Embed Size (px)
Citation preview

Archives of Disease in Childhood, 1980, 55, 331-347
Shwachman's syndromeA review of 21 cases
P J AGGETT, N P C CAVANAGH, D J MATTHEW, J R PINCOTT, J SUTCLIFFE,AND J T HARRIES
The Hospitalfor Sick Children and Institute of Child Health, London
SUMMARY 21 patients (10 male, 11 female) aged between 11 months and 29 years with Shwachman'ssyndrome are reviewed. All patients had exocrine pancreatic insufficiency. Haematological featuresincluded neutropenia in 19 (95%), anaemia in 10 (50%), and thrombocytopenia in 14 (70%); onepatient developed erythroleukaemia. Severe infections occurred in 17 (85%) from which 3 (15%)died. Only one child exceeded the 3rd centile for height, and growth retardation was particularlyevident in the older patients. All had skeletal abnormalities or delayed skeletal maturation, or both.Metaphyseal dyschondroplasia affected 13 of the older patients and was associated with skeletaldeformities. Eight of 9 children under 2j years had rib abnormalities. Respiratory function tests inchildren under 2 years demonstrated reduced thoracic gas volume and chest wall compliance.Older patients had reduced forced expiratory volume and forced vital capacity.
Neurological assessment showed developmental retardation or reduced IQ assessments, or both, in85% of patients studied. Other neurological abnormalities included hypotonia, deafness, andretinitis pigmentosa.
Neonatal problems had been present in 16 (80%) of the patients and 5 were of low birthweights.Hepatomegaly with biochemical evidence of liver involvement occurred in the younger patients andresolved with age. Other associated features included dental abnormalities, renal dysfunction, anicthyotic maculopapular rash in 13 (65%), delayed puberty, diabetes mellitus, and various dysmorphicfeatures. These findings stress the diverse manifestations of the syndrome and extend knowledge ona number of aspects. Sibship segregation ratios support an autosomal mode of inheritance and anhypothesis for the pathophysiological basis of this syndrome is advanced.
Shwachman's syndrome is a rare multiorgan diseaseof urtknown cause. The features which patientsexhibit include exocrine pancreatic insufficiency(EPI), growth retardation, metaphyseal dyschondro-plasia, bone marrow hypoplasia, neutropenia,anaemia, thrombocytopenia, and a raised level ofhaemoglobin F.' In this paper, we studied 21patients with this syndrome which represents thelargest series so far reported. The extent and
The Hospital for Sick ChildrenN P C CAVANAGH, senior registrar in neurologyD J MATTHEW, consultant paediatricianJ SUTCLIFFE, consultant radiologistInstitute of Child HealthJ R PINCOTT, senior lecturer in histopathologyJ T HARRIES, reader in paediatricsDepartment of Physiology, Marischal College, University ofAberdeenP J AGGETT, senior lecturer
frequency of the various features which comprise thesyndrome have been carefully documented, anddetailed studies of the affected systems have beenperformed.
Subjects and methods
Subjects (Table 1). There were 10 male and 11 femalepatients among whom were 3 pairs of siblings(Cases 3 and 4, 7 and 20, 17 and 18). An affectedolder sister of Case 9 had attended another hospitaland, although some of her features are described,she has been excluded from the overall analysis. Themean age of the male patients at the time of evalua-tion was 12 years (range 1 year 5 months to 28 years11 months) while that for the girls was 5 years7 months (range 11 months to 16 years 4 months. Allexcept 3 patients (Cases 11, 12,and 15) were receiving
331
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
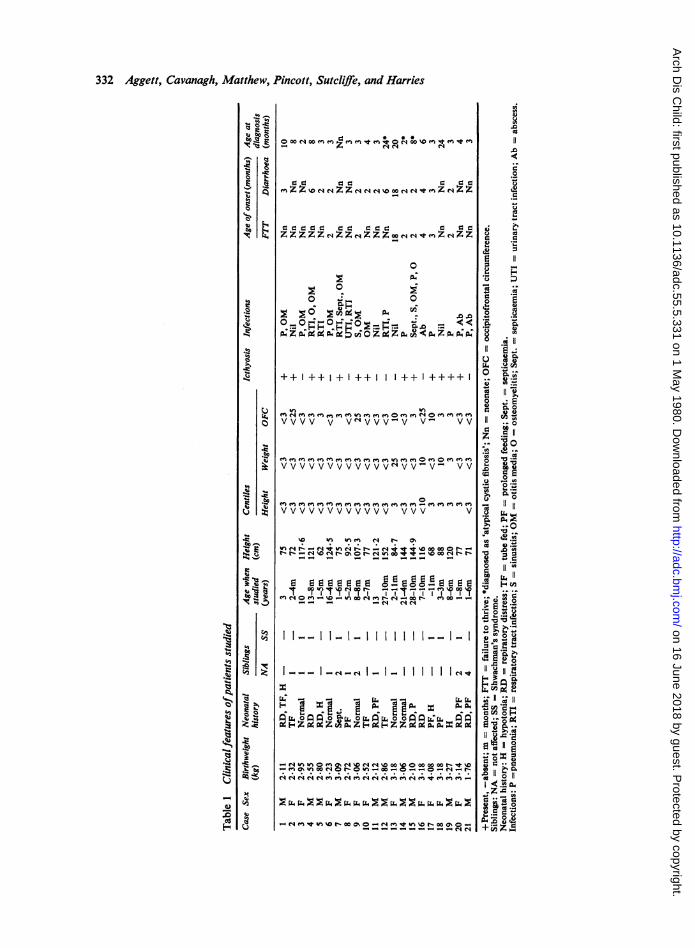
332 Aggett, Cavanagh, Matthew, ]
-0:'ts
0.k6
ICX
CO)
.It3
u
0
O9
.10
I-
k..
0
00I
I~0
U~
Pincott, Sutcliffe, and Harries
OooNoo *z *noo
Z- e Z 00qC4o o fCCleezN
C z C 00
ZZZZZNZNZZZ4'.teZcNZZ
0
0 0
++ I++ I+ I++ I++ I++++
vvvv v VNVVV-V V- VV
VVVVVVVVVVVVvNVV-V V V
e^ e eC Y t ,^ °^e^YVVVVVVVVVVVV VVV V
*O * . . .
en-%D- in -N.-o- - e0-l-N qN
I__ _e._( _ _ 1II1II1I16
7 x7 . ,g X fl
C 4A A A A A A e A00 00C A00
vs 00~ CO Oas 0-N Ov O" " - -4 -4 e" A
0
Cu
.0
to
0
Ca
0 0
C) -
Cu
0
o0
Uo .
U-*e Cu
o 'O.M
1 0
o llC11*, r0ea.
_, *^o-
o 11 of-
,4E 04
m. b3 0
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
Shwachman's syndrome 333
pancreatic and fat-soluble vitamin supplements atthe time of study. Cases 1, 3, 4, 8, 9, and 14 were on alow-fat or a medium-chain triglyceride diet, or both.Case 14 had diabetes mellitus and had receivedchlorpropamide for the last 10 years. Two patients(Cases 10 and 19) had received prednisone for themanagement of neutropenia and Case 16 received ashort course of oxymetholone when aged 2 years foraplastic anaemia.
Methods
The patients' clinical histories were analysed usingcontemporary records, and those patients availablefor follow-up were studied during optimum healthand while free of infection. Each had a full clinicalexamination, height and weight centiles weredetermined using the standards for British childrenestablished by Tanner et al.2 13 children received fullneurological examinations from one of us (N C) andhad electroencephalogram (EEG) recordings withvisually-evoked responses and electroretinography(ERG). Seven of these children also had formalpsychological assessments.Venous blood was taken for full blood count,
haemoglobin electrophoresis, plasma aspartate andalanine transaminase activities (AST and ALT), totalbilirubin, total a-amylase activity, amino-acidchromatography and, in 10 patients, leucocytekaryotyping. Urine was analysed for sugars andamino-acids and sweat tests were performed bypilocarpine iontophoresis.
Radiological assessment included review ofprevious x-rays, and skeletal surveys and bone ageestimation of patients when attending for review.Skeletal maturation was assessed using the Greulich-Pyle3 and TW2 atlases.2Respiratory function tests were performed in 9
older children using routine methods" and theresults were expressed as a percentage of that pre-dicted normal for height." In 4 children under theage of one year, thoracic gas volume (TGV) andpulmonary resistance were measured using a whole-body plethysmograph. Measurements ofcompliance7were performed in 2 patients.
Results
Clinical features (Table 1). The mean birthweight was2 82 kg (range 1 76 to 4 08 kg) and 5 patientsweighed less than 2 5 kg. All were born at termexcept Cases 11 and 15 (each 36 weeks' gestation bydates), Case 10 was a breech delivery, and Case 13was delivered by caesarean section because ofcephalopelvic disproportion. During the neonatalperiod 13 patients failed to thrive, 7 had diarrhoea,
10 had feeding dificulties which necessitated tubefeeding in 4, and 4 were markedly hypotonic; only5 had no neonatal abnormalities. Two patients(Cases 7 and 15) developed pneumonia andsepticaemia during the neonatal period.Of the 8 patients who did not have symptoms of
malabsorption in early infancy, 7 developedsymptoms by age 4 months; the exception (Case 13)presented with muscle wasting, ankle oedema, andgrowth retardation at 18 months.
