 10 mM b-glycerophosphate, 10 mMNaF, 300 nM okadaic acid, 50 mM dephostatin, 10 mM cypermethrin, 2 mM Na3VO4 and50 nM calyculin A. Sam68 was immunoprecipitated by standard procedures with anti-Sam68 polyclonal antibody C20 (Santa Cruz Biotechnology) and Protein G Plus-agarose(Calbiochem).
In vitro phosphorylation assayApproximately 0.8 mg of bacterially expressed GST–(mouse)Sam68 fusion proteins, or thesame amount of GST as a control, were mixed in 40-ml reactions with 20–100 units ofactivated recombinant ERK2 (Cell Signaling Technology) in 1 £ MAPK buffer (CellSignaling Technology), in the presence of 100 mM ATP. Reactions were split, one half wassupplemented with 5 mCi of [g-32P]ATP and reactions were incubated at 30 8C for 30 min.After resolution by SDS–PAGE, [g-32P]ATP-labelled reactions were subjected toautoradiography, non-radioactive reactions were analysed by immunoblotting withantibodies against phosphothreonine-proline (Cell Signaling Technology), against GST(2D5) or against Sam68 (clone 7-1; Santa Cruz Biotechnology).
GST fusion proteinsGST–Sam68 and GST–Sam68 mutants were expressed in Escherichia coli BL21 fromconstructs carrying corresponding Sam68 complementary DNAs (starting with thesecond amino acid) in the EcoRI site of pGEX-3X (Amersham Pharmacia). Fusionproteins were purified by the GST-purification module (Amersham Pharmacia) inaccordance with the instructions of the manufacturer. Purified proteins were dialysedagainst 20 mM Tris-HCl pH 7.5, 50 mM NaCl, 2 mM EDTA, 1 mM DTT, 50% glycerol.
In vitro splicingCapped pre-mRNA (713 nucleotides; nt) was transcribed in vitro with T3 RNA polymerasefrom linearized pSKv5DAvr/Spe. Nuclear extracts were prepared by a modified version ofthat in Dignam et al.26 Splicing reactions (18.5 ml) contained 4 fmol ml21 pre-mRNA,32.4% HeLa nuclear extract (4.6 mg ml21), 1.1 mM ATP, 2.9 mM MgCl2, 21.6 mM creatinephosphate, 0.9 U ml21 RNasin, 64.9 mM KCl, 13 mM HEPES pH 7.9, 0.13 mM EDTA, 13%glycerol, 0.65 mM DTT and 3.2% polyvinyl alcohol. After incubation at 30 8C for 90 min,reactions were deproteinized by proteinase K and extraction with phenol/chloroform.RNA was precipitated with ethanol and analysed by primer extension with a 32P-labelledsplice-junction primer complementary to the last nine nucleotides of insulin exon 2 and tothe first 21 nucleotides of CD44 exon v5. Thiophosphorylation (in the presence of 2 mMATP-gS) or mock-phosphorylation (in the absence of ATP) of recombinant Sam68 by
ERK was as described above, followed by dialysis against Dignam buffer D. Splicingreactions were performed with two different preparations of nuclear extracts and ofrecombinant Sam68 with entirely consistent results.
Morpholino antisense experimentsPre-paired morpholino oligonucleotides were obtained from GeneTools, LCC. Anantisense Sam68 sequence (samMO, 5
0-GGGATGATGGGTGGCGAGCGAACGA-3
0) or a
standard control sequence24 (cMO, 50-CCTCTTACCTCAGTTACAATTTATA-3
0) was
delivered by electroporation (10 nmol per 1.3 £ 107 cells). After 24 h, cells were eithermock-treated (DMSO) or treated with 12-O-tetradecanoylphorbol-13-acetate (TPA) for6 h; 4 £ 106 cells were used for the preparation of cell lysates (as described above) and forthat of cytoplasmic RNA5. Western blot analysis of Sam68 was performed as describedabove; heterogeneous ribonucleoprotein A1 (hnRNPA1) was detected by monoclonalantibody 4B10. RT–PCR was performed essentially as described5 with 38 cycles for CD44and 25 cycles for GAPDH amplifications. PCR amplifications were in linear phase asverified with different amounts of cDNA.
Received 25 June; accepted 6 September 2002; doi:10.1038/nature01153.
1. International human genome sequencing consortium Initial sequencing and analysis of the human
genome. Nature 409, 860–921 (2001).
2. Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
3. Sharp, P. A. Split genes and RNA splicing. Cell 77, 805–815 (1994).
4. Maniatis, T. & Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature
418, 236–243 (2002).
5. Konig, H., Ponta, H. & Herrlich, P. Coupling of signal transduction to alternative pre-mRNA splicing
by a composite splice regulator. EMBO J. 17, 2904–2913 (1998).