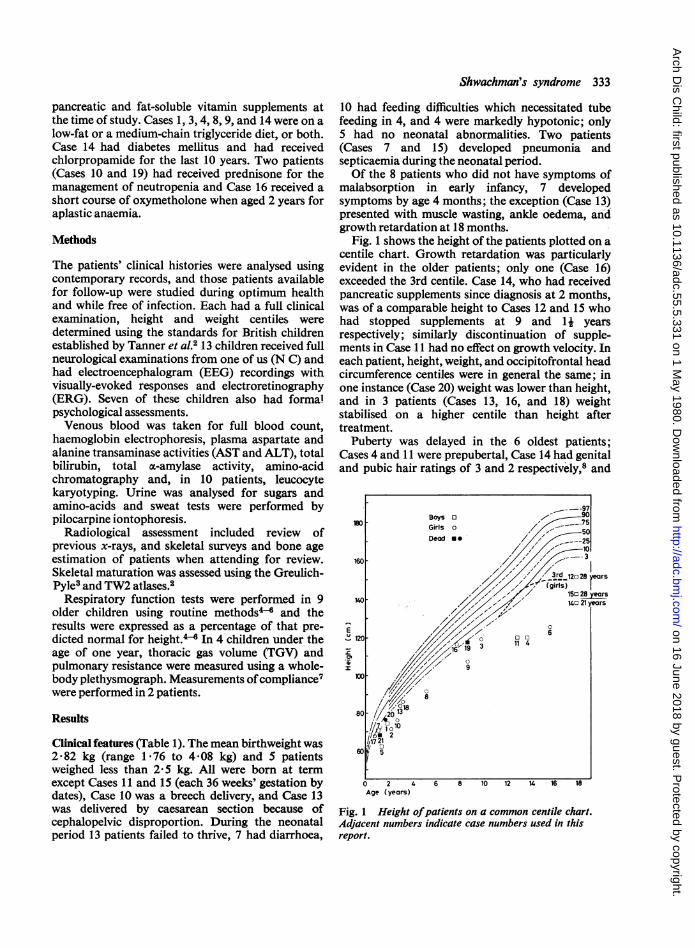
Fig. 1 shows the height of the patients plotted on acentile chart. Growth retardation was particularlyevident in the older patients; only one (Case 16)exceeded the 3rd centile. Case 14, who had receivedpancreatic supplements since diagnosis at 2 months,was of a comparable height to Cases 12 and 15 whohad stopped supplements at 9 and 11 yearsrespectively; similarly discontinuation of supple-ments in Case 11 had no effect on growth velocity. Ineach patient, height, weight, and occipitofrontal headcircumference centiles were in general the same; inone instance (Case 20) weight was lower than height,and in 3 patients (Cases 13, 16, and 18) weightstabilised on a higher centile than height aftertreatment.
Puberty was delayed in the 6 oldest patients;Cases 4 and 11 were prepubertal, Case 14 had genitaland pubic hair ratings of 3 and 2 respectively,8 and
---97- 90iso ~ Bays 0 7 75
Girls o /50Dead a: ,' --25.
-, (girls rs7'7 / ~~~150 28 years
140~~~~ 7 / 4o 21years
0
6~~~~~~~~~~~~~~~~120 ' 7 6E
I /1' 9
Fig. 1 Height ofpatients on a common centile chart.Adjacent numbers indicate case numbers used in thisreport.
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
334 Aggett, Cavanagh, Matthew, Pincott, Sutcliffe, and Harries
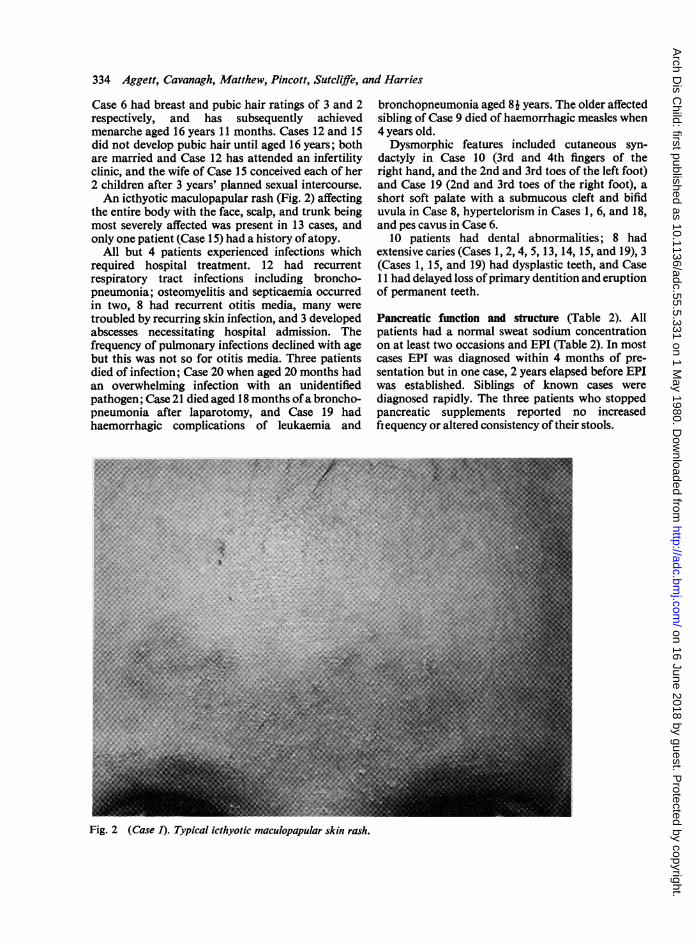
Case 6 had breast and pubic hair ratings of 3 and 2respectively, and has subsequently achievedmenarche aged 16 years 11 months. Cases 12 and 15did not develop pubic hair until aged 16 years; bothare married and Case 12 has attended an infertilityclinic, and the wife of Case 15 conceived each of her2 children after 3 years' planned sexual intercourse.An icthyotic maculopapular rash (Fig. 2) affecting
the entire body with the face, scalp, and trunk beingmost severely affected was present in 13 cases, andonly one patient (Case 15) had a history of atopy.
All but 4 patients experienced infections whichrequired hospital treatment. 12 had recurrentrespiratory tract infections including broncho-pneumonia; osteomyelitis and septicaemia occurredin two, 8 had recurrent otitis media, many weretroubled by recurring skin infection, and 3 developedabscesses necessitating hospital admission. Thefrequency of pulmonary infections declined with agebut this was not so for otitis media. Three patientsdied of infection; Case 20 when aged 20 months hadan overwhelming infection with an unidentifiedpathogen; Case 21 died aged 18 months ofa broncho-pneumonia after laparotomy, and Case 19 hadhaemorrhagic complications of leukaemia and
bronchopneumonia aged 8 years. The older affectedsibling of Case 9 died of haemorrhagic measles when4 years old.Dysmorphic features included cutaneous syn-
dactyly in Case 10 (3rd and 4th fingers of theright hand, and the 2nd and 3rd toes of the left foot)and Case 19 (2nd and 3rd toes of the right foot), ashort soft palate with a submucous cleft and bifiduvula in Case 8, hypertelorism in Cases 1, 6, and 18,and pes cavus in Case 6.
10 patients had dental abnormalities; 8 hadextensive caries (Cases 1, 2, 4, 5, 13, 14, 15, and 19), 3(Cases 1, 15, and 19) had dysplastic teeth, and Case11 had delayed loss ofprimary dentition and eruptionof permanent teeth.
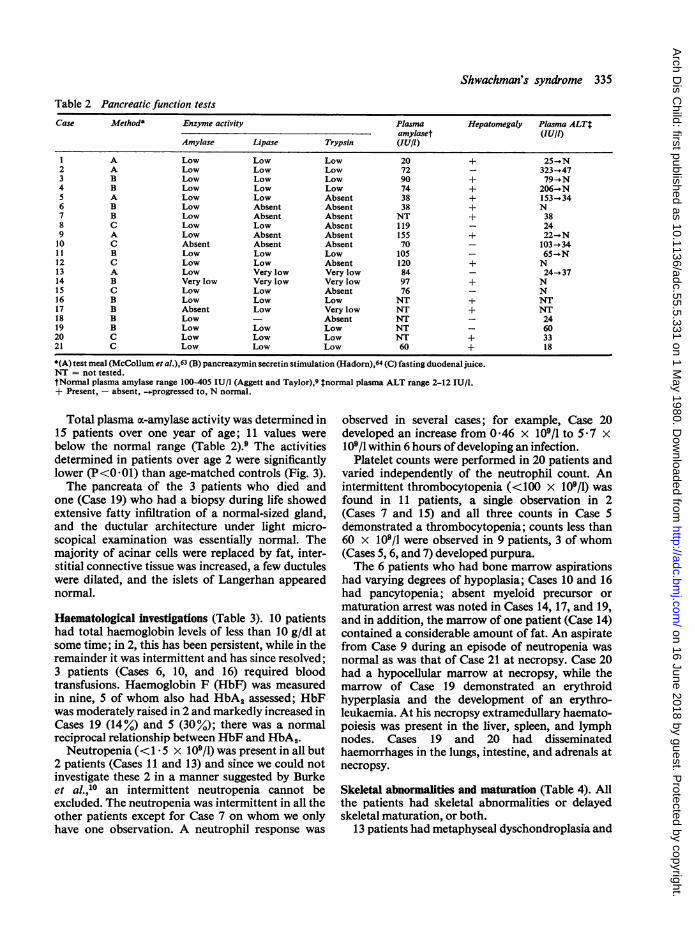
Pancreatic function and structure (Table 2). Allpatients had a normal sweat sodium concentrationon at least two occasions and EPI (Table 2). In mostcases EPI was diagnosed within 4 months of pre-sentation but in one case, 2 years elapsed before EPIwas established. Siblings of known cases werediagnosed rapidly. The three patients who stoppedpancreatic supplements reported no increasedfrequency or altered consistency of their stools.
Fig. 2 (Case 1). Typical icthyotic maculopapular skin rash.
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
Shwachman's syndrome 335
Table 2 Pancreatic function tests
Case Method Enzyme activity Plasma Hepatomegaly Plasma ALTtamylaset (lU/l)
Amylase Lipase Trypsin (lU/I)
1 A Low Low Low 20 + 25-i N2 A Low Low Low 72 - 323 - 473 B Low Low Low 90 + 79--N4 B Low Low Low 74 + 206 *N5 A Low Low Absent 38 + 153-346 B Low Absent Absent 38 + N7 B Low Absent Absent NT + 388 C Low Low Absent 119 - 249 A Low Absent Absent 155 + 22-oN10 C Absent Absent Absent 70 103-3411 B Low Low Low 105 65-)IN12 C Low Low Absent 120 + N13 A Low Very low Very low 84 - 24-_3714 B Very low Very low Very low 97 + N15 C Low Low Absent 76 - N16 B Low Low Low NT + NT17 B Absent Low Very low NT + NT18 B Low - Absent NT - 2419 B Low Low Low NT - 6020 C Low Low Low NT + 3321 C Low Low Low 60 + 18
*(A) test meal (McCollum et al.),63 (B) pancreazymin secretin stimulation (Hadorn),64 (C) fasting duodenal juice.NT = not tested.tNormal plasma amylase range 100405 IU/l (Aggett and Taylor),9 tnormal plasma ALT range 2-12 IU/l.+ Present, - absent, --progressed to, N normal.
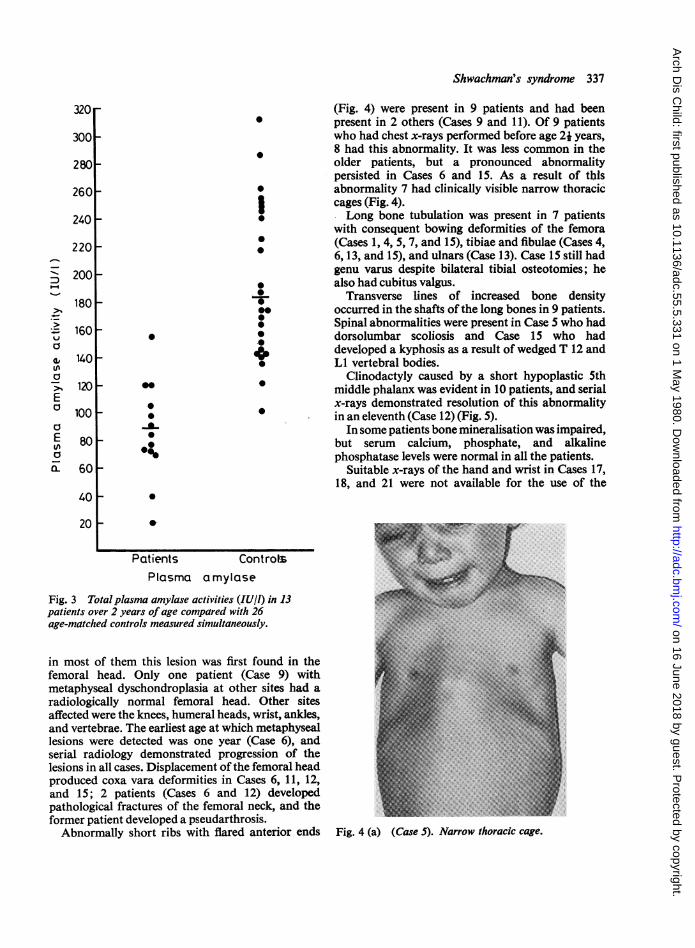
Total plasma x-amylase activity was determined in15 patients over one year of age; 11 values werebelow the normal range (Table 2).9 The activitiesdetermined in patients over age 2 were significantlylower (P<001) than age-matched controls (Fig. 3).The pancreata of the 3 patients who died and
one (Case 19) who had a biopsy during life showedextensive fatty infiltration of a normal-sized gland,and the ductular architecture under light micro-scopical examination was essentially normal. Themajority of acinar cells were replaced by fat, inter-stitial connective tissue was increased, a few ductuleswere dilated, and the islets of Langerhan appearednormal.
Haematological investigations (Table 3). 10 patientshad total haemoglobin levels of less than 10 g/dl atsome time; in 2, this has been persistent, while in theremainder it was intermittent and has since resolved;3 patients (Cases 6, 10, and 16) required bloodtransfusions. Haemoglobin F (HbF) was measuredin nine, 5 of whom also had HbA. assessed; HbFwas moderately raised in 2 and markedly increased inCases 19 (14%) and 5 (30%); there was a normalreciprocal relationship between HbF and HbA2.Neutropenia (<1 5 x 109/l) was present in all but
2 patients (Cases 11 and 13) and since we could notinvestigate these 2 in a manner suggested by Burkeet al.,10 an intermittent neutropenia cannot beexcluded. The neutropenia was intermittent in all theother patients except for Case 7 on whom we onlyhave one observation. A neutrophil response was
observed in several cases; for example, Case 20developed an increase from 0 46 x 109/l to 517 x109/l within 6 hours of developing an infection.
Platelet counts were performed in 20 patients andvaried independently of the neutrophil count. Anintermittent thrombocytopenia (<100 x 109/1) wasfound in 11 patients, a single observation in 2(Cases 7 and 15) and all three counts in Case 5demonstrated a thrombocytopenia; counts less than60 x 109/l were observed in 9 patients, 3 of whom(Cases 5, 6, and 7) developed purpura.The 6 patients who had bone marrow aspirations
had varying degrees of hypoplasia; Cases 10 and 16had pancytopenia; absent myeloid precursor ormaturation arrest was noted in Cases 14, 17, and 19,and in addition, the marrow of one patient (Case 14)contained a considerable amount of fat. An aspiratefrom Case 9 during an episode of neutropenia wasnormal as was that of Case 21 at necropsy. Case 20had a hypocellular marrow at necropsy, while themarrow of Case 19 demonstrated an erythroidhyperplasia and the development of an erythro-leukaemia. At his necropsy extramedullary haemato-poiesis was present in the liver, spleen, and lymphnodes. Cases 19 and 20 had disseminatedhaemorrhages in the lungs, intestine, and adrenals atnecropsy.
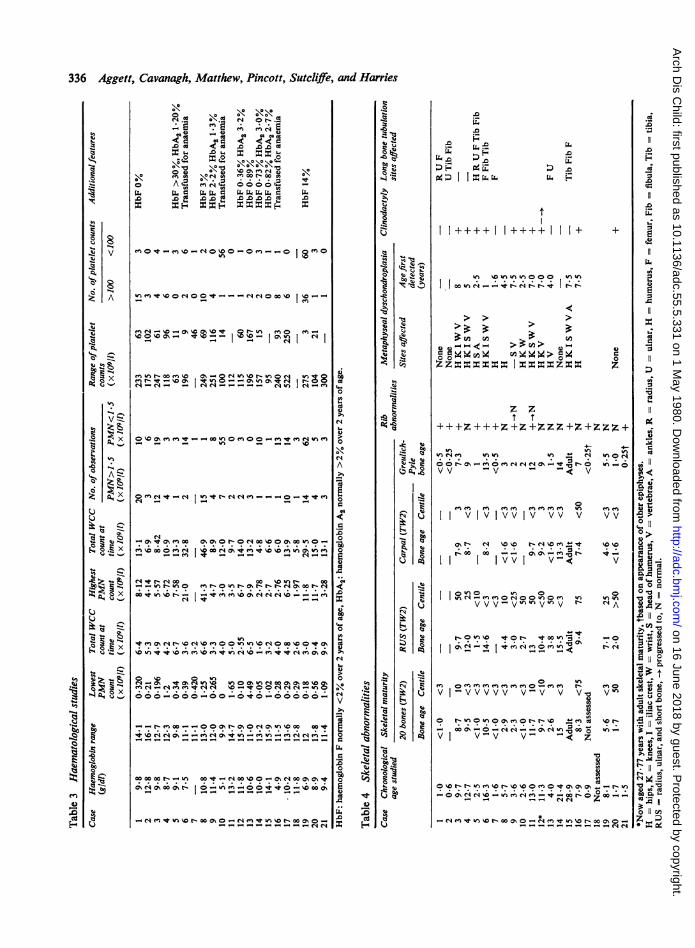
Skeletal abnormalities and maturation (Table 4). Allthe patients had skeletal abnormalities or delayedskeletal maturation, or both.
13 patients had metaphyseal dyschondroplasia and
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
336 Aggett, Cavanagh, Matthew, Pincott, Sutcliffe, and Harries
\ -o c) %CSo
D dcoYD1.0Cu 0
\
-0X0 0X00 0 %0 ~~~C- 00 Cu
0 Au ~enoooonLi. , C0 [Li.LC. 1.1.11CD0 D0 .00D .D.0.0.0CD .0
5
mcoq-M'4o%Oo-omc.-4-O,OeI
en in t- en %o _ > O \0 0n ~o t-n__o
m0e0 O eeino%X 0--
>-%0-00enN W tC eO0 fin Y%00%00- ts- I-V@_I02tbIfl
% 000ooei O j OOt0 % 0O-.^ C Ot I? C u O-C4' m f * > I-
00_qt in %O |-- "t en __ON N0 %o' 0
O _moCIn 00aO00o- 0%_ _0 _ O c ) __l_0e W~ cl 0%
0 %0> t _ > I u * r9 _ _ _ o-tr
O? -0- 0? - -- 0
-o
'
- Cl Co OC CO C 00 0- ClO O m C CO\ 0000-Ot. ... . . . . . .
009co0
0
rA
06.
9
.0
0
0
S
A
00
0
Cl
0
a
0
.00
0
C
.0
Ca
DO
0
..
la
9A
a:.0
,-0
0)c
H-
I -tC. V
0
1.01
1.010.0
15
cs
04
K
0
0. u
'- 00a 0
0t
E. O
0
.0
44.0~DPI9 :
.0
.0.0
-LJ#D
I I = v.
.0
~).0
II++++I I++++I 11+ +
Ix0l CON__Vt-Nt-t-0 t
>:, >:> r
3: m : 11 (A
4 4 =4)2 w41 42zzz=Z=ZI==zzz
0
0z
Z Zt t
+++Z+++Z +Z+ZZZ+Z+ZZZ+'C 4~~~~~~~~~- 4
... ..in In 0. ... .
ino00 CC o -0e o -o
vv -v - 1- V
0'en en en n e e enenC'ren "II VIVIVvIv vv v
~.00 0 0 --FN D^
I I IvyVVI V -.
I") m o 0s et}o soo°
V V V-V 'C V V
v v 0
VIVVVV VV, VW V.