6. van der Houven van Oordt, W. et al. The MKK3/6-p38–signaling cascade alters the subcellular
distribution of hnRNP A1 and modulates alternative splicing regulation. J. Cell Biol. 149, 307–316
(2000).
7. Xie, J. & Black, D. L. A CaMK IVresponsive RNA element mediates depolarization-induced alternative
splicing of ion channels. Nature 410, 936–939 (2001).
8. Weg-Remers, S., Ponta, H., Herrlich, P. & Konig, H. Regulation of alternative pre-mRNA splicing by
the ERK MAP-kinase pathway. EMBO J. 20, 4194–4203 (2001).
9. Arch, R. et al. Participation in normal immune response of a splice variant of CD44 that encodes a
metastasis-inducing domain. Science 257, 682–685 (1992).
10. Cooper, D. L. & Dougherty, G. J. To metastasize or not? Selection of CD44 splice sites. Nature Med. 1,
635–637 (1995).
11. Sherman, L., Wainright, D., Ponta, H. & Herrlich, P. A splice variant of CD44 expressed in the apical
ectodermal ridge presents fibroblast growth factors to limb mesenchyme and is required for limb
outgrowth. Genes Dev. 12, 1058–1071 (1998).
12. Sherman, L. et al. The CD44 proteins in embryonic development and in cancer. Curr. Top. Microbiol.
Immunol. 213, 249–269 (1996).
13. Chang, L. & Karin, M. Mammalian MAP kinase signalling cascades. Nature 410, 37–40 (2001).
14. Stoss, O. et al. The STAR/GSG family protein rSLM-2 regulates the selection of alternative splice sites.
J. Biol. Chem. 276, 8665–8673 (2001).
15. Vernet, C. & Artzt, K. STAR, a gene family involved in signal transduction and activation of RNA.
Trends Genet. 13, 479–484 (1997).
16. Taylor, S. J. & Shalloway, D. An RNA-binding protein associated with Src through its SH2 and SH3
domains in mitosis. Nature 368, 867–871 (1994).
17. Fumagalli, S., Totty, N. F., Hsuan, J. J. & Courtneidge, S. A. A target for Src in mitosis. Nature 368,
871–874 (1994).
18. Lin, Q., Taylor, S. J. & Shalloway, D. Specificity and determinants of Sam68 RNA binding. J. Biol.
Chem. 272, 27274–27280 (1997).
19. Reddy, T. R., Tang, H., Xu, W. & Wong-Staal, F. Sam68, RNA helicase A and Tap cooperate in the post-
transcriptional regulation of human immunodeficiency virus and type D retroviral mRNA. Oncogene
19, 3570–3575 (2000).
20. Downward, J., Graves, J. D., Warne, P. H., Rayter, S. & Cantrell, D. A. Stimulation of p21ras upon T-cell
activation. Nature 346, 719–723 (1990).
21. Favata, M. F. et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol.
Chem. 273, 18623–18632 (1998).
22. Mermoud, J. E., Cohen, P. & Lamond, A. I. Ser/Thr-specific protein phosphatases are required for
both catalytic steps of pre-mRNA splicing. Nucleic Acids Res. 20, 5263–5269 (1992).
23. Summerton, J. Morpholino antisense oligomers: the case for an RNase H-independent structural
type. Biochim. Biophys. Acta 1489, 141–158 (1999).
24. Nasevicius, A. & Ekker, S. C. Effective targeted gene ‘knockdown’ in zebrafish. Nature Genet. 26,
216–220 (2000).
25. Matter, N. et al. Heterogeneous ribonucleoprotein A1 is part of an exon-specific splice-silencing
complex controlled by oncogenic signaling pathways. J. Biol. Chem. 275, 35353–35360 (2000).
26. Dignam, J. D., Lebovitz, R. M. & Roeder, R. G. Accurate transcription initiation by RNA polymerase II
in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 (1983).
Acknowledgements We thank S. Stamm and O. Stoss for discussions and the gift of antibodies
against SLM-2 and Sam68; S. Richard for the murine myc-Sam68 expression construct; S. Weg-
Remers for luciferase plasmids; J. Sleeman and G. Dreyfuss for antibodies (2D5 and 4B10,
respectively); U. Rahmsdorf and H. Olinger for technical assistance; and J. Valcarcel and I. Mattaj
for advice on in vitro splicing and mRNA transport, respectively. This work was supported by the
Deutsche Forschungsgemeinschaft.
Competing interests statement The authors declare that they have no competing financial
interests.
Correspondence and requests for materials should be addressed to H.K.
(e-mail: [email protected]).
letters to nature
NATURE | VOL 420 | 12 DECEMBER 2002 | www.nature.com/nature 695© 2002 Nature Publishing Group





