0n 0en0n %en en e =en O m0tCn
VI V- v V- -< Z
V V\0 v
t-4V
0tn in
-0
c 0V
la
00%l ClCO -) A Cl C; - -00 C- O 00---- Cl4Cl4
- Cl C~) ~ 0CO Ci000%0 _l Cq 0n O - 00%rooo0-_ _ _ _ N G j4~ClC
0
0
.b V
0
0.
-~ _
}il
0
0:E .,
CZ0C.10
Cud
.0
.0
0.0
z
C1
._
E
S
1C
0
D._
CO
11
cdi
co
O._
Cu
.*z
.0 11
O 2
..0> O 0
010
0 o
I.,II0Ila0)
I0InH
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
Shwachman's syndrome 337
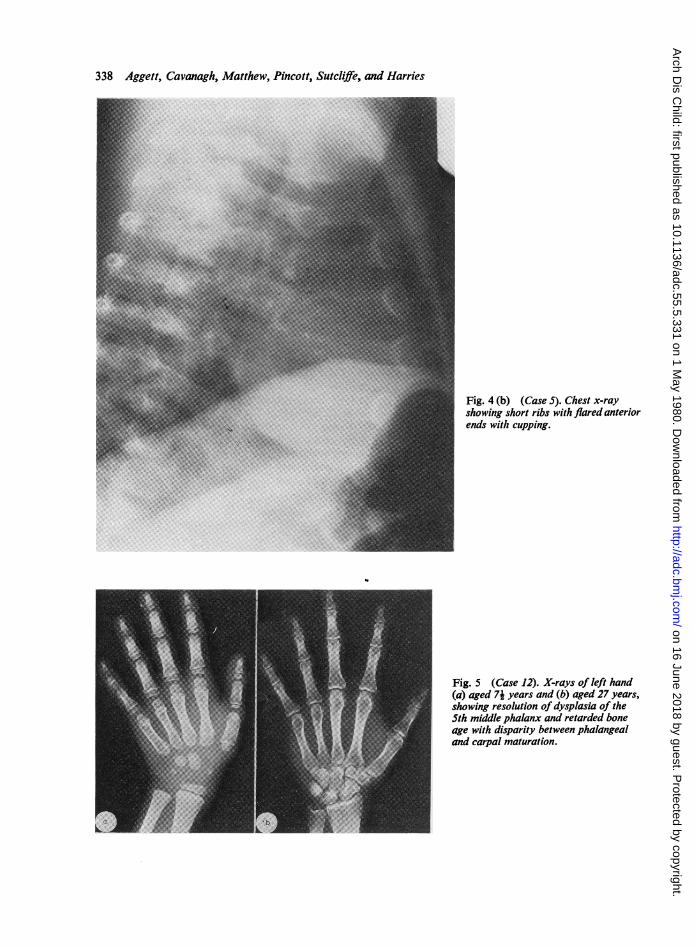
320 (Fig. 4) were present in 9 patients and had been* present in 2 others (Cases 9 and 11). Of 9 patients
300 who had chest x-rays performed before age 2j years,i 8 had this abnormality. It was less common in the
280 older patients, but a pronounced abnormalitypersisted in Cases 6 and 15. As a result of this
260 0 abnormality 7 had clinically visible narrow thoracic3 cages (Fig. 4).
240 Long bone tubulation was present in 7 patientswith consequent bowing deformities of the femora
220 0 (Cases 1, 4, 5, 7, and 15), tibiae and fibulae (Cases 4,220 9 6, 13, and 15), and ulnars (Case 13). Case 15 still had
200 genu varus despite bilateral tibial osteotomies; he0z also had cubitus valgus.
180 _+Y Transverse lines of increased bone density00 *-occurred in the shafts of the long bones in 9 patients.
> * Spinal abnormalities were present in Case S who had_ 160 * * dorsolumbar scoliosis and Case 15 who had
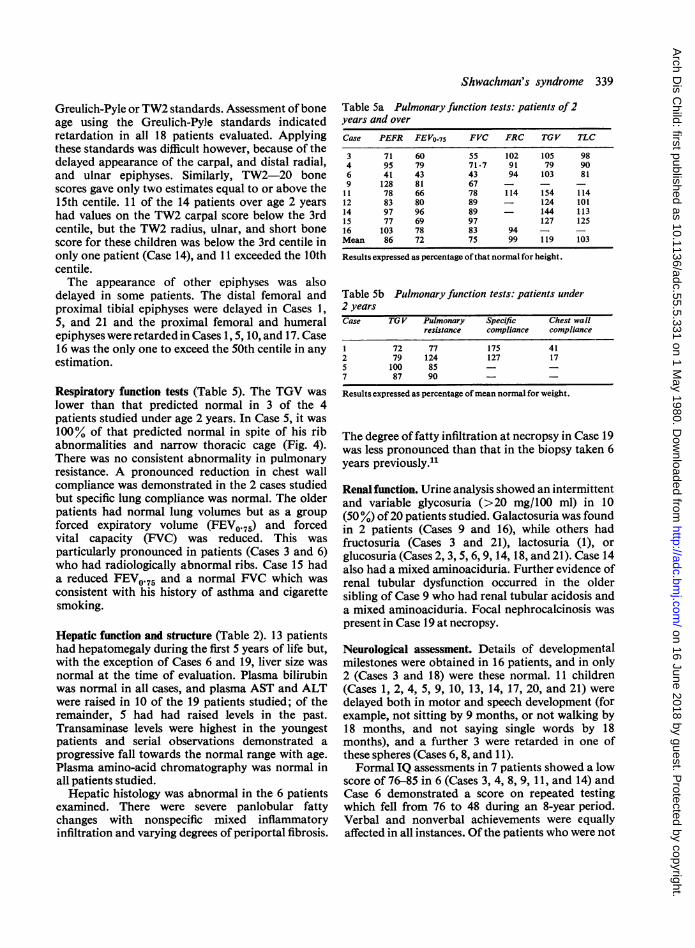
0 4 developed a kyphosis as a result of wedged T 12 and{, 140 1* LI vertebral bodies.co s Clinodactyly caused by a short hypoplastic 5th>, 120 middle phalanx was evident in 10 patients, and serial
10 x-rays demonstrated resolution of this abnormalityl00- ~ * *in an eleventh (Case 12) (Fig. 5).
EIn some patients bone mineralisation was impaired,
Efi80 ~ but serum calcium, phosphate, and alkalineO % phosphatase levels were normal in all the patients.0- 60 Suitable x-rays of the hand and wrist in Cases 17,
18, and 21 were not available for the use of the40 _
20
Patients ControtsPlasma amylase
Fig. 3 Total plasma amylase activities (IUIl) in 13patients over 2 years of age compared with 26age-matched controls measured simultaneously.
in most of them this lesion was first found in thefemoral head. Only one patient (Case 9) with ..metaphyseal dyschondroplasia at other sites had aradiologically normal femoral head. Other sitesaffected were the knees, humeral heads, wrist, ankles,and vertebrae. The earliest age at which metaphyseallesions were detected was one year (Case 6), andserial radiology demonstrated progression of thelesions in all cases. Displacement of the femoral headproduced coxa vara deformities in Cases 6, 11, 12,and 15; 2 patients (Cases 6 and 12) developedpathological fractures of the femoral neck, and theformer patient developed a pseudarthrosis.Abnormally short ribs with flared anterior ends Fig. 4 (a) (Case 5). Narrow thoracic cage.
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
338 Aggett, Cavanagh, Matthew, Pincott, Sutclife, and Harries
Fig. 4 (b) (Case S). Chest x-rayshowing short ribs with flared anteriorends with cupping.
Fig. 5 (Case 12). X-rays of left hand(a) aged 7i years and (b) aged 27 years,showing resolution of dysplasia of the5th middle phalanx and retarded boneage with disparity between phalangealand carpal maturation.
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from
Shwachman's syndrome 339
Greulich-Pyle or TW2 standards. Assessment ofboneage using the Greulich-Pyle standards indicatedretardation in all 18 patients evaluated. Applyingthese standards was difficult however, because of thedelayed appearance of the carpal, and distal radial,and ulnar epiphyses. Similarly, TW2-20 bonescores gave only two estimates equal to or above the15th centile. 11 of the 14 patients over age 2 yearshad values on the TW2 carpal score below the 3rdcentile, but the TW2 radius, ulnar, and short bonescore for these children was below the 3rd centile inonly one patient (Case 14), and 11 exceeded the 10thcentile.The appearance of other epiphyses was also
delayed in some patients. The distal femoral andproximal tibial epiphyses were delayed in Cases 1,5, and 21 and the proximal femoral and humeralepiphyses were retarded in Cases 1, 5, 10, and 17. Case16 was the only one to exceed the 50th centile in anyestimation.
Respiratory function tests (Table 5). The TGV waslower than that predicted normal in 3 of the 4patients studied under age 2 years. In Case 5, it was100% of that predicted normal in spite of his ribabnormalities and narrow thoracic cage (Fig. 4).There was no consistent abnormality in pulmonaryresistance. A pronounced reduction in chest wallcompliance was demonstrated in the 2 cases studiedbut specific lung compliance was normal. The olderpatients had normal lung volumes but as a groupforced expiratory volume (FEVO.75) and forcedvital capacity (FVC) was reduced. This wasparticularly pronounced in patients (Cases 3 and 6)who had radiologically abnormal ribs. Case 15 hada reduced FEVo.75 and a normal FVC which wasconsistent with his history of asthma and cigarettesmoking.
Hepatic function and structure (Table 2). 13 patientshad hepatomegaly during the first 5 years of life but,with the exception of Cases 6 and 19, liver size wasnormal at the time of evaluation. Plasma bilirubinwas normal in all cases, and plasma AST and ALTwere raised in 10 of the 19 patients studied; of theremainder, 5 had had raised levels in the past.Transaminase levels were highest in the youngestpatients and serial observations demonstrated aprogressive fall towards the normal range with age.Plasma amino-acid chromatography was normal inall patients studied.
Hepatic histology was abnormal in the 6 patientsexamined. There were severe panlobular fattychanges with nonspecific mixed inflammatoryinfiltration and varying degrees of periportal fibrosis.
Table 5a Pulmonary function tests: patients of2years and over
Case PEFR FEV0.75 FVC FRC TGV TLC
3 71 60 55 102 105 984 95 79 71.7 91 79 906 41 43 43 94 103 819 128 81 67 - - -11 78 66 78 114 154 11412 83 80 89 - 124 10114 97 96 89 - 144 11315 77 69 97 127 12516 103 78 83 94 - -Mean 86 72 75 99 119 103
Results expressed as percentage ofthat normal for height.
Table 5b Pulmonary function tests: patients under2 yearsCase TG V Pulmonary Specific Chest wall
resistance compliance compliance
1 72 77 175 412 79 124 127 175 100 85 - -7 87 90 - -
Results expressed as percentage ofmean normal for weight.
The degree of fatty infiltration at necropsy in Case 19was less pronounced than that in the biopsy taken 6years previously."
Renal function. Urine analysis showed an intermittentand variable glycosuria (>20 mg/100 ml) in 10(50 %) of 20 patients studied. Galactosuria was foundin 2 patients (Cases 9 and 16), while others hadfructosuria (Cases 3 and 21), lactosuria (1), orglucosuria (Cases 2, 3, 5, 6, 9, 14, 18, and 21). Case 14also had a mixed aminoaciduria. Further evidence ofrenal tubular dysfunction occurred in the oldersibling of Case 9 who had renal tubular acidosis anda mixed aminoaciduria. Focal nephrocalcinosis waspresent in Case 19 at necropsy.
Neurological assessment. Details of developmentalmilestones were obtained in 16 patients, and in only2 (Cases 3 and 18) were these normal. 11 children(Cases 1, 2, 4, 5, 9, 10, 13, 14, 17, 20, and 21) weredelayed both in motor and speech development (forexample, not sitting by 9 months, or not walking by18 months, and not saying single words by 18months), and a further 3 were retarded in one ofthese spheres (Cases 6, 8, and 11).Formal IQ assessments in 7 patients showed a low
score of 76-85 in 6 (Cases 3, 4, 8, 9, 11, and 14) andCase 6 demonstrated a score on repeated testingwhich fell from 76 to 48 during an 8-year period.Verbal and nonverbal achievements were equallyaffected in all instances. Ofthe patients who were not
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

340 Aggett, Cavanagh, Matthew, Pincott, Sutcliffe, and Harries
formally assessed, Case 12 had achieved a goodstandard of secondary education and works as acomputer data processor; Case 15 attended schoolsfor the physically-handicapped and had trained toassemble electronic circuit boards, but now works asa pop band drummer, and Case 14 works as a hotelpage boy. Cases 12 and 15 have their own house-holds but Case 14 is still dependent on his parents.
Corrected visual acuity was normal in all butCase 6, who had optic atrophy and retinitis pig-mentosa; Cases 5 and 20 had reduced retinal andchoroidal pigment. The ERG was normal in 11patients but Case 5 had a generalised reduction inamplitude consistent with her retinitis pigmentosa.
Bilateral conductive deafness was demonstratedby audiometry in 3 patients (Cases 6, 9, and 10),and Case 11 had a sensorineural loss of hearing.
Further neurological examination was normal inall but 3 patients; Case 16 in addition to her pro-gressively deteriorating IQ, retinitis pigmentosa, andhearing loss was ataxic and had a history ofpersistinghypotonia in early childhood as did Case 20; Case 5remained hypotonic and also developed truncalataxia; Case 11 who was born at 36 weeks' gestationhad a slight ataxic diplegia.Case 15 experienced bilateral parasthesiae in the
L5, S1 root distribution after prolonged standing, butneurological examination was normal.
13 patients had EEGs performed, and restingrecords showed no local abnormalities or paroxysmalfeatures. During photic stimulation 6 patients(Cases 3,4,5,9, 11, and 15) showed some symmetricalbursts posteriorly containing discharges, and in onepatient, sharp components appeared. Four otherpatients had conspicuous lambda waves. Rhythmicactivity in the Rolandic area was in keeping with age,and the a rhythm was well formed and regular inmost ofthem.
Endocrine studies. Glucose tolerance tests wereperformed in 5 patients (Cases 2,9, 14,19, and 21) andwere normal except for Case 14, who had a responsetypical of diabetes mellitus and an impaired insulinresponse which was improved by tolbutamide.Growth hormone response to Bovril stimulation wasnormal in the 3 patients (Cases 3, 11, and 13) studied.Tests of thyroid function in 3 patients (Cases 2, 5,and 16) were normal.
Genetics. There was no consanguinity in the 18families from which our patients are derived. In 2of the 4 pairs of affected siblings, the second childwas discovered as a consequence of diagnosing thefirst. In the othertwo pairs, this was not certain and all4 patients could be regarded as index cases. Segrega-tion ratios were calculated therefore with either 18 or
20 index cases. These gave values of 3 5 1 and3: 1 respectively; both of which are consistentwith an autosomal recessive mode of inheritance.The 3 half-siblings in the families studied werenormal.The leucocyte karyotypes contained no con-
sistent abnormality. Case 1 had a single abnormalcell with a 47XY + G min karyotype among 24normal cells. Case 3 hada46XY lqh + abnormality (alarge heterochromatic band next to the centromereon chromosome 1) which was not present in hersibling (Case 4) who had a normal karyotype as didall the other patients studied.
Discussion
Our results in this large series of patients withShwachman's syndrome stress the protean manifesta-tions of the condition and extend the knowledge on anumber ofaspects.
Infections. The series again confirms earlier observa-tions of the susceptibility to respiratory, cutaneous,and systemic infections in this syndrome.1 12Shmerling et al.' calculated a 25% mortality rate dueto infection from their review of cases. The abnormalthoracic configuration and hypotonia may explainthe predominance of pneumonia in younger patients.Although lower respiratory tract infections are lesscommon in older patients, the incidence of sinusitisand otitis media suggests a continuing susceptibilityto infection.The nature and frequency of infection does not
correlate with the degree of neutropenia and neutro-philic responses to infection have been demonstratedby others'° 13 as well as by ourselves. Case reports ofa variable immunoglobulin deficiency have appearedbut these are not consistent.1'16 We have recentlydemonstrated that defective neutrophil mobility isan almost invariable finding in Shwachman'ssyndrome,17 and may contribute to the susceptibilityto infections.
Pancreatic insufficiency. The degree of pancreaticinsufficiency in these patients was variable and thepersistence of some residual exocrine functionpossibly accounts for the delayed presentation ofmalabsorption in some cases.1013 The pancreatichistology described is similar to that notedbefore ;1 11-13 and the arguments in favour ofeither ahypoplastic or degenerative lesion of the pancreashave been discussed by Nezelof and Watchi.'2 Thepresence of a normal ductular architecture and isletstructure with some residual exocrine tissue in anormal-sized, though lipomatous gland, suggests a
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

fatty metamorphosis secondary to a progressivedegenerative lesion. This was essentially concludedby Burke et al.'0 who described a 5-month-oldpatient with this syndrome in whom the onlypancreatic abnormality at necropsy was inspissatedsecretions in the ducts; the exocrine tissue was
histologically normal.The low plasma total amylase may represent the
extent of EPI. Levitt et al.18 have demonstrated a
2: 1 ratio of nonpancreatic to pancreatic amylase innormal serum and the absence of the pancreaticcomponents in the serum of patients withShwachman's syndrome. We did not study theplasma x-amylase isoenzymes and can only speculateon whether one or more isoenzymes are affected, andwhether impaired secretion ofnonpancreatic amylasesuch as the salivary components may coexist. It ispossible that the salivary glands may be affected inthis syndrome; a child with EPI with both pancreaticand parotid lipomatous changes has been described."The low serum amlyase activity may also representan impaired enteropancreatic circulation.
Haematological abnormalities. An intermittentneutropenia was demonstrated in all 18 patients whowere adequately studied as proposed by Burke et al.,'0and we did not observe the constant neutropeniareported by Shmerling et al.' The incidence (60-70 %)of thrombocytopenia in this series is comparablewith that described by Shwachman et al.'3
In addition to our own observations, normal bonemarrow aspirates in neutropenic patients with thissyndrome have been described by Goldstein'5 andBurke et al.'0 This coupled with the neutrophilresponse to infections suggests that, in some patientsat least, the marrow hypoplasia is 'patchy' indistribution. We did not perform multiple marrowaspirations in our patients and thus are unable toconfirm whether this is the case.
Erythroleukaemia has not previously beenreported in Shwachman's syndrome, but 2 cases withother myeloproliferative leukaemias have beendescribed; Nezolofand Watchi'2 reported a case withmonocytic leukaemia, and Huijgens et al.20 a casewith myeloblastic leukaemia. Bone marrow hypo-plasia has been identified as a possible preleukaemiccondition, and is often associated with a raised HbFwhich may precede the bone marrow dysfunction.HbF can be moderately raised in many haemato-logical abnormalities but the highest observationshave been associated with erythroleukaemia andjuvenile chronic myeloid leukaemia where levels of30% or more may occur but since levels of 10-20%may exist with hypoplastic anaemias such asFanconi's,2' in which leukaemic transformation isnot invariable, we cannot ascribe any prognostic
Shwachman's syndrome 341
significance to the 2 grossly raised HbFs seen in thisseries.
Lymphoproliferative neoplasia has also beendescribed in this condition, and a patient withlymphoblastic leukaemia22 has been reported.Hantelmann23 described a child with Hodgkin'sdisease who was found to have a lipomatouspancreas at necropsy.
Skeletal abnormalities. Our study is the first toreview comprehensively the incidence and extent ofthe skeletal abnormalities in this syndrome. Meta-physeal dyschondroplasia affecting ribs and kneeswas first described by Burke et al.10 and Giedionet al.,24 and Stanley and Sutcliffe25 reported the moreextensive metaphyseal involvement especially of thefemoral head. Fellman et al.26 expanded the range ofskeletal abnormalities to include abnormal long bonetubulation, and genu and cubitus valgus. McLennanand Steinbach27 reported narrowing of the sciaticnotch and hypoplasia of the 5th middle phalanxcausing clinodactyly.We have shown the high frequency of rib abnor-
malities which are particularly common in theyounger patients. Their relative infrequency in theolder children can be explained by a spontaneous andcomplete resolution such as that observed in 2 of ourcases and which was occurring in a patient describedby Brueton et al.16 We have also demonstratedclinodactyly in half of our patients and the resolutionof this lesion in one case.
This report and that of Shmerling et al.' indicatesthat the hip is the earliest and most frequentlyaffected site for metaphyseal chondrodysplasia,though this may not be invariable.27 The sequence ofmetaphyses involved in this diseasewas unpredictablebut certainly progressive. The earliest age at whichwe detected metaphyseal lesions was one year, butDanks et al.28 found evidence of early femoral andhumeral involvement at 4 months in one case. Thepathogenesis of these lesions is obscure. Spycheret al.29 reported poorly vascularised and disorientatedcolumnar cartilage, and Danks et al.28 found similarfeatures of defective endochondrial ossification.These features would seem to be primary to thesyndrome rather than secondary to malnutrition.Long bone tubulation is a nonspecific feature and
may be associated with syndromes, such as Marfan'ssyndrome or homocystinuria, or with disusesecondary to immobilisation or neuromusculardisease.30 This was not the case in our patients andthe tubulation may reflect abnormal diaphysealdevelopment or a defect in membranous ossification.The transverse lines on the long bones probably
represent periods of arrested bone growth secondary
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

342 Aggett, Cavanagh, Matthew, Pincott, Sutcliffe, and Harries
to infection and other stress associated with thissyndrome.3'The pathological fractures and skeletal deformities
requiring surgical correction in 2 of our patients,illustrate the degree of disability experienced by thesepatients. Shmerling et al.1 described a case requiringbilateral osteotomies to correct coxa vara deformities,and Karjoo et al.32 reported the necessity of a sternalsplitting operation to alleviate thoracic constrictionand improve ventilatory function in an infant. Thisdid not improve the child's growth rate, however.
Skeletal maturity and growth. This study illustratesthe dissociation between the osseous centres of thehand and wrist, a feature noted by Shwachman et al.13in the 3 siblings described in their paper. The carpalbone age is more retarded than that of the phalanges,and its inclusion in a composite bone age assessment,such as the 20 bone TW2 estimate, results in a lowevaluation. The radius, ulnar, and short bone scoressuggest that there is some delay in skeletal matura-tion in some patients especially, though notexclusively, the youngest in whom delayed ulnar andradial epiphyseal appearance would reduce thescore. Four of the 5 children under 2i years, inwhom radius, ulnar, and short bone scores could bedetermined, gave values below the 10th centile.
Malnutrition and metabolic abnormalities retardossification centres equally and may influence thematurity in our younger patients but the discrepancybetween centres which is apparent in all our patientsis more characteristic of a primary bone dysplasia.The only patient (Case 16) with a skeletal maturity
above the 50th centile was also the only child toexceed the 3rd centile for height. This girl had hadoxymetholone some years before and, while theseeffects have not been documented with this particularanabolic steroid, Shmerling et aL.' noted that in oneof their patients anabolic steroids did increase thebone age without affecting height.Growth retardation was most evident in the older
children. This has been noted by others, as has itsindependence of the frequency of infections and thedegree of EPI and malnutrition." 13 Shmerling et aL.calculated that the severity of growth retardationcould not be explained by the degree of metaphysealdyschondroplasia. In this series Case 14, like thesecond of McLennan and Steinbach's two cases,27
shows that dwarfism can occur in the absence ofradiologically demonstrable metaphyseal lesions.McLennan and Steinbach suggested that theirpatient might not have been growing rapidly enoughfor metaphyseal dyschondroplasia to develop.Growth retardation is probably therefore a
primary feature and the low birthweight of some
patients in this series suggests that intrauterinegrowth retardation may be a contributory factor.The delayed onset of puberty in this condition has
not been previously documented.
Respiratory function. The reports of Taybi et al.33Karjoo et al., 32and Danks et al.28 demonstrated thatrib shortening can be sufficient to cause respiratoryand feeding problems in the neonatal period. Dankset al.28 described a patient with a low tidal volume at4 weeks which was normal at 14 weeks. Our resultsshow that impaired respiratory function may persistfor much longer. The reduced chest wall compliancein the 2 young patients is consistent with theirabnormal thoraces. It is more difficult to explain thereduced FEVo.75 and FVC in the older patients,apart from Cases 3 and 6 who still had abnormal ribs,but this may represent some continuing musculo-skeletal abnormality rather than airways obstruction.
Hepatic function and structure. Hepatic abnormalitieshave been described in many cases."1 34 Bruetonet al.'6 were the first to show raised transaminases ina patient with abnormal liver histology. Our studyincludes a number of similar cases, as well as somepatients with biochemical evidence of liver involve-ment. The tendency for the transaminases to falltowards normal limits, and the resolution of hepato-megaly was present in both groups of patients andmay reflect decreased fatty infiltration or aprogressive fibrosis. The absence of any evidence ofhepatic dysfunction in the older patients precludedinvestigative biopsies, but the necropsy findings inCase 19 suggest that reversal of the fatty infiltrationmay occur. Fatty change in the liver can be secondaryto malnutrition and infection, both ofwhich are morelikely in the newly diagnosed young patient. It ispossible, therefore, that the liver abnormalities mayrepresent a secondary rather than a primary featureofthe syndrome though the latter cannot be excluded.
Renal function. The varied glycosuria and amino-aciduria together with the renal tubular acidosis inthe affected sibling of Case 9 and in a recent casereport (S Hassell, 1979, personal communication)suggests that renal tubular dysfunction may be moreextensive than previous reports have implied.Similarly the nephrocalcinosis discovered at necropsyin Case 19 may represent an undetected abnormalityin urinary acidification.
Neurological and developmental findings. Shwachmanet al.13 specified that their patients had normalmotor and mental development. Other reportshave described developmental retardation either
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

Shwachman's syndrome 343
alone" 3 or associated with hypotonia.'2Hypotonia has also been noted to occur indepen-dently.32 37-38
This review, the first systematic neurologicalassessment in this syndrome, establishes that earlyspeech and motor delay is common and that this isprobably due to an inherent below-averageintelligence. The early overall delay may possibly beinfluenced by intercurrent infections and socialdeprivation during time in hospital, but the persistingpoor intellectual achievement during optimumhealth and the similar achievement in the absence of ahistory of gestational or perinatal insults, suggestthat psychomotor retardation is a primary feature ofthe syndrome. Malnutrition during the criticalperiod of brain growth could cause such retardationbut this degree of retardation is not seen in patientswith cystic fibrosis.39
Except in Case 8, who had a short soft palate witha submucous cleft palate, there were no structuralabnormalities or focal neurological signs which couldaccount for the delayed speech. The frequency ofotitis media and the presence of hearing loss in 4cases, stress the importance of regular hearing testsin these patients.Hypotonia was often noted particularly in infancy,
and probably contributes to the early respiratory andfeeding problems. It is possible that in most patientshypotonia is a secondary feature. The profoundhypotonia of patients in the absence of infectionhowever, suggests that in some patients the defectmay be primary to the syndrome.
Abnormalities of retinal pigmentation have notbeen noted previously in this syndrome. Retinitispigmentosa can be associated with syndromesinvolving neurosensory deafness, defective intelli-gence, or skeletal abnormalities ;40 no associationwith EPI has been reported.Nearly half the patients tested had abnormal
sensitivity to photic stimulation during EEGrecording but none gave a history of photic-inducedfits. The incidence of occipital photosensitivity inthis group of patients is higher than would beexpected in the general population41 but itssignificance is unknown.
Endocrine studies. The impaired insulin response inone patient was the only gross endocrine abnormalityfound. Shwachman and Holsclaw42 described onepatient with a diabetic glucose tolerance test andShmerling et aL.l discounted their patient with dia-betes mellitus because of his strong family history ofthe disease. Our case has no such history, so thepossibility of an association of diabetes mellitus withthis syndrome is again raised. The insulin response
was improved by tolbutamide in our patient and hehas been treated successfully with chlorpropamidefor 10 years since diagnosis.
Genetics. The karyotype abnormalities cannoteasily be interpreted. The single abnormal celldetected in Case 1 could have arisen during cellculture. The other abnormality noted is sometimesinherited and may be associated with congenitaldefects, but it is unlikely to be contributory to thissyndrome since it is absent in this patient's siblingand the other cases.The segregation ratios derived from sibship
analysis support an autosomal recessive mode ofinheritance which until now has been assumed on thebasis of a familiai incidence of the syndrome.L Thismode of inheritance is also supported by our recentdemonstration of defective neutrophil mobility bothin the patients with this syndrome and their parents.'7
Icthyosis. The icthyotic skin rash frequently observedamong our patients had some characteristics incommon with those observed in one case by Nezelofand Watchi,'2 and those described in the first case ofShwachman et al.'3 Shwachman and Holsclaw42described a varying degree of eczema among theirpatients. The presence of xerophthalmia in the casedescribed by Nezelof and Watchi'2 suggests thepossibility of hypovitaminosis A. In the presentseries the skin lesion affected patients on fat-solublevitamin supplements and with normal serum vitaminA levels, but we cannot exclude the possibility that itmay be secondary to an undetected nutritionaldeficiency.
Dental abnormalities. The prevalence of these mayagain represent another manifestation of malnutri-tion, but the occurrence of dysplastic teeth in somepatients suggests the possibility of a primaryaetiology. Frequent exposure to cariogenic antibioticsyrups may also be contributory.
Other associated features. Features such as thesubmucous cleft palate and the cutaneous syndactyly,which was present in 2 unrelated cases, may representa fortuitous association. The hypertelorism noted in3 patients has been described in another case.43Clinodactyly may be associated with Fanconi'sanaemia and again with aplastic anaemia in thesyndrome of EPI, midline ectodermal defects,microcephaly, severe mental retardation, neuro-sensory deafness, hypotonia, hypothyroidism, dentaldysplasia, rectourogenital abnormalities, anddwarfism described by Johanson and Blizzard44 andSchussheim et al.45 Some of these features have
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

344 Aggett, Cavanagh, Matthew, Pincott, Suttcliffe, and Harries
accompanied EPI in a case with congenital rubella46and in a patient with Klinefelter's syndrome.47Burke et al.10 and McLennan and Steinbach27 havenoted some similarities between Shwachman'ssyndrome and the cartilage-hair syndrome.48 Suffi-cient dissimilarities usually exist however, todistinguish between these syndromes though someoverlap may exist.
Incomplete or partial syndromes in which themanifestation of bone marrow dysfunction ormalabsorption is delayed are illustrated in thisseries, by the experience of Burke et al.10 who dis-covered EPI by specifically investigating neutropenicpatients, and by the 2 children described byHavlik6va et al.49 who had EPI, hepatomegaly,raised plasma AST and ALT, narrow chests,neonatal respiratory distress, and developmentalretardation but no haematological abnormalities.This is not surprising in such a complex conditionand the patient with neutropenia, metaphysealdyschondroplasia, tibial bowing, dental caries,moderate mental retardation, and hypogonadismdescribed by Theodorou and Adams50 may representanother example of a partial syndrome.Burke et al.10 described a patient who also had
Hirschsprung's disease, and an association withendocardial fibrosis has been reported.51 Neither ofthese was present in this series.
Diagnosis, treatment, and prognosis. Diagnosis ismore difficult in the young infant when many of thesyndrome's features are indistinguishable from themanifestations of immaturity. Some of the presentingfeatures such as hypotonia, bronchopneumonia, orrespiratory distress with thoracic dystrophy maymask other symptoms. The high incidence ofneonatal problems is shown in this paper and thediagnosis may not be apparent until the child is olderand the more classic features of the syndrome arepresent. Shwachman's syndrome should be con-sidered in any infant or child presenting withan association of any of the features described here(Table 6). We have found the presence of rib abnor-malities in the child and a neutrophil mobility defectin his parents of value in diagnosing one infant withgrowth retardation, diarrhoea, and neutropenia.Treatment is symptomatic. Infections usually
respond to prompt treatment with antibiotics.Steroids, such as prednisone and oxymetholone,may improve marrow function,"1 13 but theseand anaesthetic agents may increase the risk ofinfection.'7 Transfusion of whole blood and plateletshas been used when appropriate such as beforesurgery. The spontaneous symptomatic improvementof diarrhoea in some patients has enabled with-drawal of pancreatic supplements without adverse
Table 6 Features associated with Shwachman'ssyndromeExocrine pancreatic insufficiency
steatorrhoeaGrowth retardationSkeletal abnormalities
metaphyseal dyschondroplasia, delayed maturation, ribabnormalities, long bone tubulation, clinodactyly
Narrow thoraxHaematological abnormalitiesbone marrow hypoplasia, neutropenia, thrombocytopenia,raised HbF, lymphoproliferative and myeloproliferative neoplasia
Recurrent infectionsDefective neutrophil mobilityNeonatal problemspoor feeding, respiratory distress
Psychomotor retardationHypotoniaHepatomegaly
raised ALT and ASTRenal tubular dysfunctionIcthyosisDental abnormalitiesDelayed pubertyDiabetes mellitusDysmorphic featuresEndocardial fibrosisHirschsprung's disease
effects both in some of our patients and those inearlier reports.' 10 13 Nutritional supplements (forexample, vitamins) are probably advisable but theprogress of Cases 12 and 15 in the absence of thesehas been similar to other patients.The prognosis for this syndrome is probably better
than that for cystic fibrosis though it is accompaniedby considerable morbidity. Shmerling et aL. reporteda 25% mortality which compares with one of 15%reported here. Infections and a haemorrhagicdiathesis can prove fatal, and any prognosticationand follow-up should consider these, and the risk ofmyeloproliferative and lymphoproliferative neo-plasia.
An hypothesis for the primary abnormality inShwachman's syndromeThe primary abnormality in Shwachman's syndromehas not been defined, and we conclude by presentingan hypothesis to explain the protean manifestationsof this syndrome. Unrelated tissues appear to be bothmalfunctioning and, as the lipomatous changesimply, undergoing degenerative changes. Spycheret al.29 described large glycogen and small lipiddeposits in the chondrocytes from an area ofmetaphyseal dyschondroplasia. Their electron-microscopical studies also demonstrated dilatedendoplasmic reticulum cisternae containing anincreased amount of granules which appeared asintracellular inclusions. They proposed that thisrepresented either a failure to secrete an abnormalsynthetic product or a defect in the secretion of anormal product from the endoplasmic reticulum.
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

Shwachman's syndrome 345
Cellular secretion is dependent on the adequatefunction of microtubules and microfilaments. Theformer are thought to provide the structural andorientating system for the motive force provided bythe microfilaments.Use of agents such as colchicine which blocks
microtubular reassembly, vincristine alkaloids whichprecipitate microtubular protein, or the cytochalasinswhich disrupt microfilaments have demonstratedthat these ubiquitous subcellular components areessential for cell division, lipid absorption,52 hexoseuptake,53 neutrophil mobility,54 phagocytosis and thedisposition of cell surface receptors,55 as well as forthe secretion of histamine,56 hormones includinginsulin,57 and thyroxine,58 hepatic lipoproteins andalbumin,59 collagen,60 pancreatic amylase,61 andneurotransmitters.62
In cells exposed to colchicine, secretory productsfail to migrate to the cell periphery and accumulatewithin golgi-associated vacuoles and in the endo-plasmic reticulum cisternae,590 resulting in anelectron microscopical appearance similar to thatnoted by Spycher et al.29 The demonstration ofimpaired polymorphonuclear mobility in patientswith this syndrome and in some of their parentssuggests that this defect, since it is manifest inpresumed heterozygotes, is close to the abnormalgene product. Similar defects in polymorphonuclearmobility can be induced by colchicine and cyto-chalasin B.54We speculate that the basic defect in Shwachman's
syndrome may lie in the function of the micro-tubular and microfilament elements. Too little isknown about the co-ordination, regulation, ormetabolism of these systems to be more specific, butsuch a functional defect could account for both theabnormal appearance of the chondrocytes and theimpaired polymorphonuclear mobility. Thepancreatic cell inclusion bodies noted by Shwachmanet al03 may represent accumulating syntheticproducts which ultimately lead to degenerativechanges with subsequent lipid or glycogen deposition.Mitotically-active tissues such as the bone marrowwould be most vulnerable to such a defect, as wouldbe metabolically active tissues like the liver.
We thank Dr L J H Arthur, Dr C Boothby, Dr DLawson, Professor June K Lloyd, Dr J D Maxwell,Dr R J Pugh, Dr C Upjohn, Dr J A Walker-Smith,and Professor 0 H Wolff for letting us study theirpatients, Professsor C Carter, Dr D Hughes, Dr AHarden, Dr M Konikko, Dr E N Hey, Dr J PMcCollum, and Dr J Pritchard for helpful advice andcriticism, and the Joint Research Board of TheHospital for Sick Children and Institute of ChildHealth for financial support.
P J A thanks the Medical Research Council, andN C thanks the Neurology Fund of The H4ospital forSick Children for financial support.
References
Shmerling D H, Prader A, Hitzig W H, Giedion A,Hadorn B, Kuhni M. The syndrome ofexocrine pancreaticinsufficiency, neutropenia, metaphyseal dysostosis, anddwarfism. Helv Paediatr Acta 1969; 24: 547-75.
2 Tanner J M, Whitehouse R H, Marshall W A, HealyM J R, Goldstein H. Assessment of skeletal maturity andprediction of adult height (TW2 method). New York:Academic Press, 1976.
3 Greulich W W, Pyle S C. Radiographic atlas of skeletaldevelopment of the hand and wrist. London: OxfordUniversity Press, 1966.
4 Cogswell J J, Hull D, Milner A D, Norman A P, Taylor B.Lung function in childhood. I. The expiratory volumes inhealthy children using a spirometer and reverse plethys-mograph. BrJ Dis Chest 1975; 69: 40-50.Cogswell J J, Hull D, Milner A D, Norman A P, Taylor B.Lung function in childhood. II. Thoracic gas volumes andhelium functional residual capacity measurements inhealthy children. BrJ Dis Chest 1975; 69: 118-24.
6 Cogswell J J, Hull D, Milner A D, Norman A P, Taylor B.Lung function in childhood. III. Measurement ofairflow resistance in healthy children. BrJ Dis Chest 1975;69: 177-87.
7 Hatch D J, Taylor B W. Thoracic gas volume in earlychildhood. Arch Dis Child 1976; 51: 859-64.
8 Tanner J M. Growth at adolescence. 2nd ed. Oxford:Blackwell, 1962.
9 Aggett P J, Taylor F. A normal paediatric amylaserange. Arch Dis Child 1980; 55: 236-8.
10 Burke V, Colebatch J H, Anderson C M, Simons M J.Association of pancreatic insufficiency and chronicneutropenia in childhood. Arch Dis Child 1967; 42:147-57.
11 Bodian M, Sheldon W, Lightwood R. Congenitalhypoplasia of the exocrine pancreas. Acta PaediatrScand 1964; 53: 282-93.
1 Nezelof C, Watchi M. L'hypoplasie congenitale lipo-mateuse du pancreas exocrine chez l'enfant. Arch FrPediatr 1961; 18: 1135-72.
13 Shwachman H, Diamond L K, Oski F A, Khaw K T. Thesyndrome of pancreatic insufficiency and bone marrowdysfunction. JPediatr 1964; 65: 645-63.
14 Hudson E, Alder T. Pancreatic insufficiency and neutro-penia with associated immunoglobulin deficit. ArchIntern Med 1970; 125: 314-6.
15 Goldstein R. Congenital lipomatosis of the pancreas.Malabsorption, dwarfism, leukopenia with relativegranulocytopenia, and thrombocytopenia. Clin Pediatr(Phila) 1968; 7: 419-22.
6 Brueton M J, Mavromichalis J, Goodchild M C, AndersonC M. Hepatic dysfunction in association with pancreaticinsufficiency and cyclic neutropenia. Shwachman-Diamond syndrome. Arch Dis Child 1977; 52: 76-8.
17 Aggett P J, Harries J T, Harvey B A M, Soothill J F. Aninherited defect of neutrophil mobility in Shwachman'ssyndrome. JPediatr 1979; 94: 391-4.
18 Levitt M D, Ellis C, Engel R R. Isoelectric focusingstudies of human serum and tissue amylases. J Lab ClinMed 1977; 90: 141-52.
19 Seifert G. Die pathologie der Kindlichen Pancreas.Leipzig: Thieme, 1956.
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

346 Aggett, Cavanagh, Matthew, Pincott, Sutcliffe, and Harries
20 Huijgens P C, van der Veen E A, Meijer S, Muntinghe0 G. Syndrome of Shwachman and leukaemia. Scand JHaematol 1977; 18: 20-4.
21 Cooper H A, Hoagland H C. Fetal hemoglobin. MayoClin Proc 1972; 47: 402-14.
22 Strevens M J, Lilleyman J S, Williams R B. Shwachman'ssyndrome and acute lymphoblastic leukaemia. Br Med J1978; ii: 18.
23 Hantelmann W. Fettsucht und Atrophie der Bauch-speicheldriuse bei Jugendlichen. Virchows Arch (PatholAnat) 1931; 282: 630-42.
24 Giedion A, Prader A, Hadorn B, Shmerling D H,Auricchio S. Metaphysare Dysostose und angeborenePankreasinsuffizienz. ROEFO 1968; 108: 51-7.
25 Stanley P, Sutcliffe J. Metaphyseal chondro-dysplasia withdwarfism, pancreatic insufficiency, and neutropenia.Pediatr Radiol 1973; 1: 119-26.
26 Fellman K, Kozlowski K, Senger A. Unusual bonechanges in exocrine pancreatic insufficiency with cyclicneutropenia. Acta Radiol [Diagn] (Stockh) 1972; 12:428-32.
27 McLennan T W, Steinbach H L. Shwachman's syndrome:the broad spectrum of bony abnormalities. Radiology1974; 112: 167-73.
28 Danks D M, Haslam R, Mayne V, Kaufmann H J,Holtzapple P G. Metaphyseal chondrodysplasia, neutro-penia, and pancreatic insufficiency presenting withrespiratory distress in the neonatal period. Arch Dis Child1976; 51: 697-702.
29 Spycher M A, Giedion A, Shmerling D H, Ruttner J R.Electron microscopic examination of cartilage in thesyndrome of exocrine pancreatic insufficiency, neutro-penia, metaphyseal dysostosis, and dwarfism. HelvPaediatr Acta 1974; 29: 471-9.
30 Caffey J. Pediatric X-ray diagnosis. 6th ed, vol. 2,section 8. Chicago: Year Book Medical Publishers,1973.
31 Park E A. The imprinting of nutritional disturbances onthe growing bone. Pediatrics 1964; 33: Supplement 2,815-62.
32 Karjoo M, Koop C E, Cornfeld D, Holtzapple P G.Pancreatic exocrine enzyme deficiency associated withasphyxiating thoracic dystrophy. Arch Dis Child 1973; 48:143-6.
33 Taybi H, Mitchell A D, Friedman G D. Metaphysealdysostosis and the associated syndrome of pancreaticinsufficiency and blood disorders. Radiology 1969; 93:563-71.
34 Higashi 0, Hayashi T, Ohara K, et al, Pancreaticinsufficiency with bone marrow dysfunction. Shwachman-Diamond-Oski-Khaw's syndrome. Tohuku J Exp Med1967;92: 1-12.
35 Moller E, Olin P, Zetterstrom R. Neutropenia andinsufficiency of the exocrine pancreas. Acta PaediatrScand (Suppl) 1967; Supplement 177, 29-31.
36 Doe W F. Two brothers with congenital pancreaticexocrine insufficiency, neutropenia, and dysgamma-globulinaemia. Proc R Soc Med 1973; 66: 1125-6.
37 Saint-Martin J, Fournet J P, Charles J, et al. Insuffisancepancreatique externe avec granulopenie chronique. ArchFr Pediatr 1969; 26: 861-71.
38 Jeune M, Hermier M, Germain D, et al. L'insuffisancepancreatique externe avec insuffisance medullairehematopietique chez le nourisson et d'enfant. Syndromede Shwachman. Pediatrie 1967; 22: 551-68.
39 Valman H B. Intelligence after malnutrition caused byneonatal resection ofileum. Lancet 1974; i: 425-7.
40 Tasman W. Diseases of the retina and vitreous. In:Harley R D, ed. Pediatric ophthalmology. Philadelphia:Saunders, 1975: 354.
41 Jeavons P M, Harding G F A. Photosensitive epilepsy.Clinics in Developmental Medicine No. 56. London:Heinemann Medical, 1975: 58.
42 Shwachman H, Holsclaw D. Some clinical observationson the Shwachman syndrome (pancreatic insufficiency andbone marrow hypoplasia). Birth Defects 1972; 8: Pt 3,46-9.
43 Pringle E M, Young W F, Haworth E M. Syndrome ofpancreatic insufficiency, blood dyscrasia, and metaphysealdysostosis. Proc R Soc Med 1968; 61: 776-7.
44 Johanson A, Blizzard R. A syndrome of congenitalaplasia of the alae nasi, deafness, hypothyroidism,dwarfism, absent permanent teeth, and malabsorption. JPediatr 1971; 79: 982-7.
45 Schussheim A, Choi S J, Silverberg M. Exocrine pancreaticinsufficiency with congenital abnormalities. J Pediatr1976; 89: 782-4.
46 Donowitz M, Gryboski J D. Pancreatic insufficiency andthe congenital rubella syndrome. JPediatr 1975; 87: 241-2.
47 Grand R J, Rosen S W, Di Sant'Agnese P A, KirkhamW R. Unusual case ofXXY Klinefelter's syndrome, withpancreatic insufficiency, hypothyroidism, deafness,chronic lung disease, dwarfism, and microcephaly. Am JMed 1966; 41: 478-85.
48 McKusick V A, Eldridge R, Hostetler J A, Ruangwit V,Egeland J A. Dwarfism in the Amish. II. Cartilage-hairhypoplasia. Johns Hopkins MedJ 1965; 116: 285-326.
49 Havlik6va D, Vychytil 0, Jelinek J. The syndrome ofcongenital pancreatic insufficiency, chronic respiratorydisease, and chronic liver damage. Acta Paediatr Scand1967; 56: 676-80.
50 Theodorou S D, Adams J. An unusual case ofmetaphysealdyschondroplasia. J Bone Joint Surg (Br) 1963; 45: 364-9.
51 Sacrez R, Klein F, Hoffman B, Levy J M, Geisert J,Korn R. Hypoplasie du pancreas exocrine. Fibrosemyoendocardique associee chez l'un des deux freresobserves. Ann Pediatr 1969; 16: 43-8.
52 Reaven E P, Reaven G A. Distribution and control ofmicrotubules in relation to the transport of lipid. J CellBiol 1977; 75: 559-72.
53 Mizel S B, Wilson L. Inhibition of the transport ofseveral hexoses in mammalian cells by cytochalasin B. JBiol Chem 1974; 247: 4102-5.
54 Malech H L, Root R K, Gallin J I. Structural analysis ofhuman neutrophil migration. J Cell Biol 1977; 75: 666-93.
55 Berlin R D, Oliver J M, Ukena T E, Yin H H. The cellsurface. NEnglJMed 1975; 292: 515-20.
56 Gillespie E, Lichtenstein L M. Histamine release fromhuman leukocytes: studies with deuterium oxide,colchicine, and cytochalasin B. J Clin Invest 1972; 51:2941-7.
57 Malaisse W J, Malaisse-Lagae F, Walker M 0, Lacy P E.The stimulus-secretion coupling of glucose-inducedinsulin release. V. The participation of a microtubular-microfilamentous system. Diabetes 1971 ; 20: 257-65.
58 Wolff J, Williams J A. The role of microtubules andmicrofilaments in thyroid secretion. Recent Prog HormRes 1973; 29: 229-86.
59 Redman C M, Banerjee D, Manning C, Huang C Y,Green K. In vivo effect of colchicine on hepatic proteinsynthesis and on the conversion of proalbumin to serumalbumin. J Cell Biol 1978; 77: 400-16.
60 Ehrich H P, Ross R, Bornstein P. Effects of antimicro-tubular agents on the secretion of collagen; a biochemicaland morphological study. J Cell Biol 1974; 62: 390-405.
61 Williams J A, Lee M. Microtubules and pancreaticamylase release by mouse pancreas in vitro. J Cell Biol1976; 71: 795-806.
62 Thoa N B, Wooten G F, Axelrod J, Kopin I J. Inhibitionof release of dopamine-,-hydroxylase and norepinephrine
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from

Shwachman's syndrome 347
from sympathetic nerves by colchicine, vinblastine, orcytochalasin B. Proc Natl Acad Sci USA 1972; 69: 520-2.
63 McCollum J P K, Muller D P R, Harries J T. Test mealfor assessing intraluminal phase of absorption inchildhood. Arch Dis Child 1977; 52: 887-9.
64 Hadorn B. Diseases of the pancreas in children. ClinGastroenterol 1972; 1: 125-46.
Correspondence to Dr P J Aggett, University ofAberdeen, Department of Physiology, MarischalCollege, Aberdeen AB9 lAS.
Received 22 May 1979
on 16 June 2018 by guest. Protected by copyright.
http://adc.bmj.com
/A
rch Dis C
hild: first published as 10.1136/adc.55.5.331 on 1 May 1980. D
ownloaded from





























